 Marie Valmori
Marie Valmori Vincent Marie
Vincent Marie François Fenaille
François Fenaille Benoit Colsch
Benoit Colsch David Touboul
David Touboul
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Anal. Sci. , 10 February 2023
Sec. Omics
Volume 3 - 2023 | https://doi.org/10.3389/frans.2023.1118742
This article is part of the Research Topic Perspectives in Omics 2022 View all 7 articles
Untargeted lipidomics applied to biological samples typically involves the coupling of separation methods to high-resolution mass spectrometry (HRMS). Getting an exhaustive coverage of the lipidome with a high confidence in structure identification is still highly challenging due to the wide concentration range of lipids in complex matrices and the presence of numerous isobaric and isomeric species. The development of innovative separation methods and HRMS(/MS) acquisition workflows helped improving the situation but issues still remain regarding confident structure characterization. To overcome these issues, thoroughly optimized MS/MS acquisition methods are needed. For this purpose, different methodologies have been developed to enable MS and MS/MS acquisition in parallel. Those methodologies, derived from the proteomics, are referred to Data Dependent Acquisition (DDA) and Data Independent Acquisition (DIA). In this context, this perspective paper presents the latest developments of DDA- and DIA-based lipidomic workflows and lists available bioinformatic tools for the analysis of resulting spectral data.
Lipids represent a large class of primary metabolites playing vital biological functions to maintain homeostasis in living organisms, serving as energy storage and cell signaling processes (Züllig et al., 2020). They are also involved in a large number of heterogeneous diseases including cancer, neurodegenerative and metabolic disorders. Lipids are classified in eight categories according to the Lipid Maps classification system (Fahy et al., 2005; 2009; Liebisch et al., 2020) available online through the curated Lipid MAPS Structure Database (LMSD) gathering almost 48,000 unique lipid species in 2022 (https://www.lipidmaps.org/). Those categories can be further separated into classes, in which lipids share some building blocks but also differ by the length of alkyl chains and number of unsaturations creating a high diversity of closely related lipid species making their distinction highly challenging (Quehenberger et al., 2010; Harayama and Riezman, 2018).
Lipidomics can be defined as a system-level analysis of lipids and their interacting factors (Wenk, 2005). Lipidomics is a subset of metabolomics requiring dedicated sample preparation, as lipids are mostly soluble in organic solvents (e.g., methyl tert-butyl ether, chloroform and methanol for Bligh-Dyer and Folch extractions), as well as specific analytical tools to qualitatively and quantitatively characterize complex samples.
One of the major challenges in lipidomics is to distinguish isobaric and isomeric lipid species by implementing either optimized hyphenated mass spectrometry (MS)-based methods and/or tandem mass spectrometry approaches. The fine structure elucidation of complex lipids, such as glycerophospholipids, requires the identification of the polar head group, the chain lengths and the position of the fatty acids together with the number and position of unsaturations or other chemical modifications of the lipid backbone. The precise characterization of lipid species is mandatory to interpret their biological or chemical roles in a defined context (Wenk, 2010).
Among the technical developments performed in the last 70 years, gas chromatography coupled to electron ionization—mass spectrometry (GC-EI-MS) can be considered as the first major tools towards lipidomics. Even if the techniques required complex sample preparation including subclasses separation and chemical derivatization, GC-MS allowed efficient profiling of the fatty acids composing complex lipids (Chiu and Kuo, 2020).
The multiple ionization sources and particularly soft ionization such as Electrospray Ionization (ESI), Matrix-Assisted Laser Desorption/Ionization (MALDI) and Atmospheric Pressure Chemical/Photo-Ionization (APCI/APPI), have made mass spectrometry as the technique of preference to perform lipidome characterization and quantification. Also, Atmospheric Pressure ionization sources are mostly employed for compounds difficult to ionize in ESI, mostly represented by neutral lipids such as triacylglycerols or carotenoids (Byrdwell, 2001; Imbert et al., 2012).
The advent of liquid chromatography coupled to mass spectrometry (LC-MS) in the 80’s offered new perspectives as intact complex lipids can be directly separated, detected and isolated for tandem mass spectrometry (MS/MS) experiments. Nevertheless, the low mass resolution instruments available at that time did not offer sufficient high mass measurement accuracy and led to difficulty in distinguishing isobaric species. The commercialization of fast and robust high-resolution (HR)/high-mass accuracy tandem mass spectrometers paved the route to the analysis of high-mass lipids thanks to extended mass capacities and dynamic ranges (up to 4 decades on modern instruments), high ionization rate for increased sensitivity in both polarities and for all lipid sub-classes. Time-of-flight (TOF) mass analyzers show the highest repetition rate. In terms of mass accuracy and resolving power, Fourier Transform mass spectrometers, such as Orbitrap and Fourier-transform ion cyclotron resonance (FTICR) instruments, are overpassing the TOF performances in spite of their slower acquisition rates limiting the number of data points per chromatographic peak. The Orbitrap-based instruments, since their first commercialization with the LTQ-Orbitrap in 2006, have witnessed innovations in sensitivity, resolution and scan speed (Eliuk and Makarov, 2015). The lipidome investigation can be now achieved at high throughput by either a shotgun approach (direct introduction into the mass spectrometer) or using hyphenated MS-based techniques such as normal or reverse liquid phase or supercritical fluid chromatography (Rustam and Reid, 2018). The common analysis workflow for those methods is based on the acquisition at the MS level of robust data for biological and technical replicates together with quality control (QC) injections to correct any deviation of the analytical chain. Then the identification of relevant markers or compounds differentiating sample groups is further accomplished by tandem mass spectrometry (MS/MS) following multiple injections into the LC-MS/MS system using targeted MS/MS workflows. MS/MS spectral data are necessary for the elucidation structure of lipids. The major part of the existing lipid databases mostly includes tandem mass spectra acquired under low-energy non-resonant collision-induced dissociation (CID) or higher energy collision dissociation (HCD) conditions on hybrid TOF and Orbitrap-based instruments. But, multiple types of ion activation modes (i.e., resonant and non-resonant activation modes) exist and can be employed in MS/MS and are currently used to a lesser extent for high throughput lipidomics workflows.
As for classical proteomics approaches, the parallel acquisition of HRMS and MS/MS data can be achieved using either a Data-Dependent Analysis (DDA) or a Data-Independent Analysis (DIA) approach. In the following Perspective article, technical and practical considerations on DDA and DIA for lipidomics will be discussed to offer an overview of the latest developments and future trends in the field of lipidomics.
