 Steven Ngo
Steven Ngo Despoina Papazoglou
Despoina Papazoglou Hector Huerga Encabo
Hector Huerga Encabo Dominique Bonnet
Dominique Bonnet- Haematopoietic Stem Cell Laboratory, The Francis Crick Institute, London, United Kingdom
Haematopoietic stem cells (HSCs) reside within an intricate network of cells in the bone marrow (BM) niche. HSC crosstalk with niche compartments influences lineage determination and blood cell production, while independent niche interactions are essential for the maintenance of HSC quiescence. How different niche components influence the genetic diversity of HSCs represents an expanding field of investigation. As such, we will summarise the current knowledge of the contribution to the Darwinian evolution of mutant HSCs of both haematopoietic and non-haematopoietic cells residing in the BM. In this review, we will disentangle how somatic evolution associates with the niche at two stages: from (1) the stage of preleukaemic HSC expansion and clonal haematopoiesis (CH) to (2) leukaemia-initiating cells (LICs) and the development of myeloid malignancies with acute myeloid leukaemia (AML) being the most prevalent. We will finally describe current challenges such as limitations in models used in the field or the difficulty in studying specific genetic clones in isolation.
1 Introduction to clonal haematopoiesis and clonal AML and redefining the bone narrow niche
The bone marrow (BM) niche has been extensively studied over the last decade; however, the exact role and composition of cell types defining this highly orchestrated, complex microenvironment is constantly evolving. In its early conception, the BM niche initially referred to mesenchymal stromal cells (MSCs); however, in recent years, it has become apparent that the niche comprises a constellation of cell types, all of which play an integral and unique role in supporting the stem cell compartment. In this review, we will consider how the two different cellular components of the BM niche, haematopoietic (immune cells) and non-haematopoietic (stromal cells) cells, influences the clonal evolution of haematopoietic stem cells (HSCs). Whilst there are also extramedullary derived molecular factors that migrate to the bone marrow and regulate HSCs, here, we will only focus on cellular and molecular factors produced locally. We also recognise that non-haematopoietic niche cells can bear genetic mutations shared or not shared with haematopoietic cells. Indeed, early studies in murine models have reported that altered (mutated) niche cells actively shape HSC responses and can be directly involved in the expansion of clonal HSCs and initiation of haematological malignancies (1). More recent evidence further supports this notion of the niche as an oncogenic driver or facilitator of malignancy. For example, PTPN11-activating mutations in the mouse MSCs and osterix (Osx)-expressing osteoprogenitors lead to donor cell-derived myeloproliferating neoplasms (MPN) in BM transplantation assays through the upregulation of pro-inflammatory cytokines (2). Additionally, mutations on ASXL1 have shown to regulate the self-renewal and differentiation of bone marrow MSCs (3) and HSCs (4), while MSCs derived from chronic myelomonocytic leukaemia patients have also reduced the expression of ASXL1 (5). Across the same lines, endothelial cells bearing the JAK2V617-F mutation have been shown to promote clonal expansion in murine models of myeloproliferating neoplasms (6). Whereas the clinical evidence for niche lesions as initiating events in haematological malignancies requires future work and validation, it is now clear that malignant cells transform the niche to promote their own survival. Here, we will focus only on this aspect, where somatic mutations acquired by HSC compartment remodel their niche and drive unique interactions with the niche that are required for the expansion and development of clonal haematopoiesis and leukaemia.
1.1 Niche players in the Darwinian evolution of HSC clones
HSC crosstalk with specific niche compartments can influence lineage determination and blood cell production, while independent niche interactions are also essential for the maintenance of HSC quiescence (7). BM-residing common myeloid (CMP) and lymphoid progenitors (CLP) give rise to mature immune cells, which together form the immune microenvironment. B cells, T cells, NK cells, monocytes, granulocytes, erythroblasts, and megakaryocytes all actively contribute to the normal function of the BM and immune system, both in health and during environmental stresses (Figure 1). The non-haematopoietic microenvironment comprises a highly complex, yet intricately organised network of cells characterised by significant heterogeneity within the vascular (10) and mesenchymal stromal cell compartments (11, 12). To date, studies that address the molecular heterogeneity and functional plasticity of the BM microenvironment have been limited largely due to the low frequencies of these populations in the BM and the technical challenges associated with their isolation. Recently, three complementary studies have elegantly characterised the murine BM stromal microenvironment during homeostasis at a single-cell resolution (13–15). The key stroma subsets (in homeostasis) are summarised in Table 1. Baryawno et al. identified 17 stroma cell subsets that include mesenchymal, pericyte, fibroblast, endothelial, MSC-descendent osteolineage cells (OLCs), adipocytes, and chondrocytes (14). In another study, lineage-specific Cre-transgenic mice crossed with a knock-in reporter strain were used to broadly identify the major non-haematopoietic niche components within the BM. Using this strategy, the authors were able to isolate the vascular niche, (VE-cadherin+ cre;LoxP-tdTomato), Leptin-R (LEP-R)+ MSCs (Lepr-cre;LoxP-tdTomato), and the endosteal niche (Colagen2.3-cre;LoxP− tdTomato). Bulk and single-cell RNA sequencing (scRNASeq) was utilised to further stratify additional sub-clusters within each of these niche components (15). Baccin et al. combined single-cell and spatially resolved transcriptomics to demonstrate that Cxcl12-abundant reticular (CAR) cell subsets (Adipo-CAR and Osteo-CAR) differentially localise to sinusoidal and arteriolar surfaces, acting locally as “professional cytokine-secreting cells” (13). Beyond Adipo- and Osteo-CAR cells, they further defined Schwann cells, Ng2+ MSCs, osteoblasts, chondrocytes, fibroblast/chondrocyte progenitors, sinusoidal and arterial ECs, smooth muscle cells, and fibroblasts (myofibroblasts, stromal, arteriolar, and endosteal) (13). An integrated analysis of these published datasets revealed commonalities and differences in the subclusters identified (16). Similar subsets were also identified during aging and inflammation (17).
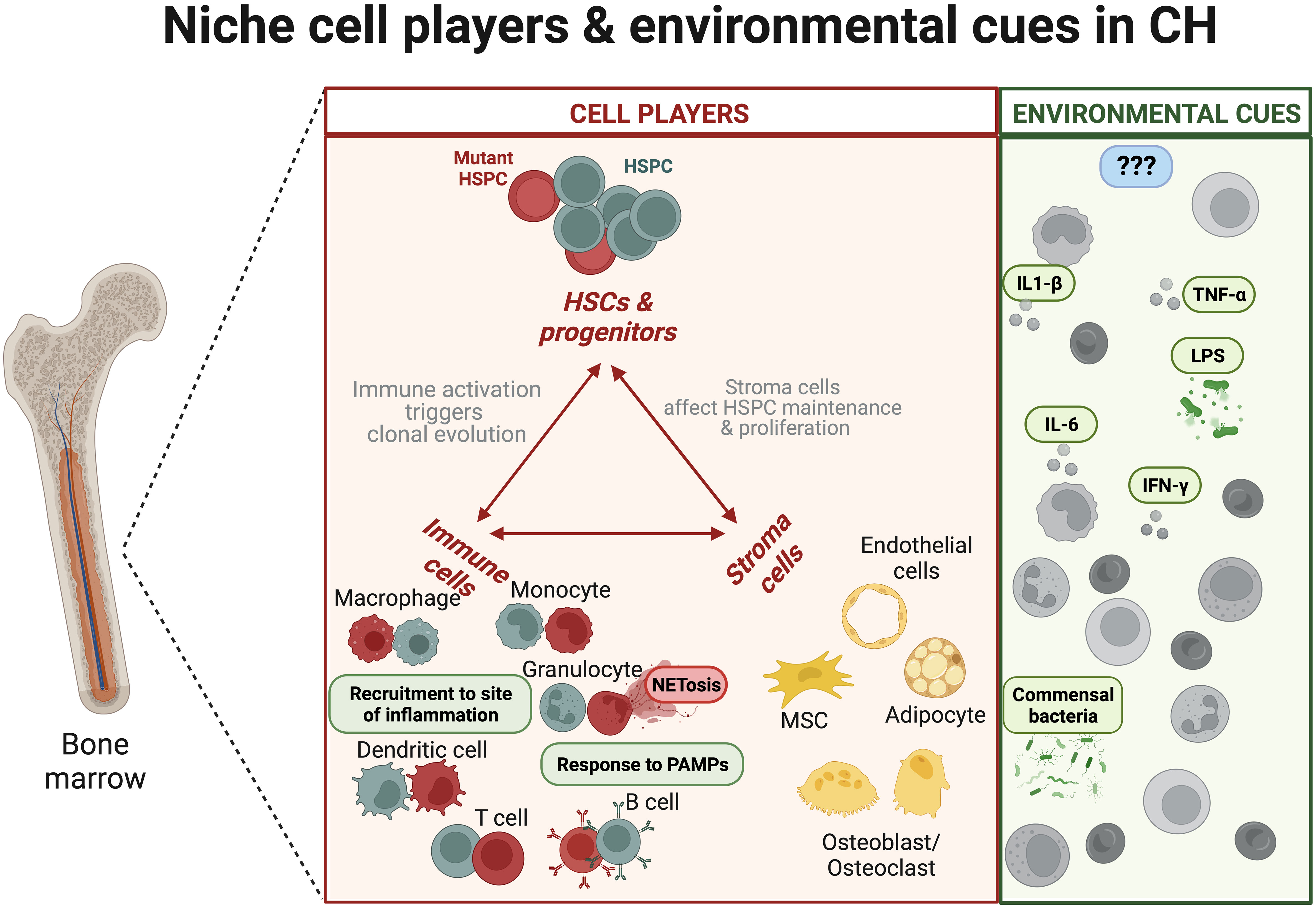
Figure 1 The niche hematopoietic and stroma cell components and environmental factors during clonal haematopoiesis. Mutant haematopoietic stem and progenitor cells (HSPCs) give rise to mature hematopoietic cells harbouring preleukaemic mutations with distinct immune response (8). Overall, the innate immune system from mutant HSPCs is characterised by changes in cell recruitment and production of inflammatory cytokines. In parallel, environmental cues, like LPS or infections, could further promote the expansion of the mutant clones [nicely reviewed in (9)]. The boxes in red show dysregulation of immune properties (e.g., mutant neutrophil NETosis). The green boxes indicate molecular factors that promote the expansion of mutant clones. The blue boxes indicate the possible existence of unknown regulators that could reduce CH.
