 Alejandro Cano-Villagrasa
Alejandro Cano-Villagrasa Francisco J. Moya-Faz
Francisco J. Moya-Faz Nadia Porcar-Gozalbo2*
Nadia Porcar-Gozalbo2* Miguel López-Zamora
Miguel López-Zamora
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Child Adolesc. Psychiatry, 22 February 2024
Sec. Autism and Other Neurodevelopmental Disorders
Volume 3 - 2024 | https://doi.org/10.3389/frcha.2024.1265081
Autism Spectrum Disorder (ASD) is a neurodevelopmental disorder characterized by deficits in social communication, repetitive behaviors, and restricted interests (1). ASD is associated with a broad range of concomitant conditions, with epilepsy being one of the most significant and frequent (2, 3). Although ASD and epilepsy exhibit different symptoms and underlying causes (4), there is a strong relationship in their physical and cognitive manifestations (4, 5), as confirmed by the fact that up to 30% of individuals with ASD may have epilepsy (6).
In a recent meta-analysis, a comprehensive exploration of seventy-three studies delved into the point prevalence of active epilepsy, ultimately distilling 67 estimates from 63 unique studies for inclusion in the meta-analysis (6, 7). The resultant synthesis revealed a pooled point prevalence of active epilepsy standing at 6.38 per 1,000 persons (7). Shifting focus to the USA, several population-based studies in the pediatric realm uncovered a notably higher incidence of epilepsy within the first year of life, ranging from 82.1 to 118 per 100,000 person-years, eclipsing the rate observed in older children, which hovered around 46 per 100,000 person-years. An additional prospective study mirrored this trend, recording an incidence of 75 per 100,000 live births before 6 months and 62 per 100,000 between 6 and 12 months—an unprecedented revelation that diverges significantly from previously published estimates (8). Beyond point prevalence, twelve studies contributed insights into the annual prevalence of active epilepsy (9). The pooled outcome showcased an annual prevalence of 2.83 per 1,000 persons, while the median annual prevalence charted at 3.91 per 1,000 persons (10). Nestled within this complex tapestry of prevalence, the pediatric population grappling with both ASD and epilepsy emerges as a multifaceted, multidimensional conundrum. This intricate landscape unfolds against a backdrop of diverse symptoms, genetic foundations, and clinical expressions (11). ASD and epilepsy embark on an evolutionary journey from childhood to adulthood, casting a growing therapeutic demand that resonates not only with patients and their families but reverberates within educational systems and society at large (11, 12).
Delving into the realm of seizures, their classification unfolds into episodes characterized by either motor or non-motor symptoms—a spectrum encompassing generalized tonic-clonic seizures, clonic seizures, tonic seizures, and myoclonic seizures (13–15). Typical absences manifest with a sudden onset, interrupting ongoing activities, featuring a fixed gaze, unresponsiveness to speech, and a duration spanning seconds to half a minute, culminating in a swift recovery (16). Crucially, it's paramount to underscore that the term “absence” doesn't equate to a fixed gaze, as this characteristic may also manifest in focal onset seizures (17). In contrast, atypical absence displays more pronounced changes in tone compared to typical absence, marked by a non-abrupt onset or termination. Myoclonic absence introduces a sudden, brief (<100 ms), involuntary, non-repetitive, and non-sustained contraction with associated absence. Adding another layer to this intricate spectrum, absence with eyelid myoclonia involves eyelid jerks occurring at a frequency of less than 3 per second, often with eyes deviated upward, typically lasting less than 10 s. This phenomenon is frequently triggered by eye closure and bears a high likelihood of photosensitivity (18).
In 2017, the ILAE Epilepsy Classification unfurled three diagnostic levels (15), enriching our understanding of this intricate landscape. The first level takes root in the seizure type, encompassing an expansive array of concepts—be they focal, generalized, or of unknown onset. This level also introduces two pivotal concepts woven into the diagnostic fabric: comorbidity (associated pathological entities) and etiology. Some patients may find a resting place at this diagnostic level due to investigational limitations, either as a valid endpoint or as the precursor in their diagnostic journey (19). Stepping into the second diagnostic level, the scene unfolds when at least one EEG and brain imaging study graces the diagnostic landscape. Here, the focus shifts to determining the type of epilepsy—focal, generalized, combined (merging both focal and generalized seizures, a common theme in various epileptic syndromes), or of unknown origin. Etiological diagnoses cast their net into structural, genetic, infectious, metabolic, immune, or unknown categories (20). In the intricate dance of medical complexities, a patient may wear more than one etiological hat. For instance, a patient with tuberous sclerosis might proudly display cortical tubers, embodying both a structural and genetic etiology (21). The final frontier, the third diagnostic level, immerses us in the realm of epileptic syndromes—a composite tapestry weaving together common characteristics such as seizure types, specific EEG findings, age-dependent imaging features, onset and remission age, specific triggering factors, daily variations, prognosis nuances, and distinctive intellectual and psychiatric comorbidities. It's a canvas where etiological and treatment implications unfurl their tales (22, 23). This paradigm-shifting classification boldly swaps out the term “benign” for the more apt descriptors “self-limited” or “drug-responsive.” The unfolding narrative beckons further exploration and inquiry into the ever-evolving lexicon of epilepsy classification.
Regarding ASD, there is significant interindividual clinical variability. Within families, it has been indicated that both genetic and environmental factors influence the phenotypic expression of ASD (12, 13). Many of the core or nuclear symptoms, while reduced to deficits in social communication and interaction and repetitive or stereotyped behaviors, do not manifest their extensive symptomatic expression (10–13).
The neural pathways proposed as the biological basis in ASD associated with epilepsy are related to: (1) imbalance between the glutamatergic (excitatory) and GABAergic (inhibitory) pathways; (2) alteration in the cholinergic pathway; and (3) disruption in synaptic plasticity and oxidative stress (23).
