 Tyler C. Cropley
Tyler C. Cropley Mengqi Chai1,2†
Mengqi Chai1,2†- 1Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL, United States
- 2Department of Chemistry, Washington University in St. Louis, Saint-Louis, MO, United States
- 3Institute of Molecular Biophysics, Florida State University, Tallahassee, FL, United States
Cellular processes are usually carried out collectively by the entirety of all proteins present in a biological cell, i.e., the proteome. Mass spectrometry-based methods have proven particularly successful in identifying and quantifying the constituent proteins of proteomes, including different molecular forms of a protein. Nevertheless, protein sequences alone do not reveal the function or dysfunction of the identified proteins. A straightforward way to assign function or dysfunction to proteins is characterization of their structures and dynamics. However, a method capable to characterize detailed structures of proteins and protein complexes in a large-scale, systematic manner within the context of cellular processes does not yet exist. Here, we discuss the potential of tandem-ion mobility/mass spectrometry (tandem-IM/MS) methods to provide such ability. We highlight the capability of these methods using two case studies on the protein systems ubiquitin and avidin using the tandem-TIMS/MS technology developed in our laboratory and discuss these results in the context of other developments in the broader field of tandem-IM/MS.
Introduction
This Perspective discusses the potential of tandem-ion mobility spectrometry/mass spectrometry (tandem-IM/MS) methods for the emerging field of Structural Proteomics. Tandem-IM/MS methods (Figure 1A) conduct two or more ion mobility separations in series, either tandem-in-space or tandem-in-time, prior to mass analysis (Tang et al., 2005; Koeniger et al., 2006c; Kurulugama et al., 2009; Simon et al., 2015; Allen et al., 2017; Poyer et al., 2017; Liu et al., 2018; Giles et al., 2019; Eldrid and Thalassinos, 2020; Eldrid et al., 2021; Liu et al., 2021). These methods also allow selection of mobility-separated ions and their energetic activation in-between individual ion mobility separation steps (Figure 1A). Hence, tandem-IM methods can be seen in analogy to tandem-MS with the difference that tandem-IM separates ions by differences in their ion mobilities instead of their mass-to-charge ratios. Subsequently, the mobility-separated compounds can be energetically-activated and characterized by the mobilities and m/z of the produced ions. We present two examples showcasing the ability of tandem-IM/MS methods to disentangle structures of otherwise unresolved protein systems to underscore the potential of tandem-IM/MS to analyze heterogenous samples such as those encountered in the field of Structural Proteomics.
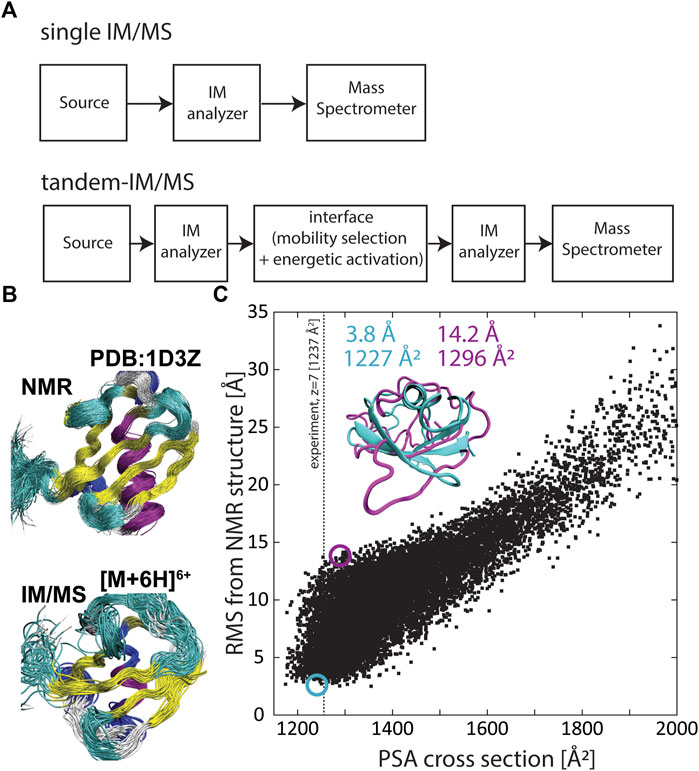
FIGURE 1. (A) Generalized schematics of a conventional IM/MS instrument coupling a single IM analyzer with a mass spectrometer (top) and a tandem-IM/MS instrument coupling two IM analyzers with a mass spectrometer (bottom). (B) The solution ensemble (PDB 1D3Z) (top) and an ensemble of structures predicted by the structural relaxation approximation (SRA) that reflect the “native-like” structure of ubiquitin (bottom). (C) Correlation between root-mean-square deviation (RMSD) and calculated cross sections for 10,000 conformations of the small protein ubiquitin. RMSD was calculated with respect to PDB 1D3Z and the projection superposition approximation (PSA) was used to calculate the cross section for each ubiquitin structure. Adapted from (Bleiholder and Liu, 2019) with permission from the American Chemical Society.