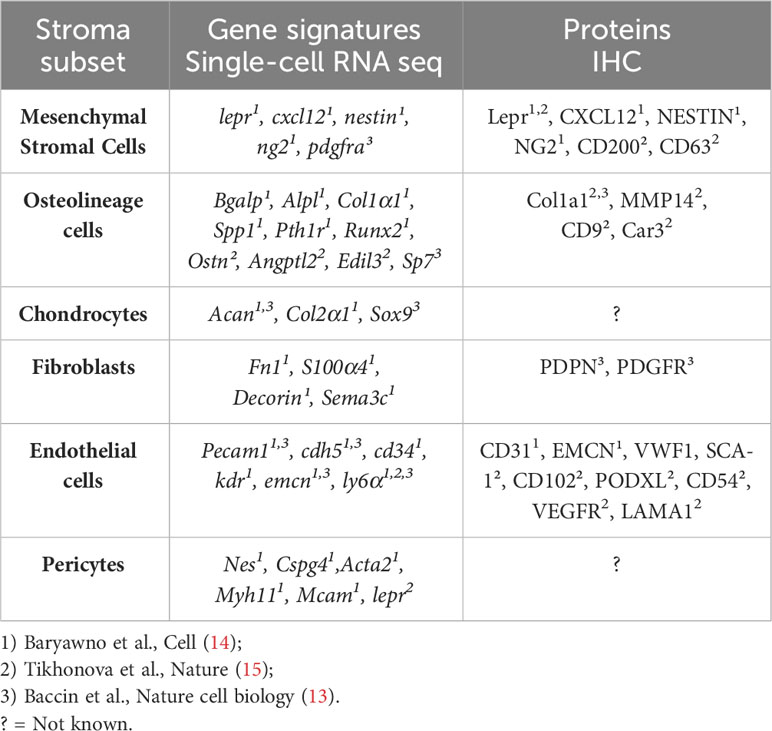
Table 1 The murine stroma cell types defined by single-cell RNA sequencing.
To date, most studies relating to the impact of the bone-marrow niche on clonal expansion in either the CH or AML context have been primarily performed on mouse models. However, in recent years, there have been seminal papers that have described the BM microenvironment in the human context (18–20). ScRNAseq was also used to characterise the non-haematopoietic components of the human bone marrow. The authors describe potential key factors that govern the transition from stem and progenitor cells to fate-committed cells, and in situ localisation analysis showed distinct spatial organisation of different niche compartments within the BM (21). A recent study in AML has shown that during the course of the disease, there is clear organisation and significant remodelling of the human bone marrow samples of NPM1-mutated AML patients. Frozen BM aspirates from AML patients showed clear increase in inflammatory signatures in BM-MSC clusters when compared to healthy BM controls. Furthermore, there is selective loss of MSCs critical for long-term HSC maintenance, thus providing evidence showing AML-mediated transformation of the BM microenvironment at the expense of healthy HSCs (22). These studies helped to characterise and define the human stroma cell populations in the BM niche in different contexts, although additional work, including functional studies, is warranted to fully understand the effect of such populations on the BM-residing haematopoietic cell subsets.
Unlike in humans, more high-throughput analysis of murine BM described the active changes of stroma cell populations during leukaemia development (18) highlighting the plasticity and importance of niche-residing stroma components for disease biology. Additional functional work evaluated how stromal cells respond to different environmental stresses like infections affecting both haematopoietic and immune cells in the BM. Endothelial and CXCL12-abundant reticular (CAR)/LepR+ cells alter the expression of cytokines and chemokines in cellular niches following activation of various pattern-recognition receptors (PRRs). In a model of lymphocytic choriomeningitis virus (LCMV) infection, CD8+-derived interferon γ (IFN-γ) activates MSC-derived interleukin 6 (IL-6) production, leading to the expansion of the immature and mature myeloid compartments (23). A similar CAR cell-derived, IL-6-induced myelopoiesis is also observed in chronic inflammation mediated through lipopolysaccharides (LPSs) (24). During infection, upregulation of granulocyte colony-stimulating factor (G-CSF) suppresses CXCL12 production by CAR cells, which, in turn, induced the mobilisation of haematopoietic stem and progenitor cells (HSPCs) to the circulation (25). Additionally, CAR/LepR+ cells increase the number of circulating monocytes by upregulating CCL2, a potent chemoattractant responsible for facilitating cell migration (26). During LPS-induced inflammation, signalling through toll-like receptor 4 in endothelial cells increases G-CSF production subsequently leading to emergency granulopoiesis (27). The interaction of DEL-1 in endothelial cells and CAR cells with β3 integrin expressed on HSCs promotes myeloid priming and supports emergency myelopoiesis in response to LPS-related inflammation (28).
1.2 Prevalent mutations in CH and AML
The gradual accumulation of somatic mutations in HSPCs leading to the outgrowth of mutant clones is known as clonal haematopoiesis (CH). Individuals with CH have a higher risk of developing haematological malignancies such as AML; therefore, mutations implicated in CH are also sometimes defined as preleukaemic (29–31). DNMT3A and TET2 are the most commonly occurring mutations in CH and are epigenetic regulators involved in DNA methylation and demethylation, respectively (32, 33). Preleukaemic mutations in HSCs have been described to confer a repopulating advantage upon stress-induced haematopoiesis in mouse models. Furthermore, recent studies have aided our understanding of how preleukaemic HSPCs expand in inflammatory environments (9, 34, 35). Here, we will describe how haematopoietic (immune cells) and non-haematopoietic cell populations residing in the BM are affected by CH and whether they further contribute to the evolution of mutated clones. Regarding CH, we will focus on DNMT3A, TET2, and JAK2 mutations given their strong association with progression to myeloid neoplasms (36). Keeping in the direction of clonal expansion and the increasing evidence that AMLs are often genetically polyclonal, we will also describe the niche involvement in AMLs driven by commonly occurring mutations. Finally, we will critically discuss the current challenges and pitfalls in the field, including the limitations in models used to investigate the niche, the characterisation of cell-specific roles of the niche, and the difficulty in studying specific genetic clones in isolation.
2 Clonal haematopoiesis and the niche
In regard to the BM immune compartment, dissecting the immune progeny contribution to clonal evolution can pose a significant challenge given the difficulty of separating cell-autonomous functions from mutation-specific effects. For example, innate immune cells present in the haematopoietic niche contribute to clonal selection by secreting pro- or anti-inflammatory cytokines based on environmental cues such as infections or underlying inflammatory disorders. Conversely, the production of pro-inflammatory cytokines has also been reported to be upregulated when monocytes and neutrophils inherit preleukaemic mutations from HSCs (Figure 1). Therefore, it has been an ongoing issue to distinguish “cause” from “consequence” (i.e., a chicken-and-egg situation) in driving CH evolution. Overall, the current paradigm suggests that stress signals drive preleukaemic mutation expansion directly by favouring mutant HSCs or indirectly via the response produced by cellular components of the niche (which, in the case of haematopoietic/immune progeny, will be influenced by the preleukaemic cell of origin). To summarise, given that preleukaemic mutations naturally occur independently of stress as well as early in age, we believe that environmental cues or underlying pathologies play an active role in shaping CH evolution from a preexisting pool of healthy and pre-malignant clones. Although CH has also been shown to increase risk in developing non-haematopoietic disorders (37–39), this review will strictly focus on the impact of CH in haematological disorders.
