
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Anal. Sci. , 12 September 2022
Sec. Environmental Analysis
Volume 2 - 2022 | https://doi.org/10.3389/frans.2022.941883
 Juliana Cristina Barreiro1,2*Anna Patrícya Florentino3Izadora Liranço Furlani4Gustavo H. R. Silva1Quezia Bezerra Cass4*
Juliana Cristina Barreiro1,2*Anna Patrícya Florentino3Izadora Liranço Furlani4Gustavo H. R. Silva1Quezia Bezerra Cass4*An analytical method was developed to quantify a mixture of acetaminophen, metoprolol, methylparaben, carbamazepine, naproxen, estrone, estradiol, diclofenac, benzophenone, ibuprofen, progesterone, and mefenamic acid from domestic wastewater samples. To match fast and efficient chromatographic separation for different classes of compounds, an automated scouting liquid chromatographic system was associated with the experimental design produced by the DryLab® software. HLB cartridges were used to extract the analytes from the sample matrix, which was followed by detection and quantitation by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The lowest detection limits were found for acetaminophen and carbamazepine (0.625 pg L−1) and metoprolol (0.4 pg L−1).
Anthropogenic pollution has a direct impact on the quality of aquatic environment systems, resulting in the contamination of the water resources, due to the high loads of organic matter, micropollutants, and pathogen concentrations (Gravilescu et al., 2016; Desiante et al., 2022). In this context, special attention can be given to the presence of micropollutants, such as pharmaceutical and personal care products (PPCPs) (Duan et al., 2021). Although these substances are continuously introduced into the environment in small amounts, their potential chronic health effects associated with long exposures are not well known. Numerous papers have summarized and discussed the occurrence of PPCPs in different compartments of the environment (e.g., groundwater and wastewater) as well as the technologies for their removal (Hena et al., 2021). Although several advanced treatment technologies are available for withdrawal of PPCPs, low removal efficiencies and/or limitations are still a drawback and, thus, better strategies are necessary. In this scenario, the use of microalgae-mediated bioremediation of PPCPs is of growing scientific interest. Its advantages include being driven by solar energy; small amounts of operational inputs; being eco-friendly; quick and efficient uptake of nitrogen, phosphorus, and PPCPs (i.e., as a carbon source); quick and efficient fixation and turnover of carbon; and the simultaneous generation of products with high-value aggregate (e.g., nutraceuticals and cosmetics) and low-value food products for aquaculture and microalgal biomass for the production of animal feed and biofuel (Gao et al., 2016; Metcalf & Eddy, 2016; Cuellar-Bermudez et al., 2017). The possibility to remove micropollutants (e.g., diclofenac, ibuprofen, paracetamol, and metoprolol) from sanitary sewage by microalgae Chlorella sorokiniana with the simultaneously recovery of nutrients from blackwater treatment system was demonstrated by Wilt et al. (2016) on the laboratory scale. In addition, Kumar et al. (2015) demonstrated their higher potential for the remediation of heavy metals.
Herein, we report an analytical method to quantify 1) acetaminophen (ACM), 2) metoprolol (MET), 3) methylparaben (MEP), 4) carbamazepine (CBZ), 5) naproxen (NAP), 6) estrone (ESN), 7) estradiol (EST), 8) diclofenac (DIC), 9) benzophenone (BZN), 10) ibuprofen (IBU), 11) progesterone (PRO), and 12) mefenamic acid (AM) from domestic wastewater samples under bioremediation by microalgae culture in a laboratory scale plant.
To select the chromatographic conditions with high efficiency, short analysis time, and minimizing the production of residues in the method development, a column and eluent scouting LC system, and DryLab® software were used to select the Design of Experiments (DoEs) parameters and predict the optimized chromatographic conditions.
All reagents and solvents obtained from commercial suppliers were used without further purification. Methanol (MeOH) and acetonitrile (ACN) HPLC grade were purchased from J.T. Baker (Philipsburg, United States). Formic acid (p.a. grade) was acquired from Fluka (Buchs, Switzerland), and ammonium formate and acetate were acquired from Sigma-Aldrich (St. Louis, MO, United States). All pharmaceutical, benzophenone, and methylparaben were purchased from Sigma-Aldrich with a high purity ≥ 95%.
The scouting UHPLC system used (Nexera®, Shimadzu) consisted of two quaternary LC-30AD pumps, a DGU-20A5R degasser, a SIL-30A autosampler, a CTO-20AC column oven, a six-column selector valve, diode array detector SPD-M30A, and CBM 20A interface. The Method Scouting Solution® system was used to develop the analysis sequence, and LabSolution® workstation software was used to control all modules and for data processing.
The analyses were carried out using an Acquity UPLC® system coupled with a Xevo® TQ-MS (Waters, Milford, MA, United States). The chromatographic equipment consisted of a binary pump (BSM), a quaternary H-Class pump (QSM), and an autosampler model 2777C with one six-port Valco® valve. MassLynx 4.1 software (Waters, Milford, MA, United States) was used for data acquisition and processing. The optimization of the ionization source and voltages on the lenses was done by IntelliStart tune mode.
For the analyses in LC-DAD system, a stock solutions of 1.00 mg mL−1 were prepared for 12 PPCPs in methanol, they were diluted to 100 µg ml−1, and analyzed by liquid chromatography-diode array detector (LC-DAD) using the Method Scouting Solution® with four different orthogonal columns (100 × 2.1 mm): XSelect® CSH (2.5 µm, Waters); Xterra® (3.0 µm, Waters); Ascentis® Express F5 (2.7 µm, Supelco), and XSelect® HSST3 (3.5 µm, Waters).