Proteins rarely exert their biological function in isolation. Instead, cellular processes are usually carried out collectively by the proteome, i.e., the entirety of all proteins present in a biological cell (Aebersold and Mann, 2016). Hence, significant efforts have been devoted to developing methods that enable large-scale, quantitative characterization of the proteome. Mass spectrometry-based methods have proven particularly successful in identifying and quantifying the constituent proteins of proteomes, including different molecular forms of a protein (“proteoforms”) produced via mechanisms such as alternative splicing of transcripts and post-translational modification of proteins (Yates and Kelleher, 2013; Catherman et al., 2014; Meier et al., 2015; Li et al., 2016; Yang et al., 2016; Aebersold et al., 2018; Smith and Kelleher, 2018; Meier et al., 2020).
Nevertheless, protein sequences alone do not reveal the function or dysfunction of the identified proteins within the context of cellular processes. In the absence of annotated gene products, which in principle applies to all newly identified proteoforms, functional information of a protein can be obtained by identifying the interaction network (“interactome”) with other proteins (Perkins et al., 2010; Havugimana et al., 2012; Mendoza et al., 2012). Hence, systematic characterization of protein-protein interaction networks, and their alterations in the context of disease phenotypes (Vidal et al., 2011; Richards et al., 2021), is one avenue to systematically introduce functional information into proteomic analyses.
Another approach to assigning function or dysfunction to proteins is characterization of their structure and dynamics. This is so because the biological activity of proteins arises from their structural heterogeneity and dynamic flexibility (Frauenfelder et al., 1991; Frauenfelder et al., 2003), described by an energy landscape comprising a hierarchy of conformational states and motional transitions between these states (Frauenfelder et al., 1991; Onuchic and Wolynes, 2004; Henzler-Wildman et al., 2007). Indeed, protein-protein interaction networks are physically mediated via (transient) formation of protein complexes (Marsh and Teichmann, 2015). The structure and dynamics of these protein complexes can be perturbed by e.g. presence of altered proteoforms which can lead to perturbation of protein-protein interaction networks and therefore result in disease phenotypes.
These above considerations underline that characterizing structures of proteins and protein complexes in a large-scale, systematic manner within the context of cellular processes can be useful to assign function or dysfunction to protein sequences determined in proteomic experiments. Traditional structural biology methods such as x-ray crystallography, NMR spectroscopy, or cryogenic electron microscopy have been applied to determine structures of biological systems (Tzeng and Kalodimos, 2012; Ho et al., 2020; Günther et al., 2021), but these methods work best with purified samples and are limited in throughput. By contrast, MS-based methods are well-suited to handle the heterogeneity arising from presence of post-translational modifications and exhibit sufficient sensitivity, sample-throughput, and dynamic range to enable large-scale systematic measurements of proteomes (Benesch et al., 2007; Kondrat et al., 2015; Meier et al., 2015; Meier et al., 2020). Moreover, traditional MS-based methods can provide indirect structural information via measurements of mass-to-charge ratios of labelled or digested protein components (Chea et al., 2021). Furthermore, hybrid ion mobility/mass spectrometry (IM/MS, Figure 1A) methods characterize atomic structures of proteins and protein complexes via their orientationally-averaged collision cross sections. Electrospray ionization (ESI) coupled to IM/MS enables protein and protein complexes to be gently transferred into the gas phase without significant structural rearrangement or dissociation (Ruotolo et al., 2005; Koeniger et al., 2006a; Breuker and McLafferty, 2008; Jurneczko and Barran, 2011; Wyttenbach and Bowers, 2011; Zhou et al., 2014; Bleiholder and Liu, 2019; Rolland and Prell, 2019). When energetic activation throughout the measurement is minimized, the structures measured by IM/MS can be similar to the structures adopted in solution. However, it is commonly accepted that transfer into the gas phase can result in compaction and some restructuring of the ions, and hence referred to as “native-like” (Figure 1B). Thus, at least in principle, IM/MS methods bear the potential to systematically characterize structures of proteins present in proteomic samples.
However, a major limitation of IM/MS methods to characterize structurally flexible molecules such as proteins is that the measured collision cross section is a structurally ambiguous quantity (Voronina et al., 2016; Bleiholder and Liu, 2019). Because a protein can adopt many three-dimensional conformations that have the same two-dimensional cross-sectional area, it is not generally possible to reliably characterize protein structures from only measuring their collision cross sections (Bleiholder and Liu, 2019). Figure 1C exemplifies the underlying problematic using the small protein ubiquitin. Here the root-mean-square deviation (RMSD) from the native protein structure is correlated with the computed cross sections for a set of 10,000 ubiquitin conformations. The plot underlines that it is not possible to unambiguously assign a specific structure to an experimentally measured cross section because typically many distinct protein structures have a cross section consistent with the measured cross section. Further, because protein side chain orientations considerably influence the cross section, it is also possible to assign the “native” backbone conformation to a wide range of measured cross sections—in the case of ubiquitin from ∼1,180 Å2 to ∼1,350 Å2. This structural ambiguity takes on increased relevance with increasing protein size or with the complexity of protein assemblies, because here the number of possible conformations and isomers increases. Hence, the structural ambiguity of collisional cross sections limits the fidelity by which IM/MS characterizes structures of proteins and protein complexes.
