 Prashant N. Jethva
Prashant N. Jethva Michael L. Gross
Michael L. Gross- Department of Chemistry, Washington University in St. Louis, St Louis, MO, United States
Antigen-antibody interactions are a fundamental subset of protein-protein interactions responsible for the “survival of the fittest.” Determining the interacting interface of the antigen, called an epitope, and that on the antibody, called a paratope, is crucial to antibody development. Because each antigen presents multiple epitopes (unique footprints), sophisticated approaches are required to determine the target region for a given antibody. Although X-ray crystallography, Cryo-EM, and nuclear magnetic resonance can provide atomic details of an epitope, they are often laborious, poor in throughput, and insensitive. Mass spectrometry-based approaches offer rapid turnaround, intermediate structural resolution, and virtually no size limit for the antigen, making them a vital approach for epitope mapping. In this review, we describe in detail the principles of hydrogen deuterium exchange mass spectrometry in application to epitope mapping. We also show that a combination of MS-based approaches can assist or complement epitope mapping and push the limit of structural resolution to the residue level. We describe in detail the MS methods used in epitope mapping, provide our perspective about the approaches, and focus on elucidating the role that HDX-MS is playing now and in the future by organizing a discussion centered around several improvements in prototype instrument/applications used for epitope mapping. At the end, we provide a tabular summary of the current literature on HDX-MS-based epitope mapping.
1 Introduction
Proteins are the ultimate working horses of cells. Under typical cellular conditions, the cellular matrix has roughly 200–400 mg/mL of protein molecules (Brown, 1991). It is remarkable that under these highly crowded conditions, proteins carry out fundamental cellular biochemistry repeatedly with high fidelity. Activities range from replication of DNA to signal transduction across cellular compartments to neutralize foreign infectious agents (Ellis and Minton, 2003; Xiao et al., 2018). It is the latter that contributes most to Darwin’s famous assertion of survival of fittest, here overcoming debilitating infection, a prime challenge of the surviving species made particularly relevant in the midst of a SARS CoV2 pandemic.
Various types of immunoglobulins (antibodies) (IgA, D, E, G, and M) form the pool of inactivating agents for the immune system (Schroeder and Cavacini, 2010). The most common immunoglobulin for immune protection, however, is G, (Vidarsson et al., 2014), explaining the IgG is the most widely studied for its structure and the nature of the antigens it targets. Because antibodies have high specificity and selectivity, they may be the “magic bullets” for treating many diseases especially cancers.
The first therapeutic monoclonal antibody (muromonab-CD3 (Orthoclone OKT3) approved for patient use was in 1986 (Reichert, 2012). Further, monoclonal antibodies are one of the fastest growing drug classes (Ecker et al., 2015; Martin, 2016; Reichert, 2023) (Figure 1) (as of this writing, 104 approved monoclonal antibodies are IgG, three Fabs, two ScFvs, one nanobody, and one DsFv (https://www.antibodysociety.org/resources/approved-antibodies/). Additionally, antibodies are widely used as analytical reagents for diagnostic tests and in research and development (Leinikki et al., 1993; Sun et al., 2022; Song et al., 2023). The introduction of monoclonal antibodies for therapeutic use was a paradigm shift in the pharma industry. As opposed to small-molecule drugs, which are mostly unknown to the human body, a large part of an antibody sequence and structure is known to our immune system. Although many small molecules can be synthesized chemically where the tools for characterization are well-established, antibodies, which are approximately 150 kDa in mass, are synthesized in cell culture and are variable starting from clone selection to final product packaging, and these variables potentially affect safety and efficacy (Birch and Racher, 2006; Shukla et al., 2007). Important factors include primary structure, disulfide linkages, post-translational modifications, and high order structure and stability over the lifetime of the antibody (Beck et al., 2013; Alt et al., 2016). All factors affect the binding of an antibody to its target antigen and hence its overall efficacy.
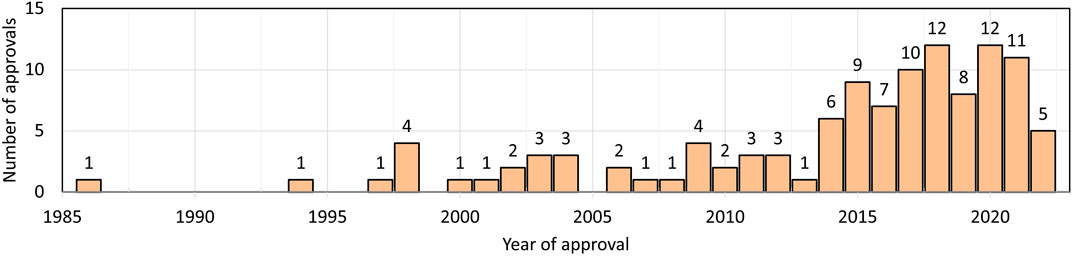
FIGURE 1. Number of therapeutic antibodies approved in each year. Data are from Antibody society. https://www.antibodysociety.org/resources/approved-antibodies/.
The first detailed structural studies of IgG resulted in the Nobel Prize in 1972 (Edelman, 1959; Porter, 1959), and the first 3D structure solved in 1973 (Poljak et al., 1973). The structure reveals the famous “Y” shape (Poljak et al., 1975) (Figure 2). IgG is technically a homodimer made up of two heavy chains and two light chains linked by disulfide bonds. The top “V” is called the Fab (fragment antigen binding) and made up of variable domains from heavy and light chains (Figure 2). The bottom “I” shaped region is called the Fc (fragment crystallizable or constant region). It is exclusively made up of heavy-chain domains. The interacting interface is called the paratope, and that on the antigen is called the epitope (Jerne, 1960). Owing to advances in structure determination and the well-characterized nature of the complementarity determining region of the antibody (outward protruding loops on variable domains of the Fab), it is relatively easy to determine the paratope. The diverse shapes and sizes and the multiple unique footprints exposed for antibody binding of the antigen make it more challenging to identify the epitope for a given antibody (Van Regenmortel, 1996).
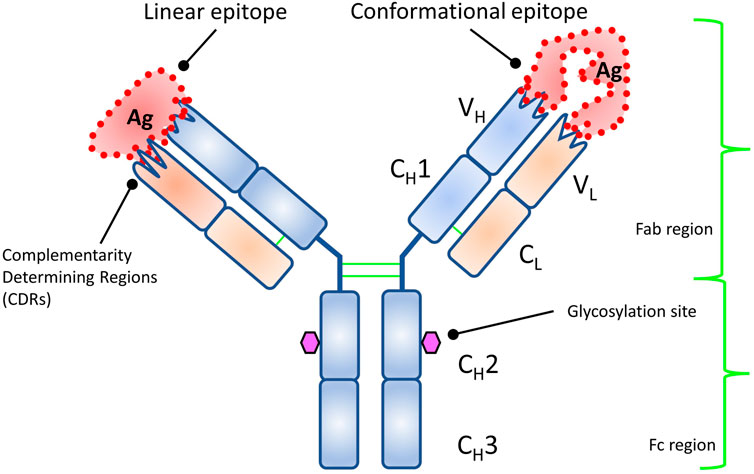
FIGURE 2. Structure of an antibody showing the structural domains. Each variable domain of heavy and light chains has multiple outward protruding loops (complementarity determining regions, CDRs) and confer binding specificity. The types of epitopes targeted by antibody are also shown.
Epitopes are divided into two categories (Barlow et al., 1986) (Figure 2): a linear epitope where the antibody binds to continuous sequence of amino acids, and a conformational or discontinuous epitope. For the latter, different residues, owing to folding of polypeptide chain(s), come in close contact structurally and form a unique footprint that is targeted by the antibody. Constituent residues of discontinuous epitopes are often far apart in the protein sequence. In general, most antibodies have conformational epitopes and need a fully functional (native) antigen for mapping its antigenic determinant (epitope) (Jemmerson, 1987; Laver et al., 1990; Rubinstein et al., 2008; Ramaraj et al., 2012).
Understanding the antigen-antibody interactions assists the characterization of the immune response against pathogens (Volk et al., 2016), not only for infections but also for agents of bioterrorism or bioweapons (Toth IV et al., 2017; Fang et al., 2022). Similarly, the design of better antigens and prophylactics or therapeutics/vaccines can evoke strong immune responses (Irving et al., 2001; Gershoni et al., 2007; Brooks et al., 2014; Sharon et al., 2014; Kwong et al., 2020; Kim et al., 2021; Suryadevara et al., 2021; Crowe Jr, 2022). The determination of a paratope-epitope not only helps secure intellectual property rights (Gresl et al., 2016; Deng et al., 2018), but also provides in-depth characterization of the antibody-antigen interaction especially of the epitope. Thus, epitope mapping is ever growing.
Many methods are in use (Figure 3) for epitope mapping including X-Ray crystallography (Poljak et al., 1973; Toride King and Brooks, 2018), Nuclear Magnetic Resonance (NMR) (Bardelli et al., 2015; Di Muzio et al., 2020; Valente and Manzano-Rendeiro, 2021), Cryo-Electron Microscopy (Cryo-EM)(Renaud et al., 2018; Wigge et al., 2020), alanine-scanning mutagenesis (Cunningham and Wells, 1989; Weiss et al., 2000), phage display libraries (Smith, 1985; Cwirla et al., 1990; Devlin et al., 1990; Scott and Smith, 1990; Böttger and Böttger, 2009), peptide library scanning (one of the oldest method for locating antigenic sites or epitopes) (Landsteiner, 1942; Geysen et al., 1984; Carter, 1996; Forsström et al., 2014), escape mutation analysis (Davidson and Doranz, 2014; Starr et al., 2021), chemical-modification based approaches (Atassi, 1964; Burnens et al., 1987; Fiedler et al., 1998; Jones et al., 2011), and others (Morris, 1996; Ladner, 2007; Rockberg and Nilvebrant, 2018; Bondarenko et al., 2021). Most of the high-resolution structural techniques (X-ray, NMR, and cryo-EM) provide atomic details of epitopes. These latter methods, however, are laborious and require complex instrumentation and data analysis (Dale et al., 2003; Simonelli et al., 2018). In this context, mass spectrometry (MS)-based approaches are becoming increasingly popular because they use little sample, have readily available sophisticated instruments and automation, can maintain solution binding, and can provide residue-resolved epitope sites. (We apologize that this review does not cover all methods for epitope mapping; the methods we cover are limited to MS-based.)
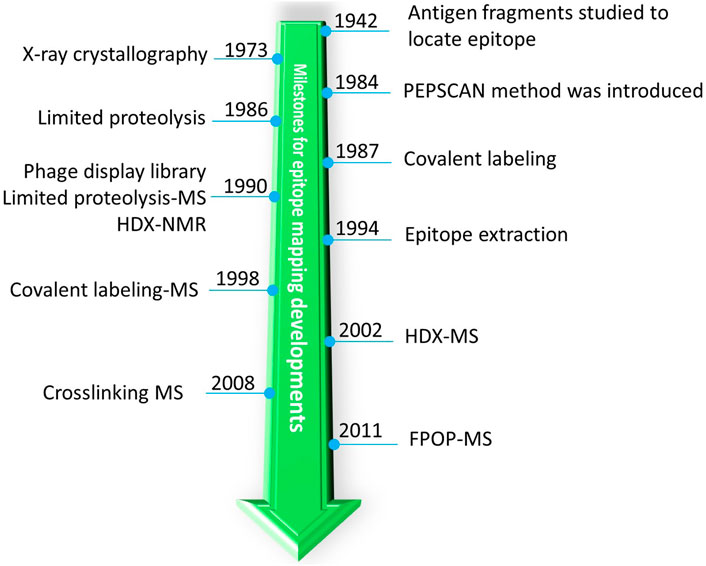
FIGURE 3. A historic timeline for introduction of different MS-related approaches for milestones of epitope mapping by MS.
2 Historical perspective: MS-based approaches for epitope mapping
2.1 Peptide level analysis
One of the earliest detailed structural studies about antigenicity of proteins (and one of the first protein-protein interaction studies) was carried out with Sperm whale myoglobin because its structure had become available (Atassi, 1964). The 3D structure of an antigen (myoglobin) allows structural rationalization of epitopes. Furthermore, an antibody raised against an antigen is not effective against homologous antigens with slightly different epitopes, suggesting that antibodies bind conformational epitopes (Benjamin et al., 1984). Owing to lack of high-resolution techniques to study large proteins, the antigen was proteolyzed, and those fragments of the antigen that bind were identified (Landsteiner, 1942; Atassi, 1964) (Figure 3).
As peptide-library synthesis became more feasible, scanning of many peptides having overlapping sequences (PEPSCAN) became an important tool for identifying epitopes (Geysen et al., 1984). Often, an epitope identified by this approach is validated by site-directed mutagenesis. During 1990s, the phage display library method for epitope mapping was introduced (Cwirla et al., 1990; Devlin et al., 1990; Scott and Smith, 1990). With this approach, each bacteriophage displays unique/overlapping sequences of an antigen, and by affinity selecting the phages and identifying the sequence of “displayed peptide,” an investigator could locate the epitope for a given antibody. Theoretically, the phage-display library approach has circumvented, by clever genetics, the enormous task of generating peptide libraries. An advantage of a library-based approach, however, is that once the library is constructed, one can use it for screening epitopes for many antibodies. Because small peptides are synthesized or displayed on the surface, only linear epitope mapping is possible. Recently, the high throughput phage display (Phage-based Representation OF ImmunoLigand Epitope Repertoire (PROFILER) method was introduced where large segments of antigens can be displayed to reveal potentially conformational epitopes (Cariccio et al., 2016).
2.2 Protein level analysis
In the 1980s, chemical modification of a protein was also applied to map an epitope on an antigen (Burnens et al., 1987) (Figure 3). An antigen was modified by selective labeling in the presence and absence of an antibody. Since binding will sterically hinder the amino acid labeling involved at epitope interface, comparing the antigen labeling in the absence and presence of the antibody can reveal the epitope (this approach is a predecessor of modern MS-based covalent footprinting). The differential labeling was initially analyzed by both High Performance Liquid Chromatography (HPLC) and sequencing of the labeled peptide to identify the epitope. This simple and elegant approach of differential labeling was later combined with MS-based detection (more below) of a differentially labeled peptide by Przybylski and coworkers (Fiedler et al., 1998) and in a modern day version by Gross and coworkers (Jones et al., 2011). Because the turnaround is rapid, the population-level information is accurate, and the assignment of peptide sequences can be nearly unambiguous (by MS/MS), such differential labeling (footprinting) is now a pillar of MS-based protein/protein interaction studies, especially epitope mapping (Liu et al., 2020).
Limited proteolysis or proteolytic footprinting is widely used for epitope mapping. One of the seminal observations that opened the door for limited proteolysis is that antibody-bound regions under native conditions are resistant to proteolytic cleavage. This property is exploited in proteolytic footprinting of antibody-antigen complexes (immune complexes). Upon immune complex formation, that part of the antigen interacting with the antibody will bury the cleavage site or stabilize it, making it inaccessible or less reactive to the protease. By examining the differential nature of the resultant peptides, an investigator can identify the potential epitope (Jemmerson and Paterson, 1986). Very early on, such differential protease digestions were analyzed by HPLC-based separation followed by peptide-sequence analysis. The introduction of limited proteolysis to epitope mapping was a major milestone in the field, and the approach also unravels discontinuous epitopes.
The next breakthrough was the coupling of limited proteolysis with MS, called “epitope excision” by Przybylski and coworkers (Suckau et al., 1990). In this method, proteolysis of the free antigen or antibody-bound antigen, was followed first by plasma desorption MS and later by MALDI MS (Papac et al., 1994; Zhao and Chalt, 1994). This was significant because MS provided rapid turnaround along with nearly unambiguous mass and sequence confirmation of peptides. Although this approach was initially used for linear epitope mapping, as MS instrumentation became more sophisticated, this approach was quickly adapted to proteins ranging from small globular proteins to large glycosylated viral antigens (Parker et al., 1996; Jeyarajah et al., 1998). Around the same time, a similar approach was introduced called “epitope extraction” by Chait and coworkers (Zhao and Chalt, 1994) where the full antigen is digested by a protease, and those peptides that bind are allowed to form an immune complex. Non-interacting peptides are washed off, and the interacting peptides released from the complex are analyzed quickly by MALDI MS. This approach was later extended to affinity proteomics where the antigen was either separated by using the antibody of interest and then digested, or a complex cellular matrix containing the antigen was digested and bound peptides were extracted and analyzed (Yu et al., 1998; Peter and Tomer, 2001).
Contemporaneously, chemical crosslinking coupled with MS was introduced by Zenobi and coworkers (Pimenova et al., 2008) to show that the combination has potential for epitope mapping. The epitope extraction method was also used in conjunction with native MS whereby the mass of the peptide bound in the antibody complex was analyzed under native conditions, and from the increase in mass, one can infer the peptide location and sequence (Lu et al., 2009). Very recently, another version of peptide extraction under native conditions was proposed by Glocker and coworkers (Yefremova et al., 2017) where bound peptides were ejected by collision energy ramping to distinguish the relative binding affinities of closely related peptides. The current status of excision and extraction methods for epitope mapping was recently summarized by Glocker and coworkers (Opuni et al., 2018).
3 Current mass spectrometry approaches for epitope mapping
As discussed earlier, several MS approaches (e.g., epitope excision, epitope extraction, crosslinking, chemical labeling) are available. Epitope excision was widely used owing to its simplicity and ability to provide conformational epitope information. This approach, however, requires digestion to be optimized. Additionally, owing to steric hinderance and blockage of the digestion site, this approach may overestimate the epitope interface. Epitope extraction is an excellent amalgamation of PEPSCAN and limited proteolysis. In epitope extraction, the antigen is predigested, and the pool of peptides is allowed to bind with the antibody. Because an investigator can conduct all digestion reactions separately or together to shorten the peptide length, one can improve the spatial resolution of the epitope. Epitope extraction, however, has a drawback; it is not suitable for identification of conformational/discontinuous epitopes.
Selective amino-acid footprinting involves labeling solvent-exposed amino acids. By comparing amino acid reactivity in the presence and absence of an antibody, an investigator one can deduce the epitope site (Tremblay et al., 2022; Kant et al., 2023). This approach suffers because only a few amino acids can be targeted at a time, and there is a need to control the extent of labeling to prevent conformational change in the proteins. Excess labeling can perturb the stability of the immune complex, causing labeling-induced unfolding and/or dissociation of the bound complex, increasing the surface accessibility and reactivity of the residues as they become solvent-exposed (Limpikirati et al., 2020).
To address these shortcomings, FPOP (Fast Photochemical Oxidation of Proteins) methodology was introduced by Gross and coworkers (Hambly and Gross, 2005). FPOP uses fast (tunable from ms to µs labeling by using radical scavengers), highly controlled reactions (only one pulse per plug of solution), and a broadly reactive •OH radical (size is very similar to the water molecule so results can be compared with water accessible surface area) to avoid the shortcomings of slow labeling (Gau et al., 2009; Vahidi and Konermann, 2016; Niu et al., 2020). Additionally, amino-acid-specific, and free-radical labeling are often irreversible as opposed to reversible labeling by HDX (Section 4), enabling the use of several digestion schemes and CID-MS for locating the binding residues. CL-MS and FPOP-MS are gaining traction, but they require high-resolution MS to handle complex mixture of labeled and unlabeled peptides and specific skills for data analysis. Furthermore, FPOP requires additional instrumentation including a laser and flow system, which is expensive for many labs and can raise safety concerns. There are, however, alternatives (Sharp et al., 2021) that utilize FPOP chemistry without the laser to alleviate safety concerns.
