 Bing Chen
Bing Chen Haochuan Liu
Haochuan Liu Zhengang Liu
Zhengang Liu Fan Yang2
Fan Yang2- 1Department of Anesthesiology, China-Japan Union Hospital of Jilin University, Changchun, China
- 2Department of Orthopaedics, China-Japan Union Hospital of Jilin University, Changchun, China
Humanized mouse models with functional human genes, cells, and tissues are typically used for in vivo studies of diseases. Decades of studies on humanized mouse models have improved our understanding of hematopoiesis, infectious diseases, cancer biology, innate and adaptive immunity, and regenerative medicine. This review discusses the establishment and development of humanized mouse models and how they are used to model red blood cell-related diseases facilitating research in several biomedical disciplines. Furthermore, we provide approaches to overcome the limitations of these models.
1. Introduction
Hematopoietic stem cell (HSC) differentiation produces various components of human blood through a complex and delicate process. The components produced vary according to the body’s needs and are flexible, requiring HSCs to have compatible properties. HSCs can differentiate and develop into myeloid and erythroid precursor cells, forming the terminal cells in the blood system. Moreover, HSCs are responsible for the production and long-term maintenance of the blood system (1). Erythrocytes are the most important components of human blood, developing mainly through the differentiation of HSCs in the bone marrow, a process called erythropoiesis. Erythropoiesis involves the differentiation of megakaryocyte-erythroid precursor cells in the bone marrow to form reticulocytes, which migrate to peripheral blood to differentiate and form mature erythrocytes (2). Erythropoiesis is a delicate and complex process in which a slight abnormality in gene regulation can lead to the development of erythroid-related severe diseases, such as β-thalassemia, hereditary hemolytic anemia, and sickle cell disease (SCD) (3–5). Species-specific differences in the erythropoietic program (6) demand in vivo animal models of human erythropoiesis (7), especially mouse models; however, no humanized immunodeficient mouse models exist with persistent, mature human red blood cell (huRBC) reconstitution in peripheral blood (8, 9). Preclinical analysis of hematopoietic and RBC disorders, such as hereditary hemolytic anemia and SCD, requires mature RBCs in circulation for pathologic manifestations and therapies research.
Over the decades, scientists have developed various humanized mouse models for studying RBC-related diseases and exploring new therapies. In vivo animal models have contributed to understanding the molecular mechanisms underlying erythropoiesis and its associated disorders (10, 11). Inbred mice have identical backgrounds and short reproductive cycles and are supported by complementary transgenic technologies, making them extremely important animal models in immunology, medicine, biology, and related research fields. However, because of the significant genetic differences between rodents and humans, the immune system of mice strongly rejects foreign cells and tissues. Therefore, some essential research objects, such as the human immune deficiency virus, cannot infect mouse cells, and the hepatitis B virus cannot replicate in ordinary mice. Consequently, humanized mouse models possessing the human immune system have been developed to mimic our immune functions and physiological and pathological manifestations. After > 30 years of experimental research and technological development, humanized mouse models have been gradually improved and applied in many fields, such as infection, immunity, and hematological system disease research.
Developing a humanized mouse model with high levels of huRBC reconstitution facilitates studying erythropoiesis and huRBC-related diseases. It has been demonstrated that the robust rejection of huRBCs mediated by mouse macrophages is the major obstacle inhibiting their reconstitution in humanized mouse models. Furthermore, we found that mouse complement C3 could directly opsonize huRBCs and mediate their phagocytosis by mouse macrophages (11). Recently, scientists established a new humanized mouse model based on huHepMISTRGFah−/− mice with a better and longer huRBC reconstitution; additionally, they used this model mimic human SCD (10).
This review provides an overview of humanized mouse development and describes these models specifically for studying huRBC-related diseases. Moreover, we discuss existing and developing approaches to further advance humanized mouse models to suit intended clinical applications.
2. Evolution of humanized mouse models
Humanized mouse models are constructed by transplanting functional human cells, tissues, or organs into mice. These animal models can be used for preclinical in vivo studies of human diseases. Humanized mouse models have been widely used in many research areas, such as cancer, infectious diseases (12), and acquired immunodeficiency syndrome (13). However, evolutionary differences cause the recipient mouse’s immune system to strongly reject the foreign cells or tissues of the human donor. Therefore, the mouse’s immune system must first be destroyed to prevent this rejection. Consequently, using immunodeficient mice as recipients is the basis for constructing humanized mouse models. Furthermore, the generation of new strains of immunodeficient mice is an essential driving force for the continuous optimization of humanized mouse models.
2.1. Development of humanized mouse models
The earliest immunodeficient mice were nude mice with a FOXN1 gene defect preventing them from developing thymus. Therefore, they lack mature T cells and their associated immune rejection but possess B and natural killer (NK) cells, consequently rejecting human-derived cells. Later, the severe combined immunodeficient (SCID) mouse model was developed. This strain of mice is defective in T and B cells (14); hence human cells can be transplanted into it. The peripheral blood lymphocytes (PBL)-SCID and SCID-Hu mouse models we will discuss are both constructed with SCID mice.
The first breakthrough was the appearance of the Prkdcscid (DNA activated, protein kinase, catalytic polypeptide; severe combined immunodeficiency, abbreviated as scid) mutation in CB17 mice (C.B17-SCID mice) (15). The first models of humanized mouse models were reported in 1988 by three independent groups who transplanted human hematopoietic cells into immunodeficient mice (16–18). Two of these studies used C.B17-SCID mice as recipients for human peripheral blood (17) or human fetal tissues (16). C.B17-SCID mice (C.B17-Prkdcscid) possess a genetic autosomal recessive mutation (scid) affecting both B and T lymphocytes. Additionally, they contain normal NK cells, macrophages, and granulocytes (15). However, engraftment occurred at a low level, and the transplanted human cells failed to generate an immune response. Limitations hampering human cell engraftment in C.B17-SCID mice include T and B cell leakiness and high levels of host NK cells, limiting the transplantation of human HSCs (19). Additionally, scid mutation resulted in defective DNA repair and increased radiosensitivity (20).
Immunodeficient mouse strains have been gradually developed and optimized, as shown in Figure 1 (TIMELINE). In 1995, the advent of non-obese diabetic SCID (NOD/SCID) mice was notably a second breakthrough involving the transfer of scid mutation onto a NOD background (NOD.CB17-Prkdcscid) (21). Backcrossing of the scid mutation onto the NOD genetic background resulted in an immunodeficient recipient in which increased levels of human engraftment were achieved (22). Animals homozygous for the scid mutation have impaired T- and B- cell development, and NOD background results in deficient NK cell function. Therefore, the NOD/SCID mice have the following characteristics (23): (a) lack of complement C5; hence, complement activation is inhibited; (b) deficient level and decreased killing function of NK cells; (c) lack of T and B cells and antibodies in body fluids. The optimal method for constructing a humanized mouse model is to irradiate NOD/SCID mice with sublethal doses and transplant the human embryonic liver and thymus tissue blocks under the kidney capsule, followed by tail vein injection of homologous human embryonic liver-derived CD34+ HSCs. The transfused human HSCs produce large numbers of B and myeloid cells, whereas T cells develop in the transplanted thymus (24). However, some human cells, such as erythrocytes, do not reconstitute well due to strong rejection by the immune system of immunodeficient mice. Notably, NOD/SCID mice have additional shortcomings; for example, they are sensitive to radiation and can only receive small irradiation doses. Moreover, leakage of T and B cells occurs in older NOD/SCID mice, and their survival period is only approximately 8 months.
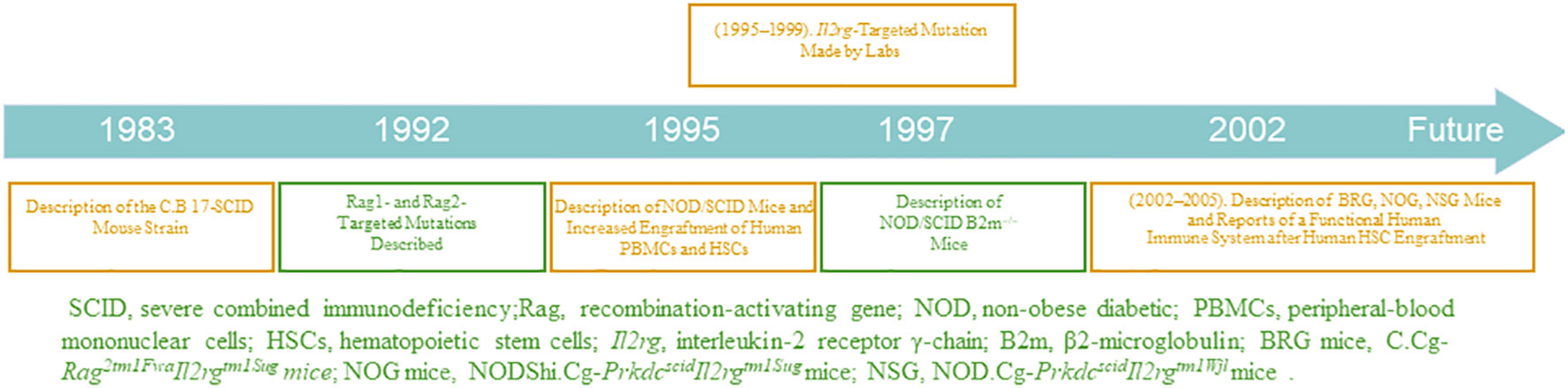
Figure 1 TIMELINE: Critical Events in the Development of Humanized Mice.