The following aqueous phases (A) were prepared with a concentration of 10 mmol L−1: ammonium formate pH 3.0, ammonium acetate pH 4.0, and pH 5.0. Three different organic modifiers were used (B) (ACN, MeOH, and MeOH:ACN (50:50, v/v)). Two linear gradients with a ΔB (5–95%) were evaluated, as followed: 0 min, 5% B; 0–6 min, 5–95% B; 6.01–8 min, 95%B and 8.01–10 min, 5% B, and the second one was: 0 min, 5% B; 0–18 min, 95% B; 18.01–20 min, 95% B and 20.01–21 min, 5% B. The flow rate was 0.3 ml min−1, and the injection volume was 2 µl. All columns were maintained at 40°C. DAD absorption was acquired in a range of 200–400 nm, with detection in 226 nm. The combination of different pH, stationary and mobile phases led to 72 different experiments, which were programmed in a single batch using Method Scouting Solution®.
The number of chromatographic peaks was the determinant parameter to choose XSelect® CSH (100 × 2.1 mm, 2.5 µm) as the column and MeOH:ACN ((50:50, v/v) as the organic modifier (B), 10 mmol L−1ammonium acetate as the aqueous phase (A) of the mobile phase. The chromatograms obtained under those conditions were exported to DryLab®4 Software (Version 4.2.1.9, Molnár-Institute, Berlin, Germany) to model the aqueous phase (A) pH, the gradient range (ΔB), and the gradient time (tG). The optimized chromatographic conditions obtained and also used for LC-MS/MS were: 0 min 15% B; 2.28–10.82 min; 59% B and 14 min, 90% B.
The MS instrument consisted of a XEVO TQ-MS (Waters, Milliford, United States), which was assisted by MassLynx 4.1 software. Data analyses are acquired on the MS fitted with an electrospray ionization (ESI) source operating in positive mode. To optimize the ionization conditions and to obtain a maximum of sensitivity, identification, and detection of all target compounds, two selected reaction monitoring (SRM) transitions were optimized for each compound. For that, standard solutions of each compound (100 μg mL−1) were continuously infused at a flow rate of 20 μl min−1 by a syringe pump into the mobile phase stream. Nitrogen was used as desolvation gas at 600 L h−1 flow rate with desolvation temperature set at 200°C and the source temperature at 150°C. The capillary voltage was set at 2.90 kV and the collision gas flow at 0.15 ml min−1. The cone voltage (CV) and collision energy (CE) for all transitions are illustrated at Table 1.
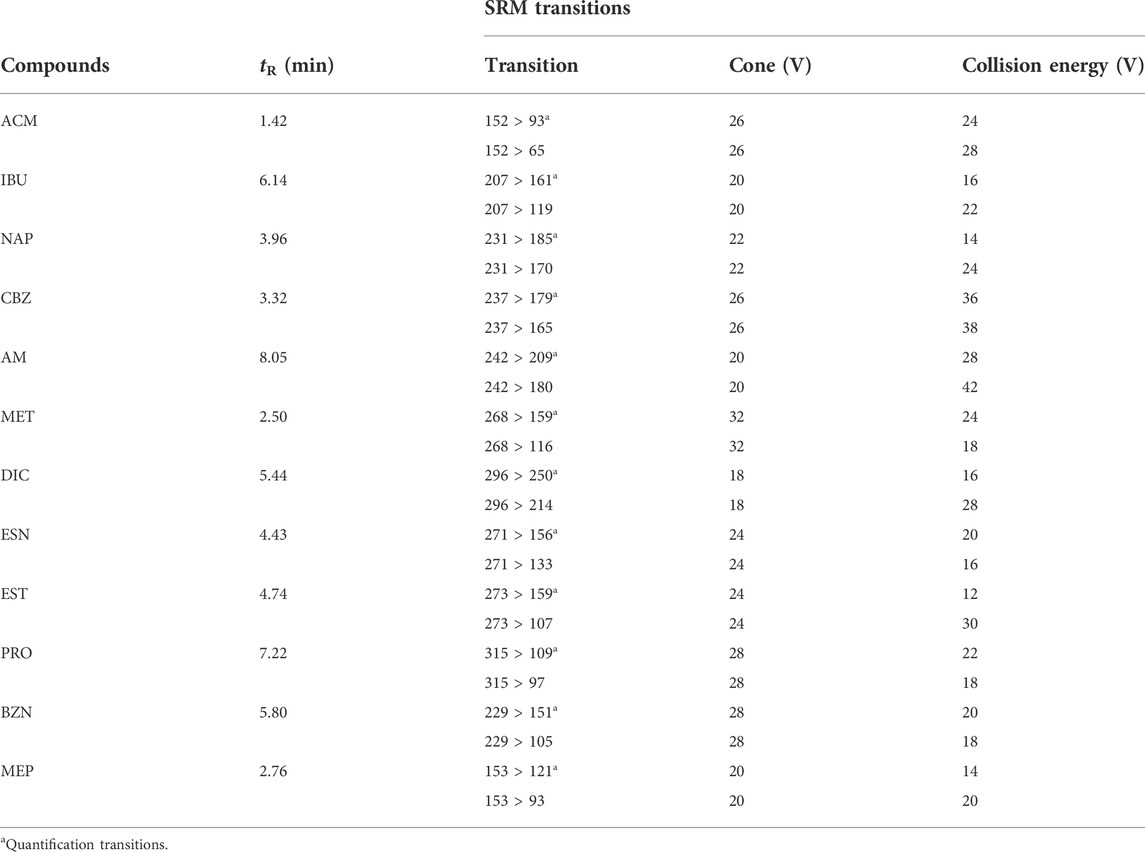
TABLE 1. MS/MS parameters for the selected compounds.