In this regard, HDX-MS enjoys widespread popularity because its chemical reaction is induced by simple dilution of the media with D2O, the reactivity of the protein backbone is broad (except proline), and there are now robotic systems for automation and several packages for data interpretation. Another important factor contributing to the growing use of HDX-MS is its non-perturbing nature (labeling reagent OH− vs. OD−) to protein and protein complexes. Yamada and coworkers (Yamada et al., 2002) and contemporaneously Komives and coworkers (Baerga-Ortiz et al., 2002) were first to use HDX-MS for epitope mapping (Figure 3). Since then, developments in the HDX-MS field have been adopted for epitope mapping to gain deeper insights (see Section 5). As for other approaches, HDX-MS has its own shortcomings. There are few proteases that are active in acidic conditions (Section 5.1), and the analysis requires quick liquid chromatography coupled to Mass Spectrometry (LC-MS) runs owing to the reversible nature of labeling. Thus, coverage of the protein may be incomplete for hard-to-digest proteins (Section 4.2). A factor complicating identification of the antibody binding, applicable to all MS based approaches, is to distinguish binding from induced allosteric changes in antigen dynamics. Another challenge is to provide residue-level information by an instrumental approach given that collisional activation is not suitable because it scrambles the H and Ds. ETD or ECD-HDX-MS (Electron Transfer Dissociation/Electron Capture Dissociation coupled to HDX-MS) are possibilities, but the need to optimize these activation methods while minimizing H/D scrambling and achieving high charging of peptides needed for efficient fragmentation limits widespread use (Section 5.3). Another important factor for HDX-MS and other MS based approaches for epitope mapping is the stability of the bound complex. If dissociation rates for the antibody in complex with the antigen are high, then on/off dynamics will dilute the differential labeling signature and make interpreting the kinetics difficult. In most cases, however, antibodies have strong binding to antigen (∼nM range), and for these systems, high off rates are not a major issue. One further drawback of all MS-based techniques is they need one or more validatory experiments such as mutational analysis to confirm or refine their outcomes. Although mutated residues are often part of surface exposed flexible loops, site-directed mutagenesis is sometime difficult to achieve for that part of a protein that is critical for stability and may lead to erroneous conclusions (Greenspan and Di Cera, 1999).
4 HDX for epitope mapping
4.1 HDX theory and use
Proteins possess dynamic personalities, and even under native conditions, they sample a wide variety of open confirmations (Henzler-Wildman and Kern, 2007). Conformational opening involves breaking and forming (polar) hydrogen bonds. A proton is mobile, and during structural opening of H-bonding, a “new” proton from the solvent usually replaces it (Eigen, 1964; Hvidt and Nielsen, 1966). This phenomenon is utilized in HDX. By diluting the protein (ideally >10 fold) with D2O, the reformation of an H-bond will occur mostly with deuterium (forward exchange). As HDX occurs, the probability of finding protons in the protein diminishes rapidly. In addition to amide hydrogens on the backbone, those attached to electronegative oxygen (O), sulfur (S), and nitrogen (N) atoms in the side chains are also involved in reversible hydrogen bonding and undergo exchange. Because peptide-bond hydrogens undergo H-bonding and in enol-keto isomerization, the backbone amide hydrogens are more stable and slower to exchange than the side-chain H-bonds.
The HDX reaction is pH- and temperature-dependent. At physiological pH, exchange is OD− catalyzed, and at low pH, it is D3O+ catalyzed. At low temperature (≤0°C) and pH −2.6 HDX, rates are minimum, and, hence, these conditions are utilized to quench HDX at specific times and minimize deuterium back exchange (exchange out those deuteriums that had exchanged in the protein with solvent protons) during LC-MS (Bai et al., 1993). In the back exchange occurring after quench and before detection, the side-chain Hs back exchange quickly, and hence, HDX predominantly measures the backbone dynamics (Skinner et al., 2012).
The ability of HDX to “footprint” backbone amide bonds of different open conformations makes HDX one of the most valuable techniques to study both protein dynamics (James et al., 2022) (Jethva and Udgaonkar, 2017; 2018) and changes accompanying protein-protein interactions (e.g., epitope mapping). Apart from structural aspects of protein dynamics that affect the exchange reaction, there are other physicochemical factors (e.g., pH, temperature, and ionic strength) that influence HDX. Thus, it is important to control carefully the physicochemical parameters for comparative analysis of protein dynamics as in epitope mapping (Hamuro, 2021).
Hydrogen and deuterium differ in several physicochemical properties including density (Hvidt and LinderstrØM-Lang, 1954), infrared absorbance (Haggis, 1957; Blout et al., 1961; Goormaghtigh, 2013), NMR absorbance (Wishnia and Saunders, 1962; Otting and Wüthrich, 1990; Paterson et al., 1990; Englander and Mayne, 1992), radioactivity (for hydrogen tritium exchange, HTX) (Leach and Springell, 1962; Englander, 1963; Ashman et al., 1971), UV absorbance (Englander et al., 1979), neutron diffraction (Kossiakoff, 1982), and molecular weight. All these properties can be exploited to follow the extent of HDX. The first use of MS was to measure the changes in exchanged water (Nabedryk-Viala et al., 1976), but subsequently MS was utilized for monitoring the exchange in peptides in a bottom-up analysis (reconstructing the protein level HDX behavior by looking at the peptide level exchange behavior) (Sethi et al., 1983; Verma et al., 1986; Katta and Chait, 1991; Mandell et al., 1998). Currently, MS analysis of exchanged peptides is a mainstream methodology because it affords medium structural resolution, high sensitivity, and practically no size limit for the target protein (antigen) (Engen and Komives, 2020; James et al., 2022). Thus, locating an epitope map is most often met by using a bottom-up HDX-MS approach (Figure 4).
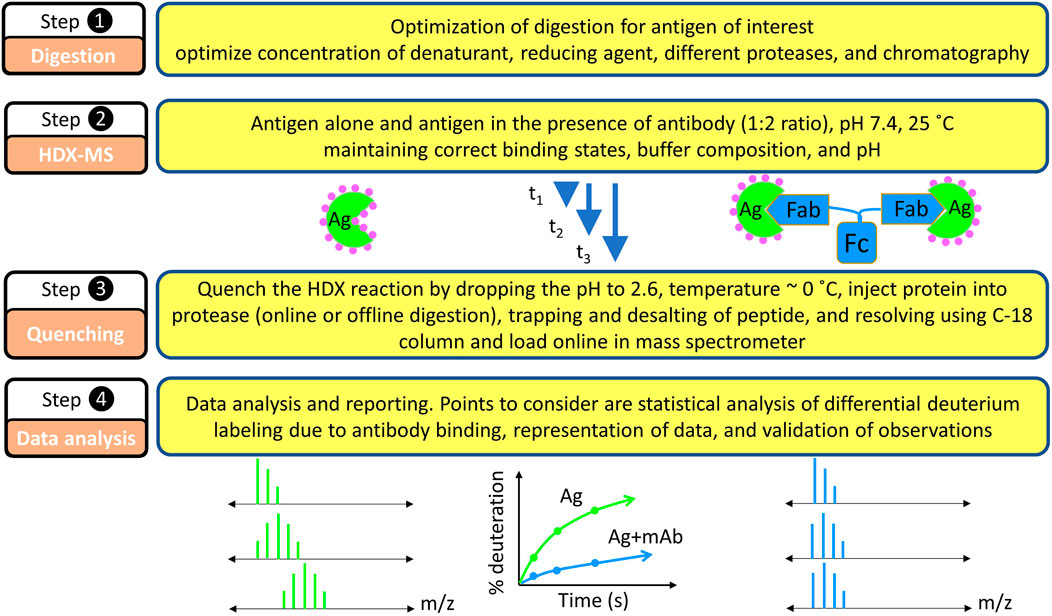
FIGURE 4. Basic steps in HDX-MS based epitope mapping. Each step requires equal consideration in the experimentation. Because the experiments are differential in nature, factors that possibly affect the HDX kinetics must be carefully considered: pH, temperature, buffer strength, binding stoichiometry during isotope dilutions, and others.
Like HDX, the sister technique of HTX, pioneered by Englander and coworkers (Englander, 1963), was among the first to be deployed for antigen-antibody interface analysis (Ashman et al., 1971; Liberti et al., 1972; Liberti et al., 1981). As other detection methods evolved, HDX was coupled with infrared spectroscopy (Závodszky et al., 1981), subsequently with NMR for epitope mapping (Paterson et al., 1990). Such studies brilliantly elucidated the role HDX can play for epitope and paratope mapping to reveal binding-induced conformational changes (Benjamin et al., 1992; Mayne et al., 1992). Although NMR provides atomic level details of the epitope map, the relatively high concentrations of protein required (often mM concentration) and the size of antigen that can be studied (<30 kDa) are shortcomings. Nevertheless, NMR is still used (Bardelli et al., 2015; Simonelli et al., 2018; Di Muzio et al., 2020; Valente and Manzano-Rendeiro, 2021). To address the shortcomings of NMR, investigators turned to MS in the early 1980s with fast atom bombardment (FAB) (Barber et al., 1981) and in the late 1980s with MALDI (Karas and Hillenkamp, 1988; Tanaka et al., 1988) and finally with ESI (Fenn et al., 1989). The first MS application of HDX to intact peptide/proteins was by Katta and Chait (Katta and Chait, 1991) followed by design of the bottom up approach by Zhang and Smith using FAB (Zhang and Smith, 1993), but the complexities of FAB were an obstacle to widespread adoption. In the 2000s, the first HDX in combination with ESI-FTICR-MS (Yamada et al., 2002) and around same time the coupling with MALDI (Baerga-Ortiz et al., 2002) were utilized for the epitope mapping. The compatibility of ESI with online LC systems has made it the most widely used soft ionization approach for HDX. Early epitope mapping studies, using both HDX-NMR and -MS, clearly indicate not only that antigen-antibody bonding primarily stabilizes side-chain interactions but also slows the backbone dynamics, affording a map of an epitope.
4.2 HDX protocol for epitope mapping
Epitope mapping by HDX-MS is comparative in nature, meaning that the HDX kinetics of the unbound antigen is compared with that of the antibody-bound antigen (Figure 4). The first step is to consider the feasibility of HDX for a given epitope mapping problem (Hamuro and Coales, 2018). HDX can be monitored at the intact protein level, but that gives no spatial resolution, motivating protein digestion post HDX and bottom-up MS analysis. As mentioned earlier, HDX, being a reversible footprinting technique, requires monitoring the isotopic exchange via fast gradient LC-MS runs. Quenching the HDX reaction at pH 2.6, approximately 0°C can preserve the labeling pattern for a longer time than at pH 7 and room temperature, hence making possible reliable detection by MS.
Forty years ago, a bottom-up protocol using porcine pepsin (an acid endo-protease) was introduced for improving structural resolution (Rosa and Richards, 1979). Although the field has grown rapidly since, most studies still utilize pepsin (Section 7, meta-analysis of literature) because it is reproducible, active at low pH and at quench conditions involving denaturant and reducing agents (Blumenfeld et al., 1960) and has broad specificity for producing many overlapping peptides (Hamuro et al., 2008; Ahn et al., 2013). The non-specific nature of pepsin also improves spatial resolution and increases confidence because results are recapitulated in several different peptides. Currently, the bottom-up protocol has essentially the same features as 40 years ago; that is, the modern protocol seamlessly interfaces with online digestion, LC, and MS (Zhang and Smith, 1993). The peptides generated by online acid protease digestion are trapped, eluted, separated by using C-18 RP-HPLC, and, after separation, submitted to the mass spectrometer usually operating with ESI.
The bottom up HDX-MS experiment starts with the generation of a peptide map for a given antigen (Figure 4, step 1) in the absence of HDX (unexchanged) where CID-MS/MS helps identify the peptides and determine their retention times. Given the labile nature of H and D positions in peptides, however, CID cannot be used for locating deuterium in the HDX experiment (Section 5.3).
The unexchanged antigen is diluted in H2O followed by a quench whereby the pH is decreased to approximately 2.6, and the protein injected onto the online protease column where it is digested. The generated peptides are captured on a trap column while salts, unfriendly to the mass spectrometer, are washed off. When an LC gradient is applied, the peptides are shifted to an analytical column where they are separated and transferred online to the mass spectrometer. One way to improve spatial resolution is to tune the conditions of proteolysis (e.g., the time for passage of the protein through protease columns and/or concentration of reducing and denaturing agents) to create many overlapping peptides (see below).
For mapping, the spectrometer is usually run in a Data Dependent Analysis (DDA) mode where ions representing predefined precursors are selected and fragmented individually. The saved data are analyzed using LC-MS/MS data search programs containing the antigen sequence. Typically, peptides that have a precursor mass within a predefined tolerance (set for the MS instrumentation) of an expected mass of a given sequence are accepted but refined in a second pass to include those peptides undergoing reliable fragmentations that are consistent with the sequence (typically 3 to 4 fragments per 10 amino acids) are considered. In a third pass, the above procedure is repeated at least three times to assess the reproducibility of the quench conditions, digestion, gradient, and LC-MS parameters.
Setting up the HDX protocol takes advantage of the unfolding of most proteins at pH 2.6 used for the quench. Often parts of proteins are disulfide-bonded and acid stable, adding difficulty for acid proteases to digest and provide full coverage. In such scenarios, addition of denaturants and reducing agents (e.g., urea, a mild denaturant, guanidinium hydrochloride, a strong denaturant, tris-carboxyethyl-phosphine hydrochloride (TCEP-HCl), an acid compatible reducing agent) or an organic solvent (Guo et al., 2020; Fang et al., 2022) can be explored to achieve good coverage and overlapping peptides. Additionally, the use of “fast” (non-scrambling fragmentation methods) in lieu of CID for the peptides is gaining some traction, giving improvement in spatial resolution of the HDX data (see below).
Once digestion is optimized and good coverage is obtained, the next step is to monitor the HDX kinetics for each peptide (Figure 4, step 2). Typically, a protonated protein is diluted 5 to 10-fold in deuterated buffer of the same pH (ideally of the same composition as well), and HDX is initiated, in the presence and absence of the antibody. At predetermined times, the HDX is quenched by decreasing the pH to 2.6, approximately 0°C followed by online protease digestion (most commonly used) or offline digestion (historically used) (Figure 4, step3). The generated peptides are trapped by using a small C-8/C-18 trap column and then eluted as resolved by the C-18 analytical column and submitted to the mass spectrometer. During HDX-MS analysis, the spectrometer is in the MS-only mode (not MS/MS) to measure the isotope distribution of the peptides. This workflow is repeated as a function of time (from seconds to hours), to cover the time scales of protein (antigen) breathing motions in the presence and absence of the antibody.
The collected HDX kinetic data are analyzed manually by first obtaining extracted ion chromatograms (EIC) for integration or by using sophisticated, automated software for the data analysis (Figure 4, step 4). Such software packages use the LC-MS/MS mapping runs to locate the correct peptide from the unexchanged controls based on retention time, ppm mass error, and expected isotopic distribution. A non-exhaustive list of software includes HXExpress (Weis et al., 2006a), DynamX (Waters, Milford, MA), HDX Workbench (Pascal et al., 2012), HDExaminer (Hamuro et al., 2003), Mass Spec Studio (Raval et al., 2021), Deuteros (Lau et al., 2019), HDX suite from PMI, and others (Wales et al., 2013). Some are open source.
Finally, the differential HDX pattern must be visualized, and this is done by either HDX kinetics plots showing patterns in the absence and presence of antibody (Figure 5A) or in a more condensed differential format such as Woods’ plots (Yamada et al., 2002) (Figure 5B), bar graphs (Figure 5C), heat maps (Zhang et al., 2010), and butterfly plots (Houde et al., 2011). Although each visualization has its own appeal, it is beyond the scope of this review to comment on each. It is the authors’ opinion that kinetic plots are simplest and most informative. As an example, the kinetic plot for the POV4 bound state of peptide 12–21 shows the most protection (decrease in rate of deuterium incorporation upon complex formation) at the earliest time point of HDX, but at the longest time point, the kinetic curve shows HDX similar to that of the unbound state (Figure 5A). Furthermore, peptide 12–24 does not show any merging of the HDX kinetic curves. Both peptides indicate that the 12–21 region remains dynamic, and that the epitope contacts are near residues 22–24. Similarly, for POWV-63 peptides, residues 73–90 and 243–253 show protection. The strong protection and non-converging HDX kinetics indicate that the main epitope contact lies in the region 73–90, and that region 243–253 remains dynamic as compared to the 73–90 region in the immune complex. Figures 5B,C show Woods’ plots and bar graph presentations where cumulative differences across all HDX timepoints (bound-unbound) are plotted along with the propagated error for each peptide (for Woods’ plot global error), respectively. The Woods’ plot is more informative as multiple dimensions of HDX data can be presented including secondary structure, sequence coverage, peptide lengths, and the extent of HDX as a function of sequence. Furthermore, the confidence interval can be color-coded in a fashion that is similar to that in volcano plots.
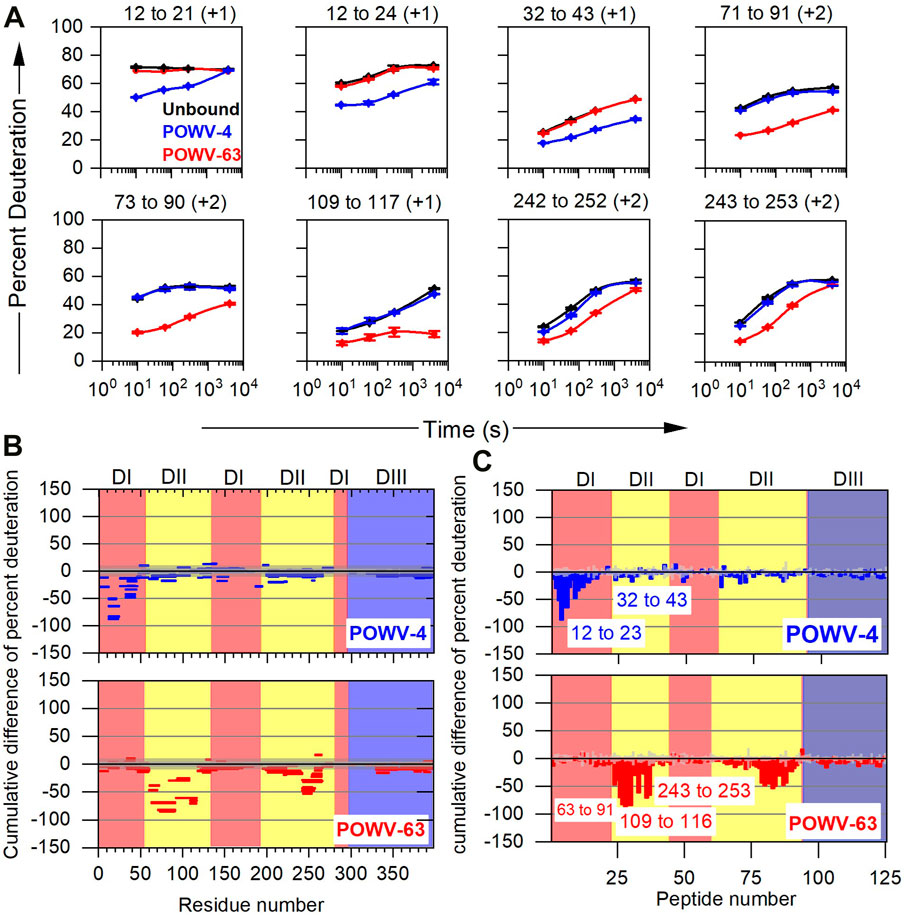
FIGURE 5. Typical example of data presentation in HDX-MS based epitope mapping. (A) Representative kinetic plots for eight peptides showing the effects of antibody binding on HDX. Black, blue, and red lines are for POWV E in the absence of antibody, in the presence of POWV-4 mAb, and in the presence of POWV-63 mAb, respectively. At the top of each panel are the residue numbers and charge states of the peptides. Error bars represent SEM from duplicate measurements. (B) Woods’ plot and (C) bar graph showing accumulated difference in percent deuteration (bound state—unbound state) across all time points for each analyzed peptide. Central and outer gray shaded areas indicate 95% and 99% confidence intervals, respectively, for the global SEM calculated for each differential HDX experiment and given in the Woods’ plot. For the bar graph, the Gy bar indicates three times the standard error of mean for significance. Republished from VanBlargan et al. (2021), with permission from Rockefeller University Press.
