 Loren Kell
Loren Kell Anna Katharina Simon3,4
Anna Katharina Simon3,4 Ghada Alsaleh
Ghada Alsaleh Lynne S. Cox
Lynne S. Cox
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging , 03 July 2023
Sec. Aging and the Immune System
Volume 4 - 2023 | https://doi.org/10.3389/fragi.2023.1202152
This article is part of the Research Topic Women in Aging and the Immune System View all 8 articles
Ageing is the biggest risk factor for the development of multiple chronic diseases as well as increased infection susceptibility and severity of diseases such as influenza and COVID-19. This increased disease risk is linked to changes in immune function during ageing termed immunosenescence. Age-related loss of immune function, particularly in adaptive responses against pathogens and immunosurveillance against cancer, is accompanied by a paradoxical gain of function of some aspects of immunity such as elevated inflammation and increased incidence of autoimmunity. Of the many factors that contribute to immunosenescence, DNA damage is emerging as a key candidate. In this review, we discuss the evidence supporting the hypothesis that DNA damage may be a central driver of immunosenescence through senescence of both immune cells and cells of non-haematopoietic lineages. We explore why DNA damage accumulates during ageing in a major cell type, T cells, and how this may drive age-related immune dysfunction. We further propose that existing immunosenescence interventions may act, at least in part, by mitigating DNA damage and restoring DNA repair processes (which we term “genoprotection”). As such, we propose additional treatments on the basis of their evidence for genoprotection, and further suggest that this approach may provide a viable therapeutic strategy for improving immunity in older people.
The past century has seen a marked increase in overall human lifespan (Oeppen and Vaupel, 2002; Dong et al., 2016), largely arising from improvements in diet, lifestyle, sanitation, and healthcare. However, this increase in life expectancy has not been met by an increase in healthy life expectancy, with ageing being the biggest risk factor for developing chronic diseases and multimorbidity (Barnett et al., 2012). Older people experience an overall decline in their responses to immune challenges such as vaccination (Lord, 2013; Wagner et al., 2018), resilience against novel pathogens [e.g., SARS-CoV2 (O'Driscoll et al., 2021; Hou et al., 2022)], and diminished antigen-specific memory responses (Vukmanovic-Stejic et al., 2018) together with reduced immunosurveillance for cancer neoantigens (Ovadya et al., 2018). Age-related immune dysfunction is also associated with an increase in low-grade inflammation, termed inflammageing (Franceschi et al., 2000). These maladaptive changes in immunity are broadly termed immunosenescence [reviewed in (Teissier et al., 2022)]. Despite major advances in the field, there are still significant gaps in our understanding of the causes of immunosenescence, and of how immunosenescence underpins the age-related increase in susceptibility to and severity of infections. As the recent COVID-19 pandemic highlighted, new therapies to alleviate immunosenescence are urgently needed, and our increasing understanding of ageing biology may provide such therapies (Cox et al., 2020).
Biological underpinnings of the ageing process have been encapsulated in the hallmarks of ageing (Lopez-Otin et al., 2013; Schmauck-Medina et al., 2022; Lopez-Otin et al., 2023). Immunosenescence is associated with many of these ageing hallmarks, including genome instability, while stromal cellular senescence and altered intercellular communication contribute to inflammageing (Aiello et al., 2019). However, though many ageing hallmarks overlap with immunosenescence, it is still unclear whether the hallmarks causally drive age-related immune decline and whether they act in a cell autonomous manner. For example, the proportion of cells which have undergone proliferative arrest and become senescent increases in both haematopoietic (immune) and non-haematopoietic compartments during ageing (Lopez-Otin et al., 2013; Tuttle et al., 2020; Mittelbrunn and Kroemer, 2021). Although many triggers for cellular senescence have been identified (van Deursen, 2014), there is accumulating evidence that these converge upon, and are further driven by, the ageing hallmark of genome instability and persistent DNA damage (Yousefzadeh et al., 2021a; Schumacher et al., 2021). Indeed, markers of DNA damage are observed to increase in the immune system during ageing, but until recently it was unknown whether this was either causal, or secondary to other age-related organismal changes. In support of a causative role, recent studies have demonstrated that inducing persistent DNA damage in haematopoietic cells alone is sufficient to recapitulate ageing-induced immunosenescence and organismal ageing (Desdin-Mico et al., 2020; Yousefzadeh et al., 2021b). While we do not know whether senescence of other cell types would have had this effect, these recent findings suggest that DNA damage in the immune system may be causal in precipitating immunosenescence and accelerating whole body ageing.
In this review, we explore the role of DNA damage in relation to age-related dysfunction of the immune system. We consider how DNA damage contributes to cellular senescence and accelerated ageing in haematopoietic cells and non-haematopoietic cells, and examine the causes of increased DNA damage in immune cells during ageing, focusing on T cells. We further discuss how senescence in non-blood-forming cells impairs immunity in older individuals. Finally, we suggest that therapies targeting immunosenescence, particularly through restoring genome stability (which we call “genoprotective” therapies), may be a promising approach for improving immunity in older people and reducing age-related risk of both chronic non-communicable and infectious diseases.
Endogenous and exogenous factors can lead to a variety of different types of genome lesions, on one or both strands of the DNA, and that occur at scales ranging from individual bases to much larger tracts of the genome (Huang and Zhou, 2021). Endogenous sources of DNA damage include reactive oxygen species (ROS), normal by-products of oxidative metabolism which are generated at abnormally high levels by dysfunctional mitochondria [a hallmark of ageing (Lopez-Otin et al., 2013)]; ROS can induce oxidative DNA damage and lead to DNA replication errors that require DNA excision repair pathways to correct. Exogenous UV light can induce lesions including (6–4) photoproducts and cyclobutane pyrimidine dimers (i.e., covalently linked adjacent cytosine or thymidine bases on one DNA strand), while interstrand crosslinks can be formed by crosslinking agents such as mitomycin C. Other exogenous sources of DNA damage, such as ionising radiation, or radiomimetic genotoxins such as etoposide or bleomycin, can induce single- or double-strand breaks (SSBs or DSBs) i.e., breaks in one or both of the DNA strands of a duplex (Mason and Cox, 2012).
