
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Hematol. , 06 March 2024
Sec. Red Cells, Iron and Erythropoiesis
Volume 3 - 2024 | https://doi.org/10.3389/frhem.2024.1331485
This article is part of the Research Topic Oxygen Sensing and Oxidative Stress in and Around Erythropoiesis View all 4 articles
 Tiziana Coppola1
Tiziana Coppola1 Hannah Hughes2
Hannah Hughes2 Paul T. Finch2
Paul T. Finch2 Joshua A. Hess2
Joshua A. Hess2 Steve Wu3,4
Steve Wu3,4 Carlos E. Prada5,6
Carlos E. Prada5,6 Alexander G. Miethke4,7
Alexander G. Miethke4,7 Wenying Zhang4,8
Wenying Zhang4,8 Theodosia A. Kalfa1,4*
Theodosia A. Kalfa1,4*We present a rare case of erythrocytosis due to a homozygous SLC30A10 mutation, causative of Hypermanganesemia with Dystonia, Polycythemia, and Cirrhosis (HMDPC). The patient presented at 7 years of age with the incidental finding of hemoglobin up to 22.3 g/dL. Despite extensive phenotypic evaluation and genetic testing for common causes of erythrocytosis, etiology remained unknown for 1.5 years until whole exome sequencing revealed the genetic diagnosis. Upon neurological evaluation, the patient was found to have very mild dystonia due to manganese deposition in the basal ganglia, midbrain, and cerebellum, as seen in brain MRI. Chelation with trientine orally was initiated, which led to resolution of his erythrocytosis along with decrease of Mn deposition in brain tissues. Recurrence of erythrocytosis occurred after 4 years due to inappropriately stored trientine and iron deficiency. This required the temporary addition of intravenous calcium disodium edetate to his treatment regimen. This case illustrates the development of secondary erythrocytosis due to hypermanganesemia leading to manganese deposition in the hepatocytes, causing a sense of tissue hypoxia and stimulating erythropoietin production. It also shows the competition between manganese and iron for absorption in the body. Moreover, it demonstrates the importance of broad genetic evaluation for the diagnosis of ultra-rare diseases. In this case, timely diagnosis and initiation of appropriate treatment prevented irreversible damage to the brain and liver with a profound improvement on his prognosis.
Biallelic SLC30A10 mutations leading to Hypermanganesemia with Dystonia, Polycythemia, and Cirrhosis (HMDPC) are ultra-rare causes of familial erythrocytosis. We describe here a unique case of this syndrome where the child was diagnosed before demonstrating neurological manifestations of HMDPC, after presenting with an incidental finding of erythrocytosis. Work-up for more common causes of erythrocytosis was unrevealing, but whole exome sequencing (WES) with trio-analysis for the patient and parents identified a homozygous pathogenic variant in the SLC30A10 gene, coding for a manganese exporter. This case highlights the competition between manganese and iron for absorption in the body and therefore the importance of iron supplementation and avoidance of phlebotomy for treatment of HMDPC.
A previously healthy 7-year-old European-American boy presented to the emergency department with left testicular torsion. He was treated successfully with prompt surgery for orchiopexy, but a complete blood count (CBC) at the time revealed, surprisingly, an elevated hemoglobin (Hgb) at 22.3 g/dL, red blood cell (RBC) count of 8.87 x 109/mL, and hematocrit (Hct) at 68%. White blood cell and platelet count were normal. He was referred to hematology for further evaluation that followed the algorithmic approach shown in Figure 1A. Pulse-oximetry showed SpO2 98%, making cardiac or pulmonary causes unlikely as primary causes for the patient’s erythrocytosis (2). History revealed no exposure to smoking or to high altitude. The parents reported that he sometimes snored in his sleep but without apnea episodes. He also had a normal sleep study that verified no nocturnal hypoxia. The family’s vacation place had a gas stove, but carboxyhemoglobin level was normal at 0.3%. Erythropoietin (Epo) level was 11 milliunits per mL which was inappropriately high for the concurrent Hct value (1) (Figure 1B). An abdominal ultrasound was performed to screen for an erythropoietin-producing tumor, and it was normal. He had a normal Hgb-O2 affinity (p50) by hemoximetry, indicating that his erythrocytosis was not due to a high-affinity hemoglobin. Hgb levels of his father and mother were 16.5 g/dL and 11.5 g/dL respectively. There was no family history of erythrocytosis, polycythemia vera, or other hematologic disorders.
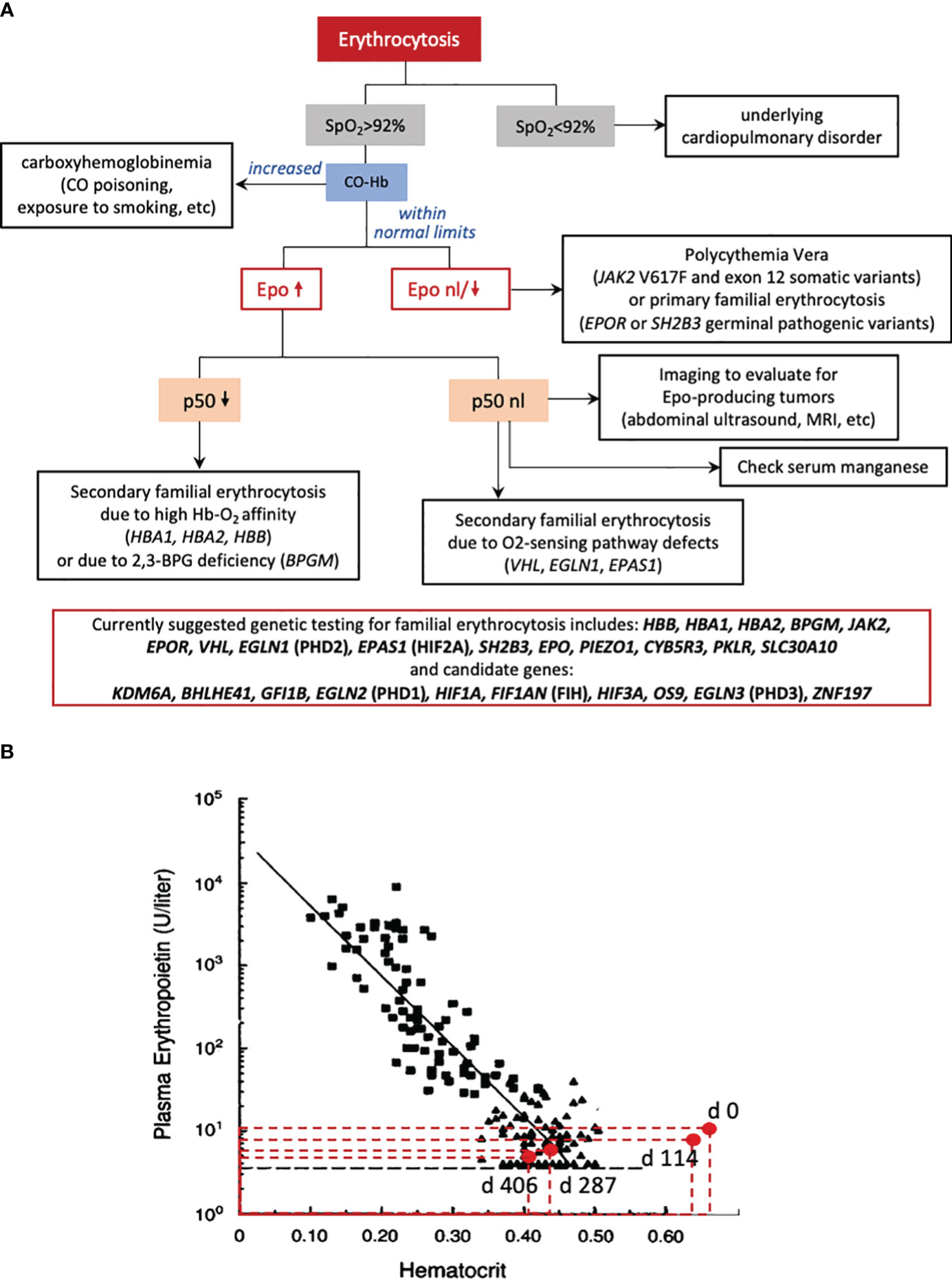