One approach to overcoming this ambiguity in characterizing protein structures is that of collisional-induced unfolding (CIU). CIU experiments (Shelimov et al., 1997; Shi et al., 2014; Zhong et al., 2014) measure how protein cross sections change as the protein ions unfold in the gas phase due to vibrational activation. Hence, such measurements characterize the energy barriers associated with the unfolding process (Clemmer and Jarrold, 1997; Zhong et al., 2014). Because these unfolding energy barriers arise from breaking of non-covalent bonds (i.e., hydrogen bonds or salt-bridges), CIU measurements thus characterize protein conformations in terms of differences in their hydrogen bonds or salt-bridges. For example, Eschweiler et al. (2017) showed that the amino acid sequence of homologous serum albumins affects the stability of unfolding intermediates, suggesting differences in their initial native-like conformation despite similar cross sections. Analogous results were observed for the binding of thyroxine to tetrameric transthyretin (TTR) (Hyung et al., 2009). While apo-TTR and its singly- and doubly liganded forms have nearly identical cross sections, apo-TTR is significantly more susceptible to energetic activation when compared to the liganded forms. These results underscore that differences in the unfolding susceptibilities or unfolding pathways can help characterize structural differences that may not be revealed by the collision cross section of the protein or protein complex alone.
However, conventional hybrid IM/MS instruments contain only one IM analyzer (Figure 1A). Hence, when analyzing complex samples such as mixtures of proteins or protein complexes, these methods are restricted to either separate the protein analytes by their mobilities without performing collisional-unfolding or, alternatively, to perform CIU measurements without first separating the protein isomers contained in the sample. By contrast, tandem-IM/MS methods contain two or more IM separation stages coupled by an interface that allows selection of mobility-separated ions and their energetic activation (Figure 1A). Hence, tandem-IM/MS methods can perform both tasks, that is, to separate the mixture of proteins by the mobilities of their individual protein species in the first IM device and to subsequently perform CIU of the mobility-separated protein species using the second IM device. In the following, we showcase the ability of tandem-IM/MS methods to characterize structures of proteins and protein complexes from a mixture of otherwise unresolved species to underscoring the potential of these methods for the study of complex, heterogenous samples.
Illustrative Example 1. Differentiation of unresolved protein conformers with identical cross sections
Our first example discusses the ability to characterize, from a distribution of unresolved protein conformations, structurally different protein conformations that have the same collision cross section by mobility-selected CIU.
Figure 2A shows the ion mobility spectrum of charge state 7 + recorded for the small protein ubiquitin (bovine erythrocytes) from native conditions on the tandem-trapped ion mobility spectrometer/mass spectrometer (tTIMS/MS) developed in our laboratory (Liu et al., 2018) operating under “soft” conditions. The ion mobility spectrum is dominated by a compact peak centered at 1,237 Å2, which had previously been associated with a native-like ubiquitin structure (Koeniger et al., 2006b; Wyttenbach and Bowers, 2011; May et al., 2018; Bleiholder and Liu, 2019). Furthermore, the feature is broad which was shown to arise from multiple protein conformations that are metastable on the experimental time-scale of ∼100–200 ms (Koeniger et al., 2006a; Koeniger et al., 2006b). Figure 2B shows the spectrum of the same charge state but after collisional activation prior to the first ion mobility separation as described elsewhere (Liu et al., 2016). While collisional activation leads to formation of two extended features at 1,515 Å2 and 1,806 Å2, respectively, a compact feature with mean cross section 1,237 Å2 remains abundant. A conventional IM/MS instrument with a single IM analyzer would suggest these compact features refer to the same protein structures because of the similarities of their cross sections.
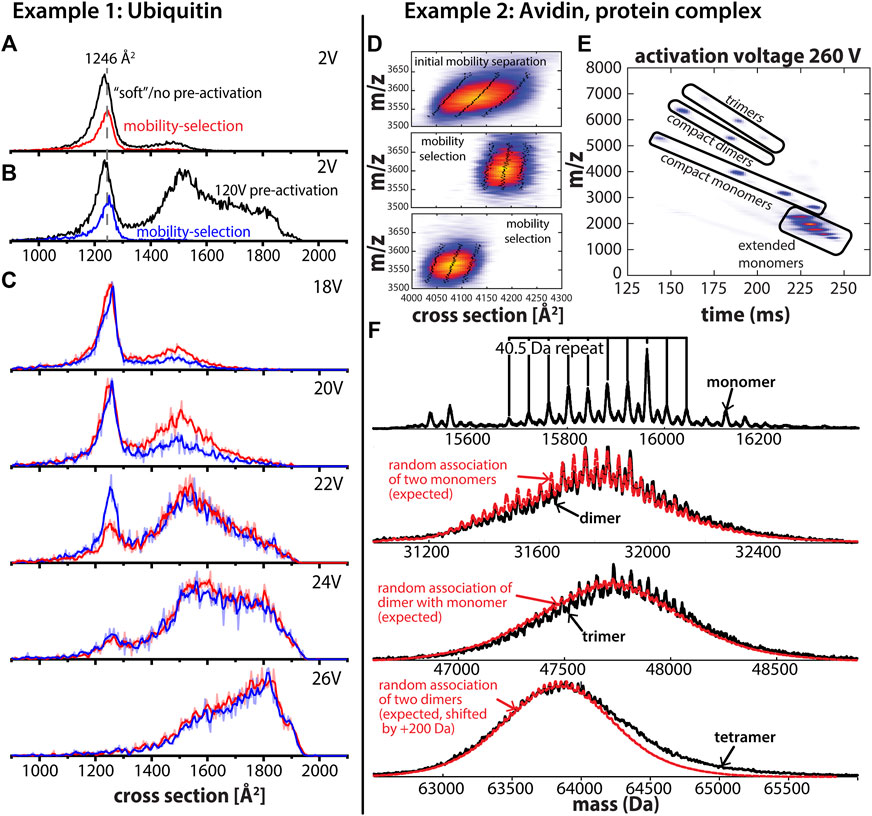
