 Sean G. Lacoursiere
Sean G. Lacoursiere Jiri Safar
Jiri Safar David Westaway3
David Westaway3 Majid H. Mohajerani
Majid H. Mohajerani Robert J. Sutherland
Robert J. Sutherland
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Dement. , 12 September 2022
Sec. Cellular and Molecular Mechanisms of Dementia
Volume 1 - 2022 | https://doi.org/10.3389/frdem.2022.941879
Alzheimer's disease (AD) is characterized by the prion-like propagation of amyloid-β (Aβ). However, the role of Aβ in cognitive impairment is still unclear. To determine the causal role of Aβ in AD, we intracerebrally seeded the entorhinal cortex of a 2-month-old AppNL−G−F mouse model with an Aβ peptide derived from patients who died from rapidly progressing AD. When the mice were 3 months of age or 1 month following seeding, spatial learning and memory were tested using the Morris water task. Immunohistochemical labeling showed seeding with the Aβ was found accelerate Aβ plaque deposition and microgliosis in the AppNL−G−F mice, but this was dependent on the presence of the knocked-in genes. However, we found no correlation between pathology and spatial performance. The results of the present study show the seeding effects in the AppNL−G−F knock-in model, and how these are dependent on the presence of a humanized App gene. But these pathological changes were not initially causal in memory impairment.
Alzheimer's disease (AD) is the most common form of dementia, affects millions of people, and has a high social and monetary cost (Alzheimer's Association, 2019). It is characterized by stereotypical pathological stages of amyloid-β (Aβ) aggregation and neurofibrillary tangle formation that are progressive (Braak and Braak, 1991; Ettcheto et al., 2018)—this protein aggregation is assumed to be central to AD pathogenesis (Friesen and Meyer-Luehmann, 2019; McAllister et al., 2020). Progression of pathology is associated with memory loss, impaired thinking skills, and, eventually, impairments in all facets of life (Matteson et al., 1996; Ferri et al., 2004; de Vugt et al., 2005). The cause of AD may be a prion-like spread of Aβ, resulting in neuroinflammation, plaque deposition, and hyperphosphorylation of tau, ultimately causing synapse loss and brain atrophy (Harper and Lansbury, 1997; Bloom, 2014; Walker et al., 2018).
Intracerebral seeding has become an effective tool to understand the role of protein aggregation in neurodegenerative diseases as it allows the control over the spatial and temporal onset of amyloidosis (Friesen and Meyer-Luehmann, 2019) as seeding Aβ accelerates the deposition of Aβ plaque in vivo in a prion-like manner (Walker et al., 2016; Olsson et al., 2018), in which native Aβ species are misfolded following the template and conformational properties of the seeded Aβ (Come et al., 1993; Eisele, 2013). The effects of seeding are also dependent on the genotype of the host: Mice without mutations in App or only possessing murine App do not show this effect, or, if they do, the required incubation time increases significantly before effects are seen (Meyer-Leuhmann et al., 2006; Eisele et al., 2009; Friesen and Meyer-Luehmann, 2019).
Much of the intracerebral seeding work has been done using first-generation mouse models using synthetic or murine Aβ (McAllister et al., 2020); however, due to the presence of APP artifacts in the first-generation mouse models, the conclusions drawn about the correlation between Aβ pathology, the effects of seeding, and the behavioral outcomes are in question (Sasaguri et al., 2017, 2022). Recently, the single knock-in AppNL−G−F mouse model has been developed (Saito et al., 2014). To develop this second-generation, knock-in model, the murine Aβ sequence was first humanized, and Swedish, Beyreuther/Iberian, and Arctic mutations were inserted. The Swedish (NL) mutation (KM670/671NL) increases the production of APPβ and the C-terminal fragment containing the entire Aβ sequence in neuronal cells (Shin et al., 2010). The Beyreuther/Iberian (F) mutation (I716F) increases the ratio of Aβ42 to Aβ40 and APP C-terminal fragments but also decreases the APP intracellular domain production; this is thought to be due to a reduction in APP proteolysis by γ-secretase due to the mutation leading to a protein that is poorly processed by γ-secretase (Guardia-Laguarta et al., 2010). The Arctic mutation (G) alters binding properties of various antibodies to Aβ for immunohistochemistry (Saito et al., 2014).
Several studies have characterized the development of pathology, functional connectivity, and behavioral phenotypes of these mice under multiple conditions (Jafari et al., 2018; Latif-Hernandez et al., 2019, 2020; Mehla et al., 2019; Upite et al., 2020). Of the studies using this for Aβ seeding, both studies found seeding accelerated Aβ deposition, but neither tested behavior of the mice following the seeding (Purro et al., 2018; Ruiz-Riquelme et al., 2018). To address the paucity of behavioral testing immediately following seeding, we seeded human Aβ into the AppNL−G−F mouse model.
Here, we wanted to determine whether seeding Aβ would cause immediate impairment in spatial learning and memory in the single App knock-in mouse model and to further understand how the presence of mutations influenced the effects of seeding on Aβ deposition but also on the response of microglia. We used young mice to determine if the initial Aβ deposition caused cognitive impairment without any other endogenously generated pathology and cognitive impairment.
First, we predicted that the mice seeded with Aβ would show a reduction in spatial learning and memory; second, it is predicted that the presence and absence of the knocked-in mutations would determine the seeding effects and the level of cognitive impairment. Finally, we predicted that the Aβ seeding would increase activated microglia. Overall, we found that the effects of seeding human HPC tissue and Aβ containing tissue were dependent on the presence of the NL G F knock-in genes.
Fifty-three single knock-in App mice were seeded in this study. Similar number of male and female mice was used (31 men, 22 women) as no difference between sexes was found in previous characterization (Mehla et al., 2019). The mice were caged in standard housing, 2–5 mice per cage, and kept on a 12-h light/dark cycle. The mice were given ad libitum food and water. The mice were handled prior to behavioral testing, which was completed at approximately the same time during the light cycle by an experimenter blinded to the conditions.
The mice were created by crossing mice from the RIKEN institute and C57Bl/6J mice. The mice were grouped based on genotype and seed. The mice were either homozygous negative (App−/−), carrying no mutations; heterozygous (App+/−), carrying only one copy of the Swedish, Beyreuther/Iberian, and Arctic mutations; and homozygous positive (App+/+), carrying two copies of the knocked-in mutations. The mice were then randomly assigned to be seeded with the control or rpAD seed. See Table 1 for final grouping.
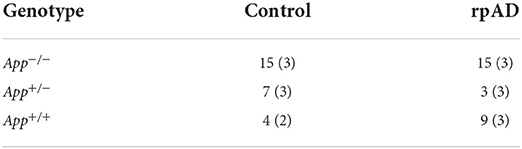
Table 1. Grouping of genotype and seed used for behavioral testing (and immunohistochemical analysis).
Punched mouse ear tissue was subjected to DNA extraction and PCR cycling using a Millipore-Sigma's RedExtract-N-Amp Tissue PCR kit (XNAT-100RXN). PCR cycler condition: 94°C for 3 min, 94°C for 30 s, 57°C for 45 s, 72°C for 1 min x 35 cycles. Stored at 4°C. Primer sequences were obtained from the Riken Institute: E16WT: 5′-ATC TCggAAgTgAAgATg-3′; E16MT: 5′-ATCTCggAAgTgAAT CTA-3′; WT: 5′-TgTAgATgAgAA CTT AAC-3′; loxP: 5′-CgT ATA ATgTATgCT ATA CgA Ag-3′. PCR products were loaded onto agarose gel electrophoresis for visualization, with a wild-type band at 700 bp and a mutant band at 400 bp.
