 Candela Ceballos1*
Candela Ceballos1* Mª Cruz Viguria1Carlos Panizo1Juan Roberto Rodríguez-Madoz2,3,4Felipe Prósper2,3,4,5
Mª Cruz Viguria1Carlos Panizo1Juan Roberto Rodríguez-Madoz2,3,4Felipe Prósper2,3,4,5- 1Haematology Department, Hospital Universitario de Donostia, San Sebastián, Spain
- 2Hemato-Oncology Program, Cima Universidad de Navarra, Instituto de Investigación Sanitaria de Navarra (IdiSNA), Pamplona, Spain
- 3Centro de Investigacion Biomedica en Red de Cancer (CIBERONC), Madrid, Spain
- 4Cancer Center Clinica Universidad de Navarra (CCUN), Pamplona, Spain
- 5Hematology and Cell Therapy Department, Clinica Universidad de Navarra, IdiSNA, Pamplona, Spain
Cell therapy utilizing chimeric antigen receptors (CARs) in conjunction with immune cells, primarily T lymphocytes, is known as CAR-T cell therapy. This innovative approach is revolutionizing the landscape of oncohaematology by precisely targeting specific antigens for elimination. However, despite its promising prospects, CAR-T therapy presents several challenges, including a notable rate of disease relapse, intricate pathologies impeding widespread adoption, prolonged manufacturing timelines, and substantial costs. Looking forward, ongoing research and progress aim to address these challenges to mitigate these constraints, underlining the continuous efforts to enhance the efficacy and accessibility of this transformative therapy
1 Introduction
CAR-T therapy has emerged as a promising strategy in oncohematology, precisely steering immune responses toward specific targets for effective treatment. However, despite its immense potential, several challenges, such as disease recurrence rates, intricate pathophysiology of target neoplasms, prolonged manufacturing times, and high costs, underscore the imperative to persevere in research and innovation within this transformative field.
2 CAR-T development: early steps and progress
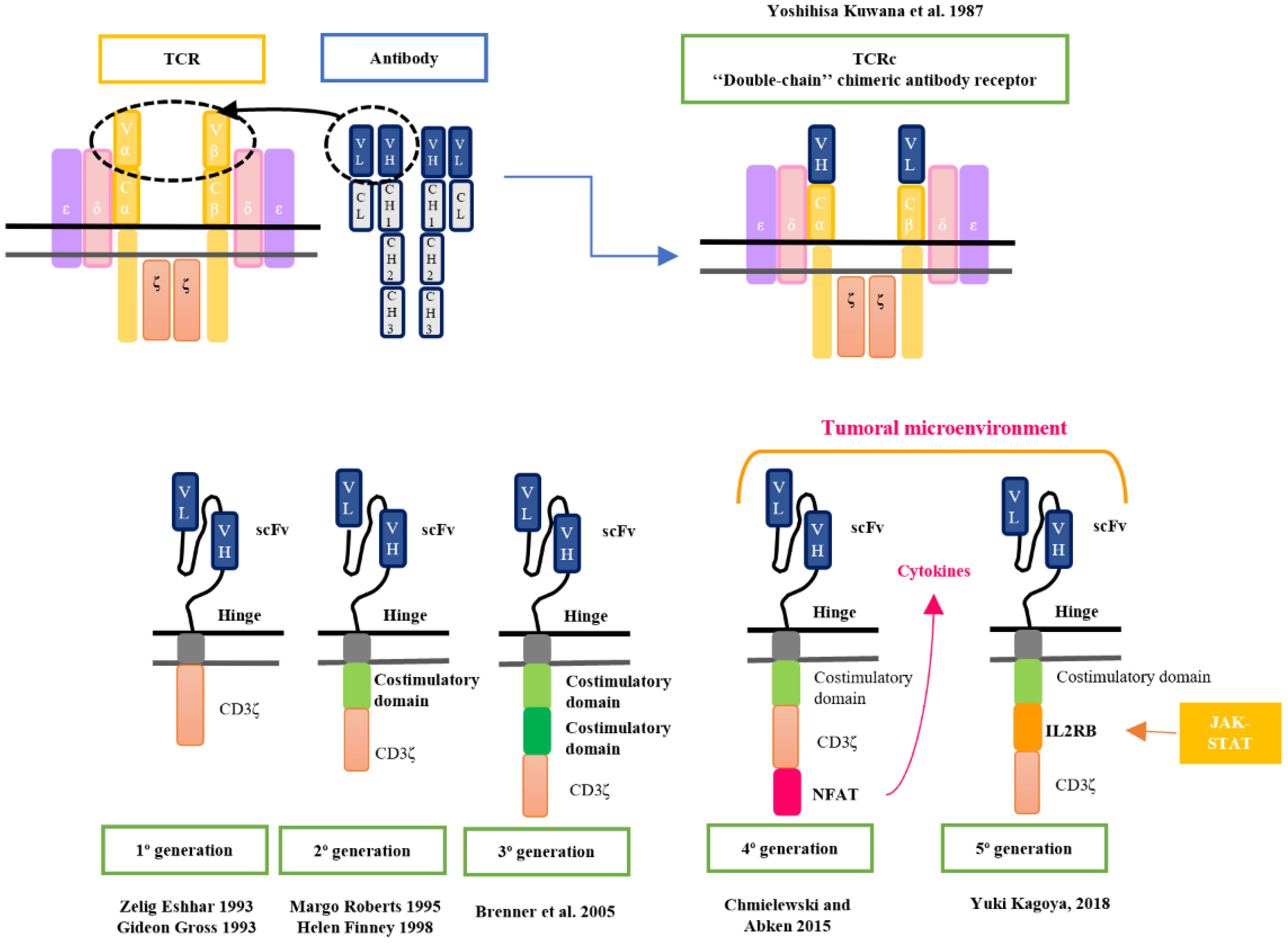
The origins of CAR-T cell therapy can be traced back to 1987 when Kuwana et al. first introduced the concept of HLA-independent antigen recognition through a chimeric receptor (1, 2). In the early 1990s, Arthur Weiss of the University of California (San Francisco) reported that chimeric receptors containing the intracellular signaling domain of CD3ζ, activated T-cell signaling. This work resulted in the binding of the intracellular domains of CD3ζ to the chimeric receptors to transfer the activation signaling to the T cell (3).