FIGURE 2. (A) Cross section distribution for ubiquitin charge state 7 + using “soft” instrument settings without mobility selection (black trace) and with mobility selection of a subset of structures with a cross section of 1,246 Å2 (red trace). (B) Cross section distribution after 120V activation in the entrance funnel of the first TIMS analyzer (“pre-activation”) without mobility selection (black trace) and with mobility selection of a subset of structures with a cross section of 1,246 Å2 (blue trace). (C) Cross section distributions after collisional activation in the interface separating the two TIMS analyzers. Mobility-selected structures from “soft” measurements (red trace) and mobility-selected structures from “pre-activated” experiments (blue trace). The comparison of their unfolding behavior shows that the selected structures differ despite identical cross sections. (D) The asymmetric peak of avidin tetramer charge state 18 + after the first stage of mobility separation and two subsets of the asymmetric peak of the avidin tetramer after mobility selection and subsequent mobility separation. Mobility selection enables fractionation of the avidin tetramer peak, which is a heterogenous mixture of (glyco) proteoforms as shown in (F). The dotted lines represent the peak width and the full width half maximum (FWHM), respectively. (E) CID of mobility-selected avidin tetramers produces monomer, dimer, and trimer subunits which are separated in the second TIMS analyzer to simplify mass spectral analysis. Contrary to conventional IM/MS instruments, ions are parked at an initial position determined by the force balance between the electric field and frictional force from the flowing buffer gas. As the electric field decreases, ions with a lower mobility elute first and ions with increasing mobility follow. (F) Charge-deconvolved mass spectrum obtained for avidin monomers reveal multiple avidin glycoforms. Comparison of experimental mass spectra (black traces) for dimer, trimer, and tetramer assembly states to those expected for random assemblies of the monomer glycoforms (red trace) indicates that avidin tetramers are most likely composed of (almost) random glycoforms combinations. Adapted from (Liu et al., 2020) with permission from the American Chemical Society.
The question we are now pursuing is whether subsets of the compact feature in Figures 2A, B with the same cross sections also have the same structure. This question is difficult to address using conventional, hybrid IM/MS instruments but straight-forward using tandem-IM/MS instruments. To exemplify, we first selected a subset of the compact feature with a cross section of 1,246 Å2 after elution from the first ion mobility analyzer for both conditions (Figures 2A, B). Subsequently, we collisionally activated the selected ions and performed mobility-analysis in the second IM analyzer (TIMS-2). The resulting spectra obtained upon collisional-activation are shown in Figure 2C which reveal significant differences in the susceptibility to unfold for the two selected ion populations. Specifically, the data show the pre-activated ion population (Figure 2B) is less susceptible to unfolding by collisional-activation than the subset selected from the “soft” experiment (Figure 2A). Note that such behavior is in line with observations reported from (Koeniger et al., 2006b) and indicates that some annealing of the protein structures has taken place upon collisional activation. Hence, the tandem-IM/MS measurements discussed in Figure 2 reveal in a straight-forward manner that the mobility-selected subsets of the broad compact feature in Figures 2A, B differ in their structures despite having the same cross section. By contrast, conventional IM/MS instruments containing only a single IM analyzer would incorrectly interpret the compact features in both spectra as the same conformation.
Illustrative Example 2. Glycoforms of the glycoprotein complex avidin
Our second example discusses the ability to characterize different structural levels of avidin, a homo-tetrameric protein complex of a 128-amino acid residue protein extracted from egg white with a single glycosylation site at Asn17 (DeLange, 1970). Several glycoforms and sequence variants have been reported for avidin (Bruch and White, 1982; Oliver et al., 1996; Yang and Orlando, 1996).