The Aβ seeds were obtained from human hippocampal tissue (The National Prion Disease Pathology Surveillance Center at Case Western Reserve University Medical School), assessed for purity, and stereotaxically injected. The University Hospitals Institutional Review Board (IRB) approval is for all autopsied (“discarded”) human tissues, and all samples are anonymized (coded) and handled in compliance with the NIH policy to protect privacy. The type of seed was determined by the rate of AD progression. The biochemical analysis for the control tissue showed effectively zero Aβ (D/N) ratio Aβ40 or Aβ42, but Aβ%, according to sedimentation velocity in a calibrated sucrose ingredient, showed between ~8 and 12% Aβ40 and Aβ42 for the controls (Cohen et al., 2015). Furthermore, the rpAD seed showed higher levels of Aβ42 particles between 30 and 100 monomers with few particles <30 monomers; therefore, the rpAD seed particles are of higher molecular weight. It is these monomers that trigger the initial phase of Aβ seeding (Katzmarski et al., 2019). All brain tissue homogenate was buffered with phosphate-buffered saline (PBS) at a pH of 7.4 and kept at −80°C. The seed was 10% w/v. Prior to possession of the tissue, the tissue underwent several selection criteria steps.
1. Referral to the National Prion Disease Pathology Surveillance Center to classify any prion disease.
2. Six or more MMSE points of decline per year.
3. Absence of autosomal dominant AD patterns.
4. Absence of mutations in human prion protein.
5. Aβ and tau proteins resembling sporadic Alzheimer's disease.
6. No other neuropathological comorbidity.
7. All results within 85% confidence interval.
Another five inclusion criteria for the classical Alzheimer's disease tissue were used, they are as follows:
1. Clear clinical diagnosis of Alzheimer's disease.
2. No autosomal dominant patterns of dementia.
3. Alzheimer's disease based on tau and Aβ proteins.
4. No comorbidity with other neuropathological diseases.
5. Results within 95% confidence interval.
The mice were subcutaneously injected with buprenorphine (Vetergesic; 0.05 mg/kg; concentration = 0.03 mg/ml) 30 min prior to anesthesia induction (isoflurane). The oxygen flow rate for induction was between 4 and 5 L/min, and isoflurane was increased in a stepwise manner to a maximum of 5 L/min. Oxygen and anesthesia flow rates were reduced to 0.9 and 1.5–3 L/min, respectively, for the duration of the surgery. After the head was shaved, the scalp was cleaned with 4% stanhexidine (Omegalab), followed by 70% isopropyl alcohol. Lidocaine (0.1 ml of 0.2%; Rafter8) was subcutaneously injected under the scalp. Bregma was used to find the stereotaxic coordinates for the medial entorhinal cortex (Allen Institute for Brain Science, 2016). The coordinates used for injection were AP: −4.48, ML: 3.00, DV: 3.44 to target the medial entorhinal cortex. A 0.5-mm diameter hole was drilled through the skull to the brain at the coordinates.
The tissue homogenate was vortexed for 30 s before being loaded into the micropipette. Each mouse received 2 μL (1 μL/hemisphere) of the Aβ or control tissue homogenates. Seeding was performed with a Nanoject II (Drummond Scientific Company, PA) set to slowly inject 50.6 nL. Prior to seeding, a test injection was done to ensure proper flow, and the micropipette was cleaned with 70% isopropyl alcohol. The micropipette was inserted into the brain at the locations described and allowed to rest for 2 min before the first injection, with all following injections 20 s apart for a total of 20 injections. The micropipette stayed in place for 2 min after the final injection before being removed. A test injection was done again once the micropipette was removed, and the micropipette was cleaned with 70% isopropyl alcohol before the next hemisphere injection. The mice were kept on the same 12-h-light/dark cycle throughout recovery.
Following the behavioral testing, the mice were overdosed with an intraperitoneal injection of sodium pentobarbital and transcardially perfused with 1X phosphate-buffered saline (PBS) and 4% paraformaldehyde (PFA). The brains were fixed for 24 h in 4% PFA before being transferred to a solution of 30% sucrose solution for at least 3 days. The brains were sectioned on a frozen-sliding microtome at 40 μm in a 1:6 series and stored in 1X PBS + 0.02% sodium azide solution until staining. Prior to staining, the brain sections were mounted on super frost positively charged slides, allowed to dry up right for 30 min, and stored overnight at 4°C.
The slides were rinsed in 4% PFA for 4 min, washed with 1X Tris Buffer Saline (TBS), and underwent a 70% Formic Acid wash. The slides were rinsed in 1X TBS, TBS-A (1X TBS + 0.1% Triton X), and TBS-B [TBS + 0.1% Triton X + 2% bovine serum albumin (BSA)]. The primaries used were: Anti-82E1 [an Anti-β-amyloid (N), IBL, 10323, mouse] 1:1,000 and Anti-Iba1 (Rabbit, 019-19741, Wako) −20C 1:1,000, 1 ml/slide in TBS-B for 2 days at RT in a dark humid chamber, sealed in a plastic wrap on the rotator at 50RPM. Following primary incubation, TBS, TBS-A, and TBS-B washes were repeated. Secondaries used: Anti-mouse-alexa-488 [IgG (H + L) goat, Abcam, ab150113] 1:1,000 and anti-rabbit-alexa-594 [IgG (H + L) goat, Invitrogen, A11037] 1:1,000 (1 ml/slide) in TBS-B overnight in a dark humid chamber sealed in a plastic wrap on the rotator at 50RPM. Secondary was washed with 1X TBS and cover slipped with Vectashield + DAPI.
Full-slide imaging was completed on a digital slide scanner (NanoZoomer 2.0-RS, HAMAMATSU, JAPAN) at 20X magnification. Slide images were exported using NDP.View 2. Quantification of Aβ plaques and microgliosis was done by pixel and object classification using iLastik (version 1.3.0-OSX) (Berg et al., 2019) and ImageJ (version 1.51 s). ILastik was trained to segment both plaque and activated microglia separately. Threshold values were between 0.3 and 0.4, with a size filter minimum of 10 pixels. Sections were processed from ~AP 1.7 to −4.77, and the count for each section was averaged within groups.
The MWT pool was 1.55 m in diameter with water temperature maintained at 21 ± 1°C with a white, 12.5-cm submerged target platform and unobstructed distal cues surrounding the edges. A camera was fixed to the ceiling connected to a laptop with HVS Image 2100, which was used to track the swim patterns of the mice.
At 2 months of age, the mice were intracerebrally injected with a human tissue or the rpAD seed. One month following the seeding, the mice were tested using the MWT to determine if this seeding resulted in an impairment in spatial learning and memory. The mice were given four 30-s trials each day for 6 days. Starting position for each day was pseudo-randomized based on the cardinal starting locations, with each sequence of starting locations being different each day. On Day 7, the mice were tested using a no-platform probe. The temperature of the pool is kept at a cool 21°C to incentivize the mice to escape. Following the testing, tissue was collected to assess the extent of Aβ deposition and microgliosis.
Four parameters were measured during MWT training: the proximity of the mouse to the hidden platform, the time to find the platform, the path length, and the swim speed. Following the training, the mice were tested on a no-platform probe trial, and the amount of time spent in the target quadrant was compared to the opposing quadrant, and to the average of the two adjacent quadrants. For the pathology analysis, the number and the size of Aβ plaque throughout the brain were measured along with the number of microglia cells.
A two-way repeated measures ANOVA with a Dunnett's multiple comparison test was used to determine whether the mice significantly reduced their swim parameters from the 1st to the last day of the training and to determine whether the time spent in the target quadrant was influenced by the seed and the genotype, using the time in the target quadrant as the dependent variable. Probe test performance was also compared to chance performance (25%) using a single-tailed-paired sample t-test as we only wanted to know if performance was better than chance.