Most data analysis software packages include data visualization as well, and many dedicated software packages are available (mostly for free). These packages help in data visualization, and they include MSTools (Kavan and Man, 2011), MEMHDX (Hourdel et al., 2016), HDfleX (Seetaloo et al., 2022), HD-eXplosion (Zhang et al., 2021), and others. As with most comparative analysis, HDX differential plots also require statistical analysis, described by (Houde et al., 2011; Masson et al., 2019; Weis et al., 2019).
In the next sections, we organize our review around the use of several improvements to complement/validate observations by HDX-MS and, in that way, provide a current view of the field. Although we center our discussions around epitope mapping, we will show how improvements in the HDX-MS technology overcome past shortcomings of the general HDX-MS methodology.
5 Improvement in HDX-MS protocols
5.1 Incorporating multiple proteases
Very early on, it was realized that digestion of a protein would improve the spatial resolution afforded by HDX. As mentioned earlier, porcine pepsin was introduced 40 years ago for digesting a protein post HDX. Its utility is due to its compatibility with the HDX quench conditions (low pH, presence of denaturants) (Blumenfeld et al., 1960). Subsequently, other proteases including acid proteases from type XIII fungus (Aspergillus saitoi) (Cravello et al., 2003; Englander et al., 2003), type XVIII fungus (Rhizopus species) (Cravello et al., 2003), nepenthesin I and II, acid proteases from a carnivorous plant (Rey et al., 2013), from rice field eel (Monopterus albus Zuiew) (Ahn et al., 2013), Rhizopuspepsin (Rey et al., 2009), and Pepsin A1 and A2 (Brier et al., 2007). Because each protease has broad but different specificities, tandem (Cravello et al., 2003; Mayne et al., 2011; VanBlargan et al., 2021; Doyle et al., 2022), parallel (Nirudodhi et al., 2017), or concurrent digestions (Hamuro and Zhang, 2019; Mullahoo et al., 2020) using multiple enzymes can greatly improve the sequence coverage of hard to digest proteins and increase the number of overlapping peptides.
Marshall and coworkers (Zhang et al., 2011) used for the first time for epitope mapping tandem protease digestion for the large nut allergen protein Ana o 2 (95 kDa) to improve coverage and give many overlapping peptides. Using a combination of an automated HDX platform, tandem protease digestion, and high-resolving power of FT-ICR-MS (Zhang et al., 2008; Kazazic et al., 2011), Marshall and coworkers (Zhang et al., 2011) investigated more than 100 peptides from the antibody-bound complex to find both linear (for 1F5) and conformational (for 2B5) epitopes. It is important to note here that Ana o 2 is a homotrimeric protein (monomer 95 kDa, trimer −285 kDa) in solution. Considering that each homotrimer will bind one antibody (150 kDa), then the solution complex is nearly half a mega Dalton (0.435 MDa).
5.2 Using new data processing
Any digestion that gives many peptides leads to other problems of chromatographic crowding, spectral overlap, and demanding data analysis. Furthermore, ESI forms multiple charge states for many peptides, adding complexity to the mixture of ions. Especially for epitope mapping, the presence of the large antibody (150 kDa MW) compounds the problem because its digestion affords many more peptides than does the antigen. Hamuro and coworkers (Coales et al., 2009) tried to minimize the chromatographic crowding by doing on-column exchange using an immobilized antibody. The antigen is deuterated in solution and then in the on-column, antibody-bound state, and the antigen is eluted under quench conditions. This approach increases the number of analyzable peptides detected post HDX. Jørgensen and coworkers (Jensen et al., 2013) introduced a modified protocol for affinity separation of biotinylated antibodies post HDX. Such affinity-based antibody separation, however, requires preparation and characterization of an immobilized antibody for its binding. Another difficulty arising from the large numbers of peptides is that deuteration causes each peptide to shift on the m/z scale, adding more signals and complicating interpretation. Although managing those shifts manually is an enormous task, this issue can be addressed in several ways. The easiest is to use automated HDX data analysis software packages (see HDX data analysis Section 4.2). This allows the analysis of hundreds of peptides in a relatively short time. Additionally, software packages also provide statistical analysis and different ways of HDX data visualization.
Englander and coworkers (Casina et al., 2015) used a multiprong strategy for high resolution mapping of epitopes for autobodies targeting MDTCS (Metalloprotease, Disintegrin, TSP1, Cys-rich, and Spacer) regions of ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13). Inhibition of ADAMTS13 by autobodies leads to a severe pathological condition called acquired thrombotic thrombocytopenic purpura (TTP). Thus, it is important to identify those epitopes that are targeted by autobodies and render ADAMTS13 ineffective. To do this, the authors first used pepsin and fungal type XIII protease digestion to obtain many overlapping fragments (Figure 6A). Following that, the relevant domain (MDTCS) of the larger antigen (ADAMTS13) was used as an antigen. Third, HDX was carried out on-column with immobilized single-chain fragments of the variable regions (scFvs) so that post quench, only the antigen would elute. All these steps reduced chromatographic complexity. Furthermore, to record the isotopic distribution post exchange, the authors used ExMS (Kan et al., 2011) software followed by HDsite (Kan et al., 2013).
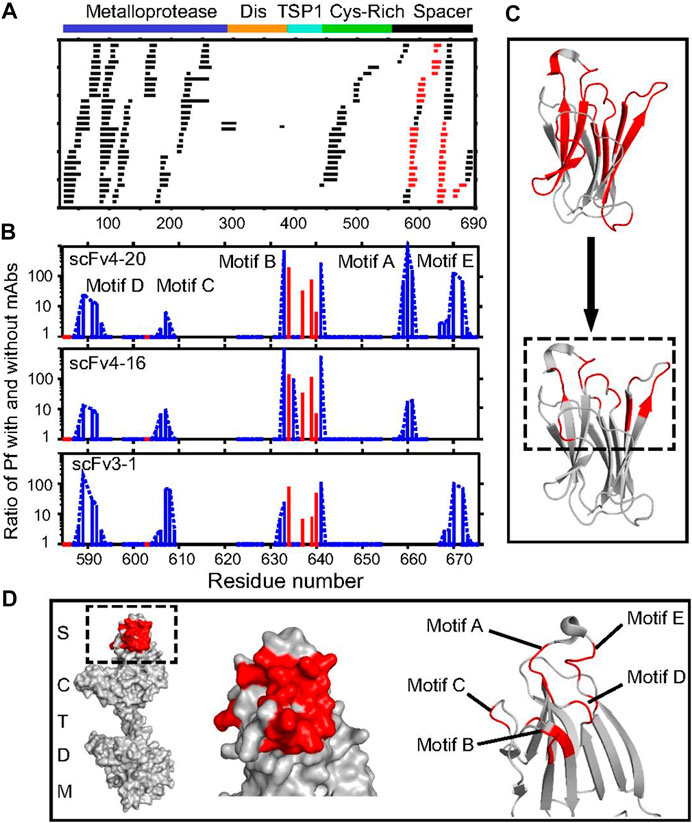
FIGURE 6. Antibody-binding sites in ADAMTS13 revealed by HDX MS. (A) Acid protease digestion of human MDTCS fragment produced 208 unique overlapping peptides with 70 derived from the spacer domain and 113 from M. The peptides shown in black show no difference in HDX rate with and without scFv bound; peptides depicted in red show decreased HDX upon scFv binding. (B) The ratio of an additional protection factor (Pf) with scFv bound. Resolved single residues are in red; switchable residues in blue are connected by dashed lines. Some otherwise-protected regions missing in motifs A and E are ambiguous owing to fewer peptides recovered and not necessarily to the absence of antigen–antibody interaction. (C) Representation of the binding epitope (red) at coarse peptide resolution (Top) and at refined residue resolution (Bottom). (D) Representations of the binding epitope (red) in the spacer domain of ADAMTS13. The slowed HX in the five-loop region identify the binding epitope but do not necessarily indicate the residues that become engaged in specific interprotein interactions, as explained in the text. Republished from Casina et al. (2015), with permission from National Academy of Sciences.
For improving the spatial resolution when overlapping peptides are available, most investigators rely on simple subtraction of centroid mass of smaller peptides from that of the larger peptide. This strategy works best when peptides have a common N-terminus. It is often not possible that there is a perfect series of overlapping peptides to obtain a deuterium location with high certainty. As another metric, the isotopic distribution envelope shape is often overlooked in HDX-MS analysis although its broadness can provide a clear signature of nature of exchanging deuteriums (Weis et al., 2006b; Zhang J. et al., 2013).
In another approach, Englander and coworkers (Kan et al., 2013) developed the HDsite algorithm that takes the isotopic distribution post HDX as input and quantifies deuterium retention solely based on envelope shape. By looking at many overlapping peptides (although not perfectly overlapped), one can validate deuterium location in employing an iterative protocol and obtain higher spatial resolution and even residue-resolved deuteration. For example, the effort by England and coworkers (Kan et al., 2013) identified five contiguous loops on ADAMTS13 that were targeted by autobodies indicating a conformational epitope (Figure 6D). Interestingly, for some of the loop regions, residue-level resolution could be obtained to define precise boundaries for the epitope (Figures 6B,C) by using the HDsite algorithm. Epitope validation studies revealed that all five loops are essential for the autobody binding as deletion of any one loop leads to complete abrogation of binding.
5.3 Using gas-phase fragmentation
The most common way of improving spatial resolution is to use of multiple proteases in tandem. This approach, however, can lead to noisy, overlapped isotopic distributions. At the same time, overlapping peptides may not back exchange similarly owing to sequence, near-neighbor effects, and different responses to the reversed-phase column (Sheff et al., 2013). Such compounding effects can lead to faulty sub-localization of deuteriums based solely on the degree of overlaps. To address this issue, rapid gas-phase fragmentation may be successful.
As mentioned earlier, CID cannot be used because it raises stepwise the vibrational energy of a peptide ion, leading to deuterium scrambling prior to fragmentation (Jørgensen et al., 2005). Electron Capture Dissociation (ECD, using electrons from heated filaments) and Electron Transfer Dissociation (ETD, transferring an electron from a radical anion) give similar outcomes where electron transfer to a protonated peptide leads to rapid (allegedly picosec) fragmentation, thereby preempting scrambling and preserving the sensitive pattern of deuterium labeling (Zubarev et al., 1998; Syka et al., 2004). Although the ETD/ECD approaches have exciting potential, an investigator needs to ensure that scrambling is minimal because “naked” gaseous ions are labile, and even the acceleration and consequent collisions while transferring ions from one instrument compartment to another can lead to redistribution of labels (scrambling) (Demmers et al., 2002; Hoerner et al., 2004; Rand and Jørgensen, 2007; Wang et al., 2019; Wollenberg et al., 2020). Fortunately, there is a well-established protocol available for checking and minimizing scrambling for HDX by using a properly designed standard peptide (Rand and Jørgensen, 2007). ETD/ECD fragmentations can be used in two ways: top-down (where the intact protein is fragmented and its regional behavior studied) and bottom up (Pan et al., 2009; Catherman et al., 2014; Karch et al., 2018). Currently, a bottom-up approach for epitope mapping (see case studies described below) is most often used. Recently, a promising approach, ultraviolet photodissociation (UVPD), was optimized for gas-phase fragmentation that gives minimal H/D scrambling (Mistarz et al., 2018; Modzel et al., 2021).
In another example, Chen and coworkers (Huang et al., 2018) identified mAb1, a strong binder with TL1A, a member of the tumor necrosis factors superfamily. In the presence of a soluble decoy protein, the death domain receptor 3 (Dr3) protein inhibits TL1A-mediated activity. By inhibiting TL1A and Dr3 interactions, the necrosis activity of TL1A can be restored. Competitive binding studies reveal that mAb1 and Dr3 have overlapping binding sites. Because TL1A is a functional trimer (90 kDa MW for monomer) and binds with mAb1 with 3:3 stoichiometry, the functional complex has a MW of ∼0.55 MDa, challenging traditional structural methods. When the authors looked at differential deuterium labeling on TL1A in the presence of mAb1, two regions, encompassing two solvent-exposed loops, and one structured region became most protected upon antibody binding (Huang et al., 2018). Protection in the structured region was hypothesized to be due to binding-induced remote conformational changes. Based on solvent accessibility docking studies, the epitope was localized within two HDX protected loop regions. Because both loop-containing peptides are long (residues 102–116 and 166–180), the investigators used ETD to fragment peptides post HDX in the presence and absence of mAb1 and located the protected sites to be 113–116 and 169–172, indicating that both loops are involved in antibody binding and that only a few residues in the large peptides contact mAb1.
Recently, the same group deployed HDX-MS for identifying epitopes for four antibodies targeting major histocompatibility complex class I chain-related A (MICA) (Huang et al., 2020a). MICA’s interaction with natural killer cells leads to cytotoxicity and antitumor activity. Proteolysis of MICA by tumor-specific proteolysis leads to inhibition of its antitumor activity. Thus, targeting a proteolytic site on MICA by monoclonal antibodies may effectively restore its antitumor activity. Three mAbs, mAb2, mAb39, mAb40, with similar binding constants, target similar epitope regions whereas mAb39, which binds 10 times stronger than the other mAbs, chooses a similar region but a different conformational epitope. HDX footprints indicate that mAb36 binding is stabilized by multiple epitope contacts, which may be responsible for its stronger binding. When two strongly protected peptides in the presence of Fab36 were subjected to ETD post HDX, only one small (8 amino acids) peptide showed complete fragmentation whereas a longer peptide (15 amino acids) did not fragment well under scrambling-minimized ETD. ETD indicates that only two residues (234 and 237) are responsible for protection, pinpointing specific contacts by the antibody.
These studies demonstrate that the spatial resolution can be improved for epitope boundaries by gas-phase, electron-based fragmentation. Further, understanding the extent of scrambling is important for successful application of ETD/ECD methods for epitope mapping. Unfortunately, few peptide fragments are seldom sufficiently long and multiply charged to be amenable to ETD/ECD (Good et al., 2007).
5.4 Using automation and new Hardware
The improvement in LC (UPLC and nanoLC) (Wu et al., 2006) systems and the availability of refrigerated chromatography for HDX (Wales et al., 2008) are pushing HDX-MS into the mainstream structural proteomics. The introduction of a complete automated system enables HDX-MS (Chalmers et al., 2006) to become more routine and be deployed as an early structural biology approach to interrogate solution dynamics of protein complexes. A recent apt example is the use of HDX-MS to characterize epitopes for nine neutralizing antibodies against the receptor-binding domain of SARS CoV2 spike protein to assist the design of antibody cocktails for efficient neutralization of viral infections (Hansen et al., 2020). A study by Weis and collaborators (Toth IV et al., 2017) determined the epitopes of Ricin binding protein A (RiVax) for nine neutralizing and one non-neutralizing antibodies. Based on regional protection found by HDX-MS upon antibody binding, the nine neutralizing antibodies were binned to four clusters.
Similarly, Gross and collaborators (Fernandez et al., 2018; Adhikari et al., 2021) characterized epitopes for five antibodies, and Simmons et al. (Simmons et al., 2021) determined epitopes for six nanobodies. In a recent study, Weis and collaborators (Haque et al., 2022; Haque et al., 2023) characterized epitopes for eleven mAbs against the recombinant outer-surface protein A (OspA) from Borrelia burgdorferi sensu stricto (ss). These studies clearly indicate that HDX-MS can characterize immune responses for vaccine candidates during different stages of development. Automation for HDX-MS (including data analysis) enables these large studies of epitope mapping for several antibodies, bringing HDX-MS more mainstream.
In contrast to running multiple comparative HDX studies, Marshall and collaborators (Zhang Q. et al., 2013) used polyclonal antibodies (pool of antibodies extracted from serum) to characterize the immune response of a nut allergen. Subsequently, others described polyclonal antibodies for study of immune response against tumor necrosis factor α (TNF-α) (Abbott et al., 2013) and factor H-binding protein (fHbp) (Ständer et al., 2021). Further, other investigators compared the immune response for a given antigen in different serum samples (Yang et al., 2016) to evaluate different diagnostic polyclonal antibodies against C-reactive protein, an important biomarker for inflammation (Sun et al., 2022) and recently against Cardiac troponin I (cTnI), a biomarker for acute myocardial infarction (Song et al., 2023). Such studies showcase the exquisite tolerance of HDX-MS for handling molecular heterogeneity, which is difficult with other structural methods.
An obvious way to decrease chromatographic crowding and spectral overlap is to increase the gradient length. Traditionally, the gradient used for chromatographic separation is <15 min to minimize back exchange. The gradient can be lengthened while minimizing back exchange by running the chromatography at low temperatures (e.g., −10°C (Fang et al., 2021), −20°C (Wales et al., 2017), and −30°C (Venable et al., 2012; Anderson and Hudgens, 2022). Fang and others (Fang et al., 2021) showed that a chromatography gradient at −10°C can be completed in 90 min, providing a 3-fold increase in number of peptides identified. Additionally, subzero temperature HDX can be coupled with gas-phase fragmentation (Amon et al., 2012; Pan et al., 2014). Recently, a low-temperature chromatographic set up was characterized for its ability to retain deuterium; at −30°C, back exchange slowed by 40-fold as compared to 0°C. At −20°C, back exchange was ∼10%, and nearly 90% of exchanged deuteriums were retained.
Venable et al. (2012) demonstrated that a −30°C chromatographic set up can be employed to map a linear epitope (3XFLAG-epitope) recombinantly attached to C-terminus of GFP protein. The 3X-FLAG peptide sequence retained ∼2.5 fold more D (close to 90%) at −20°C as compared to chromatography at 0°C (Figure 7A). Additionally, the difference in D retention in the presence and absence of the M2 antibody was approximately 2.5 fold, nearly completely preserving the epitope footprint (Figure 7B).
