 Al-Ola Abdallah1*
Al-Ola Abdallah1* Alexander Lesokhin2
Alexander Lesokhin2 Tomasz Wrobel3
Tomasz Wrobel3 Krzysztof Jamroziak4
Krzysztof Jamroziak4 Dominik Dytfeld5
Dominik Dytfeld5 Cyrille Touzeau6
Cyrille Touzeau6 Attaya Suvannasankha7
Attaya Suvannasankha7 Xavier Leleu8
Xavier Leleu8 Rebecca Silbermann9
Rebecca Silbermann9 Abdullah M. Khan10
Abdullah M. Khan10 Shaji Kumar11Morie Gertz11Jacob P. Laubach12Ying-Ming Jou13
Shaji Kumar11Morie Gertz11Jacob P. Laubach12Ying-Ming Jou13 Merav Bar13Prianka Das13
Merav Bar13Prianka Das13 Yu Wang13
Yu Wang13 Korey Demers13Nicholas Stong13Deepak Perumal13Ross La Motte-Mohs13Kylee MacLachlan2
Korey Demers13Nicholas Stong13Deepak Perumal13Ross La Motte-Mohs13Kylee MacLachlan2 Meletios-Athanasios Dimopoulos14
Meletios-Athanasios Dimopoulos14- 1Division of Hematologic Malignancies & Cellular Therapeutics, University of Kansas Cancer Center, Westwood, KS, United States
- 2Myeloma Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 3Department of Hematology, Wroclaw Medical University, Wroclaw, Poland
- 4Department of Hematology, Medical University of Warsaw, Warsaw, Poland
- 5Department of Hematology and Bone Marrow Transplantation, Poznań University of Medical Sciences, Poznań, Poland
- 6Department of Hematology, University Hospital of Nantes, Nantes, France
- 7Department of Medicine, Indiana University School of Medicine and Richard L Roudebush Veterans' Administration Medical Center (VAMC), Indianapolis, IN, United States
- 8Department of Haematology, Hôpital La Mileterie, Poitiers, France
- 9Division of Hematology/Medical Oncology, Oregon Health and Science University, Portland, OR, United States
- 10Division of Hematology, Ohio State University Comprehensive Cancer Center, Columbus, OH, United States
- 11Division of Hematology, Mayo Clinic Transplant Center, Rochester, MN, United States
- 12Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
- 13Bristol Myers Squibb Company, Princeton, NJ, United States
- 14Department of Clinical Therapeutics, National and Kapodistrian University, Athens, Greece
Purpose: The phase I/II trial CheckMate 039 (NCT01592370) evaluated the safety, tolerability, and efficacy of nivolumab-daratumumab (ND) in patients with relapsed/refractory multiple myeloma (RRMM).
Methods: Patients with RRMM were randomized to receive ND with or without pomalidomide-dexamethasone (Pd) in cohort A and ND or D monotherapy in cohort B. The primary endpoint was safety. Secondary endpoints included minimal residual disease (MRD) negativity status, overall response rate (ORR), duration of response, and progression-free survival (PFS).
Results: Cohort A (n = 11) was terminated early due to safety concerns observed with immunomodulatory agents and checkpoint inhibitor combinations in other clinical trials. In the small number of patients treated in cohort A, no new safety concerns were observed but patients who received NDPd had numerically more grade 3/4 adverse events (AEs) and serious AEs compared with ND. Grade 3/4 AEs occurring in ≥ 1 patient in the ND group was anemia (3/6 patients); in the NDPd group, these were neutropenia (3/5 patients), upper respiratory tract infection (2/6 patients), and pneumonia (2/6 patients). In cohort B (n = 63), AE rates were similar between ND and D (any-grade: 87.8% vs 95.5%; grade 3/4: 53.7% vs 45.5%). Grade 3/4 AEs occurring in ≥ 1 patient in the ND group were neutropenia (19.5%), anemia (9.8%), thrombocytopenia (9.8%), and bronchitis (7.3%); in the D group these were anemia and pneumonia (both 9.1%). Immune-mediated AEs for ND were consistent with the known safety profile of nivolumab. In cohort A, all patients (5/5) receiving NDPd and 4/6 receiving ND achieved a response. In cohort B, the ORR with ND was numerically higher than D (22/41 [53.7%] vs 9/22 [40.9%]) and both groups had a median PFS of 6.6 months. ND also showed promising MRD negativity results (next-generation sequencing 10-5, 24.0%; next-generation flow 10-5, 22.2%).
Conclusion: NDPd demonstrated no new safety signals and encouraging efficacy despite its early termination. ND was well tolerated with a manageable toxicity and few AEs leading to discontinuation, and demonstrated a numerically higher ORR but equivalent PFS compared with D. Any clinical benefits to OS require a longer follow-up.
Clinical trial registration: https://clinicaltrials.gov/, identifier NCT01592370.
1 Introduction
Improved outcomes have been observed in patients with multiple myeloma (MM) who have received combination therapy treatment. The management of newly diagnosed and early-line relapse MM has significantly improved in the past decade due to the introduction of immunomodulatory agents, proteasome inhibitors (PIs), and monoclonal antibodies (mAbs) (1, 2). Despite this, MM remains incurable and inevitably progresses to a relapsing-remitting course with remission periods becoming shorter and outcomes worsening following each relapse (3, 4). Therefore, there is a need for novel combinations to improve outcomes for patients with relapsed/refractory MM (RRMM).
Immune checkpoint inhibitors targeting programmed death-1 (PD-1) have been explored in MM, driven by unprecedented clinical outcomes in solid tumors and Hodgkin lymphoma (5). PD-1 mediates inhibitory signals on T cells upon ligand binding leading to impaired host antitumor immune response (6). Nivolumab is a fully human IgG4 mAb, optimized with an S228P mutation to prevent FAB-arm exchange, that acts as a PD-1 checkpoint inhibitor, disrupting engagement between the PD-1 receptor and its ligands PD-L1 and PD-L2 (5, 7). Disruption of the PD-1/PD-L1/-L2 axis reduces the inhibition of the immune system, including antitumor responses, mediated by the PD-1 pathway (5). Nivolumab may have therapeutic potential in MM due to frequent expression of PD-L1 by MM cells and subpopulations of immune cells, and has been shown to play a role in mediating inhibition of immunity in the bone marrow milieu of patients with MM (8–10). However, PD-1 pathway blockade in MM has not demonstrated efficacy as a monotherapy (11). The lack of clinically meaningful efficacy has multiple proposed mechanisms, including poor antigen presentation, lack of agonistic signaling, immune suppressive cells, low mutational burden of MM cells, and senescence of tumor-specific T cells in the tumor microenvironment (12). When used as combination therapy, previous early-stage trials using an alternative PD-1 inhibitor, pembrolizumab, in combination with either lenalidomide or pomalidomide and dexamethasone, demonstrated promising efficacy and safety results (13, 14). However, safety signals identified in the subsequent phase III KEYNOTE-183 and -185 trials, which used a combination checkpoint inhibitor agent plus immunomodulatory drug regimen, resulted in early termination due to an unfavorable benefit–risk profile (15, 16).
The introduction of mAbs, especially daratumumab, changed the treatment landscape for patients with MM (1). Daratumumab is a human IgG1 mAb that mediates its killing effect of CD38-expressing MM cells through antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis, and direct apoptosis via cross-linking (17, 18). An additional part of the efficacy of daratumumab may be attributed to the reduction of immune suppression from CD38 cells while enhancing T-cell immunity (19). Daratumumab is approved as part of combination regimens for patients with newly diagnosed MM, and as a monotherapy or part of combination regimens for patients with RRMM (2, 20, 21). The rationale for combining daratumumab with nivolumab is partially based on observations from other studies where the upregulation of CD38 on T cells resulted in PD-1/PD-L1 blockade resistance (19, 22), and it was hypothesized that combining nivolumab and daratumumab may overcome daratumumab and/or nivolumab resistance. As the addition of pomalidomide and dexamethasone to daratumumab has demonstrated a good efficacy response (23, 24), it was hypothesized that adding pomalidomide and dexamethasone to nivolumab and daratumumab may enhance these effects, with potential synergistic and potentiating effects that may result in improved clinical benefit.
CheckMate 039 (cohorts A and B) was designed to determine the safety and efficacy of nivolumab-daratumumab combinations in patients with RRMM. Here we report data on the safety and preliminary efficacy of nivolumab-daratumumab (ND) and nivolumab-daratumumab-pomalidomide-dexamethasone (NDPd) combination regimens for the treatment of patients with RRMM. Biomarker data from ND and daratumumab monotherapy (D) treatments are also presented.
2 Methods
2.1 Study design and patients
CheckMate 039 was a multicenter, randomized, open-label phase I/II trial (NCT01592370). In cohort A, patients were aged ≥ 18 years, had received ≥ 2 prior lines of therapy, and had disease that was refractory to lenalidomide and a PI and refractory to the last line of treatment. In cohort B, patients were aged ≥ 18 years, had received ≥ 3 prior lines of therapy including an immunomodulatory agent and a PI, or were refractory to an immunomodulatory agent and a PI. Patients also had ECOG PS scores of 0 or 1, consented to bone marrow aspirate or biopsy, and had measurable disease. Patients were excluded if they had received prior therapy with a PD-1 inhibitor, other checkpoint inhibitor or anti-CD38 mAb, had active plasma cell leukemia, or had a history of central nervous system involvement.
This trial was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines. Approval was received from the Institutional Review Board and Independent Ethics Committee, and all patients provided written informed consent.