ASD and epilepsy are associated with a high level of psychiatric comorbidity, including anxiety, self or hetero-aggression, depression, hyperactivity, obsessive-compulsive disorder, attention difficulties, tics, eating disorders, executive function impairment, and sleep disorders, which may require pharmacological therapy at some stage of evolution, as well as other relevant symptoms (24), highlighted in the following Table 1:
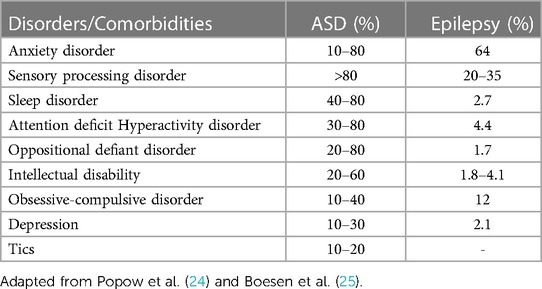
Table 1. Comorbidities associated with ASD or epilepsy condition.
There are commonly other comorbidities related to severe neurological alterations. A well-studied example of which association between ASD and epilepsy is tuberous sclerosis, in which infantile epileptic spasms appear to be a risk factor for ASD regardless of the location and number of brain tubers (26). All of this is related to the fact that, during the ontogenesis of the nervous system, certain brain areas mature chronologically before others, following a genetically determined program (27). If this maturation process is interfered with by frequent seizures, the consequences can be serious for the consolidation of emerging cognitive functions and the development of the social brain (28). Moreover, epileptiform discharges can occur in the absence of clinical seizures but still impact the brain's maturation process (29). Therefore, tuberous sclerosis represents one of the most attractive etiopathogenic models—genetic, biochemical, structural, and neurophysiological—to understand the bidirectional interaction between ASD and epilepsy (27).
Considering this information, the main objective of this opinion article is to discuss and update the primary pharmacological and surgical treatments for the care of the pediatric population diagnosed with ASD and epilepsy.
The pharmacological treatment choice in the pediatric population with ASD and epilepsy is complex and requires an interdisciplinary approach. Specialists in neurology, pediatrics, and psychiatry must design a personalized treatment plan to address the child's needs (28).
As mentioned earlier, the comorbidity of ASD and epilepsy presents a wide range of associated symptoms in individuals (29). The pharmacological care for this population becomes significantly complex. Many experts recommend treatment plan begins with the management of ASD-related symptoms using medications that help reduce or alleviate these symptoms (30). Several studies refer to the administration of drugs such as antipsychotics or antidepressants to address the semiological characteristics that appear in this disorder (31). Table 2 provides a list of the most common alterations in ASD and their pharmacological treatment.
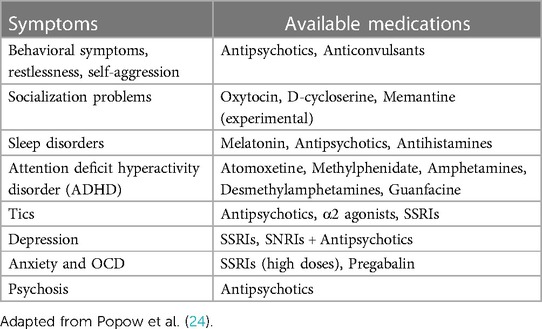
Table 2. Pharmacological Treatment of symptoms associated with ASD.
Medications administered for psychiatric symptoms work by reducing or preventing abnormal electrical activity in the brain that leads to seizures. Currently, there are different drugs available, and their selection depends on the type of seizure, the child's age, and comorbid conditions (32–35). In cases where a person with ASD does not experience seizures, the administration of medications to address psychiatric symptoms will focus on addressing behavioral, emotional, or cognitive aspects associated with ASD. These medications may be prescribed to manage issues such as anxiety, hyperactivity, aggression, sleep disorders, or other psychiatric symptoms that may arise in individuals with ASD. The choice of medication will depend on the specific nature of the symptoms and the assessment by the healthcare professional. Medications administered for psychiatric symptoms associated with ASD are often used in combination with other types of medications such as anxiolytics, antipsychotics, and antidepressants, depending on the epileptic symptoms and the severity of the epilepsy episodes the person is experiencing (35). Table 3 details the main drugs for treating epilepsy based on the symptoms presented by individuals with ASD and epilepsy.
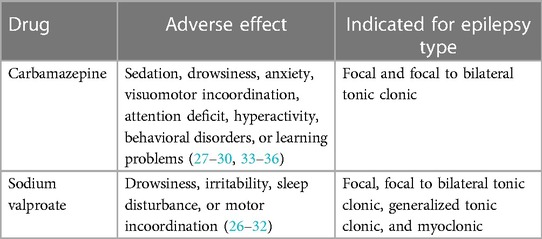
Table 3. Preferred pharmacological treatment for addressing epilepsy in the pediatric population with ASD.
Another significant challenge is the high interaction among the drugs described above (26–37). Antipsychotic drugs have significant reactions and drug interactions with anticonvulsants and mood stabilizers, including carbamazepine, though not all of these interactions occur negatively (38, 39). Identifying the consequences of such combinations is highly relevant, as, in some cases, serious adverse reactions may develop. This is exemplified by De León et al.'s (40) of interactions between anti-seizure medicines (ASMs) and second-generation antipsychotics potentially leading to exceptional cases of pancreatitis, agranulocytosis/leukopenia, and heatstroke. Additionally, Hitchings (41), in a review study, suggests that antipsychotics, by reducing the antiepileptic effect of these drugs, may reduce the efficacy of anti-seizure drugs.
Clozapine, known for reducing the seizure threshold, is linked to dose-dependent electroencephalographic alterations, impacting approximately 3%–6% of patients under clozapine treatment.
Asenjo Lobos et al. (42) highlight favorable outcomes in combined therapy involving antipsychotics for patients exhibiting partial response, as evidenced in several double-blind studies. The combinations encompass olanzapine paired with lithium or valproate, risperidone combined with lithium or valproate, haloperidol alongside lithium or valproate, and quetiapine in conjunction with lithium or valproate.
Similarly, the effectiveness of combined therapy has been demonstrated in open-label studies for patients with both ASD and epilepsy showing partial response. Such studies endorse combinations like olanzapine with lithium, valproate, or carbamazepine; risperidone paired with lithium or valproate, and quetiapine combined with lithium or valproate (43). The simultaneous administration of these medications should be approached with caution due to the potential emergence of toxic symptoms at high doses. Moreover, acute manic symptoms, extrapyramidal effects, physiological disorders, or brain damage could manifest, altering the neurological structure in which the epilepsy focus is located (44–46).