In a new immunodeficient mouse model, the recombination-activating gene (RAG)-deficient mouse, targeted mutations at RAG1 (25) and RAG2 (26) loci can prevent mature T and B cell development (27), allowing the mouse to receive transplanted human cells, without causing radiosensitivity or leakiness. However, their high NK cell activity limits the reconstitution of human HSCs. To optimize RAG-deficient mice and reduce their NK cell-killing function, scientists have modified mouse genes, further optimizing the strain. In addition, major histocompatibility complex-I molecules are essential for NK cell development, containing an important component, B2m. β2 -microglobulin is required for the expression of MHC class I molecules and the lack of MHC class I molecules in NOD/SCID B2m-/- mice prevents NK-cell development (28). Therefore, the constructed novel immunodeficient NOD/SCID B2m-/- mice do not have the functional NK cells present in NOD/SCID mice (29); hence, they can be applied for higher levels of humanized cell reconstitution.
The third breakthrough was the humanization of immunodeficient mice homozygous for targeted mutations at the interleukin (IL)-2 receptor γ-chain locus (IL2rg; also known as the common cytokine-receptorγ-chain, γc, and CD132) in the mid-2000s (30, 31). γc, is a component of the receptors for ILs-2, 4, 7, 9, 15, and 21; the IL-2 receptor gamma chain (IL2Rγ) is a common signaling component of these ILs (32). The absence of IL2Rγ blocks NK cell development owing to the ablation of ILs-7 and 15 signaling, which researchers have exploited to enhance the transplantation of humanized cells with the advancement of technology (33). In addition, this mutation results in poor lymph node development and organization.
Mice with mutations in IL2rg can be selectively bred with those with mutations in RAG1, RAG2, NOD, SCID, and other genes to generate new immunodeficient mice that are more suitable for transplantation of human cells (34). Such new immunodeficient mouse lines include NOD.Cg-PrkdcscidIl2rgtm1Wjl (NSG) mice (35), NODShi.Cg-Prkdc scidIl2rg tm1Sug (NOG) mice (30), NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl (NRG) mice (36), Rag2-/-γc-/- mice (37), C.Cg-Rag2tm1FwaIl2rgtm1Sug (BRG) mice (31), and C.Cg-Rag1 tm1MomIl2rg tm1Wjl (BRG) mice (36).
The three major mouse strain platforms used are NSG, NOG, and BRG mice. The acronym BRG usually relates to the BALB/c-Rag1 −/− or BALB/c-Rag2 −/−strains and the IL2rg null mutation. Regarding the different mice backgrounds, RG mice included the NRG and BRG. The NRG and BRG backgrounds are usually NOD and BALB/c mice, respectively. Several strains of BRG and NSG mice are available from the Jackson Laboratory, and NOG mice from Taconic. The characteristics and derivation of each immunodeficient mouse model from immunodeficient IL2rg null mice are reviewed in Table 1.
● More evidence has shown that mouse models differ in their ability to support and maintain the engraftment of functional human immune cells. For instance, NSG mice support higher levels of human HSCs engraftment in the bone marrow than NOG mice (38). Furthermore, NSG and NRG mice support higher levels of human HSCs engraftment and T cell development than BRG mice (36). Therefore, the choice of a specific mouse model affects the engraftment of transplanted human HSCs and the immune system. The major genetic factor that contributes to the capacity of NOD to support higher levels of human engraftment was identified. A strain-specific polymorphism in the NOD Sirpa gene encodes a variant of the SIRPα protein that cross-reacts with the human form of its ligand-CD47 (39, 40). The SIRPα proteins expressed by C57BL/6 and BALB/c mice have much less homology to human SIRPα (39). The NOD Sirpa polymorphism confers phagocytic tolerance for the human xenograft. Appropriate interaction of SIRPα on host macrophages with human CD47 on engrafted hematopoietic cells is important for hematopoietic cell survival, and therefore expression of human or NOD mouse (human-like) SIRPα is a major determinant for the engraftment and survival of human hematopoietic cells in mice (39). Expression of NOD SIRPα in the Balb/c background is sufficient to confer phagocytic tolerance and to support enhanced human cell engraftment (41). In BRG mice transgenically expressing human SIRPα, engraftment is increased to the levels achieved in NSG, NOG and NRG mice, indicating that SIRPα is a causal factor in the control of engraftment levels (42). One NSG mouse strain transgenically expressing human SIRPα is currently under development and will enable the identification of additional strain-specific factors important in human HSC engraftment (43).
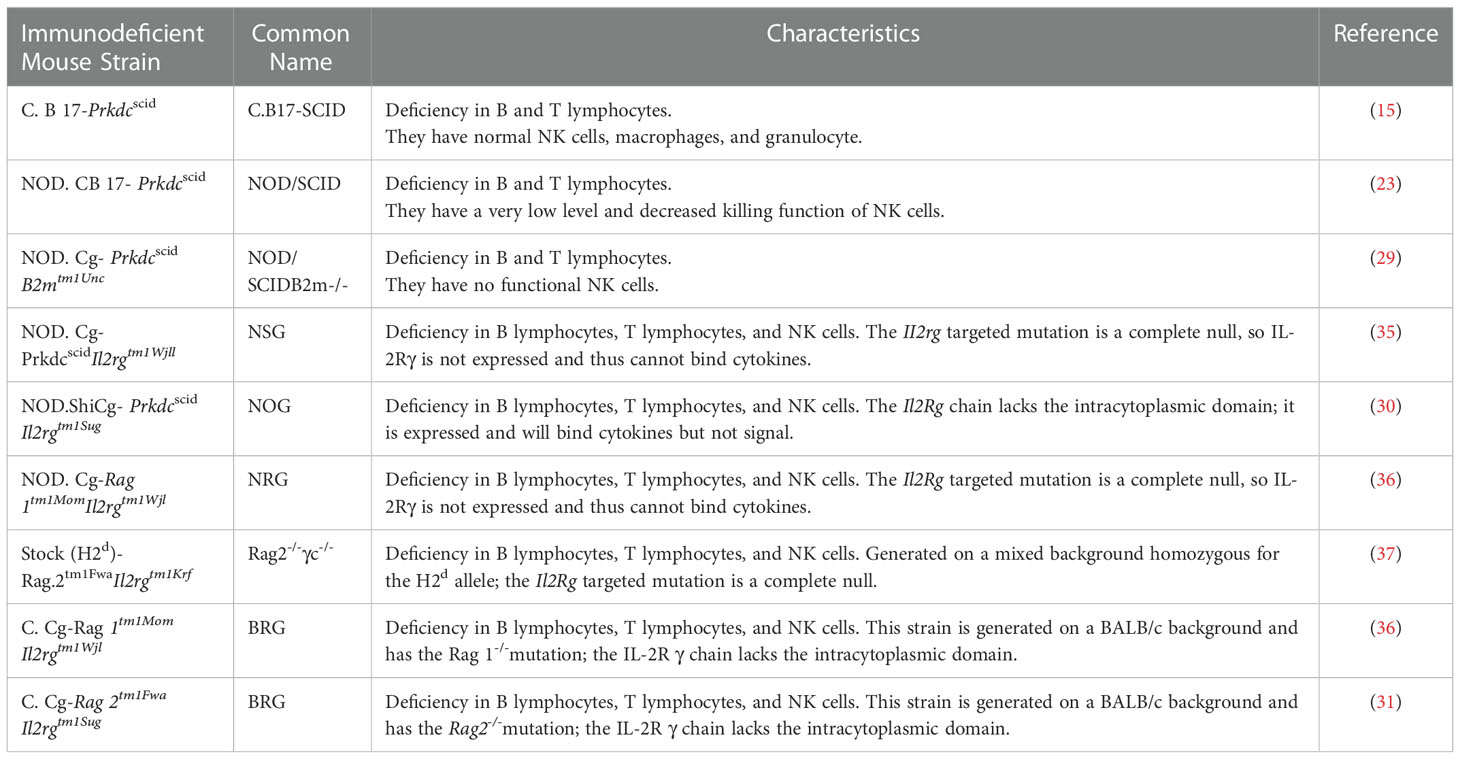
Table 1 Immunodeficient Mice for Establishing Humanized Mice.
Based on the types of human-derived cells or tissues transplanted into the strains of immunodeficient mice for humanized mouse model construction, these models can be divided into four classical types (shown in Table 2), discussed below.