2.2 Study treatments
Patients in cohort A were randomized 1:1 to receive NDPd or ND until recruitment was terminated in light of the safety concerns arising from other trials using an immunomodulatory agent with a PD-1 inhibitor (15, 16). Cohort B was subsequently opened, with patients randomly assigned 2:1 to receive ND or D (Figure 1). Randomization in both cohorts was performed using an interactive voice response system after informed consent was obtained and screening was complete. Treatment was administered in 28-day cycles until disease progression, unacceptable toxicity, withdrawal of consent, or the patient met other discontinuation criteria.
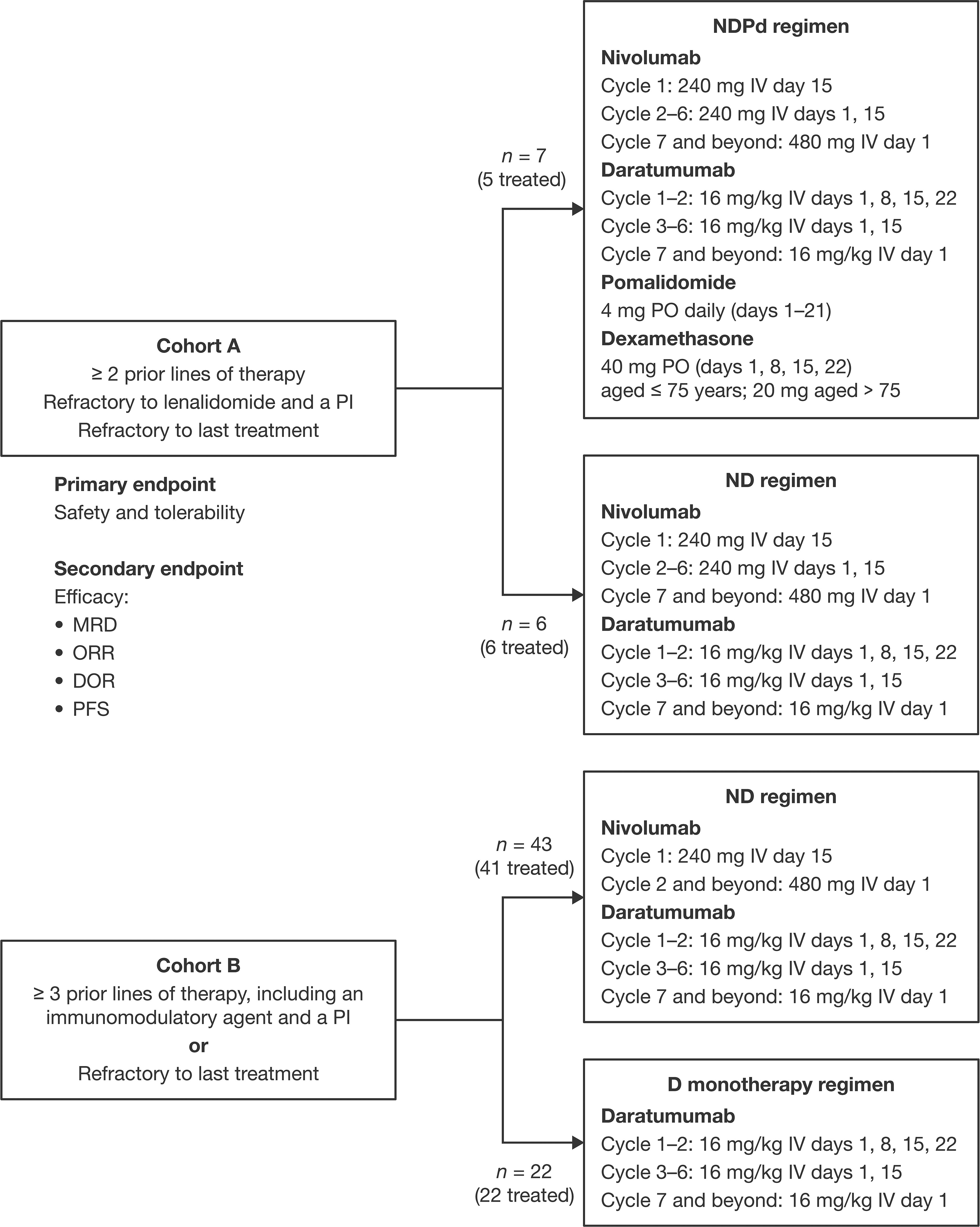
Figure 1 Study design. D, daratumumab monotherapy; DOR, duration of response; IV, intravenous; MRD, minimal residual disease; ND, nivolumab-daratumumab; NDPd, nivolumab-daratumumab-pomalidomide-dexamethasone; ORR, overall response rate; PI, proteasome inhibitor; PFS, progression-free survival; PO, orally.
In cohort A, patients in the ND group received nivolumab 240 mg IV on day 15 in cycle 1, days 1 and 15 in cycles 2–6, and 480 mg IV on day 1 of each subsequent cycle. Daratumumab 16 mg/kg IV was administered on days 1, 8, 15, and 22 in cycles 1 and 2, days 1 and 15 in cycles 3–6, and on day 1 of each subsequent cycle. Patients in the NDPd group received the same ND dosing, plus pomalidomide 4 mg orally (PO) daily on days 1–21 of each cycle and dexamethasone 40 mg PO (20 mg for patients aged > 75 years) on days 1, 8, 15, and 22, in weeks daratumumab was not administered, or 20 mg IV prior to daratumumab and 20 mg PO following daratumumab (16 mg IV and 4 mg PO for patients aged > 75 years).
In cohort B, patients in the ND group received nivolumab 240 mg IV on day 15 of cycle 1, and 480 mg IV on day 1 of cycle 2 and each subsequent cycle. Daratumumab was given as 16 mg/kg IV on days 1, 8, 15, and 22 of cycles 1 and 2, days 1 and 15 of cycles 3–6, and on day 1 of each subsequent cycle. Patients could opt to receive the first daratumumab dose split over 2 days (8 mg/kg for days 1 and 2 of cycle 1). Patients in the D group received the same daratumumab dosing as those in the ND group.
2.3 Endpoints
The primary endpoint was the number of adverse events (AEs), serious adverse events (SAEs), and immune-mediated AEs (IMAEs) from the time of informed consent up to 100 days after the last dose of the study drug. The secondary endpoints were minimal residual disease (MRD) negativity status, overall response rate (ORR), duration of response (DOR), and progression-free survival (PFS). Overall survival (OS) was considered exploratory. Biomarkers were considered exploratory and retrospective.
2.4 Assessments
Safety assessments included assessments of AEs, clinical laboratory tests (hematology, chemistry), vital sign measurements, and physical examination with assessment of ECOG PS. Vital signs, AEs, and laboratory tests were assessed on days 1, 8, 15, and 22 of cycles 1 and 2, days 1 and 15 of cycles 3–6, and day 1 of each subsequent cycle. Physical examination was performed on day 1 of each cycle.
Efficacy was assessed per International Myeloma Working Group criteria and was based on analysis of molecular and cytometry MRD, serum and urine protein electrophoresis (SPEP and UPEP) with immunofixation, serum free light chain (sFLC) (for patients with sFLC-only disease), corrected calcium (serum calcium and serum albumin), imaging, and bone marrow assessments. MRD, SPEP, UPEP, and sFLC assessments were done centrally, while bone lesion, extramedullary plasmacytoma, bone marrow disease, and corrected calcium assessments were done at local laboratories. MRD was assessed (sensitivity level of 10-5) from bone marrow aspirates collected at screening (next-generation sequencing [NGS]), day 1 of cycle 4 or at achievement of very good partial response (VGPR) or better (whichever occurred first) (NGS and next-generation flow [NGF]), and every 6 cycles until disease progression (NGS and NGF). All other assessments were done on day 1 of every cycle until progression, even if the patient was on subsequent therapy.
2.5 Biomarkers
Sanger method sequencing for Fc gamma receptor (FcγR) single-nucleotide polymorphisms (SNP), whole exome sequencing (WES), RNA-sequencing, gene signature score (GES), and flow cytometry were used to determine potential biomarkers (Supplemental Methods).
2.6 Statistical analysis
The ‘treated’ population included all patients who received ≥ 1 dose of nivolumab and/or daratumumab; the ‘biomarker’ population included all ‘treated’ patients with available biomarker data. An additional ‘pooled ND’ population was analyzed post hoc and encompassed all patients who received ND from cohorts A and B. Patient demographics and baseline characteristics were collected at screening and summarized.
AEs were coded using MedDRA 23.1 and graded according to NCI CTCAE version 4. The exact ORR, DOR, and PFS with a corresponding 95% confidence interval (CI) were determined per treatment group, with the best overall response tabulated. Kaplan–Meier methodology was used to estimate median DOR and PFS and their 95% CIs. MRD analysis included the frequency of MRD negativity, time to negativity, and persistent negativity for ≥ 6 or 12 months.
For the biomarker analysis, the association between baseline gene expression was evaluated by Spearman’s correlation coefficient in patients who received ND or D. The GES used median score as the cutoff to define GES.high and GES.low. The association between OS/PFS and GES was evaluated by Cox regression model for patients who received ND.
The sample size of both cohorts was not powered for statistical hypothesis testing as this was a phase I/II trial to evaluate the safety profile and potential clinical benefit of ND combination therapy.