As the effects of seeding the rpAD seed were unknown, it is difficult to specify the effect size of interest. Furthermore, we were testing whether the seeding would have an effect or not, and not necessarily the size of the effect. Therefore, the resource equation method was used to determine if the sample size was sufficient. The error degrees of freedom were determined to be 47: six treatment groups subtracted from the total number of experimental units, 53 putting the error degrees of freedom far above the necessary amount to detect a specified effect (Festing et al., 2002).
All statistics were done using Prism 9 (mac OS).
Seeding either the control or rpAD seed into the App+/+ mice resulted in significant increase of a plaque count [F(2, 11) = 136.1, p < 0.0001], plaque size [F(2,11) = 150.4, p < 0.0001] and activated microglia cells [F(2, 11) = 18.19, p = 0.0003] compared to both the App−/− and App+/−; both of which showed no Aβ plaque pathology or activated microglia (Figure 1A and B). The seed was found to have no significant effect on the plaque count [F(1, 11) = 0.303, p = 0.593], plaque size [F(1, 11) = 1.64, p = 0.227], or activated microglia [F(1, 11) = 0.472, p = 0.507]. See Supplementary Figure 1 for pathology assessment 4 months following the seeding.
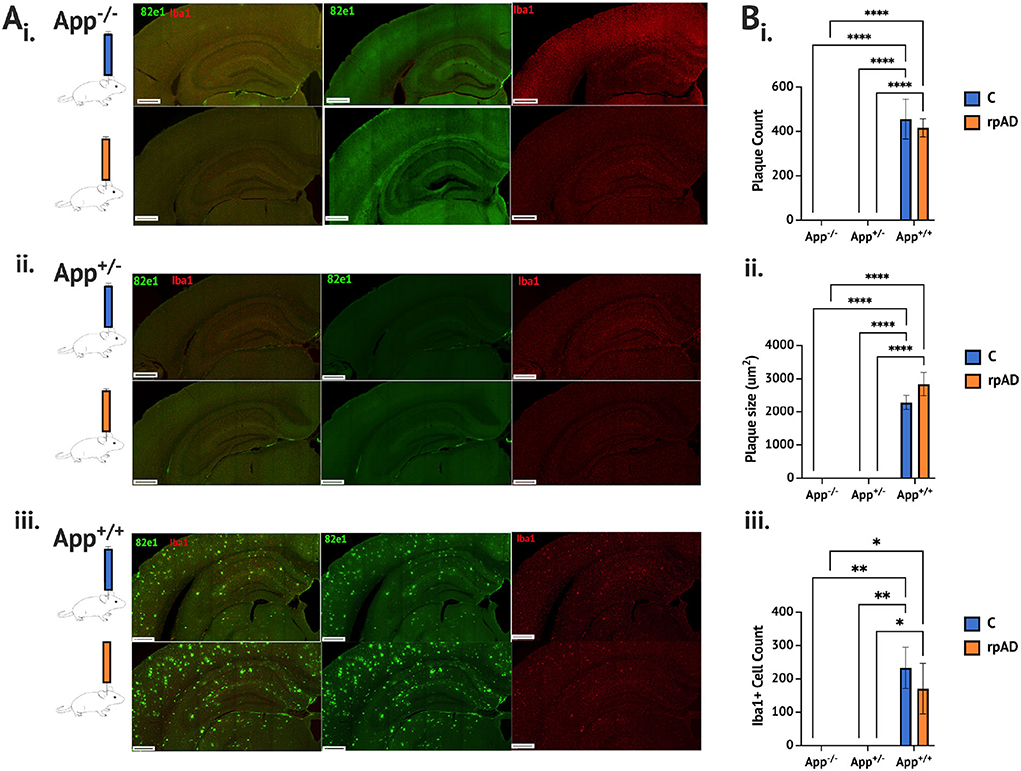
Figure 1. Aβ and microglia pathology analysis 1 month following seeding. (A) Photomicrographs of representative immunohistochemical staining of Aβ plaque (82e1, green) and microglia (Iba1, red), following seeding of the control HPC tissue (blue) and rpAD seed (orange) (B). Quantification of plaque and microglia showing (i) the plaque count (ii) the plaque size, and (iii) the microglia was significantly increased in the App+/+ mice following seeding with the C and rpAD seed. Scale bar = 1 mm. *p < 0.05; **p < 0.01, ***p < 0.001; ****p < 0.0001.
While no difference in the plaque count or size was found between the App+/+C and rpAD-seeded mice, the rpAD-seeded mice showed a significant correlation between the number of microglia and the number of plaques, whereas the App+/+ C mice did not show a significant correlation (Figure 2). A significant negative correlation was found for the number of activated microglia and the size of the plaque in the App+/+ C mice, but this correlation was not found in the rpAD mice.
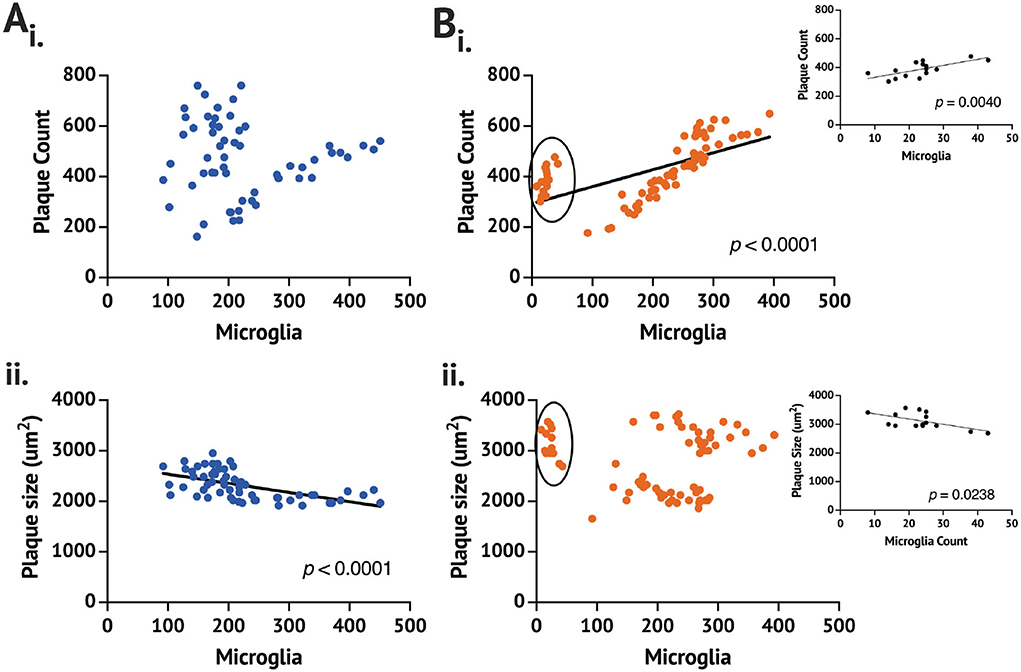
Figure 2. Correlation of the plaque and the microglia count 1 month following seeding in the App+/+ mice seeded with the (A) control (n = 2 brains,) and (B) rpAD seed (n = 3). Correlation is between the mean plaque count (i) or the plaque size (ii) and the number of counted microglia on full brain sections sampled from 1:6 sectioning series. Circled data in (Bi) and (Bii) are shown in the inset.
From the data, it is clear that one App+/+ rpAD mouse had a comparable Aβ plaque count and size but no more than 50 activated microglia counted. With removing the low-microglia data and analyzing them separately, the plaque count and microglia remain a significantly positive correlation; however, the plaque size and the microglia count become a significant positive correlation (p = 0.0047). The individual App+/+ rpAD mouse with low microglia when analyzed separately shows a significantly positive correlation between the plaque count and microglia (p = 0.004), but the correlation between the plaque size and the microglia count is found to be a significantly negative correlation (p = 0.0238), where the plaque count and microglia count increased together, but, as microglia increased, there was a reduction in the plaque size.