Distinct pathways have evolved to repair the different types of DNA lesions [reviewed recently in (Huang and Zhou, 2021)]. These include i) direct reversal of the damage, ii) base excision repair (BER), iii) nucleotide excision repair (NER) for bulkier adducts and complicated lesions, iv) mismatch repair (MMR), v) interstrand crosslink (ICL) repair, vi) single-strand break (SSB) repair and vii) double-strand break (DSB) repair, chiefly by either homologous recombination (HR) or non-homologous end joining (NHEJ). Damage to the DNA triggers the DNA damage response (DDR), which involves a tightly coordinated recruitment of damage sensors, transducers and effectors that transiently arrest the cell cycle, and drive cell fate decisions to either DNA repair, apoptosis, or senescence. Figure 1 provides a summary of DNA lesion types and their canonical repair mechanisms. Indeed, the presence of DNA damage can be probed by measuring the activation and/or localisation of proteins involved in the DDR pathways, such as γH2AX (histone variant H2AX phosphorylated at Ser139), p53-binding protein 1 (53BP1) (Huang and Zhou, 2021), and the phosphorylation of key DDR master regulators, ATM (at Ser1981) and ATR (at Thr1989). It is possible to probe for specific DNA lesion epitopes [e.g., 8-oxo-dG – e.g., (Sanderson and Simon, 2017)], while DNA fragmentation due to SSBs and DSBs can be directly measured using single cell gel electrophoresis (the comet assay); inverse comet assays can instead provide a readout of extent of interstrand crosslinking (Swift et al., 2020). Overall, cells have evolved a plethora of mechanisms to repair DNA damage; whether these are implemented successfully plays a crucial role in determining cell fate.
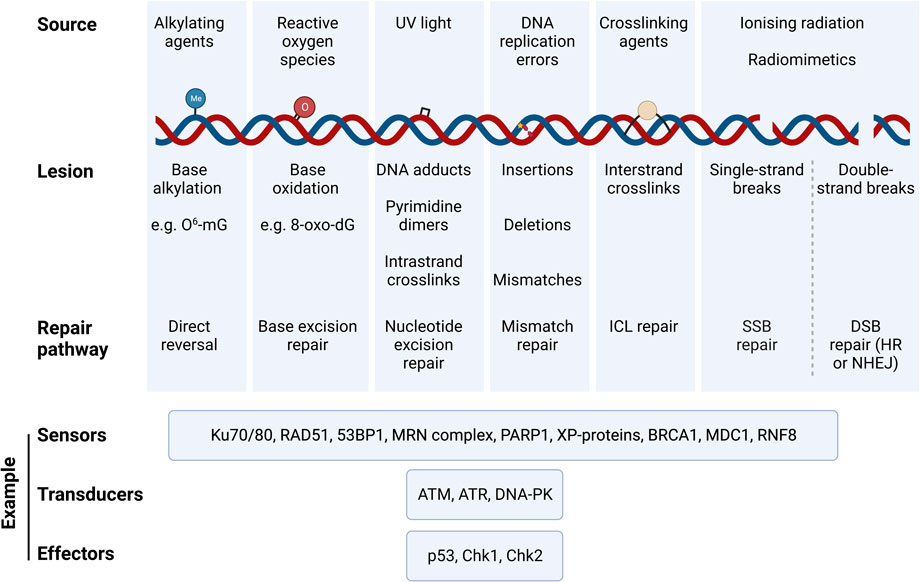
FIGURE 1. Types of DNA damaging agents, resulting lesions and canonical repair pathways that correct them. DNA experiences a variety of genotoxic insults from both endogenous and exogenous sources. These result in specific lesions that can be repaired via specific DNA repair pathways that are triggered by the DNA damage response (DDR). Damaged DNA is sensed, signalled, and repaired through a cascade of signalling mechanisms elicited by DNA sensors, tranducers, and effectors, which are specific to the DNA lesion type and repair pathway. Examples of such DDR proteins are illustrated. Abbreviations: O6-mG, O-6-methylguanine; 8-oxo-dG, 8-oxo-2′-deoxyguanosine; ICL, interstrand crosslink; HR, homologous recombination; NHEJ, non-homologous end joining; 53BP1, p53·-binding protein 1; MRN complex, Mre11/Rad50/Nbsl; PARP1, Poly [ADP-ribose] Polymerase 1; XP-proteins, Xeroderma Pigmentosum proteins; BRCA1, breast cancer gene 1; MDC1, Mediator of DNA Damage Checkpoint 1; RNF8, Ring Finger Protein 8; ATM, Ataxia-Telangiectasia Mutated; ATR, Ataxia Telangiectasia and Rad3-related; DNA-PK, DNA Protein Kinase complex; Chk1, Checkpoint Kinase 1; Chk2, Checkpoint Kinase 2.
Cellular senescence, a state of permanent proliferative arrest accompanied by a host of morphological, metabolic and functional changes, was originally discovered during long-term in vitro culture of human lung fibroblasts (Hayflick and Moorhead, 1961). Senescent cells accumulate in tissues during normal ageing (van Deursen, 2014; Tuttle et al., 2020) and increased senescent cell burden is a hallmark of ageing (Lopez-Otin et al., 2023). Senescence has been shown to be a causative factor in ageing, contributing to age-related morbidity and mortality – for instance, transplanting senescent cells into mice accelerates physical frailty and shortens lifespan (Xu et al., 2018). Moreover, deletion of senescent cells via genetic (Baker et al., 2016) or pharmacological means [e.g., fisetin (Yousefzadeh et al., 2018), or dasatinib + quercetin, “D + Q” (Xu et al., 2018)] increases median lifespan and ameliorates a range of age-related conditions in mice, thus extending healthspan. Importantly, such senolytic treatment also improved survival following coronavirus infection (Camell et al., 2021; Cox and Lord, 2021), potentially through reducing inflammatory signalling; whether such treatment also improves adaptive immunity is not yet clear. Cellular senescence therefore drives ageing and inflammation.