Until then, the chimeric receptor (TCR) was composed of an antibody’s variable region domain and the TCR’s constant region domain. However, transduction exhibited low efficiency due to the necessity of introducing two genes into the same lymphocyte using retroviral vectors to encode the chimeric receptor. In response to this issue, in 1993, Zelig Eshhar, Gideon Gross, and colleagues published the structure of a novel chimeric receptor composed of a single-stranded variable fragment (scFv) derived from an antibody bound to the CD3ζ intracellular complex (4). Both immunologists redirected this research towards the field of oncology (5, 6).
In the mid-1990s, significant progress was made by developing the first clinical trial based on CD8+ T cells modified to express a chimeric receptor structure against a surface antigen present in HIV-infected cells. This work demonstrated that the modified CD8+ T cells effectively identified and eliminated infected cells, showing a lytic capacity of 50-60%. Also, it was described that these ScFv-based receptors had a Major Histocompatibility Complex (MHC)-independent recognition, in contrast to initial cTCRs, suggesting the potential development of universal cell therapy (7). Thus, was born the first T cell designed with the chimeric receptor structure of the products currently marketed, called “T body” (8).
The hypothesis of linking the chimeric receptor to other cells of the immune system such as Natural Killer (NK) lymphocytes was proposed in 1993 to explore its HLA-independent lytic activity (4). This response is mediated by the killer immunoglobulin receptor (KIR) and triggered by detecting reduced inhibitory markers or stress markers on the surface of abnormal cells (9). However, currently, compared to CAR-T cells, CAR-NK cell-based adoptive immunotherapy is generally limited by substantial restrictions prior to large-scale practical implementation. These limitations encompass inadequate proliferation and in vivo activation capacity or limited persistence (10, 11).
3 Enhancing CAR-T cell therapy: second and third generations
In response to the non-sustained activation observed in first-generation CAR-T cells, research from 1995 to 1998 delved into the role of co-stimulatory molecules to enhance lymphocyte activation and CAR-T cell persistence. This concept is grounded in the extrapolation of the theory of dual activation signals observed in T-cell antigenic recognition.
During this period, the research published by Sadelain et al. in 1998 showed evidence that CD28 signaling had a substantial impact on the enhanced survival and proliferation of CAR-T cells and an increase of pro-inflammatory cytokines production when they recognized the target. These findings had significant implications for the potential effectiveness of T-cell therapies (12).
Margo Roberts and Helene Finney were the first to develop a 2nd generation CAR-T therapy (CD28-CARs) and achieved a patent in 1997 (Margo Robert’s patent was filed in February 1995 and Helene Finney’s patent was filed in December 1996) (13–15).
In the 2000s, several research was led to study different costimulatory molecules to improve their antileukemic effect. CAR constructs began to be developed with the 4-1BB molecule (CD137), which appeared to provide CAR-T cells with great persistence in the body (16, 17). These studies with second-generation CAR-T cells (CD137 and CD28) focused on targeting the CD19 antigen, which is specific to the B-lymphoid lineage, especially CLL (18) and ALL (16, 17).
In 2005, considering the studies published on both co-stimulatory molecules, Brenner et al. studied third-generation CAR-T consisting of two costimulatory molecules, CD28 and OX40 to improve antileukemic efficiency (19). However, these studies did not demonstrate a therapeutic benefit, but warnings regarding its use have emerged (20) (Figure 1).
4 Bridging the lab to clinical practice: FDA approvals and clinical application
In 2010, a pivotal moment arrived when Steven Rosenberg at the NCI and Carl June and David Porter at the University of Pennsylvania administered CART-19 to a patient with chronic lymphocytic leukemia (CLL), marking a turning point in CAR-T therapy’s trajectory (22).