When trained and tested on the MWT (Figures 3A–F), the mice showed a significant reduction in proximity across training [F(5, 235) = 14.41, p < 0.0001]. While no group differences were found [F(5, 47) = 0.901, p = 0.489], a significant group × day interaction was found [F(25, 235) = 1.690, p = 0.025]. The App−/− C and App−/− rpAD mice showed a significant reduction in proximity between the 1st and last days of the training (p < 0.01) and so did the App+/− rpAD (p < 0.05) and App+/+ rpAD mice (p < 0.0001)—the App+/+ rpAD-seeded mice showed a significant reduction in proximity after 1 day of the training (p = 0.01). The App+/− C mice did not show a significant reduction between the 1st and last days but did show a significant reduction by the 5th day; the App+/+ C mice did not show any significant reduction (Figure 3Ai).
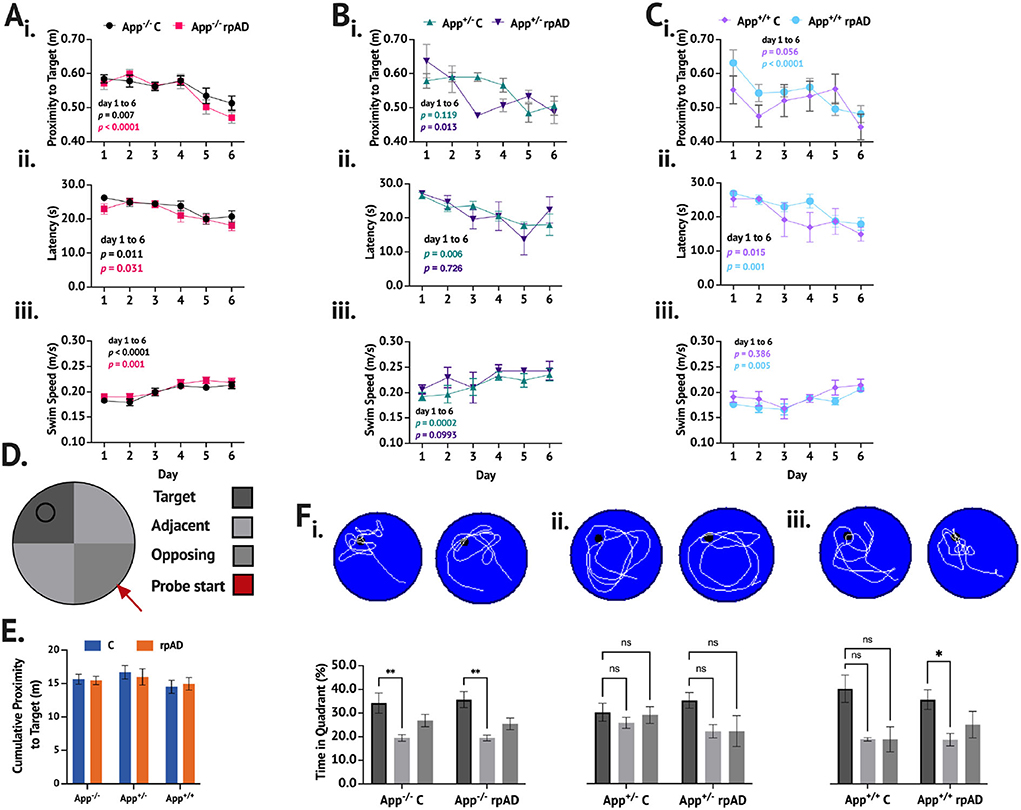
Figure 3. MWT performance following seeding. (A–C) show the performance of App−/−, App+/−, and App+/+ mice, respectively. (i.) Proximity to target, (ii.), latency to escape, and (iii.) swim speed were measured across six days of training. D. Representative schematic of pool set up. E. Cumulative proximity to target during probe trial showed no significant difference between groups. F. Representative swim path with a breakdown of time in quadrant immediately below for (i.) App−/−, (ii.) App+/−, and (iii.) App+/+ mice.
The mice also showed a significant reduction in latency to escape across training [F(5, 235) = 15.19, p < 0.0001], but no group differences [F(5, 47) = 1.190, p = 0.329] or group x day interaction [F(25, 235) = 0.969, p = 0.510] was found. All groups of the mice showed significant reduction in latency to escape. The App−/−C, App−/− rpAD, App+/− C, App+/+ C, and App+/+ rpAD all showed significant reduction in latency to escape by the final day (p < 0.05), but the App+/− rpAD only showed significant reduction by the 5th day (Figure 3Aii).
The swim speed was found to increase significantly over the training [F(5, 235) = 20.32, p < 0.0001], and significant differences between the groups were found [F(5, 47) = 3.84, p = 0.005]; no group x day interaction was found [F (25, 235) = 1.23, p = 0.218]. All the groups, except for the App+/− rpAD and App+/+ C significantly increased their swim from the 1st to last day1 of the training (p < 0.05). The App+/− C and rpAD mice were both found to have an overall significantly faster swim speed than the App+/+ rpAD mice (p < 0.05; Figure 3Aiii).
In the no-platform probe trial within the genotype, comparisons were used to determine if the mice spent significantly more time in the target quadrant compared to the average of the two adjacent quadrants and the opposing quadrant (the opposing quadrant was the starting quadrant). Both the App−/− C and rpAD mice spent significantly more time in the target quadrant compared to the non-target quadrants [F(2, 56) = 10.83, p = 0.0001], but the seed had no effect [F(1, 28) = 0.0003, p = 0.985]. Neither the App+/− C or rpAD mice spent significantly more time in the target quadrant [F(2, 16) = 1.98 p = 0.170], but, again, the seed had no significant effect on performance [F(1,8) = 0.556, p = 0.477]. Within the App+/+ mice, a significant preference for the target was found [F(2, 22) = 5.59, p = 0.012], with an overall preference for the target quadrant compared to the opposing (p = 0.0479) and adjacent (p = 0.0150) quadrants. The App+/+ rpAD mice showed significant preference to the target quadrant compared to the adjacent quadrants (p < 0.05), but not the opposing quadrant. The App+/+C mice showed no significant preference for the target quadrant (Figure 3Bi). Furthermore, no significant effect of seed [F(1, 47) = 0.0283, p = 0.867] or genotype [F(2, 47) = 0.892, p = 0.417] was found on the cumulative distance from the target location.
When comparing the time spent in the target quadrant to chance performance, the App−/− C [t(14) = 2.144, p = 0.025], App−/− rpAD [t(14) = 3.191, p = 0.003], App+/− rpAD [t(2) =2.925, p = 0.0498], App+/+ C [t(3) = 2.769, p = 0.035], and App+/+ rpAD [t(8) = 2.732, p = 0.0129]-seeded mice spent significantly more time in the target quadrant compared to what would be predicted by chance. The App+/− C mice did not show performance above chance [t(6) = 1.244, p = 0.130]. Furthermore, when looking at whether the mice crossed the platform location during the probe trial, 83% of the App−/− C mice, 92% of the App−/− rpAD, 83% of the App+/− C, 100% of the App+/− rpAD, 50% of the App+/+ C, and 57% of the App+/+ rpAD mice showed at least one platform crossing. The swim pattern also shows that the App−/− and App+/+ paths were much more directed in the target quadrant, whereas the App+/− mice showed a much more diffuse pattern of swimming, with the path following the edge of the pool.
Lastly, no effect of sex was found on time in the target quadrant [F(1, 41) = 0.345, p = 0.560].
Despite not all the mice showing preference for the target quadrant compared to the non-target quadrants, no significant effect of seed [F(1, 47) = 0.0141, p = 0.906] or genotype [F(2, 47) = 0.692, p = 0.506] was found on the time in the target quadrant.