Although no unique biomarkers for senescence have yet been described, it is associated with high expression of the tumour suppressor p53 (and also at times p-Rb), and the cyclin kinase inhibitors CDKN1A (p21Cip1) and CDKN2A (p16INK4a) which establish and maintain proliferative arrest (van Deursen, 2014). Features of senescent cells include stabilised actin stress fibres (Walters et al., 2016), increased activity of lysosomal senescence-associated β-galactosidase (SAβG) at suboptimal pH 6.0 (Dimri et al., 1995), enlarged nucleoli (Buchwalter and Hetzer, 2017), chromatin rearrangements [e.g., SAHFs (Narita et al., 2003)], nuclear lamina breakdown leading to cytoplasmic chromatin fragments [CCFs (Ivanov et al., 2013)] and persistent expression of DNA damage markers such as γH2AX and 53BP1 (Rodier et al., 2011). Senescent cells possess a unique secretome consisting of inflammatory cytokines, matrix-remodelling proteins, and extracellular vesicles, collectively termed the senescence-associated secretory phenotype (SASP) (Coppe et al., 2008; Rodier et al., 2009). Senescence markers in the immune system are similar to those in other cell types, and a variety of tools, including flow cytometry, has proven useful in identifying senescent immune cells (Table 1) (Zhou et al., 2021).
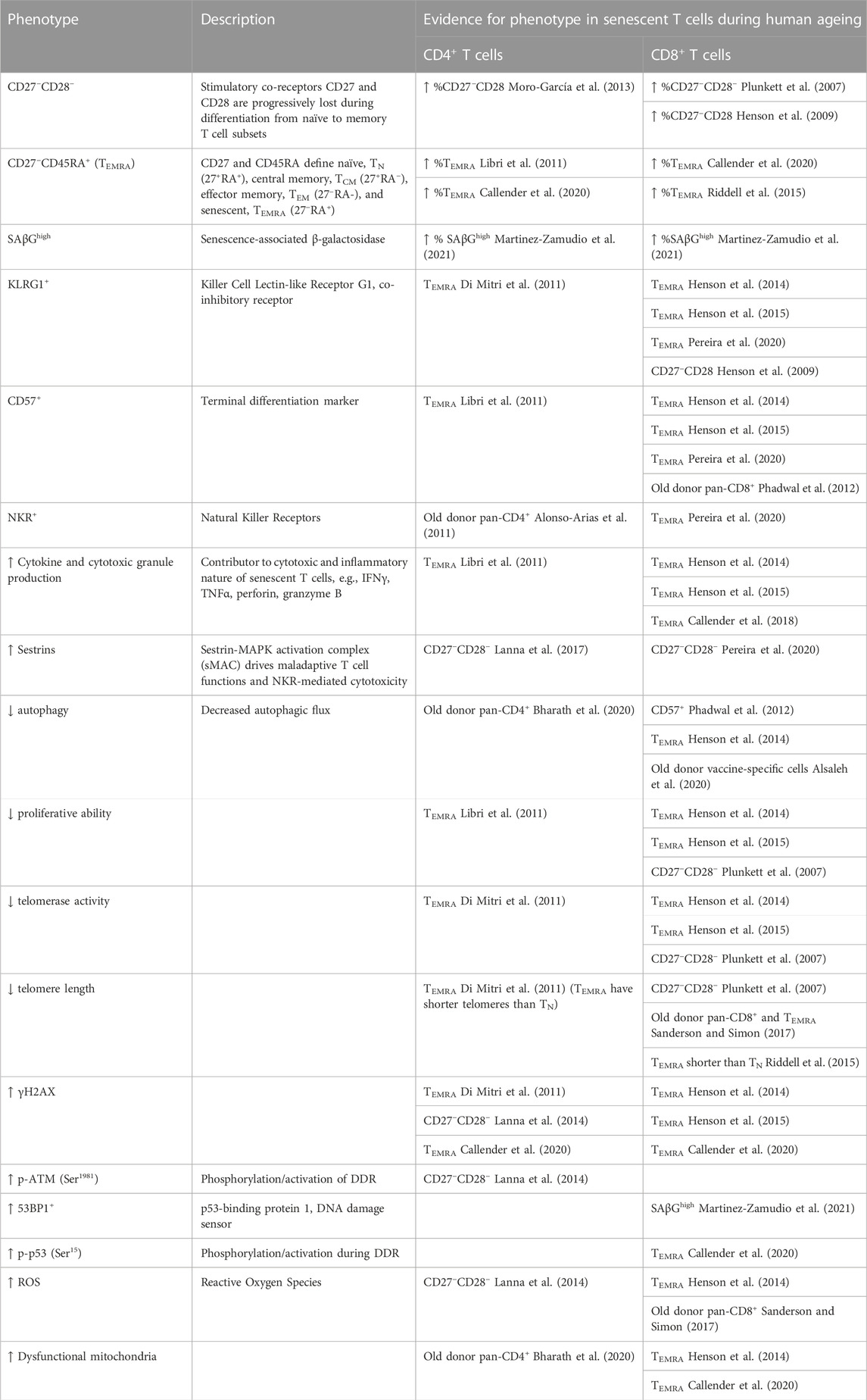
TABLE 1. Characteristics of senescent T cells.
A variety of senescence inducers have been characterised, including oncogene activation leading to oncogene-induced senescence (OIS) and some viruses, such as SARS-CoV2, leading to virus-induced senescence (VIS) (Lin et al., 1998; Lee et al., 2021); major DNA damage is also a trigger leading to senescence, and persistent DNA damage is often a feature of other senescence types (Fumagalli et al., 2014). Indeed, an imbalance between DNA damage and repair is a primary driver of cell cycle proliferative arrest resulting in one of three key cell fates: persistent arrest, cell death, or cell senescence. How this imbalance arises depends on the type of damage signal involved.