Medical treatments for ASD and epilepsy typically focus on managing the core symptoms of both disorders (ASD and epilepsy), addressing concurrent physical conditions such as sleep disorders, gastrointestinal issues, and sensory difficulties (47). However, unresolved issues persist in understanding the comorbidity between ASD and epilepsy. One fundamental challenge lies in the relationship between seizures and the central social symptoms of ASD. Although there is a high prevalence of epilepsy in individuals with ASD, the exact nature of how seizures contribute to the social aspects of ASD remains an actively researched area. Significant variability in the frequency of seizures also raises questions. While some children with ASD and epilepsy experience recurrent episodes, others may have a single episode in their lifetime. This variability not only complicates the understanding of the relationship between seizures and ASD but also presents challenges in identifying predictive patterns and personalized treatment strategies. Epilepsy itself represents a significant clinical problem, but the decision to resort to surgery must be approached with caution and is reserved for cases of refractory epilepsy with clearly defined cortical abnormalities that require surgical intervention (48). This decision entails the need for an accurate diagnosis and the identification of suitable candidates for surgical procedures, which remains an area in development. The comorbidity between ASD and epilepsy remains an unresolved clinical issue, exacerbated by a substantial lack of data and the complexity of interactions between both disorders (49). Ongoing research is essential to unravel the underlying mechanisms, better understand the variability in clinical presentation, and develop more effective and personalized treatment approaches for those affected by this comorbidity (50).
Upon receiving a referral, it is customary for the patient to undergo a thorough preoperative evaluation with the aim of identifying the epileptogenic zone and evaluating the patient's suitability for surgery (51). This evaluation typically includes a detailed medical history (analyzing clinical semiology, seizure frequency, severity, and prior treatments), video electroencephalography (EEG), magnetic resonance imaging (MRI) using an epilepsy protocol, and neuropsychological testing (52). Depending on the clinical context, additional tests such as positron emission tomography, single-photon emission computed tomography, magnetoencephalography, functional magnetic resonance imaging, and Wada testing may be considered (52, 53). The results of these assessments are then reviewed by a multidisciplinary team, which provides treatment recommendations. In certain cases, supplementary information is gathered through the surgical placement of intracranial electrodes to enhance the localization of the epileptogenic focus and identify critical brain areas for preservation during surgery (53).
For focal-onset seizures that can be localized in a surgically manageable area, available options encompass lesionectomy, temporal lobectomy, or extratemporal cortical resection. These procedures may be executed as a single intervention or in two stages, occasionally preceded by invasive EEG monitoring (54). In more severe epilepsies, alternatives may include functional disconnection (hemispherotomy) or anatomical hemispherectomy (complete removal of a hemisphere) (53, 54). Patients experiencing drop seizures, ineligible for resective options, may be candidates for palliative disconnection through a callosotomy of the corpus callosum. Palliative interventions like vagus nerve stimulation (VNS), and more recent approaches such as responsive neurostimulation (RNS) and deep brain stimulation (DBS), are also viable for patients not suitable for resection (55). Lastly, advancing technologies like laser ablation and gamma knife radiosurgery (GK) are continuously emerging, fostering ongoing exploration of minimally invasive approaches to addressing epileptogenic lesions (55, 56).
Lesionectomy stands out as a viable choice for addressing diverse lesions that provoke seizures, including neoplasms (such as gangliogliomas, oligodendrogliomas, astrocytomas, and dysembryonic neuroepithelial tumors) and vascular malformations (cavernomas and arteriovenous malformations) (57–59). In situations where there is alignment among seizure semiology, EEG monitoring findings, and lesion localization on magnetic resonance imaging (MRI), lesionectomy without the need for invasive monitoring is regarded as a favorable option (55–59). While the ultimate objective is complete lesion removal, this can prove challenging in eloquent regions. In many instances, the resection is confined to a portion of the lesion, such as retaining surrounding hemosiderin in the case of cavernomas and arteriovenous malformations (60).
Results after lesionectomy are generally excellent, particularly in pediatric patients. The extent of resection is critical, as total resection has been positively correlated with seizure freedom, reaching approximately 80%, while subtotal resection has a success rate of around 50%. Intraoperative electrocorticography guides resection and often results in the removal of perilesional tissue, with the potential to improve long-term seizure freedom rates (60, 61).
As for temporal lobectomy, one of the most studied surgical options, randomized controlled trials have shown that 58% of patients are seizure-free one year after surgery, compared to only 8% with medical management. The complication rate is relatively low, although side effects such as contralateral upper quadrantanopsia, loss of verbal memory, and, rarely, depression may arise (60–62). The success rate of temporal lobectomy in children is reported to be around 76% (62).
Extratemporal resection typically requires prior implantation of intracranial electrodes to map the epileptogenic focus and preserve eloquent brain areas. Although the effectiveness of extratemporal resection is lower than that of temporal foci, it significantly surpasses continuous medical management, with seizure freedom rates close to 56% in pediatric patients, according to a meta-analysis. Variability in results is associated with seizure duration, the presence of epileptogenic lesions, and/or partial seizures (63).
In extreme cases of catastrophic epilepsy in children and adolescents, such as hemimegalencephaly, hemispherectomy is presented as an option. This intervention aims to functionally disconnect or, in more aggressive cases, physically remove an affected cerebral hemisphere (64). “Hemispherotomy,” a less resective variant, has demonstrated similar success rates, with an overall seizure freedom rate of 73.4%. Functional disconnection, known as “disconnective hemispherectomy,” reduces intraoperative blood loss and minimizes anatomical resection, albeit with slightly higher risks of hydrocephalus (14%) and superficial hemosiderosis (65). Other less resective modifications have emerged under the term “hemispherotomy.” It is emphasized that up to approximately 14 years of age, pediatric and adolescent patients experience greater brain plasticity, contributing to substantially higher neurological recovery compared to adults undergoing more aggressive surgical interventions (64–66). Systematic studies have reviewed various hemispherectomy techniques in the pediatric/adolescent population, reporting an overall seizure freedom rate of 73.4%, with no significant differences in outcomes based on the type of hemispherectomy. Hemispherotomies, however, are associated with a lower risk of complications compared to other techniques. Additional research supports similar outcomes, highlighting seizure freedom rates of 63% or higher in patients five years post-surgical intervention (67).