The DDA fragmentation method allows successive acquisitions of MS and MS/MS data. The most widely used DDA acquisition method is often designated as the “DDA Top N”. It consists of a cycle of a single HRMS scan followed by the automatic selection and further fragmentation of the N most intense signals in a defined m/z range (Figure 1). The first HRMS scan offers the unique possibility of getting high mass accuracy measurements, typically below 1–2 ppm. Unambiguous chemical formula can be easily determined for small lipids, below m/z 300–400 but much higher mass accuracy (<0.1 ppm) would be required to determine the elemental composition of complex lipids. Then automatic MS/MS scans are programmed based on rules such as peak intensity and mass range on HRMS data. This method enables fragmenting a high number of ions and leads to high-quality and high-purity MS/MS spectra since the selection window is restricted to monoisotopic ions. Nevertheless, precursor ions selection is a (semi-) stochastic event suffering from low analytical reproducibility and favoring selection of the most abundant (but sometimes biologically irrelevant) precursor ions to the detriment of other co-eluting relevant ones (Fenaille et al., 2017). These issues can be partially overcome through the implementation of exclusion rules, typical after one or several consecutive MS/MS experiments. The number of selected ions must be thoroughly optimized considering the MS/MS overall duty cycle (usually few tens to hundreds of milliseconds per MS/MS spectrum) and the LC peak width. A too high number of precursor ions will limit the number of complete DDA cycles recorded per chromatographic peak, thus downgrading the quality of peak integration and quantification at the MS level due to insufficient number of data points. This clearly advantages fast MS/MS scanning instruments such as hybrid TOF, with MS acquisition rate up to 100 Hz in both the MS and MS/MS modes (at 50 K resolution) for the TripleTOF 6,600 and timsTOF instruments, compared to the latest generation of quadrupole-Orbitrap instruments (Orbitrap Exploris series) working at 40 Hz at 7.5 K resolution at m/z 200 (Perez de Souza et al., 2021).
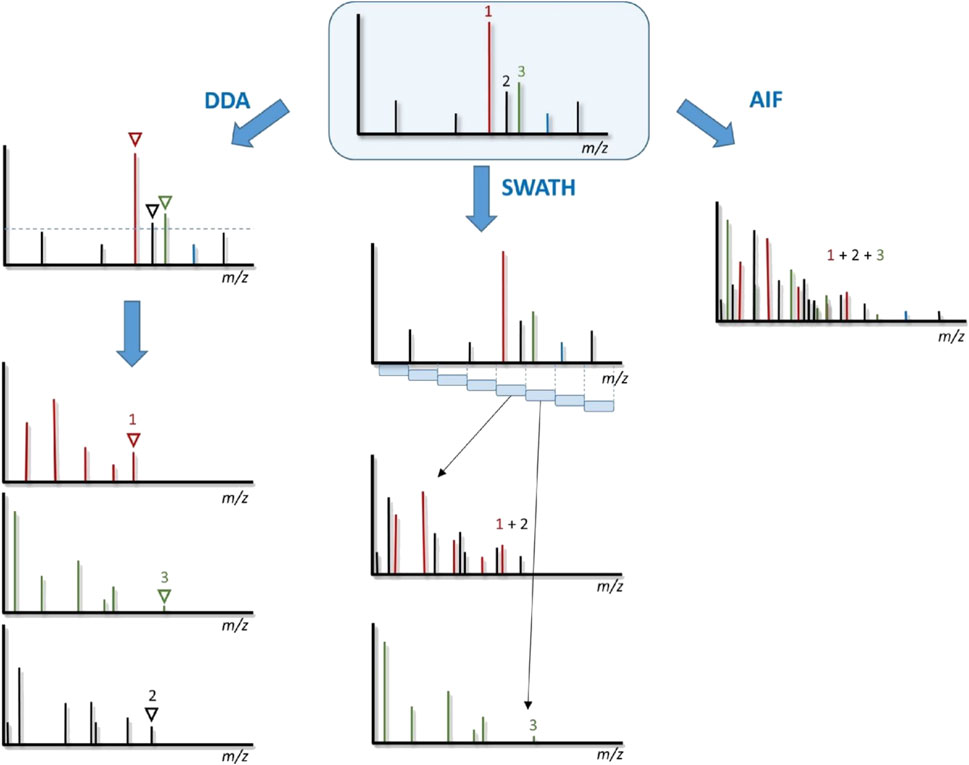
FIGURE 1. Scheme illustrating the DDA, SWATH, and AIF acquisition workflows reproduced with the permission from Fenaille et al. (2017) with permission from Elsevier, copyright (2017).
Neumann et al. proposed a protocol to realize a better sampling in MS/MS mode by focusing on biologically relevant and meaningful features (Neumann et al., 2013). First, a LC run at the MS level is performed, with the direct analysis of the data by preprocessing software XCMS for automated annotation and statistical analysis to select significant features. Then methods with inclusion lists are created and performed on pooled samples using targeted MS/MS (tMS2) acquisition mode in order to confirm previous annotations while also limiting the number of LC-MS/MS experiments. This method enables a better coverage of the sample by selecting lower abundance relevant features, while losing the unbiased nature of DDA.
Koelmel et al. developed an Iterative Exclusion IE-Omics approach whose goal is to create several non-redundant inclusion lists (Koelmel et al., 2017). First, a classic DDA acquisition is performed, and all the fragmented ions are added to an exclusion list. The workflow is repeated up to 5 times until the intensity of the precursor ions becomes too low for getting exploitable MS/MS data for automated annotation. When compared to five independent DDA scans performed on the same sample, the IE-Omics method shows an increase of 30% in the coverage of the lipidome for a plasma sample.
A strategy based on an automated DDA inclusion list workflow, called Data-Set-Dependent Acquisition (DsDA), was developed by Broeckling et al. (Broeckling et al., 2018). This method reports the specific combination of MS-data processing and target prioritization to enable a more comprehensive MS/MS coverage of the metabolome. The workflow consists of Top4 DDA scan, then data is automatically treated and a score is attributed to each MS/MS spectra to determine its importance to be fragmented. A module, written in R programming language, forces the workflow to fragment precursor ions not previously fragmented in order to improve the metabolome coverage. This novel method offered 45% more MS/MS features. In the recently introduced BoxCar method, the MS spectrum is segmented in several windows to improve detection sensitivity and the dynamic range of the MS acquisition (Meier et al., 2018). Under these conditions, less abundant ions can be isolated during DDA, thus providing unprecedented structural information.