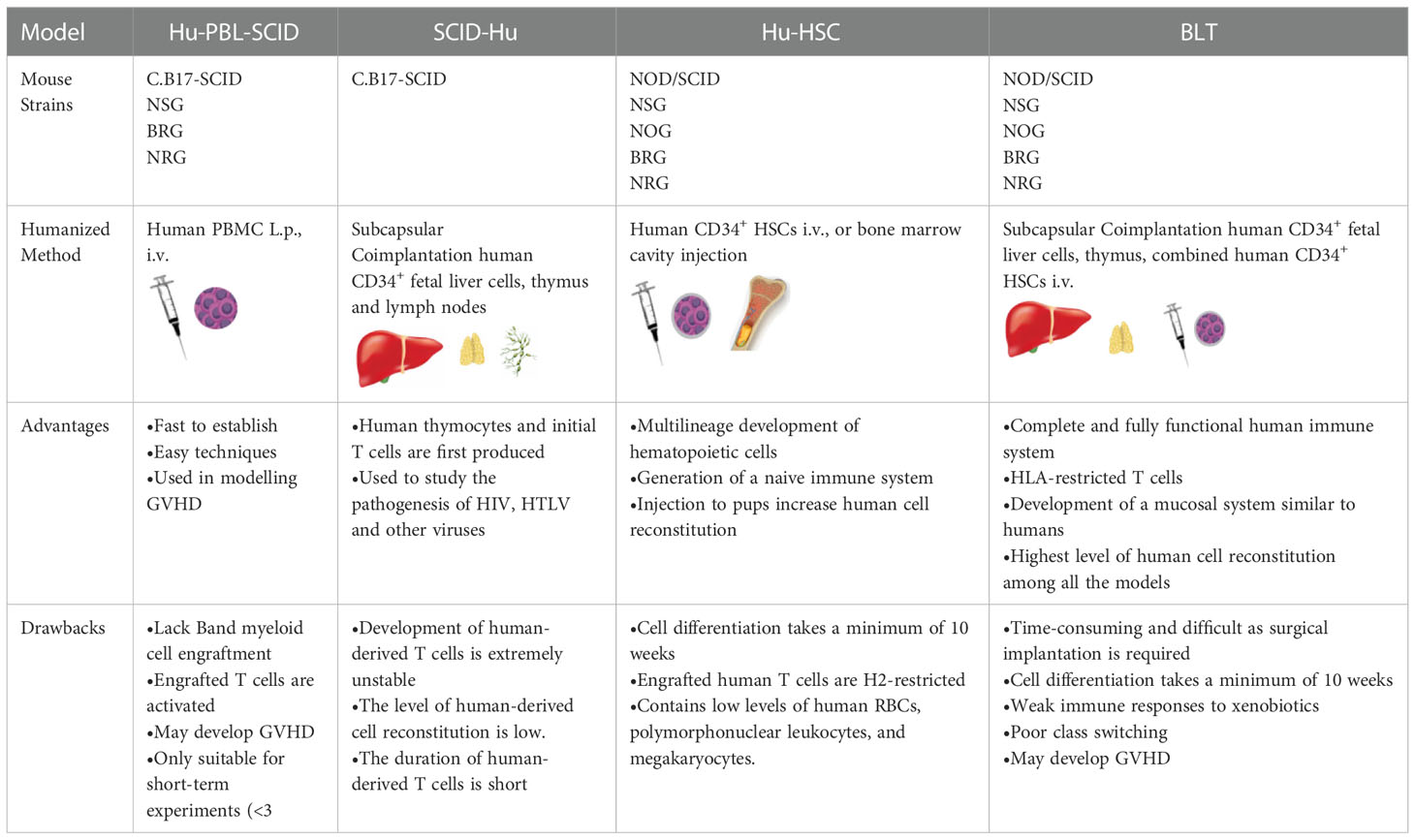
Table 2 Four Classical Types of Humanized Mouse Models.
2.1.1. Hu-PBL-SCID mouse model
The Hu-PBL-SCID mouse model is constructed by injecting mature human PBLs intraperitoneally or via the tail vein into adult SCID mice (44). Furthermore, this model can be generated using non-irradiated mice or mice that have received a sublethal irradiation dose; additionally, clonal expansion of pre-existing T cells injected into the mice can be found. After 1-week, small numbers of human myeloid, B, and other immune cells are detected in these mice. The transplanted human-derived immune cells in the mice survive for several weeks, during which they exhibit their normal activities. The Hu-PBL-SCID mouse model was first constructed using C.B17-SCID mice (17), which do not produce strong graft-versus-host reactions and have the following characteristics: (a) the mice can secrete specific antibodies; (b) human B, T, and mononuclear cells can be detected in mice; and (c) B cell lymphomas caused by Epstein-Barr virus infection can develop in mice. Currently, optimized forms of Hu-PBL-SCID mice are being used: NSG or RG immunodeficient mice. The materials required to construct the Hu-PBL-SCID mouse model (including the mature human PBLs) are readily available. However, this model has some obvious disadvantages: (a) the reconstitution of human-derived lymphocytes is unstable and occurs at a low level; (b) mice injected with a large number of human-derived cells can develop Epstein-Barr virus-related B-cell lymphoma; (c) there is no normal lymphoid tissue in the mice after the reconstitution of human-derived cells, and the spleen has a defective vesicular germinative structure; (d) there is significant allograft rejection in the mice. Furthermore, within a few weeks after transplantation of human-derived cells, many mice die due to graft-versus-host disease; hence, the experimental and observation periods are short. These disadvantages limit the widespread use of this model, although it is ideal for studying graft-versus-host disease in vivo.
2.1.2. SCID-Hu mouse model
In 1988, a team led by McCune et al. achieved the first xenotransplantation of human cells or tissues without destroying the immune system of recipient mice. This was performed by injecting human CD34+ embryonic liver cells into mice and transplanting human embryonic thymus and lymph nodes. This model is called the SCID-Hu mouse model. In this model, a human-derived embryonic thymus provides a site for developing human T cells, human-derived embryonic liver cells provide hematopoietic precursor cells, and human-derived lymph nodes provide a site for T and B cell interaction. The human-derived embryonic thymus transplanted into the subrenal capsule of the recipient immunodeficient mice gradually grows, and over time, human T cells appear in the peripheral blood, and significant levels of human IgG antibody secretion occur, all indicating the presence of human T and B cell interaction and, consequently, T cell-dependent antibody production in this humanized mouse model.
In summary, this model is constructed by transplanting an embryonic thymus containing human HSCs under the kidney capsule of SCID mice and injecting human CD34+ embryonic liver cells into mice to establish a humanized mouse model with a functional human thymus (45). In SCID-Hu mice, human thymocytes and premature T cells are first produced and mainly colonize the human thymus/liver-like organs, inhibiting peripheral circulation of T cells. As the first humanized mouse model, SCID-Hu mice have the following shortcomings: (a) the development of human-derived T cells is extremely unstable, (b) the level of human-derived cell reconstitution is low, and (c) the lifespan of human-derived T cells is short. However, this model can still be used to study the pathogenesis of the human immune deficiency virus, the human T-lymphotropic virus, and other viruses. Furthermore, the emergence of the SCID-Hu mouse model laid an excellent foundation for optimizing new humanized mouse models (46).
2.1.3. Hu-HSC mouse model
SCID-Hu and Hu-PBL-SCID mouse models can be constructed using C.B17-SCID mice. However, in the absence of human cytokines, the level of human bone marrow cell reconstitution in these models is low. With technological advances, NOD/SCID mice have been introduced to effectively solve this problem. The Hu-HSC mouse model is constructed by injecting human CD34+ HSCs into newborn or adult immunodeficient mice via the tail vein or bone marrow cavity. Human CD34+ HSCs can be obtained from cord blood, bone marrow, embryonic liver tissue cells mobilized by granulocyte colony-stimulating factor, or peripheral blood cells. Cord blood and embryonic liver are the most commonly used, as they are more likely to produce cells that can colonize immunodeficient mice. In the Hu-HSC humanized mouse model, human-derived T cells can differentiate and develop in the mouse thymus and undergo positive and negative selection with mouse major histocompatibility complex (H2) restriction. However, this limits the human leukocyte antigen (HLA)-restricted interactions of human antigen-presenting cells with human T cells in peripheral tissues. This model can be constructed in two main ways, with significant differences between them. In the first approach, adult NOD/SCID, NSG, or NOG mice are irradiated with sublethal doses and then injected with human HSCs, which subsequently produce a variety of human hematopoietic precursor cells but rarely produce human-derived T cells. The transplantation can be optimized using neonatal IL-2γc-/- mice, including NOG, RG, and NSG mice. A second approach in which neonatal NSG, NOG, or RG mice are irradiated at sublethal doses and intrahepatically injected with human HSCs allows for better human cell transplantation with the concomitant production of human B, T, NK, and dendritic cells and macrophages (47). Hu-HSC humanized mouse models are widely used in studies of cell-mediated immune responses, human hematopoietic system development, human immune deficiency virus, Epstein-Barr virus, and other viral infectious diseases.
2.1.4. FLC (BLT) mouse model
Scientists optimized SCID-Hu and Hu-HSC-SCID to construct a new humanized mouse model, the FLC mouse model. In the SCID-Hu mouse model, human T cells can differentiate and develop because of the transplantation of a human embryonic thymus; however, T cell development is unstable and weak. Moreover, in the Hu-HSC-SCID mouse model, human CD34+ HSCs are transplanted into mice after sublethal irradiation doses, generating many human myeloid and B cells; however, the model lacks T cell development. Therefore, these two models can complement each other. Some researchers have combined the advantages of the two by irradiating NOD/SCID mice with sublethal irradiation doses, transplanting human embryonic thymus and homologous embryonic liver tissue under the kidney capsule, and injecting human CD34+ HSCs isolated from the homologous embryonic liver through the tail vein of mice, thus constructing a new humanized mouse model (24). They found that the spleen and lymph nodes of mice in this humanized mouse model were enlarged and that the reconstitution of human T cells was significantly increased to approximately 20% after 12 weeks. Additionally, other cells, such as human B and mononuclear cells, were present in large numbers. Furthermore, in the spleens of the mice, human T cells were distributed around the central splenic artery, and there were scattered clusters of human B cells near the human T cells. Moreover, they found that human IgM and IgG were produced (at approximately 150 µg/mL after 16 weeks) in the FLC mouse model. These results indicate that human T and B cells can interact in the mice’s secondary immune tissues. Furthermore, they observed antibodies switching from IgG1 to IgG4, with time and frequency extremely similar to those observed in the natural state human immune system. This study was the first to report a humoral immune response in a humanized mouse model, and Melkus et al. replicated this result in a subsequent study; they named the new model the BLT mouse model, derived by transplanting bone marrow, liver, and thymus. In early BLT mouse models, the immunodeficient mice used were NOD/SCID mice; however, with technical optimization and improvement, NOG, NSG, or RG mice are now more often used (48, 49). In the modified BLT mouse model, more humanized T/B cells, dendritic cells, macrophages, and NK cells are generated, and the transplantation of an autologous human thymus allows for improved development of homologous human T cells with appropriate restriction. Compared with the SCID-Hu humanized mouse model, the BLT mouse model has an improved capacity to develop humanized hematopoietic and immune systems. Additionally, T cells develop in the human thymus with HLA restriction, thus optimizing transplantation effects.