A stock solution of 200 μg ml−1 was prepared in methanol for each PPCPs. From this, we prepared the following working solutions, 5.00, 10.0, 25.0, 50.0, 100, 150, and 200 μg L−1 for benzophenone (BZN), acetaminophen, (ACM) carbamazepine (CBZ), naproxen (NAP), progesterone (PRO), mefenamic acid (AM), and metoprolol (MET) (to calibration) and 6.00, 80.0 e 160 μg L−1 (to quality control (QC) samples); 250, 375, 500, 625, 750, 875, and 1,000 μg L−1 for estradiol, (EST) and estrone (ESN) (to calibration), and 300, 400, and 800 μg L−1 (to QC samples); 100, 250, 500, 750, 1,000, 1,500 and 2000 μg L−1 for diclofenac (DIC) (to calibration) and 120, 800, and 1,600 μg L−1 (to QC samples); 50.0, 100, 200, 300, 400, 450, and 500 μg L−1 for methylparaben (MEP) (to calibration), and 60.0, 250, and 375 μg L−1 (to QC samples) and 1,000, 1,500, 2000, 2,500, 3,000, 3,500, and 4,000 μg L−1 for ibuprofen (IBU) (to calibration), and 1,200, 2,400, and 3,200 μg L−1 (to QC samples). All of the stock and working solutions were stored at −20°C. The calibrators and quality controls samples were prepared from the working solutions and diluted using the appropriated volume with 50:50 (v/v) - aqueous phase (A) (10 mmol L−1ammonium acetate pH 4.0)/organic modifier mobile phase (B) (MeOH:ACN (50:50, v/v)).
The sample preparation was carried out in triplicate, and the solid-phase extraction (SPE) was a modification of Zhang et al. (2020) procedure. To meet this end, the samples (10.0 ml) were centrifuged for 5 min at 10,000g and 4°C previously to SPE. The centrifuged supernatants were then added to Oasis® HLB (1 ml) cartridges preconditioned with 3.00 ml of MeOH (× 2) and 3.00 ml of water (× 2). After loading, the samples were washed with 3.00 ml of water (× 2) before eluting the analytes at 1 drop s−1 with 1.00 ml of MeOH (2.00 × 0.50 ml). The extracts were dried for 10 min under vacuum at room temperature. For analysis, the extracts were reconstituted in 100 μl of mobile phase (ammonium acetate (pH 4,0): MeOH/ACN (50:50, v/v)–(50:50, v/v)).
To prepare the calibration standards and quality control samples, 20 μl of aliquots of the appropriate working solutions were spiked in Eppendorf containing the matrix (domestic wastewater cultivated with microalgae) previously extracted by SPE procedure. In total, 80 μL of mobile phase 50:50 (v/v) - aqueous phase (A) (10 mmol L−1: ammonium acetate pH 4.0)/organic modifier mobile phase (B) (MeOH:ACN (50:50, v/v)) was added. The spiked samples were vortex-mixed for 30 s, transferred to the autosampler vials, and injected into the LC–MS/MS system.
The effluent used in this research was collected at the Tibiriçá Wastewater Treatment Plant (WWTP), which is located in the city of Bauru-SP, Brazil. The WWTP has an average flow rate of 6.00 L s−1 and the treatment consists of railing, sanding box, primary decanter, and biologic anaerobic filter with ascending flow. The native culture was obtained by enriching the effluent of the WWTP by serial inoculation of 10% (v/v) when in early stationary phase, which was performed in 1.00 L Erlenmeyer, and stored under photoperiod of 12/12h, average temperature 36 ± 2°C and luminosity of 160 mmol s m−1 C. sorokiniana was obtained from the microalgae collection of the Algal Biotechnology Laboratory, Department of Botany, UFSCar-São Carlos/SP, Brazil. The effluent was previously autoclaved to prevent the growth of other microorganisms that are naturally present in the system, and also to maintain concentrations of nitrogen, phosphorus, and BOD similar to those used in native cultivation. Batches were performed in biological triplicates. At the starting time, microalgae concentration in the cultures was stated at the order of 105 cells mL−1 in cylindrical reactors with 1.50 L working-volume and kept under the same cultivation conditions. The reactors were connected to the aeration system, which was composed of an air compressor and polyurethane distribution hoses with a flow rate of 1.00 L min−1 per reactor. Cultures were daily monitored along the whole cycle of growth and photosynthetic activity. Control groups with no addition of inoculum were carried out for 10 days. Analyses were performed for the beginning and end of incubation period, with collection in different periods of days of the cycle. A mix of 12 PPCPs contaminants commonly found in WWTP all over the world was prepared and the adopted concentrations were based on the maxima values reported in the literature (Dias, 2014; Lima et al., 2017; Bisognin et al., 2018; Derisso et al., 2020): ACM (60.0 µg L− ), MET (1.00 µg L−1), MEP (1.00 µg L−1), CBZ (4.00 µg L−1), NAP (15.0 µg L−1), ESN (0.10 µg L−1), EST (4.00 µg L−1), DIC (15.0 µg L−1), BZN (1.00 µg L−1), IBU (300 µg L−1), PRO (0.10 µg L−1), and AM (1.00 µg L−1). Samples were collected in different periods during 7 days using a syringe connected to a glass tube inside the reactor (Supplementary Figure S1). Growth cycle and photosynthetic activity were monitored daily. Samples were analyzed for dry weight, chlorophyll-a, ammoniacal nitrogen, nitrate, total phosphorus, number of cells, pH, dissolved oxygen, and optical density (American Public Health Association et al., 2005). The analyses were carried out at the Environmental Sanitation Laboratory in the Department of Civil and Environmental Engineering at UNESP in Bauru- SP, Brazil.