Another recently introduced gas phase fractionation approach is the so-called Parallel Accumulation Serial Fragmentation (PASEF, available on TimsTOF Pro instruments) that synchronizes Trapped Ion Mobility Spectrometry (TIMS) with MS/MS precursor selection and fragmentation. This acquisition mode drastically multiplies the fragmentation speed and detection sensitivity thereby approaching almost 100% duty cycle, thus annotating up to 1,100 unique lipids in 1 μL of human plasma (Vasilopoulou et al., 2020).
All the presented methods of DDA are suffering from the same issue. As the sample often needs to be injected several times (up to five in both positive and negative ionization modes), its volume needs to be sufficient and the global analysis time is significantly extended making it impossible to apply these strategies for large cohort analysis in the field of medicine or biology.
DIA performance was first described in the proteomic field 18 years ago (Venable et al., 2004) and have been implemented for lipidomics (Schwudke et al., 2006). The application of DIA is essentially motivated by the wider accessible dynamic range (4 orders of magnitude) (Gillet et al., 2012), higher sensitivity and reproducibility and, more importantly, the unbiased characterization of complex biological matrices (Venable et al., 2004).
Analyzing the lipidome by direct injection into the mass spectrometer without prior chromatographic separation steps (shotgun approach) is highly attractive owing to the short acquisition time, the low solvent consumption, and its ease of implementation even if ion suppression effects are usually reported (Schwudke et al., 2011; Yang and Han, 2016). Shotgun DIA mode is mainly based on MSALL workflow (Simons et al., 2012) and aims at monitoring MS/MS spectral information with 1 Da mass isolation on a large mass range for a run time lasting less than 6 min (Simons et al., 2012). MSALL mode was efficient to identify and quantify lipids belonging to different classes in various biological matrices (Simons et al., 2012; Gao et al., 2016; 2018; 2021).
MSE for Q-TOF or All Ion Fragmentation (AIF) for Orbitrap-based instruments consist in acquiring alternating scans of all ions without mass filtering at two or three collision energies (Plumb et al., 2006) (Figure 1). Precursor information are retrieved at low collision energy whereas specific product ions are given by higher collision energy, facilitating the annotation step by combining all the MS/MS data into consensus MS/MS spectra (Plumb et al., 2006; Diedrich et al., 2013).
One of the first approaches of DIA-AIF method was implemented on a LC coupled to a single-stage Orbitrap mass analyzer using polarity switching in a single analytical run and using either in source CID or HCD fragmentation (Gallart-Ayala et al., 2013). Annotation of both phospholipids and triacylglycerols from serum samples for diagnosis of canine mammary cancer thanks to known retention times and typical diagnostic product ions was performed. Similarly, Castro-Perez et al. reported the advantages of applying alternated low and high collision energies in positive and negative ionization modes to identify lipids from human plasma (Castro-Perez et al., 2010). More recently, lipids from human plasma and dermal fibroblasts extracts were annotated using DIA-AIF and HCD in both polarities (Ventura et al., 2020a; 2020b).
In order to improve the association of precursor ions with their corresponding fragments, SWATH (Sequential Window Acquisition of All THeoretical) has been developed (Gillet et al., 2012). The principle is to define sequential MS/MS experiments in reduced m/z windows, typically 10–20 Da to cover the whole mass range, all ions within these windows are then fragmented simultaneously (Figure 1). It has been initially developed on hybrid TOF instruments since a high-speed acquisition is mandatory, but can be now implemented on Orbitrap-based instruments (Fenaille et al., 2017). SWATH experiments were performed to characterize 214 and 140 lipids from immortalized keratinocytes in both positive and negative ionization modes, respectively. As statistical treatment of the data is mainly based on retention times, authors strongly advise to add internal standards to correct any deviation (Calderón et al., 2020; Cebo et al., 2021). Interestingly, SWATH can be employed for compound annotation using MS/MS library search but also quantification at the MS/MS level because, unlike classic DDA, it offers continuous MS/MS spectra acquisition for particular fragment ions.
One of the main drawbacks of DIA workflows is related to the obtained (highly) convoluted tandem mass spectra. Indeed, isobaric or isomeric lipid species from different classes can produce overlapping isotopic clusters that will be co-isolated and fragmented MS/MS thus yielding highly convoluted and thereby difficult-to-match MS/MS spectra. For instance, protonated phosphatidylcholine (PC 33:1) and phosphatidylethanolamine (PE 36:1) share the same elemental composition (Züllig and Köfeler, 2021). The frequent presence of sodium adducts can bring additional spectral complexity with lipid sodiated species isobaric to protonated unsaturated ones differing only by ∼2 mDa in some extreme cases (Höring et al., 2020; Züllig and Köfeler, 2021).
In DIA workflows, such as SWATH, the direct link between a given precursor ion and its corresponding fragments is often lost, which renders MS/MS data interpretation of unknown lipid species much more difficult than that of MS/MS data resulting from DDA protocols. Data treatment step, i.e. the concatenation and alignment of precursor and fragment ions, is indeed often considered as the critical point. This can be performed according to retention times (Castro-Perez et al., 2010) or by specific data filtering through mass defect calculation (Stagliano et al., 2010). A correlation-based deconvolution (CorrDec) method that uses correlation of ion abundances between precursor and product ions has been developed to improve the metabolite annotation rate (Tada et al., 2020).
While SWATH techniques provide a better coverage of the lipidome, it comes at the cost of lower spectral quality. Therefore, DIA SWATH method has been implemented together with DDA leading to the hybrid DaDIA mode (Guo et al., 2021). The sequence is composed of blanks and samples analyzed in DIA SWATH mode and quality controls (QCs) in DDA mode. DDA MS/MS data provided high quality spectra for the highest intensity features and are used to improve the deconvolution of SWATH data for better quality. This hybrid method enables a better lipidome coverage while improving the quality of deconvoluted SWATH spectra to a level similar to DDA spectra. Combination of 2D-LC, DIA MSE and DDA Top3, called HDDIDDA, outperforms DDA with a slightly higher annotation rate and MSE with a slightly higher coverage at the cost of a complex data treatment (Wang et al., 2022).