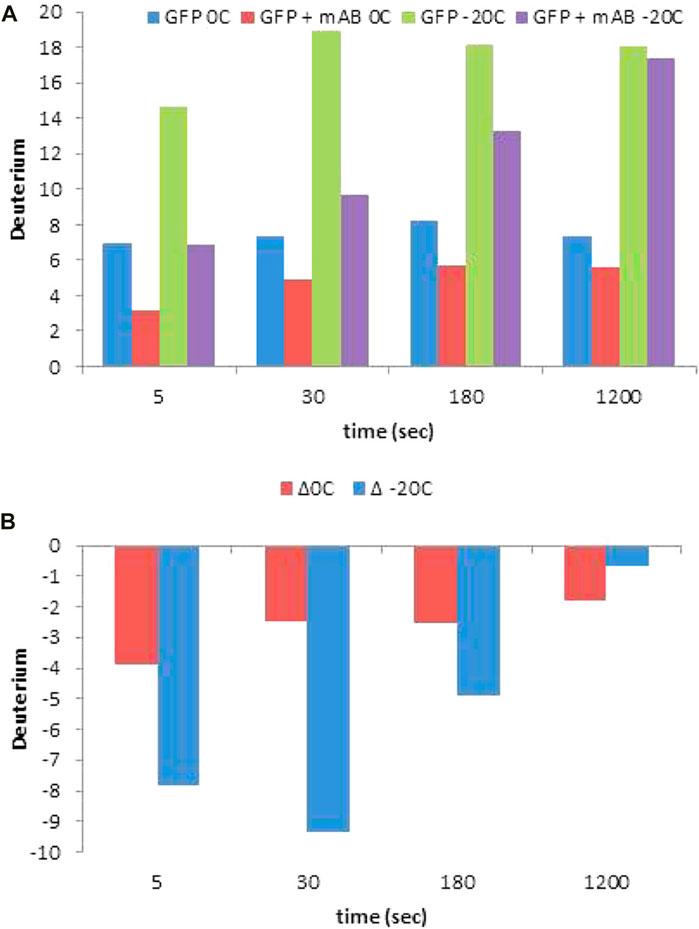
FIGURE 7. (A) HDX levels and measured protection (B) for each time point of an epitope mapping experiment for the 3X FLAG-GFP/M2 anti-FLAG complex for the +5-charge state of the peptide YFQGDYKDHDGDYKDHDIDYKDDDDKMVSKGEE, containing the entire 3X FLAG tag. Reprinted with permission from Venable et al. (2012), Copyright 2012, American Chemical Society.
In another study, Zhang et al. (Zhang et al., 2018) used chromatography at −9°C and increased the gradient length, reduced spectral overlap, and improved overall sequence coverage for the Birch Pollen Allergen. Fang et al. (Fang et al., 2022) used a −10°C chromatographic set up with a 30 min gradient for epitope mapping of Anthrax Protective Antigen and four monoclonal antibodies.
The above studies indicate that lowering the chromatography temperature can significantly expand the time window for the gradient and allow more and larger protein assemblies/antigens to be interrogated for epitope mapping. There is also growing interest in use of Ion mobility (Iacob et al., 2008; Cryar et al., 2017) and capillary electrophoresis (CE) (Black et al., 2015) separation for tackling chromatographic crowding. Wider adoption of automation (Section 7) could further integrate low temperature (∼30°C) chromatography, and with many studies utilizing the Waters Synapt G2 system, it may be possible to see routine incorporation of ion mobility and ETD (for obtaining residue-level resolution) for not only epitope mapping but also for other HDX-MS studies. Ultra-low temperature, however, has its own challenges in terms of back pressure and the need to redesign the chromatographic system. Nevertheless, it remains a promising approach.
5.5 Reducing disulfide bonds
Many surface-exposed viral antigens (e.g., SARS CoV2 spike protein) and soluble serum antibodies are rich in disulfide bonds. As described earlier, successful HDX-MS requires good sequence coverage of the antigen. Given that proteases cannot cleave the tightly packed regions of a protein, reduction of disulfide bonds becomes imperative. TCEP-HCl is a widely used reducing agent as it is odorless and works under acidic conditions. For each new protein, the concentration of TCEP-HCl, time, and pH of the quench must be optimized. An important consideration is the reduction potential of TCEP because its reactivity is affected at low pH and temperature. Most investigators add denaturants to facilitate protein unfolding so that efficient reduction can be achieved.
We did a meta-analysis of >80 papers for this review (see below) and found that approximately 60% of the epitope mapping studies use TCEP-HCl denaturant for disulfide bond reduction (Section 7). Some highly disulfide-bonded proteins are difficult to reduce in the short times needed for HDX-MS. Furthermore, the presence of TCEP also hampers the chromatographic and MS performance owing to formation of ion clusters (Zhang et al., 2010; Mysling et al., 2014). To address the shortcomings and “widen the net” for HDX-MS, online electrochemical reduction was implemented by Jørgensen and collaborators (Mysling et al., 2014). Subsequently, an online electrochemical reduction workflow was evaluated by the same group for highly disulfide-bonded proteins under different solvent conditions (Trabjerg et al., 2015), and for epitope mapping (Comamala et al., 2020b; Sheff et al., 2021). An ECD-based approach for improved gas-phase reduction of disulfides gave improved sequence coverage and localization of the deuterium label (Zubarev et al., 1999; Bobst and Kaltashov, 2014). ECD/ETD based approaches, however, still require optimization of instrument conditions such that H/D scrambling is minimal.
5.6 Dealing with glycosylation
Glycosylation (N (Asp)- or O (Ser/Thr)-linked), with its usual high heterogeneity, leads to poor MS signal and inadequate fragmentation, causing incorrect or no identification in HDX of glycosylated proteins. Furthermore, bulky glycans sterically hinder protease digestion, often leading to poor sequence coverage around the glycosylation site. The glycan acetamido group can lead to errors in locating deuterium (Guttman et al., 2011), whereas pre-HDX removal of glycans raises concerns that the native state of protein is not being studied. To address these issues, HDX protocols incorporating N-deglycosylation enzymes such as PNGase A (Jensen et al., 2016; Wagner et al., 2021) and PNGase H+ (acid compatible) (Comamala et al., 2020a) were introduced. Recently, Rand and collaborators (Comamala et al., 2021) integrated online electrochemical reduction along with chip-based “Immobilized Enzyme Reactors (IMERs)” with pepsin and PNGase Dj (newly introduced acid-compatible N-deglycosylation enzyme) for disulfide-bond reduction, digestion, and deglycosylation of peptides for epitope mapping of the Sema domain of c-Met (SD c-Met), a proto-oncogene (refer to figure 2 in original reference Comamala et al., 2021). Using a conventional HDX-MS protocol with TCEP as a reducing agent, the investigators observed only −68% sequence coverage. Missing were cysteine-containing and glycan-containing regions. With the introduction of online EC, the use of pepsin and PNGase Dj chip columns gave >80% sequence coverage, and all the Cys-containing and most of glycan-containing peptides were detected. With better sequence coverage, the investigators assessed the epitope regions and identified the antibody-induced conformational changes on SD c-Met. This study highlights the value of a flexible HDX-MS platform that incorporates modular functionality in tandem for addressing improvements for epitope mapping. Currently, no enzymatic counterpart exists for the removal of O-glycans, and hence, similar strategies cannot be utilized for addressing poor sequence coverage around O-glycosylation sites (Wilkinson and Saldova, 2020). An ETD/ECD based approach was shown to be promising for fragmenting glycosylated peptides, and, hence, scrambling optimized conditions can also be utilized for improving sequence coverage (Mikesh et al., 2006).
5.7 Distinguishing binding and allostery
One drawback of using HDX for epitope mapping and other protein-ligand interaction studies is the difficulty to distinguish protection induced by binding vs. that due to allosteric or remote conformational changes. Distinguishing these effects becomes more difficult for epitope mapping as epitopes can be conformational. During the equilibration stage, binding-induced structural changes (decreases in solvent accessibility) occur before allosteric changes (Ramirez-Sarmiento and Komives, 2018). Therefore, at short times, protection is seen at a binding site (orthosteric) as well as at distant sites (allosteric sites). Often allosteric protection in HDX can be seen from changes in protection and confirmed by site-directed mutagenesis (Hamuro et al., 2006). If structural details are not available, however, such assignments become tenuous (Sowole and Konermann, 2014; Konermann, 2016). Because most antibodies target solvent-exposed dynamic loop regions, protection achieved at the earliest times would be indicative of binding (Rob et al., 2013). Monitoring exchange in the millisecond time region can decouple protection induced by binding from that resulting from allostery. By adding the antibody (viewed as a ligand) in D2O itself, one can initiate binding concurrent with HDX and then separate in time the two phenomena.
For monitoring millisecond exchange, a custom-built, time-resolved electrospray ionization MS source for HDX (TRESI-HDX) was introduced by Konermann and coworkers (Wilson and Konermann, 2003; Rob et al., 2012) (refer to figure 1 in original reference Deng et al., 2017). Subsequently, Wilson and coworkers (Deng et al., 2017) used TRESI-HDX to reveal the allosteric changes in myoglobin upon antibody binding. They compared exchange when the antibody and antigen are pre-equilibrated with that when the antibody was introduced in the D2O. The results show, as predicted, that the epitope site developed protection earlier than the sites of allosteric changes (refer to figure 6 in original reference Deng et al., 2017, kinetic experiment). At approximately 4 s of HDX, the allosteric and binding changes are complete, motivating the need for millisecond time resolution. Recently, these investigators used the millisecond platform to evaluate the biosimilarity of a mAb under development and the original (Brown et al., 2020).
Millisecond time resolution for HDX is not new (Lento and Wilson, 2022). Traditionally, fast-pulse labeling was achieved in HDX with a quenched flow for studying fast protein folding (Roder et al., 1988; Udgaonkar and Baldwin, 1988; Miranker et al., 1993). For monitoring allosteric changes, a careful design of mixing sequences on the commercially available LEAP PAL robotic system enables sub-second HDX (Chalmers et al., 2006).
5.8 Using HDX for large antigen complexes
Increasingly the ongoing technological developments in HDX position it to tackle even larger protein complexes, as in viral particles, to understand their dynamics under physiologically relevant conditions. Flaviviruses are highly pathogenic, vector-borne, single-stranded RNA viruses (Pierson and Diamond, 2020). Transmission between cold-blooded mosquito (Aedes sp.) to warm-blooded humans occurs with structural changes even before the virus attaches to host cells (Zhang et al., 2015). During late endocytosis, the acidic pH of the endosome leads to structural reorganization of the protein, fusion of the virus to the membrane, and release (Mukhopadhyay et al., 2005). Although X-ray structures are available for different physicochemical perturbations and facilitate understanding of the structural changes in viral particles, at best they are static pictures, endpoints that miss the dynamics of the change. For HDX, the effects of pH (Matthew and Richards, 1983; Bai et al., 1993), temperature, and ionic strength (Kim and Baldwin, 1982; Christoffersen et al., 1996) can be deconvolved from the overall change, leaving behind the changes on the viral-particle and providing new insights on the nature of human immune response against deadly viruses. Flavivirus on its surface has 180 copies of an envelope protein, each with approximately 53 kDa MW (total approximately 10 MDa complex). Despite the size, HDX-MS of the intact virus-like particles of the proteolytic peptides could be measured (Wang et al., 2001; Wang and Smith, 2005).
Anand and coworkers (Lim et al., 2017b) first deployed HDX-MS to understand the temperature-dependent structural rearrangement of Dengue virus strains (DENV1 and DENV2) particles and revealed that only the DENV2 envelope is expanded at 37°C (human body temperature) as compared to 28°C (vector temperature). DENV1 viral particles exhibit structural expansion only at 40°C (conditions of high fever in humans). The difference can explain how changes in temperature facilitate early stages of DENV strain-specific infection. In a subsequent study, the same group (Lim et al., 2017a) measured the dynamics of the virus-like DENV2 particle by using HDX-MS and located temperature-specific changes in epitope and paratope dynamics. Importantly, the study revealed that an antibody (2D22) primarily targets an envelope protein (E) and the E protein dimer interface (refer to figure 6 in original reference Lim et al., 2017a). By targeting the interface, 2D22 can dampen the temperature-associated changes in the viral envelope during early stages of infection. Paratope mapping revealed that heavy-chain contacts are maintained irrespective of temperature whereas the light chain contacts are lost upon temperature-mediated expansion. Such epitope and paratope mapping of the intact infectious virus in a pathophysiologically relevant state represents a quantum leap in epitope and paratope mapping compared to the study of constituent viral proteins and offers clear insights into antibody action at the target viral surface.
6 Case studies: HDX-MS combined with other structural-proteomics and complementary approaches
As we have seen, developments in the field are addressing some of the shortcomings for HDX-MS epitope mapping. These advances suggest the efficacy of combining different MS-based footprinting to alleviate or even to complement HDX-MS and thereby to understand better the epitope interface. In this section, we will discuss some epitope-mapping studies where HDX-MS was used with complementary techniques for epitope mapping.
6.1 Providing complementarity with X-ray crystallography
Factor H binding protein (fHbp) is the main virulence factor for the Neisseria meningitides, a leading cause for meningitides and sepsis. Owing to high sequence variability of the fHbp protein, the goal of developing a broad-based universal vaccine remains a challenge. Epitope mapping on such a variable antigen via a quick but reliable method is a pressing need. Enrico and coworkers (Malito et al., 2013) used several epitope mapping methods to reveal finer details of the epitope targeted by the anti fHbp antibody. Scanning a synthetic library comprised of 64 dodecameric peptide sequences of fHbp revealed only one binding peptide, 238–249. Scanning a phage-display library containing on average 55 amino acid length segments reinforced the conclusion of the PEPSCAN results and pointed to three peptides encompassing region 238–249. SPR binding analysis using PEPSCAN, however, identified a peptide from the C-terminal region of fHbp that showed poor binding (KD > 1 mM) as compared to the full-length protein (KD < 0.05 nM). The poor binding of the identified peptides clearly shows that PEPSCAN and a phage-display library failed to reveal a complete epitope for 12C1. To resolve, the investigators deployed HDX-MS (using a similar protocol to that discussed in Section 4.2). They found protection not only at the C-terminal but also in the N-terminal domains. HDX-MS results clearly reveal the conformational epitope for 12C1. To corroborate, the investigators also conducted X-ray crystallographic analysis to verify that the interface is the same as that determined by HDX-MS. Furthermore, mutational analysis supports a broad binding interface also revealed by HDX-MS and X-ray crystallography. This study underscores not only the importance of using multiple techniques to determine the full epitope but also the ability of HDX-MS to provide peptide-level information, yielding results similar to X-ray crystallography but with more efficiency. Thus, deploying HDX-MS very early on during lead optimization is less resource-intensive, recommending that resource-intensive X-ray and Cryo-EM be deployed later when the problem has some definition.
6.2 Combining MS-based approaches for epitope mapping
There are other complementary footprinting techniques to HDX, and they often utilize an advantage of irreversible labeling (Limpikirati et al., 2018; Liu et al., 2020; Ralston and Sharp, 2022). Conceptually, the process remains the same; the differential labeling of an antigen in the presence and absence of binding partner is compared. Such chemical labeling can be specific for one or a few amino acids (glycine ethyl ester (GEE) or diethylpyrocarbonate (DEPC)) or it could be more broadly reactive (•OH or •CF3 radicals as in FPOP). Irreversible labeling has an advantage compared to HDX-MS that CID-MS can be used for gas-phase fragmentation to locate sites of labeling. Such covalent labeling was applied during the late 1980s for epitope mapping but not with MS detection (Burnens et al., 1987). As MS capabilities grew, studies began to utilize MS for locating covalent modification sites (Fiedler et al., 1998). Investigators employed several reagents including GEE (labels “D” and “E” residues) (Wecksler et al., 2015; Lin et al., 2018), DEPC (Tremblay et al., 2022), •OH (Jones et al., 2011; Yan et al., 2014; Zhang et al., 2017; Lin et al., 2018) and others (Dhungana et al., 2009; Liu et al., 2020) to map the epitope. It is •OH, generated by Fast Photochemical Oxidation of Proteins (FPOP), that enjoys some acceptance owing to its ability to control labeling and take advantage of the broad and high reactivity of free radicals and other reactive species (e.g., carbenes and carbocations), of the near pH independence of labeling, and of the ability to provide complementary information with respect to HDX-MS. FPOP depends on side-chain dynamics whereas HDX-MS uncovers backbone dynamics. These advantages make FPOP a good complement to alleviate some of the shortcomings of HDX-MS for epitope mapping. The Gross lab and their collaborators at Bristol Myers Squibb (BMS) have taken a lead in pushing the use of several mass spectrometry-based approaches to fine map the epitopes. In the following section, we discuss some of them.
6.2.1 Combining HDX-MS and covalent labeling
Gross and collaborators at BMS (Li J. et al., 2017) studied the epitope for 7B7 (a marketed therapeutic Fab) on IL-23 by using combined HDX-MS, FPOP, alanine-scanning mutagenesis, and binding studies. IL-23 (interleukin 23) is a pro-inflammatory cytokine composed of two domains linked via disulfide bonds, p19 and p40. An antibody against IL-23 has the potential to treat autoimmune disorders. When binding-induced changes in backbone dynamics were followed using HDX-MS, the discontinuous epitope for 7B7, covering five-peptide regions showed differential extents of HDX upon binding. Further interrogation with FPOP revealed protection of five peptides overlapping the five protected regions identified by HDX-MS, providing evidence of complementarity among the two approaches (Figure 8B). Looking at residue-specific labeling, the investigators saw that residues W29, M35, L97/L96/P98 show large difference in labeling. Based on HDX, FPOP, and structural analysis, the investigators generated panels of mutants to test binding. That mutational analysis revealed that residues M35, D36, E93, and L97 (Figure 8B) are the main contributors to stabilize the IL-23-7B7 fab interaction whereas the other regions become protected owing to allostery.
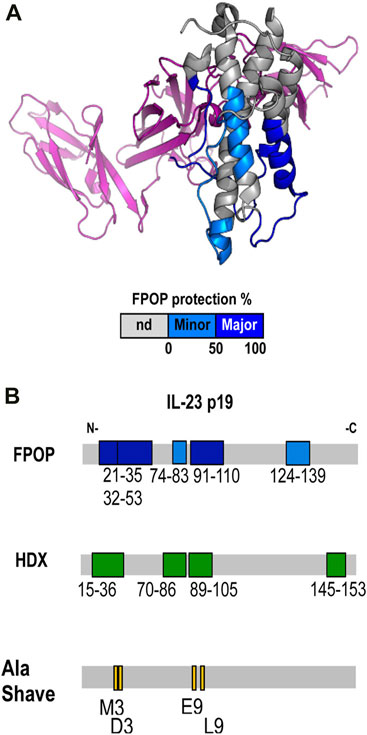
FIGURE 8. (A) Epitope regions determined by FPOP mapped on the crystal structure of IL-23. Color code: no significant difference (gray), minor epitope region (cyan), and major epitope region (blue). The p40 subunit is colored in purple. (B) Epitope regions determined by FPOP, HDX, and Ala Shave Energetics as mapped on the linear sequence of the IL-23 p19. Republished from Li J. et al. (2017). Copyright 2017, American Chemical Society.