Figure 1 (A) Suggested diagnostic algorithm for patients presenting with erythrocytosis. A pulse oximeter reading (SpO2) is the simplest first step: if SpO2 is less than 92%, the patient should be evaluated for cardiac or pulmonary disorders causing hypoxia and secondary erythrocytosis. If SpO2 is greater than 92%, exposure to carbon monoxide, indicated by increased carboxyhemoglobin in a venous blood gas, needs to be excluded, as pulse oximetry cannot differentiate between carboxyhemoglobin and oxyhemoglobin. The patient’s serum erythropoietin (Epo) should be measured and the value should be compared to the patient’s concurrent Hct or Hgb in a nomogram such as the one in panel (B). If the erythropoietin is low or normal, relative to the patient’s concurrent Hct, then the concern is for primary erythrocytosis. Primary erythrocytosis can include polycythemia vera or primary familial erythrocytosis, where there is increased Epo-independent activity of Epo-signaling, downstream of the erythropoietin receptor (gain-of-function variants in EPOR, SH2B3, and JAK2). If the erythropoietin is elevated related to the patient’s concurrent Hct, then the concern is for secondary erythrocytosis. Decreased p50 (i.e., O2 pressure where Hgb is 50% saturated with oxygen) by hemoximetry indicates secondary erythrocytosis due to high oxygen-affinity hemoglobin, which may be caused by certain HBA1, HBA2, or HBB missense mutations or by 2,3-bisphosphoglycerate (2,3-BPG) deficiency, caused by biallelic pathogenic variants of BPG mutase (BPGM). Alternatively normal p50 in this algorithmic approach, should trigger imaging to evaluate for Epo-producing tumors and evaluating the genes known to affect the hypoxia sensing pathway (VHL, EGLN1, EPAS1), as well as checking a serum manganese level, as a screen for hypermanganesemia. Our patient had an inappropriately elevated erythropoietin level with a normal p50 level and no known mutation in the oxygen sensing pathway. WES-trio or genetic analysis of the genes included in the red box is a less targeted but more comprehensive approach to facilitate genetic diagnosis of familial erythrocytosis. (B) Epo-Hct correlation in our patient at the time of genetic diagnosis and initiation of chelation with trientine (day 0) up to the time Hct normalized (days 287 and 406). The values of Epo and Hct are plotted against a classic Epo-Hct nomogram (1). Epo level on days 0 and 114 were inappropriately elevated with respect to his hematocrit indicating secondary erythrocytosis.
He had a bone marrow aspirate and biopsy revealing no pathology except of erythroid hyperplasia. Cytogenetic evaluation for BCR-ABL1 fusion by FISH were negative. Sequencing for the most common genetic causes of polycythemia and familial erythrocytosis (JAK2 Val617Phe and exon 12, EGLN1, EPAS1 exons 9 and 12, EPOR exon 8 and VHL), BPGM, HBA, HBB did not reveal any pathogenic variants.
The patient reported decreased energy level, headaches, numbness and tingling in his fingers at least once weekly, but no pruritus even with exposure to hot water. He had frequent episodes of vomiting with no identifiable trigger. On exam, he was a well-nourished, well-developed boy, at 75th percentile for weight and height, with a “ruddy” skin complexion, evoking in memory the term “polycythemia rubra”. He had been started on aspirin for thromboprophylaxis since diagnosis. The first year after presentation, he had 4 phlebotomies with removal of 5mL blood per kg and replacement with same volume of normal saline, roughly 3 months apart, mainly because of increased headaches, which however recurred just 2-3 days after phlebotomy. Iron deficiency was noted, and he was given iron supplementation by mouth for about a month until this was stopped for concerns of increasing his RBC count further.
The diagnostic odyssey for this family lasted almost two years until a third-opinion hematology consult in a tertiary care center and enrollment in a study offering whole exome sequencing (WES) with trio-analysis to patients with undiagnosed disorders and their parents (Genetics For Undiagnosed Disorders (GFUD) study, approved by Cincinnati Children’s IRB). WES-trio revealed homozygous SLC30A10 c.585del, p.(Thr196Profs*17), expected to be pathogenic for a rare inborn error of manganese (Mn) metabolism, namely, hypermanganesemia with dystonia, polycythemia, and cirrhosis (HMDPC), also called Hypermanganesemia with dystonia 1 (HMNDYT1) (3). Measurement of serum Mn level after this genetic diagnosis revealed an elevated level of 10.9 mcg/L (reference range 0.0-2.0 mcg/L). There was no evidence of significant liver disease based on liver function testing, including Alanine Transaminase (ALT), Aspartate Transaminase (AST), γ-Glutamyl Transferase (γGT), Prothrombin Time (PT), and activated Partial Thromboplastin Time (aPTT). Liver ultrasound with Acoustic Radiation Force Impulse (ARFI) Imaging to evaluate for tissue fibrosis was also normal.
On neurological exam, there was no tremor, dysrhythmia, dysmetria, chorea, or myoclonus. When doing rapid movements in left hand, his right arm pronated. Gait was normal (narrow-based). He was able to toe-walk, heel-walk, and tandem walk. Romberg sign was negative. When running and walking backwards, he had occasional internal rotation of the right foot. Neuropsychological testing revealed a diagnosis of reading disability but otherwise normal development. However, brain MRI revealed an abnormal T1 hyperintense signal in the deep gray structures, brainstem, and cerebellum indicating manganese deposition (Figure 2).