3 Results
3.1 Patient disposition
Thirteen patients from 5 US sites were randomized in cohort A and 11 were treated (ND, n = 6; NDPd, n = 5); 2 patients failed to meet eligibility criteria (history of primary biliary cirrhosis, n = 1; administrative reason by sponsor, n = 1) and were not treated. At data cutoff (September 25, 2020) there was 1 patient in each group continuing with treatment; reasons for discontinuing treatment included disease progression (ND, n = 3; NDPd, n = 3), patient withdrawal from study (ND, n = 1; NDPd, n = 1), and maximum clinical benefit (ND, n = 1).
A total of 65 patients from 10 sites in the USA, Poland, France, Belgium, and Greece were randomized in cohort B; 63 were treated (ND, n = 41; D, n = 22). Two randomized patients were not treated due to failing to meet eligibility criteria (ECOG PS > 1, n = 1) and investigator decision (patient not eligible for further treatment after bronchopneumonia, n = 1). At data cutoff, 7 patients receiving ND and 6 receiving D were on active study treatment. Most patients in cohort B discontinued treatment due to disease progression (ND, n = 31, 75.6%; D, n = 14, 63.6%).
3.2 Baseline patient demographics and disease characteristics
Baseline demographics and disease characteristics were generally balanced between treatment groups in both cohorts (Table 1; Table S1). In cohort A, patients had a median time from diagnosis of 3.2 years, received a median of 2 (range, 2–7) prior lines of therapy, and 63.6% were double refractory to lenalidomide and a PI. In cohort B, patients had a median time from diagnosis of 5.4 years, received a median of 3 (range, 1–7) prior lines of therapy, and 71.4% were double refractory to lenalidomide and a PI.
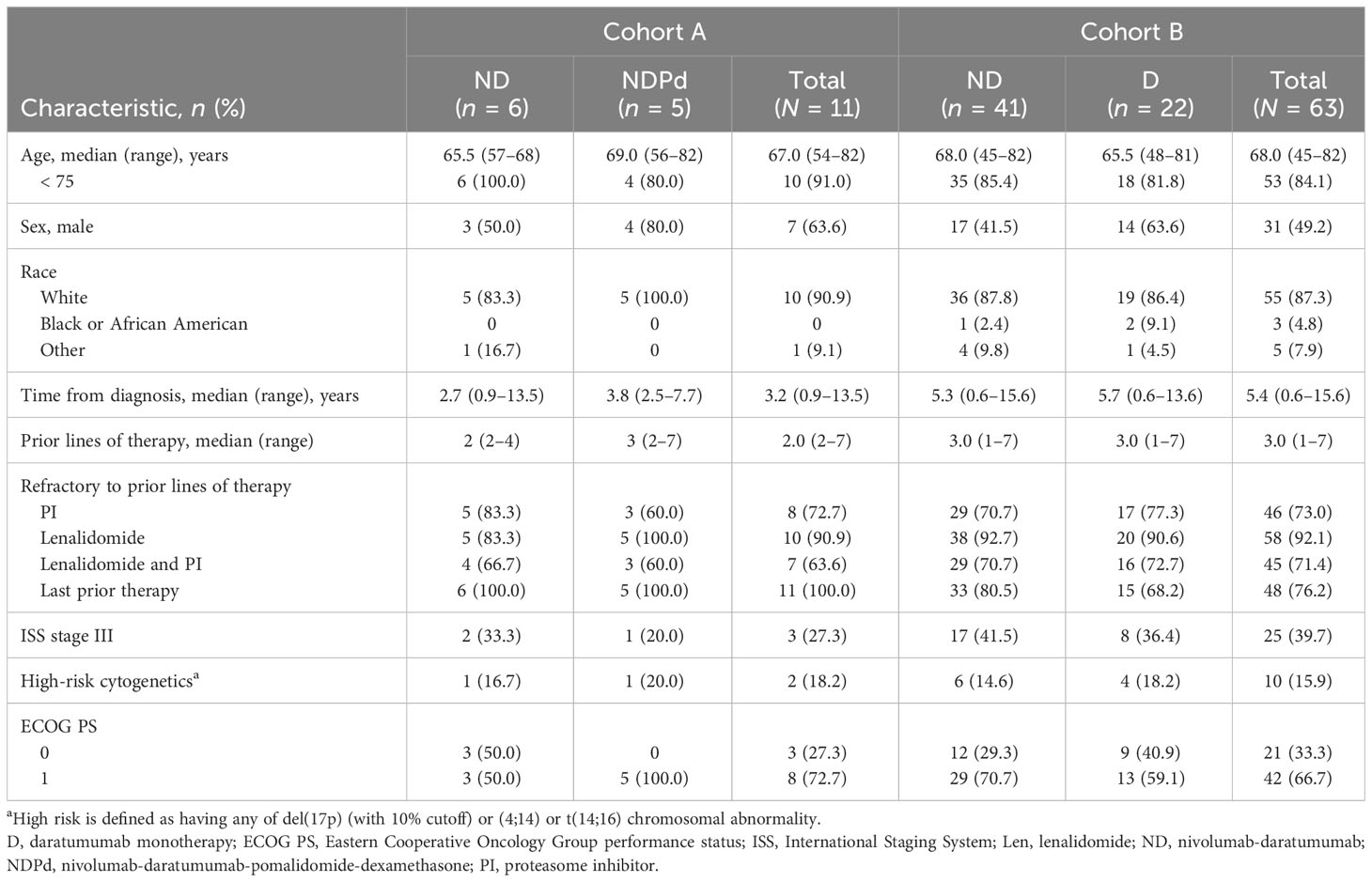
Table 1 Baseline demographics and disease characteristics.
Among pooled patients who received ND (n = 47), median time since diagnosis was 4.8 (range, 0.6–15.6) years, and 70.2% were double refractory to lenalidomide and a PI (Table S2).
3.3 Duration of treatment
In cohort A, patients in the ND group had a median duration of treatment of 26.1 and 28.1 weeks for nivolumab and daratumumab, respectively. The duration of treatment was longer for patients in the NDPd group, with a median of 68.3, 73.0, and 71.3 weeks of treatment for nivolumab, daratumumab, and pomalidomide, respectively. In cohort B, the ND group had a median duration of treatment of 30.1 and 32.1 weeks for nivolumab and daratumumab, respectively, and the D group had a median duration of treatment of 22.6 weeks.
3.4 Safety
In cohort A, all patients who received ND (n = 6) or NDPd (n = 5) experienced an AE of any grade (Table 2). Grade 3/4 AEs were reported in 3/6 patients who received ND and all 5 patients who received NDPd. The most common grade 3/4 AEs reported were anemia (3/6 patients) in the ND group and neutropenia (3/5 patients) in the NDPd group. Drug-related grade 3/4 AEs were reported for 2/6 patients in the ND group, who experienced 3 AEs (anemia, n = 1; neutropenia, n = 1; thrombocytopenia, n = 1) and all 5 patients in the NDPd group (including neutropenia, n = 3; upper respiratory tract infection, n = 2; and reduced neutrophil count, n = 2). SAEs were reported for 2/6 patients in the ND group, all of which were grade 3/4, and included pneumonia (n = 1), MM progression (n = 1), and dyspnea (n = 1); these were not considered drug related. All 5 patients who received NDPd experienced SAEs, of which 4 patients experienced grade 3/4 SAEs with only pneumonia (n = 2) occurring in more than 1 patient. None of these SAEs led to treatment discontinuation. Two patients experienced drug-related SAEs, which included peripheral edema (n = 1), pneumonia (n = 1), and upper respiratory tract infection (n = 1). IMAEs were experienced by patients in both treatment groups, the most common being 2 patients in each group experiencing infusion-related reactions. No AEs leading to discontinuation and no grade 5 AEs were reported in cohort A. One patient in the ND group died due to disease progression.