This therapy underwent a revolution with the case of Emily Whitehead, a 7-year-old diagnosed with acute lymphoblastic leukemia (ALL) refractory to standard treatments. She participated in a phase 1 clinical trial (NCT01626495), receiving an infusion of autologous CTL019, a CAR-T therapy with 4-1BB as a co-stimulatory molecule, similar to the previous studies published in CLL (23). An example of the influence of this case is the foundation created by Emily Whitehead’s family to encourage donations to support oncology research for children’s treatment (24).
Between 2013 and 2014, the first clinical trial results were published and it was not until 2017 that the Food and Drug Administration (FDA) approved Kymriah (tisagenlecleucel) (25) for use in relapsed or refractory ALL after two prior lines for children and young adults up to 25 years old, and Yescarta (axicabtagene ciloleucel) for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) after two or more lines of treatment. The latter study included DLBCL, primary mediastinal lymphoma, high-grade B lymphoma, and DLBCL arising from follicular lymphoma. Axicabtagene ciloleucel is an anti-CD19 CAR-T like tisagenlecleucel but differs in the co-stimulatory molecule as it is made up of CD28 rather than 4-1BB (26, 27).
Since that time until now, various regulatory agencies such as the FDA or the European Medicine Agency (EMA) have approved different products for oncohematological pathologies. For instance, Tecartus (brexucabtagene autoleucel) has been approved for mantle cell lymphoma treatment (28), Breyanzi (lisocabtagene maraleucel) for DLBCL treatment (29) or Abecma (Idecabtagene vicleucel) (30) and Carvykti (ciltacabtagene autoleucel) (31) for multiple myeloma treatment. Additionally, tisagenlecleucel and axicabtagene ciloleucel are seeking to expand their indications to follicular lymphoma treatment through the ELARA (32) and ZUMA-5 (33) clinical trials, respectively. These products have been approved for these pathologies in the relapsed or refractory setting after two or more lines of treatment (Figure 2).
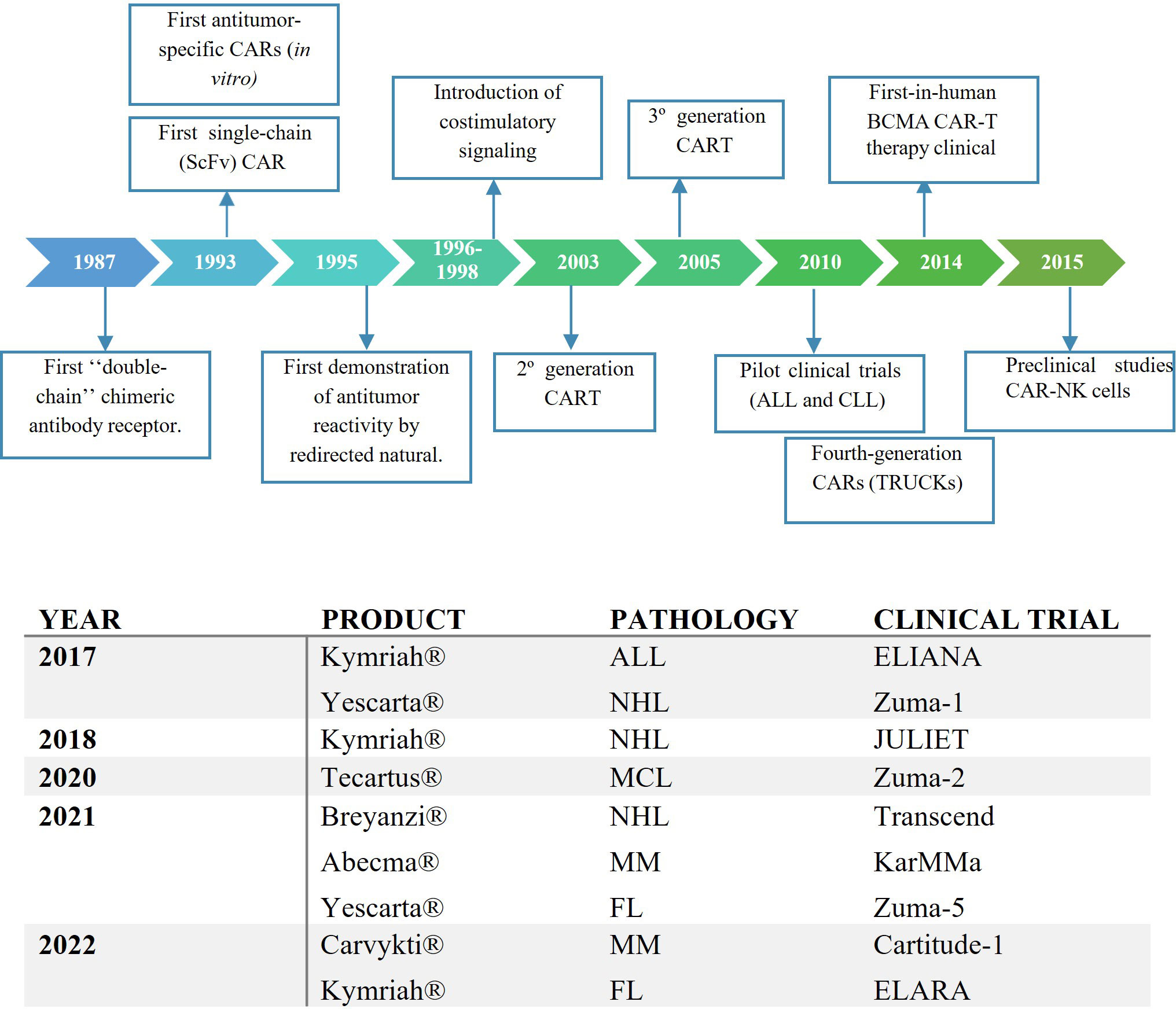
Figure 2 (A) Key preclinical events in the history of the development of CAR-T therapy. (B) The First FDA Approvals for CAR-T Cell Therapies. ALL, acute lymphoblastic leukaemia; NHL, non-Hodgkin lymphoma; ML, Mantle lymphoma; MM, multiple myeloma; FL, follicular lymphoma.