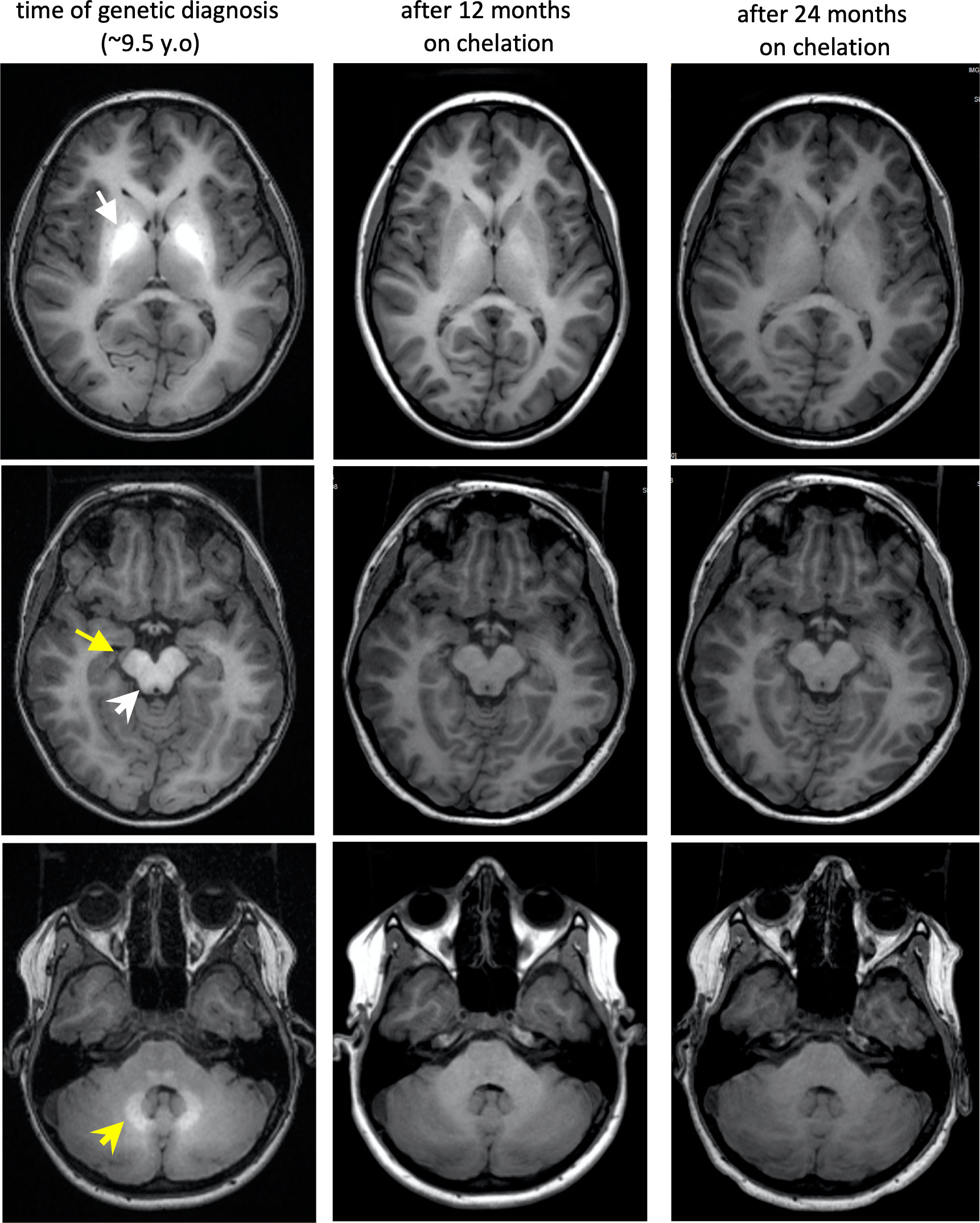
Figure 2 Brain MRI findings of our patient at diagnosis and through chelation treatment with trientine. T1 bright hyperintensities are present at the first brain MRI, soon after genetic diagnosis, in bilateral globi pallidi (white arrow), cerebral peduncles (yellow arrow), dorsal brainstem (white arrowhead), and deep cerebellar white matter (yellow arrowhead) indicating manganese deposition. With progression of chelation treatment, Mn deposition in the brain resolves indicated by decrease in T1 hyperintensities. Although, urinary excretion of Mn was much lower with trientine rather than during IV chelation with calcium disodium edetate, this imaging and the resolution of polycythemia with trientine chelation, demonstrates that this is also a reasonable choice for HMDPC treatment with the appropriate formulation, iron supplementation as needed, and close monitoring.
Treatment was initiated with trientine, an oral chelator that is typically used in Wilson’s disease to chelate copper, but can also chelate other metal ions, such as iron and zinc (4, 5). Iron supplementation was also started to reduce manganese absorption as manganese and iron utilize the same transporter proteins (6, 7). Trientine was given twice daily (morning and evening time), while iron was given at least four hours apart from both Trientine doses to avoid complex formation. After eight months, hemoglobin normalized, while serum manganese trended to borderline high (Figure 3). Repeat brain MRI revealed gradual decrease in T1 hyperintense signal within the globi pallidi and complete resolution of signal abnormality into the brainstem, superior cerebellar peduncles, and cerebellum by 24 months after treatment initiation (Figure 2). Aspirin thromboprophylaxis (81 mg once daily) was discontinued after Hgb decreased to less than 18 g/dL, since the patient also has mild Type I von Willebrand disease and had epistaxis while on aspirin.