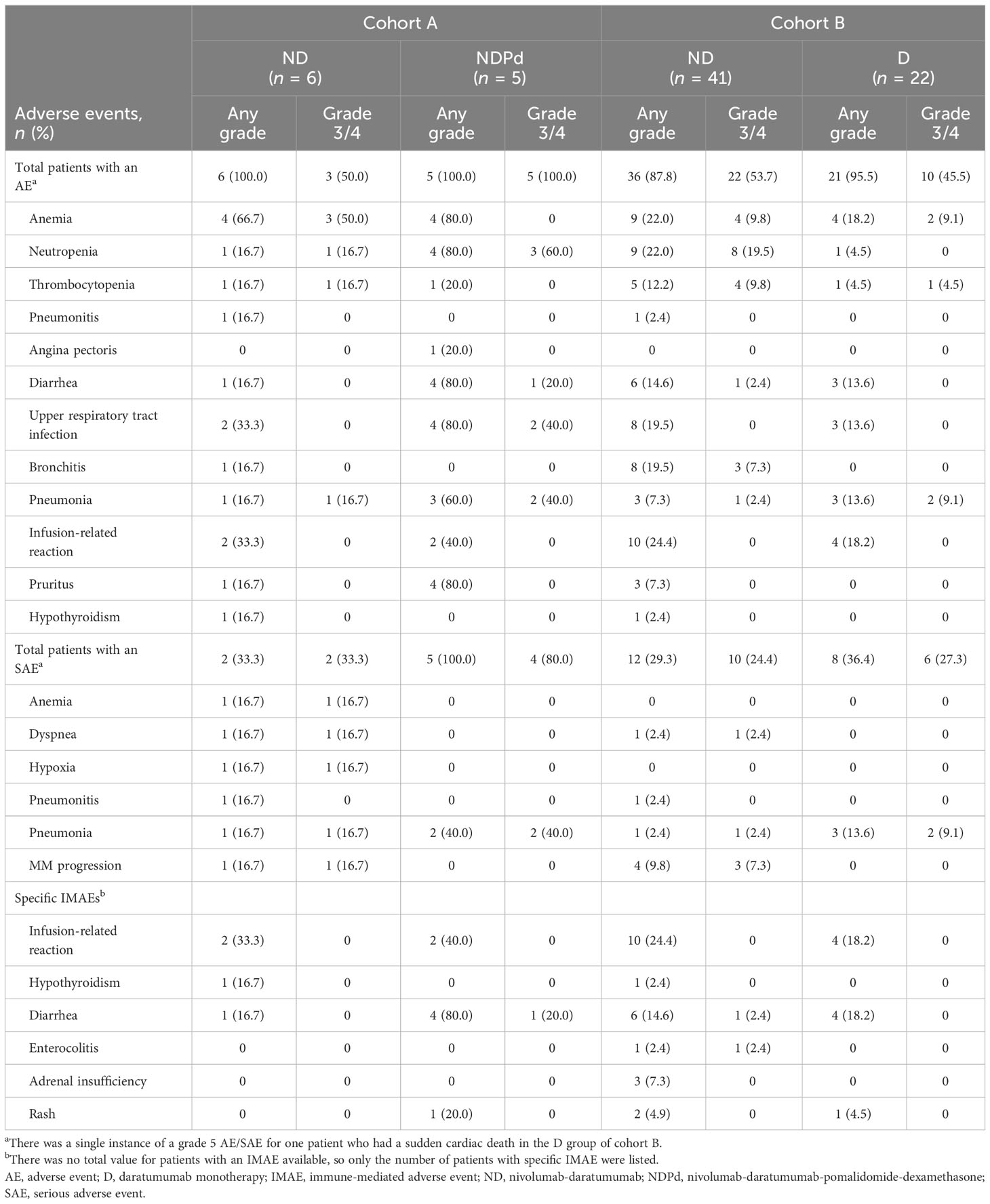
Table 2 Select adverse events reported for patients in cohorts A and B.
In cohort B, 36 (87.8%) patients who received ND and 21 (95.5%) who received D experienced AEs of any grade. Grade 3/4 AEs were reported in 22 (53.7%) patients in the ND group and 10 (45.5%) patients in the D group. The most common grade 3/4 AEs reported for the ND group were neutropenia (19.5%), thrombocytopenia (9.8%), and anemia (9.8%); in the D group these were anemia and pneumonia (each 9.1%). There were 11 (26.8%) patients in the ND group and 2 (9.1%) in the D group with drug-related grade 3/4 AEs. With ND, these included blood disorders (17.1% [9.8% neutropenia, 7.3% thrombocytopenia, 2.4% anemia, 2.4% granulocytopenia, and 2.4% lymphopenia]), reduced neutrophil count (2.4%), and vertigo (2.4%); with D, these included pneumonia (4.5%) and reduced lymphocyte count (4.5%). Grade 3/4 SAEs were reported in 24.4% of patients in the ND group and 27.3% of patients in the D group. The most common grade 3/4 SAEs reported in the ND and D group were infections (ND group, 14.6% [7.3% bronchitis]; D group, 13.6% [9.1% pneumonia]). There was one grade 3/4 drug-related SAE in each group (ND, vertigo; D, pneumonia). One patient in the D group had a grade 5 sudden cardiac death. IMAEs were recorded in both groups, with 1 grade 3/4 IMAE of enterocolitis reported for the ND group initially treated with antibiotics and an antifungal, followed by steroids. Treatment was not reattempted as enterocolitis developed about 6 weeks after coming off treatment due to disease progression. IMAEs of infusion-related reactions (grade 1/2) were reported for 10 patients in the ND group and 2 patients in the D group, and included chills, cough, shortness of breath, and flushing. AEs for 4 patients in the ND group (due to disease progression) and 2 patients in the D group (pneumonia, n = 1; pulmonary embolism, n = 1) led to study drug discontinuation. There were 8 deaths in the ND group, 7 from disease progression and 1 from influenza A, and 6 deaths in the D group due to disease progression (n = 3), sudden cardiac death (n = 1), multi-organ failure (n = 1), and pulmonary embolism (n = 1). None of the deaths were considered related to the study treatment, as assessed by the investigator.
The safety data for the pooled ND-treated population reflected the AE rates reported for the individual cohorts (Table S4).
3.5 Efficacy
In cohort A, the median follow-up duration was 30.6 months for ND and 41.5 months for NDPd. Four out of 6 treated patients in the ND group achieved a response, while all 5 patients in the NDPd group responded, with 1 patient in each group having achieved a complete response (CR) (Table 3). A median PFS (mPFS) of 7.6 months was achieved with ND and 17.0 months with NDPd; median DOR and OS were not reached, and the 1-year OS was 100% in both treatment groups. However, due to the small patient numbers, interpretation of the PFS and OS data should be undertaken with caution. With ND, 1 patient died at 22.5 months, and the other 5 patients were censored at 2.0, 15.4, 38.7, 42.5, and 43.7 months. With NDPd, no deaths were reported, and all were censored after 3 years (range, 37.3–44.5 months).
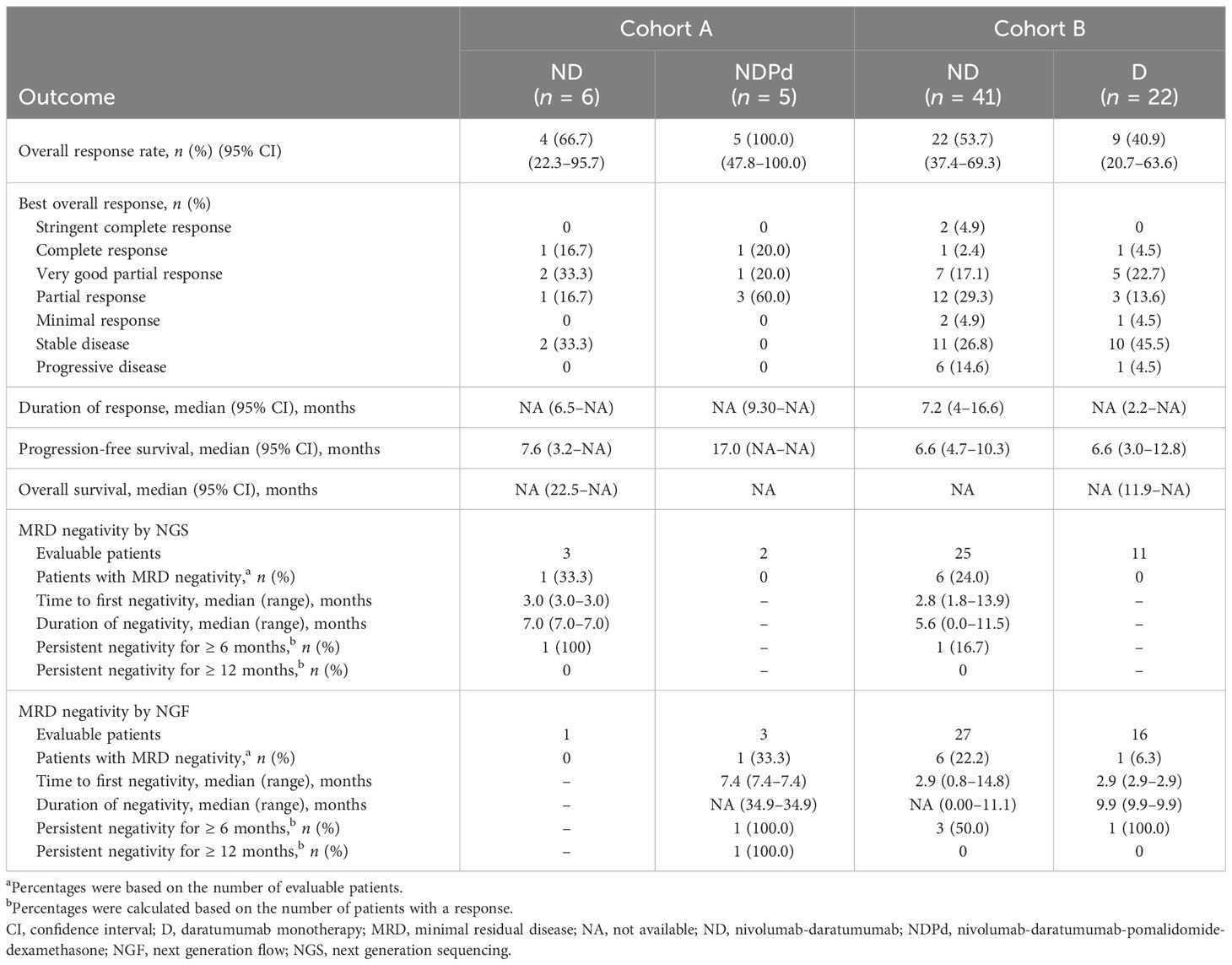
Table 3 Efficacy results for cohort A and cohort B.
In the ND group, MRD negativity was observed in 1 of 3 NGS MRD-evaluable patients but was not observed in the single NGF MRD-evaluable patient. In the NDPd group, MRD negativity was not observed in either of the 2 NGS MRD-evaluable patients but was observed in 1 of 3 NGF MRD-evaluable patients. MRD negativity persisted for 7 months in the ND patient and 35 months in the NDPd patient.