2.1 TET2
Over the last decade, there has been a growing body of evidence detailing the cellular and molecular mechanisms driving the evolution of TET2-mutant HSCs. Initial characterisation of TET2 detailed its important role in supressing the upregulation of pro-inflammatory cytokines IL-6 and IL-1-β upon immune activation of macrophages (40, 41). Other reports further characterised TET2 loss of function (LoF) mutations, whereby monocytes and macrophages confer a hyperinflammatory phenotype upon acquisition of the mutation (41–45). In tumour-associated macrophages (TAMs), TET2 is reported to dampen immune activation and maintain the TAM-immunosuppressive properties (46). Whilst the impact of TET2 loss on the monocytic compartment has been extensively characterised, new evidence suggests significant implications of TET2 LoF mutations in other immune cell types. Our group recently described defective NETosis, granule content and hyperinflammatory phenotypes in TET2-mutant neutrophils (8). In dendritic cells (DCs), TET2 has been reported to cooperate with vitamin D receptor and STAT3 to establish tolerogenesis (47). In plasmacytoid DCs, TET2 regulates Irf7 expression and IFN response and has been known to confer resistance to UV-induced death (48, 49). TET2-mutated HSPCs also lead to myeloid bias in various mouse models (50–52). Of note, recent works using genetically engineered TET2 mutations in human cord blood HSCs identify clonal advantage along with myeloid and granulocyte bias in the context of reconstituting human haematopoiesis upon transplantation in immunodeficient mice (8, 53). Finally, with regard to molecular mechanisms directly involved in the evolution of TET2-mutant HSCs, the Rauh Lab Team first reported clonal advantage of mutant HSPCs via tumour necrosis factor-alpha (TNF-α) signalling (34). This subsequently initiated a series of discoveries including LPS or microbial translocation promoting TET2-driven CH expansion (54). Caiado et al. investigated the contribution of aging-associated extrinsic factors as drivers of clonal haematopoiesis of intermediate potential (CHIP). In a TET2 CHIP mouse model, they observed an association between increased TET2+/− clonal expansion and higher BM levels of the inflammatory cytokine IL-1 upon aging. This expansion of TET2+/− HSCs, HSPCs, and mature blood cells was dependent on the IL-1 receptor. Interestingly, the genetic deletion of IL-1R1 in TET2+/− HPSCs or the pharmacological inhibition of IL-1 signalling impaired TET2+/− clonal expansion establishing the IL-1 pathway as a relevant and therapeutically targetable driver of TET2+/− CHIP progression during aging (40). Little is known about the interactions between the non-haematopoietic niche components and CH, and studies investigating how the expansion of mutated or preleukaemic HSCs affects the stroma subsets are lacking. Interestingly, Li et al. have recently reported using in situ imaging and found that more TET2-deficient AML cells migrate into the osteopontin (OPN)-positive BM-derived stromal cells compared to the TET2 wild-type group. They further reported that TET2-deficient AML cells show increased migration, growth, and self-renewal ability when co-cultured with stromal cells in vitro (55).
2.2 DNMT3A
In comparison to TET2, less is known regarding the cellular and molecular factors driving DNMT3A-related CH. Unlike the nature of TET2 mutations that are predicted to inhibit or abolish the enzyme’s catalytic activity, DNMT3A clone variants involved in AML or other haematological malignancies affect the R882 hotspot residue in the methyltransferase domain. Reports characterising specific R882 variants and their impact on haematopoiesis are lacking compared with those reporting LoF mutations in DNMT3A. Nonetheless, in CH, DNMT3A mutations are distributed across the three functional domains of the gene and predicted to cause LoF. Hence, models characterising DNMT3A LoF clones are helpful to gain understanding of CH, whereas R882 variants are more appropriate for studies focusing on evolution to malignancy. Regarding studies on the impact of innate immune cells upon DNMT3A mutations, the Glass’s group reported that DNMT3A LoF in human macrophages induces type I interferon response due to impaired mitochondrial DNA integrity and activation of cGAS signalling (56). In mouse models, it has been shown that bone marrow DNMT3A deletion blocks effective efferocytosis and mast cell inflammatory responses (57, 58). Regarding the impact of molecular players on the direct evolution of DNMT3A mutant HSCs, it has been shown that chronic IFN-γ signalling via Mycobacterium avium infection confers advantage to LoF DNMT3A HSCs (35). In the context of R882 variants, TNF-α signalling promotes the selective advantage of mutant HSCs (59). Additionally, it has been recently reported that a significant increase in self-renewal is detected in both human and mouse DNMT3AMut-HSCs when exposed to fatty bone marrow (FBM), which is often seen in aged individuals. Furthermore, the authors, via single-cell RNA sequencing, show a 6- to 10-fold increase in DNMT3AMut-HSCs after exposure to FBM in vivo. Secretome analysis of the BM fluid and in vitro BM adipocyte cultures showed increased IL-6 levels under FBM conditions. Consequently, treatment with neutralising IL-6 antibody reduced the selective advantage of DNMT3AMut-HSCs exposed to FBM. DNMT3AMut-HSCs derived from older mice interacting with FBM have a higher self-renewal capacity compared to DNMT3AMut-HSCs from younger mice suggesting that the BM niche can actively shape CH (60). Lastly, using murine models, Kim et al. have shown that haematopoietic-specific mutations in DNMT3A decrease bone mass via osteoclastogenesis. This bone loss was driven by inflammatory cytokines including IL-20 derived from DNMT3A mutant macrophages (61).
2.3 JAK2
Mutations in the epigenetic regulators TET2 and DNMT3A account for almost 70% of the CH cases (62, 63). The remaining drivers of CH consist of a heterogenous group of mutations associated with chromatin remodelling (ASXL1), splicing (SF3B1, SRSF2), DNA damage (PP1MD), or cell signalling (JAK2). Among these, mutations in JAK2 locus V617F have been reported to have the highest frequency in pre-AML and CH cases (64). Regarding the impact of JAK2V617F mutations in innate immune cells, macrophages displayed increased expression of pro-inflammatory cytokines (65), and neutrophils increased neutrophil extracellular trap formation providing a molecular explanation for the increased thrombosis in patients with MPN (66). In DCs, inhibition of JAK2 reverts vitamin D-induced tolerogenesis (47). Regarding the impact of JAK2V617F mutations on the non-haematopoietic compartment, conditional JAK2V617F knock-in murine models show that the transplantation of JAK2V617F long-term HSCs results in the development of MPN and myelofibrotic transformation showcasing the role of JAK2Mut HSCs in remodelling the BM niche (67). In the endosteal niche, monocytes derived from JAK2V617F-MPN cells have enhanced osteoclast-formation ability compared to that of WT monocytes. This enriched osteoclast environment further supports the proliferation and survival of the mutated MPN cells (68). Loss of TGF-β signalling in Osx-Cre-targeted MSCs can prevent the development of myelofibrosis in a JAK2V617F in vivo model (69). Transcriptomic comparisons between BM-MSCs derived from healthy individuals or MPN patients harbouring the JAK2V617F mutation identified distinct gene signatures in MPN-MSCs including the overexpression of SPP1 and NF-kB. This was coupled with a downregulation of ANGPT1 and THPO; however, the exact mechanisms underlying the JAK2Mut HSC : MSC interactions are still unknown (70). A reduction in Nestin+ BM-MSCs and CXCL12 expression with an expansion of JAK2+ HSCs has been detected as a consequence of a local neuropathy that occurs in MPN-BM (71). Co-culture models have also identified that cytokines secreted by BM-stroma cells protect JAK2V617F-MPN cells from the JAK2 inhibitor therapy (72). In addition to the involvement of MSC and MSC-derived stroma cell contribution to MPN progression, clonal–MPN cells have the capacity to disrupt the vascular niche (73). In patients with MPN, BM vascularisation has been shown to correlate with JAK2 allele burden (74). Interestingly, using murine models, it was found that despite all HSCs carrying the JAK2V617F mutation, disease progression varied and is highly dependent on the localisation in specific BM niche compartments. The authors identified distinct BM remodelling (endosteal and central BM vessels) in essential thrombocythemia (ET) versus polycythemia vera (PV) settings. For example, the bone (OPN+) area was 25-fold higher in mice engrafted with ET compared to that of PV hHSPCs, even though hHSPCs carried the same oncogenic driver (75). Such studies therefore highlight the importance of detailed investigations of clonal/mutant HSC interactions with the niche, thereby determining both the intrinsic and extrinsic factors that favour CH.
3 Clonal AML and the niche
Whereas studies relating to the importance of the niche during clonal expansion and CH are scarce, the integral role of BM-residing cells has been extensively studied in the context of AML. The involvement of the various niche components during leukaemia development has been broadly reviewed (76, 77). Our group has previously reported alterations in the vascular components of the niche that contribute to AML progression and drug response using patient-derived xenograft (PDX) models (78). Importantly, however, many of the studies pertaining to the AML–niche crosstalk have focused primarily on genetically monoclonal mouse models. Hence, given the genetic homogeneity of such models, it is difficult to determine the niche contributions to the selection of a given leukaemia-initiating clone. With recent advancements in next-generation sequencing, it has become increasingly evident that AMLs often comprise multiple genetically distinct subclones. This therefore raises the question of whether genetically distinct subclones may differ in their dependency on the BM microenvironment, and more importantly, whether these unique interactions may contribute to the fitness of a clone during AML progression or in response to therapy. Whilst studies dissecting the subclone-specific interactions with BM cellular components from multi-clonal AMLs are currently lacking, current mouse models utilising commonly occurring oncogenic lesions in AML provide insight as to how genetically distinct AMLs may interact with the BM niche (Figure 2).