To probe if the avidin tetramer exists as different combinations of glycoforms, we disassembled the intact avidin tetramers into their subunits by means of collision-induced dissociation (CID) of mobility-selected avidin tetramers in the interface region of the tandem-TIMS instrument (Liu et al., 2020). Mobility selection, only possible with tandem-IM/MS methods, enables isolation of a subpopulation of proteoforms from a heterogenous mixture of avidin homotetramers (Figure 2D). Figure 2E shows that CID of the avidin tetramer in the interface region of tandem-TIMS/MS produces various charge states of monomeric, dimeric, as well as trimeric subunits of the avidin tetramer that can be mobility-separated in the second TIMS analyzer of tandem-TIMS/MS. Further, Figure 2E underlines that the second IM separation after CID is needed to simplify mass spectral analysis by separating different assembly states with the same mass-to-charge ratio. Moreover, as discussed (Liu et al., 2020), the data reveal that the avidin tetramers can be disassembled into their subunits without noticeable cleavage of the avidin backbone or glycan components. Figure 2F shows the recorded charge-deconvolved mass spectra for the identified avidin monomer, dimer, and trimer subunits produced from CID. As described (Liu et al., 2020), all spectra display a ∼40.5 Da repeat pattern corresponding to the mass differences between an N-acetyl glucosamine (203.20 Da) and a mannose residue (162.10 Da) of the glycans on the corresponding avidin protomers. The deconvolved monomer spectrum shows presence of multiple glycoforms of the avidin monomer. Figure 2E further compares the deconvolved mass spectra to those expected for random combinations of avidin monomer glycoforms, which reveals a strong agreement between the experimental dimer, trimer, and tetramer spectra and those expected for their random-assembly from monomer glycoforms. This agreement between the experimental and expected spectra holds with respect to both the position and the width of the mass spectral envelope as well as with respect to the 40.5 Da repeat between the peaks of the various glycoforms. Hence, these results allowed us to conclude in a straight-forward manner that avidin assemblies are most likely composed of (almost) random glycoforms combinations (Liu et al., 2020). This example thus highlights the ability of tandem-IM/MS methods to investigate samples composed of different proteoforms.
Discussion
The case studies discussed here demonstrate the ability of tandem-IM/MS methods to characterize subsets of structures from a heterogenous population of different conformations (case 1) and composition of specific protein species from a heterogenous sample of different proteoforms (case 2), even when these cannot be separated in the first ion mobility dimension. Hence, these examples highlight the ability of tandem-IM/MS methods to characterize protein and protein complexes otherwise hidden among unresolved features of ion mobility/mass spectra and thus underline the power of tandem-IM/MS methods to characterize protein structures from heterogenous samples.
The measurements on ubiquitin and avidin discussed above were conducted on the tandem-TIMS/MS instruments developed in our own laboratory (Liu et al., 2018; Kirk et al., 2019; Bleiholder et al., 2020; Liu et al., 2021), for which a recent review is available (Liu et al., 2022). There are, however, multiple other, currently ongoing efforts to develop instruments with the capability to carry out multiple ion mobility separations, selection, and activation steps in series. These instruments include tandem-drift tube instruments (Koeniger et al., 2006c; Gaye et al., 2015), cyclic travelling wave ion mobility instruments (Ollivier et al., 2021), and also tandem-ion mobility spectrometers based on the structures for lossless ion manipulations (SLIM) technology (Allen et al., 2017; Bansal et al., 2020). Particularly interesting, in our view, is the coupling of tandem-IM/MS instruments with IR spectroscopy because it adds another dimension to the structural characterization of the measured ions in addition to their ion mobilities and m/z (Bansal et al., 2020). The case studies discussed in this Perspective highlight the ability of such tandem-IM/MS-based methods to reveal structure and composition of proteins and protein complexes that remain “hidden” to conventional IM/MS-based technologies. For these reasons, tandem-IM/MS methods will, in our opinion, highly likely contribute significantly to the field of Structural Proteomics.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Institutes of Health under grant R01GM135682 (CB) and by the National Science Foundation under grant CHE-1654608 (CB).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aebersold, R., Agar, J. N., Amster, I. J., Baker, M. S., Bertozzi, C. R., Boja, E. S., et al. (2018). How many human proteoforms are there? Nat. Chem. Biol. 14, 206–214. doi:10.1038/nchembio.2576