Callosotomy is frequently reserved as a surgical intervention for managing medically resistant atonic seizures. Its primary objective is to reduce the occurrence of drop attacks, subsequently minimizing injuries resulting from falls. In contrast to procedures aiming for complete seizure freedom, callosotomy is considered a palliative measure (60–62).
Within this intricate landscape, the decision-making process between opting for a partial callosotomy, involving the anterior two-thirds of the corpus callosum, and a complete callosotomy remains a nuanced endeavor without a definitive consensus. The pursuit of superior seizure control leans decisively towards a complete callosotomy, boasting a remarkable 91% achievement of Engel class I–III, in stark comparison to the relatively lower 75% observed in partial callosotomies (59–61). The inclination towards the latter procedure may stem from a strategic intent to mitigate the occurrence of disconnection syndromes. While transient acute disconnection syndromes find a place in relative commonality, the occurrence of permanent disconnection syndromes is a rare phenomenon. Furthermore, strategic measures such as restricting the extent of callosal transection and orchestrating the procedure at a younger age or in staged interventions appear promising avenues to curtail the risk of disconnection syndromes (64).
On a divergent note, Vagus Nerve Stimulation (VNS), having received FDA approval in 1997, emerges as a compelling alternative for grappling with intractable partial-onset epilepsy, particularly in patients aged 12 and older. Despite the enigma surrounding its mechanism of action, VNS is postulated to exert its effects through thalamocortical projections. While the surgical complications of VNS are notably infrequent, boasting an infection rate of 4%–6%, and accompanied by generally well-tolerated side effects like hoarseness and voice changes, the occurrence of asystole is an exceedingly rare event, reported in less than 0.1% of patients. A recent meta-analysis paints a compelling picture, revealing a 50% reduction rate in seizures for half of the patients undergoing VNS therapy. Notably, this therapeutic avenue extends its benefits to both pediatric and adult populations with generalized epilepsy, despite its off-label application in both cohorts (58, 64).
In tandem with VNS, Responsive Neurostimulation (RNS) emerges as a notable player, securing FDA approval in 2013. Current indications cast a broad net, encompassing individuals aged 18 or older who grapple with at least three partial-onset seizures monthly, have experienced treatment failures, and have undergone comprehensive diagnostic tests to pinpoint a seizure focus amenable to stimulation (65). While the current approval lends credence to its clinical utility, ongoing research endeavors strive to illuminate its long-term efficacy, particularly in the context of treating medically resistant epilepsies in the pediatric population. A recent multicenter, double-blind, randomized controlled trial involving 191 patients paints an optimistic picture, indicating a median reduction of 44% in seizures at 1 year and an even more encouraging 53% at the 2-year mark within an open-label treatment interval (68). These strides in neuromodulatory therapies underscore a continuously evolving landscape, offering renewed hope and options for patients ensnared by the clutches of treatment-resistant epilepsy.
Finally, the precise mechanism by which Deep Brain Stimulation (DBS) exerts its anticonvulsant effect is not precisely known, but the most widely accepted explanation is that it induces an acute electrical disruption of synchronous activity at its origin, necessary for the propagation of ictal activity (48). It is noteworthy that, in DBS, most efferents of a target can follow action potentials at the stimulation frequency (49, 50). However, the generation of action potentials may be interfered with by the stimulation of afferent fibers towards the dendrites and somas of the target neurons, which can be excitatory or inhibitory, affecting the soma (49, 50). Nevertheless, the palliative option of deep brain stimulation for patients with poorly localized focal or generalized-onset epilepsy has been proposed for analysis, considering it as an emerging treatment in pediatrics, although it lacks FDA approval (49, 61).
In the realm of cutting-edge therapeutic approaches, interstitial laser thermal therapy guided by MRI emerges as a promising, less invasive method for ablating epileptic foci. This innovative procedure involves the meticulous placement of an optical fiber catheter, encased in a cooling sheath, within an identified epileptogenic focus. The subsequent application of laser energy orchestrates thermal ablation in the region of interest, unfolding a novel frontier in epilepsy treatment (67). Executed within the confines of the MRI suite, this procedure leverages real-time thermal imaging to meticulously monitor the treatment's progression, estimating the size and final location of the lesion with unparalleled precision. Currently, the narrative of its application in epilepsy treatment is predominantly woven through the fabric of case reports and small case series (68–70). The roster of treated lesions encompasses periventricular heterotopias, hypothalamic hamartomas, cortical dysplasia, and tubers. A groundbreaking study featuring 268 consecutively treated patients with Magnetic Resonance-guided Laser Interstitial Thermal Therapy (MRgLITT) in the medial temporal lobe has unveiled compelling rates of seizure freedom over time: 55.8% at 1 year, 52.5% at 2 years, and 49.3% at the last follow-up of ≥1 year (median of 47 months). Further bolstering these findings, combined results of Engel I or II showcase encouraging rates of 74.2% at 1 year, 75.0% at 2 years, and 66.0% at the last follow-up. Notably, an independent association has been discerned between preoperative focal to bilateral tonic-clonic seizures and the recurrence of seizures. In a striking turn of events, among patients grappling with seizure recurrence, a significant cohort attained seizure freedom post subsequent surgeries—be it anterior temporal lobectomy (ATL) or a repeat of MRgLITT—underscoring the profound effectiveness of these supplementary approaches in the realm of long-term seizure control (71).