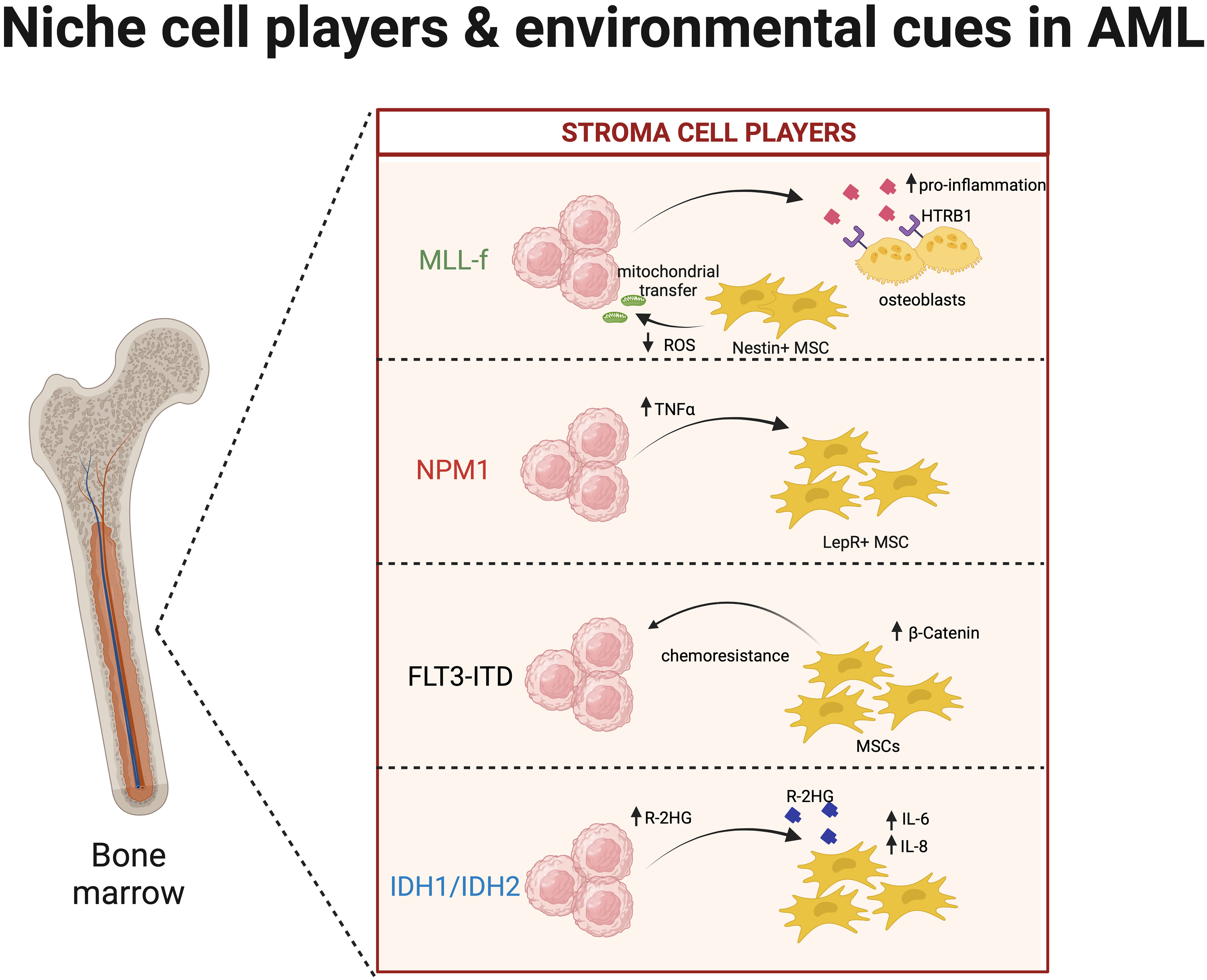
Figure 2 Interaction of acute myeloid leukaemia (AML) clones and the niche microenvironment. Similar to clonal haematopoiesis (CH), mutated clones during AML development highly interact with niche cells like mesenchymal stromal cells (MSCs), osteoblasts, endothelial cells, and macrophages. Here, we summarise the key interactions of AML cells harbouring the following mutations: MLL-f (79–81), FLT3-ITD (82), NPM1 (22), and IDH1/IDH2 (83, 84) with key bone marrow (BM) cell types.
3.1 MLL-F
MLL-rearranged fusion oncogenes (including MLL-AF9, MLL-ENL) are commonly used for in vivo studies of AML in part due to the aggressive nature of the disease. Single-cell sequencing characterising the BM harvested from MLL-AF9 knock-in mice demonstrates widespread remodelling of the BM in AML (14). MLL-AF9 cells induce a shift in the osteolineages (OLC), from haematopoiesis supporting OLCs to OLCs that promote calcification, ossification, and degradation of the bone (14, 85). Endosteal remodelling has also been observed in other studies using MLL-AF9-driven AMLs, where the presence of AML cells causes a pro-inflammatory and anti-angiogenic switch in endosteal OLCs resulting in loss of the MSCs, endothelial cells, and osteoblasts. The endosteal remodelling by the AML clone is also at the expense of non-leukaemic HSPCs resulting in loss of normal haematopoiesis (79). Interestingly, this remodelling is not observed in the central regions of the bone suggesting a localised impact of the niche where AML cells reside (79). MLL-F-driven AMLs can also drive significant metabolomic changes to the endosteal niche (80). Osteoblasts promote leukaemia cell proliferation through serotonin receptor signalling triggered by MLL-AF9 blast secretion of kynurenine. Kynurenine functions as an oncometabolite that interacts with the serotonin receptor HTR1B expressed on osteoblasts and induces a pro-inflammatory signature in osteoblasts (22, 80). This positive feedback loop between AML cells and the osteoblasts is required for AML progression in both MLL-AF9-driven mouse models as well as human AML cell lines containing the MLL-AF9 mutation (80). Whilst osteoblast support of AML cells may not be unique to MLL-AF9-driven AMLs (86, 87), some osteoblast/AML cell crosstalk may be MLL-AF9 specific. Activation of the parathyroid hormone (PTH) receptor on the osteoblast niche enhances MLL-AF9-driven AML progression but not BCR-ABL1-driven MPN models (88) demonstrating genotype-specific interactions with the niche. Nestin+ MSCs are uniquely important in their ability to support the high bioenergetic capacity of AML clones (81). AML cells from MLL-AF9-driven models rely on mitochondrial oxidative phosphorylation (OXPHOS) to meet their high metabolic demand; however, high OXPHOS also causes abnormally high reactive oxygen species (ROS) levels in AML cells. Nestin+ MSCs not only provide substantial metabolic support for AML cells through mitochondrial transfer but also reduce ROS levels in co-cultured MLL-AF9 AML cells (81). Nestin+ MSCs increase glutathione (GSH) availability for neighbouring AML cells. Uptake of GSH and subsequent oxidation result in cell detoxification and a significant decrease in ROS in AML cells. Although this Nestin+ MSC-AML cell crosstalk is crucial for leukaemia progression, the same mechanisms of support are also crucial for chemoresistance (81) emphasising the context-specific importance of MSCs for MLL-AF9-driven AMLs.
3.2 FLT3-ITD
Although FLT3-ITD mutations alone are rarely potent drivers of AML, co-mutated mouse models have been used to demonstrate the impact of FLT3-ITDmut AML blasts on the BM niche. FLT3-ITDmut TET2 knock-out mice show significant changes to the cellular composition of the mouse BM including increased abundance of endothelial cells and CAR cells (89). The CXCL12–CXCR4 axis provides a protective niche and regulates adaptation to the hypoxic environment, cellular migration, and survival of mutant clones during leukaemogenesis (90), as well as provides resistance to chemotherapy (89, 91). Disruption of this interaction between FLT3-ITDmut LICs (which phenotypically resemble CD34+CD38- HSPCs) and BM-MSC results in a delay in AML progression and also sensitises FLT3-ITDmut LICs to both chemotherapy and tyrosine kinase inhibitors (TKIs) through the downregulation of p38 MAPK signalling (89). Elevated levels of β-catenin induced by MSCs contribute to drug resistance and maintenance of FLT3-ITDmut LICs (82, 92) through the activation of Wnt signalling, which plays a critical role for AML cell survival. The dependency of FLT3-ITDmut LICs on BM-MSC-derived β-catenin is also corroborated in an independent study where FLT3-ITDmut-driven AML showed higher upregulation of β-catenin in BM-MSCs compared to BM-MSCs derived from mice transplanted with AML1-ETO or MLL-ENL-driven AMLs (93). However, upregulation of β-catenin itself is not unique to FLT3-ITDmut AMLs as elevated β-catenin is also seen in MSCs co-cultured with FLT3-ITD wild-type AMLs (82). Whether or not non-FLT3-ITDmut AMLs rely on β-catenin for their survival has yet to be elucidated.
Interestingly, the BM-MSCs may also convey resistance to FLT3-ITDmut subclones directly via metabolism of the drugs themselves. Cytochrome p450 enzymes (CYPs) is a drug-metabolising enzyme expressed in BM-MSCs and has been previously shown to provide resistance for FLT3-ITDmut cells through the local metabolism of FLT3-ITD inhibitors such as sorafenib and gilteritinib (94).