In another collaborative work, Gross and BMS collaborators (Li K. S. et al., 2017) used multiple orthogonal footprinting techniques to study epitopes for adnectins-1 and 2 (adnectins are small domain proteins designed based on 10th human fibronectin type III domain) on the Interleukin-6 (IL-6) receptor. The team first deployed HDX-MS for its ability to monitor backbone dynamics and found one peptide (130–141) showing small but statistically significant protection for both adnectin1/2-IL-6R complexes. FPOP revealed peptide region 135–148 was protected in adenectin1/2 bound states. Overlap between HDX (131–141) and FPOP (135–148) indicates that the contacting residues lie in the region of 135–141 (50% improvement in resolution). Oxidative labeling has a bias for targeting sulfur and aromatic side chains. At the same time, quantification of the differential labeling extent becomes difficult if a peptide contains multiple reactive sites and/or generates isomeric labeling for a given residue. Such is the case for the peptide 135–148, preventing residue-level quantification. The presence of multiple Asp/Glu residues in the target sequence prompted the use of GEE labeling to reveal that only Glu140 of three potential sites Glu140, Asp141, and Glu144 shows differential labeling. Although other contact residues in 135–141 cannot be ruled out, the results indicate Pro138 (silent to HDX) and Glu140 are involved in binding.
Recently, investigators at BMS (Huang et al., 2020b) utilized multiple MS approaches to characterize epitopes on the bispecific antibody 1 (BiAb1). BiAb1 can engage with clusters of differentiation 3 (CD3) and B-cell maturation antigens (BCMA), leading to T-cell mediated killing of tumor cells. The investigators first used SEC-MALS and native MS to confirm the formation of binary and ternary complexes. HDX showed that protection on CD3 was localized in residues one to seven in the presence of BiAb1 whereas FPOP and GEE labeling indicated residues M7 and E5/E6 are primarily engaged by BsAb1. BCMA is a glycosylated antigen and, hence, poor sequence coverage was obtained. Yet the potential binding site on BCMA was shortened to residues 14–32 and 46–63. FPOP protection demonstrated that residues M7, Y13, F14, D15, S16, L17, H19, and P33/P34/L35 are among the key sites for interaction.
6.2.2 Combining HDX-ETD-MS, crosslinking, and docking
Another use of footprinting combinations for epitope mapping, Gross and BMS collaborators (Zhang et al., 2020) mapped the epitope for the Nivolumab (marketed anticancer mAb) on the PD1 (programmed cell death-1, an immune checkpoint receptor). They first used HDX-MS as a broad “scout” for contours of the binding interfaces, establishing that three peptides covering an N loop, FG, and CD loops in PD1 showed protection. Because the peptides are large, the investigators turned to gas-phase fragmentation by ETD-MS to identify residue-specific protection. Poor fragmentation of N and CD loops limited conclusions to one of three peptides in the FG-loop, which was ETD amenable. This shows the limitation discussed earlier of ETD-MS for obtaining residue-resolved protection (Section 5.3). Nevertheless, ETD-MS experiments identified five critical points among the 14-residue peptide. To dig deeper, the authors used two multiple crosslinking reagents to map the contact residues and found that only the N-terminus, BC, and FG loops are the targets for the crosslinks in the complex, suggesting changes in the CD loop are binding-induced conformational changes where the BC loop was missed in HDX-MS studies. In addition to fast HDX, combinations of FPOP and crosslinking can distinguish binding and remote conformational changes. The two approaches give data that match well with the X-ray crystal structure, more evidence for the value of using orthogonal approaches. Utilizing crosslinking distances and contacting residues guided their docking experiment to afford the best scoring complex that recapitulates overall HDX-MS results. We should mention that HDX-MS for docking studies in epitope mapping is not new (Pandit et al., 2012) and is summarized in (Tran et al., 2022)
7 Conclusion and future prospects
Meta analysis of >80 papers reviewed (Supplementary Table S1) in the preparation of this article indicates interesting trends for HDX-MS in epitope mapping (Figure 9). 1) Immobilized pepsin columns now enjoy widespread popularity in the field. 2) In addition to pepsin, other acid proteases are entering the mainstream often used in series with pepsin. 3) Dealing with disulfide bonds in the antigen is most commonly done with approximately 0.5 M TCEP as the reducing agent. 4) As discussed earlier, there are many automated software packages available for HDX-MS data analysis. Most epitope mapping studies utilized DynamX, followed by HDExaminer. 5) Interestingly, nearly 50% of epitope mapping studies made use of LEAP PAL robotic HDX systems, presumably for improved precision and speed. 6) Although most studies map the epitope for one antibody, increasingly more studies are identifying epitopes for multiple monoclonal/polyclonal antibodies, including more than five samples. 7) Finally, most antigens studied are ≤300 amino acids in length.
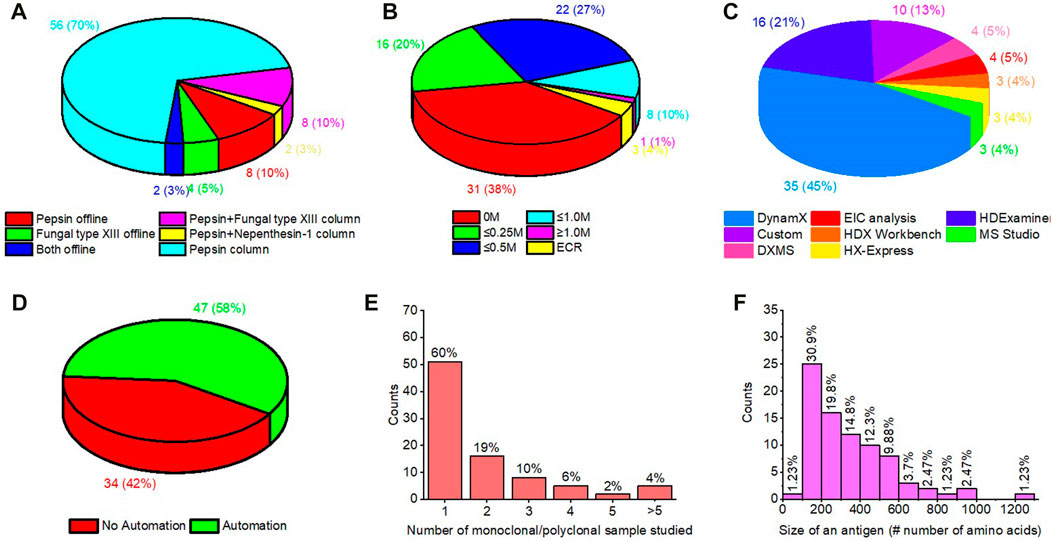
FIGURE 9. Meta analysis of literature reporting epitope mapping using HDX-MS. Panel (A) is for nature and way protease used. (B) reported concentration of reducing agent (TCEP-HCl) in quench buffer. Usually quench buffer is diluted 1:1 with labeling reaction. (C) data analysis software used. (D) use of automated system. (E) number of sample (mAbs/polyclonal samples) analyzed/compared in each reported study. (F) size of antigen in terms of number of amino acids.
It will be interesting to see whether these trends will change in the future, and larger and more complex antigens will come under the HDX-MS umbrella. A determining factor is that antigens targeted by antibodies are multidomain proteins. The ability to identify a specific domain of interaction quickly, even with low spatial resolution, provides a relatively quick guide for designing subsequent studies that can focus on that protein region containing the epitope.
To close this review, we present a table (Supplementary Table S1) of all the HDX epitope mapping studies published thus far to provide an index of antigens that were studied, the types of experiments conducted, and the antibodies or surrogates that were investigated.
Author contributions
The review was conceived by MG. PJ, and MG did literature searches. The organization of the review was done by PJ, who also drafted the article. PJ and MG analyzed the data. PJ wrote the review, which was edited by MG. All authors contributed to the article and approved the submitted version.
Funding
Preparation of this review was supported by NIH contract (HHSN272201400018C), Daved Fremont PI, and by the NIH NIGM (R24GM136766) to MG).
Conflict of interest
MG is an unpaid member of the scientific advisory boards of GenNext and Protein Metrics, two companies seeking to commercialize approaches in protein footprinting. With that exception, the authors declare that the research was conducted in the absence of any other commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frans.2023.1118749/full#supplementary-material
Abbreviations
IgG, Immunoglobulin; mAb, Monoclonal antibody; Fab, Fragment of antibody binding; MS, Mass Spectrometry; HDX-MS, Hydrogen Deuterium Exchange Mass Spectrometry; ETD, Electron Transfer Dissociation; CID, Collisional Induced Dissociation.
References
Abbott, W. M., Snow, M., Eckersley, S., Renshaw, J., Davies, G., Norman, R. A., et al. (2013). Characterization of the complex formed between a potent neutralizing ovine-derived polyclonal anti-TNFα Fab fragment and human TNFα. Biosci. Rep. 33 (4), e00060–e00664. doi:10.1042/BSR20130044
Adhikari, J., Zhao, H., Fernandez, E., Huang, Y., Diamond, M. S., Fremont, D. H., et al. (2021). Hydrogen–deuterium exchange mass spectrometry identifies spatially distinct antibody epitopes on domain III of the Zika virus envelope protein. J. Mass Spectrom. 56 (1), e4685. doi:10.1002/jms.4685
Ahn, J., Cao, M.-J., Yu, Y. Q., and Engen, J. R. (2013). Accessing the reproducibility and specificity of pepsin and other aspartic proteases. Biochimica Biophysica Acta (BBA) - Proteins Proteomics 1834 (6), 1222–1229. doi:10.1016/j.bbapap.2012.10.003
Alt, N., Zhang, T. Y., Motchnik, P., Taticek, R., Quarmby, V., Schlothauer, T., et al. (2016). Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals 44 (5), 291–305. doi:10.1016/j.biologicals.2016.06.005
Amon, S., Trelle, M. B., Jensen, O. N., and Jørgensen, T. J. D. (2012). Spatially resolved protein hydrogen exchange measured by subzero-cooled chip-based nanoelectrospray ionization tandem mass spectrometry. Anal. Chem. 84 (10), 4467–4473. doi:10.1021/ac300268r
Anderson, K. W., and Hudgens, J. W. (2022). Chromatography at −30 °C for reduced back-exchange, reduced carryover, and improved dynamic range for hydrogen–deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 33 (7), 1282–1292. doi:10.1021/jasms.2c00096
Ashman, R. F., Kaplan, A. P., and Metzger, H. (1971). A search for conformational change on ligand binding in a human γM macroglobulin—I: Circular dichroism and hydrogen exchange. Immunochemistry 8 (7), 627–641. doi:10.1016/0019-2791(71)90203-5
Atassi, M. (1964). Properties of components of myoglobin of the sperm whale. Nature 202 (4931), 496–498. doi:10.1038/202496a0
Baerga-Ortiz, A., Hughes, C. A., Mandell, J. G., and Komives, E. A. (2002). Epitope mapping of a monoclonal antibody against human thrombin by H/D-exchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein. Protein Sci. 11 (6), 1300–1308. doi:10.1110/ps.4670102
Bai, Y., Milne, J. S., Mayne, L., and Englander, S. W. (1993). Primary structure effects on peptide group hydrogen exchange. Proteins Struct. Funct. Bioinforma. 17 (1), 75–86. doi:10.1002/prot.340170110
Barber, M., Bordoli, R., Sedgwick, R., and Tyler, A. (1981). Fast atom bombardment of solids as an ion source in mass spectrometry. Nature 293 (5830), 270–275. doi:10.1038/293270a0
Bardelli, M., Livoti, E., Simonelli, L., Pedotti, M., Moraes, A., Valente, A., et al. (2015). Epitope mapping by solution NMR spectroscopy. J. Mol. Recognit. 28 (6), 393–400. doi:10.1002/jmr.2454
Barlow, D., Edwards, M., and Thornton, J. (1986). Continuous and discontinuous protein antigenic determinants. Nature 322 (6081), 747–748. doi:10.1038/322747a0
Beck, A., Wagner-Rousset, E., Ayoub, D., Van Dorsselaer, A., and Sanglier-Cianférani, S. (2013). Characterization of therapeutic antibodies and related products. Anal. Chem. 85 (2), 715–736. doi:10.1021/ac3032355
Benjamin, D. C., Berzofsky, J. A., East, I. J., Gurd, F. R., Hannum, C., Leach, S. J., et al. (1984). The antigenic structure of proteins: A reappraisal. Annu. Rev. Immunol. 2, 67–101. doi:10.1146/annurev.iy.02.040184.000435
Benjamin, D. C., Williams, D. C., Smith-Gill, S. J., and Rule, G. S. (1992). Long-range changes in a protein antigen due to antigen-antibody interaction. Biochemistry 31 (40), 9539–9545. doi:10.1021/bi00155a005
Birch, J. R., and Racher, A. J. (2006). Antibody production. Adv. drug Deliv. Rev. 58 (5-6), 671–685. doi:10.1016/j.addr.2005.12.006
Black, W. A., Stocks, B. B., Mellors, J. S., Engen, J. R., and Ramsey, J. M. (2015). Utilizing microchip capillary electrophoresis electrospray ionization for hydrogen exchange mass spectrometry. Anal. Chem. 87 (12), 6280–6287. doi:10.1021/acs.analchem.5b01179
Blout, E. R., De Loze, C., and Asadourian, A. (1961). The deuterium exchange of water-soluble polypeptides and proteins as measured by infrared spectroscopy. J. Am. Chem. Soc. 83 (8), 1895–1900. doi:10.1021/ja01469a028
Blumenfeld, O. O., Leonis, J., and Perlmann, G. E. (1960). The effect of guanidine hydrochloride on crystalline pepsin. J. Biol. Chem. 235, 379–382. doi:10.1016/s0021-9258(18)69532-8
Bobst, C. E., and Kaltashov, I. A. (2014). Enhancing the quality of H/D exchange measurements with mass spectrometry detection in disulfide-rich proteins using electron capture dissociation. Anal. Chem. 86 (11), 5225–5231. doi:10.1021/ac500904p
Bondarenko, P. V., Nichols, A. C., Xiao, G., Shi, R. L., Chan, P. K., Dillon, T. M., et al. (2021). Identification of critical chemical modifications and paratope mapping by size exclusion chromatography of stressed antibody-target complexes. mAbs 13 (1), 1887629. doi:10.1080/19420862.2021.1887629
Böttger, V., and Böttger, A. (2009). “Epitope mapping using phage display peptide libraries,” in Epitope mapping protocols (Springer), 181–201.
Brier, S., Maria, G., Carginale, V., Capasso, A., Wu, Y., Taylor, R. M., et al. (2007). Purification and characterization of pepsins A1 and A2 from the Antarctic rock cod Trematomus bernacchii. Febs J. 274 (23), 6152–6166. doi:10.1111/j.1742-4658.2007.06136.x
Brooks, B. D., Miles, A. R., and Abdiche, Y. N. (2014). High-throughput epitope binning of therapeutic monoclonal antibodies: Why you need to bin the fridge. Drug Discov. Today 19 (8), 1040–1044. doi:10.1016/j.drudis.2014.05.011
Brown, G. C. (1991). Total cell protein concentration as an evolutionary constraint on the metabolic control distribution in cells. J. Theor. Biol. 153 (2), 195–203. doi:10.1016/s0022-5193(05)80422-9
Brown, K. A., Lento, C., Rajendran, S., Dowd, J., and Wilson, D. J. (2020). Epitope mapping for a preclinical bevacizumab (Avastin) biosimilar on an extended construct of vascular endothelial growth factor a using millisecond hydrogen–deuterium exchange mass spectrometry. Biochemistry 59 (30), 2776–2781. doi:10.1021/acs.biochem.0c00308
Burnens, A., Demotz, S., Corradin, G., Binz, H., and Bosshard, H. R. (1987). Epitope mapping by chemical modification of free and antibody-bound protein antigen. Science 235 (4790), 780–783. doi:10.1126/science.2433768
Cariccio, V. L., Domina, M., Benfatto, S., Venza, M., Venza, I., Faleri, A., et al. (2016). Phage display revisited: Epitope mapping of a monoclonal antibody directed against Neisseria meningitidis adhesin A using the PROFILER technology. mAbs 8 (4), 741–750. doi:10.1080/19420862.2016.1158371
Carter, J. M. (1996). Epitope mapping of a protein using the Geysen (PEPSCAN) procedure. Methods Mol. Biol. 36, 207–223. doi:10.1385/0-89603-274-4:207
Casina, V. C., Hu, W., Mao, J. H., Lu, R. N., Hanby, H. A., Pickens, B., et al. (2015). High-resolution epitope mapping by HX MS reveals the pathogenic mechanism and a possible therapy for autoimmune TTP syndrome. Proc. Natl. Acad. Sci. U. S. A. 112 (31), 9620–9625. doi:10.1073/pnas.1512561112
Catherman, A. D., Skinner, O. S., and Kelleher, N. L. (2014). Top down proteomics: Facts and perspectives. Biochem. Biophysical Res. Commun. 445 (4), 683–693. doi:10.1016/j.bbrc.2014.02.041
Chalmers, M. J., Busby, S. A., Pascal, B. D., He, Y., Hendrickson, C. L., Marshall, A. G., et al. (2006). Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 78 (4), 1005–1014. doi:10.1021/ac051294f
Christoffersen, M., Bolvig, S., and Tüchsen, E. (1996). Salt effects on the amide hydrogen exchange of bovine pancreatic trypsin inhibitor. Biochemistry 35 (7), 2309–2315. doi:10.1021/bi951711q
Coales, S. J., Tuske, S. J., Tomasso, J. C., and Hamuro, Y. (2009). Epitope mapping by amide hydrogen/deuterium exchange coupled with immobilization of antibody, on-line proteolysis, liquid chromatography and mass spectrometry. Rapid Commun. Mass Spectrom. 23 (5), 639–647. doi:10.1002/rcm.3921
Comamala, G., Krogh, C. C., Nielsen, V. S., Kutter, J. P., Voglmeir, J., and Rand, K. D. (2021). Hydrogen/deuterium exchange mass spectrometry with integrated electrochemical reduction and microchip-enabled deglycosylation for epitope mapping of heavily glycosylated and disulfide-bonded proteins. Anal. Chem. 93 (49), 16330–16340. doi:10.1021/acs.analchem.1c01728
Comamala, G., Madsen, J. B., Voglmeir, J., Du, Y.-M., Jensen, P. F., Østerlund, E. C., et al. (2020a). Deglycosylation by the acidic glycosidase PNGase H+ enables analysis of N-linked glycoproteins by Hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 31 (11), 2305–2312. doi:10.1021/jasms.0c00258
Comamala, G., Wagner, C., de la Torre, P. S., Jakobsen, R. U., Hilger, M., Brouwer, H. J., et al. (2020b). Hydrogen/deuterium exchange mass spectrometry with improved electrochemical reduction enables comprehensive epitope mapping of a therapeutic antibody to the cysteine-knot containing vascular endothelial growth factor. Anal. Chim. Acta 1115, 41–51. doi:10.1016/j.aca.2020.04.014
Cravello, L., Lascoux, D., and Forest, E. (2003). Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom. 17 (21), 2387–2393. doi:10.1002/rcm.1207
Crowe, J. E. (2022). Human antibodies for viral infections. Annu. Rev. Immunol. 40, 349–386. doi:10.1146/annurev-immunol-042718-041309
Cryar, A., Groves, K., and Quaglia, M. (2017). Online hydrogen-deuterium exchange traveling wave ion mobility mass spectrometry (hdx-im-ms): A systematic evaluation. Journal of the American Society for Mass Spectrometry 28 (6), 1192–1202. doi:10.1007/s13361-017-1633-z
Cunningham, B. C., and Wells, J. A. (1989). High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244 (4908), 1081–1085. doi:10.1126/science.2471267
Cwirla, S. E., Peters, E. A., Barrett, R. W., and Dower, W. J. (1990). Peptides on phage: A vast library of peptides for identifying ligands. Proc. Natl. Acad. Sci. 87 (16), 6378–6382. doi:10.1073/pnas.87.16.6378
Dale, G. E., Oefner, C., and D’Arcy, A. (2003). The protein as a variable in protein crystallization. J. Struct. Biol. 142 (1), 88–97. doi:10.1016/S1047-8477(03)00041-8
Davidson, E., and Doranz, B. J. (2014). A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 143 (1), 13–20. doi:10.1111/imm.12323
Demmers, J. A., Rijkers, D. T., Haverkamp, J., Killian, J. A., and Heck, A. J. (2002). Factors affecting gas-phase deuterium scrambling in peptide ions and their implications for protein structure determination. J. Am. Chem. Soc. 124 (37), 11191–11198. doi:10.1021/ja0125927
Deng, B., Zhu, S., Macklin, A. M., Xu, J., Lento, C., Sljoka, A., et al. (2017). Suppressing allostery in epitope mapping experiments using millisecond hydrogen/deuterium exchange mass spectrometry. mAbs 9 (8), 1327–1336. doi:10.1080/19420862.2017.1379641
Deng, X., Storz, U., and Doranz, B. J. (2018). Enhancing antibody patent protection using epitope mapping information. MAbs 10, 204–209. doi:10.1080/19420862.2017.1402998
Devlin, J. J., Panganiban, L. C., and Devlin, P. E. (1990). Random peptide libraries: A source of specific protein binding molecules. Science 249 (4967), 404–406. doi:10.1126/science.2143033
Dhungana, S., Fessler, M. B., and Tomer, K. B. (2009). “Epitope mapping by differential chemical modification of antigens,” in Epitope mapping protocols: Second edition. Editors M. Schutkowski, and U. Reineke (Totowa, NJ: Humana Press), 119–134.