In replicative senescence (RS), the type of cell senescence thought to arise most commonly during normal ageing, damage persistence is a consequence of telomere attrition (Harley et al., 1990). Loss of telomeric DNA at every round of DNA replication gradually erodes telomeres until they become critically short, which impairs the stabilisation of the protective shelterin complex (Rossiello et al., 2014). Uncapping of eroded telomeres is sensed as a DSB, triggering a persistent DDR and permanent exit from the cell cycle. The shortening of telomeres towards this critical length after each mitotic cell cycle therefore fundamentally limits the replicative lifespan of cells, i.e., drives replicative senescence. Maintaining telomere length and capping is therefore important for chromosomal stability and to prevent early onset of senescence. Indeed, ectopic expression of telomerase, which extends telomeres, overcomes premature senescence phenotypes in progeroid Werner syndrome cells in vitro (Wyllie et al., 2000), while experimental telomerase reactivation promotes tissue rejuvenation in mice (Jaskelioff et al., 2011). Unlike most somatic cells where telomerase is repressed, adaptive immune T and B cells can upregulate telomerase expression, which prolongs their replicative potential; this is likely to be an evolved response to permit multiple rounds of clonal proliferation upon antigenic stimulation without inducing replicative exhaustion (Akbar et al., 2016). Remarkably, T cells from an immunised mouse could be transferred and expanded in 51 successive recipient mice, over 10 years, without showing any proliferative dysfunction or telomere shortening (Soerens et al., 2023). However, even where telomere length has not reached the point of shelterin destabilisation, DNA damage – particularly 8-oxo-dG – within the telomeric regions is effective at signalling for cell cycle arrest and senescence (von Zglinicki, 2000). Stress-induced premature senescence (SIPS) can occur, e.g., in response to oxidising environments and can result in telomere oxidation. Notably up to half of all DNA damage foci (as measured by a combination of immunofluorescence and ChIP) have been reported to localise at telomeres (Hewitt et al., 2012) suggesting either that telomeres are particularly prone to damage, or that their heterochromatic nature precludes effective repair (Fumagalli et al., 2012; von Zglinicki, 2002). Telomeric vulnerability due to their high guanine content remains the most likely explanation.
As well as oxidative damage, exposure to genotoxic agents can drive DNA damage-induced cellular senescence (DIS). Following transient cell cycle arrest immediately after DNA damage, in cases where DNA lesions are successfully repaired (Figure 1), the cell may re-enter the proliferative cell cycle. However, in situations where the damaging agent persists, where the lesion burden is high, or where the cell is unable to repair the DNA lesions (e.g., through loss of function of critical DNA repair factors), persistent DDR signalling can result in irreversible cell cycle arrest and DIS. DIS is considered to have evolved as a tumour-suppressive strategy to limit cancerous transformation in the presence of a high lesion burden (van Deursen, 2014).
DNA damage is therefore well established as a driver of senescence. It is also strongly associated with pathology during ageing (Schumacher et al., 2021). Indeed, the majority of human premature ageing (progeroid) syndromes (e.g., Werner syndrome, Rothmund-Thomson syndrome, Bloom syndrome, Cockayne’s syndrome, Fanconi’s anaemia, and Ataxia-Telangiectasia) result from mutations in DNA repair factors (WRN, RECQL4, BLM, CSB, FANC proteins and ATM respectively); patients show premature ageing across multiple organs often accompanied by greatly elevated cancer incidence, and patient-derived cells senesce prematurely in culture (Cox and Faragher, 2007). In Hutchinson-Gilford progeria, where the driver mutation occurs in the lamin A/C gene, i.e., not a DNA repair factor, the resulting instability of the nuclear lamina leads to significant genome instability and DNA breakage (Liu et al., 2005). Hence progeroid syndromes provide strong evidence that DNA damage is a major driver of senescence and ageing (Yousefzadeh et al., 2021a).
Further to DNA damage being an inducer of senescence, unrepaired DNA lesions propagate further cellular dysfunction and hallmarks of ageing, thereby positioning DNA damage as a major node in senescence and ageing (Schumacher et al., 2021). For example, free DNA ends at sites of damage are recognised by poly [ADP-ribose] polymerase 1 (PARP1) which uses NAD+ as a substrate for sensing the damaged DNA (Yoshino et al., 2018). Continuous and persistent DNA damage therefore leads to increased PARP1 activity and depletion of NAD+ stores, resulting in reduced function of other NAD+-requiring enzymes such as the sirtuins, a family of NAD-dependent deacetylases, some of which are important regulators of chromatin organisation and mitochondrial health, associated with longevity (Fang et al., 2014). Maintaining sirtuin activity limits senescence: indeed, the age-related loss of SIRT6 activity results in heterochromatin decompaction and expression of retrovirus cDNA in the cytosol, triggering innate cGAS-STING signalling and an interferon-induced SASP response (De Cecco et al., 2019; Simon et al., 2019). Similarly, loss of sirtuin activity in DNA repair-deficient models [e.g., XPA (Fang et al., 2014)] demonstrate that DNA damage can induce mitochondrial dysfunction and additional ROS production, and that ROS is requisite for further deepening senescence (Passos et al., 2010). DNA damage can thus drive a positive feedback mechanism to propagate further dysfunction and senescence.
Since DNA damage is an important trigger and propagator of senescence and ageing pathology, supporting and enhancing cells’ ability to repair DNA lesions and limit senescence may be a valuable strategy for promoting healthy ageing.
Immunosenescence describes organism-level age-related dysfunction of both innate and adaptive immunity, which leads to increased susceptibility to pathogens [most recently exemplified by the COVID-19 pandemic (Cox et al., 2020)], poor vaccine responses (Lord, 2013), elevated cancer risk, and increased autoimmunity (Goronzy and Weyand, 2012). Additionally, immunosenescence is associated with a low-grade inflammatory environment that is generated, at least in part, by the SASP of senescent cells of both immune and non-immune origin (Teissier et al., 2022). Indeed, immunosenescence is partially underpinned by the age-related changes in immune cells, leading to a variety of dysfunctional phenotypes, several of which are characteristic of senescence (Zhou et al., 2021). However, not all age-related changes to immune cells are indicative of cellular senescence - for example, T cells with an exhausted phenotype accumulate with age and/or chronic antigenic stimulation, such as CMV exposure (Fletcher et al., 2005). Furthermore, the age-related shifts in other immune cell subsets, particularly with a bias from anti-inflammatory to pro-inflammatory cells, may contribute to immune dysfunction consistent with immunosenescence (Marquez et al., 2020).
Here we discuss the unique function of physiological DNA damage in lymphocytes. We then discuss how persistent immune DNA damage is associated with ageing and immunosenescence, with a particular focus on T cell senescence. We further describe the known sources of T cell DNA damage, and how genotoxic stress may cause the maladaptive phenotypes of senescent T cells that promote immunosenescence. Finally, we discuss how the senescence of non-haematopoietic cells can suppress adequate antigen-specific responses and thus reduce older people’s ability to respond effectively to neoantigens, leading to risk of severe illness or death from infectious disease.