3.3 NPM1
Mutation to nucleophosmin 1 (NPM1) is the most common mutation found in adult AML and frequently co-occurs with FLT3-ITD mutations to drive disease (95). Although studies relating specifically to NPM1 mutations and the BM microenvironment are scarce, Chen et al. were able to perform scRNAseq analysis on frozen BM aspirates of AML patients with the NPM1 mutation (NPM1m). Here, they showed that TNFα mediated inflammatory remodelling of LepR+ BM-MSCs and extracellular matrix (ECM) remodelling in NPM1m AML patients (22). Interestingly, AML-mediated remodelling of the BM-MSCs resulted in loss of MSC populations heavily associated with HSPC maintenance of short-term (ST), but not long-term (LT), “healthy” HSCs. Furthermore, whilst inflammatory remodelling of the BM-MSCs was associated with a loss of ST-HSCs, such remodelling was also associated with a favourable outcome, likely due to the dependency of residual relapse initiating on niche support for survival from chemotherapy. Of note, further analysis revealed that inflammatory remodelling of the BM-MSCs may not be specific to NPM1m AMLs; however, niche remodelling impacting patient outcome may be mutation-specific given that AML patients with NPM1m have a favourable outcome in comparison to other AML-driving mutations.
3.4 IDH1/IDH2
IDH1 and IDH2 play a protective role against oxidative stress by regulating intracellular NADP+/NADPH ratio (96). Both IDH1 and IDH2 epigenetic modulators are commonly mutated in AML with up to 20% of adult AMLs being driven by either mutation (95). IDH1/2 mutant cells produce the oncometabolite R-2-hydroxyglutarate (R-2HG), which is primarily responsible for epigenetic reprogramming of the malignant clone resulting in a differentiation block (97). Although IDH1/2 mutations keep AML blasts in a highly proliferative state, the production of R-2HG also remodels the local BM niche into a pro-AML state. Uptake of R-2HG by BM-MSCs induces NFKB phosphorylation. Stabilisation of NFKB results in enhanced expression of important cytokines, such as IL-6 and IL-8, which are not only important for AML clone proliferation (98) but also in promoting chemoresistance (83). BM-MSC metabolism of R-2HG also induces CXCR4 upregulation enhancing AML cell–MSC crosstalk, AML progression, and chemoprotection (98).
3.5 The influence of the immune niche on AML
Thus far, most studies relating to AML cell–niche crosstalk have focused primarily on the non-haematopoietic BM microenvironment. This is not to say, however, that the immune niche does not have an impact on AML transformation and expansion. For example, studies on leukaemia-associated macrophages (LAMs) have highlighted their importance in leukaemia development and resistance to chemotherapy (84). Using PDX models, it was found that co-injection of primary human PMLRARα-mutated AML cells with M2-polarised macrophages induced a high acute promyelocytic leukaemia (APL) engraftment in xenograft mice suggesting a vital role of the immune niche in APL (99). Interestingly, M2 macrophages, inducing metabolic changes to AML cells and direct mitochondrial transfer from macrophages to AML cells, facilitating their increased metabolic activity, was reported. Adding to the complexity, partial maturation of AML cells into monocyte/macrophage lineage is observed clinically, as well as in in vivo models of AML (20). Therefore, it is enticing to speculate whether AML-derived macrophages also play a supportive role for the LICs, thereby creating their own immune niche for leukaemia propagation. Indeed, macrophages harbouring AML mutations have been shown to have reduced phagocytic abilities (99); however, its potential function as a supportive LAM has yet to be explored.
4 Current limitations and future questions when investigating the BM niche influence on clonal evolution
4.1 Cell of origin
Whilst the oncogenic mutation may influence the LICs’ interaction with the BM niche, the LIC “cell type” may also contribute to this process. This is particularly the case in AML, where scRNAseq has showed significant cellular heterogeneity in AML cells between different patient samples (20). However, given that some leukaemogenic mutations often occur in specific cell subtypes, it is difficult to discern whether specific niche interactions are due to the oncogenic mutation or the cell of origin of the LIC. For example, MLL-F-driven AMLs are typically classified as myelomonocytic leukaemias (100), whereas FLT3-ITD mutations are particularly prevalent in M1/M2 leukaemias with a more immature immunophenotype (101). Hence, whether the AML cell interaction with the niche is specific to the mutation or the cell type can, at times, be unclear. Interestingly, Li et al. in their MLL-AF9 murine model, showed the co-existence of immature (LMPP-like) and more mature (GMP-like) subpopulations, both having the capacity to initiate/propagate the disease when transplanted into secondary mice (102). Consequently, both AML subpopulations required different cytokine signals to propagate disease (SCF and FLT3L for LMPP-like and IL-3 and IL-6 for GMP-like cells) demonstrating that, in addition to oncogenic mutations, dependency on the BM niche may differ depending on the “cell of origin” of the AML cells themselves.
4.2 The right models
Advancements in next-generation sequencing and the ever-improving sensitivity of such platforms have revealed the prevalence of genetically distinguishable clones in both the normal and malignant settings. Exome sequencing can monitor previously undetectable subclones in patient samples with precision. Furthermore, it is increasingly evident that the process of clonal selection and expansion is dynamic, influenced by both cell and non-cell autonomous mechanisms (78, 81, 103). To date, most functional studies relating the BM microenvironment and clonal expansion rely on the use of transgenic mouse models driven by commonly occurring CH or AML-driving mutations. Such studies have provided invaluable insight into the niche–CH/AML crosstalk; however, a clear concern is the relevance of these interactions in the human context. Often, validation of these in vivo findings in human cohorts is missing. PDX models address one-half of the issue allowing the investigation of the niche crosstalk with human-derived clones, however, still relying on the murine BM microenvironment (intra-species interactions). More recently, surgically implanted scaffolds pre-seeded with human MSCs have been used to mimic a humanised niche (104, 105). Multiple groups have described the development of such niche models, which can support normal and malignant haematopoiesis (106–108). Whilst these models may lack the spatial organisation of the niche components that is inherent in a given tissue, such humanised models not only allow the interrogation of clonal expansion in notoriously difficult diseases to model (such as MDS) (105) but also provide a useful tool to investigate niche–clonal interactions across the haematological disease spectrum in a more clinically relevant setting.
4.3 Studying the subclones in isolation and in competition
Studying the dynamics of clonal populations can be challenging in many ways. Much of the research relating to niche-mediated clonal expansion uses murine models that are genetically homogenous. Consequently, they inherently neglect the competition between distinguishable subclones (i.e., mutated vs. non-mutated clones), which would better reflect the true biological context of clonal expansion. With a few exceptions (93), these questions may be beyond the scope of the studies mentioned in this review; however, without direct comparisons between clones or models driven by different mutations, it is difficult to discern whether a given interaction with the niche may be mutation specific or common amongst many mutations related to a given disease. Indeed, of the limited head-to-head studies that do characterise oncogenic lesion-specific interaction with the BM, they do provide compelling evidence to suggest that the mutational profile can influence the transformation of the BM niche (93). In a similar direction and mostly related to CH, another drawback is the limited access to primary human samples that bear only the mutation of interest to validate the findings from mouse in vivo studies.
4.4 Technical limitations
Certainly, the technical improvement and accessibility to scRNASeq analyses are able to guide our understanding of MSC biology, and their exploration can inspire creativity in our scientific inquiry. However, they cannot replace functional experiments required to answer complex biological questions. scRNAseq has been able to showcase the significant heterogeneity amongst subpopulations within the BM niche (16). Our understanding of the niche heterogeneity by scRNAseq, however, is not reflected in our ability to characterise and identify such subclusters immunophenotypically. Additionally, on a functional level, reliable antibody panels to identify such subpopulations through immunohistochemistry is lacking making it difficult to interrogate the BM spatial organisation in a subpopulation level within the bone. Encouragingly, the panel of markers to identify subclusters within different niche components is growing (109, 110). A major limitation to the aforementioned studies, however, is the loss of spatial organisation and BM cellular architecture inherent to these experiments. This could prove to be highly important given that there is growing evidence to suggest significant changes to spatial organisation of HSPCs as the bone marrow ages (13, 111–113). In recent years, innovative multiplex imaging techniques, such as CODEX (114, 115) or MACSima (110), have emerged, allowing for in-depth characterisation of cell types in situ. Such techniques may be proven vital in investigating the interplay between the niche and subclones, as detailed characterisation of the cellular neighbourhoods surrounding the clone of interest can faithfully describe the multi-cellular contribution of the BM in the expansion of a given clone.
Similarly, spatial RNAseq technologies, such as SpatialScope (116), STAPL-3D (117), and CelEry (118), have emerged in recent years, providing a possible means to investigate the transcriptional profile of relevant BM-residing cells whilst retaining the cellular architecture of the bone (110, 119, 120). However, consistent and successful use of such technologies on bone samples has yet to be achieved in part due to the technical difficulties with imaging of the bone (i.e., bone decalcification is essential for bone sectioning; however, it significantly compromises the quality of immunostaining). Even though such techniques open avenues to define and investigate the involvement of niche cells on clonal expansion, a great challenge remains: our ability to isolate these cells and their subclusters to study them functionally is limited. Therefore, it is very challenging to define their distinct roles and confirm that they are all functionally distinct populations (even though they do have distinct gene signatures). In a similar direction, given the heterogeneity and high plasticity of many immune progenitor and stroma cell components, longitudinal studies are warranted to confirm that these subsets/subclusters are the same/similar through time and through the expansion of the clone of interest.