Aebersold, R., and Mann, M. (2016). Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355. doi:10.1038/nature19949
Allen, S. J., Eaton, R. M., and Bush, M. F. (2017). Structural dynamics of native-like ions in the gas phase: Results from tandem ion mobility of cytochrome c. Anal. Chem. 89, 7527–7534. doi:10.1021/acs.analchem.7b01234
Bansal, P., Yatsyna, V., Abikhodr, A. H., Warnke, S., Ben Faleh, A., Yalovenko, N., et al. (2020). Using SLIM-based IMS-IMS together with cryogenic infrared spectroscopy for glycan analysis. Anal. Chem. 92, 9079–9085. doi:10.1021/acs.analchem.0c01265
Benesch, J. L. P., Ruotolo, B. T., Simmons, D. A., and Robinson, C. V. (2007). Protein complexes in the gas phase: Technology for structural genomics and proteomics. Chem. Rev. 107, 3544–3567. doi:10.1021/cr068289b
Bleiholder, C., Liu, F. C., and Chai, M. (2020). Comment on effective temperature and structural rearrangement in trapped ion mobility spectrometry. Anal. Chem. 92, 16329–16333. doi:10.1021/acs.analchem.0c02052
Bleiholder, C., and Liu, F. C. (2019). Structure relaxation approximation (SRA) for elucidation of protein structures from ion mobility measurements. J. Phys. Chem. B 123, 2756–2769. doi:10.1021/acs.jpcb.8b11818
Breuker, K., and McLafferty, F. W. (2008). Stepwise evolution of protein native structure with electrospray into the gas phase, 10-12 to 102 s. Proc. Natl. Acad. Sci. 105, 18145–18152. doi:10.1073/pnas.0807005105
Bruch, R. C., and White, H. B. (1982). Compositional and structural heterogeneity of avidin glycopeptides. Biochemistry 21, 5334–5341. doi:10.1021/bi00264a033
Catherman, A. D., Skinner, O. S., and Kelleher, N. L. (2014). Top down proteomics: Facts and perspectives. Biochem. Biophys. Res. Commun. 445, 683–693. doi:10.1016/j.bbrc.2014.02.041
Chea, E. E., Prakash, A., and Jones, L. M. (2021). The utilization of the search engine, Bolt, to decrease search time and increase peptide identifications in hydroxyl radical protein footprinting-based workflows. Proteomics 21, 2000295. doi:10.1002/pmic.202000295
Clemmer, D. E., and Jarrold, M. F. (1997). Ion mobility measurements and their applications to clusters and biomolecules. J. Mass Spectrom. 32, 577–592. doi:10.1002/(sici)1096-9888(199706)32:6<577::aid-jms530>3.0.co;2-4
DeLange, R. (1970). Egg white avidin. J. Biol. Chem. 245, 907–916. doi:10.1016/s0021-9258(18)63268-5
Eldrid, C., Ben-Younis, A., Ujma, J., Britt, H., Cragnolini, T., Kalfas, S., et al. (2021). Cyclic ion mobility–collision activation experiments elucidate protein behavior in the gas phase. J. Am. Soc. Mass Spectrom. 32, 1545–1552. doi:10.1021/jasms.1c00018
Eldrid, C., and Thalassinos, K. (2020). Developments in tandem ion mobility mass spectrometry. Biochem. Soc. Trans. 48, 2457–2466. doi:10.1042/BST20190788
Eschweiler, J. D., Martini, R. M., and Ruotolo, B. T. (2017). Chemical probes and engineered constructs reveal a detailed unfolding mechanism for a solvent-free multidomain protein. J. Am. Chem. Soc. 139, 534–540. doi:10.1021/jacs.6b11678
Frauenfelder, H., McMahon, B. H., and Fenimore, P. W. (2003). Myoglobin: The hydrogen atom of biology and a paradigm of complexity. Proc. Natl. Acad. Sci. 100, 8615–8617. doi:10.1073/pnas.1633688100
Frauenfelder, H., Sligar, S. G., and Wolynes, P. G. (1991). The energy landscapes and motions of proteins. Science 254, 1598–1603. doi:10.1126/science.1749933
Gaye, M. M., Kurulugama, R., and Clemmer, D. E. (2015). Investigating carbohydrate isomers by IMS-CID-IMS-MS: Precursor and fragment ion cross-sections. Analyst 140, 6922–6932. doi:10.1039/c5an00840a
Giles, K., Ujma, J., Wildgoose, J., Pringle, S., Richardson, K., Langridge, D., et al. (2019). A cyclic ion mobility-mass spectrometry system. Anal. Chem. 91, 8564–8573. doi:10.1021/acs.analchem.9b01838
Günther, S., Reinke, P. Y. A., Fernández-García, Y., Lieske, J., Lane, T. J., Ginn, H. M., et al. (2021). X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 372, 642–646. doi:10.1126/science.abf7945
Havugimana, P. C., Hart, G. T., Nepusz, T., Yang, H., Turinsky, A. L., Li, Z., et al. (2012). A census of human soluble protein complexes. Cell 150, 1068–1081. doi:10.1016/j.cell.2012.08.011
Henzler-Wildman, K. A., Lei, M., Thai, V., Kerns, S. J., Karplus, M., and Kern, D. (2007). A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 450, 913–916. doi:10.1038/nature06407
Ho, C.-M., Li, X., Lai, M., Terwilliger, T. C., Beck, J. R., Wohlschlegel, J., et al. (2020). Bottom-up structural proteomics: cryoEM of protein complexes enriched from the cellular milieu. Nat. Methods 17, 79–85. doi:10.1038/s41592-019-0637-y