DNA damage within the immune system is not always deleterious. In fact, the introduction of DNA lesions plays an evolved role in generating receptor diversity in the B and T cells of the adaptive immune system; such physiological genome alterations do not trigger the irreversible cell cycle arrest characteristic of cell senescence (Gullickson et al., 2022). V(D)J recombination, employed by both cell types, relies on the induction of DSBs that allow the rearrangement of individual Variable, Diversity, and Joining segments to produce a unique repertoire of immune receptors. B cell receptor genes undergo the critical processes of somatic hypermutation and class-switch recombination, both of which require the introduction of DNA lesions by the enzyme activation-induced cytidine deaminase, and modification by canonical DNA repair pathways and factors - including DNA-PK, Ku, Artemis, DNA ligase IV, and ERCC4 as well as recombination-inducing RAG proteins (Gellert, 2002). Crucially, loss of key DNA repair proteins, particularly those involved in DSB repair, leads to severe combined immunodeficiency characterised by a reduction of T and B cell populations and poor responses to immune challenge (Gullickson et al., 2022). Maintaining proper DNA damage and repair processes is therefore central for normal immune function.
The hypothesis that increased DNA damage in the immune system drives accelerated ageing and immunosenescence is supported by a growing body of literature. A recent meta-analysis which evaluated DNA damage in the circulating immune cells of 2,403 individuals, as measured by the comet assay, reported that higher levels of damage were associated with increased mortality (Bonassi et al., 2021). Further to this correlative association, immune DNA damage has been linked causally to increased mortality and ageing. For instance, immune-specific genetic knockout of Ercc1, an endonuclease involved in multiple DNA repair pathways, is sufficient to phenocopy many aspects of immunosenescence and drive age-related changes in mice, including accelerated ageing of solid organs of non-immune lineages (Yousefzadeh et al., 2021b). Ercc1-deficient immune cells showed increased levels of the DNA damage marker γH2AX, and DNA oxidation product 8-oxo-dG, and their high expression of the cell cycle suppressor, Cdkn2a, indicated permanent exit from the cell cycle and senescence (Yousefzadeh et al., 2021b). Although it is not clear which haematopoietic cells drive this phenotype, the abundance of T cells in the immune system could contribute to a large extent, especially given that DNA damage accumulates in these cells and is associated with normal human ageing (discussed later). Supporting the view that T cells may be the major cell type responsible is a recent study using Tfamfl/fl Cd4Cre mice, in which the nuclear-encoded mitochondrial transcription factor A (TFAM) was knocked out in T cells only (Tfam encodes a protein involved in mitochondrial DNA stability, replication, and transcription) (Desdin-Mico et al., 2020). These mice had significantly shorter lifespans compared to controls with functional TFAM, and exhibited the premature onset of age-related muscular, cardiovascular, and cognitive dysfunctions, as well as age-related susceptibility to an acute viral infection, indicating premature immunosenescence (Desdin-Mico et al., 2020).
Though these studies utilise mice which harbour non-physiological mutations driving a severe pro-ageing phenotype, they suggest that DNA damage and senescence in immune cells alone is sufficient to drive age-related dysfunction. It does not exclude the possibility that DNA repair deficiency in other individual non-haematopoietic cell types may have progeroid effects. However, given that increased DNA damage in the immune system is observed during physiological ageing in T cells, and T cells are both numerous and readily available from the blood of older adults, targeting DNA repair capabilities in these cells may serve as a valuable axis in designing therapeutics to promote healthy longevity.
Increased DNA damage during immunosenescence has been best characterised in the senescence of T cells during ageing. Naïve and memory T cells are long-lived cells of the adaptive immune system that become activated via their T cell receptor (TCR) to induce clonal proliferation and cytokine secretion. The specific functions of T cells are determined by their CD4+ or CD8+ subtype. Human senescent T cells are terminally differentiated, antigen experienced cells that are defined by their successive loss of expression of the co-stimulatory molecules, CD27 and CD28, together with high expression of CD57 and KLRG1 (Henson et al., 2014). T cell differentiation subsets can be determined by their relative expression of CD27 and CD45RA, thus defining four subsets: naïve (TN, CD27+CD45RA+), central memory (TCM, CD27+CD45RA−), effector memory (TEM, CD27−CD45RA−), and effector memory re-expressing CD45RA, known as TEMRA (CD27−CD45RA+). TEMRA cells are considered a senescent memory subset of T cells (Henson et al., 2014). Senescent T cells can also be identified by their high activity of senescence-associated β-galactosidase (SAβG), a classical senescence biomarker (Martinez-Zamudio et al., 2021). Thus, senescent T cells have been defined in the literature as being CD27−CD28−, CD27−CD45RA+ (TEMRA), or more recently as SAβGhigh cells within the CD4+ or CD8+ subsets.
Though defined by differing markers, CD27−CD28−, TEMRA, and SAβGhigh cells crucially share the characteristics both of being greatly expanded during chronological ageing and of displaying many features of senescence. The characteristics of these senescent T cell subsets are summarised in Table 1. SAβGhigh senescent T cells accumulate in both CD4+ and CD8+ T cell compartments during ageing and are particularly enriched in the TEMRA terminally-differentiated subpopulation (Martinez-Zamudio et al., 2021). Compared to non-senescent SAβGlow cells, senescent SAβGhigh CD8+ T cells have impaired proliferative ability and high p16/INK4A expression. Furthermore, increased T cell expression of the cell cycle arrest gene p16/INK4A is an accurate predictor of chronological age in humans (Liu et al., 2009). Compared with non-senescent cells, the functions of senescent TEMRA cells are severely impaired and include the inability both to stimulate proper downstream TCR signalling and to proliferate after activation - though, unlike senescent fibroblasts, their proliferative ability is not fully lost (Henson et al., 2014; Akbar et al., 2016). Senescent CD8+ TEMRA cells are highly cytotoxic and, much like senescent fibroblasts, produce a SASP with immune-active components (namely, TNF-α and IFN-γ) after short-term activation, that may contribute to the low-grade inflammatory environment in older people (Henson et al., 2014; Henson et al., 2015; Akbar et al., 2016). T cells from older adults have lower autophagic flux (Phadwal et al., 2012), contain dysfunctional mitochondria, and have higher levels of ROS (Henson et al., 2014; Sanderson and Simon, 2017) than their younger counterparts, all of which are hallmarks of ageing (Lopez-Otin et al., 2023). It is not clear if the accumulation of TEMRA (or senescent T cells) with age may be adaptive or maladaptive–for example, their hyper-cytotoxic and -secretory phenotypes may beneficially compensate for the increased burden of malignant cells during ageing, but bear the disadvantages of increased destruction of healthy self-tissues (Pereira and Akbar, 2016). Nevertheless, many of these age-related dysfunctional characteristics have been linked to the impaired function of senescent T cells, and reversing these characteristics may promote immune resilience in old people.