To summarize, no significant effect of genotype or seed or overall significant differences were found in MWT performance between groups of the mice despite not all the groups showing significant learning or memory of the target location. The App−/− mice showed learning and memory, the App+/− mice did not, and only the App+/+ rpAD mice showed preference for the target quadrant despite the App+/+ C mice showing some evidence of learning, although all the groups showed significantly greater performance compared to chance, except for the App+/− C mice.
The results presented show that 1 month following seeding both human tissue and Aβ protein increased Aβ plaque pathology, and this was dependent on the presence of the NL G F mutations on the App gene. When tested on the MWT, the mice showed they were able to learn and remember the location of the hidden platform except for the App+/−C mice. Despite the most extensive pathology, the App+/+ mice were able to successfully learn the task. However, the App+/+ mice showed the smallest proportion of the mice, showing at least one platform crossing, whereas the App+/− mice showed relatively high occurrences of the mice crossing the platform at least once.
Here, we show that the initial pathology of AD—microglia activation and Aβ plaque deposition—is dependent on the combination of knocked-in genes and the seed. Each combination resulted in unique phenotypic expression of behavior, Aβ deposition, and the microglia activation pattern in young AppNL−G−F mice. One month following intracerebral seeding, prior to when the natural endogenous development of Aβ plaque deposition and microgliosis occurred, we found that Aβ plaque deposition and microgliosis increased in the App+/+ mice, but not in the App−/− and App+/− mice. It was not until 4 months following seeding did minimal Aβ plaque deposition occur in App+/−-seeded mice (Supplementary Figure 1). But no Aβ plaque pathology or microgliosis was found in the App−/− mice (Supplementary Figure 1). The difference in Aβ deposition and microgliosis between the mice tested could be that the genes promote Aβ deposition or reduce the ability to slow Aβ deposition. Here we provide evidence suggesting the difference in pathology is due to a reduced ability of the App+/+ mice to slow Aβ deposition; likely due to the prion - like properties of Aβ and the role microglia play in the development and growth of Aβ plaque.
In the test of spatial learning and memory used, both the App−/− and App+/+ mice showed evidence of learning the location of the hidden target. Due to only using 30-s trials, the learning curves were not as strong as those seen using 60-s trials, but the performance of the mice by the 6th day of the training was similar to tasks using longer trials; however, the increased days of training do result in better probe test performance (Mehla et al., 2019), suggesting that the task parameters resulted in similar patterns of learning compared to age matched, non-seeded controls, but, due to the reduced training volume before the testing, the no-platform probe trial becomes more difficult. However, no significant difference was found in the time spent in the target quadrant between the different groups in our study despite the App+/− C mice, showing no preference for the target quadrant; this may be due to the small sample size, but the swim path suggests these mice did not learn the task. The impairment found in the App+/− C mice is an effect that will have to be further investigated as well as an in-depth characterization of the App+/− mice.
The App knock-in mice have humanized Aβ, but if one allele was producing murine Aβ, a disruption in the processing and function of Aβ during development may occur, specifically impairing the cerebrovascular system (Luna et al., 2013). This may not be occurring when only murine Aβ is present (App−/−) or when only human Aβ is present (App+/+). Unfortunately, we did not assess cerebrovascular health in these mice, but other work has shown that cerebrovascular dysfunction is a risk-factor AD (Esiri et al., 1999; Zhai et al., 2016). But understanding how the cerebrovascular system changes in the brain during AD progression is important for understanding AD pathobiology.
One facet of AD that is still not well-understood is the pathogenetic mechanisms leading to the difference in progression and phenotypic expression of AD (Schellenberg and Montine, 2012; Cohen et al., 2015). Our results and others' suggest that two general factors influence the difference in phenotypic expression: the type of Aβ and the underlying genetics. The underlying mechanism leading to these phenotypic differences may be the activation pattern of microglia in the brain.
Here, we showed that the relationship of microglia to Aβ plaque pathology was influenced by the type of seed and the presence of the three knock-in genes described. For example, in the App+/+ C mice, no significant correlation was found between the plaque count and the activated microglia, but, as the number of the microglia increased, there was a concomitant decrease in the average size of the plaque. In the App+/+-rpAD mice, as the plaque count increased so did the microglia; however, the plaque size did not correlate with the number of activated microglia. Despite the differences in correlation between the size of the plaque and the number of microglia cells, the overall number of microglia was not found to be significantly different between the control and the rpAD-seeded App+/+ mice.
The App−/− mice did not develop plaque or microgliosis up to 7 months following seeding—or 9 months of age (not shown). The App+/− mice did develop plaque 4 months following seeding but to a significantly lesser degree than the App+/+ mice at the same age (Supplementary Figure 1).
Despite finding that microglia and the plaque size and the count were significantly related, we were not able to directly discern the direction of this relationship. But, from the work of others, the elimination of microglia results in a reduction of Aβ plaque production early in the disease, but not in the later portions of the disease (Spangenberg et al., 2019; Saucken et al., 2020). Microglia are thought to internalize neuronally derived Aβ and begin the initial aggregating phase of Aβ before being deposited into the extracellular space (Spangenberg et al., 2019), suggesting that the activation of microglia may initially be causing the plaque deposition to protect the brain from the exogenously introduced agents, such as bacteria, viruses, such as the currently relevant SARS-COV2 virus, and exogenous Aβ (Eimer et al., 2018; Dominy et al., 2019; Montalvan et al., 2020; Wu et al., 2020); however, Aβ deposition could also occur due to an autoimmune response (Meier-Stephenson et al., 2022). We show that this plaque/microglia response is dependent specifically on the brain environment and the genetics underlying this phenotype as we found no microglia activation in the control or the rpAD-seeded App−/− mice.
Microglia are known as the resident immune cells of the CNS and are known to interact with plaque to create a barrier. These innate immune cells (Webers et al., 2020) interact with plaque to control growth, development, and plaque morphology (Bolmont et al., 2008; Baik et al., 2016; Casali et al., 2020) and to prevent the toxic effects of Aβ42 (Condello et al., 2015). However, excessive uptake of Aβ can result in microglial death, resulting in the release of accumulated Aβ into the extracellular space and contributing to plaque growth (Baik et al., 2016). The activation of microglia cells with Aβ plaque pathology may explain why the App+/+ mice did not show impairment.
It is not uncommon for a peripheral inflammatory response to have effects on CNS function (Block, 2019). A poor microbiome, which is associated with inflammation, is associated with lower cognitive scores (Fröhlich et al., 2016; Gareau, 2016; Komanduri et al., 2021) and can also lead to AD (Thaiss et al., 2016; Shi et al., 2017; Lin et al., 2018). One potential mechanism that could explain why a poor microbiome is associated with lower cognitive could be due to disruption in tryptophan metabolism. Tryptophan metabolism is a key regulator of brain innate immunity (Meier-Stephenson et al., 2022) and a poor microbiota can lead to impaired tryptophan metabolism and serotonin signaling (Jenkins et al., 2016; Dinan and Cryan, 2017).
Further evidence that the etiology may be an immune response arises from fecal microbiota transplant experiments. The microbiota of the 6-month-old AppNL−G−F mice when transplanted into wild-type controls resulted in a disease phenotype; this effect was also sex and genotype specific (Kundu et al., 2022). The APP/PS1 mice transplanted with healthy fecal microbiota were found to have alleviated AD symptoms, such as a reduction in Aβ production and increased short chain fatty acid butyrate (Sun et al., 2019). Aged microbiota when transplanted was found to accelerate age and drive a pathological phenotype in young mice (Parker et al., 2022).
Lipid metabolism of microglia appears central to the functioning of microglia (Chausse et al., 2021), suggesting that disruption of these processes may impair microglia function and, therefore, their influence on plaque morphology and the innate immune system. As described above, the microbiota influences the immune system, and this appears to be through the maturation, function, and lipid metabolism of microglia in the CNS (Erny et al., 2015). Therefore, an impairment of the gut microbiome may be a central point in which AD begins. Its disruption leads to increased inflammation, disrupts microglia function and maturation, and, potentially, the metabolic processes of microglia, such as the processes related to managing Aβ plaque growth and toxicity (Bolmont et al., 2008; Condello et al., 2015; Baik et al., 2016).