Di Muzio, M., Wildner, S., Huber, S., Hauser, M., Vejvar, E., Auzinger, W., et al. (2020). Hydrogen/deuterium exchange memory NMR reveals structural epitopes involved in IgE cross-reactivity of allergenic lipid transfer proteins. J. Biol. Chem. 295 (51), 17398–17410. doi:10.1074/jbc.RA120.014243
Doyle, M. P., Genualdi, J. R., Bailey, A. L., Kose, N., Gainza, C., Rodriguez, J., et al. (2022). Isolation of a potently neutralizing and protective human monoclonal antibody targeting yellow fever virus. mBio 13 (3), 00512222–e100522. doi:10.1128/mbio.00512-22
Ecker, D. M., Jones, S. D., and Levine, H. L. (2015). The therapeutic monoclonal antibody market. MAbs 7, 9–14. doi:10.4161/19420862.2015.989042
Edelman, G. M. (1959). Dissociation of γ-globulin. J. Am. Chem. Soc. 81 (12), 3155–3156. doi:10.1021/ja01521a071
Eigen, M. (1964). Proton transfer, acid-base catalysis, and enzymatic hydrolysis. Part I: Elementary processes. Angewandte Chemie Int. Ed. Engl. 3 (1), 1–19. doi:10.1002/anie.196400011
Ellis, R. J., and Minton, A. P. (2003). Cell biology: Join the crowd. Nature 425 (6953), 27–28. doi:10.1038/425027a
Engen, J. R., and Komives, E. A. (2020). Complementarity of hydrogen/deuterium exchange mass spectrometry and cryo-electron microscopy. Trends Biochem. Sci. 45 (10), 906–918. doi:10.1016/j.tibs.2020.05.005
Englander, J. J., Calhoun, D. B., and Englander, S. W. (1979). Measurement and calibration of peptide group hydrogen-deuterium exchange by ultraviolet spectrophotometry. Anal. Biochem. 92 (2), 517–524. doi:10.1016/0003-2697(79)90693-6
Englander, J. J., Del Mar, C., Li, W., Englander, S. W., Kim, J. S., Stranz, D. D., et al. (2003). Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 100 (12), 7057–7062. doi:10.1073/pnas.1232301100
Englander, S. W. (1963). A hydrogen exchange method using tritium and sephadex: Its application to ribonuclease. Biochemistry 2, 798–807. doi:10.1021/bi00904a030
Englander, S. W., and Mayne, L. (1992). Protein folding studied using hydrogen-exchange labeling and two-dimensional NMR. Annu. Rev. Biophys. Biomol. Struct. 21, 243–265. doi:10.1146/annurev.bb.21.060192.001331
Fang, M., Wang, Z., Cupp-Sutton, K. A., Welborn, T., Smith, K., and Wu, S. (2021). High-throughput hydrogen deuterium exchange mass spectrometry (HDX-MS) coupled with subzero-temperature ultrahigh pressure liquid chromatography (UPLC) separation for complex sample analysis. Anal. Chim. Acta 1143, 65–72. doi:10.1016/j.aca.2020.11.022
Fang, M., Wang, Z., Norris, K., James, J. A., Wu, S., and Smith, K. (2022). Hydrogen-deuterium exchange mass spectrometry reveals a novel binding region of a neutralizing fully human monoclonal antibody to Anthrax protective antigen. Toxins 14 (2), 92. doi:10.3390/toxins14020092
Fenn, J. B., Mann, M., Meng, C. K., Wong, S. F., and Whitehouse, C. M. (1989). Electrospray ionization for mass spectrometry of large biomolecules. Science 246 (4926), 64–71. doi:10.1126/science.2675315
Fernandez, E., Kose, N., Edeling, M. A., Adhikari, J., Sapparapu, G., Lazarte, S. M., et al. (2018). Mouse and human monoclonal antibodies protect against infection by multiple genotypes of Japanese encephalitis virus. mBio 9 (1), e00008–e00018. doi:10.1128/mBio.00008-18
Fiedler, W., Borchers, C., Macht, M., Deininger, S.-O., and Przybylski, M. (1998). Molecular characterization of a conformational epitope of hen egg white lysozyme by differential chemical modification of immune complexes and mass spectrometric peptide mapping. Bioconjugate Chem. 9 (2), 236–241. doi:10.1021/bc970148g
Forsström, B., Axnäs, B. B., Stengele, K.-P., Bühler, J., Albert, T. J., Richmond, T. A., et al. (2014). Proteome-wide epitope mapping of antibodies using ultra-dense peptide arrays. Mol. Cell. Proteomics 13 (6), 1585–1597. doi:10.1074/mcp.M113.033308
Gau, B. C., Sharp, J. S., Rempel, D. L., and Gross, M. L. (2009). Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 81 (16), 6563–6571. doi:10.1021/ac901054w
Gershoni, J. M., Roitburd-Berman, A., Siman-Tov, D. D., Tarnovitski Freund, N., and Weiss, Y. (2007). Epitope mapping: The first step in developing epitope-based vaccines. BioDrugs 21 (3), 145–156. doi:10.2165/00063030-200721030-00002
Geysen, H. M., Meloen, R. H., and Barteling, S. J. (1984). Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc. Natl. Acad. Sci. 81 (13), 3998–4002. doi:10.1073/pnas.81.13.3998
Good, D. M., Wirtala, M., McAlister, G. C., and Coon, J. J. (2007). Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 6 (11), 1942–1951. doi:10.1074/mcp.M700073-MCP200
Goormaghtigh, E. (2013). “Infrared spectroscopy of protein dynamics: H/D exchange,” in Encyclopedia of biophysics. Editor G. C. K. Roberts (Berlin, Heidelberg: Springer), 1081–1083.
Greenspan, N. S., and Di Cera, E. (1999). Defining epitopes: It's not as easy as it seems. Nat. Biotechnol. 17 (10), 936–937. doi:10.1038/13590
Gresl, T., Storz, U., and Sandercock, C. (2016). An update on obtaining and enforcing therapeutic antibody patent claims. Nat. Biotechnol. 34 (12), 1242–1244. doi:10.1038/nbt.3735
Guo, C., Steinberg, L. K., Henderson, J. P., and Gross, M. L. (2020). Organic solvents for enhanced proteolysis of stable proteins for hydrogen–deuterium exchange mass spectrometry. Anal. Chem. 92 (17), 11553–11557. doi:10.1021/acs.analchem.0c02194
Guttman, M., Scian, M., and Lee, K. K. (2011). Tracking hydrogen/deuterium exchange at glycan sites in glycoproteins by mass spectrometry. Anal. Chem. 83 (19), 7492–7499. doi:10.1021/ac201729v
Haggis, G. H. (1957). Proton-deuteron exchange in protein and nucleoprotein molecules surrounded by heavy water. Biochimica Biophysica Acta 23, 494–503. doi:10.1016/0006-3002(57)90368-2
Hambly, D. M., and Gross, M. L. (2005). Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 16 (12), 2057–2063. doi:10.1016/j.jasms.2005.09.008
Hamuro, Y., Coales, S. J., Molnar, K. S., Tuske, S. J., and Morrow, J. A. (2008). Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22 (7), 1041–1046. doi:10.1002/rcm.3467
Hamuro, Y., Coales, S. J., Morrow, J. A., Molnar, K. S., Tuske, S. J., Southern, M. R., et al. (2006). Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 15 (8), 1883–1892. doi:10.1110/ps.062103006
Hamuro, Y., and Coales, S. J. (2018). Optimization of feasibility stage for hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 29 (3), 623–629. doi:10.1007/s13361-017-1860-3
Hamuro, Y., Coales, S. J., Southern, M. R., Nemeth-Cawley, J. F., Stranz, D. D., and Griffin, P. R. (2003). Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J. Biomol. Tech. JBT 14 (3), 171–182.
Hamuro, Y. (2021). Tutorial: Chemistry of hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 32 (1), 133–151. doi:10.1021/jasms.0c00260
Hamuro, Y., and Zhang, T. (2019). High-resolution HDX-MS of cytochrome c using pepsin/fungal protease type XIII mixed bed column. J. Am. Soc. Mass Spectrom. 30 (2), 227–234. doi:10.1007/s13361-018-2087-7
Hansen, J., Baum, A., Pascal, K. E., Russo, V., Giordano, S., Wloga, E., et al. (2020). Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 369 (6506), 1010–1014. doi:10.1126/science.abd0827
Haque, H. M. E., Ejemel, M., Vance, D. J., Willsey, G., Rudolph, M. J., Cavacini, L. A., et al. (2022). Human B cell epitope map of the lyme disease vaccine antigen, OspA. ACS Infect. Dis. 8, 2515–2528. doi:10.1021/acsinfecdis.2c00346
Haque, H. M. E., Mantis, N. J., and Weis, D. D. (2023). High-throughput epitope mapping by hydrogen exchange-mass spectrometry. J. Am. Soc. Mass Spectrom. 34 (1), 123–127. doi:10.1021/jasms.2c00255
Henzler-Wildman, K., and Kern, D. (2007). Dynamic personalities of proteins. Nature 450 (7172), 964–972. doi:10.1038/nature06522
Hoerner, J. K., Xiao, H., Dobo, A., and Kaltashov, I. A. (2004). Is there hydrogen scrambling in the gas phase? Energetic and structural determinants of proton mobility within protein ions. J. Am. Chem. Soc. 126 (24), 7709–7717. doi:10.1021/ja049513m
Houde, D., Berkowitz, S. A., and Engen, J. R. (2011). The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100 (6), 2071–2086. doi:10.1002/jps.22432
Hourdel, V., Volant, S., O’Brien, D. P., Chenal, A., Chamot-Rooke, J., Dillies, M.-A., et al. (2016). Memhdx: An interactive tool to expedite the statistical validation and visualization of large HDX-MS datasets. Bioinformatics 32 (22), 3413–3419. doi:10.1093/bioinformatics/btw420
Huang, R. Y. C., Krystek, S. R., Felix, N., Graziano, R. F., Srinivasan, M., Pashine, A., et al. (2018). Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/TL1A Interaction. mAbs 10 (1), 95–103. doi:10.1080/19420862.2017.1393595
Huang, R. Y. C., Kuhne, M., Deshpande, S., Rangan, V., Srinivasan, M., Wang, Y., et al. (2020a). Mapping binding epitopes of monoclonal antibodies targeting major histocompatibility complex class I chain-related A (MICA) with hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry. Anal. Bioanal. Chem. 412 (7), 1693–1700. doi:10.1007/s00216-020-02409-x
Huang, R. Y. C., Wang, F., Wheeler, M., Wang, Y., Langish, R., Chau, B., et al. (2020b). Integrated approach for characterizing bispecific antibody/antigens complexes and mapping binding epitopes with SEC/MALS, native mass spectrometry, and protein footprinting. Anal. Chem. 92 (15), 10709–10716. doi:10.1021/acs.analchem.0c01876
Hvidt, A., and LinderstrØM-Lang, K. (1954). Exchange of hydrogen atoms in insulin with deuterium atoms in aqueous solutions. Biochim. Biophys. Acta 14 (4), 574–575. doi:10.1016/0006-3002(54)90241-3
Hvidt, A., and Nielsen, S. O. (1966). Hydrogen exchange in proteins. Adv. Protein Chem. 21, 287–386. doi:10.1016/s0065-3233(08)60129-1
Iacob, R. E., Murphy III, J. P., and Engen, J. R. (2008). Ion mobility adds an additional dimension to mass spectrometric analysis of solution-phase hydrogen/deuterium exchange. Rapid Communications in Mass Spectrometry 22 (18), 2898–2904. doi:10.1002/rcm.3688
Irving, M. B., Pan, O., and Scott, J. K. (2001). Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Curr. Opin. Chem. Biol. 5 (3), 314–324. doi:10.1016/s1367-5931(00)00208-8
James, E. I., Murphree, T. A., Vorauer, C., Engen, J. R., and Guttman, M. (2022). Advances in hydrogen/deuterium exchange mass spectrometry and the pursuit of challenging biological systems. Chem. Rev. 122 (8), 7562–7623. doi:10.1021/acs.chemrev.1c00279
Jemmerson, R. (1987). Antigenicity and native structure of globular proteins: Low frequency of peptide reactive antibodies. Proc. Natl. Acad. Sci. 84 (24), 9180–9184. doi:10.1073/pnas.84.24.9180
Jemmerson, R., and Paterson, Y. (1986). Mapping epitopes on a protein antigen by the proteolysis of antigen-antibody complexes. Science 232 (4753), 1001–1004. doi:10.1126/science.2422757
Jensen, P. F., Comamala, G., Trelle, M. B., Madsen, J. B., Jørgensen, T. J. D., and Rand, K. D. (2016). Removal of N-linked glycosylations at acidic pH by PNGase A facilitates hydrogen/deuterium exchange mass spectrometry analysis of N-linked glycoproteins. Anal. Chem. 88 (24), 12479–12488. doi:10.1021/acs.analchem.6b03951
Jensen, P. F., Jørgensen, T. J., Koefoed, K., Nygaard, F., and Sen, J. W. (2013). Affinity capture of biotinylated proteins at acidic conditions to facilitate hydrogen/deuterium exchange mass spectrometry analysis of multimeric protein complexes. Anal. Chem. 85 (15), 7052–7059. doi:10.1021/ac303442y
Jerne, N. K. (1960). Immunological speculations. Annu. Rev. Microbiol. 14 (1), 341–358. doi:10.1146/annurev.mi.14.100160.002013
Jethva, P. N., and Udgaonkar, J. B. (2017). Modulation of the extent of cooperative structural change during protein folding by chemical denaturant. J. Phys. Chem. B 121 (35), 8263–8275. doi:10.1021/acs.jpcb.7b04473
Jethva, P. N., and Udgaonkar, J. B. (2018). The osmolyte TMAO modulates protein folding cooperativity by altering global protein stability. Biochemistry 57 (40), 5851–5863. doi:10.1021/acs.biochem.8b00698
Jeyarajah, S., Parker, C. E., Summer, M. T., and Tomer, K. B. (1998). Matrix-assisted laser desorption ionization/mass spectrometry mapping of human immunodeficiency virus-gp120 epitopes recognized by a limited polyclonal antibody. J. Am. Soc. Mass Spectrom. 9 (2), 157–165. doi:10.1016/s1044-0305(97)00247-x
Jones, L. M., Sperry, J. B., Carroll, J. A., and Gross, M. L. (2011). Fast photochemical oxidation of proteins for epitope mapping. Anal. Chem. 83 (20), 7657–7661. doi:10.1021/ac2007366
Jørgensen, T. J. D., Gårdsvoll, H., Ploug, M., and Roepstorff, P. (2005). Intramolecular migration of amide hydrogens in protonated peptides upon collisional activation. J. Am. Chem. Soc. 127 (8), 2785–2793. doi:10.1021/ja043789c
Kan, Z.-Y., Mayne, L., Sevugan Chetty, P., and Englander, S. W. (2011). ExMS: Data analysis for HX-MS experiments. J. Am. Soc. Mass Spectrom. 22 (11), 1906–1915. doi:10.1007/s13361-011-0236-3
Kan, Z.-Y., Walters, B. T., Mayne, L., and Englander, S. W. (2013). Protein hydrogen exchange at residue resolution by proteolytic fragmentation mass spectrometry analysis. Proc. Natl. Acad. Sci. 110 (41), 16438–16443. doi:10.1073/pnas.1315532110
Kant, R., Moyle, A. B., Jethva, P. N., and Gross, M. L. (2023). “Chapter 9 - protein footprinting by mass spectrometry: H/D exchange, specific amino acid labeling, and fast photochemical oxidation of proteins,” in Advanced spectroscopic methods to study biomolecular structure and dynamics. Editors P. Saudagar, and T. Tripathi (Academic Press), 227–270.