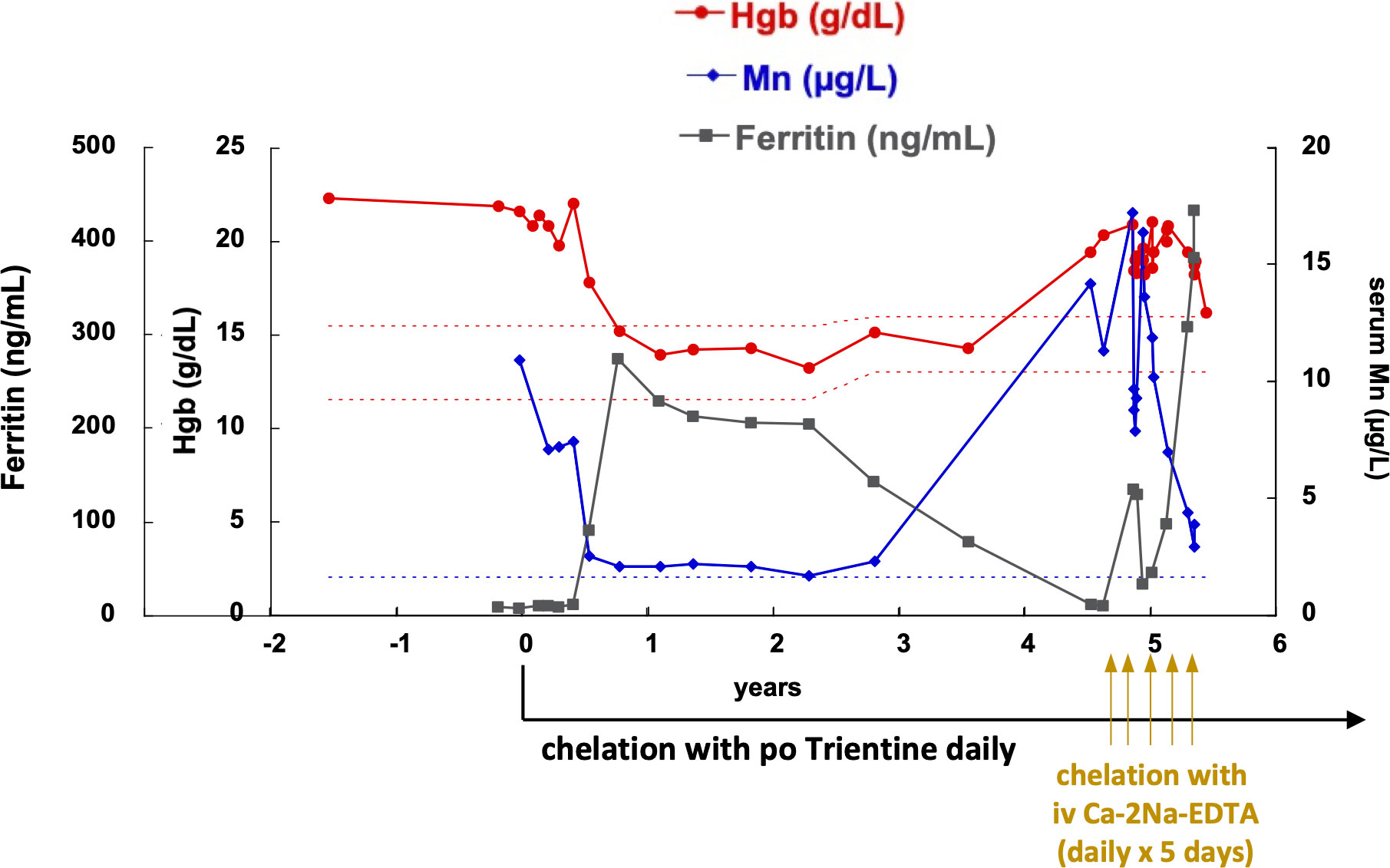
Figure 3 Manganese, hemoglobin, and ferritin values throughout diagnosis and treatment course. After chelation with oral trientine was initiated, serum Mn levels decreased gradually to just above the normal range, and hemoglobin also decreased in parallel to normal. Iron supplementation was also prescribed to treat iron deficiency and compete manganese absorption and cellular uptake. However, due to inactivation of trientine as it was not refrigerated and poor compliance with iron supplementation, hemoglobin levels and manganese levels rose again. In order to resolve erythrocytosis and decrease thrombophilia risk faster, IV chelation courses with calcium disodium edetate (Ca-2Na-EDTA) were added as daily infusion for 5 days every 4-6 weeks. After five such courses, his hemoglobin is now back to normal range and serum manganese level has improved to borderline high levels. His ferritin did increase due to iron supplementation and recycling of iron from the excess red blood cells. The patient continues treatment with oral trientine.
At the time of the COVID-19 pandemic, the family missed several clinic visits for follow-up hematology care in the hospital with the fear of being exposed to the virus. Early during the pandemic, the patient’s insurance dictated the change of trientine to a generic formulation, that we retrospectively realized was not refrigerated in storage by the distributing pharmacy per manufacturer’s recommendations. At the same time, iron supplementation with ferrous sulfate was very inconsistent due to nausea and upset stomach, unfortunately not an unusual side effect for this iron formulation.
Upon return to the clinic, with a lapse in monitoring for about a year, Hgb was up to 19.4 g/dL, serum manganese up to 14.2 mcg/L, and ferritin down to 11 ng/mL (Figure 3). We restarted aspirin thromboprophylaxis, changed his iron supplementation to polysaccharide iron complex to improve compliance and also changed the trientine generic formulation to one that was kept refrigerated from the time of production to the time of administration, as recommended by the American Association for the Study of Liver Diseases (8). However, the patient was now 14 years old with higher risk for thrombotic complications than the time he was a prepubertal boy, and whole blood viscosity at shear rate of 187.5 s-1 was up to 7.6-9.18 centipoise (reference range 3.4-5.1 centipoise). Brain MRI again showed increasing T1 hyperintensities reflecting manganese deposition in his cerebellum, dorsal pons, substantia nigra, and lentiform nuclei.
Therefore, we elected to proceed to IV chelation with calcium disodium edetate (Ca-2Na-EDTA) to accelerate erythrocytosis resolution. Chelation therapy with calcium disodium edetate has been shown to enhance urinary Mn excretion, decrease serum Mn level, improve neurologic symptoms and brain MRI changes, and prevent further liver damage in other cases with HMDPC previously described (7, 9–11). Indeed, urine Mn excretion was up to 24.6 mcg/24h urine collection while on chelation with trientine and up to 940 mcg/24h urine collection while on IV chelation with calcium disodium edetate (1g/m2 administered over 16 hrs daily for 5 days every 4-6 weeks). After 5 such courses, serum Mn decreased down to 2.9 mcg/L and Hgb down to 16.2 mg/dL. Erythropoietin also normalized; of note, we attempted phlebotomy during these admissions of 300 mL blood, followed by infusion of 300 grams iron sucrose IV, to avoid aggravating iron deficiency. However, Hgb was fast increasing back to pre-phlebotomy value within 1-2 days while Mn levels remained high and erythropoietin levels were also inappropriately increased.
Urinary excretion and serum levels of other essential heavy metals (zinc, copper, chromium) were also monitored as they may be affected by chelation with calcium disodium edetate (12). No supplementation of these metals was required. Along with the improvement in serum manganese levels and Hgb values, brain MRI also showed significant improvement of the manganese deposition. While on oral iron supplementation at home and IV iron supplementation while in the hospital, with the goal to compete with Mn intestinal absorption, ferritin has increased up to 430 ng/mL. Additional iron storage attained after normal splenic destruction of the excess, aging RBCs without replacing them at the same levels likely also contributed to these high ferritin values. The patient now continues only on treatment with trientine, now appropriately refrigerated; aspirin and iron supplementation have been discontinued.
Close monitoring with laboratory evaluation every 3 months is planned to assure maintaining high normal ferritin values, with further iron supplementation when needed, and adequate Mn chelation with trientine, or if needed additional courses of IV chelation with calcium disodium edetate.