Even if DIA approach looks appealing in terms of numbers of annotated lipid species, the annotation steps required the most exhaustive and reliable MS/MS databases including normalized retention times if available. Acquiring MS/MS spectra from pure authentic standards is the best way to generate spectral databases, but this is clearly unrealistic due to the cost and viability of pure compounds. The most feasible strategy is to generate in silico low-energy fragmentation spectra thanks to the well-documented mechanisms of lipid fragmentation (Murphy, 2015) as defined in LipidBlast database. As already documented in the literature, MS/MS data are strongly dependent on the type of tandem mass spectrometer and the collision energy used that is challenging to normalize especially in the case of small molecule analysis (Murphy, 2015). Thus, the information related to the relative intensities of the fragments cannot be easily employed to refine structure annotation making the match scoring process less robust.
Several software solutions (Kitata et al., 2021) include modules capable of achieving the complete pipeline from the conversion of vendor raw files to statistical analysis and through the identification from integrated and/or implementable lipid libraries as exemplified in Table 1. While attributing ion products to the precursor ion is unproblematic in DDA mode, to reconstruct MS/MS data in DIA mode remains highly challenging. To the best of our knowledge, only two robust software tools, i.e., MS-DIAL and DecoMetDIA, offer a deconvolution step for untargeted analysis in the DIA mode (Tsugawa et al., 2019; Yin et al., 2019; Tada et al., 2020). Multiple processing strategies for DIA deconvolution emerge in proteomics (Kitata et al., 2021) and are expected to be applied to small compounds in the near future.
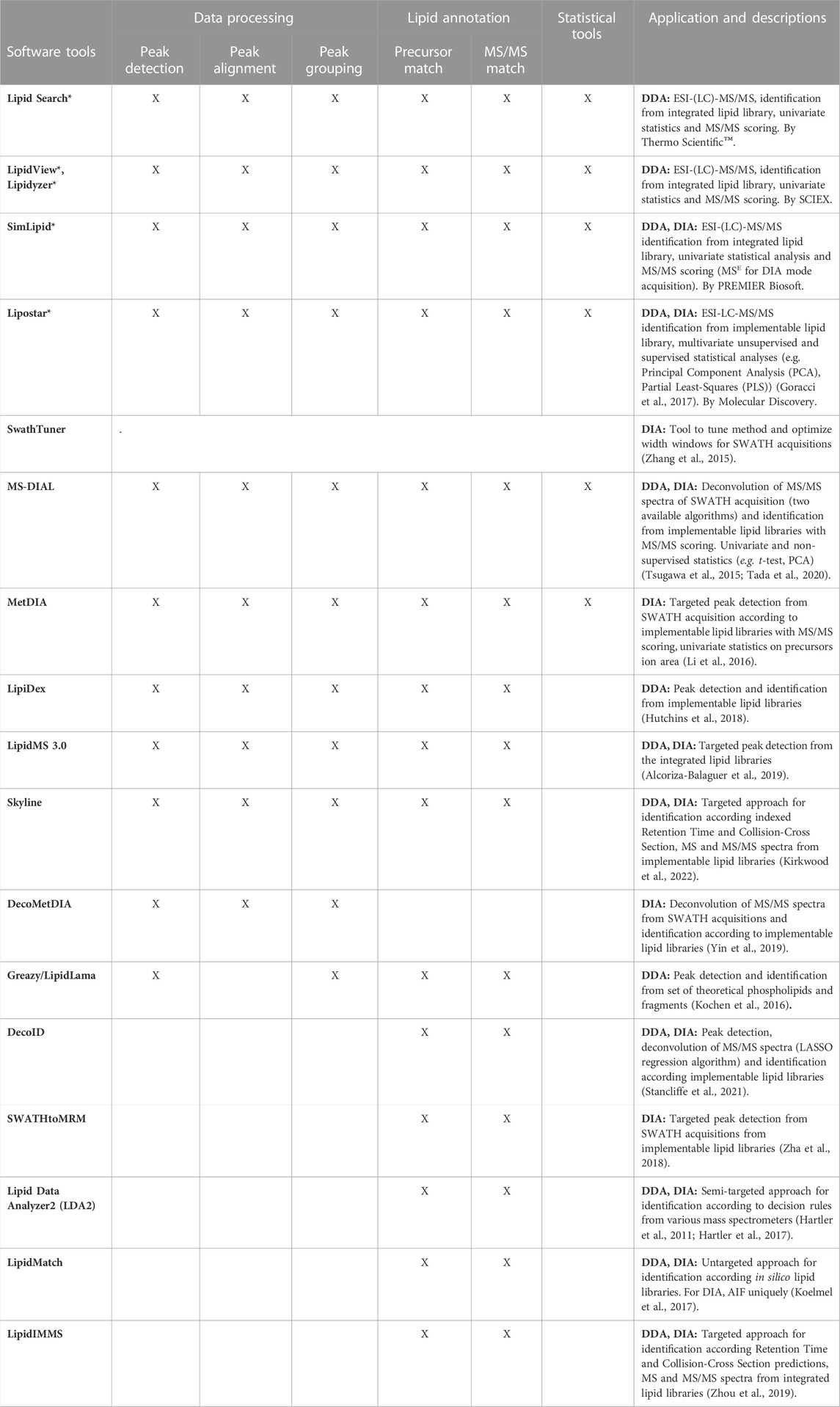
TABLE 1. Overview of software and tools for the processing of lipid identification step from spectral data acquired by tandem mass spectrometry in DDA and DIA. Remark: Software specified by * are commercial.
Considering the rapid development of mass spectrometers in terms of scan speed, resolving power and mass accuracy, the analytical strategies for lipidomics have widely expanded during the last two decades. Beyond the use of high mass accuracy at MS level to suggest molecular formulas, MS/MS experiments are fully integrated in lipidomics workflows to provide successful semi-quantification and structural characterization. Both DDA and DIA methodologies require high acquisition rate tandem mass spectrometers to access deep lipidome. The quality of MS/MS data is needed for facilitating annotation but the total number of MS/MS scans on a unique feature is limited. Iterative DDA allows to exceed this limitation but significantly increases the sample consumption and the overall experimental time making this approach not compatible with the analysis of large cohorts. On the contrary, DIA significantly increases the number of annotated lipid species but is still far limited by the available software solutions (notably regarding spectra deconvolution).