Hyung, S.-J., Robinson, C. V., and Ruotolo, B. T. (2009). Gas-phase unfolding and disassembly reveals stability differences in ligand-bound multiprotein complexes. Chem. Biol. 16, 382–390. doi:10.1016/j.chembiol.2009.02.008
Jurneczko, E., and Barran, P. E. (2011). How useful is ion mobility mass spectrometry for structural biology? The relationship between protein crystal structures and their collision cross sections in the gas phase. Analyst 136, 20–28. doi:10.1039/C0AN00373E
Kirk, S. R., Liu, F. C., Cropley, T. C., Carlock, H. R., and Bleiholder, C. (2019). On the preservation of non-covalent peptide assemblies in a tandem-trapped ion mobility spectrometer-mass spectrometer (TIMS-TIMS-MS). J. Am. Soc. Mass Spectrom. 30, 1204–1212. doi:10.1007/s13361-019-02200-y
Koeniger, S. L., Merenbloom, S. I., and Clemmer, D. E. (2006a). Evidence for many resolvable structures within conformation types of electrosprayed ubiquitin ions. J. Phys. Chem. B 110, 7017–7021. doi:10.1021/jp056165h
Koeniger, S. L., Merenbloom, S. I., Sevugarajan, S., and Clemmer, D. E. (2006b). Transfer of structural elements from compact to extended states in unsolvated ubiquitin. J. Am. Chem. Soc. 128, 11713–11719. doi:10.1021/ja062137g
Koeniger, S. L., Merenbloom, S. I., Valentine, S. J., Jarrold, M. F., Udseth, H. R., Smith, R. D., et al. (2006c). An IMS−IMS analogue of MS−MS. Anal. Chem. 78, 4161–4174. doi:10.1021/ac051060w
Kondrat, F. D. L., Struwe, W. B., and Benesch, J. L. P. (2015). “Native mass spectrometry: Towards high-throughput structural proteomics,” in Structural proteomics, methods in molecular biology. Editor R. J. Owens (New York, NY: Springer New York), 349–371. doi:10.1007/978-1-4939-2230-7_18
Kurulugama, R. T., Nachtigall, F. M., Lee, S., Valentine, S. J., and Clemmer, D. E. (2009). Overtone mobility spectrometry: Part 1. Experimental observations. J. Am. Soc. Mass Spectrom. 20, 729–737. doi:10.1016/j.jasms.2008.11.022
Li, Y. I., van de Geijn, B., Raj, A., Knowles, D. A., Petti, A. A., Golan, D., et al. (2016). RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604. doi:10.1126/science.aad9417
Liu, F. C., Cropley, T. C., Ridgeway, M. E., Park, M. A., and Bleiholder, C. (2020). Structural analysis of the glycoprotein complex avidin by tandem-trapped ion mobility spectrometry–mass spectrometry (Tandem-TIMS/MS). Anal. Chem. 92, 4459–4467. doi:10.1021/acs.analchem.9b05481
Liu, F. C., Kirk, S. R., and Bleiholder, C. (2016). On the structural denaturation of biological analytes in trapped ion mobility spectrometry – mass spectrometry. Analyst 141, 3722–3730. doi:10.1039/C5AN02399H
Liu, F. C., Ridgeway, M. E., Park, M. A., and Bleiholder, C. (2018). Tandem trapped ion mobility spectrometry. Analyst 143, 2249–2258. doi:10.1039/C7AN02054F
Liu, F. C., Ridgeway, M. E., Park, M. A., and Bleiholder, C. (2022). Tandem-trapped ion mobility spectrometry/mass spectrometry (t TIMS/MS): A promising analytical method for investigating heterogenous samples. Analyst 147, 2317–2337. doi:10.1039/D2AN00335J
Liu, F. C., Ridgeway, M. E., Winfred, J. S. R. V., Polfer, N. C., Lee, J., Theisen, A., et al. (2021). Tandem-trapped ion mobility spectrometry/mass spectrometry coupled with ultraviolet photodissociation. Rapid Commun. Mass Spectrom. 35, e9192. doi:10.1002/rcm.9192
Marsh, J. A., and Teichmann, S. A. (2015). Structure, dynamics, assembly, and evolution of protein complexes. Annu. Rev. Biochem. 84, 551–575. doi:10.1146/annurev-biochem-060614-034142
May, J. C., Jurneczko, E., Stow, S. M., Kratochvil, I., Kalkhof, S., and McLean, J. A. (2018). Conformational landscapes of ubiquitin, cytochrome c, and myoglobin: Uniform field ion mobility measurements in helium and nitrogen drift gas. Int. J. Mass Spectrom. 427, 79–90. doi:10.1016/j.ijms.2017.09.014
Meier, F., Beck, S., Grassl, N., Lubeck, M., Park, M. A., Raether, O., et al. (2015). Parallel accumulation–serial fragmentation (PASEF): Multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J. Proteome Res. 14, 5378–5387. doi:10.1021/acs.jproteome.5b00932
Meier, F., Brunner, A.-D., Frank, M., Ha, A., Bludau, I., Voytik, E., et al. (2020). diaPASEF: parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 17, 1229–1236. doi:10.1038/s41592-020-00998-0
Mendoza, J. L., Schmidt, A., Li, Q., Nuvaga, E., Barrett, T., Bridges, R. J., et al. (2012). Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell 148, 164–174. doi:10.1016/j.cell.2011.11.023
Oliver, R. W. A., Green, B. N., and Harvey, D. J. (1996). The use of electrospray ionization MS to determine the structure of glycans in intact glycoproteins. Biochem. Soc. Trans. 24, 917–927. doi:10.1042/bst0240917
Ollivier, S., Tarquis, L., Fanuel, M., Li, A., Durand, J., Laville, E., et al. (2021). Anomeric retention of carbohydrates in multistage cyclic ion mobility (IMSn): De novo structural elucidation of enzymatically produced mannosides. Anal. Chem. 93, 6254–6261. doi:10.1021/acs.analchem.1c00673