Senescent T cells display markers of persistent DNA damage (Table 1). They have high expression of γH2AX, low telomerase expression, and critically short telomeres (Di Mitri et al., 2011; Henson et al., 2014; Sanderson and Simon, 2017). Even though T cells retain telomerase expression for far longer than other somatic cells, telomere erosion is observed in T cells (and peripheral blood mononuclear cells, PBMCs, more generally) during ageing (Lin et al., 2015; Sanderson and Simon, 2017). Similarly, compared to non-senescent SAβGlow cells, SAβGhigh senescent T cells are enriched for the DNA damage marker, 53BP1, especially at telomeres (Martinez-Zamudio et al., 2021). Further evidence for the relationship between DNA damage and ageing T cells comes from DNA repair-deficient mice (Ercc1−/Δ), which experience an accumulation of highly-differentiated memory T cells akin to naturally aged wildtype mice (Pieren et al., 2021). Overall, senescent T cells with persistent DNA damage accumulate in older people. Due to their dysfunctional phenotype and cytotoxic nature, senescent T cells are therefore thought to contribute both to immunopathology and immunosenescence during ageing (Akbar et al., 2016; Covre et al., 2020).
The causes of DNA damage in aged T cells may be attributed both to endogenous and exogenous sources of genotoxins, and to the endogenous decline of DNA repair factor expression. In this section we discuss the sources of DNA damage which T cells experience across the lifespan. These are summarised in Figure 2.
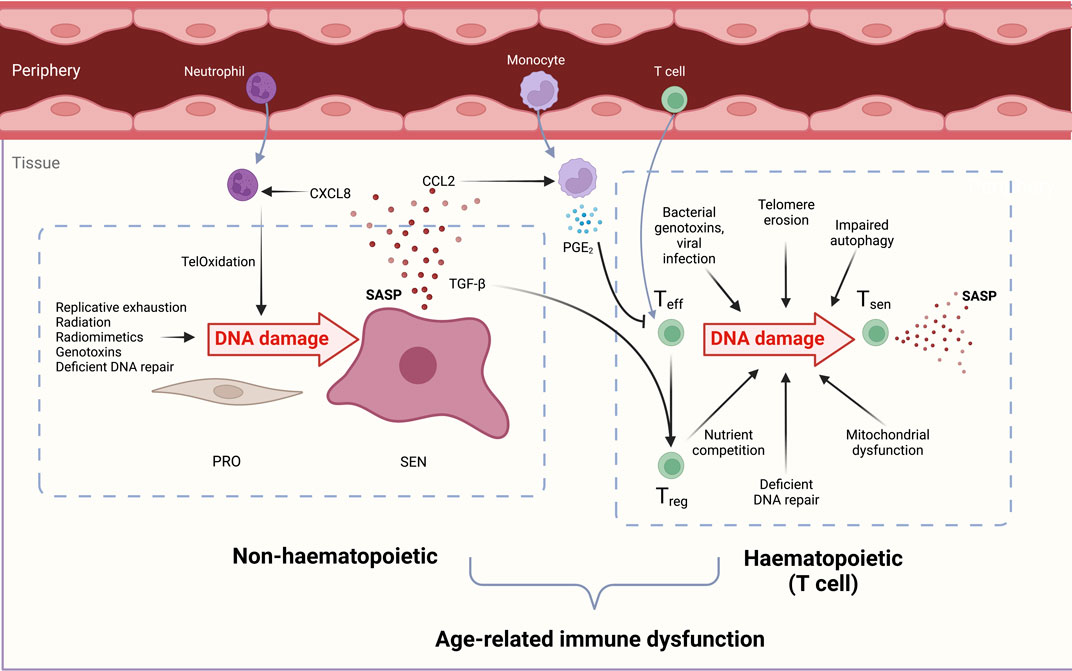
FIGURE 2. Overview of the causes and consequences of DNA damage in both haematopoietic (T cell) and non-haematopoietic cells which contribute to immunosenescence. DNA-damaged immune and non-haematopoietic cell senescence are linked. During ageing, senescent cells increase in both compartments due to reduced senescent cell clearance, deficient DNA repair processes, and increased genotoxin abundance. Non-haematopoietic cells: Proliferating cells (PRO) experience genotoxicity from radiation, excessive replication, chemotherapeutic drugs, and radiomimetics to drive persistent DNA damage, proliferation arrest, and cellular senescence (SEN). The senescence-associated secretory phenotype (SASP) contains chemoattractants such as CXCL8 that draw in peripheral neutrophils. These induce oxidative damage in stromal cell telomeric DNA, called TelOxidation, propagating further paracrine senescence. The SASP component CCL2 attracts monocytes which secrete PGE2 and suppress T cell functions. TGF-β in the SASP favours Treg generation and a pro-suppressive, inadequate immune response during ageing. Haematopoietic cells: Effector T cells (Teff) experience genotoxic injury from bacterial genotoxins viral infections short telomeres, reactive oxygen species (ROS) from dysfunctional mitochondria, and nutrient competition with regulatory T cells (Tregs). DNA-damaged T cells manifest senescent phentotypes in a sestrin- and ATM-dependent manner. Senescent T cells (Tsen) are dysfunctional and contribute to inflammageing with their inflammatory SASP, tissue cytotoxicity, and immune dysfunction during ageing.
Due to the highly proliferative nature of T cells upon activation, chronic stimulation over the lifecourse leads to telomere erosion (Lin et al., 2015; Sanderson and Simon, 2017). Although T cells can upregulate telomerase in order to maintain their telomeres, this ability is impaired during ageing, especially in the CD27−CD28− senescent T cell population (Plunkett et al., 2007; Patrick et al., 2019). This leads to critically short telomeres which, as previously discussed, triggers a persistent DNA damage response and subsequent senescence. A recent study identified a novel mechanism by which human T cells may acquire telomeres delivered in extracellular vesicles from antigen-presenting cells, which increases their replicative capacity and prevents senescence (Lanna et al., 2022). It would be particularly interesting to determine whether this intercellular telomere transfer occurs as efficiently in old as it does in young individuals, as failure with ageing could potentially contribute to telomere attrition in T cells.