Free software tools are reviewed in the Table 1 and three of them, namely MS-DIAL (Tsugawa et al., 2015), DecoMetDIA (Yin et al., 2019) and DecoID (Stancliffe et al., 2021) propose efficient bioinformatic tools for the deconvolution of mixed tandem mass spectra obtained from SWATH workflows.
Also, as exposed by Barbier Saint Hilaire et al., well-designing SWATH isolation windows can impact significantly the detection rate of targeted compounds (Barbier Saint Hilaire et al., 2020). The free software tool SwathTUNER (Table 1) permits the automatized design of the variable isolations windows according either the Total Ion Current (TIC) or precursors density (Zhang et al., 2015) to significantly lower the complexity of tandem mass spectra obtained in the DIA-SWATH mode. Also, the commercial vendor Sciex proposes the SWATH® Acquisition module available on their TripleTOF® systems for the automatically designing of variable isolation windows, permitting the all-in-one acquisition workflow (https://sciex.com/technology/swath-acquisition).
Since a decade, ion mobility (IM) offers the unique possibility to separate ions not only on their m/z values but also on their shape in the gas phase, thus enabling the measure of collision cross-sections (CCSs). The IM time is fully compatible with the chromatographic time and the fast acquisition time of modern mass spectrometers. Several groups demonstrated that lipid isomers bearing different fatty chains but the same total number of carbons, including Z/E isomers, can be efficiently separated by IM-MS (Paglia et al., 2015). MSE experiments by coupling LC to IM-QTOF were performed for the characterization of non-purified glycolipids extracts from Caenorhabditis elegans (Witting et al., 2021). The multidimensional information collected from chromatographic separation (Retention Time), ion mobility (CCS) and MSE acquisition permits extended lipid annotation. Ion mobility coupled to DIA or DDA approaches paves the way for a rather complete structural annotation and (semi-) quantification of deep lipidome including all minor species, while also significantly improving the quality of both DDA and DIA tandem mass spectra by separating isobaric/isomeric lipid ion species prior to their isolation and fragmentation. The file sizes, from several hundreds of Mo to several Go per injection, the limited software availability for efficiently handling resulting datasets and the difficulty to generate absolute collision cross-sections for highly flexible molecules, such as lipids in the gas phase, strongly restrict the full exploitation of the data acquired by DIA- or DDA-ion mobility.
A last issue in MS/MS is the efficient location of double bonds on lipid structures as dissociations at low-energy are not efficient (Ma et al., 2016). Prior chemical derivatization of carbon-carbon double bonds either directly by a chemical agent (e.g., ozonolysis, epoxidation, hydroxylation) or facilitated by UV irradiation (e.g., Paternò-Büchi reaction) can lead to reaction products that can be further fragmented under low-energy dissociation conditions to pinpoint the location of double bonds (Cao et al., 2018; Harris et al., 2018). Nonetheless, the routine and broad application of these methods remain limited due to the required long reaction time and the inadequate utilization with online LC-MS/MS workflows (Poad et al., 2017). Other fragmentation techniques can be also implemented. Electron-capture Dissociation (ECD) on a divalent metal complex of glycerophospholipids was proposed for double bond location (James et al., 2011). Other electron-activated dissociation (EAD) types have been devised such as Electron-induced dissociation (EID) and Electron-Impact Excitation of Ions from Organics (EIEIO) compatible with singly-charged precursor ions (Baba et al., 2021). Ultraviolet photodissociation (UVPD) activation mode has recently been used to distinguish sn-positional isomers and to determine carbon-carbon double bonds location (Williams et al., 2017). Only few commercial instruments are now equipped with EAD, EID, EIEO, or UVPD while relatively low yields of fragmentation are usually reported for those methods restricting their applications to abundant species even if the fragmentation process is fully compatible with LC peak width.
Even if major progresses in lipidomics have been reported in the last years, improvements of each step of the workflow, from the sample preparation, the chromatography, the MS and MS/MS acquisition to the automated data treatment, are needed to expand lipidome coverage.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study was supported by the MetaboHUB infrastructure (ANR-11-INBS-0010 grant). Marie Méjean thanks the ministère de l’Enseignement supérieur, de la Recherche et de l’Innovation (MESRI) for his PhD fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alcoriza-Balaguer, M. I., Garcia-Cañaveras, J. C., López, A., Conde, I., Juan, O., Carretero, J., et al. (2019). LipidMS: An R package for lipid annotation in untargeted liquid chromatography-data independent acquisition-mass spectrometry lipidomics. Anal. Chem. 91 (1), 836–845. doi:10.1021/acs.analchem.8b03409
Baba, T., Ryumin, P., Duchoslav, E., Chen, K., Chelur, A., Loyd, B., et al. (2021). Dissociation of biomolecules by an intense low-energy electron beam in a high sensitivity time-of-flight mass spectrometer. J. Am. Soc. Mass Spectrom. 32 (8), 1964–1975. doi:10.1021/jasms.0c00425