In cohort B, the median duration of follow-up was 14.3 months for ND and 12.6 months for D. Patients who received ND achieved an ORR of 53.7%, with 10/41 (24.4%) having achieved deep responses of ≥ VGPR (Table 3). The median DOR among the 22 responders was 7.2 (95% CI, 4.0–16.6) months. Patients in the D group achieved an ORR of 40.9%, with 6/22 (27.3%) having achieved deep responses of ≥ VGPR. The median DOR among the 9 responders was not reached. mPFS was 6.6 months in both groups (Table 3; Figure 2). Median OS was not reached, and the 1-year OS was 81.3% (95% CI, 64.6–90.6) and 71.2% (95% CI, 46.6–86.0) in the ND and D groups, respectively. Survival data beyond 1 year are not reported as the minimum follow-up for cohort B was only 1 year. There were 8/41 (19.5%) patients who received ND and 6/22 (27.3%) who received D who had died at the time of the data cutoff.
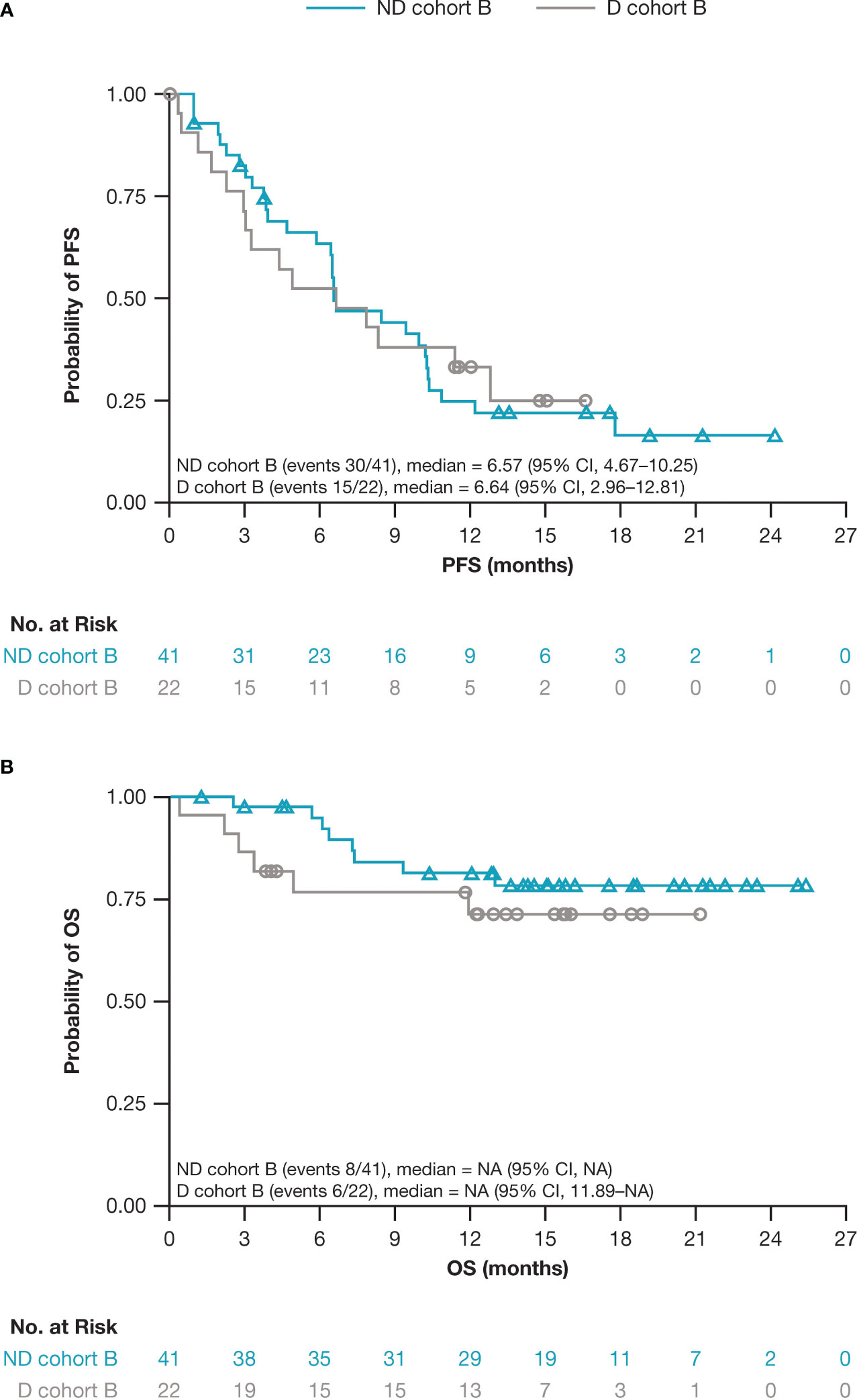
Figure 2 Kaplan–Meier estimates for (A) PFS and (B) OS for patients who received ND versus D in cohort B. CI, confidence interval; D, daratumumab monotherapy; NA, not available; ND, nivolumab-daratumumab; OS, overall survival; PFS, progression-free survival.
In the ND group, MRD negativity was observed in 6/25 (24.0%) NGS MRD-evaluable patients and in 6/27 (22.2%) NGF MRD-evaluable patients (MRD negativity by both NGS and NGF was documented in 3 patients). In the D group, MRD negativity was observed in 0/11 NGS MRD-evaluable patients and in 1/16 (6.3%) NGF MRD-evaluable patients (Table 3). Persistent MRD negativity for ≥ 6 months was observed in 4 patients (1 via NGS, 3 via NGF) in the ND group and 1 patient in the D group (via NGF).
For the pooled ND-treated population, ORR was 55.3%, with 4 patients (8.5%) having achieved a CR/stringent CR (sCR) (Table S3). The median DOR and PFS among the pooled ND population were 7.2 months and 6.6 months, respectively. MRD negativity for evaluable patients was 25.0% when determined by NGS and 21.4% by NGF.
3.6 Biomarkers
Biomarker data were available for a subset of patients treated with ND (n = 47; cohort A, n = 6; cohort B, n = 41) or D (n = 22, cohort B only). Somatic mutation data, obtained from 19 of 47 ND-treated patients, showed that KRAS was the most frequently mutated gene, with more KRAS mutations detected in responders (5/11, 45.5%) than in non-responders (2/8, 25%) (Figure 3A). No KRAS mutations were detected in the D group (data available for 9 of 22 patients). Analysis of RNA gene expression by evaluating gene set enrichment of HALLMARK gene sets from the Molecular Signatures Database (MSigDB; Broad Institute, Inc., Cambridge, MA) in the ND group indicated that HALLMARK.KRAS.UP-high patients showed a trend of better OS compared with HALLMARK.KRAS.UP-low patients, but no trends were associated with PFS (Figure S1). There were no trends identified in the D group. In a similar analysis on pre-treatment tumor samples (ND, n = 14; D, n = 6), statistically significant gene sets that were differentially enriched based on response were analyzed (Figure 3B). Two of the most enriched gene sets in samples from patients who achieved VGPR or better response were the HALLMARK_KRAS_SIGNALLING_UP and HALLMARK_MYC_TARGETS_V2 gene sets. The HALLMARK_KRAS_SIGNALLING_UP gene set was enriched among patients who achieved VGPR or better compared with patients who achieved partial response (PR) or less, which is consistent with the association observed between KRAS mutation and response (Figures 3B, C). The gene set most enriched in VGPR or better pre-treatment samples was HALLMARK_MYC_TARGETS_V2 (Figures 3B, D), while the gene set with the largest negative enrichment score in PR or less pre-treatment samples was ISG_RS (Figures 3B, E).