2.2. Human growth factors and cytokines involved in humanized mouse models establishment
Due to the differences between mice and humans in the growth factors and cytokines required for hematopoietic development, there are several major limitations inherent in humanized mice. Scientists are currently focused on ensuring the development and maintenance of human cells in the murine microenvironment, and human growth factors and cytokines are essential to achieve this.
Technologies to deliver human species-specific factors into mice to enhance human hematopoiesis and immune system development are diverse. The simplest approach is to inject human cytokines as recombinant proteins or through the hydrodynamic injection of plasmids. Erythropoietin is an important cytokine in terminal RBCs differentiation; however, mouse erythropoietin cannot sufficiently cross-react with the respective human receptor (50). IL-3 is a growth factor that stimulates hematopoiesis; the administration of human IL-3 to patients increases the number of reticulocytes and platelets (51, 52). In a humanized mouse model, human erythroid lineage cells can develop in the mouse bone marrow; however, mature huRBCs are deficient in peripheral blood. Notably, the provision of human erythropoietin and IL-3 can improve human erythropoiesis (53). Several factors contribute to adequate T cell homeostasis in circulation, including human T cell survival in the presence of mouse macrophages, such as ILs-7 and 15 (54). IL-7 is a central cytokine involved in hematopoietic cell development; the injection of its recombinant form transiently affects thymic lymphopoiesis but does not improve peripheral T cell homeostasis (35, 55). Higher levels of human DCs can be obtained by the hydrodynamic injection of plasmids encoding human granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-4, and Fms-like tyrosine kinase 3 ligand (Flt3L) (56).
With the increasing feasibility of genetic engineering, different protocols have been used to deliver a variety of human cytokines in humanized mice, especially NSG and BRG mice. Three main genetic engineering technologies have been applied: transgenic expression of bacterial artificial chromosomes, transgenic expression of cDNA constructs driven by tissue-specific or ubiquitous promoters, and knock-in technology. Furthermore, stem cell factor (SCF) is critical for HSC engraftment, proliferation, development, and survival. Scientists found that human membrane-bound SCF increases the levels of HSC engraftment in non-irradiated NSG mice. The transgenic expression of this SCF under a cytomegalovirus promoter in NSG mice resulted in a higher engraftment level after human Hematopoietic stem and progenitor cell (HSPC) transplantation, even without recipient pretreatment (57, 58). Notably, GM-CSF was initially classified as a hematopoietic growth factor (59). The transgenic expression of cytokines, known to support human myelopoiesis, can improve the major deficiencies in the development of human myeloid cells in humanized mice. Transgenic mice expressing human GM-CSF, SCF, and IL-3 under the control of a ubiquitous cytomegalovirus promoter are generated from an NSG background (60). Furthermore, the knock-in humanization of the two adjacent genes encoding GM-CSF and IL-3 has a critical effect on the lung alveolar macrophages, primarily due to the humanization of GM-CSF (61). CSF-1 and macrophage-CSF(M-CSF) are early cytokines that promote hematopoiesis, and in the hematopoietic system, CSF-1/M-CSF is believed to act specifically on myeloid progenitors.
Similarly, the knock-in replacement of the gene encoding mouse M-CSF by its human counterpart leads to improved engraftment of functional CD33+CD14+ monocytes in the tissues of recipient mice (62). Furthermore, the most noticeable effect of IL-3 transgenesis in NSG mice is in human T cell improvement, especially the regulatory T cells (60). Additionally, the transgenic expression of membrane human SCF has a positive effect on myelopoiesis, particularly on the development of mast cells (58). Studies on dengue virus showed that human T cells developed after engraftment of HLA-A2 transgenic NSG mice with HLA-A2(+) human cord blood HSCs and were able to secrete IFN-γ, IL-2, and TNF-α in response to stimulation with dengue virus (63). Furthermore, MISTRG mice carry knock-ins for human cytokines granulocyte-monocyte and M-CSF, IL-3(I), thrombopoietin, signal regulatory protein α, and the receptor for the “do not eat me” signal regulatory protein CD47 in the Rag2 −/− Il2rg −/−(RG) background (64). Based on these mice, scientists deleted the fumarylacetoacetate hydrolase (FAH) gene using CRISPR-Cas9 technology in MISTRG to generate MISTRGFah −/−(MISTRGFah) mice and then humanized the liver of this model. Scientists have established a new humanized mouse model based on huHepMISTRGFah−/−mice with a better and longer huRBC reconstitution and used the model to mimic human SCD (10).
Table 3 lists the cytokines delivered exogenously into humanized mice by injection of recombinant cytokines and hydrodynamic injection of plasmid DNA. Table 4 shows the genetic expression of human growth factors and cytokines in the genomes of the immunodeficient mice.
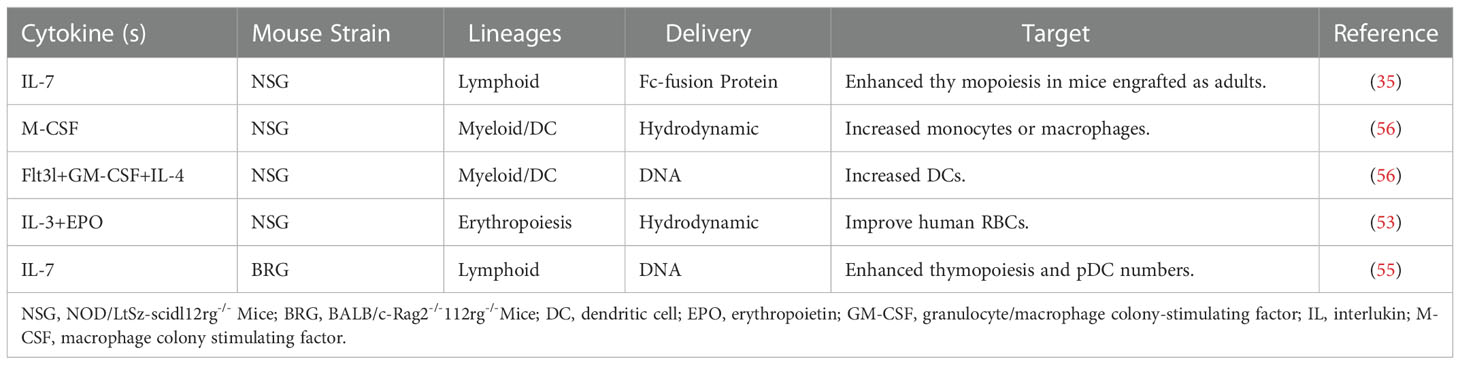
Table 3 Improvement of Humanized Mice by Exogenous Cytokine Administration.
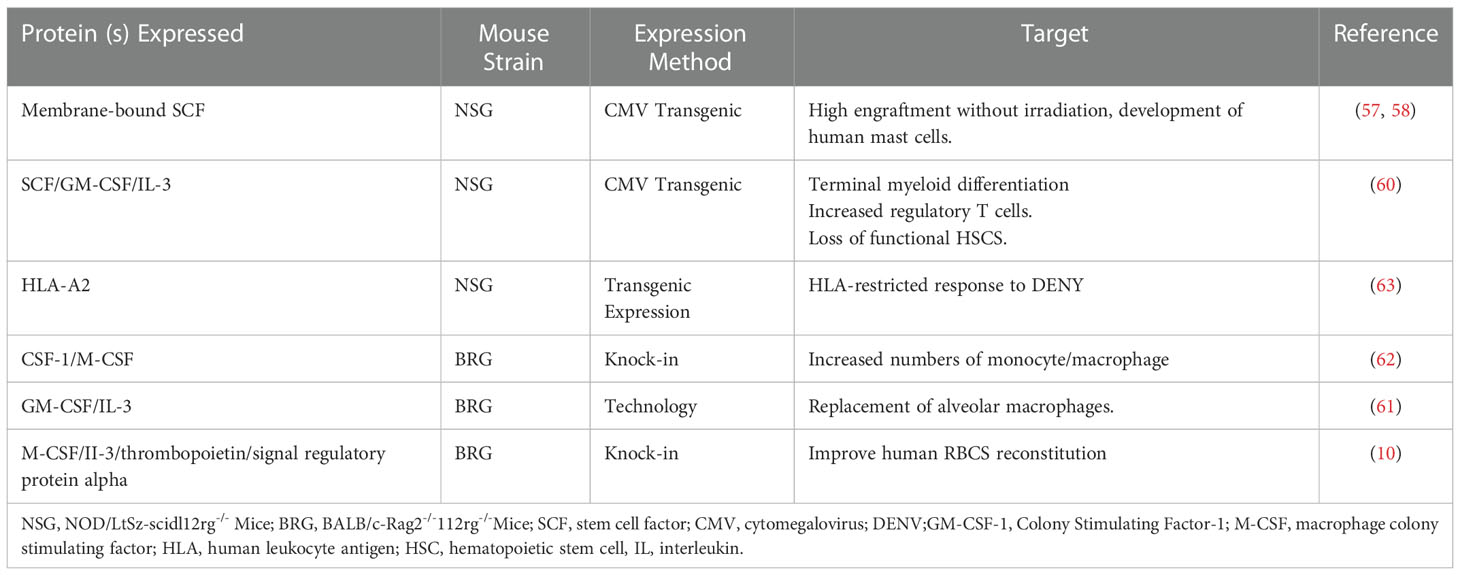
Table 4 Improvement of Humanized mice by genetic expression of cytokines or HLA molecules.