The evaluation of matrix effect (ME) (EMA, 2011) and validation of the analytical method was carried out following the European Medicines Agency guideline procedure. The QC spiked samples (see Section 2.5) were prepared in triplicate and employed to evaluate the recovery.
The MEs from suppression or enhancement of analyte ionization were expressed as: ME (%) = B/A × 100 in accordance with the equation proposed by Matuszewski et al. (1998). The analyte responses areas in spiked (500 μg L−1) real samples after extraction (A) were compared with the areas obtained for a solvent solution at the same concentration level (B). The experimental procedure was based on published works (Madureira et al., 2009). The absence of absolute matrix effect is indicated by a value of 100%, a value > 100% indicates an ionization enhancement, and a value < 100% corresponds to ionization suppression.
The analyte recovery was calculated as the ratio of the response after extraction to the response of a calibration standard generated at the concentration corresponding to 100% recovery during SPE.
To determine linearity, six blanks of domestic wastewater cultivated with microalgae were spiked in triplicate at seven different concentration levels and then analyzed (see Section 2.4). For all of the 12 compounds, external calibration curve was constructed by plotting the peak area versus concentration. For selectivity evaluation, domestic wastewater samples were compared with samples spiked with a mixture of the compounds at limit of quantitation (LOQ) concentration, which were individually analyzed and evaluated for interference.
Intra- and inter-batch precision and accuracy were determined by analysis at five replicates of each of the QC spiked samples. Precision was expressed as CV of the replicate measurements. The accuracy of the method was evaluated as the percentage between the calculated and the nominal concentration of each compound.
The limit of detection (LOD) and LOQ values were determined using spiked domestic wastewater samples in triplicate. The LODs were calculated as the minimum detectable amount of compound, with a signal-to-noise (S/N) ratio of three. A variability of less than 20% for precision and accuracy was the accepted criteria for the LOQ. However, for the analytes ACM, CBZ, NAP, AM, and MET, the first calibrate level used was superior to the LOQ to facilitate the preparation of the calibrators.
The chemical stability of the drugs was evaluated using the QC samples, at room temperature, as freshly prepared samples, and after 24, and 48 h (autosampler stability). A CV of less than 15% was the criterion for the stability evaluation.
The utility of the method was evaluated by determining the selected compounds in domestic wastewater samples, which were bioremediated by microalgae on a laboratory scale plant. For the analysis, external calibration curve and quality control samples were run between the environmental samples. For comparison, analysis of samples of the control group was also carried out (see Section 2.7).
Analysis of complex mixtures is still a great challenge in liquid chromatography, mainly due to the different physical-chemical characteristics of the compounds (Snyder et al., 2004). The optimization of a chromatographic condition could take a long time for the analyst, and could cause wasted solvents and samples. Thus, the association of automated LC scouting system and the use of DryLab® software for the DoE parameters and to predict the optimized chromatographic conditions has become an interesting workflow for getting high chromatographic efficiency in short analysis time, which can overcome these drawbacks (Krisko et al., 2006; Racz et al., 2018; Guichard et al., 2019; Radic et al., 2020).
To select the chromatographic conditions for the separation and analysis of a mixture of 12 PPCPs, an automated column and eluent scouting workflow was carried out (as illustrated at Figure 1). Four orthogonal columns were selected based on Snyder’s hydrophobic-subtraction model (Snyder et al., 2004), in accordance with their Fs at pH 3.0 (The United States Pharmacopeial Convention, 2022). The separation was also evaluated in terms of pH and organic phase composition, under a linear gradient of AB (5–95%). A 2D resolution map for pH and tG variables was generated by the software and is presented in Supplementary Figure S2. The generated data corresponded to 72 chromatographic runs.