Seven years after the diagnosis, the patient is doing well without any neurological sequalae. Early diagnosis and intensive chelation and monitoring, even with the lapses and challenges described above, were critical in maintaining a good outcome. Key points derived from this patient’s course were: 1) Phlebotomy is contraindicated for treatment of erythrocytosis in HMDPC, since it aggravates iron deficiency which increases manganese absorption. 2) Chelation therapy with trientine was effective along with iron supplementation. 3) Improper storage of trientine and decreased iron intake rendered the oral chelation therapy ineffective, requiring IV chelation with calcium disodium edetate in 5 day-courses. 4) After near normalization of serum manganese and resolution of iron deficiency, oral chelation with trientine appears to be effective again to prevent hypermanganesemia and its associated complications.
This case highlights a rare case of familial erythrocytosis due to biallelic pathogenic variants of SLC30A10, which codes for a channel present in plasma membrane, Golgi, and endosomes, and exports Mn out of cells, preventing Mn overload in all tissues (13). The patient presented here had a homozygous frameshift variant, predicted to cause loss-of-function; both parents and the patient’s sister were asymptomatic carriers of the same variant. There was no known consanguinity of the parents; the family is from a relatively small community and there may be a distant common ancestor. Of note, the same homozygous variant has been reported by Tuschl et al. in 2012 (14), in patient E within their cohort (Table 1). That patient had presented at 14 years of age with erythrocytosis with Hgb at 22.5 g/dL, but also spastic paraparesis, hepatomegaly, and liver cirrhosis on biopsy (24).
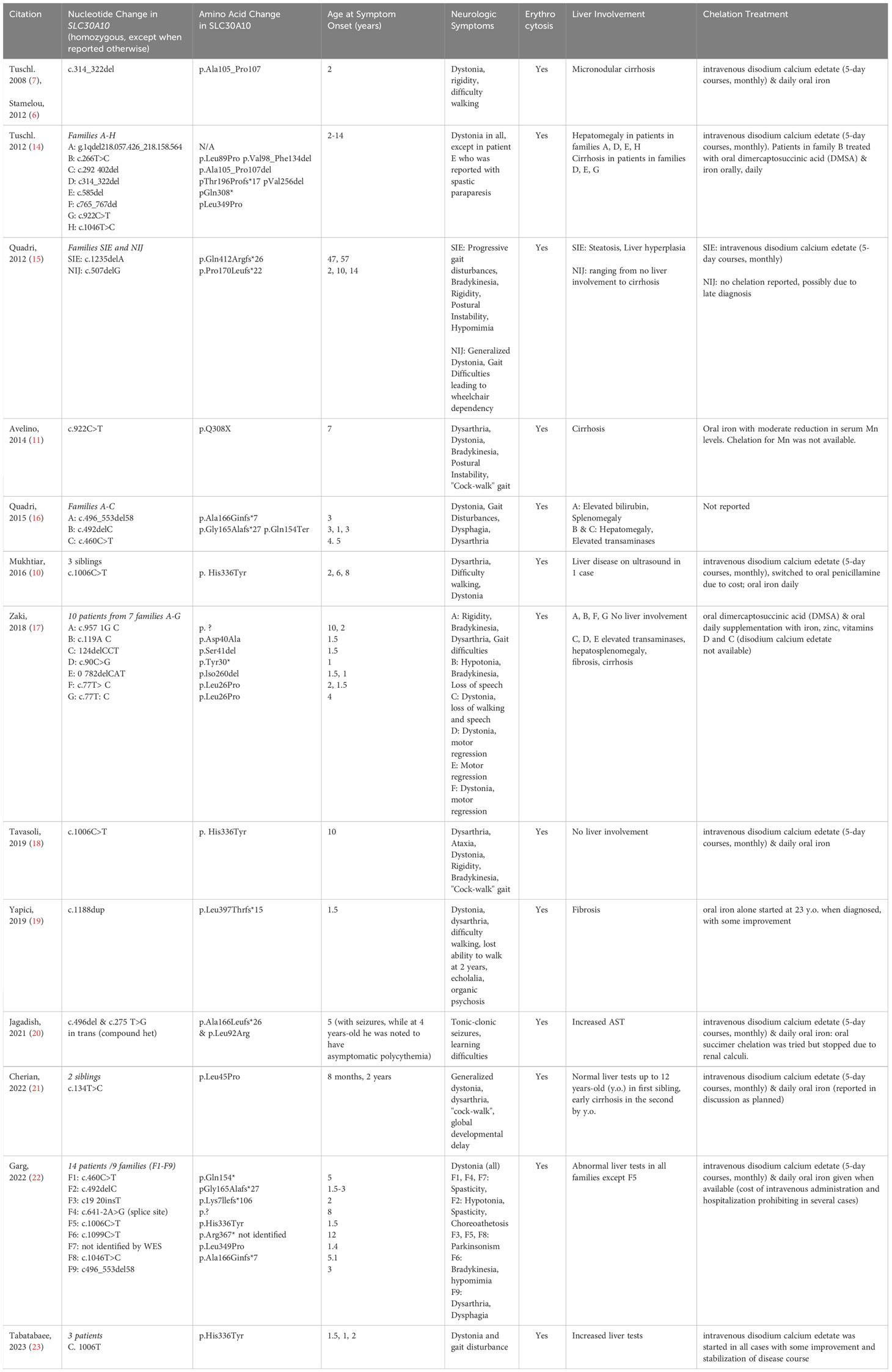
Table 1 Clinical presentation and symptoms of patients with SLC30A10 mutations.
Mn is an essential trace metal that plays a critical role as a cofactor for a variety of enzymes involved in amino acid, lipid, and carbohydrate metabolism, immune function, bone and connective tissue growth, and blood clotting (25). Mn transport is tightly regulated in the body to avoid toxicity. It is ingested through our diet and absorbed into the small intestine by a not yet completely defined path. Mn is a transition element like iron (Fe); they both exist in a reduced state as divalent, water-soluble ions (Mn2+ and Fe2+) and in an oxidized trivalent state (Mn3+ and Fe3+). Mn3+ and Fe3+ compete for transferrin, their main ligand-protein in the plasma (13). It is not completely understood how Mn+2 enters the enterocytes. The divalent metal ion transporter 1 (DMT1) has been shown to participate in the transport of both Fe2+ and Mn2+ into the intestinal epithelial cells (enterocytes), but studies with DMT1-deficient mice demonstrated that DMT1 is required for iron but not for manganese absorption (26). However, in iron deficiency DMT1 is upregulated and may significantly increase Mn2+ intestinal absorption (Figure 4). Once Mn2+ enters the blood circulation, it circulates unbound or bound to a carrier protein, such as albumin or α2-macroglobulin. As it goes through the portal circulation, it enters the hepatocytes via the SLC39A14 transporter; any excess Mn in these cells is excreted into the bile via SLC30A10, which is present on their canalicular membrane. The bile containing Mn will then circulate back to the small intestine where it can be reabsorbed back to the enterohepatic circulation or excreted with feces. Absorbed Mn will enter the systemic circulation, traveling as Mn3+ bound to transferrin, or as Mn2+ circulating unbound or bound to albumin or α2-macroglobulin. A very small amount is excreted in the urine, even in conditions of excess, in the absence of therapeutic chelation. Mn can enter cells through transferrin mediated endocytosis via the transferrin receptor (TFRC or CD71) or via SLC39A14 (13). With iron deficiency, transferrin is also upregulated and is more available to bind and transfer Mn3+. In hypermanganesemia, unbound Mn2+ enters the brain via the epithelial cells of the choroid plexus; Mn transport into the brain cells also increases during iron deficiency (27).