Barbier Saint Hilaire, P., Rousseau, K., Seyer, A., Dechaumet, S., Damont, A., Junot, C., et al. (2020). Comparative evaluation of data dependent and data independent acquisition workflows implemented on an Orbitrap fusion for untargeted metabolomics. Metabolites 10, 158. doi:10.3390/metabo10040158
Broeckling, C. D., Hoyes, E., Richardson, K., Brown, J. M., and Prenni, J. E. (2018). Comprehensive tandem-mass-spectrometry coverage of complex samples enabled by data-set-dependent acquisition. Anal. Chem. 90, 8020–8027. doi:10.1021/acs.analchem.8b00929
Byrdwell, W. C. (2001). Atmospheric pressure chemical ionization mass spectrometry for analysis of lipids. Lipids 36 (4), 327–346. doi:10.1007/s11745-001-0725-5
Calderón, C., Rubarth, L., Cebo, M., Merfort, I., and Lämmerhofer, M. (2020). Lipid atlas of keratinocytes and betulin effects on its lipidome profiled by comprehensive UHPLC–MS/MS with data independent acquisition using targeted data processing. PROTEOMICS 20 (11), e1900113. doi:10.1002/pmic.201900113
Cao, W., Ma, X., Li, Z., Zhou, X., and Ouyang, Z. (2018). Locating carbon–carbon double bonds in unsaturated phospholipids by epoxidation reaction and tandem mass spectrometry. Anal. Chem. 90 (17), 10286–10292. doi:10.1021/acs.analchem.8b02021
Castro-Perez, J. M., Kamphorst, J., DeGroot, J., Lafeber, F., Goshawk, J., Yu, K., et al. (2010). Comprehensive LC−MSE lipidomic analysis using a shotgun approach and its application to biomarker detection and identification in osteoarthritis patients. J. Proteome Res. 9 (5), 2377–2389. doi:10.1021/pr901094j
Cebo, M., Calderón Castro, C., Schlotterbeck, J., Gawaz, M., Chatterjee, M., and Lämmerhofer, M. (2021). Untargeted UHPLC-ESI-QTOF-MS/MS analysis with targeted feature extraction at precursor and fragment level for profiling of the platelet lipidome with ex vivo thrombin-activation. J. Pharm. Biomed. Anal. 205, 114301. doi:10.1016/j.jpba.2021.114301
Chiu, H.-H., and Kuo, C.-H. (2020). Gas chromatography-mass spectrometry-based analytical strategies for fatty acid analysis in biological samples. J. Food Drug Analysis 28, 60–73. doi:10.1016/j.jfda.2019.10.003
Diedrich, J. K., Pinto, A. F. M., and Yates, J. R. (2013). Energy dependence of HCD on peptide fragmentation: Stepped collisional energy finds the sweet spot. J. Am. Soc. Mass Spectrom. 24 (11), 1690–1699. doi:10.1007/s13361-013-0709-7
Eliuk, S., and Makarov, A. (2015). Evolution of Orbitrap mass spectrometry instrumentation. Annu. Rev. Anal. Chem. 8 (1), 61–80. doi:10.1146/annurev-anchem-071114-040325
Fahy, E., Subramaniam, S., Brown, H. A., Glass, C. K., Merrill, A. H., Murphy, R. C., et al. (2005). A comprehensive classification system for lipids. J. Lipid Res. 46, 839–861. doi:10.1194/jlr.E400004-JLR200
Fahy, E., Subramaniam, S., Murphy, R. C., Nishijima, M., Raetz, C. R. H., Shimizu, T., et al. (2009). Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 50, S9–S14. doi:10.1194/jlr.R800095-JLR200
Fenaille, F., Barbier Saint-Hilaire, P., Rousseau, K., and Junot, C. (2017). Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand? J. Chromatogr. A 1526, 1–12. doi:10.1016/j.chroma.2017.10.043
Gallart-Ayala, H., Courant, F., Severe, S., Antignac, J.-P., Morio, F., Abadie, J., et al. (2013). Versatile lipid profiling by liquid chromatography–high resolution mass spectrometry using all ion fragmentation and polarity switching. Preliminary application for serum samples phenotyping related to canine mammary cancer. Anal. Chim. Acta 796, 75–83. doi:10.1016/j.aca.2013.08.006
Gao, F., McDaniel, J., Chen, E. Y., Rockwell, H. E., Nguyen, C., Lynes, M. D., et al. (2018). Adapted MS/MSALL shotgun lipidomics approach for analysis of cardiolipin molecular species. Lipids 53 (1), 133–142. doi:10.1002/lipd.12004
Gao, F., McDaniel, J., Chen, E. Y., Rockwell, H., Lynes, M. D., Tseng, Y.-H., et al. (2016). Monoacylglycerol analysis using MS/MSALL quadruple time of flight mass spectrometry. Metabolites 6 (3), 25. doi:10.3390/metabo6030025
Gao, Y., Qi, Y., Huang, Y., Li, X., Zhou, L., and Zhao, S. (2021). Lipidomics analysis of the tears in the patients receiving LASIK, FS-lasik, or SBK surgery. Front. Med. 8, 731462. doi:10.3389/fmed.2021.731462
Gillet, L. C., Navarro, P., Tate, S., Röst, H., Selevsek, N., Reiter, L., et al. (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11 (6), O111.016717. doi:10.1074/mcp.O111.016717
Goracci, L., Tortorella, S., Tiberi, P., Pellegrino, R. M., Di Veroli, A., Valeri, A., et al. (2017). Lipostar, a comprehensive platform-neutral cheminformatics tool for lipidomics. Anal. Chem. 89 (11), 6257–6264. doi:10.1021/acs.analchem.7b01259
Guo, J., Shen, S., Xing, S., and Huan, T. (2021). DaDIA: Hybridizing data-dependent and data-independent acquisition modes for generating high-quality metabolomic data. Anal. Chem. 93, 2669–2677. doi:10.1021/acs.analchem.0c05022
Harayama, T., and Riezman, H. (2018). Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 19, 281–296. doi:10.1038/nrm.2017.138
Harris, R. A., May, J. C., Stinson, C. A., Xia, Y., and McLean, J. A. (2018). Determining double bond position in lipids using online ozonolysis coupled to liquid chromatography and ion mobility-mass spectrometry. Anal. Chem. 90 (3), 1915–1924. doi:10.1021/acs.analchem.7b04007
Hartler, J., Triebl, A., Ziegl, A., Trötzmüller, M., Rechberger, G. N., Zeleznik, O. A., et al. (2017). Deciphering lipid structures based on platform-independent decision rules. Nat. Methods 14 (12), 1171–1174. doi:10.1038/nmeth.4470
Hartler, J., Trötzmüller, M., Chitraju, C., Spener, F., Köfeler, H. C., and Thallinger, G. G. (2011). Lipid data analyzer: Unattended identification and quantitation of lipids in LC-MS data. Bioinformatics 27 (4), 572–577. doi:10.1093/bioinformatics/btq699