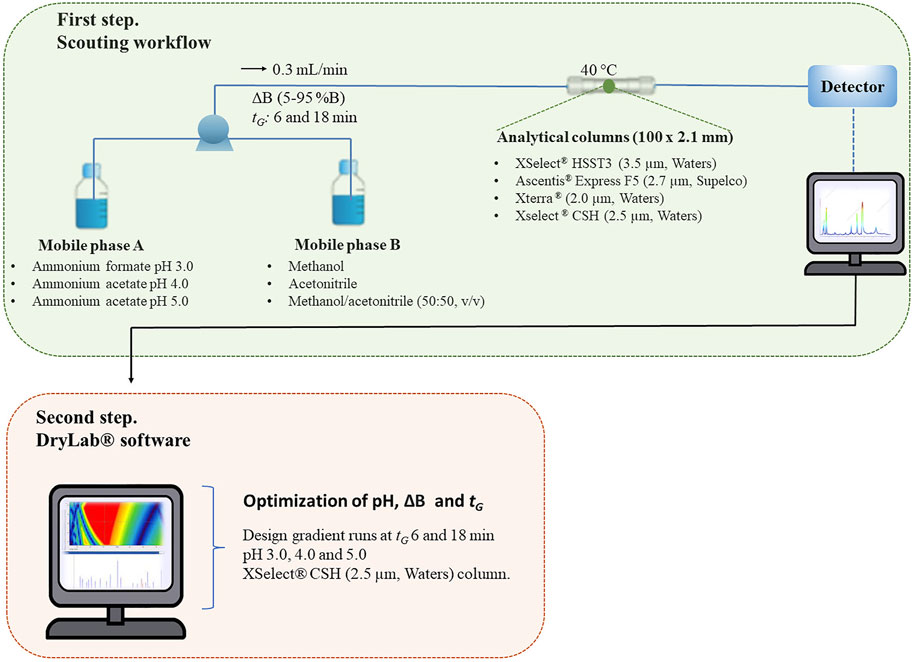
FIGURE 1. Schematic workflow for the chromatographic method development.
In all cases, the combination of ACN and MeOH (50:50 v/v) as the organic modifier furnished higher selectivity. Throughout the evaluated conditions, the columns XSelect® HSST3 (3.5 µm, Waters) and Ascentis® Express F5 (2.7 µm, Supelco) gave the lowest resolution for the drugs mixture, and was not considered in the next step of the workflow. The columns Xterra® (2.0 µm, Waters) and XSelect® CSH (2.5 µm, Waters) showed resolution similarities for the polar drugs, but the separation of the nonpolar drugs was achieved only with the latter column, which was selected for the next step of the workflow (Supplementary Figures S3–S6). Figure 1 shows the schematic workflow for the method development.
Six LC scouting chromatograms obtained from three different pH at two gradient runs (6 and 18 min) using XSelect® CSH and ACN/MeOH (50:50 v/v) as modifier (B) and 10 mmol L−1ammonium acetate as the aqueous phase (A) at λ = 226 nm were exported to the DryLab® software for optimization of the mobile phase pH, (Figure 1), and ΔB (as illustrated in Supplementary Table S1).
The correlation of chromatogram peaks was adjusted for the two gradients, based on the area and confirmed by UV spectra of each drug. After data processing, a 2D resolution map for pH and (Figure 1) variables was generated by the software and the highest selectivity was predicted at the following chromatographic conditions—mobile phase pH 4.2, (Figure 1) 14 min, at the following gradient steps: 0 min 20% B; 2.28–10.82 min; 59% B and 14 min, 90% B. The predicted (A) and experimental (B) chromatograms are illustrated in Figure 2.
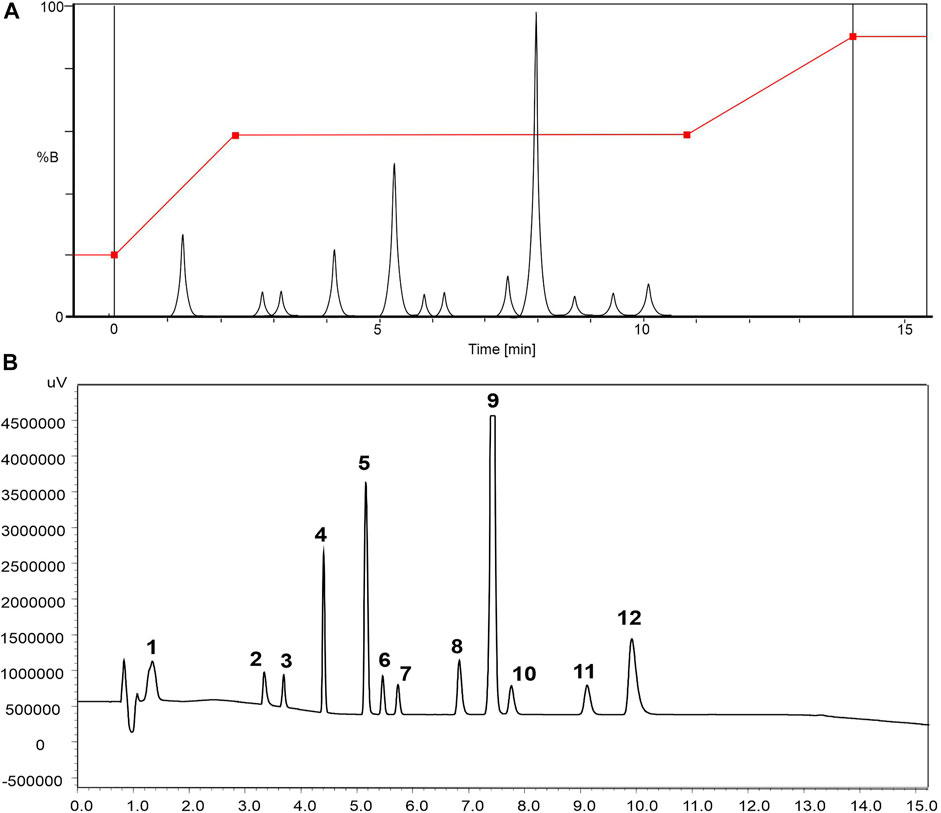
FIGURE 2. Chromatograms (LC-DAD), using XSelect CSH column (100 × 2.1 mm, 2.7 µm), ammonium acetate 10 mmol L−1 pH 4.0/ACN:MeOH (50:50 v/v) as mobile phase, at 0.3 ml min−1, λ = 226 nm. Gradient elution mode of 0 min, 20% B; 2.28–10.82 min; 59% B and 10.82–14 min, 90% B, and reconditioning (2 min). (A) Simulated chromatogram and (B) experimental chromatogram for the mixture of compounds (100 mg ml−1): 1) ACM, 2) MET, 3) MEP; 4) CBZ, 5) NAP, 6) ESN, 7) EST, 8) DIC, 9) BZN, 10) IBU, 11) PRO, and 12) AM.