Considering the promising results, several pivotal trials of CAR-T products with extensive experience have been published, aiming to administer them in earlier lines of therapy. The ZUMA-7 (34) and ZUMA-12 (35) trials have reported results of administering axicabtagene ciloleucel in the 2nd and 1st line of treatment, respectively.
However, currently, several limitations related to this therapy have been described.
5 Limitations of CAR-T therapy: an in-depth analysis and exploring solutions
5.1 “On-target/Off tumor” side effect
In the first place, the coexistence of the target antigen in the tumor and healthy cells results in an “on-target/off-tumor” activity. In the case of B cell lymphoid pathologies, CART anti-CD19 cells cause an aplasia in this line, leading to a break in the humoral immune system and, consequently, hypogammaglobulinemia. This condition increases the risk of infection due to reduced levels of antibodies but can be resolved through intravenous immunoglobulin supplementation and antiviral, antibacterial, and antifungal prophylaxis (36, 37), although an increase in fungal infection has not been documented in these patients (38).
An obstacle in CAR-T cell development for myeloid and T-cell malignancies is the concern surrounding “on-target/off-tumor” toxicity. This toxicity in the case of these pathologies produces aplasia in granulocytes and T-cell lineage. However, managing this aplasia presents a significant challenge as effective strategies to mitigate infectious risks remain unidentified. Allogeneic transplantation following CAR-T cell therapy stands as the most explored option (39, 40).
One of the barriers to overcome in T-lymphoid neoplasms is the shared presence of tumor T cells and CAR-T cells, which could lead to CAR-T fratricide at the time of expansion and reduce T cell activity (41, 42). To avoid this, Li et al. designed an anti-CD7 CART, with a built-in command to retain its own CD7 in the endoplasmic reticulum so that it is not expressed on the membrane (39).
5.2 Manufacturing limitations
Currently, the manufacturing process of an autologous CAR-T product takes approximately 15 days (43, 44), although the timeline we handle with the industry from the patient’s apheresis to administration is around 30 days (45).
Many groups are studying methods to minimize manufacturing times without compromising anti-tumor activity (46, 47) by reducing the ex-vivo phase or developing a universal CAR-T therapy (48, 49).
The development of an allogeneic CAR-T or a universal CAR-T would allow administering the product with greater flexibility, as it would not rely on the patient’s apheresis process, and its manufacturing could be initiated in advance.
5.3 Relapse after CAR-T therapy
After CAR-T therapy, disease relapse in the case of ALL or lymphoma can occur in 40-60% of patients at 12 months (50) and the overall survival and event-free survival at 5 years is around 35-45% (51). Among these relapses, 60% still express the target antigen and it is considered that the issue lies in a low persistence or suboptimal functionality of the infused product (25).
5.3.1 Loss of therapeutic efficacy
A decreased functionality or persistence may be caused by exhausted CAR-T cells, defined as more differentiated lymphocytes that exhibit increased expression of cell exhaustion molecules (PD-1, TIM3, LAG-3, TRAIL), making them more susceptible to antigen-induced cell death (AICD) following antigen exposure. Consequently, exhausted T cells experience reduced activity and proliferative capacity, potentially functioning as a detrimental mechanism in the functionality of the CAR-T product (52, 53).
A retrospective study conducted on patients who received CART-19 (both tisagenlecleucel and axicabtagene ciloleucel) reveals that a lower presence of Treg (54) and CAR-T cells predominantly composed of memory T lymphocytes is indicative of a favourable treatment response (55).
Another cause of reduced functionality resulting from an increase in AICD is the high density of CAR on the membrane. This can lead to an increase in tonic signalling and facilitate its elimination (56).
To avoid this kind of relapse there are several projects aimed at selecting less differentiated CAR-T lymphocyte populations (Naïve, Stem Central Memory, and Central Memory) to improve CAR-T persistence (57–59). In addition, allogeneic post-CAR-T transplantation is being considered as a consolidation therapy for patients at higher risk of relapse following this treatment.
5.3.2 Immune evasion
However, around 30-40% of disease relapses present malignant cells that do not express the target antigen and may occur despite detecting the presence of CAR-T cells in peripheral blood. In these cases, there are various hypotheses to explain this mechanism of treatment resistance while being an immune escape from the CART-CD19 attack (41).