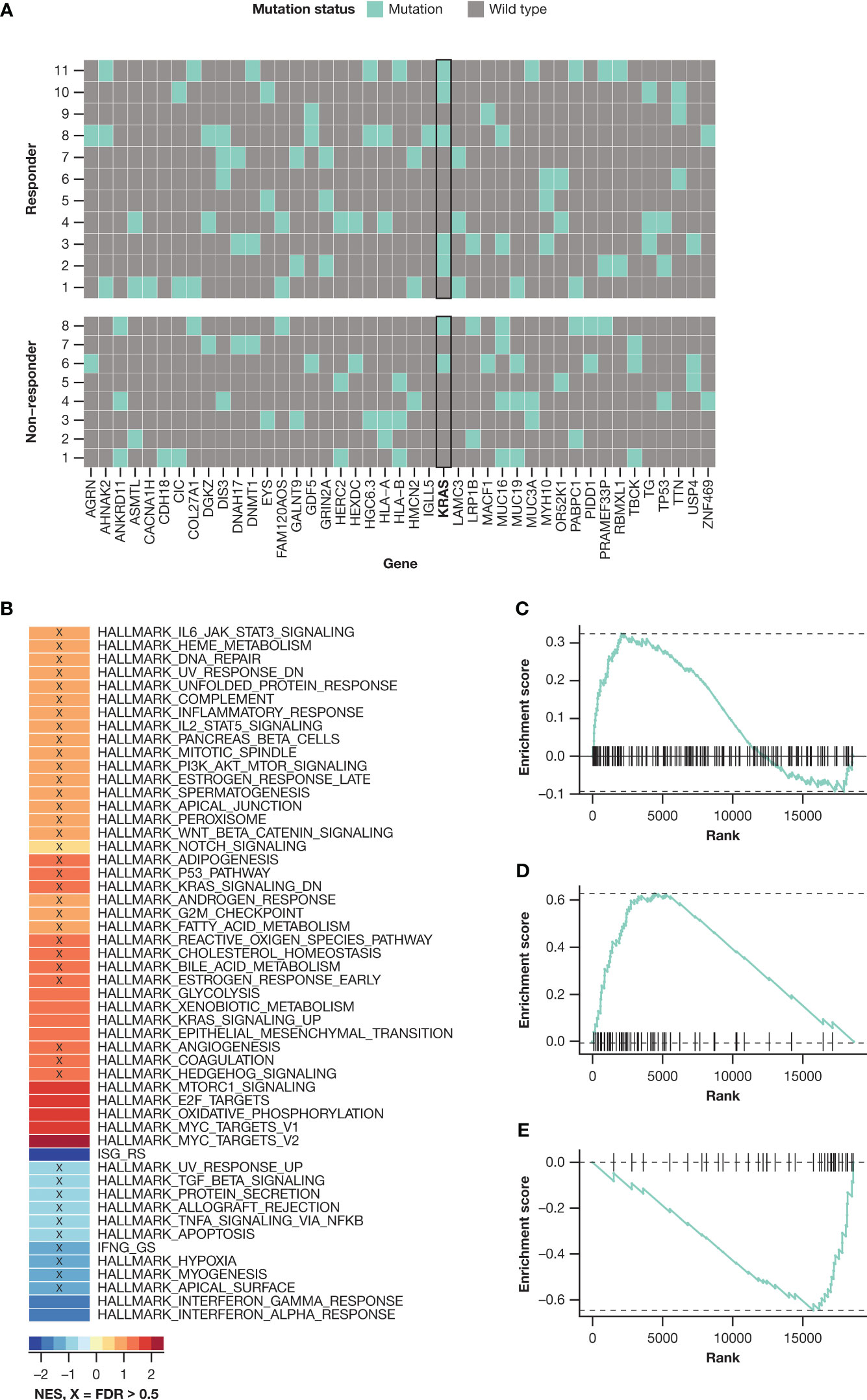
Figure 3 KRAS activation: (A) KRAS mutation prevalence in the ND group; (B) gene set enrichment analysis evaluating Hallmark gene set collections from MSigDB on CD138-selected cells collected at baseline, comparing patients achieving ≥ VGPR with those achieving < VGPR; gene set enrichment graphs for (C) KRAS_SIGNALLING_UP gene set, (D) MYC_TARGETS_V2 gene set, and (E) ISG_RS gene set. FDR, false discovery rate; ND, nivolumab-daratumumab; NES, normalized enrichment score; VGPR, very good partial response.
FcγR polymorphisms showed that patients in the ND group with FCGR2A-131 H/R or FCGR3A-158 F/F genotypes had a slight trend towards better ORR than other FCGR2A-131 or FCGR3A-158 genotypes (Figure S2). However, the analysis was not powered to evaluate associations between FcγR polymorphisms and PFS. No similar ORR trend was observed in the D group. Immunophenotyping analysis by flow cytometry indicated a trend towards natural killer (NK) cell depletion in response to daratumumab in the D and ND groups (data not shown). Single cell RNAseq (scRNAseq) data from peripheral blood mononuclear cells (PBMCs) taken at baseline indicated a higher number of NK cells in responders compared with non-responders in both the ND and D groups (data not shown). There were no significant associations observed between fluorescence in situ hybridization and prognosis, tumor mutational burden and prognosis or other factors, or serum cytokine and clinical response (data not shown).
4 Discussion
In this phase I/II trial of patients with RRMM, ND therapy showed numerically higher ORR and comparable PFS versus D, with an encouraging level of MRD negativity achieved in the ND group of cohort B. Combination therapies were well tolerated, with few AEs leading to discontinuation of treatment and toxicities being manageable and within the expected profile of the individual agents. Despite the small sample size, the addition of pomalidomide and dexamethasone to ND demonstrated an efficacy signal comparable to ND. Although a small number of patients were treated with an immune checkpoint inhibitor and immunomodulatory agent combination (i.e., NDPd) in this study, no new safety concerns were observed in the limited number of patients who were enrolled prior to cohort closure, even with the long follow-up of 41.5 months, despite patients having a numerically higher number of grade 3/4 AEs and SAEs compared with ND.
The ND combination regimens were well tolerated in both cohorts. In cohort A there were no patient discontinuations due to AEs; a single death due to disease progression occurred in the ND group. There were numerically more AEs and SAEs in the NDPd group, most notably cytopenias, infections, diarrhea, and pruritus, compared with the ND group. These differences were likely a reflection of pomalidomide being part of this combination regimen, as these AEs are consistent with its known safety profile (25, 26). While excess cardiac AEs and mortality were identified in the KEYNOTE-183 and -185 trials, there did not appear to be any unifying causes of death or unique AE patterns that could be attributed to the experimental arms of these studies (15, 16, 27). Furthermore, there was no evidence in the KEYNOTE-183 or -185 studies demonstrating that adding pembrolizumab to an immunomodulatory agent accelerated MM progression. Although a limited number of patients were treated with NDPd in cohort A of this study, with a minimum of 37 months, there were no excess grade ≥ 3 cardiac events or deaths reported compared with the immunomodulatory agent-free combination of ND. In this cohort, the safety profile of NDPd was manageable. Overall, these findings do not support a mechanism of action effect due to the checkpoint inhibitor and immunomodulatory agent combination, but due to the small numbers within the NDPd group, interpretation is limited.
In cohort B, the rates of AEs and SAEs were similar between treatment groups, and reflective of the known safety profile of nivolumab and daratumumab. Further, the rate of discontinuations due to the study drugs was low and none of the deaths were related to the study drugs.
All patients in the NDPd group of cohort A (n = 5) responded to treatment, compared with 4/6 (66.7%) patients in the ND group. While these efficacy data are encouraging, no definitive conclusions can be drawn due to the small cohort size (n = 11) and no DPd group as a comparison.
In cohort B, response rates were numerically higher in the ND group compared with the D group, and the ND group had encouraging MRD negativity levels. However, mPFS was comparable between the two groups. The lack of difference in the mPFS, despite the higher ORR with the ND group, was potentially due to only 10/22 (45.5%) patients in the ND group compared with 6/9 (66.7%) in the D group achieving a ≥ VRPR, resulting in a shorter PFS in those who had a PR. The longer mPFS observed in the D group compared with that in the daratumumab monotherapy registrational SIRIUS trial may be attributable to the more favorable baseline disease characteristics of the patients in this study (CheckMate 039 vs SIRIUS: median lines of prior therapy, 3 vs 5; double-refractory patients, 73% vs 95%) (28). In general, this study did not demonstrate that combining nivolumab and daratumumab improves clinical outcome compared with daratumumab monotherapy, despite prior data indicating PD-1/PD-L1 combined with daratumumab could increase responses (29, 30).
In the pooled ND population, ORR and safety data were in line with the individual cohorts, with only minor differences in the number of previous lines of therapy and the dosing of nivolumab. The ORR in the combined ND group in this trial was 55.3%, while the ORR in the D group was 40.9%. A previous trial that evaluated nivolumab monotherapy in 27 patients with MM reported stable disease as the best response achieved (in 63% of patients) (11). These data suggest potential better response rates for the ND combination compared with each agent alone. However, no conclusions can be drawn from this small trial, which was not designed to compare the two regimens.
The biomarker analyses were exploratory and performed retrospectively to assess multiple objectives including disease monitoring, surrogate clinical endpoints, risk stratification, identification of potential subgroups, and response predictions. The main biomarker findings of interest focused on KRAS mutations and RNA expression. With respect to RNA gene expression, HALLMARK_KRAS_SIGNALLING was enriched among responders, with a trend of better OS in HALLMARK.KRAS.UP-high patients compared with HALLMARK.KRAS.UP-low patients in the ND group. Similarly, KRAS was shown to be the most frequently mutated gene, and KRAS mutations were more prevalent in ND responders compared with non-responders. This suggested that either a more heavily mutated KRAS population was controlled just as effectively with ND compared with D alone, the 2:1 randomization for ND:D presented opportunities for more patients with KRAS mutations to enroll to the ND group at trial start, or the data were skewed due to having limited sample numbers. While the slight differences in response rates between ND and D could be related to KRAS mutations detected in the ND group, this is speculative. However, this interpretation is interesting given the role that neoantigens play in generating antitumor activity. Specifically, it has been demonstrated that shared neoantigens are detectable in KRAS in patients with RRMM, with neoantigen-specific T-cell expansion having been associated with antitumor activity in vitro and clinical response in vivo (31). Additionally, a recent study in lung cancer demonstrated that KRAS mutations correlated with superior efficacy of PD-1 inhibitors (32). In addition to HALLMARK_KRAS_SIGNALLING, the enrichment of HALLMARK_MYC_TARGETS_V2 in pre-treatment samples for patients who achieved a VGPR or better is consistent with published data describing MYC regulation of the antitumor immune response through anti-CD47 and PD-L1 (33). In contrast, enrichment of ISG_RS in pre-treatment samples in patients who achieved a PR or less is consistent with data in melanoma and lung cancer demonstrating an association between interferon-stimulated genes and resistance to checkpoint blockade (34).
FCGR2A-131 H/R or FCGR3A-158 F/F FcγR polymorphisms were associated with better ORR in the ND group compared with D. However, the analysis was not powered to evaluate associations between FcγR polymorphisms and PFS. Although FCGR2A/FCGR3A polymorphisms have been demonstrated to influence clinical outcome in patients with MM treated with daratumumab monotherapy (35), and other cancers and interactions with some cancer treatments (36–38), the significance of the association between FCGR2A-131 H/R or FCGR3A-158 F/F FcγR polymorphisms and improved ORR in our study and its effect on PFS are unclear.