Onuchic, J. N., and Wolynes, P. G. (2004). Theory of protein folding. Curr. Opin. Struct. Biol. 14, 70–75. doi:10.1016/j.sbi.2004.01.009
Perkins, J. R., Diboun, I., Dessailly, B. H., Lees, J. G., and Orengo, C. (2010). Transient protein-protein interactions: Structural, functional, and network properties. Structure 18, 1233–1243. doi:10.1016/j.str.2010.08.007
Poyer, S., Comby-Zerbino, C., Choi, C. M., MacAleese, L., Deo, C., Bogliotti, N., et al. (2017). Conformational dynamics in ion mobility data. Anal. Chem. 89, 4230–4237. doi:10.1021/acs.analchem.7b00281
Richards, A. L., Eckhardt, M., and Krogan, N. J. (2021). Mass spectrometry-based protein protein interaction networks for the study of human diseases. Mol. Syst. Biol. 17, e8792. doi:10.15252/msb.20188792
Rolland, A. D., and Prell, J. S. (2019). Computational insights into compaction of gas-phase protein and protein complex ions in native ion mobility-mass spectrometry. Trac. Trends Anal. Chem. 116, 282–291. doi:10.1016/j.trac.2019.04.023
Ruotolo, B. T., Giles, K., Campuzano, I., Sandercock, A. M., Bateman, R. H., and Robinson, C. V. (2005). Evidence for macromolecular protein rings in the absence of bulk water. Science 310, 1658–1661. doi:10.1126/science.1120177
Shelimov, K. B., Clemmer, D. E., Hudgins, R. R., and Jarrold, M. F. (1997). Protein structure in vacuo: Gas-phase conformations of BPTI and cytochrome c. J. Am. Chem. Soc. 119, 2240–2248. doi:10.1021/ja9619059
Shi, H., Atlasevich, N., Merenbloom, S. I., and Clemmer, D. E. (2014). Solution dependence of the collisional activation of ubiquitin [M + 7H]7+ ions. J. Am. Soc. Mass Spectrom. 25, 2000–2008. doi:10.1007/s13361-014-0834-y
Simon, A.-L., Chirot, F., Choi, C. M., Clavier, C., Barbaire, M., Maurelli, J., et al. (2015). Tandem ion mobility spectrometry coupled to laser excitation. Rev. Sci. Instrum. 86, 094101. doi:10.1063/1.4930604
Smith, L. M., and Kelleher, N. L. (2018). Proteoforms as the next proteomics currency. Science 359, 1106–1107. doi:10.1126/science.aat1884
Tang, K., Li, F., Shvartsburg, A. A., Strittmatter, E. F., and Smith, R. D. (2005). Two-dimensional gas-phase separations coupled to mass spectrometry for analysis of complex mixtures. Anal. Chem. 77, 6381–6388. doi:10.1021/ac050871x
Tzeng, S.-R., and Kalodimos, C. G. (2012). Protein activity regulation by conformational entropy. Nature 488, 236–240. doi:10.1038/nature11271
Vidal, M., Cusick, M. E., and Barabási, A.-L. (2011). Interactome networks and human disease. Cell 144, 986–998. doi:10.1016/j.cell.2011.02.016
Voronina, L., Masson, A., Kamrath, M., Schubert, F., Clemmer, D., Baldauf, C., et al. (2016). Conformations of prolyl–peptide bonds in the bradykinin 1–5 fragment in solution and in the gas phase. J. Am. Chem. Soc. 138, 9224–9233. doi:10.1021/jacs.6b04550
Wyttenbach, T., and Bowers, M. T. (2011). Structural stability from solution to the gas phase: Native solution structure of ubiquitin survives analysis in a solvent-free ion mobility–mass spectrometry environment. J. Phys. Chem. B 115, 12266–12275. doi:10.1021/jp206867a
Yang, X., Coulombe-Huntington, J., Kang, S., Sheynkman, G. M., Hao, T., Richardson, A., et al. (2016). Widespread expansion of protein interaction capabilities by alternative splicing. Cell 164, 805–817. doi:10.1016/j.cell.2016.01.029
Yang, Y., and Orlando, R. (1996). Identifying the glycosylation sites and site-specific carbohydrate heterogeneity of glycoproteins by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 10, 932–936. doi:10.1002/(SICI)1097-0231(19960610)10:8<932::AID-RCM595>3.0.CO;2-X
Yates, J. R., and Kelleher, N. L. (2013). Top down proteomics. Anal. Chem. 85, 6151. doi:10.1021/ac401484r
Zhong, Y., Han, L., and Ruotolo, B. T. (2014). Collisional and coulombic unfolding of gas-phase proteins: High correlation to their domain structures in solution. Angew. Chem. Int. Ed. 53, 9209–9212. doi:10.1002/anie.201403784
Keywords: ion mobility, tandem ion mobility, protein structure, mass spectrometry, cross section, collision induced unfolding, TIMS
Citation: Cropley TC, Chai M, Liu FC and Bleiholder C (2023) Perspective on the potential of tandem-ion mobility/mass spectrometry methods for structural proteomics applications. Front. Anal. Sci. 3:1106752. doi: 10.3389/frans.2023.1106752
Received: 24 November 2022; Accepted: 06 January 2023;
Published: 18 January 2023.
Edited by:
Ian K. Webb, Indiana University, Purdue University Indianapolis, United StatesReviewed by:
Suk-Joon Hyung, Genentech Inc., United StatesJames Prell, University of Oregon, United States
Copyright © 2023 Cropley, Chai, Liu and Bleiholder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Bleiholder, Y2JsZWlob2xkZXJAZnN1LmVkdQ==
†These authors have contributed equally to this work