One of them is the negative selection that occurred after the CART-CD19 administration. This selection can promote the survival of cellular subclones exhibiting diminished CD19 expression. These subclones acquire a competitive advantage for survival and proliferation in the presence of CAR-T cells, leading to relapse (60).
Another hypothesis is that the loss of CD19 antigen expression may also be associated with changes in the epigenetic regulation of genes. Alterations in DNA methylation or histone modifications could influence the expression of CD19 and contribute to its reduction or changes in the isoform (61).
To address this issue, dual-targeting CAR-T cells are being developed, capable of recognizing different antigens in the tumor, thus preventing cancer cells from escaping therapy by eliminating one of the target antigens. A well-known approach in this field is the use of CAR-T cells targeting both CD19 and CD22 antigens, providing a more effective strategy for treating malignant diseases and reducing the possibility of treatment resistance or evasion (42, 48).
5.4 Early side effects after administration
Rather than these limitations, there are two commonly described side effects related to this therapy in the literature: cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS).
The previously described case of Emily Whitehead not only marked a milestone in demonstrating CAR-T efficacy but also represented the first instance of using tocilizumab in the treatment of CRS. This patient experienced severe side effects, primarily a persistent fever with hemodynamic instability. These inflammatory effects were accompanied by significantly elevated IL-6 levels. The patient’s condition improved after the administration of tocilizumab (62), an anti-IL6 monoclonal antibody recently approved by the FDA for rheumatoid arthritis (63).
CRS is the most common side effect (6, 64, 65) and affects approximately 80% of patients, with roughly 20% experiencing severe cases (45, 51). This condition results from the release of effector cytokines (IFN-g, TNF-a, IL-2) that can trigger the release of proinflammatory cytokines (IL-1, IL-6, IFN-g, IL-10, and monocyte chemoattractant protein-1). Clinical features are fever, hemodynamic instability, and hypoxemia. The treatment for this clinical condition is protocolized and severity-dependent, with tocilizumab (anti-IL-6 monoclonal antibody) proving to be the most effective option.
The most severe cases of CRS, which can even lead to multiorgan failure, are often confused with Macrophage Activation Syndrome (MAS) due to shared clinical features stemming from hyperinflammation. Unlike CRS, these patients do not respond to Tocilizumab administration and require other treatments such as Anakinra (anti-IL1) or corticosteroids. Therefore, it represents a significant diagnostic challenge, and an early diagnosis of this condition has a notable impact on prognosis.
Its pathophysiology is akin to that described in Hemophagocytic Lymphohistiocytosis, characterized by hemophagocytosis-related features such as hepatomegaly, cytopenia, coagulopathy, and multiorgan failure. In response to this challenge, a proposed name change to “Immune Effector Cell-Associated Hemophagocytic Lymphohistiocytosis-Like Syndrome” (IEC-HS) has been suggested, acknowledging its resemblance to hemophagocytic lymphohistiocytosis (HLH) (66).
ICANS affects 20-60% of patients and is characterized by neurological symptoms of varying intensity, including confusion, altered speech, dysarthria, emotional lability, seizures, delirium, or changes in consciousness level. The pathophysiology of ICANS is likely due to the release of inflammatory cytokines that increase vascular permeability and endothelial activation, potentially leading to disruption of the blood-brain barrier, resulting in elevated cytokines in the cerebrospinal fluid and even cerebral edema (36).
Several factors that might contribute to the manifestation of clinical conditions associated with CAR-T cell therapy have been identified. One of the most studied factors is the type of CAR-T product infused. Studies on products like Yescarta, Kymriah, and lisocabtagene maraleucel have revealed incidence rates of cytokine release syndrome (CRS) around 92% (26), 60% (67), and 42% (29), respectively. Furthermore, researchers are actively investigating ways to improve the effectiveness of CAR-T therapy and reduce its side effects after the manufacturing process. These studies cover a range of strategies, such as optimizing the dosage of fludarabine or cyclophosphamide (68) for lymphodepletion therapy and exploring the use of CAR-T administered in divided doses, among other innovative approaches. For instance, at the Hospital Clinic in Barcelona (Spain) they’ve introduced a protocol for administering their academic CAR-T therapy (ARI-002) in two or three divided doses to minimize the risk of side effects (69).
5.5 Solid tumors: microenvironment’s role
Developing CAR-T cells for certain diseases like T-ALL, AML, and solid tumors is posing the greatest challenge. The tumor microenvironment has been identified as a significant factor contributing to tumor escape and resistance in solid organ neoplasms. Strategies involving CAR-T cells that can effectively interact with the tumor microenvironment are being explored. These strategies include the development of CAR-T cells capable of producing cytokines (such as IL12 or IL18) or enhancing their proliferation and activation through specific cytokines present in the tumor stroma. These advanced CAR-T cell designs are often referred to as TRUCKs (T cells Redirected for Universal Cytokine Killing) or “fourth-generation CART cells” and “fifth-generation or next-generation CAR-T”, respectively (70–72).