Karas, M., and Hillenkamp, F. (1988). Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 60 (20), 2299–2301. doi:10.1021/ac00171a028
Karch, K. R., Coradin, M., Zandarashvili, L., Kan, Z.-Y., Gerace, M., Englander, S. W., et al. (2018). Hydrogen-deuterium exchange coupled to top- and middle-down mass spectrometry reveals histone tail dynamics before and after nucleosome assembly. Structure 26 (12), 1651–1663.e3. doi:10.1016/j.str.2018.08.006
Katta, V., and Chait, B. T. (1991). Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 5 (4), 214–217. doi:10.1002/rcm.1290050415
Kavan, D., and Man, P. (2011). MSTools—web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 302 (1-3), 53–58. doi:10.1016/j.ijms.2010.07.030
Kazazic, S., Zhang, H.-M., Schaub, T. M., Emmett, M. R., Hendrickson, C. L., Blakney, G. T., et al. (2011). Automated data reduction for hydrogen/deuterium exchange experiments, enabled by high-resolution Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 21 (4), 550–558. doi:10.1016/j.jasms.2009.12.016
Kim, A. S., Kafai, N. M., Winkler, E. S., Gilliland, T. C., Cottle, E. L., Earnest, J. T., et al. (2021). Pan-protective anti-alphavirus human antibodies target a conserved E1 protein epitope. Cell 184 (17), 4414–4429.e19. doi:10.1016/j.cell.2021.07.006
Kim, P. S., and Baldwin, R. L. (1982). Influence of charge on the rate of amide proton exchange. Biochemistry 21 (1), 1–5. doi:10.1021/bi00530a001
Konermann, L. (2016). Heavy lessons in protein allostery. Nat. Struct. Mol. Biol. 23 (6), 511–512. doi:10.1038/nsmb.3234
Kossiakoff, A. A. (1982). Protein dynamics investigated by the neutron diffraction-hydrogen exchange technique. Nature 296 (5859), 713–721. doi:10.1038/296713a0
Kwong, P. D., DeKosky, B. J., and Ulmer, J. B. (2020). Antibody-guided structure-based vaccines. Seminars Immunol. 50, 101428. doi:10.1016/j.smim.2020.101428
Ladner, R. C. (2007). Mapping the epitopes of antibodies. Biotechnol. Genet. Eng. Rev. 24 (1), 1–30. doi:10.1080/02648725.2007.10648092
Landsteiner, K. (1942). Serological reactivity of hydrolytic products from silk. J. Exp. Med. 75 (3), 269–276. doi:10.1084/jem.75.3.269
Lau, A. M., Ahdash, Z., Martens, C., and Politis, A. (2019). Deuteros: Software for rapid analysis and visualization of data from differential hydrogen deuterium exchange-mass spectrometry. Bioinformatics 35 (17), 3171–3173. doi:10.1093/bioinformatics/btz022
Laver, W. G., Air, G. M., Webster, R. G., and Smith-Gill, S. J. (1990). Epitopes on protein antigens: Misconceptions and realities. Cell 61 (4), 553–556. doi:10.1016/0092-8674(90)90464-p
Leach, S., and Springell, P. (1962). Tritium-hydrogen exchange in studies of protein structure. Aust. J. Chem. 15 (2), 350–364. doi:10.1071/ch9620350
Leinikki, P., Lehtinen, M., Hyöty, H., Parkkonen, P., Kantanen, M.-L., and Hakulinen, J. (1993). Synthetic peptides as diagnostic tools in virology. Adv. virus Res. 42, 149–186. doi:10.1016/s0065-3527(08)60085-8
Lento, C., and Wilson, D. J. (2022). Subsecond time-resolved mass spectrometry in dynamic structural biology. Chem. Rev. 122 (8), 7624–7646. doi:10.1021/acs.chemrev.1c00222
Li, J., Wei, H., Krystek, S. R., Bond, D., Brender, T. M., Cohen, D., et al. (2017). Mapping the energetic epitope of an antibody/interleukin-23 interaction with hydrogen/deuterium exchange, fast photochemical oxidation of proteins mass spectrometry, and alanine Shave mutagenesis. Anal. Chem. 89 (4), 2250–2258. doi:10.1021/acs.analchem.6b03058
Li, K. S., Chen, G., Mo, J., Huang, R. Y. C., Deyanova, E. G., Beno, B. R., et al. (2017). Orthogonal mass spectrometry-based footprinting for epitope mapping and structural characterization: The IL-6 receptor upon binding of protein therapeutics. Anal. Chem. 89 (14), 7742–7749. doi:10.1021/acs.analchem.7b01748
Liberti, P. A., Bausch, D. M., and Chu, M. (1981). Hydrogen exchange analysis of ligand-induced conformational changes in Fab. Biochemistry 20 (4), 1012–1019. doi:10.1021/bi00507a056
Liberti, P. A., Stylos, W. A., and Maurer, P. H. (1972). Conformational change(s) induced in sheep calcium-dependent antibody upon interaction with homologous polypeptide antigen. I. Hydrogen-exchange studies of immunoglobulin G and (Fab') 2 fragment. Biochemistry 11 (18), 3312–3320. doi:10.1021/bi00768a002
Lim, X.-X., Chandramohan, A., Lim, X.-Y. E., Crowe, J. E., Lok, S.-M., and Anand, G. S. (2017a). Epitope and paratope mapping reveals temperature-dependent alterations in the dengue-antibody interface. Structure 25 (9), 1391–1402.e3. doi:10.1016/j.str.2017.07.007
Lim, X.-X., Chandramohan, A., Lim, X. Y. E., Bag, N., Sharma, K. K., Wirawan, M., et al. (2017b). Conformational changes in intact dengue virus reveal serotype-specific expansion. Nat. Commun. 8 (1), 14339–14413. doi:10.1038/ncomms14339
Limpikirati, P. K., Zhao, B., Pan, X., Eyles, S. J., and Vachet, R. W. (2020). Covalent labeling/mass spectrometry of monoclonal antibodies with diethylpyrocarbonate: Reaction kinetics for ensuring protein structural integrity. J. Am. Soc. Mass Spectrom. 31 (6), 1223–1232. doi:10.1021/jasms.0c00067
Limpikirati, P., Liu, T., and Vachet, R. W. (2018). Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods 144, 79–93. doi:10.1016/j.ymeth.2018.04.002
Lin, M., Krawitz, D., Callahan, M. D., Deperalta, G., and Wecksler, A. T. (2018). Characterization of ELISA antibody-antigen interaction using footprinting-mass spectrometry and negative staining transmission electron microscopy. J. Am. Soc. Mass Spectrom. 29 (5), 961–971. doi:10.1007/s13361-017-1883-9
Liu, X. R., Zhang, M. M., and Gross, M. L. (2020). Mass spectrometry-based protein footprinting for higher-order structure analysis: Fundamentals and applications. Chem. Rev. 120 (10), 4355–4454. doi:10.1021/acs.chemrev.9b00815
Lu, X., DeFelippis, M. R., and Huang, L. (2009). Linear epitope mapping by native mass spectrometry. Anal. Biochem. 395 (1), 100–107. doi:10.1016/j.ab.2009.08.018
Malito, E., Faleri, A., Surdo, P. L., Veggi, D., Maruggi, G., Grassi, E., et al. (2013). Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proc. Natl. Acad. Sci. U. S. A. 110 (9), 3304–3309. doi:10.1073/pnas.1222845110
Mandell, J. G., Falick, A. M., and Komives, E. A. (1998). Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal. Chem. 70 (19), 3987–3995. doi:10.1021/ac980553g
Martin, F. (2016). Antibodies as leading tools to unlock the therapeutic potential in human disease. Immunol. Rev. 270 (1), 5–7. doi:10.1111/imr.12410
Masson, G. R., Burke, J. E., Ahn, N. G., Anand, G. S., Borchers, C., Brier, S., et al. (2019). Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. methods 16 (7), 595–602. doi:10.1038/s41592-019-0459-y
Matthew, J. B., and Richards, F. M. (1983). The pH dependence of hydrogen exchange in proteins. J. Biol. Chem. 258 (5), 3039–3044. doi:10.1016/s0021-9258(18)32826-6
Mayne, L., Kan, Z. Y., Chetty, P. S., Ricciuti, A., Walters, B. T., and Englander, S. W. (2011). Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J. Am. Soc. Mass Spectrom. 22 (11), 1898–1905. doi:10.1007/s13361-011-0235-4
Mayne, L., Paterson, Y., Cerasoli, D., and Englander, S. W. (1992). Effect of antibody binding on protein motions studied by hydrogen-exchange labeling and two-dimensional NMR. Biochemistry 31 (44), 10678–10685. doi:10.1021/bi00159a006
Mikesh, L. M., Ueberheide, B., Chi, A., Coon, J. J., Syka, J. E., Shabanowitz, J., et al. (2006). The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta 1764 (12), 1811–1822. doi:10.1016/j.bbapap.2006.10.003
Miranker, A., Robinson, C. V., Radford, S. E., Aplin, R. T., and Dobson, C. M. (1993). Detection of transient protein folding populations by mass spectrometry. Science 262 (5135), 896–900. doi:10.1126/science.8235611
Mistarz, U. H., Bellina, B., Jensen, P. F., Brown, J. M., Barran, P. E., and Rand, K. D. (2018). UV photodissociation mass spectrometry accurately localize sites of backbone deuteration in peptides. Anal. Chem. 90 (2), 1077–1080. doi:10.1021/acs.analchem.7b04683
Modzel, M., Wollenberg, D. T. W., Trelle, M. B., Larsen, M. R., and Jørgensen, T. J. D. (2021). Ultraviolet photodissociation of protonated peptides and proteins can proceed with H/D scrambling. Anal. Chem. 93 (2), 691–696. doi:10.1021/acs.analchem.0c02957
Mukhopadhyay, S., Kuhn, R. J., and Rossmann, M. G. (2005). A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3 (1), 13–22. doi:10.1038/nrmicro1067
Mullahoo, J., Zhang, T., Clauser, K., Carr, S. A., Jaffe, J. D., and Papanastasiou, M. (2020). Dual protease type XIII/pepsin digestion offers superior resolution and overlap for the analysis of histone tails by HX-MS. Methods 184, 135–140. doi:10.1016/j.ymeth.2020.01.016
Mysling, S., Salbo, R., Ploug, M., and Jørgensen, T. J. (2014). Electrochemical reduction of disulfide-containing proteins for hydrogen/deuterium exchange monitored by mass spectrometry. Anal. Chem. 86 (1), 340–345. doi:10.1021/ac403269a
Nabedryk-Viala, E., Thiéry, C., Calvet, P., and Thiéry, J. M. (1976). Hydrogen-isotope exchange of oxidized and reduced cytochrome c: A comparison of mass spectrometry and infrared methods. Eur. J. Biochem. 61 (1), 253–258. doi:10.1111/j.1432-1033.1976.tb10018.x
Nirudodhi, S. N., Sperry, J. B., Rouse, J. C., and Carroll, J. A. (2017). Application of dual protease column for HDX-MS analysis of monoclonal antibodies. J. Pharm. Sci. 106 (2), 530–536. doi:10.1016/j.xphs.2016.10.023
Niu, B., Appleby, T. C., Wang, R., Morar, M., Voight, J., Villaseñor, A. G., et al. (2020). Protein footprinting and X-ray crystallography reveal the interaction of PD-L1 and a macrocyclic peptide. Biochemistry 59 (4), 541–551. doi:10.1021/acs.biochem.9b00822
Opuni, K. F. M., Al-Majdoub, M., Yefremova, Y., El-Kased, R. F., Koy, C., and Glocker, M. O. (2018). Mass spectrometric epitope mapping. Mass Spectrom. Rev. 37 (2), 229–241. doi:10.1002/mas.21516
Otting, G., and Wüthrich, K. (1990). Heteronuclear filters in two-dimensional [1H,1H]-NMR spectroscopy: Combined use with isotope labelling for studies of macromolecular conformation and intermolecular interactions. Q. Rev. Biophys. 23 (1), 39–96. doi:10.1017/s0033583500005412
Pan, J., Han, J., Borchers, C. H., and Konermann, L. (2009). Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. J. Am. Chem. Soc. 131 (35), 12801–12808. doi:10.1021/ja904379w
Pan, J., Zhang, S., Parker, C. E., and Borchers, C. H. (2014). Subzero temperature chromatography and top-down mass spectrometry for protein higher-order structure characterization: Method validation and application to therapeutic antibodies. J. Am. Chem. Soc. 136 (37), 13065–13071. doi:10.1021/ja507880w
Pandit, D., Tuske, S. J., Coales, S. J., Yen, E. S., Liu, A., Lee, J. E., et al. (2012). Mapping of discontinuous conformational epitopes by amide hydrogen/deuterium exchange mass spectrometry and computational docking. J. Mol. Recognit. 25 (3), 114–124. doi:10.1002/jmr.1169
Papac, D. I., Hoyes, J., and Tomer, K. B. (1994). Epitope mapping of the gastrin-releasing peptide/anti-bombesin monoclonal antibody complex by proteolysis followed by matrix-assisted laser desorption ionization mass spectrometry. Protein Sci. 3 (9), 1485–1492. doi:10.1002/pro.5560030914
Parker, C. E., Papac, D. I., Trojak, S. K., and Tomer, K. B. (1996). Epitope mapping by mass spectrometry: Determination of an epitope on HIV-1 IIIB p26 recognized by a monoclonal antibody. J. Immunol. 157 (1), 198–206. doi:10.4049/jimmunol.157.1.198
Pascal, B. D., Willis, S., Lauer, J. L., Landgraf, R. R., West, G. M., Marciano, D., et al. (2012). HDX workbench: Software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 23 (9), 1512–1521. doi:10.1007/s13361-012-0419-6
Paterson, Y., Englander, S. W., and Roder, H. (1990). An antibody binding site on cytochrome c defined by hydrogen exchange and two-dimensional NMR. Science 249 (4970), 755–759. doi:10.1126/science.1697101
Peter, J. F., and Tomer, K. B. (2001). A general strategy for epitope mapping by direct MALDI-TOF mass spectrometry using secondary antibodies and cross-linking. Anal. Chem. 73 (16), 4012–4019. doi:10.1021/ac010258n
Pierson, T. C., and Diamond, M. S. (2020). The continued threat of emerging flaviviruses. Nat. Microbiol. 5 (6), 796–812. doi:10.1038/s41564-020-0714-0
Pimenova, T., Nazabal, A., Roschitzki, B., Seebacher, J., Rinner, O., and Zenobi, R. (2008). Epitope mapping on bovine prion protein using chemical cross-linking and mass spectrometry. J. Mass Spectrom. 43 (2), 185–195. doi:10.1002/jms.1280
Poljak, R., Amzel, L., Avey, H., Chen, B., Phizackerley, R., and Saul, F. (1973). Three-dimensional structure of the Fab′ fragment of a human immunoglobulin at 2.8-Å resolution. Proc. Natl. Acad. Sci. 70 (12), 3305–3310. doi:10.1073/pnas.70.12.3305
Poljak, R., Amzel, L., Chen, B., Phizackerley, R., and Saul, F. (1975). Structure and specificity of antibody molecules. Philosophical Trans. R. Soc. Lond. B, Biol. Sci. 272 (915), 43–51. doi:10.1098/rstb.1975.0069
Porter, R. (1959). The hydrolysis of rabbit y-globulin and antibodies with crystalline papain. Biochem. J. 73 (1), 119–126. doi:10.1042/bj0730119
Ralston, C. Y., and Sharp, J. S. (2022). Structural investigation of therapeutic antibodies using hydroxyl radical protein footprinting methods. Antibodies 11 (4), 71. doi:10.3390/antib11040071
Ramaraj, T., Angel, T., Dratz, E. A., Jesaitis, A. J., and Mumey, B. (2012). Antigen–antibody interface properties: Composition, residue interactions, and features of 53 non-redundant structures. Biochimica Biophysica Acta (BBA)-Proteins Proteomics 1824 (3), 520–532. doi:10.1016/j.bbapap.2011.12.007
Ramirez-Sarmiento, C. A., and Komives, E. A. (2018). Hydrogen-deuterium exchange mass spectrometry reveals folding and allostery in protein-protein interactions. Methods 144, 43–52. doi:10.1016/j.ymeth.2018.04.001
Rand, K. D., and Jørgensen, T. J. (2007). Development of a peptide probe for the occurrence of hydrogen (1H/2H) scrambling upon gas-phase fragmentation. Anal. Chem. 79 (22), 8686–8693. doi:10.1021/ac0710782
Raval, S., Sarpe, V., Hepburn, M., Crowder, D. A., Zhang, T., Viner, R., et al. (2021). Improving spectral validation rates in hydrogen-deuterium exchange data analysis. Anal. Chem. 93 (9), 4246–4254. doi:10.1021/acs.analchem.0c05045
Reichert, J. M. (2023). Antibody therapeutics approved or in regulatory review in the EU or US. Available at: https://www.antibodysociety.org/resources/approved-antibodies/(Accessed.
Reichert, J. M. (2012). Marketed therapeutic antibodies compendium. MAbs 4 (3), 413–415. doi:10.4161/mabs.19931
Renaud, J.-P., Chari, A., Ciferri, C., Liu, W.-t., Rémigy, H.-W., Stark, H., et al. (2018). Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 17 (7), 471–492. doi:10.1038/nrd.2018.77
Rey, M., Man, P., Brandolin, G., Forest, E., and Pelosi, L. (2009). Recombinant immobilized rhizopuspepsin as a new tool for protein digestion in hydrogen/deuterium exchange mass spectrometry. Rapid Commun. Mass Spectrom. 23 (21), 3431–3438. doi:10.1002/rcm.4260
Rey, M., Yang, M., Burns, K. M., Yu, Y., Lees-Miller, S. P., and Schriemer, D. C. (2013). Nepenthesin from monkey cups for hydrogen/deuterium exchange mass spectrometry. Mol. Cell Proteomics 12 (2), 464–472. doi:10.1074/mcp.M112.025221
Rob, T., Gill, P. K., Golemi-Kotra, D., and Wilson, D. J. (2013). An electrospray ms-coupled microfluidic device for sub-second hydrogen/deuterium exchange pulse-labelling reveals allosteric effects in enzyme inhibition. Lab a Chip 13 (13), 2528–2532. doi:10.1039/C3LC00007A
Rob, T., Liuni, P., Gill, P. K., Zhu, S., Balachandran, N., Berti, P. J., et al. (2012). Measuring dynamics in weakly structured regions of proteins using microfluidics-enabled subsecond H/D exchange mass spectrometry. Anal. Chem. 84 (8), 3771–3779. doi:10.1021/ac300365u
Roder, H., Elöve, G. A., and Englander, S. W. (1988). Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature 335 (6192), 700–704. doi:10.1038/335700a0
Rosa, J. J., and Richards, F. M. (1979). An experimental procedure for increasing the structural resolution of chemical hydrogen-exchange measurements on proteins: Application to ribonuclease S peptide. J. Mol. Biol. 133 (3), 399–416. doi:10.1016/0022-2836(79)90400-5
Rubinstein, N. D., Mayrose, I., Halperin, D., Yekutieli, D., Gershoni, J. M., and Pupko, T. (2008). Computational characterization of B-cell epitopes. Mol. Immunol. 45 (12), 3477–3489. doi:10.1016/j.molimm.2007.10.016
Schroeder, H. W., and Cavacini, L. (2010). Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 125 (2), S41–S52. doi:10.1016/j.jaci.2009.09.046
Scott, J. K., and Smith, G. P. (1990). Searching for peptide ligands with an epitope library. Science 249 (4967), 386–390. doi:10.1126/science.1696028
Seetaloo, N., Kish, M., and Phillips, J. J. (2022). HDfleX: Software for flexible high structural resolution of hydrogen/deuterium-exchange mass spectrometry data. Anal. Chem. 94 (11), 4557–4564. doi:10.1021/acs.analchem.1c05339
Sethi, S. K., Smith, D. L., and McCloskey, J. A. (1983). Determination of active hydrogen content by fast atom bombardment mass spectrometry following hydrogen-deuterium exchange. Biochem. Biophys. Res. Commun. 112 (1), 126–131. doi:10.1016/0006-291x(83)91806-5
Sharon, J., Rynkiewicz, M. J., Lu, Z., and Yang, C. Y. (2014). Discovery of protective B-cell epitopes for development of antimicrobial vaccines and antibody therapeutics. Immunology 142 (1), 1–23. doi:10.1111/imm.12213
Sharp, J. S., Chea, E. E., Misra, S. K., Orlando, R., Popov, M., Egan, R. W., et al. (2021). Flash oxidation (fox) system: A novel laser-free fast photochemical oxidation protein footprinting platform. J. Am. Soc. Mass Spectrom. 32 (7), 1601–1609. doi:10.1021/jasms.0c00471
Sheff, J. G., Rey, M., and Schriemer, D. C. (2013). Peptide-column interactions and their influence on back exchange rates in hydrogen/deuterium exchange-MS. J. Am. Soc. Mass Spectrom. 24 (7), 1006–1015. doi:10.1007/s13361-013-0639-4
Sheff, J., Wang, P., Xu, P., Arbour, M., Masson, L., van Faassen, H., et al. (2021). Defining the epitope of a blood–brain barrier crossing single domain antibody specific for the type 1 insulin-like growth factor receptor. Sci. Rep. 11 (1), 4284. doi:10.1038/s41598-021-83198-w
Shukla, A. A., Hubbard, B., Tressel, T., Guhan, S., and Low, D. (2007). Downstream processing of monoclonal antibodies—Application of platform approaches. J. Chromatogr. B 848 (1), 28–39. doi:10.1016/j.jchromb.2006.09.026
Simmons, Z. R., Sharma, S., Wayne, J., Li, S., Vander Kooi, C. W., and Gentry, M. S. (2021). Generation and characterization of a laforin nanobody inhibitor. Clin. Biochem. 93, 80–89. doi:10.1016/j.clinbiochem.2021.03.017
Simonelli, L., Pedotti, M., Bardelli, M., Jurt, S., Zerbe, O., and Varani, L. (2018). “Mapping antibody epitopes by solution NMR spectroscopy: Practical considerations,” in Epitope mapping protocols. Editors J. Rockberg, and J. Nilvebrant (New York, NY: Springer New York), 29–51.