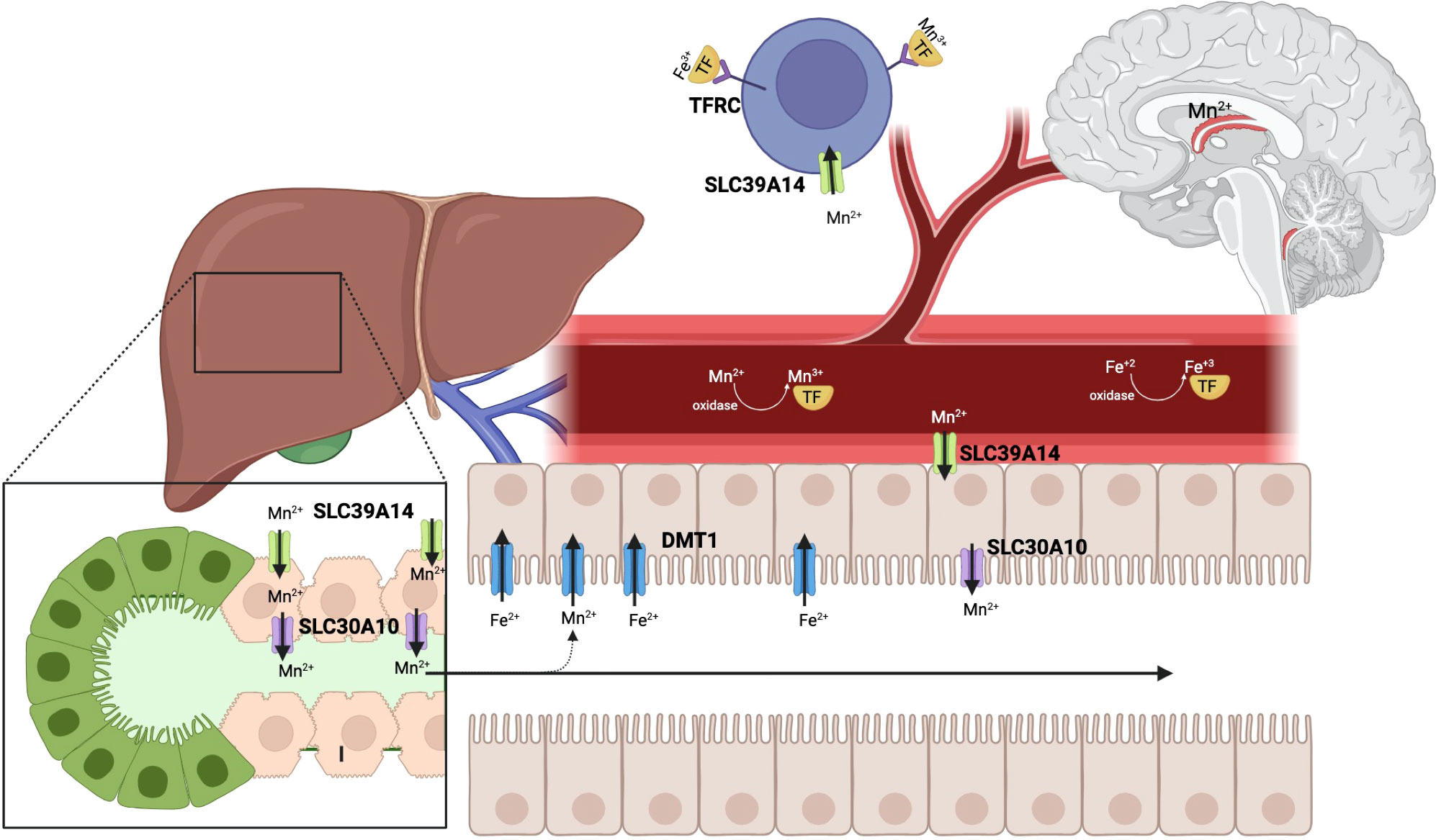
Figure 4 The SLC30A10 protein is an efflux transporter, present on hepatocytes, enterocytes, and neural cells, that decreases cellular Mn and prevents Mn toxicity. Manganese (Mn) through the human body and its competition with iron (Fe) transport is depicted in this sketch. Mn and Fe are taken in through our diet. Although DMT1 is shown not to be required for Mn2+ uptake into the enterocytes, it likely facilitates intestinal absorption, especially at times of iron deficiency, when DMT1 is upregulated. From the enterocytes Mn moves through the portal circulation to the liver, where it enters the hepatocytes via SLC39A14. Excess Mn is exported from the hepatocytes into the bile via SLC30A10 and returns to the intestine, where it is excreted with the feces, while a small amount may re-enter the entero-hepatic circulation. When Mn2+ is oxidized to Mn+3, it needs to be bound to a carrier protein in the circulation, such as transferrin. It is then taken up via transferrin receptor mediated endocytosis or via SLC39A14 into cells. SLC39A14 also facilitates Mn transport from the circulation to enterocytes, from which it can be exported to the intestinal lumen via SLC30A10. In hypermanganesemia, unbound Mn2+ enters the brain via the epithelial cells of the choroid plexus. Manganese competes with iron for its transporters and carrier proteins, so in times of iron deficiency, manganese transport and deposition into cells, without a functional exporter SLC30A10, is exacerbated (Figure created with BioRender.com).
When Mn cannot be excreted from the liver and digestive system, as is the case in patients with SLC30A10 pathogenic loss-of-function variants, it will be deposited into the liver, kidney, pancreas, bone, and brain (13). Within the brain, Mn will accumulate into the globus pallidus, putamen, caudate, midbrain, cerebellum, subthalamic and dentate nucleus, while typically sparing the ventral pons and thalamus. The excess Mn in the cells leads to oxidative stress, endoplasmic reticulum stress, mitochondrial dysfunction, and apoptosis (28). Consequently, patients with Mn overload due to mutations in SLC30A10, develop neurological deficits, liver dysfunction, and erythrocytosis.