Initiated in the 1980s, research in this field gained momentum with promising results from pivotal clinical trials of various CAR-T cell therapy products in oncohematology. These scientific advancements represent a significant milestone in cancer treatment, potentially leading to more effective and less side effect therapies in the future. However, it’s crucial to emphasize that there is still much to explore and investigate to address the current limitations.
Author contributions
The article has been written by CC and reviewed by MCV, CP, JRR-M, and FP. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR-T cells. Br. J. Cancer (2019) 120:26–37 doi: 10.1038/s41416-018-0325-1.
2. Kuwana Y, Asakura Y, Nakanishi M, Utsunomiya N, Arata Y, Nagase F, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor derived C regions. Biochem Biophys Res Commun (1987) 149(3):960–8. doi: 10.1016/0006-291X(87)90502-X
3. Irving’ EA, Weisst A. The cytoplasmic domain of the T cell receptor 6 chain is sufficient to couple to receptor-associated signal transduction pathways. Cell (1991) 64(5):891–901. doi: 10.1016/0092-8674(91)90314-O
4. Eshhar Z, Waks T, Grosst G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the y or C subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A (1993) 90(2):720–4. doi: 10.1073/pnas.90.2.720
5. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity (chimeric genes/antibody variable region). Proc Natl Acad Sci U S A (1989) 86(24):10024–8. doi: 10.1073/pnas.86.24.10024
6. Braendstrup P, Levine BL, Ruella M. The long road to the first FDA-approved gene therapy: chimeric antigen receptor T cells targeting CD19. Cytotherapy (2020) 22:57–69. doi: 10.1016/j.jcyt.2019.12.004
7. Roberts MR, Qin L, Zhang D, Smith DH, Tran A-C, Dull TJ, et al. Targeting of human immunodeficiency virus-infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood (1994) 84:2878–89. doi: 10.1182/blood.V84.9.2878.2878
8. Eshhar Z. From the mouse cage to human therapy: A personal perspective of the emergence of T-bodies/Chimeric antigen receptor T cells. Hum. Gene Ther. (2014) 25:773–8. doi: 10.1089/hum.2014.2532
9. Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front. Immunol. (2015) 6:578. doi: 10.3389/fimmu.2015.00578
10. Daher M, Melo Garcia L, Li Y, Rezvani K. CAR-NK cells: the next wave of cellular therapy for cancer. Clin. Transl. Immunol. (2021) 10(4):e1274. doi: 10.1002/cti2.1274
11. Zhang L, Meng Y, Feng X, Han Z. CAR-NK cells for cancer immunotherapy: from bench to bedside. biomark. Res. (2022) 10(1):12. doi: 10.1186/s40364-022-00364-6
12. Krause A, Guo H-F, Latouche J-B, Tan C, Cheung N-KV, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med (1998) 188(4):619–26. doi: 10.1084/jem.188.4.619
13. Roberts M. United States Patent. Chimeric receptor molecules for delivery of co-stimulatory signals. (1997). Available from: https://pubchem.ncbi.nlm.nih.gov/patent/US-5686281-A.
14. Finney H. United States Patent. Cell activation process and reagents therefore. (1997). Available from: https://pubchem.ncbi.nlm.nih.gov/patent/WO-9900494-A3.
15. Finney HM, Lawson ADG, Bebbington CR, Weir ANC. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. (1950) (1998) 161:2791–7.
16. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui C-H, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
17. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. (2009) 17:1453–64 doi: 10.1038/mt.2009.83.
18. Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. (2003) 9:279–86 doi: 10.1038/nm827.
19. Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. (2005) 12:933–41 doi: 10.1016/j.ymthe.2005.04.016.
20. Bourbon E, Ghesquieres H, Bachy E. CAR-T cells, from principle to clinical applications. Bull. Cancer (2021) 108(10S):S4–17. doi: 10.1016/j.bulcan.2021.02.017
21. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR-T cell therapy. Nat. Rev. Clin. Oncol. (2020) 17:147–67 doi: 10.1038/s41571-019-0297-y.
22. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. New Engl. J. Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
23. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. New Engl. J. Med. (2013) 368:1509–18. doi:10.1056/NEJMoa1215134
24. Emily Whitehead foundation. Available at: https://emilywhiteheadfoundation.org/.
25. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Engl. J. Med. (2018) 378:439–48 doi: 10.1056/NEJMoa1709866.
26. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR-T-cell therapy in refractory large B-cell lymphoma. New Engl. J. Med. (2017) 377:2531–44 doi: 10.1056/NEJMoa1707447.
27. Subklewe M, Von Bergwelt-Baildon M, Humpe A. Chimeric antigen receptor T cells: A race to revolutionize cancer therapy. Transfusion Med. Hemother (2019) 46:15–24. doi: 10.1159/000496870
28. Frey NV. Approval of brexucabtagene autoleucel for adults with relapsed and refractory acute lymphocytic leukemia. Blood (2022) 140(1):11–15. doi: 10.1182/blood.2021014892
29. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
30. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. New England Journal of Medicine (2021) 384(8):705–16. doi: 10.1056/NEJMoa2024850
31. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet (2021) 398:314–24. doi: 10.1016/S0140-6736(21)00933-8
32. Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat. Med. (2022) 28:325–32 doi: 10.1038/s41591-021-01622-0.