Turning the narrative lens to another groundbreaking frontier, Gamma Knife (GK) radiosurgery has ascended in recent years as a precision-driven, stereotactic powerhouse for delivering focal radiation to intracranial epileptogenic targets, all while sparing surrounding tissues from appreciable radiation damage (72). This sophisticated tool offers a ray of hope in the treatment of epilepsy, presenting the capability to administer targeted radiation doses to specific regions of the affected brain. By elegantly sidestepping the need for whole-brain irradiation, the GK minimizes the collateral damage and toxicity associated with conventional radiotherapy. Its prowess in delivering focused and precise treatments positions it as a valuable asset in the nuanced landscape of epilepsy management, particularly when pharmacological avenues have proven futile. Beyond the realm of epilepsy, this technology currently finds widespread application in focal radiosurgical ablation, ranging from intracranial metastatic diseases and pituitary lesions to acoustic neuromas, refractory trigeminal neuralgia, and various other neurosurgical conditions (73). Its efficacy has been scrutinized in the context of mesial temporal lobe epilepsy, particularly when there is telltale evidence of hippocampal sclerosis in magnetic resonance imaging—a scenario traditionally addressed through selective amygdalohippocampectomy (74). For those navigating the treacherous waters of surgical risks or having faced disappointments in prior surgical interventions for epilepsy, Gamma Knife radiosurgery emerges as a beacon of hope—a non-invasive alternative boasting successful long-term seizure freedom outcomes of 60% or more (75–77). However, the saga continues with an earnest call for further research and clinical studies to meticulously evaluate the effectiveness and potential enduring benefits of stereotactic radiosurgery, notably the kind bestowed by the Gamma Knife, in the expansive tapestry of epilepsy treatment. This quest extends to comparing its outcomes with other therapeutic modalities, charting the course for the future of epilepsy care.
This opinion article aimed to describe and analyze the most effective pharmacological and surgical treatments for addressing the symptoms in the pediatric population with ASD and epilepsy.
The analysis conducted in this study identified the challenge of treating the clinical picture experienced by individuals with ASD and epilepsy. Approaching this comorbidity from a pharmacological perspective is a complex task. Firstly, pharmacological treatment does not allow for the elimination of the aversive symptoms of ASD and epilepsy; instead, it focuses on alleviating and reducing the patient's symptoms. Furthermore, the heterogeneity of symptoms in individuals with ASD and epilepsy significantly complicates the selection of combined drugs to reduce behavioral and physiological symptoms of ASD, as well as epilepsy episodes. Additionally, drug interactions contribute to unwanted effects in patients with this comorbidity.
Secondly, medical interventions focus on alleviating symptoms by addressing concurrent physical conditions. For those resistant to pharmacotherapy, surgery, such as temporal resection or functional disconnection, is considered effective, with careful attention to thorough preoperative evaluation. Additionally, palliative procedures like callosotomy and neuromodulatory therapies such as VNS, RNS, and DBS offer promising treatments for patients who are not resection' candidates. Emerging therapies, like laser ablation and GK, present minimally invasive approaches. These surgical and therapeutic options constitute comprehensive strategies to address the complexity of the comorbidity between ASD and epilepsy, emphasizing the need to consider approaches beyond conventional medical options to enhance the quality of life for patients.
Current scientific literature does not present significant results that allow determining the most effective treatment choice for the pediatric population with ASD and epilepsy (37–42, 44, 53, 55–57). In this regard, (I) the clinical, genetic, and physiopathological heterogeneity, the multiple possible targets, and the different pathways involved in the pathogenesis; (II) the lack of case-control and cohort follow-up studies from the onset of symptoms in preschoolers (2 years or less) that differentiate between the natural evolution of the condition and the evolution of the condition with intervention; (III) the difficulty in conducting studies with patient and family consent; (IV) the absence of biomarkers that allow grouping similar patients with more homogeneous, evaluable, and reproducible clinical characteristics; (V) the lack of randomized, placebo-controlled, and double-blind studies; (VI) the difficulty in determining the duration of studies, as the evolution of children with ASD and epilepsy undergoes modifications not only attributable to pharmacological interventions but also inherent to development; and (VI) the lack of family adherence to research significantly limit the treatment plan for individuals with this comorbidity (11–14).
In conclusion, the treatment of children with ASD and epilepsy requires an interdisciplinary approach that includes rehabilitation, pharmacology, and ultimately, surgical intervention (23). It is important to mention that these treatments can be costly and may not always be available to all children with ASD and epilepsy (2–5). Accessibility to treatments is a significant concern for parents, caregivers, and healthcare professionals (6), which can greatly influence the choice of the most suitable treatment modality (24). Additionally, pharmacological, and surgical treatments may have side effects, emphasizing the importance of closely monitoring children during treatment to minimize them as much as possible (37). Finally, it is essential to consider the warning signs of ASD and epilepsy to provide early attention to comorbidity, allowing for a differential diagnosis and designing evidence-based personalized interventions to enhance children's development and ensure an improvement in their quality of life (13).
AC-V: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. FM-F: Conceptualization, Supervision, Writing – review & editing. NP-G: Writing – original draft, Writing – review & editing. ML-Z: Conceptualization, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington, VA: American Psychiatric Publishing (2013).