While the excess Mn deposition in the brain and liver explains the neurological deficits and liver dysfunction, the mechanism causing erythrocytosis is not yet clear. We found that the patient’s erythropoietin was inappropriately increased for his concurrent hematocrit before treatment (Figure 1B) indicating that his erythrocytosis was secondary, likely due to disrupted hypoxia-sensing pathway. Some in vitro data support this hypothesis: the human hepatoma cell line Hep3B produces erythropoietin upon incubation with manganese (29). Moreover, high Mn in culture of primary human hepatocytes activates HIF1/HIF2 by blocking their prolyl hydroxylation and consequently their degradation (30). Further insights into the mechanism connecting SLC30A10 deficiency to erythrocytosis were recently provided by work in Slc30a10-/- mice available currently in a preprint (31). Although this data may be considered as preliminary, since still in a preprint, provide an interesting possibility for the mechanism of erythrocytosis in SLC30A10 deficiency: erythropoietin (Epo) mRNA was shown to be increased in the liver but decreased in the kidneys of these mice with Mn overload. Hif2α antisense oligonucleotides (ASOs) and hepatocyte-specific Hif2α deficiency, via an albumin promoter-driven Cre recombinase transgene (Alb-Cre), were shown to decrease liver Epo mRNA levels in Slc30a10-/- mice, indicating that erythropoietin is induced by Hif2α. In contrast, Hif1α ASOs and Alb-Cre mediated Hif1α deficiency did not affect the increased Epo expression in the liver of these mice. It is proposed that SLC30A10 deficiency leading to excess Mn deposition in the liver, induces increased HIF2α activity, compatible with a sense of tissue hypoxia, stimulating liver erythropoietin production and consequently erythrocytosis (31).
In 2008, the first two patients with hypermanganesemia, dystonia, polycythemia, and cirrhosis (HMDPC) were described in the literature (7). By 2012, the etiology of HMDPC was discovered to be loss-of-function biallelic mutations of the manganese transporter SLC30A10, causing excess Mn deposition in liver and brain. This autosomal recessive disease is the first known inherited disorder of manganese metabolism (16). Patients typically present between 1-15 years of age with symptoms of dystonia, dysarthria, or bradykinesia (6, 7, 10, 11, 14–23) as summarized in Table 1. A patient with the same homozygous SLC30A10 variant as our patient presented at 14 years of age with spastic paraparesis without extrapyramidal symptoms (14, 24). Liver impairment varies between patients ranging from no involvement to transaminitis and direct hyperbilirubinemia to cirrhosis and typically follows the neurological presentation, so occasionally patients are identified with dystonia without any liver involvement (18). Erythrocytosis is a consistent finding early in the disease course, before any liver or brain manifestations (16), and is associated with elevated erythropoietin and low iron stores as indicated by low ferritin and elevated total iron binding capacity (14). The presentation of symptoms appears to follow a similar pattern with a variability in the age of presentation ranging from 1 to 15 years of age. A significant exception was noted in two siblings homozygous for SLC30A10 p.Gln412Argfs∗26, who presented in their late 40s and 50s with progressive gait disturbance, postural instability, rigidity, and adult-onset parkinsonism (15). The pathogenic variant in this family caused frameshift fairly close to the carboxyterminal end of this 486-aminoacid-protein, possibly allowing some preservation of the function of the transporter.
It was fortunate that our patient presented at age 7 with incidentally found erythrocytosis and without any symptoms of dystonia or liver disease on presentation. Despite an extensive standard workup for erythrocytosis (Figure 1A), the etiology of his erythrocytosis was unclear until WES-trio revealed homozygous SLC30A10 mutations. We now screen for hypermanganesemia in young children presenting with erythrocytosis and the erythrocytosis gene sequencing panel in Cincinnati Children’s includes SLC30A10 as one of the genes that causes erythrocytosis. Although a rare disease, it is important to keep hypermagnesemia, dystonia, polycythemia, and cirrhosis in the differential diagnosis of genetic causes for erythrocytosis as early recognition and treatment can prevent devastating morbidity and early mortality. Phlebotomy should be avoided in these patients as iron deficiency will only worsen manganese absorption and deposition in tissues. Instead, patients should be treated with chelation therapy early in their disease course to avoid irreversible damage to their liver and brain.
Our case report highlights a rare cause of erythrocytosis due to mutations in SLC30A10, a manganese efflux transporter that prevents excess manganese deposition in organs. Our erythrocytosis gene sequencing panel now includes SLC30A10 to be tested for patients with erythrocytosis. Treatment should focus on chelation of the manganese and iron supplementation. Iron supplementation is needed to avoid further manganese absorption. Phlebotomy should be avoided in these patients as it will worsen the manganese absorption. Patients will need close monitoring of their hemoglobin and serum manganese to track response to treatment.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Cincinnati Children’s Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
TC: Conceptualization, Data curation, Visualization, Writing – original draft, Writing – review & editing. HH: Data curation, Writing – original draft, Writing – review & editing. PF: Data curation, Writing – original draft, Writing – review & editing, Visualization. JH: Writing – original draft, Writing – review & editing. SW: Data curation, Formal analysis, Visualization, Writing – review & editing. CP: Data curation, Formal analysis, Writing – review & editing. AM: Data curation, Formal analysis, Writing – review & editing. WZ: Conceptualization, Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. TK: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by the National Institutes of Health, National Heart, Lung, and Blood Institute grant R01HL152099 and by the Cincinnati Children’s Hospital Center for Pediatric Genomics.
We acknowledge Dr. Janice Staber from the University of Iowa Stead Family Children’s Hospital, Dr. Nicholas Newman and Dr. William Balistreri from Cincinnati Children’s for useful discussions and valuable input on chelation choices and hypermanganesemia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Cario H, McMullin MF, Bento C, Pospisilova D, Percy MJ, Hussein K, et al. Erythrocytosis in children and adolescents-classification, characterization, and consensus recommendations for the diagnostic approach. Pediatr Blood Cancer. (2013) 60:1734–8. doi: 10.1002/pbc.24625
3. Tuschl K, Clayton PT, Gospe SM Jr, Mills PB. Hypermanganesemia with dystonia 1. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, editors. GeneReviews((R)). Seattle (WA (1993).