3. Erythrocyte-related diseases
Erythropoiesis is a delicate and complex process in which a small abnormality in gene regulation can lead to the development of erythroid-related severe diseases such as β-thalassemia, hereditary hemolytic anemia, and SCD (3–5). Although allogeneic HSC transplantation can cure erythroid-related severe diseases caused by abnormality in gene regulation, immune-related complications, such as graft-versus-host disease, limit their clinical application (3, 4). Theoretically, gene therapy that modifies HSCs can be performed for certain hematological diseases (3, 5). However, gene therapy in humans is associated with numerous adverse effects and risks, such as the induction of in vivo mutations by gene editing techniques, and ultimately, death (65, 66). Therefore, an in vivo animal model for preclinical studies of human erythrocyte-related diseases is essential.
Malaria is another RBC-related disease caused by Plasmodium parasites, threatening hundreds of millions in underdeveloped countries yearly. Natural Plasmodium infection involves the hepatic and RBC phases, and human liver chimeric mice can be used to study these phases (67, 68). Human HSC-transplanted mice have few circulating huRBCs due to phagocytosis by mouse macrophages, thereby limiting Plasmodium falciparum infection; consequently, RBCs should be administered daily (69). Human liver chimeric Fah-/-Rag2-/-IL2rg-/-NOD and thymidine kinase NOG mice engrafted with human hepatocytes and transfused with human RBCs enable complete replication of P. falciparum infection (70, 71). In these studies, exogenous huRBC transfusion was required. Therefore, a humanized mouse model with continuous reconstitution of huRBCs is urgently needed.
3.1. HuRBC reconstitution in humanized mouse models
In 2011, Yang et al. found that macrophages in immunodeficient mice are a key factor in the rejection of human erythrocytes and that clodronate liposomes application to remove macrophages made it possible to optimize the level of human erythrocyte reconstitution in humanized mice; the proportion of reconstituted human erythrocytes in this model was approximately 3% (53). Furthermore, in 2013, Chen et al. applied plasmids encoding human IL-15 and Flt3L to enhance human NK cells in mice and administered a sufficient amount of human erythrocytes daily to study erythrocyte-related diseases, such as malaria (72). However, on stopping the administration, mice did not produce human erythrocytes; hence, this method cannot be used to study the differentiation of human HSCs into erythrocytes and their development in humanized mice. Furthermore, in a 2014 study on malaria, Chen et al. used clodronate liposomes to remove mouse macrophages and antibodies to neutralize mouse neutrophils, thereby inhibiting human erythrocyte rejection. Additionally, they applied high-pressure injection of plasmids expressing human erythropoietin and IL-3, promoting human erythrocytes reconstitution in humanized mice (73). However, the percentage of reconstituted human erythrocytes in this humanized mouse model was not very high (1.6–4%). In 2014, Wathsala et al. used gene editing to construct humanized mice by transplanting HLA-adapted human HSCs into HLA-DR4. RagKO.IL2RγcKO.NOD (DRAG) mice and the percentage of reconstituted human erythrocytes in this model was low (0.2–1%) (74). In 2017, Bing’s research showed that macrophages, neutrophils, endothelial cells, and complement C3 play important roles in rejecting human erythrocytes in NOD/SCID mice. Complement C3 depletion in the serum of mice that had already been depleted of macrophages with cobra venom factor (CVF) increased the proportion of reconstituted human erythrocytes in human HSC-transplanted mice. We found that the simultaneous application of CVF to deplete mouse complement C3 and clodronate liposomes to deplete mouse macrophages resulted in a two-fold increase in the proportion of reconstituted human erythrocytes in humanized mice compared with the application of clodronate liposomes alone (11). In 2021, Song et al. reported a new humanized mouse model in which combined human liver, and cytokine humanization improved erythropoiesis and RBC reconstitution in the circulation based on MISTRG mice. They used this model to mimic human SCD (10); however, the reconstitution rate was not ideal. The human erythrocyte reconstitution rate in these humanized mouse models is insufficient; therefore, further clarification of the mechanism of human erythrocyte rejection in humanized mice is necessary to develop a better-humanized mouse model.
Platelets are produced by the fragmentation of megakaryocytes and the differentiation of megakaryocyte-erythroid precursor cells in the bone marrow (75); Thrombopoietin(TPO) plays a major role in this process. Similar to erythrocytes, human megakaryocytes are detectable in the bone marrow of humanized mice; however, human platelets are not detectable in the peripheral blood of humanized mice. Moreover, TPO-based humanization did not significantly increase the number of platelets in the peripheral blood; however, applying clodronate liposomes to remove phagocytes significantly increased the platelet level (58, 76).
3.2. Factors affecting human erythrocyte reconstitution in humanized mouse models
The lack of human erythrocyte reconstitution in humanized mice has plagued researchers in erythrocyte-related diseases for many years. The reason why human T, B, NK, and myeloid cells can be reconstituted, but human erythrocytes are absent in humanized mice remains to be determined. The rejection of human erythrocytes by the mouse immune system is the primary factor affecting their reconstitution in humanized mice. The mouse immune system mainly includes the complement system and innate immune cells, including macrophages, neutrophils, and endothelial cells. Our previous studies showed that the rejection of human erythrocytes by mouse macrophages was a key factor in the defective reconstitution of human erythrocytes in humanized mice. In recent research, we found that another key factor is the allogeneic rejection of human erythrocytes by neutrophils and endothelial cells in immunodeficient mice (11).
3.2.1. Complement
Complement is an important component of the innate immune system that recruits immune cells, participates in the clearance of exogenous microorganisms and necrotic cells by antibodies and phagocytes (77), and is essential for maintaining host homeostasis. Complement activation in the human body occurs mainly through three traditional pathways: the classical, the lectin, and the alternative complement activation pathways. The common product of all three pathways is complement C3, which plays an important role in complement activation and regulation (77). Bing’s research has shown that mouse complement C3 can bind directly to the surface of human erythrocytes, condition human erythrocytes, and promote their adhesion to/phagocytosis by mouse phagocytes in the absence of antibodies (11). Furthermore, combined sequential injections of clodronate liposomes and CVF increased the proportion of reconstituted human erythrocytes in humanized mice. This suggests that the effect of mouse complement C3 on human erythrocytes plays an important role in promoting human erythrocyte rejection in immunodeficient mice.
3.2.2. Macrophages
As innate immune cells, macrophages have strong phagocytic functions and are primarily responsible for clearing cell debris, apoptotic, necrotic, and cancerous cells. Additionally, they are involved in exogenous cell rejection, especially foreign cells (78). Notably, macrophages, neutrophils, and immature dendritic cells are all professional phagocytic cells. CD47 is a molecule expressed on the surface of almost all cells and binds specifically to the SIRPα receptor expressed on the phagocyte’s surface (including macrophages and dendritic cells), thereby serving as an immunosuppressive”do not eat me” signal to phagocytes (79). When there is no or weak cross-reactivity between CD47 molecules on xenogeneic donor cells and SIRPα molecules on recipient macrophages, xenogeneic donor cell rejection by macrophages occurs (80). However, SIRPα gene polymorphisms in different strains of mice and cross-reactivity between SIRPα molecules on NOD mouse-derived cells and CD47 on human cells prevent macrophages in NOD/SCID and NOD/SCID Il2rg-/- mice from rejecting most human nucleated cells (39). Furthermore, our previous studies demonstrated that NOD/SCID mouse macrophages strongly reject human erythrocytes by a mechanism independent of the CD47-SIRPα interaction and that the removal of mouse macrophages in humanized mice using clodronate liposomes is sufficient to reconstitute mature human erythrocytes in mice (58). Bing’s recent findings suggest that complement C3 plays an important mediating role in human erythrocytes rejection by macrophages in humanized mice and that the removal of complement C3 by CVF injection after clodronate liposome treatment further increases the proportion of huCD235a+ human erythrocytes in the peripheral blood by approximately two to three-fold (11). These studies suggest that mouse macrophages reject human erythrocytes in a complement-dependent manner and that this rejection is a key factor in the defective reconstitution of human erythrocytes in humanized mice.
3.2.3. Neutrophils
In addition to macrophages, neutrophils are professional phagocytes with certain phagocytic functions (81). In a recent study, Bing showed that CD11b+F4/80-LY6G+ neutrophils from mice also adhere to human erythrocytes in the presence of mouse serum, and this adhesion disappears when the serum is treated with heat shock (11), suggesting that mouse neutrophils reject human erythrocytes in a complement-dependent manner.
3.2.4. Endothelial cells
Endothelial cells are widely distributed in organisms in areas, such as the blood-brain barrier, liver, spleen, kidney, heart, small capillaries, lymph nodes, and lymphatic vessels. Furthermore, they are arranged throughout the circulatory and lymphatic systems and vital organs (82). In addition to maintaining homeostasis, regulating blood flow, and exchanging nutrients (82), endothelial cells are part-time phagocytes with adhesive phagocytic functions that can adhere to abnormal erythrocytes, platelets, and leukocytes (83–85). Impairment of endothelial cell function leads to several human disorders, including hypertension and atherosclerosis (82), atypical hemolytic uremic syndrome, and acute and chronic kidney injury (86).
Endothelial cells have structural and functional differences in different organs and tissues; the ones easily isolated are generally selected for relevant experiments in vitro. Researchers have assumed that the basic properties of all endothelial cells are similar, which is sufficient to ensure that the endothelial cells used in in vitro experiments have similar activity to in vivo endothelial cells (87). In humanized mice, the blood supply to the lungs and liver is uninterrupted; therefore, they are constantly in contact with human erythrocytes, resulting in rejection. The endothelial cells of the lungs and liver can express vascular cell (CD106), endothelial-leukocyte (E-selectin, CD62E), and intercellular cell adhesion molecules-1 (CD54), among many other markers of endothelial cell activation, resulting in a rejection response to foreign antigens. Furthermore, the liver is the main endocytosis site in the organism (85), and hepatic sinusoidal endothelial cells can recognize and process antigens through scavenger receptors and other means. Additionally, these play an important phagocytic role in rejecting xenogeneic red cells (88).