2. Berg AT, Plioplys S. Epilepsy and autism: is there a special relationship? Epilepsy Behav. (2012) 23(3):193–8. doi: 10.1016/j.yebeh.2011.12.026
3. Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveillance Summaries. (2018) 67(6):1–23. doi: 10.15585/mmwr.ss6706a1
4. Li M, Wang Y, Tachibana M, Rahman S, Kagitani-Shimono K. Atypical structural connectivity of language networks in autism spectrum disorder: a meta-analysis of diffusion tensor imaging studies. Autism Res. (2022) 15(9):1585–602. doi: 10.1002/aur.2789
5. Lord C, Brugha TS, Charman T, Cusack J, Dumas G, Frazier T, et al. Autism spectrum disorder. Nat Rev Dis Primers. (2020) 6(1):5. doi: 10.1038/s41572-019-0138-4
6. Xu G, Strathearn L, Liu B, O’Brien M, Kopelman TG, Zhu J, et al. Prevalence and treatment patterns of autism spectrum disorder in the United States, 2016. JAMA Pediatr. (2019) 173(2):153–9. doi: 10.1001/jamapediatrics.2018.4208.3
7. Dietz PM, Rose CE, McArthur D, Maenner M. National and state estimates of adults with autism spectrum disorder. J Autism Dev Disord. (2020) 50(12):4258–66. doi: 10.1007/s10803-020-04494-4
8. Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE task force on nosology and definitions. Epilepsia. (2022) 63(6):1349–97. doi: 10.1111/epi.17239
9. Brugha TS, McManus S, Bankart J, Scott F, Purdon S, Smith J, et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch Gen Psychiatry. (2011) 68(5):459–65. doi: 10.1001/archgenpsychiatry.2011.38
10. Sasayama D, Kuge R, Toibana Y, Honda H. Trends in autism Spectrum disorder diagnoses in Japan, 2009 to 2019. JAMA Netw Open. (2021) 4(5):e219234. doi: 10.1001/jamanetworkopen.2021.9234
11. Ghosh A, Michalon A, Lindemann L, Fontoura P, Santarelli L. Drug discovery for autism spectrum disorder: challenges and opportunities. Nat Rev Drug Discov. (2013) 12(10):777–90. doi: 10.1038/nrd4102
12. Lee YJ, Oh SH, Park C, Hong M, Lee AR, Yoo HJ, et al. Advanced pharmacotherapy evidenced by pathogenesis of autism spectrum disorder. Clin Psychopharmacol Neurosci. (2014) 12(1):19–30. doi: 10.9758/cpn.2014.12.1.19
13. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the international league against epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia. (2017) 58:522–30. doi: 10.1111/epi.13670
14. Fisher RS, Cross JH, D’Souza C, French JA, Haut SR, Higurashi N, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia. (2017) 58:531–42. doi: 10.1111/epi.13671
15. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
16. Fisher R, Acevedo C, Arzimanoglou A, Bogacz A, Cross H, Elger CE, et al. Definición clínica práctica de la epilepsia. Separata en Español Reproducida de Epilepsia. (2014) 55:475–82.
17. Thurman DJ, Beghi E, Begley CE, Berg AT, Buchhalter JR, Ding D, et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia. (2011) 52(Suppl 7):2–6. doi: 10.1111/j.1528-1167.2011.03121.x
18. Bancaud J, Rubio-Donnadieu F, Seino M, Dreifuss F, Penry K. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. From the commission on classification and terminology of the international league against epilepsy. Epilepsia. (1981) 22:489–501. doi: 10.1111/j.1528-1157.1981.tb06159.x
19. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology, 2005–2009. Epilepsia. (2010) 51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x
20. Rubio F, Resendiz JC, Alonso MA, Senties H. Epilepsia, 1st ed. México: Editorial Alfil (2016). p. 27–42.
21. Volpe JJ. Neonatalseizures:currentconceptsandrevisedclassification. Pediatrics. (1989) 84:422–8. doi: 10.1542/peds.84.3.422
22. Mizrahi EM, Kellaway P. Characterization and classification of neonatal seizures. Neurology. (1987) 37:1837–44. doi: 10.1212/WNL.37.12.1837
23. Liu X, Lin J, Zhang H, Khan NU, Zhang J, Tang X, et al. Oxidative stress in autism spectrum disorder-current progress of mechanisms and biomarkers. Front Psychiatry. (2022) 13:813304. doi: 10.3389/fpsyt.2022.813304
24. Popow C, Ohmann S, Plener P. Practitioner’s review: medication for children and adolescents with autism spectrum disorder (ASD) and comorbid conditions. Neuropsychiatr. (2021) 35(3):113–34. doi: 10.1007/s40211-021-00395-9
25. Boesen MS, Børresen ML, Christensen SK, Klein-Petersen AW, El Mahdaoui S, Sagar MV, et al. School performance and psychiatric comorbidity in childhood absence epilepsy: a Danish cohort study. Eur J Paediatr Neurol. (2023) 42:75–81. doi: 10.1016/j.ejpn.2022.12.008
26. Rapin I, Tuchman RF. Autism: definition, neurobiology, screening, diagnosis. Pediatr Clin North Am. (2008) 55:1129–46. doi: 10.1016/j.pcl.2008.07.005
27. Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. (2010) 1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x
28. Weichhart T. Mammalian target of rapamycin: a signaling kinase for every aspect of cellular life. Methods Mol Biol. (2012) 821:1–14. doi: 10.1007/978-1-61779-430-8_1
29. Toriello HV. Approach to the genetic evaluation of the child with autism. Pediatr Clin North Am. (2012) 59:113–28. doi: 10.1016/j.pcl.2011.10.014
30. Löscher W, Potschka H, Sisodiya SM, Vezzani A. Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol Rev. (2020) 72(3):606–38. doi: 10.1124/pr.120.019539
31. Kanner AM, Bicchi MM. Antiseizure medications for adults with epilepsy: a review. JAMA. (2022) 327(13):1269–81. doi: 10.1001/jama.2022.3880
32. Sills GJ, Rogawski MA. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology. (2020) 168:107966. doi: 10.1016/j.neuropharm.2020.107966