Author contributions
SN: Writing – original draft. DP: Writing – original draft. HHE: Writing – original draft. DB: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (CC1045), the UK Medical Research Council (CC1045), and the Wellcome Trust (CC1045). HE was funded by The Kay Kendall Leukaemia Foundation (KKL1397 to HE); SN was funded by the Gilead Foundation; and DP was funded by a Blood Cancer UK project grant (22008). For the purpose of Open Access, the authors have applied a CC BY public copyright license to any author-accepted manuscript version arising from this submission.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2024.1334807/full#supplementary-material
References
1. Mendez-, Bonnet D, Steensma DP, Hasserjian RP, Ghobrial IM, Gribben JG, et al. Bone marrow niches in haematological Malignancies. Nat. Rev. Cancer. (2020) 20:285–98. doi: 10.1038/s41568-020-0245-2.
2. Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. (2016) 539:304–8. doi: 10.1038/nature20131.
3. Zhang P, Xing C, Rhodes SD, He Y, Deng K, Li Z, et al. Loss of asxl1 alters self-renewal and cell fate of bone marrow stromal cell, leading to bohring-opitz-like syndrome in mice. Stem Cell Rep. (2016) 6:914–25. doi: 10.1016/j.stemcr.2016.04.013.
4. Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. (2013) 210:2641–59. doi: 10.1084/jem.20131141.
5. Zhang P, Chen Z, Li R, Guo Y, Shi H, Bai J, et al. Loss of ASXL1 in the bone marrow niche dysregulates hematopoietic stem and progenitor cell fates. Cell Discovery. (2018) 4:4. doi: 10.1038/s41421-017-0004-z.
6. Zhan H, Lin CHS, Segal Y, Kaushansky K. The JAK2V617F-bearing vascular niche promotes clonal expansion in myeloproliferative neoplasms. Leukemia. (2018) 32:462–9. doi: 10.1038/leu.2017.233.
7. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. (2014) 505:327–34. doi: 10.1038/nature12984.
8. Huerga Encabo H, Aramburu IV, Garcia-Albornoz M, Piganeau M, Wood H, Song A, et al. Loss of TET2 in human hematopoietic stem cells alters the development and function of neutrophils. Cell Stem Cell. (2023) 30:781–99 e9. doi: 10.1016/j.stem.2023.05.004.
9. Florez MA, Tran BT, Wathan TK, DeGregori J, Pietras EM, King KY. Clonal hematopoiesis: Mutation-specific adaptation to environmental change. Cell Stem Cell. (2022) 29:882–904. doi: 10.1016/j.stem.2022.05.006.
10. Kusumbe AP, Ramasamy SK, Itkin T, Mae MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. (2016) 532:380–4. doi: 10.1038/nature17638.
11. Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, et al. Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev. Cell. (2014) 29:340–9. doi: 10.1016/j.devcel.2014.03.013.
12. Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. (2014) 15:154–68. doi: 10.1016/j.stem.2014.06.008.
13. Baccin C, Al-Sabah J, Velten L, Helbling PM, Grunschlager F, Hernandez-Malmierca P, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. (2020) 22:38–48. doi: 10.1038/s41556-019-0439-6.
14. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. (2019) 177:1915–32 e16. doi: 10.1016/j.cell.2019.04.040.
15. Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Dominguez A, et al. The bone marrow microenvironment at single-cell resolution. Nature. (2019) 569:222–8. doi: 10.1038/s41586-019-1104-8.
16. Dolgalev I, Tikhonova AN. Connecting the dots: resolving the bone marrow niche heterogeneity. Front. Cell Dev. Biol. (2021) 9:622519. doi: 10.3389/fcell.2021.622519.
17. Helbling PM, Pineiro-Yanez E, Gerosa R, Boettcher S, Al-Shahrour F, Manz MG, et al. Global transcriptomic profiling of the bone marrow stromal microenvironment during postnatal development, aging, and inflammation. Cell Rep. (2019) 29:3313–30 e4. doi: 10.1016/j.celrep.2019.11.004.
18. Mumme H, Thomas BE, Bhasin SS, Krishnan U, Dwivedi B, Perumalla P, et al. Single-cell analysis reveals altered tumor microenvironments of relapse- and remission-associated pediatric acute myeloid leukemia. Nat. Commun. (2023) 14:6209. doi: 10.1038/s41467-023-41994-0.
19. Rutella S, Vadakekolathu J, Mazziotta F, Reeder S, Yau TO, Mukhopadhyay R, et al. Immune dysfunction signatures predict outcomes and define checkpoint blockade-unresponsive microenvironments in acute myeloid leukemia. J. Clin. Invest. (2022) 132(21):e159579. doi: 10.1172/JCI159579.
20. van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. (2019) 176:1265–81 e24. doi: 10.1016/j.cell.2019.01.031.
21. Li H, Braunig S, Dhapolar P, Karlsson G, Lang S, Scheding S. Identification of phenotypically, functionally, and anatomically distinct stromal niche populations in human bone marrow based on single-cell RNA sequencing. Elife. (2023) 12:e81656. doi: 10.7554/eLife.81656.
22. Chen L, Pronk E, van Dijk C, Bian Y, Feyen J, van Tienhoven T, et al. A single-cell taxonomy predicts inflammatory niche remodeling to drive tissue failure and outcome in human AML. Blood Cancer Discovery. (2023) 4:394–417. doi: 10.1158/2643-3230.BCD-23-0043.
23. Schurch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell. (2014) 14:460–72. doi: 10.1016/j.stem.2014.01.002.
24. Gerosa RC, Boettcher S, Kovtonyuk LV, Hausmann A, Hardt WD, Hidalgo J, et al. CXCL12-abundant reticular cells are the major source of IL-6 upon LPS stimulation and thereby regulate hematopoiesis. Blood Adv. (2021) 5:5002–15. doi: 10.1182/bloodadvances.2021005531.
25. Day RB, Bhattacharya D, Nagasawa T, Link DC. Granulocyte colony-stimulating factor reprograms bone marrow stromal cells to actively suppress B lymphopoiesis in mice. Blood. (2015) 125:3114–7. doi: 10.1182/blood-2015-02-629444.
26. Shi C, Jia T, Mendez-, Hohl TM, Serbina NV, Lipuma L, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. (2011) 34:590–601. doi: 10.1016/j.immuni.2011.02.016.
27. Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood. (2014) 124:1393–403. doi: 10.1182/blood-2014-04-570762.
28. Mitroulis I, Chen LS, Singh RP, Kourtzelis I, Economopoulou M, Kajikawa T, et al. Secreted protein Del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J. Clin. Invest. (2017) 127:3624–39. doi: 10.1172/JCI92571.
29. Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, Lepine G, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. (2017) 130:753–62. doi: 10.1182/blood-2017-04-777029.
30. Chan SM, Majeti R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. (2013) 98:648–57. doi: 10.1007/s12185-013-1407-8.
31. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. (2015) 126:9–16. doi: 10.1182/blood-2015-03-631747.
32. Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic Malignancies. Cell Stem Cell. (2018) 22:157–70. doi: 10.1016/j.stem.2018.01.011.
33. Watson CJ, Papula AL, Poon GYP, Wong WH, Young AL, Druley TE, et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science. (2020) 367:1449–54. doi: 10.1126/science.aay9333.
34. Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. (2018) 59:60–5. doi: 10.1016/j.exphem.2017.11.002.
35. Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell. (2021) 28:1428–42 e6. doi: 10.1016/j.stem.2021.03.002.
36. Gu M, Kovilakam SC, Dunn WG, Marando L, Barcena C, Mohorianu I, et al. Multiparameter prediction of myeloid neoplasia risk. Nat. Genet. (2023) 55:1523–30. doi: 10.1038/s41588-023-01472-1.
37. Heyde A, Rohde D, McAlpine CS, Zhang S, Hoyer FF, Gerold JM, et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell. (2021) 184:1348–61 e22. doi: 10.1016/j.cell.2021.01.049.
38. Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. (2019) 366(6465):eaan4673. doi: 10.1126/science.aan4673.
39. Miller PG, Qiao D, Rojas-Quintero J, Honigberg MC, Sperling AS, Gibson CJ, et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood. (2022) 139:357–68. doi: 10.1182/blood.2021013531
40. Caiado F, Kovtonyuk LV, Gonullu NG, Fullin J, Boettcher S, Manz MG. Aging drives Tet2+/- clonal hematopoiesis via IL-1 signaling. Blood. (2023) 141:886–903. doi: 10.1182/blood.2022016835.
41. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. (2015) 525:389–93. doi: 10.1038/nature15252.
42. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J. Am. Coll. Cardiol. (2018) 71:875–86. doi: 10.1016/j.jacc.2017.12.037.
43. Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight. (2020) 5(6):e135204. doi: 10.1172/jci.insight.135204.
44. Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of inflammatory signaling in tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. (2018) 23:833–49 e5. doi: 10.1016/j.stem.2018.10.013.
45. Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. (2017) 55:56–70 e13. doi: 10.1016/j.exphem.2017.08.001.
46. Pan W, Zhu S, Qu K, Meeth K, Cheng J, He K, et al. The DNA methylcytosine dioxygenase tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity. (2017) 47:284–97 e5. doi: 10.1016/j.immuni.2017.07.020.
47. Catala-Moll F, Ferrete-Bonastre AG, Godoy-Tena G, Morante-Palacios O, Ciudad L, Barbera L, et al. Vitamin D receptor, STAT3, and TET2 cooperate to establish tolerogenesis. Cell Rep. (2022) 38:110244. doi: 10.1016/j.celrep.2021.110244.