Despite the predicted selectivity, minor adjustment in the mobile phase pH and on the gradient format was necessary due to the lower retention of the first peak. Thus, two others initial gradients were evaluated with 10 and 15% B. The association of scouting for selecting the column and the mobile phase composition, followed by DryLab® Software, furnished a chromatographic profile with high selectivity and short analysis time for the 12 PPCPs that we studied. To this end, 72 experiments were carried out, which represented 18.6 h of total analysis time and 334.8 ml of mobile phase. The experimental chromatographic conditions that we used were transferred to LC-MS/MS system and the selected chromatographic conditions were: ammonium acetate 10 mmol L−1 pH 4.0, and ΔB initiating in 15% B.
SPE preliminary evaluation were performed with addition of Na4EDTA (500 mg L−1). However, no difference in the matrix effect was observed. The pH of the samples was around of 6.0 and no adjustment were carried out. The recovery results for the selected drugs are reported at three different concentrations (QCs) (Supplementary Table S2). The low recoveries obtained for some of the analytes can be related to their pKa and are in agreement to the data reported by Zhang et al. (2020).
Method validation was considered in terms of the linear calibration range, LODs and LOQs, analyte recovery, intra- and inter-day accuracy and precision, and MEs (Commission Decision, 2002). These parameters are summarized for all of the 12 compounds in Supplementary Tables S2, S3.
The calibration ranges were linear with mean (n = 3) correlation coefficient (R2) values > 0.990. However, three compounds displayed lower R2 values-estradiol and estrone (0.986), and methylparaben (0.984). LODs in the order of pg L−1 was found for three drugs (ACM, CBZ, and MET), for four compounds in the order of ng L−1 (BZN, NAP, AM, and PRO), and five compounds exhibited LODs among 10 and 100 μg L−1. The accepted LOQ for all the analytes were the first concentrations of the calibration curves. Precision and accuracy were determined by analyzing the replicates (five times) of each of the three QC of domestic wastewater samples on three consecutive batches. Accuracy values were between 87.5 and 120% (Supplementary Table S3). These values were considered to be suitable once the values obtained cover the complete sample preparation procedure and not just a consecutive sequence of injections of the same sample.
The intra- and inter-day precision was assessed the same way as the accuracy, and it was found to be below 15%. The extraction recovery for all 12 PPCPs is reported in Supplementary Table S3 and was determined by comparing the peak areas of methanol solutions at three different levels with those of the QC samples.
Recovery values was determined by spiked solutions at QCs concentrations for each PPCP. The range was from 33.4 to 96.9% with RSD values less than 20%. The QC standards were stable in the autosampler tray for 24 and 48 h at room temperature (approximately 23°C). Figure 3 illustrates the SRM chromatograms at quantification transitions of the 12 PPCPs at low QC control.

FIGURE 3. SRM chromatograms at quantification transitions of the 12 PPCPs at low QC samples acetaminophen, benzophenone, carbamazepine, naproxen, progesterone, mefenamic acid, and metoprolol (6.00 (μg L−1)); estradiol and estrone (300 (μg L−1)); diclofenac (120 (μg L−1)); ibuprofen (1,200 (μg L−1)), and methylparaben (60 (μg L−1)).
To avoid erroneous quantifications, MEs were calculated (Taylor, 2005). For that, a microalgae inoculum in a Tibiriçá effluent, previously autoclaved, was used to prepare the calibrators, QC samples, and non-spiked blank samples. Co-extracted matrix components can affect analytes ionization, which might result in either enhancement or more frequently in suppression of the signals in quantitative analysis by LC-MS/MS (Madureira et al., 2009).
The analytical response was suppressed for most of the analytes. However, an enhancement of the effects was observed for ACM (43%), MET (59%), EST (13%), ESN (4%), and IBU (15%). In fact, the PPCPs physicochemical properties of the analytes can influence the degree of enhancement or suppression of their ionization (Borecka et al., 2014). Highly polar compounds are usually more affected by ion suppression than less polar molecules (Bonfiglio et al., 1999). However, this behavior depends on the components present on the matrix (Barreiro et al., 2011).
The analytical method was shown to be precise and accurate for the analysis of the samples. Meanwhile, the use of the isotopic IS approach could probably minimize the matrix effect. However, it is difficult to find a suitable IS for each compound in a series of compounds with distinct physical–chemical properties (Madureira et al., 2009).
The developed method was used to monitoring the bioremediation experiment mediated by the presence of microalgae. For that, samples of the control group (see Section 2.7) were also used. Except for estrone (which was used in the experiments below LOQ of the analytical method), all of the other analytes were monitored along the experiment period in the control samples. Meanwhile, in the samples from the algae experiment, even in the beginning of the incubation period, only five of them were quantified (BZN, NAP, CBZ, AM, and MET). However, more analysis is required to estimate the efficiency of the wastewater treatment samples. The sample collection regime should also be evaluated. The LC-MS/MS method that is described in this article has demonstrated its utility in as much as some of the selected compounds were quantified in this type of sample.