Normal human erythrocytes can deform and do not adhere to resting endothelial cells. However, when erythrocytes are abnormal, or xenografts of RBCs are performed, activation and adhesion of endothelial cells to RBCs may occur (84, 88). Markers of endothelial cell activation include vascular cell adhesion molecule-1, E-selectin, and intercellular cell adhesion molecule-1. Under normal conditions, endothelial cells are in a quiescent state (the average lifespan of endothelial cells is approximately 1 year (87). However, in the presence of foreign antigens or disease conditions, endothelial cells are activated, resulting in elevated expression of activation markers, which initiate and mediate the rejection and adhesion/phagocytosis of antigens or abnormal cells (89, 90). Furthermore, endothelial cells can adhere to/or phagocytose abnormal human erythrocytes via the following pathways. In SCD, abnormally elevated intercellular cell adhesion molecule-4 on human erythrocytes can bind directly to αvβ3 integrins on endothelial cells, thus mediating endothelial cell adhesion to abnormal human erythrocytes (91). Lutheran/basal cell adhesion molecule(Lu/BCAM) on human erythrocytes can bind to laminin-5 on endothelial cells to produce adhesion in patients with SCD and erythroblastosis (92, 93). Although Lu/BCAM is expressed on human erythrocytes, it is not found on mature erythrocytes in mice, indicating species variability (93). When human erythrocytes have abnormal morphology, such as during apoptosis, phosphatidylserine, normally located inside the erythrocyte membrane, is exposed on the erythrocyte’s surface and acts as a regulatory “eat me” signal. Phosphatidylserine binds to the lactic agglutinin MFG-E8, which binds at the other end to αvβ3 and αvβ5 integrins on the endothelial cells. The other end of MFG-E8 binds to αvβ3 and αvβ5 integrins on endothelial cells, thereby mediating endothelial cell adhesion and phagocytosis of human erythrocytes (94, 95). MFG-E8, also known as SED1, is a secreted glycoprotein produced by a variety of phagocytes, including activated macrophages and immature dendritic cells (94). Furthermore, CXCL16/SR-PSOX acts as a chemokine (in its free form and on endothelial cells) and as a scavenger receptor that binds directly to phosphatidylserine on erythrocytes, thereby promoting endothelial cell adhesion to abnormal human erythrocytes (96).
3.3. Development challenges in huRBC disease research
Humanized mouse models with huRBC reconstitution can be applied to malaria, SCD, aplastic anemia, and many other huRBC-related diseases (67, 97). For example, malaria is mainly transmitted through the bites of mosquitoes infected with Plasmodium, which reproduces in the human liver and is released into the bloodstream, infecting human erythrocytes. Therefore, the optimal choice is a humanized mouse model that simulates the stages of Plasmodium reproduction and its release into the bloodstream with human liver tissue and erythrocyte reconstitution (68). Currently, two main approaches are combined by scientists to construct mice with huRBCs: continuous infusion of huRBCs and transplantation of human HSCs in mice. For example, one group used plasmids encoding human IL-15 and Flt3L to enhance human NK cells in mice, followed by continuous daily infusion of large amounts of huRBCs to study malaria (72). However, once human erythrocytes were withdrawn, mice no longer had human erythrocytes in their bodies. Although we improved human erythrocyte reconstitution in humanized mouse models, clodronate liposomes injection was required, which has certain toxic side effects in immunodeficient mice. Furthermore, high doses of clodronate liposomes cause the death of immunodeficient mice in a short period; hence the observation window is insufficient. Therefore, a humanized erythrocyte mouse model that can consistently carry high proportions of human erythrocytes at different stages is urgently needed, and scientists are working to develop such a model.
4. Discussion
Over the past decades, humanized mouse models have developed rapidly, making experimental operations more convenient and economical. These mouse models have become an important animal model in biomedicine for the preclinical detection of diseases, thereby benefiting humans.
Recently, technology related to humanized mouse models has rapidly progressed, with newer models being easier and less expensive to use in experiments. Consequently, humanized mouse models have become important for the preclinical assessment of human diseases in biomedicine. However, these models have certain limitations, such as cross-reactivity between rodents and humans and limited development, differentiation, and migration of human HSCs in a heterogeneous environment in mice.
Genetic modification of immunodeficient mice can greatly improve the extent to which humanized mouse models can mimic human diseases; however, many shortcomings still exist to overcome. Nevertheless, to some extent, optimization of xenograft methods, transplantation strategies, and human cell sources have been achieved. Furthermore, additional modifications to key pathways in recipient mice during the transplantation process may improve humanized mouse model construction adaptability. For example, immunodeficient mice can express human cytokines, such as MISTRG mice expressing human IL-6 (98). Similarly, humanized mice expressing human IL-8 can promote myeloid cell production (99).
Irradiation of immunodeficient mice can produce bone marrow suppression; however, it induces damage, leading to inflammatory responses and death. Fundamentally, the cytokine receptor c-kit is essential for normal bone marrow hematopoietic function and facilitates the self-renewal of HSCs in the microenvironment. Mutations partially disrupting the c-kit-mediated signaling pathway affect HSCs and erythroid precursor cells, allowing erythropoiesis to occur without irradiation (9, 100–102).
Presently, huRBC reconstitution in humanized mice is still not ideal, and the exploration of the mechanisms underlying these reconstitution defects is ongoing. A humanized mouse model with stable and highly reconstituted RBCs is the goal of future research. The development of mice with humanized RBCs will rely on gene editing techniques, such as the knockout of certain targeted genes or the breeding of new strains of immunodeficient mice. Experts in humanized mouse model research worldwide are committed to optimizing mouse strains so that these models can benefit humankind soon.
In summary, an increasing number of immunodeficient mouse strains can be used to construct humanized mice; therefore, it is crucial to select the strain according to specific needs. Furthermore, rigorous analysis, adequate evaluation, and reasonable elimination of barriers can effectively improve the efficiency of humanized mouse model construction and guide the subsequent construction of better, more suitable, and durable models.
Author contributions
HL and BC wrote the main manuscript text. BC and ZL prepared TIMELINE, Tables 1 and Table 2, FY prepared Tables 3 and Table 4. All authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No: 82000120) and China Postdoctoral Science Foundation (Grant No: 2020 M681049).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
huRBC, human red blood cell; HSC, hematopoietic stem cell; SCD, sickle cell disease; PBL, peripheral blood lymphocyte; SCID, severe combined immunodeficiency; IL, interleukins; HLA, human leukocyte antigen; SCF, stem cell factor; GM-CSF, granulocyte-macrophage-colony stimulating factor; M-CSF, macrophage-colony stimulating factor; CVF, cobra venom factor; ICAM, intercellular cell adhesion molecules; NOD, non-obese diabetic; RAG, recombination-activating gene.
References
1. Laurenti E, Göttgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature (2018) 553:418–26. doi: 10.1038/nature25022
2. Nandakumar SK, Ulirsch JC, Sankaran VG. Advances in understanding erythropoiesis: evolving perspectives. Br. J. Haematol (2016) 173:206–18. doi: 10.1111/bjh.13938
3. Romero Z, DeWitt M, Walters MC. Promise of gene therapy to treat sickle cell disease. Expert Opin. Biol. Ther. (2018) 18:1123–36. doi: 10.1080/14712598.2018.1536119
4. Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat. Med. (2015) 21:221–30. doi: 10.1038/nm.3814
5. Makis A, Hatzimichael E, Papassotiriou I, Voskaridou E. 2017 Clinical trials update in new treatments of β-thalassemia. Am. J. Hematol. (2016) 91:1135–45. doi: 10.1002/ajh.24530
6. Dzierzak E, Philipsen S. Erythropoiesis: development and differentiation. Cold Spring Harb. Perspect. Med. (2013) 3:a011601. doi: 10.1101/cshperspect.a011601
7. Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, et al. Humanized mouse models of clinical disease. Annu. Rev. Pathol. (2017) 12:187–215. doi: 10.1146/annurev-pathol-052016-100332
8. Yurino A, Takenaka K, Yamauchi T, Nunomura T, Uehara Y, Jinnouchi F, et al. Enhanced reconstitution of human erythropoiesis and thrombopoiesis in an immunodeficient mouse model with Kit(Wv) mutations. Stem Cell Rep. (2016) 7:425–38. doi: 10.1016/j.stemcr.2016.07.002
9. Rahmig S, Kronstein-Wiedemann R, Fohgrub J, Kronstein N, Nevmerzhitskaya A, Bornhäuser M, et al. Improved human erythropoiesis and platelet formation in humanized NSGW41 mice. Stem Cell Rep. (2016) 7:591–601. doi: 10.1016/j.stemcr.2016.08.005
10. Song Y, Shan L, Gbyli R, Liu W, Strowig T, Patel A, et al. Combined liver-cytokine humanization comes to the rescue of circulating human red blood cells. Science (2021) 371:1019–25. doi: 10.1126/science.abe2485
11. Chen B, Fan W, Zou J, Zhang S, He J, Shu C, et al. Complement depletion improves human red blood cell reconstitution in immunodeficient mice. Stem Cell Rep. (2017) 9:1034–42. doi: 10.1016/j.stemcr.2017.08.018
12. Dash PK, Gorantla S, Poluektova L, Hasan M, Waight E, Zhang C, et al. Humanized mice for infectious and neurodegenerative disorders. Retrovirology (2021) 18:13. doi: 10.1186/s12977-021-00557-1
13. Zhang L, Su L. HIV-1 immunopathogenesis in humanized mouse models. Cell Mol. Immunol. (2012) 9:237–44. doi: 10.1038/cmi.2012.7
14. Vladutiu AO. The severe combined immunodeficient (SCID) mouse as a model for the study of autoimmune diseases. Clin. Exp. Immunol. (1993) 93:1–8. doi: 10.1111/j.1365-2249.1993.tb06488.x
15. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature (1983) 301:527–30. doi: 10.1038/301527a0
16. McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: Murine model for the analysis of human hematolymphoid differentiation and function. Science (1988) 241:1632–9. doi: 10.1126/science.241.4873.1632
17. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature (1988) 335:256–9. doi: 10.1038/335256a0
18. Kamel-Reid S, Dick JE. Engraftment of immune-deficient mice with human hematopoietic stem cells. Science (1988) 242:1706–9. doi: 10.1126/science.2904703
19. Greiner DL, Hesselton RA, Shultz LD. SCID mouse models of human stem cell engraftment. Stem Cells (1998) 16:166–77. doi: 10.1002/stem.160166
20. Fulop GM, Phillips RA. The scid mutation in mice causes a general defect in DNA repair. Nature (1990) 347:479–82. doi: 10.1038/347479a0
21. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. (1995) 154:180–91. doi: 10.4049/jimmunol.154.1.180
22. Hesselton RM, Greiner DL, Mordes JP, Rajan TV, Sullivan JL, Shultz LD. High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-scid/scid mice. J. Infect. Dis. (1995) 172:974–82. doi: 10.1093/infdis/172.4.974
23. Yu WJ, Yang WH, Shi ZX, Yang XD, Wang HJ. [Application of NOD/SCID mice in research of experimental hematology - review]. Zhongguo Shi Yan Xue Ye Xue Za Zhi (2008) 16:964–8.
24. Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood (2006) 108:487–92. doi: 10.1182/blood-2005-11-4388
25. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature b and T lymphocytes. Cell (1992) 68:869–77. doi: 10.1016/0092-8674(92)90030-g
26. Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell (1992) 68:855–67. doi: 10.1016/0092-8674(92)90029-c
27. Karo JM, Sun JC. Novel molecular mechanism for generating NK-cell fitness and memory. Eur. J. Immunol. (2015) 45:1906–15. doi: 10.1002/eji.201445339
28. Christianson SW, Greiner DL, Hesselton RA, Leif JH, Wagar EJ, Schweitzer IB, et al. Enhanced human CD4+ T cell engraftment in beta2-microglobulin-deficient NOD-scid mice. J. Immunol. (1997) 158:3578–86. doi: 10.4049/jimmunol.158.8.3578
29. Lapidot T, Kollet O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia (2002) 16:1992–2003. doi: 10.1038/sj.leu.2402684
30. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood (2002) 100:3175–82. doi: 10.1182/blood-2001-12-0207
31. Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science (2004) 304:104–7. doi: 10.1126/science.1093933
32. Asao H. [Analysis of gammac-dependent cytokines-mediated immunoregulation]. Rinsho Byori (2007) 55:51–8.
33. Akkina R. Human immune responses and potential for vaccine assessment in humanized mice. Curr. Opin. Immunol. (2013) 25:403–9. doi: 10.1016/j.coi.2013.03.009
34. Shultz LD, Brehm MA, Bavari S, Greiner DL. Humanized mice as a preclinical tool for infectious disease and biomedical research. Ann. N Y Acad. Sci. (2011) 1245:50–4. doi: 10.1111/j.1749-6632.2011.06310.x
35. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. (2005) 174:6477–89. doi: 10.4049/jimmunol.174.10.6477
36. Brehm MA, Cuthbert A, Yang C, Miller DM, DiIorio P, Laning J, et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rgamma(null) mutation. Clin. Immunol. (2010) 135:84–98. doi: 10.1016/j.clim.2009.12.008
37. Gimeno R, Weijer K, Voordouw A, Uittenbogaart CH, Legrand N, Alves NL, et al. Monitoring the effect of gene silencing by RNA interference in human CD34+ cells injected into newborn RAG2-/- gammac-/- mice: functional inactivation of p53 in developing T cells. Blood (2004) 104:3886–93. doi: 10.1182/blood-2004-02-0656
38. McDermott SP, Eppert K, Lechman ER, Doedens M, Dick JE. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood (2010) 116:193–200. doi: 10.1182/blood-2010-02-271841
39. Takenaka K, Prasolava TK, Wang JC, Mortin-Toth SM, Khalouei S, Gan OI, et al. Polymorphism in sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. (2007) 8:1313–23. doi: 10.1038/ni1527
40. Takizawa H, Manz MG. Macrophage tolerance: CD47-SIRP-alpha-mediated signals matter. Nat. Immunol. (2007) 8:1287–9. doi: 10.1038/ni1207-1287
41. Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, et al. Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc. Natl. Acad. Sci. U.S.A. (2011) 108:13224–9. doi: 10.1073/pnas.1101398108
42. Strowig T, Rongvaux A, Rathinam C, Takizawa H, Borsotti C, Philbrick W, et al. Transgenic expression of human signal regulatory protein alpha in Rag2-/-gamma(c)-/- mice improves engraftment of human hematopoietic cells in humanized mice. Proc. Natl. Acad. Sci. U.S.A. (2011) 108:13218–23. doi: 10.1073/pnas.1109769108
43. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. (2012) 12:786–98. doi: 10.1038/nri3311
44. Mosier DE. Human xenograft models for virus infection. Virology (2000) 271:215–9. doi: 10.1006/viro.2000.0336
45. McCune JM. Development and applications of the SCID-hu mouse model. Semin. Immunol. (1996) 8:187–96. doi: 10.1006/smim.1996.0024
47. Akkina R. New generation humanized mice for virus research: Comparative aspects and future prospects. Virology (2013) 435:14–28. doi: 10.1016/j.virol.2012.10.007
48. Biswas S, Chang H, Sarkis PT, Fikrig E, Zhu Q, Marasco WA. Humoral immune responses in humanized BLT mice immunized with West Nile virus and HIV-1 envelope proteins are largely mediated via human CD5+ b cells. Immunology (2011) 134:419–33. doi: 10.1111/j.1365-2567.2011.03501.x
49. Stoddart CA, Maidji E, Galkina SA, Kosikova G, Rivera JM, Moreno ME, et al. Superior human leukocyte reconstitution and susceptibility to vaginal HIV transmission in humanized NOD-scid IL-2Rγ(-/-) (NSG) BLT mice. Virology (2011) 417:154–60. doi: 10.1016/j.virol.2011.05.013
50. Manz MG. Human-hemato-lymphoid-system mice: opportunities and challenges. Immunity (2007) 26:537–41. doi: 10.1016/j.immuni.2007.05.001
51. Mangi MH, Newland AC. Interleukin-3 in hematology and oncology: current state of knowledge and future directions. Cytokines Cell Mol. Ther. (1999) 5:87–95.