48. Ma S, Wan X, Deng Z, Shi L, Hao C, Zhou Z, et al. Epigenetic regulator CXXC5 recruits DNA demethylase Tet2 to regulate TLR7/9-elicited IFN response in pDCs. J. Exp. Med. (2017) 214:1471–91. doi: 10.1084/jem.20161149.
49. Griffin GK, Booth CAG, Togami K, Chung SS, Ssozi D, Verga JA, et al. Ultraviolet radiation shapes dendritic cell leukaemia transformation in the skin. Nature. (2023) 618:834–41. doi: 10.1038/s41586-023-06156-8.
50. Ito K, Lee J, Chrysanthou S, Zhao Y, Josephs K, Sato H, et al. Non-catalytic roles of tet2 are essential to regulate hematopoietic stem and progenitor cell homeostasis. Cell Rep. (2019) 28:2480–90 e4. doi: 10.1016/j.celrep.2019.07.094.
51. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. (2011) 20:11–24. doi: 10.1016/j.ccr.2011.06.001.
52. Ostrander EL, Kramer AC, Mallaney C, Celik H, Koh WK, Fairchild J, et al. Divergent effects of dnmt3a and tet2 mutations on hematopoietic progenitor cell fitness. Stem Cell Rep. (2020) 14:551–60. doi: 10.1016/j.stemcr.2020.02.011.
53. Nakauchi Y, Azizi A, Thomas D, Corces MR, Reinisch A, Sharma R, et al. The cell type-specific 5hmC landscape and dynamics of healthy human hematopoiesis and TET2-mutant preleukemia. Blood Cancer Discovery. (2022) 3:346–67. doi: 10.1158/2643-3230.BCD-21-0143.
54. Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature. (2018) 557:580–4. doi: 10.1038/s41586-018-0125-z.
55. Li Y, Xue M, Deng X, Dong L, Nguyen LXT, Ren L, et al. TET2-mediated mRNA demethylation regulates leukemia stem cell homing and self-renewal. Cell Stem Cell. (2023) 30:1072–90 e10. doi: 10.1016/j.stem.2023.07.001.
56. Cobo I, Tanaka TN, Chandra Mangalhara K, Lana A, Yeang C, Han C, et al. DNA methyltransferase 3 alpha and TET methylcytosine dioxygenase 2 restrain mitochondrial DNA-mediated interferon signaling in macrophages. Immunity. (2022) 55:1386–401 e10. doi: 10.1016/j.immuni.2022.06.022.
57. Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C, et al. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. U S A. (2017) 114:E1490–E9. doi: 10.1073/pnas.1616420114.
58. Ampomah PB, Cai B, Sukka SR, Gerlach BD, Yurdagul A Jr., Wang X, et al. Macrophages use apoptotic cell-derived methionine and DNMT3A during efferocytosis to promote tissue resolution. Nat. Metab. (2022) 4:444–57. doi: 10.1038/s42255-022-00551-7.
59. SanMiguel JM, Eudy E, Loberg MA, Young KA, Mistry JJ, Mujica KD, et al. Distinct tumor necrosis factor alpha receptors dictate stem cell fitness versus lineage output in dnmt3a-mutant clonal hematopoiesis. Cancer Discovery. (2022) 12:2763–73. doi: 10.1158/2159-8290.CD-22-0086.
60. Zioni N, Bercovich AA, Chapal-Ilani N, Bacharach T, Rappoport N, Solomon A, et al. Inflammatory signals from fatty bone marrow support DNMT3A driven clonal hematopoiesis. Nat. Commun. (2023) 14:2070. doi: 10.1038/s41467-023-36906-1.
61. Kim PG, Niroula A, Shkolnik V, McConkey M, Lin AE, Slabicki M, et al. Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. J. Exp. Med. (2021) 218(12):e20211872. doi: 10.1084/jem.20211872.
62. Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Author Correction: Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. (2021) 591:E27. doi: 10.1038/s41586-020-2819-2
63. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. (2018) 24:1015–23. doi: 10.1038/s41591-018-0081-z.
64. Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. (2018) 559:400–4. doi: 10.1038/s41586-018-0317-6
65. Wang W, Liu W, Fidler T, Wang Y, Tang Y, Woods B, et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in jak2 (V617F) mice. Circ. Res. (2018) 123:e35–47. doi: 10.1161/CIRCRESAHA.118.313283.
66. Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. (2018) 10(436):eaan8292. doi: 10.1126/scitranslmed.aan8292.
67. Mullally A, Poveromo L, Schneider RK, Al-Shahrour F, Lane SW, Ebert BL. Distinct roles for long-term hematopoietic stem cells and erythroid precursor cells in a murine model of Jak2V617F-mediated polycythemia vera. Blood. (2012) 120:166–72. doi: 10.1182/blood-2012-01-402396.
68. Spanoudakis E, Papoutselis M, Bazdiara I, Lamprianidi E, Kordella X, Tilkeridis C, et al. The JAK2V617F Point Mutation Increases the Osteoclast Forming Ability of Monocytes in Patients with Chronic Myeloproliferative Neoplasms and Makes their Osteoclasts more Susceptible to JAK2 Inhibition. Mediterr J. Hematol. Infect. Dis. (2018) 10:e2018058. doi: 10.4084/mjhid.2018.058.
69. Yao JC, Oetjen KA, Wang T, Xu H, Abou-Ezzi G, Krambs JR, et al. TGF-beta signaling in myeloproliferative neoplasms contributes to myelofibrosis without disrupting the hematopoietic niche. J. Clin. Invest. (2022) 132(11):e154092. doi: 10.1172/JCI154092.
70. Ramos TL, Sanchez-Abarca LI, Roson-Burgo B, Redondo A, Rico A, Preciado S, et al. Mesenchymal stromal cells (MSC) from JAK2+ myeloproliferative neoplasms differ from normal MSC and contribute to the maintenance of neoplastic hematopoiesis. PloS One. (2017) 12:e0182470. doi: 10.1371/journal.pone.0182470.
71. Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. (2014) 512:78–81. doi: 10.1038/nature13383.
72. Manshouri T, Estrov Z, Quintas-Cardama A, Burger J, Zhang Y, Livun A, et al. Bone marrow stroma-secreted cytokines protect JAK2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. (2011) 71:3831–40. doi: 10.1158/0008-5472.CAN-10-4002.
73. Korn C, Mendez-Ferrer S. Myeloid Malignancies and the microenvironment. Blood. (2017) 129:811–22. doi: 10.1182/blood-2016-09-670224.
74. Medinger M, Skoda R, Gratwohl A, Theocharides A, Buser A, Heim D, et al. Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: correlation with clinical parameters and JAK2-V617F mutational status. Br. J. Haematol. (2009) 146:150–7. doi: 10.1111/j.1365-2141.2009.07726.x.
75. Grockowiak E, Korn C, Rak J, Lysenko V, Hallou A, Panvini FM, et al. Different niches for stem cells carrying the same oncogenic driver affect pathogenesis and therapy response in myeloproliferative neoplasms. Nat. Cancer. (2023) 4:1193–209. doi: 10.1038/s43018-023-00607-x.
76. Batsivari A, Grey W, Bonnet D. Understanding of the crosstalk between normal residual hematopoietic stem cells and the leukemic niche in acute myeloid leukemia. Exp. Hematol. (2021) 95:23–30. doi: 10.1016/j.exphem.2021.01.004.
77. Batsivari A, Haltalli MLR, Passaro D, Pospori C, Celso CL, Bonnet D. Author Correction: Dynamic responses of the haematopoietic stem cell niche to diverse stresses. Nat. Cell Biol. (2020) 22:257. doi: 10.1038/s41556-020-0469-0.
78. Passaro D, Di Tullio A, Abarrategi A, Rouault-Pierre K, Foster K, Ariza-McNaughton L, et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell. (2017) 32:324–41 e6. doi: 10.1016/j.ccell.2017.08.001.
79. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell. (2018) 22:64–77.e6. doi: 10.1016/j.stem.2017.11.006.
80. Galán-Díez M, Borot F, Ali AM, Zhao J, Gil-Iturbe E, Shan X, et al. Subversion of serotonin receptor signaling in osteoblasts by kynurenine drives acute myeloid leukemia. Cancer Discover. (2022) 12:1106–27. doi: 10.1158/2159-8290.CD-21-0692.
81. Forte D, García-Fernández M, Sánchez-Aguilera A, Stavropoulou V, Fielding C, Martín-Pérez D, et al. Bone marrow mesenchymal stem cells support acute myeloid leukemia bioenergetics and enhance antioxidant defense and escape from chemotherapy. Cell Metab. (2020) 32:829–43.e9. doi: 10.1016/j.cmet.2020.09.001.
82. Jiang X, Mak PY, Mu H, Tao W, Mak DH, Kornblau S, et al. Disruption of wnt/β-catenin exerts antileukemia activity and synergizes with FLT3 inhibition in FLT3-mutant acute myeloid leukemia. Clin. Cancer Res. (2018) 24:2417–29. doi: 10.1158/1078-0432.CCR-17-1556.
83. Hou D, Wang B, You R, Wang X, Liu J, Zhan W, et al. Stromal cells promote chemoresistance of acute myeloid leukemia cells via activation of the IL-6/STAT3/OXPHOS axis. Ann. Trans. Med. (2020) 8:1346. doi: 10.21037/atm.