The present work reports the development, validation, and application of an LC-MS/MS for quantification of 12 PPCPs in an experiment managed in the presence of domestic wastewater and microalgae treatment plant. For that, an automated scouting liquid chromatographic system was associated with the DryLab® software to attain selectivity and short analysis time, thus saving time and the consumption of solvents. The recoveries were appropriated considering the distinct physical-chemical properties of the selected PPCPs. However, values below 40% (33–45%) were found for the hormone class. The applicability of the analytical method was demonstrated by monitoring the PPCPs during the experiment. In monitoring the bioremediation experiment, MET was quantified in samples up to the third collection cycle while BZN, NAP, CBZ, and AM were quantified in samples of the whole incubation cycle.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
JB: methodology, validation, formal analysis, writing—original draft, writing—review and editing. AF: investigation, formal analysis. IF: methodology, formal analysis, investigation. GHRS: supervision, funding acquisition, project administration, resource. QC: validation, visualization, supervision, conceptualization, writing—review and editing.
The authors thank the Agency for the Improvement of Higher Education Personnel (CAPES) [Finance Code 001] in the scope of the Program CAPES-PrInt [grant number 88887.468868/2019-00]; National Council for Scientific and Technological Development (CNPq) [grant numbers 309064/2018-0 and 427936/2018-7]; and São Paulo Research Foundation (FAPESP) [grant numbers 2018/18367-1 and 2021/10569-7] for financial support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frans.2022.941883/full#supplementary-material
American Public Health AssociationAmerican Water Works Association, Water Environment Federation – APHAAWWAWEF (2005). Standard methods for the examination of water and wastewater. 19.ed. Washington, DC, USA: American Public Health Association.
Barreiro, J. C., Vanzolini, K. L., and Cass, Q. B. (2011). Direct injection of native aqueous matrices by achiral–chiral chromatography ion trap mass spectrometry for simultaneous quantification of pantoprazole and lansoprazole enantiomers fractions. J. Chromatogr. A 1218, 2865–2870. doi:10.1016/j.chroma.2011.02.064
Bisognin, R., Wolff, D., and Carissimi, E. (2018). Revisão sobre fármacos no ambiente. Rev. DAE 67, 78–95. doi:10.4322/dae.2018.009
Bonfiglio, R., King, R. C., Olah, T. V., and Merkle, K. (1999). The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun. Mass Spectrom. 13, 1175–1185. doi:10.1002/(SICI)1097-0231(19990630)13:12<1175::AID-RCM639>3.0.CO;2-0
Borecka, M., Białk-Bielińska, A., Siedlewicz, G., Stepnowski, P., and Pazdro, K. (2014). In book: Insights on Environmental Changes, Chapter: The influence of matrix effects on trace analysis of pharmaceutical residues in aqueous environmental samples. Geopl. Earth Planet. Sci. 14, 1–16. doi:10.1007/978-3-319-03683-0_1
Commission Decision (2002). Implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Brussels, Belgium: Official Journal of the European Communities L221, 8–36.
Cuellar-Bermudez, S. P., Aleman-Nava, G. S., Chandra, R., Garcia-Perez, J. S., Contreras-Ângulo, J. R., Markou, G., et al. (2017). Nutrients utilization and contaminants removal. A review of two approaches of algae and cyanobacteria in wastewater. Algal Res. 24, 438–449. doi:10.1016/j.algal.2016.08.018
Derisso, C. R., Pompei, C. M. E., Spadoto, M., Pinto, T., da, S., and Vieira, E. M. (2020). Occurrence of parabens in surface water, wastewater treatment plant in southeast of Brazil and assessment of their environmental risk. Water Air Soil Pollut. 231, 468. doi:10.1007/s11270-020-04835-0
Desiante, W., Carles, L., Wullschleger, S., Joss, A., Stamm, C., and Fenner, K. (2022). Wastewater microorganisms impact the micropollutant biotransformation potential of natural stream biofilms. Water Res. 217, 118413–13. doi:10.1016/j.watres.2022.118413
Dias, R. V. A. (2014). Avaliação da ocorrência de microcontaminantes emergentes em sistemas de abastecimento de água e da atividade estrogênica do estinilestradiol. 158p. Dissertação (Mestrado em Saneamento, Meio Ambiente e Recursos Hídricos). Belo Horizonte: Universidade Federal de Minas Gerais.
Duan, L., Zhang, Y., Wang, B., Zhou, Y., Wang, F., Sui, Q., et al. (2021). Seasonal occurrence and source analysis of pharmaceutically active compounds (PhACs) in aquatic environment in a small and medium-sized city, China. Sci. Total Environ. 769, 144272–144311. doi:10.1016/j.scitotenv.2020.144272
EMA (2011). European Medicines Agency - guideline on bioanalytical method validation. London, United Kingdom.