4. Walshe JM. Treatment of Wilson’s disease with trientine (triethylene tetramine) dihydrochloride. Lancet. (1982) 1:643–7. doi: 10.1016/S0140-6736(82)92201-2
5. Park HK, Kim SM, Choi CG, Lee MC, Chung SJ. Effect of trientine on manganese intoxication in a patient with acquired hepatocerebral degeneration. Mov Disord. (2008) 23:768–70. doi: 10.1002/mds.21957
6. Stamelou M, Tuschl K, Chong WK, Burroughs AK, Mills PB, Bhatia KP, et al. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: a new treatable disorder. Mov Disord. (2012) 27:1317–22. doi: 10.1002/mds.25138
7. Tuschl K, Mills PB, Parsons H, Malone M, Fowler D, Bitner-Glindzicz M, et al. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia–a new metabolic disorder. J Inherit Metab Dis. (2008) 31:151–63. doi: 10.1007/s10545-008-0813-1
8. Schilsky ML, Roberts EA, Bronstein JM, Dhawan A, Hamilton JP, Rivard AM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: Executive summary of the 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. (2023) 77:1428–55. doi: 10.1002/hep.32805
9. Tuschl K, Clayton PT, Gospe SM Jr., Gulab S, Ibrahim S, Singhi P, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. (2016) 99:521. doi: 10.1016/j.ajhg.2016.07.015
10. Mukhtiar K, Ibrahim S, Tuschl K, Mills P. Hypermanganesemia with Dystonia, Polycythemia and Cirrhosis (HMDPC) due to mutation in the SLC30A10 gene. Brain Dev. (2016) 38:862–5. doi: 10.1016/j.braindev.2016.04.005
11. Avelino MA, Fusao EF, Pedroso JL, Arita JH, Ribeiro RT, Pinho RS, et al. Inherited manganism: the “cock-walk” gait and typical neuroimaging features. J Neurol Sci. (2014) 341:150–2. doi: 10.1016/j.jns.2014.03.057
12. Sata F, Araki S, Murata K, Aono H. Behavior of heavy metals in human urine and blood following calcium disodium ethylenediamine tetraacetate injection: observations in metal workers. J Toxicol Environ Health A. (1998) 54:167–78. doi: 10.1080/009841098158881
13. Liu Q, Barker S, Knutson MD. Iron and manganese transport in mammalian systems. Biochim Biophys Acta Mol Cell Res. (2021) 1868:118890. doi: 10.1016/j.bbamcr.2020.118890
14. Tuschl K, Clayton PT, Gospe SM Jr., Gulab S, Ibrahim S, Singhi P, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. (2012) 90:457–66. doi: 10.1016/j.ajhg.2012.01.018
15. Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. (2012) 90:467–77. doi: 10.1016/j.ajhg.2012.01.017
16. Quadri M, Kamate M, Sharma S, Olgiati S, Graafland J, Breedveld GJ, et al. Manganese transport disorder: novel SLC30A10 mutations and early phenotypes. Mov Disord. (2015) 30:996–1001. doi: 10.1002/mds.26202
17. Zaki MS, Issa MY, Elbendary HM, El-Karaksy H, Hosny H, Ghobrial C, et al. Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: Six novel SLC30A10 mutations and further phenotype delineation. Clin Genet. (2018) 93:905–12. doi: 10.1111/cge.13184
18. Tavasoli A, Arjmandi Rafsanjani K, Hemmati S, Mojbafan M, Zarei E, Hosseini S. A case of dystonia with polycythemia and hypermanganesemia caused by SLC30A10 mutation: a treatable inborn error of manganese metabolism. BMC Pediatr. (2019) 19:229. doi: 10.1186/s12887-019-1611-7
19. Yapici Z, Tuschl K, Eraksoy M. Hypermanganesemia with dystonia 1: A novel mutation and response to iron supplementation. Mov Disord Clin Pract. (2020) 7:94–6. doi: 10.1002/mdc3.12861
20. Jagadish S, Howard L, Thati Ganganna S. Atypical presentation of SLC30A10 gene mutation with hypermanganesemia, seizures and polycythemia. Epilepsy Behav Rep. (2021) 16:100505. doi: 10.1016/j.ebr.2021.100505
21. Cherian A, Priya L, Divya KP. “Cock-walk” gait and “horseshoe moustache” sign on MRI in inherited hypermanganesemia. Neurol Sci. (2022) 43:1441–5. doi: 10.1007/s10072-021-05793-z
22. Garg D, Yoganathan S, Shamim U, Mankad K, Gulati P, Bonifati V, et al. Clinical profile and treatment outcomes of hypermanganesemia with dystonia 1 and 2 among 27 Indian children. Mov Disord Clin Pract. (2022) 9:886–99. doi: 10.1002/mdc3.13516
23. Tabatabaee SN, Effat Nejad S, Nikkhah A, Hashemi N, Alavi A, Lang AE, et al. Familial hypermanganesemia in Iran. Mov Disord Clin Pract. (2023) 10:850–3. doi: 10.1002/mdc3.13723
24. Gospe SM Jr., Caruso RD, Clegg MS, Keen CL, Pimstone NR, Ducore JM, et al. Paraparesis, hypermanganesaemia, and polycythaemia: a novel presentation of cirrhosis. Arch Dis Child. (2000) 83:439–42. doi: 10.1136/adc.83.5.439
25. Aschner M, Guilarte TR, Schneider JS, Zheng W. Manganese: recent advances in understanding its transport and neurotoxicity. Toxicol Appl Pharmacol. (2007) 221:131–47. doi: 10.1016/j.taap.2007.03.001
26. Shawki A, Anthony SR, Nose Y, Engevik MA, Niespodzany EJ, Barrientos T, et al. Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am J Physiol Gastrointest Liver Physiol. (2015) 309:G635–47. doi: 10.1152/ajpgi.00160.2015
27. Ye Q, Park JE, Gugnani K, Betharia S, Pino-Figueroa A, Kim J. Influence of iron metabolism on manganese transport and toxicity. Metallomics. (2017) 9:1028–46. doi: 10.1039/C7MT00079K
28. Chen P, Bornhorst J, Aschner M. Manganese metabolism in humans. Front Biosci (Landmark Ed). (2018) 23:1655–79. doi: 10.2741/4665
29. Goldberg MA, Dunning SP, Bunn HF. Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science. (1988) 242:1412–5. doi: 10.1126/science.2849206
30. Liu C, Jursa T, Aschner M, Smith DR, Mukhopadhyay S. Up-regulation of the manganese transporter SLC30A10 by hypoxia-inducible factors defines a homeostatic response to manganese toxicity. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2107673118
Keywords: erythrocytosis, SLC30A10, trientine, hypoxia-sensing pathway, case report
Citation: Coppola T, Hughes H, Finch PT, Hess JA, Wu S, Prada CE, Miethke AG, Zhang W and Kalfa TA (2024) Case report: Childhood erythrocytosis due to hypermanganesemia caused by homozygous SLC30A10 mutation. Front. Hematol. 3:1331485. doi: 10.3389/frhem.2024.1331485
Received: 01 November 2023; Accepted: 05 February 2024;
Published: 06 March 2024.
Edited by:
Anna Rita Migliaccio, Campus Bio-Medico University, ItalyReviewed by:
Paola Bianchi, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, ItalyCopyright © 2024 Coppola, Hughes, Finch, Hess, Wu, Prada, Miethke, Zhang and Kalfa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theodosia A. Kalfa, dGhlb2Rvc2lhLmthbGZhQGNjaG1jLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.