Höring, M., Ekroos, K., Baker, P. R. S., Connell, L., Stadler, S. C., Burkhardt, R., et al. (2020). Correction of isobaric overlap resulting from sodiated ions in lipidomics. Anal. Chem. 92, 10966–10970. doi:10.1021/acs.analchem.0c02408
Hutchins, P. D., Russell, J. D., and Coon, J. J. (2018). LipiDex: An integrated software package for high-confidence lipid identification. cels 6 (5), 621–625. doi:10.1016/j.cels.2018.03.011
Imbert, L., Gaudin, M., Libong, D., Touboul, D., Abreu, S., Loiseau, P. M., et al. (2012). Comparison of electrospray ionization, atmospheric pressure chemical ionization and atmospheric pressure photoionization for a lipidomic analysis of Leishmania donovani. J. Chromatogr. A 1242, 75–83. doi:10.1016/j.chroma.2012.04.035
James, P. F., Perugini, M. A., and O’Hair, R. A. J. (2011). Electron capture dissociation of complexes of diacylglycerophosphocholine and divalent metal ions: Competition between charge reduction and radical induced phospholipid fragmentation. J. Am. Soc. Mass Spectrom. 19, 978–986. doi:10.1016/j.jasms.2008.03.006
Kirkwood, K. I., Pratt, B. S., Shulman, N., Tamura, K., MacCoss, M. J., MacLean, B. X., et al. (2022). Utilizing Skyline to analyze lipidomics data containing liquid chromatography, ion mobility spectrometry and mass spectrometry dimensions. Nat. Protoc. 17, 2415–2430. doi:10.1038/s41596-022-00714-6
Kitata, R. B., Yang, J.-C., and Chen, Y.-J. (2021). Advances in data-independent acquisition mass spectrometry towards comprehensive digital proteome landscape. Mass Spectrom. Rev., e21781. doi:10.1002/mas.21781
Kochen, M. A., Chambers, M. C., Holman, J. D., Nesvizhskii, A. I., Weintraub, S. T., Belisle, J. T., et al. (2016). Greazy: Open-Source software for automated phospholipid tandem mass spectrometry identification. Anal. Chem. 88, 5733–5741. doi:10.1021/acs.analchem.6b00021
Koelmel, J. P., Kroeger, N. M., Gill, E. L., Ulmer, C. Z., Bowden, J. A., Patterson, R. E., et al. (2017). Expanding lipidome coverage using LC-MS/MS data-dependent acquisition with automated exclusion list generation. J. Am. Soc. Mass Spectrom. 28, 908–917. doi:10.1007/s13361-017-1608-0
Li, H., Cai, Y., Guo, Y., Chen, F., and Zhu, Z.-J. (2016). MetDIA: Targeted metabolite extraction of multiplexed MS/MS spectra generated by data-independent acquisition. Anal. Chem. 88, 8757–8764. doi:10.1021/acs.analchem.6b02122
Liebisch, G., Fahy, E., Aoki, J., Dennis, E. A., Durand, T., Ejsing, C. S., et al. (2020). Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 61 (12), 1539–1555. doi:10.1194/jlr.S120001025
Ma, X., Chong, L., Tian, R., Shi, R., Hu, T. Y., Ouyang, Z., et al. (2016). Identification and quantitation of lipid C=C location isomers: A shotgun lipidomics approach enabled by photochemical reaction. Proc. Natl. Acad. Sci. 113 (10), 2573–2578. doi:10.1073/pnas.1523356113
Meier, F., Geyer, P. E., Virreira Winter, S., Cox, J., and Mann, M. (2018). BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 15, 440–448. doi:10.1038/s41592-018-0003-5
Murphy, R. C. (2015). Tandem mass spectrometry of lipids: Molecular analysis of complex lipids. Cambridge: Royal Society of Chemistry.
Neumann, S., Thum, A., and Böttcher, C. (2013). Nearline acquisition and processing of liquid chromatography-tandem mass spectrometry data. Metabolomics 9, 84–91. doi:10.1007/s11306-012-0401-0
Paglia, G., Angel, P., Williams, J. P., Richardson, K., Olivos, H. J., Thompson, J. W., et al. (2015). Ion mobility-derived collision cross section as an additional measure for lipid fingerprinting and identification. Anal. Chem. 87, 1137–1144. doi:10.1021/ac503715v
Perez de Souza, L., Alseekh, S., Scossa, F., and Fernie, A. R. (2021). Ultra-high-performance liquid chromatography high-resolution mass spectrometry variants for metabolomics research. Nat. Methods 18 (7), 733–746. doi:10.1038/s41592-021-01116-4
Plumb, R. S., Johnson, K. A., Rainville, P., Smith, B. W., Wilson, I. D., Castro-Perez, J. M., et al. (2006). UPLC/MSE; a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun. Mass Spectrom. 20 (13), 1989–1994. doi:10.1002/rcm.2550
Poad, B. L. J., Green, M. R., Kirk, J. M., Tomczyk, N., Mitchell, T. W., and Blanksby, S. J. (2017). High-Pressure ozone-induced dissociation for lipid structure elucidation on fast chromatographic timescales. Anal. Chem. 89 (7), 4223–4229. doi:10.1021/acs.analchem.7b00268
Quehenberger, O., Armando, A. M., Brown, A. H., Milne, S. B., Myers, D. S., Merrill, A. H., et al. (2010). Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 51, 3299–3305. doi:10.1194/jlr.M009449
Rustam, Y. H., and Reid, G. E. (2018). Analytical challenges and recent advances in mass spectrometry based lipidomics. Anal. Chem. 90 (1), 374–397. doi:10.1021/acs.analchem.7b04836
Schwudke, D., Oegema, J., Burton, L., Entchev, E., Hannich, J. T., Ejsing, C. S., et al. (2006). Lipid profiling by multiple precursor and neutral loss scanning driven by the data-dependent acquisition. Anal. Chem. 78 (2), 585–595. doi:10.1021/ac051605m
Schwudke, D., Schuhmann, K., Herzog, R., Bornstein, S. R., and Shevchenko, A. (2011). Shotgun lipidomics on high resolution mass spectrometers. Cold Spring Harb. Perspect. Biol. 3, a004614. doi:10.1101/cshperspect.a004614
Simons, B., Kauhanen, D., Sylvänne, T., Tarasov, K., Duchoslav, E., and Ekroos, K. (2012). Shotgun lipidomics by sequential precursor ion fragmentation on a hybrid quadrupole time-of-flight mass spectrometer. Metabolites 2 (1), 195–213. doi:10.3390/metabo2010195