Skinner, J. J., Lim, W. K., Bédard, S., Black, B. E., and Englander, S. W. (2012). Protein dynamics viewed by hydrogen exchange. Protein Sci. 21 (7), 996–1005. doi:10.1002/pro.2081
Smith, G. P. (1985). Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 228 (4705), 1315–1317. doi:10.1126/science.4001944
Song, D., Sun, H., Ma, L., Liu, J., Gao, Y., Zhang, Q., et al. (2023). In-vitro diagnostic reagent evaluation of commercially available cardiac troponin I assay kits using H/D exchange mass spectrometry for antibody-epitope mapping. Anal. Chem. 95 (4), 2278–2284. doi:10.1021/acs.analchem.2c03946
Sowole, M. A., and Konermann, L. (2014). Effects of protein–ligand interactions on hydrogen/deuterium exchange kinetics: Canonical and noncanonical scenarios. Anal. Chem. 86 (13), 6715–6722. doi:10.1021/ac501849n
Ständer, S., Grauslund, R. L., Scarselli, M., Norais, N., and Rand, K. (2021). Epitope mapping of polyclonal antibodies by hydrogen-deuterium exchange mass spectrometry (HDX-MS). Anal. Chem. 93 (34), 11669–11678. doi:10.1021/acs.analchem.1c00696
Starr, T. N., Greaney, A. J., Addetia, A., Hannon, W. W., Choudhary, M. C., Dingens, A. S., et al. (2021). Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 371 (6531), 850–854. doi:10.1126/science.abf9302
Suckau, D., Köhl, J., Karwath, G., Schneider, K., Casaretto, M., Bitter-Suermann, D., et al. (1990). Molecular epitope identification by limited proteolysis of an immobilized antigen-antibody complex and mass spectrometric peptide mapping. Proc. Natl. Acad. Sci. 87 (24), 9848–9852. doi:10.1073/pnas.87.24.9848
Sun, H., Liu, J., Xiao, P., Zhou, Y., Li, H., Shen, M., et al. (2022). Epitope mapping of antibodies in C-reactive protein assay kits by hydrogen-deuterium exchange mass spectrometry explains differential results across kits. Anal. Bioanal. Chem. 414 (13), 3875–3884. doi:10.1007/s00216-022-04029-z
Suryadevara, N., Shrihari, S., Gilchuk, P., VanBlargan, L. A., Binshtein, E., Zost, S. J., et al. (2021). Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 184 (9), 2316–2331.e15. doi:10.1016/j.cell.2021.03.029
Syka, J. E., Coon, J. J., Schroeder, M. J., Shabanowitz, J., and Hunt, D. F. (2004). Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. 101 (26), 9528–9533. doi:10.1073/pnas.0402700101
Tanaka, K., Waki, H., Ido, Y., Akita, S., Yoshida, Y., Yoshida, T., et al. (1988). Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. mass Spectrom. 2 (8), 151–153. doi:10.1002/rcm.1290020802
Toride King, M., and Brooks, C. L. (2018). Epitope mapping of antibody-antigen interactions with X-ray crystallography. Methods Mol. Biol. 1785, 13–27. doi:10.1007/978-1-4939-7841-0_2
Toth IV, R. T., Angalakurthi, S. K., Van Slyke, G., Vance, D. J., Hickey, J. M., Joshi, S. B., et al. (2017). High-definition mapping of four spatially distinct neutralizing epitope clusters on RiVax, a candidate ricin toxin subunit vaccine. Clin. Vaccine Immunol. 24 (12), e00237–e00217. doi:10.1128/CVI.00237-17
Trabjerg, E., Jakobsen, R. U., Mysling, S., Christensen, S., Jørgensen, T. J. D., and Rand, K. D. (2015). Conformational analysis of large and highly disulfide-stabilized proteins by integrating online electrochemical reduction into an optimized H/D exchange mass spectrometry workflow. Anal. Chem. 87 (17), 8880–8888. doi:10.1021/acs.analchem.5b01996
Tran, M. H., Schoeder, C. T., Schey, K. L., and Meiler, J. (2022). Computational structure prediction for antibody-antigen complexes from hydrogen-deuterium exchange mass spectrometry: Challenges and outlook. Front. Immunol. 13, 859964. doi:10.3389/fimmu.2022.859964
Tremblay, C. Y., Kirsch, Z. J., and Vachet, R. W. (2022). Epitope mapping with diethylpyrocarbonate covalent labeling-mass spectrometry. Anal. Chem. 94 (2), 1052–1059. doi:10.1021/acs.analchem.1c04038
Udgaonkar, J. B., and Baldwin, R. L. (1988). NMR evidence for an early framework intermediate on the folding pathway of ribonuclease A. Nature 335 (6192), 694–699. doi:10.1038/335694a0
Vahidi, S., and Konermann, L. (2016). Probing the time scale of FPOP (fast photochemical oxidation of proteins): Radical reactions extend over tens of milliseconds. J. Am. Soc. Mass Spectrom. 27 (7), 1156–1164. doi:10.1007/s13361-016-1389-x
Valente, A. P., and Manzano-Rendeiro, M. (2021). Mapping conformational epitopes by NMR spectroscopy. Curr. Opin. Virology 49, 1–6. doi:10.1016/j.coviro.2021.04.001
Van Regenmortel, M. H. V. (1996). Mapping epitope structure and activity: From one-dimensional prediction to four-dimensional description of antigenic specificity. Methods 9 (3), 465–472. doi:10.1006/meth.1996.0054
VanBlargan, L. A., Errico, J. M., Kafai, N. M., Burgomaster, K. E., Jethva, P. N., Broeckel, R. M., et al. (2021). Broadly neutralizing monoclonal antibodies protect against multiple tick-borne flaviviruses. J. Exp. Med. 218 (5), e20210174. doi:10.1084/jem.20210174
Venable, J. D., Okach, L., Agarwalla, S., and Brock, A. (2012). Subzero temperature chromatography for reduced back-exchange and improved dynamic range in amide hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 84 (21), 9601–9608. doi:10.1021/ac302488h
Verma, S., Pomerantz, S. C., Sethi, S. K., and McCloskey, J. A. (1986). Fast atom bombardment mass spectrometry following hydrogen-deuterium exchange. Anal. Chem. 58 (14), 2898–2902. doi:10.1021/ac00127a002
Vidarsson, G., Dekkers, G., and Rispens, T. (2014). IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 5, 520. doi:10.3389/fimmu.2014.00520
Volk, A.-L., Hu, F. J., Berglund, M. M., Nordling, E., Strömberg, P., Uhlen, M., et al. (2016). Stratification of responders towards eculizumab using a structural epitope mapping strategy. Sci. Rep. 6 (1), 31365–31410. doi:10.1038/srep31365
Wagner, N. D., Huang, Y., Liu, T., and Gross, M. L. (2021). Post-HDX deglycosylation of Fc gamma receptor IIIa glycoprotein enables HDX characterization of its binding interface with IgG. J. Am. Soc. Mass Spectrom. 32 (7), 1638–1643. doi:10.1021/jasms.1c00003
Wales, T. E., Eggertson, M. J., and Engen, J. R. (2013). “Considerations in the analysis of hydrogen exchange mass spectrometry data,” in Mass spectrometry data analysis in proteomics. Editor R. Matthiesen (Totowa, NJ: Humana Press), 263–288.
Wales, T. E., Fadgen, K. E., Eggertson, M. J., and Engen, J. R. (2017). Subzero Celsius separations in three-zone temperature controlled hydrogen deuterium exchange mass spectrometry. J. Chromatogr. A 1523, 275–282. doi:10.1016/j.chroma.2017.05.067
Wales, T. E., Fadgen, K. E., Gerhardt, G. C., and Engen, J. R. (2008). High-speed and high-resolution UPLC separation at zero degrees celsius. Anal. Chem. 80 (17), 6815–6820. doi:10.1021/ac8008862
Wang, L., Lane, L. C., and Smith, D. L. (2001). Detecting structural changes in viral capsids by hydrogen exchange and mass spectrometry. Protein Sci. 10 (6), 1234–1243. doi:10.1110/ps.100101
Wang, L., and Smith, D. L. (2005). Capsid structure and dynamics of a human rhinovirus probed by hydrogen exchange mass spectrometry. Protein Sci. 14 (6), 1661–1672. doi:10.1110/ps.051390405
Wang, Q., Borotto, N. B., and Håkansson, K. (2019). Gas-phase hydrogen/deuterium scrambling in negative-ion mode tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 30 (5), 855–863. doi:10.1007/s13361-019-02143-4
Wecksler, A. T., Kalo, M. S., and Deperalta, G. (2015). Mapping of fab-1:VEGF interface using carboxyl group footprinting mass spectrometry. J. Am. Soc. Mass Spectrom. 26 (12), 2077–2080. doi:10.1007/s13361-015-1273-0
Weis, D. D. (2019). Comment on houde, D.; berkowitz, S. A.; engen, J. R., the utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 2011, 100, 2071-2086. J. Pharm. Sci. 108 (2), 807–810. doi:10.1016/j.xphs.2018.10.010
Weis, D. D., Engen, J. R., and Kass, I. J. (2006a). Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17 (12), 1700–1703. doi:10.1016/j.jasms.2006.07.025
Weis, D. D., Wales, T. E., Engen, J. R., Hotchko, M., and Ten Eyck, L. F. (2006b). Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass Spectrom. 17 (11), 1498–1509. doi:10.1016/j.jasms.2006.05.014
Weiss, G. A., Watanabe, C. K., Zhong, A., Goddard, A., and Sidhu, S. S. (2000). Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. 97 (16), 8950–8954. doi:10.1073/pnas.160252097
Wigge, C., Stefanovic, A., and Radjainia, M. (2020). The rapidly evolving role of cryo-EM in drug design. Drug Discov. Today Technol. 38, 91–102. doi:10.1016/j.ddtec.2020.12.003
Wilkinson, H., and Saldova, R. (2020). Current methods for the characterization of O-glycans. J. Proteome Res. 19 (10), 3890–3905. doi:10.1021/acs.jproteome.0c00435
Wilson, D. J., and Konermann, L. (2003). A capillary mixer with adjustable reaction chamber volume for millisecond time-resolved studies by electrospray mass spectrometry. Anal. Chem. 75 (23), 6408–6414. doi:10.1021/ac0346757
Wishnia, A., and Saunders, M. (1962). The nature of the slowly exchanging protons of ribonuclease. J. Am. Chem. Soc. 84 (22), 4235–4239. doi:10.1021/ja00881a008
Wollenberg, D. T. W., Pengelley, S., Mouritsen, J. C., Suckau, D., Jørgensen, C. I., and Jørgensen, T. J. D. (2020). Avoiding H/D scrambling with minimal ion transmission loss for HDX-MS/MS-ETD analysis on a high-resolution Q-TOF mass spectrometer. Anal. Chem. 92 (11), 7453–7461. doi:10.1021/acs.analchem.9b05208
Wu, Y., Engen, J. R., and Hobbins, W. B. (2006). Ultra performance liquid chromatography (UPLC) further improves hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 17 (2), 163–167. doi:10.1016/j.jasms.2005.10.009
Xiao, K., Zhao, Y., Choi, M., Liu, H., Blanc, A., Qian, J., et al. (2018). Revealing the architecture of protein complexes by an orthogonal approach combining HDXMS, CXMS, and disulfide trapping. Nat. Protoc. 13 (6), 1403–1428. doi:10.1038/nprot.2018.037
Yamada, N., Suzuki, E.-i., and Hirayama, K. (2002). Identification of the interface of a large protein–protein complex using H/D exchange and Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 16 (4), 293–299. doi:10.1002/rcm.579
Yan, Y., Chen, G., Wei, H., Huang, R. Y. C., Mo, J., Rempel, D. L., et al. (2014). Fast photochemical oxidation of proteins (FPOP) maps the epitope of EGFR binding to adnectin. J. Am. Soc. Mass Spectrom. 25 (12), 2084–2092. doi:10.1007/s13361-014-0993-x
Yang, D., Frego, L., Lasaro, M., Truncali, K., Kroe-Barrett, R., and Singh, S. (2016). Efficient qualitative and quantitative determination of antigen-induced immune responses. J. Biol. Chem. 291 (31), 16361–16374. doi:10.1074/jbc.M116.736660
Yefremova, Y., Opuni, K. F. M., Danquah, B. D., Thiesen, H. J., and Glocker, M. O. (2017). Intact transition epitope mapping (ITEM). J. Am. Soc. Mass Spectrom. 28 (8), 1612–1622. doi:10.1007/s13361-017-1654-7
Yu, L., Gaskell, S. J., and Brookman, J. L. (1998). Epitope mapping of monoclonal antibodies by mass spectrometry: Identification of protein antigens in complex biological systems. J. Am. Soc. Mass Spectrom. 9 (3), 208–215. doi:10.1016/s1044-0305(97)00250-x
Závodszky, P., Jaton, J. C., Venyaminov, S. Y., and Medgyesi, G. A. (1981). Increase of conformational stability of homogeneous rabbit immunoglobulin G after hapten binding. Mol. Immunol. 18 (1), 39–46. doi:10.1016/0161-5890(81)90046-8
Zhang, H.-M., Kazazic, S., Schaub, T. M., Tipton, J. D., Emmett, M. R., and Marshall, A. G. (2008). Enhanced digestion efficiency, peptide ionization efficiency, and sequence resolution for protein hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 80 (23), 9034–9041. doi:10.1021/ac801417d
Zhang, J., Ramachandran, P., Kumar, R., and Gross, M. L. (2013). H/D exchange centroid monitoring is insufficient to show differences in the behavior of protein states. J. Am. Soc. Mass Spectrom. 24 (3), 450–453. doi:10.1007/s13361-012-0555-z
Zhang, M. M., Huang, R. Y. C., Beno, B. R., Deyanova, E. G., Li, J., Chen, G., et al. (2020). Epitope and paratope mapping of PD-1/nivolumab by mass spectrometry-based hydrogen-deuterium exchange, cross-linking, and molecular docking. Anal. Chem. 92 (13), 9086–9094. doi:10.1021/acs.analchem.0c01291
Zhang, N., Yu, X., Zhang, X., and D’Arcy, S. (2021). HD-eXplosion: Visualization of hydrogen–deuterium exchange data as chiclet and volcano plots with statistical filtering. Bioinformatics 37 (13), 1926–1927. doi:10.1093/bioinformatics/btaa892
Zhang, Q., Noble, K. A., Mao, Y., Young, N. L., Sathe, S. K., Roux, K. H., et al. (2013). Rapid screening for potential epitopes reactive with a polycolonal antibody by solution-phase H/D exchange monitored by FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 24 (7), 1016–1025. doi:10.1007/s13361-013-0644-7
Zhang, Q., Willison, L. N., Tripathi, P., Sathe, S. K., Roux, K. H., Emmett, M. R., et al. (2011). Epitope mapping of a 95 kDa antigen in complex with antibody by solution-phase amide backbone hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 83 (18), 7129–7136. doi:10.1021/ac201501z
Zhang, Q., Yang, J., Bautista, J., Badithe, A., Olson, W., and Liu, Y. (2018). Epitope mapping by HDX-MS elucidates the surface coverage of antigens associated with high blocking efficiency of antibodies to Birch pollen allergen. Anal. Chem. 90 (19), 11315–11323. doi:10.1021/acs.analchem.8b01864
Zhang, X., Chien, E. Y. T., Chalmers, M. J., Pascal, B. D., Gatchalian, J., Stevens, R. C., et al. (2010). Dynamics of the beta2-adrenergic G-protein coupled receptor revealed by hydrogen-deuterium exchange. Anal. Chem. 82 (3), 1100–1108. doi:10.1021/ac902484p
Zhang, X., Sun, L., and Rossmann, M. G. (2015). Temperature dependent conformational change of dengue virus. Curr. Opin. Virol. 12, 109–112. doi:10.1016/j.coviro.2015.04.006
Zhang, Y., Wecksler, A. T., Molina, P., Deperalta, G., and Gross, M. L. (2017). Mapping the binding interface of VEGF and a monoclonal antibody fab-1 fragment with fast photochemical oxidation of proteins (FPOP) and mass spectrometry. J. Am. Soc. Mass Spectrom. 28 (5), 850–858. doi:10.1007/s13361-017-1601-7
Zhang, Z., and Smith, D. L. (1993). Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 2 (4), 522–531. doi:10.1002/pro.5560020404
Zhao, Y., and Chalt, B. T. (1994). Protein epitope mapping by mass spectrometry. Anal. Chem. 66 (21), 3723–3726. doi:10.1021/ac00093a029
Zubarev, R. A., Kelleher, N. L., and McLafferty, F. W. (1998). Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 120 (13), 3265–3266. doi:10.1021/ja973478k
Zubarev, R. A., Kruger, N. A., Fridriksson, E. K., Lewis, M. A., Horn, D. M., Carpenter, B. K., et al. (1999). Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. J. Am. Chem. Soc. 121 (12), 2857–2862. doi:10.1021/ja981948k
Keywords: epitope mapping, mass spectrometry (MS), hydrogen deuterium exchange (HDX), antibody-antigen interactions, polyclonal antibodies, fast photochemical oxidation of protein (FPOP), crosslinking, footprinting
Citation: Jethva PN and Gross ML (2023) Hydrogen deuterium exchange and other mass spectrometry- based approaches for epitope mapping. Front. Anal. Sci. 3:1118749. doi: 10.3389/frans.2023.1118749
Received: 07 December 2022; Accepted: 27 March 2023;
Published: 18 May 2023.
Edited by:
Nicholas B. Borotto, University of Nevada, Reno, United StatesReviewed by:
Milkos Guttman, University of Washington, United StatesGuanbo Wang, Peking University, China
Copyright © 2023 Jethva and Gross. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prashant N. Jethva, cGpldGh2YUB3dXN0bC5lZHU=; Michael L. Gross, bWdyb3NzQHd1c3RsLmVkdQ==