Immunophenotyping by flow cytometry did not demonstrate appreciable differences between ND and D (data not shown), consistent with the clinical narrative. However, the differences observed were consistent with the activity of daratumumab on NK cells with no additive effect of nivolumab to daratumumab. Higher baseline NK cell numbers in PBMCs from scRNAseq data also seemed to trend with responders in both ND and D cohorts compared to non-responders, consistent with the activity of daratumumab.
The trial was limited by its small sample size and that it was not powered for statistical comparisons between treatment groups in either cohort. While the results in cohort A suggest promising efficacy with no new safety signals for the NDPd combination, the small sample size and lack of a DPd control group did not allow robust assessment. Additionally, while the median follow-up duration for cohort A was over 30 months, cohort B had a relatively short median follow-up period of just over 12 months. A longer duration of follow-up and further analyses would allow more accurate assessment of PFS, calculation of median DOR and OS, and determine any long-term cumulative side effects. The median OS was not reached for all treatment groups, which limits the ability to compare the regimens to similar combination therapy trials. Finally, the biomarker data were not sufficient to evaluate the potential mechanism of action via which nivolumab may overcome daratumumab resistance.
In conclusion, although recruitment of cohort A was halted due to safety concerns observed in other trials investigating the combination of a PD-1–targeted immune checkpoint inhibitor agent plus an immunomodulatory agent, there were no new safety signals reported with the NDPd combination despite a higher number of grade 3/4 and serious AEs compared with ND. The efficacy observed in the NDPd treatment group was encouraging but limited by the small sample size and lack of a control arm. The ND combinations in both cohorts were well tolerated with manageable toxicity and few AEs leading to discontinuation. The ND combination showed numerically higher ORR but equivalent PFS compared with D, while the OS was not reached. A longer follow-up period will be required to determine any clinical benefits to OS.
Data availability statement
The datasets presented in this article are not readily available in order to protect patient confidentiality and maintain privacy. Bristol Myers Squibb policy on data sharing and how to request access to the datasets may be found at https://www.bms.com/researchers-and-partners/clinical-trials-and-research/disclosure-commitment.html. Requests to access the datasets should be directed to https://www.bms.com/researchers-and-partners/clinical-trials-and-research/disclosure-commitment.html.
Ethics statement
Approval was received from the Institutional Review Board and Independent Ethics Committee at each study site. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
A-OA: Investigation, Writing – review/editing. AL: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review/editing. TW: Investigation, Writing – review/editing. KJ: Investigation, Writing – review/editing. DD: Investigation, Methodology, Writing – review/editing. CT: Investigation, Writing – review/editing. AS: Investigation, Data curation, Formal analysis, Writing – original draft, Writing – review/editing. XL: Conceptualization, Investigation, Writing – review/editing. RS: Investigation, Writing – review/editing. AK: Investigation, Supervision, Writing – review/editing. SK: Investigation, Methodology, Supervision, Writing – review/editing. MG: Investigation, Writing – review/editing. JL: Investigation, Writing – original draft, Writing – review/editing. Y-MJ: Data curation, Formal analysis, Methodology, Validation, Visualization, Writing – review/editing. MB: Data curation, Writing – original draft, Writing – review/editing. PD: Data curation, Writing – original draft, Writing – review/editing. YW: Data curation, Formal analysis, Methodology, Validation, Visualization, Writing – original draft, Writing – review/editing. KD: Data curation, Formal analysis, Methodology, Visualization, Writing – review/editing. NS: Data curation, Formal analysis, Investigation, Methodology. DP: Data curation, Formal analysis, Methodology, Validation, Writing – original draft, Writing – review/editing. RM-M: Data curation, Formal analysis, Methodology, Validation, Writing – original draft, Writing – review/editing. KM: Formal analysis, Visualization, Writing – review/editing. MD: Investigation, Writing – review/editing. All authors contributed to the article and approved the submitted version.
Funding
Support for this study and support for preparation of this manuscript was provided by Bristol Myers Squibb.
Acknowledgments
The authors thank the patients and families who made this study possible and the clinical study teams who participated in the trial. The study was supported by Bristol Myers Squibb. All the authors contributed to and approved the manuscript. Professional medical writing support for this manuscript was provided by Joels Wilson-Nieuwenhuis, PhD, of Caudex, funded by Bristol Myers Squibb.
Conflict of interest
Author AL has received grants from Bristol Myers Squibb to institution and lab and Pfizer; royalties or licenses from Serametrix Inc; served in a consulting or advisory role for Bristol Myers Squibb, Efranat, Genmab, Iteos, Janssen, and Sanofi; personal fees honoraria from Janssen; has a patent planned US20150037346A1; and has received equipment, materials, drugs, medical writing, gifts or other services from Pfizer. Author TW has received grants from Amgen and Roche; personal fees honoraria from AbbVie, Amgen, Beigene, Bristol Myers Squibb, Celgene a Bristol-Myers Squibb company, Gilead, GlaxoSmithKline, Janssen-Cilag, Novartis, Roche, Sanofi, and Takeda; and participated on a data safety monitoring or advisory board for AbbVie, Amgen, Beigene, Bristol Myers Squibb, Celgene a Bristol-Myers Squibb company, Gilead, Janssen-Cilag, Novartis, Roche, Sanofi, and Takeda. Author KJ has received personal fees honoraria from Amgen, Beigene, Bristol Myers Squibb, GlaxoSmithKline, Janssen, Pfizer, Sandoz, and Takeda; payment for expert testimony from Janssen, travel or congress fees from AbbVie; and participated on a data safety monitoring or advisory board for AbbVie, Amgen, Bristol Myers Squibb, GlaxoSmithKline, Janssen, and Takeda. Author DD has received personal fees honoraria from Amgen, Bristol Myers Squibb/Celgene a Bristol-Myers Squibb company, Gilead, Janssen, Novartis, Sandoz, Sanofi, and Takeda; payment from expert testimony from Janssen, travel or congress fees from Amgen, Bristol Myers Squibb/Celgene a Bristol-Myers Squibb company, Gilead, and Janssen; and participated on a data safety monitoring or advisory board for Bristol Myers Squibb/Celgene a Bristol-Myers Squibb company, Janssen, and Takeda. Author CT has received grants, consulting fees, and personal fees honoraria from Bristol Myers Squibb. Author AS has received grants from Bristol Myers Squibb, GlaxoSmithKline, Janssen, Regeneron, and Sutro; consulting fees from Bristol Myers Squibb, GlaxoSmithKline, and Janssen; and participated on a data safety monitoring or advisory board for Janssen. Author XL has received consulting fees from Amgen, Bristol Myers Squibb, GlaxoSmithKline, Janssen, Karyopharm, Pfizer, Merck, Novartis, Roche, Sanofi, and Takeda; and personal fees honoraria from AbbVie, Amgen, Bristol Myers Squibb, Gilead, GlaxoSmithKline, Janssen, Karyopharm, Pfizer, Merck, Novartis, Oseus, Roche, Sanofi, and Takeda. Author RS has received research funding from and participated on a data safety monitoring or advisory board for Janssen and Sanofi-Aventis. Author AK has received grants and consulting fees from SecuraBio and personal fees honoraria from Amgen, Janssen, and Sanofi. Author SK has received grants from AbbVie, Adaptive, Celgene a Bristol-Myers Squibb company, Janssen, KITE, MedImmune/Astra Zeneca, Merck, Novartis, Roche, Sanofi, and Takeda; participated on a data safety monitoring or advisory board or advisory board for AbbVie, Adaptive, Celgene a Bristol-Myers Squibb company, Janssen, KITE, MedImmune/Astra Zeneca, and Takeda; and received payment for participation as part of an independent review committee for Oncopeptides. Author MG has received personal fees from Abbvie, Aptitude Healthgrants, Ashfield, Celgene, i3Health, Ionis/Akcea, Janssen, Johnson & Johnson, Juno, Physicians Education Resource, Prothera, Research for Practice, Sanofi, and Sorrento; and honoraria from Alnylym. Author JL has received presentation honoraria for Great Debates & Updates in Hematologic Malignancies and Grand Rounds. Author Y-MJ is employed by and owns stock or stock options in Bristol Myers Squibb. Author MB is employed by, received travel or congress fees from, and owns stock or stock options in Bristol Myers Squibb. Authors PD and YW were employed by the company Bristol Myers Squibb. Authors KD, NS, and DP were employed by and owns stock or stock options in Bristol Myers Squibb. Author RM-M was employed by the company Bristol Myers Squibb and owns stock or stock options in Bristol Myers Squibb and MacroGenics. Author MD has received honoraria for participation in advisory boards from Amgen, Beigene, Bristol Myers Squibb, Janssen, and Takeda.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author AS declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2023.1244494/full#supplementary-material
References
1. Dimopoulos MA, Moreau P, Terpos E, Mateos MV, Zweegman S, Cook G, et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. (2021) 32:309–22. doi: 10.1016/j.annonc.2020.11.014
2. National Comprehensive Cancer Network. Cancer Network. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Multiple Myeloma v.5.2022. © National Comprehensive Cancer Network, Inc. 2021. All rights reserved. Accessed June 17, 2022. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
3. Mikhael J, Ismaila N, Cheung MC, Costello C, Dhodapkar MV, Kumar S, et al. Treatment of multiple myeloma: ASCO and CCO joint clinical practice guideline. J. Clin. Oncol. (2019) 37:1228–63. doi: 10.1200/JCO.18.02096
4. Zaidi A, Lai M, Cavenagh J. Long-term stabilisation of myeloma with curcumin. BMJ Case Rep. (2017) 2017:bcr2016218148. doi: 10.1136/bcr-2016-218148
5. Bristol Myers Squibb. Opdivo® (nivolumab) prescribing information (2021). Available at: http://packageinserts.bms.com/pi/pi_opdivo.pdf.