Despite the described role of microglia in the etiology of AD, the role of Aβ oligomers cannot be overlooked. It is known that Aβ oligomers instead of fibrils and plaques are the most pathogenic Aβ species (Ashe, 2020). However, the toxicity of Aβ oligomers appears to be dependent on the temporal, spatial, and structural relationship to amyloid fibrils and dense core plaques (Liu et al., 2015). Liu et al. (2015) describe two classes of Aβ oligomers: Type 1 and Type 2. Type 2, while more abundant than Type 1, appears confined to the vicinity of plaque and does not impair cognition at levels relevant to AD. The Type-1 oligomers, however, are unrelated to amyloid fibrils and may, therefore, have a greater potential for neural dysfunction throughout the brain. It was concluded that, due to the containment of Type-2 oligomers to the Aβ plaques, the Type-2 oligomers are rendered functionally innocuous. Therefore, due to the intracerebral seeding of Aβ accelerating the nucleation and deposition of Aβ in the brain, a large portion of the Aβ oligomers in the brain may have been sequestered to the plaque, reducing the toxic effects of these oligomers, and preserving cognitive function. Yet microglia may be playing a role to contain the Type-2 Aβ oligomers inside the plaque, and it is not until later in the disease progression does the Type-2 Aβ oligomers leak out and begin damaging neurons throughout the brain (Liu et al., 2015). Unfortunately, we did not measure the levels of these two types of Aβ oligomers and, therefore, cannot determine if seeding altered the spatial or temporal characteristics of these oligomers. However, we do provide further evidence that Aβ monomers found in the seeds used can initiate Aβ aggregation in the AppNL−G−F mice.
We do acknowledge the limitations to this study. First, we focused only on spatial navigation learning and memory. In only testing spatial navigation, we were unable to make conclusions on the effects of seeding, or genotype in different cognitive domains. However, the MWT was originally designed to test the function of the HPC in memory, and one of the earliest impairments in AD is found in HPC memory. In future studies, additional behavioral tests should be included or a novel home cage-based assessment of rodent's behavior (Singh et al., 2019; Contreras et al., 2022) may offer insights into phenotypical expression of mouse genotypes and seeding and across time not found in traditional rodent behavioral testing. Furthermore, we did not measure soluble Aβ and, instead, focused on Aβ plaque load and characteristics throughout the brain and how this was associated with microglia. We, therefore, cannot make any conclusions on how seeding affected soluble Aβ. Lastly, while the control tissue had no rpAD Aβ, Aβ from the HPC where the tissue was collected could have induced the seeding response, which could explain the lack of difference in Aβ pathology 1 month following the seeding.
Given the recent failures of Aβ-targeted therapies to treat AD (Kurkinen, 2021), it is clear that the etiology of AD is not understood. The role of the immune system in the etiology of AD has been gaining interest (Jevtic et al., 2017). Recent hypotheses put forth have described that the initial Aβ plaque deposition that ultimately leads to AD occurs as a means to protect the brain from infection (Kumar et al., 2016; Eimer et al., 2018; Moir et al., 2018). While still speculative, our results along with others', suggest that the underlying genetic factors that contribute to AD may be closely related to the innate immune response and, specifically, the role of microglia. This immune response is determined by genetic and potentially epigenetic predisposition to the development of Aβ42 and the ratio of Aβ42 and Aβ40 but also the type of Aβ present in the brain, whether endogenous (Cohen et al., 2015, 2016) or exogenously introduced. It is well-known that both genetic and environmental factors influence the etiology of AD (McDonald et al., 2010), but, specifically, factors involving the immune system may be a novel approach to treat AD and age-related cognitive decline. Future studies should focus on microglia metabolism in health and disease and how this is influenced by the microbiome and the immune system.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by Animal Ethics Committee of the University of Lethbridge.
RS, MM, JS, DW, and SL: conceptualization. RS, SL, and DW: methodology. SL: software, validation, formal analysis, investigation, data curation, writing—original draft preparation, and visualization. RS, MM, DW, and JS: resources. SL, RS, MM, and DW: writing—review and editing and funding acquisition. RS and MM: supervision. SL and RS: project administration. All the authors have read and agreed to the published version of the manuscript.
This research was funded by a CIHR grant awarded to RS (452424).
We would like to thank T. Saito and T. C. Saido of the RIKEN Brain Science Institute for providing the original AppNL−G−F mice used to establish a colony at our institution, J. Safar and D. Westaway for supplying the Aβ homogenate, B. McAllister for statistical and data analysis assistance, Canadian Center for Behavioral Neuroscience Animal Care Staff, D. Shao and B. Mirza Agha for experimental help, V. Lapointe for immunohistochemistry assistance, and I. Q. Whishaw and J. Faraji for assistance with behavioral analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frdem.2022.941879/full#supplementary-material
Allen Institute for Brain Science. (2016). Injection sites and stereotaxic coordinates for anterograde projectome (brain-wide). Technical White Paper. Available online at: https://help.brain-map.org/download/attachments/2818171/InjectionSites_and_StereotaxicCoordinates.pdf
Alzheimer's Association (2019). 2019 Alzheimer's disease facts and figures. Alzheimers Dement. 15, 321–387. doi: 10.1016/j.jalz.2019.01.010
Ashe, K. (2020). The biogenesis and biology of amyloid β oligomers in the brain. Alzheimers Dement. 16, 1561–1567. doi: 10.1002/alz.12084
Baik, S., Kang, S., Son, S., and Mook-Jung, I. (2016). Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer's disease mouse model. Glia 64, 2274–2290. doi: 10.1002/glia.23074
Berg, S., Kutra, D., Kroeger, T., Straehle, C., Kausler, B., Haubold, C., et al. (2019). ilastik: Interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232. doi: 10.1038/s41592-019-0582-9
Block, J. (2019). Alzheimer's disease might depend on enabling pathogens which do not necessarily cross the blood-brain barrier. Med. Hypotheses 125, 129–136. doi: 10.1016/j.mehy.2019.02.044
Bloom, G. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Bolmont, T., Haiss, F., Eicke, D., Radde, R., Mathis, C. A., Klunk, W. E., et al. (2008). Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Casali, B. T., MacPherson, K. P., Reed-Geaghan, E. G., Landreth, G. E. (2020). Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol. Dis. 142, 104956. doi: 10.1016/j.nbd.2020.104956