Autophagy is a process by which cells degrade excess or damaged intracellular proteins and larger cargo such as organelles; altered autophagy is recognised as a hallmark of ageing (Schmauck-Medina et al., 2022; Lopez-Otin et al., 2023). Autophagic flux decreases in both B and T cells during ageing (Zhang et al., 2019; Alsaleh et al., 2020), which correlates with increased DNA damage, as measured by γH2AX expression (Phadwal et al., 2012). Autophagy is required for an adequate memory response to antigen challenge (Puleston et al., 2014). Reduced autophagy partly underlies the defective T cell response in old people: much like senescent T cells, Atg7-deficient T cells, which are autophagy-deficient, are impaired in their proliferative capacity after antigen exposure (Zhou et al., 2022). Ineffective autophagy in T cells from old donors also underlies other senescent T cell phenotypes that are capable of driving T cell DNA damage, such as dysfunctional mitochondrial biogenesis.
Senescent T cells from older people harbour large mitochondria with disrupted bioenergetics that release excessive reactive oxygen species (ROS) (Henson et al., 2014; Sanderson and Simon, 2017; Bharath et al., 2020). Excessive ROS release can cause oxidative damage of both genomic and mitochondrial DNA (Henson et al., 2014). This leads to a vicious cycle: since the mitochondrial genome encodes key components of the electron transport chain (ETC.), oxidative damage, combined with low levels of DNA repair operational for mtDNA, leads to further disruption in oxidative phosphorylation and hence higher ROS levels (Rong et al., 2021). ROS also contribute to telomere damage and attrition observed in T cells cultured long term in vitro, which can be rescued by addition of a ROS scavenger, N-acetyl-L-cysteine (Sanderson and Simon, 2017). Since autophagy is important in removing damaged mitochondria (termed mitophagy), the decline in basal autophagy observed in old lymphocytes is thought to contribute to accumulation of dysfunctional mitochondria that produce genotoxic ROS (Bektas et al., 2019). Accordingly, stimulating autophagy in T cells using either spermidine, metformin, or a p38 MAPK inhibitor, reduced the presence of giant dysfunctional mitochondria and restored mitochondrial bioenergetics, indicating an overall improvement in mitochondrial quality (Henson et al., 2014; Alsaleh et al., 2020; Bharath et al., 2020). It is unclear, however, whether these effects are due solely to the specific induction of mitophagy or other autophagy-induced processes, though this could be explored further - for example, with autolysosome-dependent fluorescent mitochondrial dyes such as that employed by Bektas and others (Bektas et al., 2019).
The ability of T cells to respond to DNA damage may determine their susceptibility to undergo senescence. After DNA damage, AMPK activation causes CD4+ T cells to increase their expression of the transcription factor, GATA3 (Callender et al., 2021). By complexing with ATR, PGC1α, and NRF2, GATA3 promotes the transcription of genes involved in mitochondrial biogenesis, thereby limiting ROS-mediated oxidative stress and preventing senescence (Callender et al., 2021). However, CD8+ T cells, unlike CD4+ T cells, do not increase GATA3 expression and mitochondrial biogenesis after DNA damage, which may partly explain their increased susceptibility to senescence and the higher proportion of senescent CD8+ than CD4+ T cells in peripheral blood (Czesnikiewicz-Guzik et al., 2008; Callender et al., 2020). This difference between CD8+ T cells and CD4+ T cells may be related to the fact that CD4+ T cells can develop into a variety of subsets (e.g.,.Th1, Th2, Th9, Th17, Tregs) and mitochondrial respiration may play a role in this lineage flexibility, whereas CD8+ T cells are mostly of the same subtype (cytotoxic) and therefore also more limited to one type of energy production. Overall, autophagy may limit genotoxic ROS production by removing dysfunctional mitochondria and ensuring mitochondrial quality is maintained. Hence age-related decreases in autophagy may contribute to both telomere attrition and oxidative DNA damage during T cell ageing and senescence.
While failure of mitochondrial biogenesis may be key to CD8+ T cell senescence, nutrient competition has been suggested to induce DNA damage and senescence in CD4+ T cells. Regulatory T cells (Tregs) play a role in restricting immune activation by inhibiting immune cell proliferation to prevent pathological hyperinflammatory responses. Naïve CD4+ T cells, when co-cultured in vitro with CD25highFoxP3+ CD4+ Tregs, displayed increased DNA damage markers (γH2AX and 53BP1) and active DDR signalling (p-ATM and p-Chk2) as well as other senescence phenotypes, including cell cycle arrest and high senescence-associated β-galactosidase activity (Ye et al., 2012; Liu et al., 2018). Since co-culturing in high glucose prevented these DNA damage phenotypes, this indicates that insufficient glucose, potentially due to the high consumption by Tregs, may be involved. Indeed, in naïve T cells co-cultured with Tregs, the resulting low glucose levels and subsequent p-AMPK activation may stimulate mitochondrial biogenesis and a shift from glycolysis to oxidative phosphorylation, driving ROS production and DNA damage (as discussed earlier). Treg-mediated immune regulation in this way could plausibly be a short-term advantageous mechanism, as induction of senescence in the effector T cells would limit their functions and therefore an overexuberant immune response. However, as senescent T cells display an inflammatory SASP and poor proliferative ability, this regulation may have longer-term drawbacks by impairing immune function. Furthermore, this process may be exacerbated in an ageing context where tissues are enriched with Tregs (Agius et al., 2009), which could plausibly lead to excessive induction of senescence in effector T cells.
Infections can directly and indirectly induce DNA damage and cellular senescence. Since immune cells accumulate at sites of infection, these cell types are ‘first responders’ which are repeatedly exposed to pathogen-derived genotoxic insults. Bacterial genotoxins, such as the typhoid toxin from Salmonella Typhi induces a γH2AX response induced by a genotoxin, or RING, in human fibrosarcoma cells, which is dependent on ATR-mediated hyperphosphorylation of RPA (Ibler et al., 2019). These RINGs comprise a non-canonical and persistent DDR that induces cellular senescence. Toxin-induced senescent cells have a unique SASP (txSASP) that further drives paracrine senescence and promotes Salmonella infection (Ibler et al., 2019). Overall, bacterial genotoxins may have evolved to enhance bacterial infection and transmissibility due to their pro-senescence effects.