52. Eder M, Geissler G, Ganser A. IL-3 in the clinic. Stem Cells (1997) 15:327–33. doi: 10.1002/stem.150327
53. Hu Z, Van Rooijen N, Yang YG. Macrophages prevent human red blood cell reconstitution in immunodeficient mice. Blood (2011) 118:5938–46. doi: 10.1182/blood-2010-11-321414
54. Takada K, Jameson SC. Naive T cell homeostasis: From awareness of space to a sense of place. Nat. Rev. Immunol. (2009) 9:823–32. doi: 10.1038/nri2657
55. van Lent AU, Dontje W, Nagasawa M, Siamari R, Bakker AQ, Pouw SM, et al. IL-7 enhances thymic human T cell development in "human immune system" Rag2-/-IL-2Rgammac-/- mice without affecting peripheral T cell homeostasis. J. Immunol. (2009) 183:7645–55. doi: 10.4049/jimmunol.0902019
56. Chen Q, Khoury M, Chen J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc. Natl. Acad. Sci. U.S.A. (2009) 106:21783–8. doi: 10.1073/pnas.0912274106
57. Brehm MA, Racki WJ, Leif J, Burzenski L, Hosur V, Wetmore A, et al. Engraftment of human HSCs in nonirradiated newborn NOD-scid IL2rγ null mice is enhanced by transgenic expression of membrane-bound human SCF. Blood (2012) 119:2778–88. doi: 10.1182/blood-2011-05-353243
58. Takagi S, Saito Y, Hijikata A, Tanaka S, Watanabe T, Hasegawa T, et al. Membrane-bound human SCF/KL promotes in vivo human hematopoietic engraftment and myeloid differentiation. Blood (2012) 119:2768–77. doi: 10.1182/blood-2011-05-353201
59. Becher B, Tugues S, Greter M. GM-CSF: From growth factor to central mediator of tissue inflammation. Immunity (2016) 45:963–73. doi: 10.1016/j.immuni.2016.10.026
60. Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, Ploss A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rγ(null) humanized mice. Blood (2011) 117:3076–86. doi: 10.1182/blood-2010-08-301507
61. Willinger T, Rongvaux A, Takizawa H, Yancopoulos GD, Valenzuela DM, Murphy AJ, et al. Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proc. Natl. Acad. Sci. U.S.A. (2011) 108:2390–5. doi: 10.1073/pnas.1019682108
62. Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, et al. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood (2011) 118:3119–28. doi: 10.1182/blood-2010-12-326926
63. Jaiswal S, Pearson T, Friberg H, Shultz LD, Greiner DL, Rothman AL, et al. Dengue virus infection and virus-specific HLA-A2 restricted immune responses in humanized NOD-scid IL2rgammanull mice. PloS One (2009) 4:e7251. doi: 10.1371/journal.pone.0007251
64. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. (2014) 32:364–72. doi: 10.1038/nbt.2858
65. Wilson RF. The death of Jesse gelsinger: new evidence of the influence of money and prestige in human research. Am. J. Law Med. (2010) 36:295–325. doi: 10.1177/009885881003600202
66. Schaefer KA, Wu WH, Colgan DF, Tsang SH, Bassuk AG, Mahajan VB. Unexpected mutations after CRISPR-Cas9 editing in vivo. Nat. Methods (2017) 14:547–8. doi: 10.1038/nmeth.4293
67. Good MF, Hawkes MT, Yanow SK. Humanized mouse models to study cell-mediated immune responses to liver-stage malaria vaccines. Trends Parasitol. (2015) 31:583–94. doi: 10.1016/j.pt.2015.06.008
68. Siu E, Ploss A. Modeling malaria in humanized mice: Opportunities and challenges. Ann. N Y Acad. Sci. (2015) 1342:29–36. doi: 10.1111/nyas.12618
69. Jiménez-Díaz MB, Mulet T, Viera S, Gómez V, Garuti H, Ibáñez J, et al. Improved murine model of malaria using plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob. Agents Chemother. (2009) 53:4533–6. doi: 10.1128/aac.00519-09
70. Soulard V, Bosson-Vanga H, Lorthiois A, Roucher C, Franetich JF, Zanghi G, et al. Plasmodium falciparum full life cycle and plasmodium ovale liver stages in humanized mice. Nat. Commun. (2015) 6:7690. doi: 10.1038/ncomms8690
71. Vaughan AM, Mikolajczak SA, Wilson EM, Grompe M, Kaushansky A, Camargo N, et al. Complete plasmodium falciparum liver-stage development in liver-chimeric mice. J. Clin. Invest. (2012) 122:3618–28. doi: 10.1172/jci62684
72. Chen Q, Amaladoss A, Ye W, Liu M, Dummler S, Kong F, et al. Human natural killer cells control plasmodium falciparum infection by eliminating infected red blood cells. Proc. Natl. Acad. Sci. U.S.A. (2014) 111:1479–84. doi: 10.1073/pnas.1323318111
73. Amaladoss A, Chen Q, Liu M, Dummler SK, Dao M, Suresh S, et al. De novo generated human red blood cells in humanized mice support plasmodium falciparum infection. PloS One (2015) 10:e0129825. doi: 10.1371/journal.pone.0129825
74. Wijayalath W, Majji S, Villasante EF, Brumeanu TD, Richie TL, Casares S. Humanized HLA-DR4.RagKO.IL2RγcKO.NOD (DRAG) mice sustain the complex vertebrate life cycle of plasmodium falciparum malaria. Malar J. (2014) 13:386. doi: 10.1186/1475-2875-13-386
75. Kaushansky K. Historical review: megakaryopoiesis and thrombopoiesis. Blood (2008) 111:981–6. doi: 10.1182/blood-2007-05-088500
76. Rongvaux A, Willinger T, Takizawa H, Rathinam C, Auerbach W, Murphy AJ, et al. Human thrombopoietin knockin mice efficiently support human hematopoiesis in vivo. Proc. Natl. Acad. Sci. U.S.A. (2011) 108:2378–83. doi: 10.1073/pnas.1019524108
77. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. (2009) 9:729–40. doi: 10.1038/nri2620
78. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
79. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science (2000) 288:2051–4. doi: 10.1126/science.288.5473.2051
80. Ide K, Wang H, Tahara H, Liu J, Wang X, Asahara T, et al. Role for CD47-SIRPalpha signaling in xenograft rejection by macrophages. Proc. Natl. Acad. Sci. U.S.A. (2007) 104:5062–6. doi: 10.1073/pnas.0609661104
81. Yu Z, Chen T, Cao X. Neutrophil sensing of cytoplasmic, pathogenic DNA in a cGAS-STING-independent manner. Cell Mol. Immunol. (2016) 13:411–4. doi: 10.1038/cmi.2015.34
82. Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, et al. The vascular endothelium and human diseases. Int. J. Biol. Sci. (2013) 9:1057–69. doi: 10.7150/ijbs.7502
83. Ji S, Dong W, Qi Y, Gao H, Zhao D, Xu M, et al. Phagocytosis by endothelial cells inhibits procoagulant activity of platelets of essential thrombocythemia in vitro. J. Thromb. Haemost. (2020) 18:222–33. doi: 10.1111/jth.14617
84. Shiu YT, McIntire LV. In vitro studies of erythrocyte-vascular endothelium interactions. Ann. BioMed. Eng. (2003) 31:1299–313. doi: 10.1114/1.1630320
85. Aird WC. Phenotypic heterogeneity of the endothelium: II. representative vascular beds. Circ. Res. (2007) 100:174–90. doi: 10.1161/01.RES.0000255690.03436.ae
86. Jourde-Chiche N, Fakhouri F, Dou L, Bellien J, Burtey S, Frimat M, et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. (2019) 15:87–108. doi: 10.1038/s41581-018-0098-z
87. Aird WC. Phenotypic heterogeneity of the endothelium: I. structure, function, and mechanisms. Circ. Res. (2007) 100:158–73. doi: 10.1161/01.RES.0000255691.76142.4a
88. Peng Q, Yeh H, Wei L, Enjyoj K, Machaidze Z, Csizmad E, et al. Mechanisms of xenogeneic baboon platelet aggregation and phagocytosis by porcine liver sinusoidal endothelial cells. PloS One (2012) 7:e47273. doi: 10.1371/journal.pone.0047273
89. El-Assaad F, Wheway J, Mitchell AJ, Lou J, Hunt NH, Combes V, et al. Cytoadherence of plasmodium berghei-infected red blood cells to murine brain and lung microvascular endothelial cells in vitro. Infect. Immun. (2013) 81:3984–91. doi: 10.1128/iai.00428-13
90. Graham SM, Rajwans N, Jaoko W, Estambale BB, McClelland RS, Overbaugh J, et al. Endothelial activation biomarkers increase after HIV-1 acquisition: plasma vascular cell adhesion molecule-1 predicts disease progression. Aids (2013) 27:1803–13. doi: 10.1097/QAD.0b013e328360e9fb
91. Zhang J, Abiraman K, Jones SM, Lykotrafitis G, Andemariam B. Regulation of active ICAM-4 on normal and sickle cell disease RBCs via AKAPs is revealed by AFM. Biophys. J. (2017) 112:143–52. doi: 10.1016/j.bpj.2016.11.3204
92. Maciaszek JL, Andemariam B, Abiraman K, Lykotrafitis G. AKAP-dependent modulation of BCAM/Lu adhesion on normal and sickle cell disease RBCs revealed by force nanoscopy. Biophys. J. (2014) 106:1258–67. doi: 10.1016/j.bpj.2014.02.001
93. Colin Y, Rahuel C, Wautier MP, El Nemer W, Filipe A, Cartron JP, et al. Red cell and endothelial Lu/BCAM beyond sickle cell disease. Transfus Clin. Biol. (2008) 15:402–5. doi: 10.1016/j.tracli.2008.07.011
94. Motegi S, Leitner WW, Lu M, Tada Y, Sárdy M, Wu C, et al. Pericyte-derived MFG-E8 regulates pathologic angiogenesis. Arterioscler. Thromb. Vasc. Biol. (2011) 31:2024–34. doi: 10.1161/atvbaha.111.232587
95. Fens MH, Storm G, Pelgrim RC, Ultee A, Byrne AT, Gaillard CA, et al. Erythrophagocytosis by angiogenic endothelial cells is enhanced by loss of erythrocyte deformability. Exp. Hematol. (2010) 38:282–91. doi: 10.1016/j.exphem.2010.02.001
96. Lauf PK. Eryptotic red blood cell adhesion to vascular endothelium: CXCL16/SR-PSOX, a pathological amplifier. focus on "Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX". Am. J. Physiol. Cell Physiol. (2012) 302:C642–643. doi: 10.1152/ajpcell.00453.2011
97. White JC, Pawar A, Fu G, Cui S, Tavernier F, Hamid M, et al. TR2/TR4 overexpression in a humanized sickle cell disease mouse model decreases RBC adhesion to VCAM-1. Blood Cells Mol. Dis. (2015) 55:316–7. doi: 10.1016/j.bcmd.2015.07.003
98. Yu H, Borsotti C, Schickel JN, Zhu S, Strowig T, Eynon EE, et al. A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood (2017) 129:959–69. doi: 10.1182/blood-2016-04-709584
99. Asfaha S, Dubeykovskiy AN, Tomita H, Yang X, Stokes S, Shibata W, et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology (2013) 144:155–66. doi: 10.1053/j.gastro.2012.09.057
100. McIntosh BE, Brown ME, Duffin BM, Maufort JP, Vereide DT, Slukvin II, et al. B6.SCID Il2rγ-/- Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Rep. (2015) 4:171–80. doi: 10.1016/j.stemcr.2014.12.005
101. Cosgun KN, Rahmig S, Mende N, Reinke S, Hauber I, Schäfer C, et al. Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell (2014) 15:227–38. doi: 10.1016/j.stem.2014.06.001
Keywords: humanized mouse models, RBC-related disease, erythrocytes, hematopoietic stem cell, innate immunity
Citation: Chen B, Liu H, Liu Z and Yang F (2023) Benefits and limitations of humanized mouse models for human red blood cell-related disease research. Front. Hematol. 1:1062705. doi: 10.3389/frhem.2022.1062705
Received: 06 October 2022; Accepted: 29 December 2022;
Published: 16 January 2023.
Edited by:
Louis Pelus, Indiana University Bloomington, United StatesCopyright © 2023 Chen, Liu, Liu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing Chen, Y2hlbmJpbmcyMDIwQGpsdS5lZHUuY24=