33. Salazar de Pablo G, Pastor Jordá C, Vaquerizo-Serrano J, Moreno C, Cabras A, Arango C, et al. Systematic review and meta-analysis: efficacy of pharmacological interventions for irritability and emotional dysregulation in autism spectrum disorder and predictors of response. J Am Acad Child Adolesc Psychiatry. (2023) 62(2):151–68. doi: 10.1016/j.jaac.2022.03.033
34. Sierra-Arregui T, Llorente J, Giménez Minguez P, Tønnesen J, Peñagarikano O. Neurobiological mechanisms of autism spectrum disorder and epilepsy, insights from animal models. Neuroscience. (2020) 445:69–82. doi: 10.1016/j.neuroscience.2020.02.043
35. Canitano R, Palumbi R, Scandurra V. Autism with epilepsy: a neuropsychopharmacology update. Genes (Basel). (2022) 13(10):1821. Published 2022 Oct 8. doi: 10.3390/genes13101821
36. Sharma AR, Batra G, Saini L, Sharma S, Mishra A, Singla R, et al. Valproic acid and propionic acid modulated mechanical pathways associated with autism Spectrum disorder at prenatal and neonatal exposure. CNS Neurol Disord Drug Targets. (2022) 21(5):399–408. doi: 10.2174/1871527320666210806165430
37. Liu G, Slater N, Perkins A. Epilepsy: treatment options. Am Fam Physician. (2017) 96(2):87–96.28762701
38. Gözükızıl ST, Aydın Z, Yalçın AD. Relationship between bone density and levetiracetam monotherapy in epilepsy patients. Clin Neurol Neurosurg. (2022) 218:107270. doi: 10.1016/j.clineuro.2022.107270
39. Sharma SR, Gonda X, Tarazi FI, Atmaca M. Major classes of antidepressants and their potential mechanisms of action. Int J Mol Sci. (2018) 19(7):2030. doi: 10.3390/ijms19072030
40. de Leon J, Santoro V, D'Arrigo C, Spina E. Interactions between antiepileptics and second-generation antipsychotics. Expert Opin Drug Metab Toxicol. (2012) 8(3):311–34. doi: 10.1517/17425255.2012.660918
41. Hitchings AW. Drugs that lower the seizure threshold. Adverse Drug Reactions Bulletin. (2016) 298:1151–4. doi: 10.1097/FAD.0000000000000016
42. Asenjo Lobos C, Komossa K, Rummel-Kluge C, Hunger H, Schmid F, Schwarz S, et al. Clozapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. (2010) (11):CD006633. Published 2010 Nov 10. doi: 10.1002/14651858.CD006633.pub2
43. Tuchman R. What is the relationship between autism spectrum disorders and epilepsy? Semin Pediatr Neurol. (2017) 24(4):292–300. doi: 10.1016/j.spen.2017.10.004
44. Zahra A, Wang Y, Wang Q, Wu J. Shared etiology in autism spectrum disorder and epilepsy with functional disability. Behav Neurol. (2022) 2022:5893519. doi: 10.1155/2022/5893519
45. Kokoszka MA, McGoldrick PE, La Vega-Talbott M, Raynes H, Palmese CA, Wolf SM, et al. Epilepsy surgery in patients with autism. J Neurosurg Pediatr. (2017) 19(2):196–207. doi: 10.3171/2016.7.PEDS1651
46. Parker JJ, Zhang Y, Fatemi P, Halpern CH, Porter BE, Grant GA. Antiseizure medication use and medical resource utilization after resective epilepsy surgery in children in the United States: a contemporary nationwide cross-sectional cohort analysis. Epilepsia. (2022) 63(4):824–35. doi: 10.1111/epi.17180
47. Zangiabadi N, Ladino LD, Sina F, Orozco-Hernández JP, Carter A, Téllez-Zenteno JF. Deep brain stimulation and drug- resistant epilepsy: a review of the literature. Front Neurol. (2019) 10:601. doi: 10.3389/fneur.2019.00601
48. Pollo C, Villemure JG. Rationale, mechanisms of efficacy, anatomical targets and future prospects of electrical deep brain stimulation for epilepsy. Acta Neurochir Suppl. (2007) 97:311–20. doi: 10.1007/978-3-211-33081-4_34
49. Veerakumar A, Berton O. Cellular mechanisms of deep brain stimulation: activity-dependent focal circuit reprogramming? Curr Opin Behav Sci. (2015) 4:48–55. doi: 10.1016/j.cobeha.2015.02.004
50. Jakobs M, Fomenko A, Lozano AM, Kiening KL. Cellular, molecular, and clinical mechanisms of action of deep brain stimulation—a systematic review on established indications and outlook on future developments. EMBO Mol Med. (2019) 11:e9575. doi: 10.15252/emmm.201809575
51. Serletis D, Bulacio J, Alexopoulos A, Najm I, Bingaman W, González-Martínez J. Tailored unilobar and multilobar resections for orbitofrontal-plus epilepsy. Neurosurgery. (2014) 75(4):388–97. doi: 10.1227/NEU.0000000000000481
52. Liava A, Francione S, Tassi L, Lo Russo G, Cossu M, Mai R, et al. Individually tailored extratemporal epilepsy surgery in children: anatomo-electro-clinical features and outcome predictors in a population of 53 cases. Epilepsy Behav. (2012) 25(1):68–80. doi: 10.1016/j.yebeh.2012.05.008
53. Cossu M, Cardinale F, Castana L, Nobili L, Sartori I, Lo Russo G. Stereo-EEG in children. Childs Nerv Syst. (2006) 22(8):766–78. doi: 10.1007/s00381-006-0127-2
54. Cossu M, Cardinale F, Colombo N, Mai R, Nobili L, Sartori I, et al. Stereoelectroencephalography in the presurgical evaluation of childrenwith drug-resistant focal epilepsy. J Neurosurg. (2005) 103(4, Suppl):333–43. doi: 10.3171/ped.2005.103.4.0333
55. Bourgeois M, Di Rocco F, Sainte-Rose C. Lesionectomy in the pediatric age. Childs Nerv Syst. (2006) 22(8):931–5. doi: 10.1007/s00381-006-0146-z
56. von der Brelie C, Malter MP, Niehusmann P, Elger CE, von Lehe M, Schramm J. Surgical management and long-term seizure outcome after epilepsy surgery for different types of epilepsy associated with cerebral cavernous malformations. Epilepsia. (2013) 54(9):1699–706. doi: 10.1111/epi.12327