33. Neelapu SS, Dickinson M, Munoz J, Ulrickson ML, Thieblemont C, Oluwole OO, et al. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: the phase 2 ZUMA-12 trial. Nat. Med. (2022) 28:735–42 doi: 10.1038/s41591-022-01731-4.
34. Locke FL, Miklos DB, Jacobson CA, Perales M-A, Kersten M-J, Oluwole OO, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. New Engl. J. Med. (2022) 386:640–54. doi: 10.1056/NEJMoa2116133
35. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. (2022) 23:91–103. doi: 10.1016/S1470-2045(21)00591-X
36. Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, et al. Management of adults and children receiving CAR-T-cell therapy: 2021 best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann. Oncol. (2022) 33:259–75. doi: 10.1016/j.annonc.2021.12.003
37. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. (2014) 371:1507–17 doi: 10.1056/NEJMoa1407222.
38. Gavriilaki E, Dolgyras P, Dimou-Mpesikli S, Poulopoulou A, Evangelidis P, Evangelidis N, et al. Risk factors, prevalence, and outcomes of invasive fungal disease post hematopoietic cell transplantation and cellular therapies: A retrospective monocenter real-life analysis. Cancers (Basel) (2023) 15:3529. doi: 10.3390/cancers15133529
39. Li Z, An N, Yang K, Meng F, Xu T, Peng X, et al. Donor CD7 chimeric antigen receptor T cell bridging to allogeneic hematopoietic stem cell transplantation for T cell hematologic Malignancy. Transplant. Cell Ther. (2023) 29:167–73 doi: 10.1016/j.jtct.2022.11.013.
40. Fried S, Shouval R, Walji M, Flynn JR, Yerushalmi R, Shem-Tov N, et al. Allogeneic hematopoietic cell transplantation after chimeric antigen receptor T cell therapy in large B cell lymphoma. Transplant. Cell Ther. (2023) 29:99–107. doi: 10.1016/j.jtct.2022.10.026
41. Xu X, Sun Q, Liang X, Chen Z, Zhang X, Zhou X, et al. Mechanisms of relapse after CD19 CAR-T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front. Immunol. (2019) 10:2664. doi: 10.3389/fimmu.2019.02664
42. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR-T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nat. (London) (2019) 568:112–6. doi: 10.1038/s41586-019-1054-1
43. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia (2015) 29:1637–47. doi: 10.1038/leu.2015.52
44. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR-T cell therapy. Mol. Ther. Methods Clin. Dev. (2017) 4:92–101. doi: 10.1016/j.omtm.2016.12.006
45. Westin JR, Kersten MJ, Salles G, Abramson JS, Schuster SJ, Locke FL, et al. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am. J. Hematol. (2021) 96:1295–312. doi: 10.1002/ajh.26301
46. de Macedo Abdo L, Barros LRC, Saldanha Viegas M, Vieira Codeço Marques L, de Sousa Ferreira P, Chicaybam L, et al. Development of CAR-T cell therapy for B-ALL using a point-of-care approach. Oncoimmunology (2020) 9(1):1752592. doi: 10.1080/2162402X.2020.1752592
47. Ghassemi S, Nunez-Cruz S, O’Connor RS, Fraietta JA, Patel PR, Scholler J, et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol. Res. (2018) 6:1100–9 doi: 10.1158/2326-6066.CIR-17-0405.
48. Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin. Cancer Res. (2021) 27:2764–72. doi: 10.1158/1078-0432.CCR-20-3863
49. Li W, Zhu X, Xu Y, Chen J, Zhang H, Yang Z, et al. Simultaneous editing of TCR, HLA-I/II and HLA-E resulted in enhanced universal CAR-T resistance to allo-rejection. Front. Immunol. (2022) 13:1052717. doi: 10.3389/fimmu.2022.1052717
50. Zinzi A, Gaio M, Liguori V, Cagnotta C, Paolino D, Paolisso G, et al. Late relapse after CAR-T cell therapy for adult patients with hematologic Malignancies: A definite evidence from systematic review and meta-analysis on individual data. Pharmacol. Res. (2023) 190:106742. doi: 10.1016/j.phrs.2023.106742
51. Elsallab M, Ellithi M, Hempel S, Abdel-Azim H, Abou-El-Enein M. Long-term response to autologous anti-CD19 chimeric antigen receptor T cells in relapsed or refractory B cell acute lymphoblastic leukemia: a systematic review and meta-analysis. Cancer Gene Ther. (2023) 30:845–54. doi: 10.1038/s41417-023-00593-3
52. Fan M, Li M, Gao L, Geng S, Wang J, Wang Y, et al. Chimeric antigen receptors for adoptive T cell therapy in acute myeloid leukemia. J. Hematol. Oncol. (2017) 10(1):151. doi: 10.1186/s13045-017-0519-7
53. Jain MD, Zhao H, Wang X, Atkins R, Menges M, Reid K, et al. Tumor interferon signaling and suppressive myeloid cells are associated with CAR-T-cell failure in large B-cell lymphoma. Blood (2021) 137(19):2621–2633. doi: 10.1182/blood.2020007445
54. Good Z, Spiegel JY, Sahaf B, Malipatlolla MB, Ehlinger ZJ, Kurra S, et al. Post-infusion CAR-TReg cells identify patients resistant to CD19-CAR-Therapy. Nat. Med. (2022) 28:1860–71 doi: 10.1038/s41591-022-01960-7.