84. Li Y, You MJ, Yang Y, Hu D, Tian C. The role of tumor-associated macrophages in leukemia. Acta Haematol. (2020) 143:112–7. doi: 10.1159/000500315.
85. Krevvata M, Silva BC, Manavalan JS, Galan-Diez M, Kode A, Matthews BG, et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood. (2014) 124:2834–46. doi: 10.1182/blood-2013-07-517219.
86. Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood. (2012) 119:540–50. doi: 10.1182/blood-2011-04-348151.
87. Battula VL, Le PM, Sun JC, Nguyen K, Yuan B, Zhou X, et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. (2017) 2(13):e90036. doi: 10.1172/jci.insight.90036.
88. Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. (2013) 19:1513–7. doi: 10.1038/nm.3364.
89. Anderson NR, Sheth V, Li H, Harris MW, Qiu S, Crossman DK, et al. Microenvironmental CXCL12 deletion enhances Flt3-ITD acute myeloid leukemia stem cell response to therapy by reducing p38 MAPK signaling. Leukemia. (2023) 37:560–70. doi: 10.1038/s41375-022-01798-5.
90. Ladikou EE, Chevassut T, Pepper CJ, Pepper AG. Dissecting the role of the CXCL12/CXCR4 axis in acute myeloid leukaemia. Br. J. Haematol. (2020) 189:815–25. doi: 10.1111/bjh.16456.
91. Cho BS, Kim HJ, Konopleva M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: from bench to bedside. Korean J. Intern. Med. (2017) 32:248–57. doi: 10.3904/kjim.2016.244.
92. Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R, et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J. Exp. Med. (2004) 199:221–9. doi: 10.1084/jem.20031615.
93. Jacamo R, Davis RE, Ling X, Sonnylal S, Wang Z, Ma W, et al. Tumor Trp53 status and genotype affect the bone marrow microenvironment in acute myeloid leukemia. Oncotarget. (2017) 8:83354–69. doi: 10.18632/oncotarget.v8i48.
94. Chang YT, Hernandez D, Alonso S, Gao M, Su M, Ghiaur G, et al. Role of CYP3A4 in bone marrow microenvironment-mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv. (2019) 3:908–16. doi: 10.1182/bloodadvances.2018022921.
95. Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl. J. Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689.
96. Han SJ, Jang HS, Noh MR, Kim J, Kong MJ, Kim JI, et al. Mitochondrial NADP(+)-dependent isocitrate dehydrogenase deficiency exacerbates mitochondrial and cell damage after kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. (2017) 28:1200–15. doi: 10.1681/ASN.2016030349.
97. Cerchione C, Romano A, Daver N, DiNardo C, Jabbour EJ, Konopleva M, et al. IDH1/IDH2 inhibition in acute myeloid leukemia. Front. Oncol. (2021) 11:639387. doi: 10.3389/fonc.2021.639387.
98. Chen J-Y, Lai Y-S, Tsai H-J, Kuo C-C, Yen BL, Yeh S-P, et al. The oncometabolite R-2-hydroxyglutarate activates NF-κB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci. Rep. (2016) 6:32428. doi: 10.1038/srep32428.
99. Weinhäuser I, Pereira-Martins DA, Almeida LY, Hilberink JR, Silveira DRA, Quek L, et al. M2 macrophages drive leukemic transformation by imposing resistance to phagocytosis and improving mitochondrial metabolism. Sci. Adv. (2023) 9:eadf8522. doi: 10.1126/sciadv.adf8522.
100. Stavropoulou V, Kaspar S, Brault L, Sanders Mathijs A, Juge S, Morettini S, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. (2016) 30:43–58. doi: 10.1016/j.ccell.2016.05.011.
101. Syampurnawati M, Tatsumi E, Furuta K, Takenokuchi M, Nakamachi Y, Kawano S, et al. (M1 and M2): FLT3 mutations (ITD and D835) and cell-surface antigen expression. Leukemia Res. (2007) 31:921–9. doi: 10.1016/j.leukres.2006.09.017.
102. Li Y, Seet CS, Mack R, Joshi K, Runde AP, Hagen PA, et al. Distinct roles of hematopoietic cytokines in the regulation of leukemia stem cells in murine MLL-AF9 leukemia. Stem Cell Rep. (2023). doi: 10.1016/j.stemcr.2023.11.003.
103. Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia. (2018) 32:575–87. doi: 10.1038/leu.2017.259.
104. Abarrategi A, Foster K, Hamilton A, Mian SA, Passaro D, Gribben J, et al. Versatile humanized niche model enables study of normal and Malignant human hematopoiesis. J. Clin. Invest. (2017) 127:543–8. doi: 10.1172/JCI89364.
105. Mian SA, Abarrategi A, Kong KL, Rouault-Pierre K, Wood H, Oedekoven CA, et al. Ectopic humanized mesenchymal niche in mice enables robust engraftment of myelodysplastic stem cells. Blood Cancer Discovery. (2021) 2:135–45. doi: 10.1158/2643-3230.BCD-20-0161.
106. Antonelli A, Noort WA, Jaques J, de Boer B, de Jong-Korlaar R, Brouwers-Vos AZ, et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow-like scaffold-based niches. Blood. (2016) 128:2949–59. doi: 10.1182/blood-2016-05-719021.
107. Chen Y, Jacamo R, Shi YX, Wang RY, Battula VL, Konoplev S, et al. Human extramedullary bone marrow in mice: a novel in vivo model of genetically controlled hematopoietic microenvironment. Blood. (2012) 119:4971–80. doi: 10.1182/blood-2011-11-389957.
108. Reinisch A, Thomas D, Corces MR, Zhang X, Gratzinger D, Hong WJ, et al. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat. Med. (2016) 22:812–21. doi: 10.1038/nm.4103.
109. Al-Matary YS, Botezatu L, Opalka B, Hönes JM, Lams RF, Thivakaran A, et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a Growth factor independence 1 dependent manner. Haematologica. (2016) 101:1216–27. doi: 10.3324/haematol.2016.143180.
110. Koldej RM, Ritchie DS. High multiplex analysis of the immune microenvironment in bone marrow trephine samples using GeoMX™ digital spatial profiling. Immuno-Oncol Technol. (2020) 5:1–9. doi: 10.1016/j.iotech.2020.02.001.
111. Aguilar-Navarro AG, Meza-León B, Gratzinger D, Juárez-Aguilar FG, Chang Q, Ornatsky O, et al. Human aging alters the spatial organization between CD34+ Hematopoietic cells and adipocytes in bone marrow. Stem Cell Rep. (2020) 15:317–25. doi: 10.1016/j.stemcr.2020.06.011.
112. Gomariz A, Helbling PM, Isringhausen S, Suessbier U, Becker A, Boss A, et al. Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat. Commun. (2018) 9:2532. doi: 10.1038/s41467-018-04770-z.
113. Sivaraj KK, Jeong H-W, Dharmalingam B, Zeuschner D, Adams S, Potente M, et al. Regional specialization and fate specification of bone stromal cells in skeletal development. Cell Rep. (2021) 36:109352. doi: 10.1016/j.celrep.2021.109352.
114. Schürch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, Zlobec I, et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell. (2020) 182:1341–59.e19. doi: 10.1016/j.cell.2020.07.005
115. Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. (2018) 174:968–81.e15. doi: 10.1016/j.cell.2018.07.010
116. Wan X, Xiao J, Tam SST, Cai M, Sugimura R, Wang Y, et al. Integrating spatial and single-cell transcriptomics data using deep generative models with SpatialScope. Nat. Commun. (2023) 14:7848. doi: 10.1038/s41467-023-43629-w.
117. van Ineveld RL, Kleinnijenhuis M, Alieva M, de Blank S, Barrera Roman M, van Vliet EJ, et al. Revealing the spatio-phenotypic patterning of cells in healthy and tumor tissues with mLSR-3D and STAPL-3D. Nat. Biotechnol. (2021) 39:1239–45. doi: 10.1038/s41587-021-00926-3.
118. Zhang Q, Jiang S, Schroeder A, Hu J, Li K, Zhang B, et al. Leveraging spatial transcriptomics data to recover cell locations in single-cell RNA-seq with CeLEry. Nat. Commun. (2023) 14:4050. doi: 10.1038/s41467-023-39895-3.
119. Longo SK, Guo MG, Ji AL, Khavari PA. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. (2021) 22:627–44. doi: 10.1038/s41576-021-00370-8.
Keywords: bone marrow niche, myeloid leukaemia, clonal haematopoiesis, microenvironment, stromal cells
Citation: Ngo S, Papazoglou D, Huerga Encabo H and Bonnet D (2024) Exploring the intricate cross-talk between clonal expansion and the bone marrow niche. Front. Hematol. 3:1334807. doi: 10.3389/frhem.2024.1334807
Received: 07 November 2023; Accepted: 02 February 2024;
Published: 01 March 2024.
Edited by:
Jason M. Butler, University of Florida, United StatesReviewed by:
Kostandin Pajcini, University of Illinois Chicago, United StatesRobert A. J. Oostendorp, Technical University of Munich, Germany
Copyright © 2024 Ngo, Papazoglou, Huerga Encabo and Bonnet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominique Bonnet, ZG9taW5pcXVlLmJvbm5ldEBjcmljay5hYy51aw==
†These authors have contributed equally to this work