Chausse, B., Kakimoto, P. A., and Kann, O. (2021). Microglia and lipids: how metabolism controls brain innate immunity. Semi. Cell Dev. Biol. 112, 137–144. doi: 10.1016/j.semcdb.2020.08.001
Cohen, M., Appleby, B., and Safar, J. G. (2016). Distinct prion-like strains of amyloid beta implicated in phenotypic diversity of Alzheimer's disease. Prion 10, 9–17. doi: 10.1080/19336896.2015.1123371
Cohen, M., Kim, C., Haldiman, T., ElHag, M., Mehndiratta, P., Pichet, T., et al. (2015). Rapidly progressive Alzheimer's disease features distinct structures of amyloid-β. Brain 138, 1009–1022. doi: 10.1093/brain/awv006
Come, J., Fraser, P., and Lansbury, P. Jr. (1993). A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc. Natl. Acad. Sci. U.S.A. 90, 5959–5963. doi: 10.1073/pnas.90.13.5959
Condello, C., Yuan, P., Schain, A., and Grutzendler, J. (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 6, 6176. doi: 10.1038/ncomms7176
Contreras, E. B., Sutherland, R. J., Mohajerani, M. H., and Whishaw, I. Q. (2022). Challenges of a small world analysis for the continuous monitoring of behavior in mice. Neurosci. Biobehav. Rev. 136, 104621. doi: 10.1016/j.neubiorev.2022.104621
de Vugt, M., Stevens, F., Aalten, P., Lousberg, R., Jaspers, N., and Verhey, F. (2005). A prospective study of the effects of behavioral symptoms on the institutionalization of patients with dementia. Int. Psychogeriatr. 17, 577–589. doi: 10.1017/S1041610205002292
Dinan, T. G., and Cryan, J. F. (2017). The microbiome-gut-brain axis in health and disease. Gastroenterol. Clin. North Am. 46, 77–89. doi: 10.1016/j.gtc.2016.09.007
Dominy, S., Lynch, C., Ermini, F., Benedyk, M., Marczyk, A., Kondradi, A., et al. (2019). Porphyromonas gingivalis in Alzheimer's disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 5, eaau3333. doi: 10.1126/sciadv.aau3333
Eimer, W., Kumar, D., Shanmugam, N., Rodriguez, A., Mitchell, T., Washicosky, K., et al. (2018). Alzheimer's Disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99, 56–63. doi: 10.1016/j.neuron.2018.06.030
Eisele, Y. (2013). From soluble Aβ to progressive Aβ aggregation: could prion-like templated misfolding play a role? Brain Pathol. 23, 333–341. doi: 10.1111/bpa.12049
Eisele, Y., Bolmont, T., Heikenwalder, M., Langer, F., Jacobson, L., Yan, Z., et al. (2009). Induction of cerebral β-amyloidosis: intracerebral versus systemic Aβ inoculation. Proc. Natl. Acad. Sci. U.S.A. 106, 12926–12931. doi: 10.1073/pnas.0903200106
Erny, D., Hrabě de Angelis, A. L., Jaitin, D., Wieghofer, P., Staszewski, O., David, E., et al. (2015). Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977. doi: 10.1038/nn.4030
Esiri, M., Nagy, Z., Smith, M., Barnetson, L., and Smith, A. (1999). Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer's disease. Lancet 354, 919–920. doi: 10.1016/S0140-6736(99)02355-7
Ettcheto, M., Abad, S., Petrov, D., Pedrós, I., Busquets, O., Sánchez-López, E., et al. (2018). Early preclinical changes in hippocampal CREB-Binding protein expression in a mouse model of familial Alzheimer's disease. Mol. Neurobiol. 55, 4885–4895. doi: 10.1007/s12035-017-0690-4
Ferri, C., Ames, D., Prince, M., 10/66 Dementia Research Group (2004). Behavioral and psychological symptoms of dementia in developing countries. Int. Psychogeriatr. 16, 441–459. doi: 10.1017/S1041610204000833
Festing, M., Overend, P., Gaines Das, R., Borja, M., and Berdoy, M. (2002). The Design of Animal Experiments: Reducing the Use of Animals in Research Through Better Experimental Design, Vol. 14. London, United Kingdom: The Royal Society of Medicine Press.
Friesen, M., and Meyer-Luehmann, M. (2019). aβ seeding as a tool to study cerebral amyloidosis and associated pathology. Front. Mol. Neurosci. 12, 233. doi: 10.3389/fnmol.2019.00233
Fröhlich, E. E., Farzi, A., Mayerhofer, R., Reichmann, F., Jačan, A., Wagner, B., et al. (2016). Cognitive impairment by antibiotic-induced gut dysbiosis: analysis of gut microbiota-brain communication. Brain Behav Immunity 56, 140–155. doi: 10.1016/j.bbi.2016.02.020
Gareau, M. G. (2016). Cognitive function and the microbiome. Int. Rev. Neurobiol. 131, 227–246. doi: 10.1016/bs.irn.2016.08.001
Guardia-Laguarta, C., Pera, M., Clarimón, J., Molinuevo, J. L., Sànchez-Valle, R., Lladó, A., et al. (2010). Clinical, neuropathologic, and biochemical profile of the amyloid precursor protein I716F mutation. J. Neuropathol. Exp. Neurol. 69, 53–59. doi: 10.1097/NEN.0b013e3181c6b84d
Harper, J., and Lansbury, P. (1997). Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Ann. Rev. Biochem. 66, 385–407. doi: 10.1146/annurev.biochem.66.1.385
Jafari, Z., Mehla, J., Kolb, B., and Mohajerani, M. (2018). Gestational stress augments postpartum β-amyloid pathology and cognitive decline in a mouse model of Alzheimer's disease. Cereb. Cortex 29, 3712–3724. doi: 10.1093/cercor/bhy251
Jenkins, T. A., Nguyen, J. C. D., Polglaze, K. E., and Bertrand, P. P. (2016). Influence of tryptophan and serotonin on mood and cognition with a possible role of the gut-brain axis. Nutrients 8, 56. doi: 10.3390/nu8010056
Jevtic, S., Sengar, A., Salter, M., and McLaurin, J. (2017). The role of the immune system in Alzheimer disease: etiology and treatment. Age Res. Rev. 40, 84–94. doi: 10.1016/j.arr.2017.08.005
Katzmarski, N., Ziegler-Waldkirch, S., Scheffler, N., Witt, C., Abou-Ajram, C., Nuscher, B., et al. (2019). Aβ oligomers trigger and accelerate Aβ seeding. Brain Pathol. 30, 36–45. doi: 10.1111/bpa.12734
Komanduri, M., Savage, K., Lea, A., McPhee, G., Nolidin, K., Deleuil, S., et al. (2021). The relationship between gut microbiome and cognition in older Australians. Nutrients 14, 64. doi: 10.3390/nu14010064
Kumar, D., Choi, S., Washicosky, K., Eimer, W., Tucker, S., Ghofrani, J., et al. (2016). Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Alzheimers Dis. 8, 340ra72. doi: 10.1126/scitranslmed.aaf1059
Kundu, P., Stagaman, K., Kasschau, K., Holden, S., Shulzhenko, N., Sharpton, T. J., et al. (2022). Fecal implants from app (NL-G-F) and app (NL-G-F/E4) donor mice sufficient to induce behavioral phenotypes in germ-free mice. Front. Behav. Neurosci. 16, 791128. doi: 10.3389/fnbeh.2022.791128
Kurkinen, M. (2021). Alzheimer's trials: a cul-de-sac with no end in sight. Adv. Clin. Exp. Med. 30, 653–654. doi: 10.17219/acem/139501
Latif-Hernandez, A., Sabanov, V., Ahmed, T., Craessaerts, K., Saito, T., Saido, T., et al. (2020). The two faces of synaptic failure in AppNL-G-F knock-in mice. ALzheimers Res. Ther. 12, 100. doi: 10.1186/s13195-020-00667-6
Latif-Hernandez, A., Shah, D., Craessaerts, K., Saido, T., Saito, T., De Stropper, B., et al. (2019). Subtle behavioral changes and increased prefrontal-hippocampal network T synchronicity in APPNL–G–F mice before prominent plaque deposition. Behav. Brain Res. 364, 431–441. doi: 10.1016/j.bbr.2017.11.017
Lin, L., Zheng, L. J., and Zhang, L. J. (2018). Neuroinflammation, gut microbiome, and Alzheimer's disease. Mol. Neurobiol. 55, 8243–8250. doi: 10.1007/s12035-018-0983-2