Gao, F., Li, C., Yang, Z. H., Zeng, G. M., Feng, L. J., Liu, J., et al. (2016). Continuous microalgae cultivation in aquaculture wastewater by a membrane photobioreactor for biomass production and nutrients removal. Ecol. Eng. 92, 55–61. doi:10.1016/j.ecoleng.2016.03.046
Gravilescu, M., Demnerová, K., Aamand, J., Agathos, S., and Fava, F. (2016). Emerging pollutants in the environment: Present and future challenges in biomonitoring, ecological risks and bioremediation. N. Biotechnol. 32, 147–156. doi:10.1016/j.nbt.2014.01.001
Guichard, N., Fekete, S., Guillarme, D., Bonnabry, P., and Fleury-Souverain, S. (2019). Computer-assisted UHPLC-MS method development and optimization for the determination of 24 antineoplastic drugs used in hospital pharmacy. J. Pharm. Biomed. Anal. 164, 395–401. doi:10.1016/J.JPBA.2018.11.014
Hena, S., Gutierrez, L., and Croué, J. P. (2021). Removal of pharmaceutical and personal care products (PPCPs) from wastewater using microalgae: A review. J. Hazard. Mat. 403, 124041–124126. doi:10.1016/j.jhazmat.2020.124041
Krisko, R. M., McLaughlin, K., Koenigbauer, M. J., and Lunte, C. E. (2006). Application of a column selection system and DryLab software for high-performance liquid chromatography method development. J. Chromatogr. A 1122, 186–193. doi:10.1016/J.CHROMA.2006.04.065
Kumar, K. S., Dahms, H. U., Won, E. J., Lee, J. S., and Shin, K. H. (2015). Microalgae–A promising tool for heavy metal remediation. Ecotoxicol. Environ. Saf. 13, 329–352. doi:10.1016/j.ecoenv.2014.12.019
Lima, D. R. S., Tonucci, M. C., Libânio, M., and de Aquino, S. F. (2017). Fármacos e desreguladores endócrinos em águas brasileiras: Ocorrência e técnicas de remoção. Eng. Sanit. Ambient. 22, 1043–1054. doi:10.1590/s1413-41522017165207
Madureira, T. V., Barreiro, J. C., Rocha, M. J., Cass, Q. B., and Tiritan, M. E. (2009). Pharmaceutical trace analysis in aqueous environmental matrices by liquid chromatography-ion trap tandem mass spectrometry. J. Chromatogr. A 1216, 7033–7042. doi:10.1016/j.chroma.2009.08.060
Matuszewski, B. K., Constanzer, M. L., and Chavez-Eng, C. M. (1998). Matrix effect in quantitative LC/MS/MS analyses of biological fluids: A method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 70, 882–889. doi:10.1021/ac971078+
Metcalf, L., and Eddy, H. P. (2016). Tratamento de Efluentes e Recuperação de Recursos. Porto Alegre: AMGH.
Racz, N., Molnar, I., Zoldhegyi, A., Rjeger, H. J., and Kormany, R. (2018). Simultaneous optimization of mobile phase composition and pH using retention modeling and experimental design. J. Pharm. Biomed. Anal. 160, 336–343. doi:10.1016/j.jpba.2018.07.054
Radic, I., Runje, M., and Babic, S. (2020). Development of an analytical method for the determination of pimavanserin and its impurities applying analytical quality by design principles as a risk-based strategy. J. Pharm. Biomed. Anal. 201, 114091–114102. doi:10.1016/j.jpba.2021.114091
Snyder, L. R., Dolan, J. W., and Carr, P. W. (2004). The hydrophobic-subtraction model of reversed-phase column selectivity. J. Chromatogr. A 1060, 77–116. doi:10.1016/j.chroma.2004.08.121
Taylor, P. J. (2005). Matrix effects: The achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin. Biochem. 38, 328–334. doi:10.1016/j.clinbiochem.2004.11.007
The United States Pharmacopeial Convention (2022). PQRI approach for selecting columns of equivalent selectivity. Available at: https://apps.usp.org/app/USPNF/columnsDB.html (Accessed May, 2022).
Wilt, A., Butkovskyi, A., Tuantet, K., Leal, L. H., Fernandes, T. V., Langenhoff, A., et al. (2016). Micropollutant removal in an algal treatment system fed with source separated wastewater streams. J. Hazard. Mat. 304, 84–92. doi:10.1016/j.jhazmat.2015.10.033
Zhang, Y., Duan, L., Wang, B., Liu, C. S., Jia, Y., Zhai, N., et al. (2020). Efficient multiresidue determination method for 168 pharmaceuticals and metabolites: Optimization and application to raw wastewater, wastewater effluent, and surface water in Beijing, China. Environ. Pollut. 261, 114113–114123. doi:10.1016/j.envpol.2020.114113
Keywords: pharmaceutical compounds, liquid chromatography, wastewater, chromatographic modeling and simulation, DryLab®
Citation: Barreiro JC, Florentino AP, Furlani IL, Silva GHR and Cass QB (2022) An LC-MS/MS method for quantitation of methylparaben, benzophenone, and pharmaceutical compounds from domestic wastewater. Front. Anal. Sci. 2:941883. doi: 10.3389/frans.2022.941883
Received: 11 May 2022; Accepted: 16 August 2022;
Published: 12 September 2022.
Edited by:
Núria Fontanals, University of Rovira i Virgili, SpainReviewed by:
Maria Celeiro, University of Santiago de Compostela, SpainCopyright © 2022 Barreiro, Florentino, Furlani, Silva and Cass. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juliana Cristina Barreiro, juliana.barreiro@gmail.com; Quezia Bezerra Cass, qcass@ufscar.br
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.