55. Haradhvala NJ, Leick MB, Maurer K, Gohil SH, Larson RC, Yao N, et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med. (2022) 28(9):1848–59. doi: 10.1038/s41591-022-01959-0
56. Rodriguez-Marquez P, Calleja-Cervantes ME, Serrano G, Oliver-Caldes A, Palacios-Berraquero ML, Martin-Mallo A, et al. CAR density influences antitumoral efficacy of BCMA CAR-T cells and correlates with clinical outcome. Sci Adv (2022) 8(39):eabo0514. doi: 10.1101/2022.01.19.22269515
57. Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD81 memory stem cells for the treatment of human B-cell Malignancies. Blood (2016) 128:519–28. doi: 10.1182/blood-2015-11-683847
58. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory selftumor-reactive CD8 T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA (2005) 102(27):9571–6. doi: 10.1073/pnas.0503726102.
59. Magraner X. El Virgen del Rocío arranca el ensayo de su CAR-T para linfoma no-Hodgkin. Redacción Med (2023).
60. Fischer J, Paret C, El Malki K, Alt F, Wingerter A, Neu MA, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J. Immunother (1997) (2017) 40:187–95. doi: 10.1097/CJI.0000000000000169
61. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discovery (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
62. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, He B. From bench to bedside: the history and progress of CAR-T cell therapy. Front. Immunol. (2023) 14:1188049–1188049. doi: 10.3389/fimmu.2023.1188049
63. Thompson CA. FDA approves tocilizumab to treat rheumatoid arthritis. Am. J. Health-system Pharm. (2010) 67:254–4. doi: 10.2146/news100012
64. Styczyński J. A brief history of car-t cells: From laboratory to the bedside. Acta Haematol Pol. (2020) 51:2–5. doi: 10.2478/ahp-2020-0002
65. Morán A. Carl June y el descubrimiento de la inmunoterapia CAR-T. Available at: https://www.dciencia.es/carl-june-y-el-descubrimiento-de-la-inmunoterapia-car-t/.
66. Hines MR, Knight TE, McNerney KO, Leick MB, Jain T, Ahmed S, et al. Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome. Transplant. Cell Ther. (2023) 29:438.e1–438.e16. doi: 10.1016/j.jtct.2023.03.006
67. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New Engl. J. Med. (2019) 380:45–56 doi: 10.1056/NEJMoa1804980
68. Fabrizio VA, Boelens JJ, Mauguen A, Baggott C, Prabhu S, Egeler E, et al. Optimal fludarabine lymphodepletion is associated with improved outcomes after CAR-T-cell therapy. Blood Adv (2022) 6(7):1961–8. doi: 10.1182/bloodadvances.2021006418
69. Oliver-Caldés A, González-Calle V, Cabañas V, Español-Rego M, Rodríguez-Otero P, Reguera JL, et al. Fractionated initial infusion and booster dose of ARI0002h, a humanised, BCMA-directed CAR-T-cell therapy, for patients with relapsed or refractory multiple myeloma (CARTBCMA-HCB-01): a single-arm, multicentre, academic pilot study. Lancet Oncol. (2023) 24:913–24. doi: 10.1016/S1470-2045(23)00222-X
70. Chmielewski M, Abken H. CAR-T cells releasing IL-18 convert to T-bethigh foxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. (2017) 21:3205–19. doi: 10.1016/j.celrep.2017.11.063
71. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. (2018) 24:352–9 doi: 10.1038/nm.4478.
72. Khanali J, Azangou-Khyavy M, Boroomand-Saboor M, Ghasemi M, Niknejad H. JAK/STAT-dependent chimeric antigen receptor (CAR) expression: A design benefiting from a dual AND/OR gate aiming to increase specificity, reduce tumor escape and affect tumor microenvironment. Front. Immunol. (2021) 12:638639. doi: 10.3389/fimmu.2021.638639
Keywords: car t therapy, CAR T development, FDA approval, limitations and barriers, immunohematology
Citation: Ceballos C, Viguria MC, Panizo C, Rodríguez-Madoz JR and Prósper F (2023) Mini review: Advances and challenges in CAR-T cell therapy: from early chimeric antigen receptors to future frontiers in oncology. Front. Hematol. 2:1217775. doi: 10.3389/frhem.2023.1217775
Received: 09 May 2023; Accepted: 26 October 2023;
Published: 28 November 2023.
Edited by:
Bhavana Bhatnagar, West Virginia University, United StatesReviewed by:
Natalie Grover, University of North Carolina at Chapel Hill, United StatesCopyright © 2023 Ceballos, Viguria, Panizo, Rodríguez-Madoz and Prósper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Candela Ceballos, Y2FuZGVsYWNlYmFsbG9zOTNAZ21haWwuY29t