Liu, P., Reed, M., Kotilinek, L., Grant, M., Forster, C., Qiang, W., et al. (2015). Quaternary structure defines a large class of amyloid-β oligomers neutralized by sequestration. Cell Rep. 11, 1760–1771. doi: 10.1016/j.celrep.2015.05.021
Luna, S., Cameron, D., Ethell, D. (2013). Amyloid-β and APP deficiencies cause severe cerebrovascular defects: important work for an old villain. PLoS ONE 8, e75052. doi: 10.1371/journal.pone.0075052
Matteson, M., Linton, A., Barnes, S., Cleary, B., Lichtenstein, M. (1996). The relationship between Piaget and cognitive levels in persons with Alzheimer's disease and related disorders. Aging Clin. Exp. Res. 8, 61–69. doi: 10.1007/BF03340117
McAllister, B. B., Lacoursiere, S. G., Sutherland, R. J., Mohajerani, M. H. (2020). Intracerebral seeding of amyloid-β and tau pathology in mice: factors underlying prion-like spreading and comparisons with α-synuclein. Neurosci. Biobehav. Rev. 112, 1–27. doi: 10.1016/j.neubiorev.2020.01.026
McDonald, R., Craig, L., and Hong, N. (2010). The etiology of age-related dementia is more complicated than we think. Behav. Brain Res. 214, 3–11. doi: 10.1016/j.bbr.2010.05.005
Mehla, J., Lacoursiere, S., Lapointe, V., McNaughton, B., Sutherland, R., McDonald, R., et al. (2019). Age-dependent behavioral and biochemical characterization of single APP knock-in mouse (APPNL-G-F/NL-G-F) model of Alzheimer's disease. Neurobiol. Aging 75, 25–37. doi: 10.1016/j.neurobiolaging.2018.10.026
Meier-Stephenson, F. S., Meier-Stephenson, V. C., Carter, M. D., Meek, A. R., Wang, Y., Pan, L., et al. (2022). Alzheimer's disease as an autoimmune disorder of innate immunity endogenously modulated by tryptophan metabolites. Alzheimers Dement. 8, e12283. doi: 10.1002/trc2.12283
Meyer-Leuhmann, M., Coomaraswamy, J., Bolmont, T., Kaeser, S., Schaefer, C., Kilger, E., et al. (2006). Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 313, 1781–1784. doi: 10.1126/science.1131864
Moir, R., Lathe, R., and Tanzi, R. (2018). The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement. 14, 1602–1614. doi: 10.1016/j.jalz.2018.06.3040
Montalvan, V., Lee, J., Bueso, T., De Toledo, J., Rivas, K. (2020). Neurological manifestations of COVID-19 and other coronavirus infections: a systematic review. Clin. Neurol. Neurosurg. 194, 105921. doi: 10.1016/j.clineuro.2020.105921
Olsson, T., Klementieva, O., and Gouras, G. (2018). Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 113, 1–10. doi: 10.1016/j.nbd.2018.01.015
Parker, A., Romano, S., Ansorge, R., Aboelnour, A., Le Gall, G., Savva, G. M., et al. (2022). Fecal microbiota transfer between young and aged mice reverses hallmarks of the aging gut, eye, and brain. Microbiome 10, 68. doi: 10.1186/s40168-022-01243-w
Purro, S., Farrow, M., Linehan, J., Nazari, T., Thomas, D., Chen, Z., et al. (2018). Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 564, 415–419. doi: 10.1038/s41586-018-0790-y
Ruiz-Riquelme, A., Lau, H., Stuart, E., Goczi, A., Wang, Z., Schmitt-Ulms, G., et al. (2018). Prion-like propagation of β-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol. Commun. 6, 26. doi: 10.1186/s40478-018-0529-x
Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., et al. (2014). Single app knock-in mouse models of Alzheimer's disease. Nat. Neurosci. 17, 661–663. doi: 10.1038/nn.3697
Sasaguri, H., Hashimoto, S., Watamura, N., Sato, K., Takamura, R., Nagata, K., et al. (2022). Recent advances in the modeling of Alzheimer's disease. Front. Neurosci. 16, 807473. doi: 10.3389/fnins.2022.807473
Sasaguri, H., Nilsson, P., Hashimoto, S., Nagata, K., Saito, T., De Strooper, B., et al. (2017). APP mouse models for Alzheimer's disease preclinical studies. EMBO J. 36, 2473–2487. doi: 10.15252/embj.201797397
Saucken, V. E., von Jay, T. R., and Landreth, G. E. (2020). The effect of amyloid on microglia-neuron interactions before plaque onset occurs independently of TREM2 in a mouse model of Alzheimer's disease. Neurobiol. Dis. 145, 105072. doi: 10.1016/j.nbd.2020.105072
Schellenberg, G. D., and Montine, T. J. (2012). The genetics and neuropathology of Alzheimer's disease. Acta Neuropathol. 124, 305–323. doi: 10.1007/s00401-012-0996-2
Shi, N., Li, N., Duan, X., and Niu, H. (2017). Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 4, 14. doi: 10.1186/s40779-017-0122-9
Shin, J., Yu, S.-B., Yu, U. Y., Jo, S. A., and Ahn, J.-H. (2010). Swedish mutation within amyloid precursor protein modulates global gene expression towards the pathogenesis of Alzheimer's disease. BMB Rep. 43, 704–709. doi: 10.5483/BMBRep.2010.43.10.704
Singh, S., Bermudez-Contreras, E., Nazari, M., Sutherland, R. J., and Mohajerani, M. H. (2019). Low-cost solution for rodent home-cage behaviour monitoring. PLoS ONE 14, e0220751. doi: 10.1371/journal.pone.0220751
Spangenberg, E., Severson, P. L., Hohsfield, L. A., Crapser, J., Zhang, J., Burton, E. A., et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat. Commun. 10, 3758. doi: 10.1038/s41467-019-11674-z
Sun, J., Xu, J., Ling, Y., Wang, F., Gong, T., Yang, C., et al. (2019). Fecal microbiota transplantation alleviated Alzheimer's disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 9, 189. doi: 10.1038/s41398-019-0525-3
Thaiss, C. A., Zmora, N., Levy, M., and Elinav, E. (2016). The microbiome and innate immunity. Nature 535, 65–74. doi: 10.1038/nature18847
Upite, J., Kadish, I., van Groen, T., and Jansone, B. (2020). Subchronic administration of auranofin reduced amyloid-β plaque pathology in a transgenic APP(NL-G-F/NL-G-F) mouse model. Brain Res. 1746, 147022. doi: 10.1016/j.brainres.2020.147022
Walker, L., Schelle, J., and Jucker, M. (2016). The Prion-Like properties of amyloid-β assemblies: implications for Alzheimer's disease. Cold Spring Harb. Perspect. Med. 6, a024398. doi: 10.1101/cshperspect.a024398
Walker, L. C., Lynn, D. G., and Chernoff, Y. O. (2018). A standard model of Alzheimer's disease? Prion 12, 261–265. doi: 10.1080/19336896.2018.1525256
Webers, A., Heneka, M. T., and Gleeson, P. A. (2020). The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer's disease. Immunol. Cell Biol. 98, 28–41. doi: 10.1111/imcb.12301
Wu, Y., Xu, X., Chen, Z., Duan, J., Hashimoto, K., Yang, L., et al. (2020). Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immunity 87, 18–22. doi: 10.1016/j.bbi.2020.03.031
Keywords: seeding, Alzheimer's disease, microglia, mice, memory, amyloid-β
Citation: Lacoursiere SG, Safar J, Westaway D, Mohajerani MH and Sutherland RJ (2022) The effect of Aβ seeding is dependent on the presence of knock-in genes in the AppNL−G−F mice. Front. Dement. 1:941879. doi: 10.3389/frdem.2022.941879
Received: 11 May 2022; Accepted: 08 August 2022;
Published: 12 September 2022.
Edited by:
Nathaniel G. N. Milton, Leeds Trinity University, United KingdomReviewed by:
Jacob Raber, Oregon Health and Science University, United StatesCopyright © 2022 Lacoursiere, Safar, Westaway, Mohajerani and Sutherland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert J. Sutherland, cm9iZXJ0LnN1dGhlcmxhbmRAdWxldGguY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.