57. Gelinas JN, Battison AW, Smith S, Connolly MB, Steinbok P. Electrocorticography and seizure outcomes in children with lesional epilepsy. Childs Nerv Syst. (2011) 27(3):381–90. doi: 10.1007/s00381-010-1279-7
58. Gump WC, Skjei KL, Karkare SN. Seizure control after subtotal lesional resection. Neurosurg Focus. (2013) 34(6):E1. doi: 10.3171/2013.3.FOCUS1348
59. Wiebe S, Blume WT, Girvin JP, Eliasziw M. Effectiveness and efficiency of surgery for temporal lobe epilepsy study group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med. (2001) 345(5):311–8. doi: 10.1056/NEJM200108023450501
60. Englot DJ, Rolston JD, Wang DD, Sun PP, Chang EF, Auguste KI. Seizure outcomes after temporal lobectomy in pediatric patients. J Neurosurg Pediatr. (2013) 12(2):134–41. doi: 10.3171/2013.5.PEDS12526
61. Dallas J, Englot DJ, Naftel RP. Neurosurgical approaches to pediatric epilepsy: Indications, techniques, and outcomes of common surgical procedures. Seizure. (2020) 77:76–85. doi: 10.1016/j.seizure.2018.11.007
62. Englot DJ, Breshears JD, Sun PP, Chang EF, Auguste KI. Seizure outcomes after resective surgery for extra-temporal lobe epilepsy in pediatric patients. J Neurosurg Pediatr. (2013) 12(2):126–33. doi: 10.3171/2013.5.PEDS1336
63. D’Argenzio L, Colonnelli MC, Harrison S, Jacques TS, Harkness W, Scott RC, et al. Seizure outcome after extratemporal epilepsy surgery in childhood. DevMed Child Neurol. (2012) 54(11):995–1000. doi: 10.1111/j.1469-8749.2012.04381.x
64. Vachhrajani S, de Ribaupierre S, Otsubo H, Ochi A, Weiss SK, Donner EJ, et al. Neurosurgical management of frontal lobe epilepsy in children. J Neurosurg Pediatr. (2012) 10(3):206–16. doi: 10.3171/2012.6.PEDS11125
65. Ibrahim GM, Morgan BR, Fallah A. A partial least squares analysis of seizure outcomes following resective surgery for tuberous sclerosis complex in children with intractable epilepsy. Childs Nerv Syst. (2015) 31(2):181–4. doi: 10.1007/s00381-014-2600-7
66. Kovanda TJ, Rey-Dios R, Travnicek J, Cohen-Gadol AA. Modified periinsular hemispherotomy: operative anatomy and technical nuances. J Neurosurg Pediatr. (2014) 13(3):332–8. doi: 10.3171/2013.12.PEDS13277
67. Bradley LH, Bahgat D, Sharp G, Willis E, Ocal E, Albert G, et al. Disconnective hemispherotomy for medically intractable status epilepticus in an 8 year-old child. J Ark Med Soc. (2015) 112(6):86–7. PMID: 26552284.26552284
68. Velasco M, Velasco F, Velasco AL. Centromedian-thalamic and hippocampal electrical stimulation for the control of intractable epileptic seizures. J Clin Neurophysiol. (2001) 18:495–513. doi: 10.1097/00004691-200111000-00001
69. Fisher RS, Uematsu S, Krauss GL, Cysyk BJ, McPherson R, Lesser RP, et al. Placebo-controlled pilot study of centromedian thalamic stimulation in treatment of intractable seizures. Epilepsia. (1992) 33(5):841–51. doi: 10.1111/j.1528-1157.1992.tb02192.x
70. Valentín A, García-Navarrete E, Chelvarajah R, Torres C, Navas M, Vico L, et al. Deep brain stimulation of the centro- median thalamic nucleus for the treatment of generalized and frontal epilepsies. Epilepsia. (2013) 54:1823–33. doi: 10.1111/epi.12352
71. Youngerman BE, Banu MA, Khan F, McKhann GM, Schevon CA, Jagid JR, et al. Long-term outcomes of mesial temporal laser interstitial thermal therapy for drug-resistant epilepsy and subsequent surgery for seizure recurrence: a multi-centre cohort study. J Neurol Neurosurg Psychiatry. (2023) 94(11):879–86. doi: 10.1136/jnnp-2022-330979
72. Téllez-Zenteno JF, McLachlan RS, Parrent A, Kubu CS, Wiebe S. Hippocampal electrical stimulation in mesial temporal lobe epilepsy. Neurology. (2006) 66:1490–4. doi: 10.1212/01.wnl.0000209300.49308.8f
73. Koeppen JA, Nahravani F, Kramer M, Voges B, House PM, Gulberti A, et al. Electrical stimulation of the anterior thalamus for epilepsy: clinical outcome and analysis of efficient target. Neuromodulation. (2019) 22:465–71. doi: 10.1111/ner.12865
74. Barbaro NM, Quigg M, Ward MM, Chang EF, Broshek DK, Langfitt JT, et al. Radiosurgery versus open surgery for mesial temporal lobe epilepsy: the randomized, controlled ROSE trial. Epilepsia. (2018) 59(6):1198–207. doi: 10.1111/epi.14045
75. Lehtimäki K, Möttönen T, Järventausta K, Katisko J, Tähtinen T, Haapasalo J, et al. Outcome based definition of the anterior thalamic deep brain stimulation target in refractory epilepsy. Brain Stimul. (2016) 9:268–75. doi: 10.1016/j.brs.2015.09.014
76. Beric A, Kelly PJ, Rezai A, Sterio D, Mogilner A, Zonenshayn M, et al. Complications of deep brain stimulation surgery. Stereotact Func Neurosurg. (2001) 77:73–8. doi: 10.1159/000064600
Keywords: ASD, epilepsy, surgical treatment, children, pharmacotherapy
Citation: Cano-Villagrasa A, Moya-Faz FJ, Porcar-Gozalbo N and López-Zamora M (2024) Treatment options in autism with epilepsy. Front. Child Adolesc. Psychiatry 3:1265081. doi: 10.3389/frcha.2024.1265081
Received: 21 July 2023; Accepted: 13 February 2024;
Published: 22 February 2024.
Edited by:
Roberto Canitano, Siena University Hospital, ItalyReviewed by:
Alessandra Carta, University of Sassari, Italy© 2024 Cano-Villagrasa, Moya-Faz, Porcar-Gozalbo and López-Zamora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alejandro Cano-Villagrasa YWxlamFuZHJvLmNhbm8udkBwcm9mZXNzb3IudW5pdmVyc2lkYWR2aXUuY29t Nadia Porcar-Gozalbo bmFkaWEucG9yY2FyQHByb2Zlc3Nvci51bml2ZXJzaWRhZHZpdS5jb20= Miguel López-Zamora bWlnbG9wemFtQHVtYS5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.