6. Boussiotis VA, Chatterjee P, Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. (Sudbury Mass). (2014) 20:265–71. doi: 10.1097/PPO.0000000000000059
7. Silva J-P, Vetterlein O, Jose J, Peters S, Kirby H. The S228P mutation prevents in vivo and in vitro IgG4 Fab-arm exchange as demonstrated using a combination of novel quantitative immunoassays and physiological matrix preparation. J. Biol. Chem. (2015) 290:5462–9. doi: 10.1074/jbc.M114.600973
8. Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia (2013) 27:464–72. doi: 10.1038/leu.2012.213
9. Görgün G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin. Cancer Res. (2015) 21:4607–18. doi: 10.1158/1078-0432.CCR-15-0200
10. Ray A, Das DS, Song Y, Richardson P, Munshi NC, Chauhan D, et al. Targeting PD1-PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia (2015) 29:1441–4. doi: 10.1038/leu.2015.11
11. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic Malignancy: preliminary results of a phase Ib study. J. Clin. Oncol. (2016) 34:2698–704. doi: 10.1200/JCO.2015.65.9789
12. Costa F, Das R, Kini Bailur J, Dhodapkar K, Dhodapkar MV. Checkpoint inhibition in myeloma: opportunities and challenges. Front. Immunol. (2018) 9:2204. doi: 10.3389/fimmu.2018.02204
13. Mateos MV, Orlowski RZ, Siegel DSD, Reece DE, Moreau P, Ocio EM, et al. Pembrolizumab in combination with lenalidomide and lowdose dexamethasone for relapsed/refractory multiple myeloma (RRMM): final efficacy and safety analysis. J. Clin. Oncol. (2016) 34:8010. doi: 10.1200/JCO.2016.34.15_suppl.8010
14. Badros AZ, Kocoglu MH, Ma N, Rapoport AP, Lederer RN, Philip S, et al. A Phase II study of anti PD-1 antibody pembrolizumab, poMalidomide and dexamethasone in patients with relapsed/refractory multiple myeloma (RRMM). Blood (2015) 126:506. doi: 10.1182/blood.V126.23.506.506
15. Usmani SZ, Schjesvold F, Oriol A, Karlin L, Cavo M, Rifkin RM, et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): a randomised, open-label, phase 3 trial. Lancet Haematol. (2018) 6:E448–E58. doi: 10.1016/S2352-3026(19)30109-7
16. Mateos MV, Blacklock H, Schjesvold F, Oriol A, Simpson D, George A, et al. Pembrolizumab plus poMalidomide and dexamethasone for patients with relapsed or refractory multiple myelome (KEYNOTE-183): a randomised, open-label, phase 3 trial. Lancet Haematol. (2019) 379:e459–69. doi: 10.1016/S2352-3026(19)30110-3
17. Van Bueren JL, Jakobs D, Kaldenhoven N, Roza M, Hiddingh S, Meesters J, et al. Direct in vitro comparison of daratumumab with surrogate analogs of CD38 antibodies MOR03087, SAR650984 and Ab79. Blood (2014) 124:3474. doi: 10.1182/blood.V124.21.3474.3474
18. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. (2011) 186:1840–8. doi: 10.4049/jimmunol.1003032
19. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood (2016) 128:384–94. doi: 10.1182/blood-2015-12-687749
20. Janssen. Daratumumab (Darzalex®) prescribing information2021. Available at: https://www.janssenmd.com/pdf/darzalex/DARZALEX_PI.pdf (Accessed April 6, 2021).
21. Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl. J. Med. (2015) 373:1207–19. doi: 10.1056/NEJMoa1506348
22. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discovery (2018) 8:1156–75. doi: 10.1158/2159-8290.CD-17-1033
23. Chari A, Suvannasankha A, Fay JW, Arnulf B, Kaufman JL, Ifthikharuddin JJ, et al. Daratumumab plus poMalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood (2017) 130:974–81. doi: 10.1182/blood-2017-05-785246
24. Siegel DS, Schiller GJ, Samaras C, Sebag M, Berdeja J, Ganguly S, et al. PoMalidomide, dexamethasone, and daratumumab in relapsed refractory multiple myeloma after lenalidomide treatment. Leukemia (2020) 34:3286–97. doi: 10.1038/s41375-020-0813-1
25. Celgene. Imnovid® (poMalidomide) Summary of Product Characteristics2020. Available at: https://www.ema.europa.eu/en/documents/product-information/imnovid-epar-product-information_en.pdf (Accessed June 4, 2021).
26. Celgene. Imnovid® (poMalidomide) Package Leaflet: Information for the patient2020. Available at: https://www.ema.europa.eu/en/documents/product-information/imnovid-epar-product-information_en.pdf (Accessed June 4, 2021).
27. Gormley NJ, Pazdur R. Immunotherapy combination in multiple myeloma - known unknowns. N Engl. J. Med. (2018) 379:1791–5. doi: 10.1056/NEJMp1803602
28. Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet (2016) 387:1551–60. doi: 10.1016/S0140-6736(15)01120-4
29. Franssen LE, Stege CAM, Zweegman S, van de Donk NWCJ, Nijhof IS. Resistance mechanisms towards CD38–directed antibody therapy in multiple myeloma. J. Clin. Med. (2020) 9:1195. doi: 10.3390/jcm9041195
30. Saltarella I, Desantis V, Melaccio A, Solimando AG, Lamanuzzi A, Ria R, et al. Mechanisms of resistance to anti-CD38 daratumumab in multiple myeloma. Cells (2020) 9:167. doi: 10.3390/cells9010167
31. Perumal D, Imai N, Laganà A, Finnigan J, Melnekoff D, Leshchenko VV, et al. Mutation-derived neoantigen-specific T-cell responses in multiple myeloma. Clin. Cancer Res. (2020) 26:450–64. doi: 10.1158/1078-0432.CCR-19-2309
32. Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Letters. (2020) 470:95–105. doi: 10.1016/j.canlet.2019.10.027
33. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Sci. (New York NY). (2016) 352:227–31. doi: 10.1126/science.aac9935
34. Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell (2019) 178:933–48.e14. doi: 10.1016/j.cell.2019.07.019
35. van de Donk NWCJ, Casneuf T, Di Cara A, Parren PW, Zweegman S, van Kessel B, et al. Impact of Fc gamma receptor polymorphisms on efficacy and safety of daratumumab in relapsed/refractory multiple myeloma. Br. J. Haematol. (2019) 184:475–9. doi: 10.1111/bjh.15122
36. Norton N, Olson RM, Pegram M, Tenner K, Ballman KV, Clynes R, et al. Association studies of Fcγ receptor polymorphisms with outcome in HER2+ breast cancer patients treated with trastuzumab in NCCTG (Alliance) Trial N9831. Cancer Immunol. Res. (2014) 2:962–9. doi: 10.1158/2326-6066.CIR-14-0059
37. Roca L, Diéras V, Roché H, Lappartient E, Kerbrat P, Cany L, et al. Correlation of HER2, FCGR2A, and FCGR3A gene polymorphisms with trastuzumab related cardiac toxicity and efficacy in a subgroup of patients from UNICANCER-PACS04 trial. Breast Cancer Res. Treat. (2013) 139:789–800. doi: 10.1007/s10549-013-2587-x
38. Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor–expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J. Clin. Oncol. (2007) 25:3712–8. doi: 10.1200/JCO.2006.08.8021
Keywords: relapsed/refractory, multiple myeloma, phase I/II, nivolumab, daratumumab, pomalidomide, checkpoint inhibition, immunotherapy
Citation: Abdallah A-O, Lesokhin A, Wrobel T, Jamroziak K, Dytfeld D, Touzeau C, Suvannasankha A, Leleu X, Silbermann R, Khan AM, Kumar S, Gertz M, Laubach JP, Jou Y-M, Bar M, Das P, Wang Y, Demers K, Stong N, Perumal D, La Motte-Mohs R, MacLachlan K and Dimopoulos M-A (2023) Nivolumab and daratumumab combination regimens for the treatment of relapsed and refractory multiple myeloma: results of a randomized phase I/II clinical trial. Front. Hematol. 2:1244494. doi: 10.3389/frhem.2023.1244494
Received: 22 June 2023; Accepted: 10 August 2023;
Published: 12 September 2023.
Edited by:
Tamar Tadmor, Technion Israel Institute of Technology, IsraelReviewed by:
Noa Lavi, Rambam Health Care Campus, IsraelEyal Lebel, Hadassah Medical Center, Israel
Reut Harel, Ha’Emek Medical Center, Israel
Copyright © 2023 Abdallah, Lesokhin, Wrobel, Jamroziak, Dytfeld, Touzeau, Suvannasankha, Leleu, Silbermann, Khan, Kumar, Gertz, Laubach, Jou, Bar, Das, Wang, Demers, Stong, Perumal, La Motte-Mohs, MacLachlan and Dimopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Al-Ola Abdallah, QWFiZGFsbGFoQGt1bWMuZWR1