Stagliano, M. C., DeKeyser, J. G., Omiecinski, C. J., and Jones, A. D. (2010). Bioassay-directed fractionation for discovery of bioactive neutral lipids guided by relative mass defect filtering and multiplexed collision-induced dissociation. Rapid Commun. Mass Spectrom. 24 (24), 3578–3584. doi:10.1002/rcm.4796
Stancliffe, E., Schwaiger-Haber, M., Sindelar, M., and Patti, G. J. (2021). DecoID improves identification rates in metabolomics through database-assisted MS/MS deconvolution. Nat. Methods 18, 779–787. doi:10.1038/s41592-021-01195-3
Tada, I., Chaleckis, R., Tsugawa, H., Meister, I., Zhang, P., Lazarinis, N., et al. (2020). Correlation-based deconvolution (CorrDec) to generate high-quality MS2 spectra from data-independent acquisition in multisample studies. Anal. Chem. 92 (16), 11310–11317. doi:10.1021/acs.analchem.0c01980
Tsugawa, H., Cajka, T., Kind, T., Ma, Y., Higgins, B., Ikeda, K., et al. (2015). MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12 (6), 523–526. doi:10.1038/nmeth.3393
Tsugawa, H., Satoh, A., Uchino, H., Cajka, T., Arita, M., and Arita, M. (2019). Mass spectrometry data repository enhances novel metabolite discoveries with advances in computational metabolomics. Metabolites 9, 119. doi:10.3390/metabo9060119
Vasilopoulou, C. G., Sulek, K., Brunner, A.-D., Meitei, N. S., Schweiger-Hufnagel, U., Meyer, S. W., et al. (2020). Trapped ion mobility spectrometry and PASEF enable in-depth lipidomics from minimal sample amounts. Nat. Commun. 11 (1), 331. doi:10.1038/s41467-019-14044-x
Venable, J. D., Dong, M.-Q., Wohlschlegel, J., Dillin, A., and Yates, J. R. (2004). Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 1 (1), 39–45. doi:10.1038/nmeth705
Ventura, G., Bianco, M., Calvano, C. D., Losito, I., and Cataldi, T. R. I. (2020a). HILIC-ESI-FTMS with all ion fragmentation (AIF) scans as a tool for fast lipidome investigations. Molecules 25 (10), 2310. doi:10.3390/molecules25102310
Ventura, G., Calvano, C. D., Porcelli, V., Palmieri, L., De Giacomo, A., Xu, Y., et al. (2020b). Phospholipidomics of peripheral blood mononuclear cells (PBMCs): The tricky case of children with autism spectrum disorder (ASD) and their healthy siblings. Anal. Bioanal. Chem. 412 (25), 6859–6874. doi:10.1007/s00216-020-02817-z
Wang, H., Wang, H., Wang, X., Xu, X., Hu, Y., Li, X., et al. (2022). A novel hybrid scan approach enabling the ion-mobility separation and the alternate data-dependent and data-independent acquisitions (HDDIDDA): Its combination with off-line two-dimensional liquid chromatography for comprehensively characterizing the multicomponents from Compound Danshen Dripping Pill. Anal. Chim. Acta 1193, 339320. doi:10.1016/j.aca.2021.339320
Wenk, M. R. (2010). Lipidomics: New tools and applications. Cell 143 (6), 888–895. doi:10.1016/j.cell.2010.11.033
Wenk, M. R. (2005). The emerging field of lipidomics. Nat. Rev. Drug Discov. 4 (7), 594–610. doi:10.1038/nrd1776
Williams, P. E., Klein, D. R., Greer, S. M., and Brodbelt, J. S. (2017). Pinpointing double bond and sn-positions in glycerophospholipids via hybrid 193 nm ultraviolet photodissociation (UVPD) mass spectrometry. J. Am. Chem. Soc. 139 (44), 15681–15690. doi:10.1021/jacs.7b06416
Witting, M., Schmidt, U., and Knoelker, H.-J. (2021). UHPLC-IM-Q-ToFMS analysis of maradolipids, found exclusively in Caenorhabditis elegans dauer larvae. Anal. Bioanal. Chem. 413 (8), 2091–2102. doi:10.1007/s00216-021-03172-3
Yang, K., and Han, X. (2016). Lipidomics: Techniques, applications, and outcomes related to biomedical sciences. Trends Biochem. Sci. 41, 954–969. doi:10.1016/j.tibs.2016.08.010
Yin, Y., Wang, R., Cai, Y., Wang, Z., and Zhu, Z.-J. (2019). DecoMetDIA: Deconvolution of multiplexed MS/MS spectra for metabolite identification in SWATH-MS-based untargeted metabolomics. Anal. Chem. 91 (18), 11897–11904. doi:10.1021/acs.analchem.9b02655
Zha, H., Cai, Y., Yin, Y., Wang, Z., Li, K., and Zhu, Z.-J. (2018). SWATHtoMRM: Development of high-coverage targeted metabolomics method using SWATH technology for biomarker discovery. Anal. Chem. 90, 4062–4070. doi:10.1021/acs.analchem.7b05318
Zhang, Y., Bilbao, A., Bruderer, T., Luban, J., Strambio-De-Castillia, C., Lisacek, F., et al. (2015). The use of variable Q1 isolation windows improves selectivity in LC–SWATH–MS acquisition. J. Proteome Res. 14 (10), 4359–4371. doi:10.1021/acs.jproteome.5b00543
Zhou, Z., Shen, X., Chen, X., Tu, J., Xiong, X., and Zhu, Z.-J. (2019). LipidIMMS analyzer: Integrating multi-dimensional information to support lipid identification in ion mobility—mass spectrometry based lipidomics. Bioinformatics 35 (4), 698–700. doi:10.1093/bioinformatics/bty661
Züllig, T., and Köfeler, H. C. (2021). High resolution mass spectrometry in lipidomics. Mass Spectrom. Rev. 40 (3), 162–176. doi:10.1002/mas.21627
Keywords: lipidomics, data-dependent acquisition, data-independent acquisition, high-resolution mass spectrometry, lipid annotation
Citation: Valmori M, Marie V, Fenaille F, Colsch B and Touboul D (2023) Recent methodological developments in data-dependent analysis and data-independent analysis workflows for exhaustive lipidome coverage. Front. Anal. Sci. 3:1118742. doi: 10.3389/frans.2023.1118742
Received: 07 December 2022; Accepted: 19 January 2023;
Published: 10 February 2023.
Edited by:
Sophie Ayciriex, Université Claude Bernard Lyon 1, FranceReviewed by:
Yohann Clement, Université de Lyon, FranceCopyright © 2023 Valmori, Marie, Fenaille, Colsch and Touboul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benoit Colsch, YmVub2l0LmNvbHNjaEBjZWEuZnI=; David Touboul, ZGF2aWQudG91Ym91bEBjbnJzLmZy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.