Like some bacteria, virus infection is also an inducer of DNA damage and senescence (Lee et al., 2021; Gioia et al., 2023). Virus-induced senescence (VIS) may have evolved as an innate mechanism to inhibit viral exploitation of host replication machinery. However, some viruses evolved to exploit their pro-senescence effects to increase their infection ability. For example, coronavirus (e.g., SARS-CoV2, which causes COVID-19) induces cellular senescence and the SASP; this virus-induced SASP promotes upregulation of viral entry proteins, ACE2 and TMPRSS2, and downregulation of antiviral factors in adjacent cells, thus enhancing further infection susceptibility (Camell et al., 2021; Lee et al., 2021). SARS-CoV2 infection causes DNA damage in vitro and in vivo in COVID-19 patients: distinct SARS-CoV2 viral proteins both promote degradation of the DDR enzyme, Chk1, and outcompete 53BP1 for binding to DNA damage-induced long non-coding RNAs, thus preventing repair of DSBs (Gioia et al., 2023). Importantly, removing senescent cells improved the survival of mice after beta-coronavirus infection, indicating the core role of VIS in the pathology of viral infection (Camell et al., 2021; Cox and Lord, 2021).
Since the immune system is intimately involved with infection resolution, it is likely that immune cells are frequently exposed to bacterial genotoxins and viral senescence inducers. Indeed, exposure of activated T cells to cytolethal distending toxin, a genotoxin produced by many pathogenic Gram-negative bacteria, induces DNA damage, senescence, and ATM-induced SASP (Mathiasen et al., 2021). Suppression of effector T cell function by inducing senescence may be an evolved strategy by pathogens to suppress the adaptive immune response (Mathiasen et al., 2021). Other pathogens, such as the parasites belonging to the Leishmania genus, have exploited this to induce senescent T cells with DNA damage, in order to hijack their hyper-cytotoxic and hyper-secretory functions and further drive immunopathology (Covre et al., 2018; Covre et al., 2020). Furthermore, IFN-α, as part of the antiviral Type I interferon response, may induce senescent phenotypes in T cells by inhibiting telomerase and driving telomere erosion (Reed et al., 2004; Lanna et al., 2013). Therefore, infections may drive immune DNA damage by both direct effects of the infectious agent, and by secondary effects of cytokine release on immune cell biology.
Heightened inflammation with increasing age, termed ‘inflammageing’, is now recognised as a major hallmark of ageing (Schmauck-Medina et al., 2022), and has been heavily implicated in driving immunosenescent phenotypes. Chronic activation of the immune system, such as in chronic viral infections and autoimmune diseases (e.g., rheumatoid arthritis), fosters a highly inflammatory environment. These disease conditions feature many of the ageing hallmarks, and have therefore been proposed to be conditions of accelerated ageing (Alsaleh et al., 2022). These conditions also mimic the accumulation of senescent CD28− T cells observed during normal ageing, and so they may provide valuable contexts to understand immunosenescence (Alsaleh et al., 2022).
T cells from donors with a chronic virus infection (e.g., HBV, HCV or HIV) have short telomeres and display high levels of DNA damage (Ji et al., 2019); indeed, the severity of liver fibrosis negatively correlates with T cell telomere length in chronic HCV infection (Hoare et al., 2010; Hohensinner et al., 2011). T cells from chronically virus-infected donors are hypersensitive to DNA damage agents, such as topoisomerase I (TOP1) inhibition by camptothecin (Ji et al., 2019). The decrease in T cell genome stability in these patients can be attributed to 1) chronic activation-induced proliferation leading to telomere erosion and 2) decreased expression and activity of DNA repair factors, such as TOP1, ATM, and the telomere shelterin complex component, TRF2 (Nguyen et al., 2018; Zhao et al., 2018; Ji et al., 2019). Importantly, restoring expression of the DDR regulator, ATM, was able to rescue IL-2 production by the T cells of patients with chronic HCV infection (Zhao et al., 2018).
Patients with autoimmune diseases, such as rheumatoid arthritis (RA), display an expansion of senescent T cells with a high burden of DNA damage (Shao et al., 2009; Hohensinner et al., 2011). These cells display critically short telomeres, which may be related to impaired upregulation of telomerase upon activation in naïve CD4+ T cells (Fujii et al., 2009). Furthermore, CD4+ T cells from RA patients display an increased burden of DNA damage, both at baseline and in response to ionising radiation (Shao et al., 2009). As with chronic HCV infection, the genome instability in T cells from RA patients is partly attributed to insufficiency of the DDR enzyme, ATM, and can be rescued by ectopic ATM overexpression (Shao et al., 2009). Furthermore, senescent CD4+ T cells from RA patients are deficient in their expression of the DNA repair nuclease, MRE11A, leading to their increased telomeric damage and ability to injure tissues (Li et al., 2016).
Thus, in the contexts of chronic immune activation where accelerated immune ageing is observed, genome instability arises because of insufficiency in key DNA repair pathway enzymes. It is unclear why DNA repair factor expression decreases in these conditions, and whether boosting DNA repair processes may be therapeutically advantageous. It remains to be assessed whether the phenotype of reduced DNA repair factor expression is present in normal human immune system ageing.
T cells experience DNA damage across the lifespan. Chronic DNA damage leads to persistent DDR signalling but without completion of DNA repair. This sustained DDR experienced by senescent T cells contributes to their maladaptive phenotypes. Activation of ATM during the DDR drives activation of metabolic master regulator AMPK, which complexes with Sestrin 2, a stress-response protein, to activate several downstream MAPKs (ERK, JNK and p38) (Lanna et al., 2017). Activation of these individual MAPKs induces distinct senescent phenotypes, such as impaired proliferation, telomerase insufficiency, hypersecretion, low autophagic flux, and high ROS (Lanna et al., 2017). Knockdown of Sestrin 2 or pharmacological inhibition of MAPKs rejuvenated senescent T cell phenotypes and, interestingly, reduced γH2AX expression (Henson et al., 2014; Lanna et al., 2017). This indicates a two-way relationship between DNA damage-ATM activation, and AMPK-Sestrin 2-MAPK activation, potentially mediated by the effects of reducing ROS levels through MAPK inhibition (Lanna et al., 2017).
DNA damage and ATM activation in senescent T cells elicits gain-of-function characteristics, via Sestrin 2, that may drive immunopathology in inflammatory contexts, such as ageing and autoimmune diseases. Senescent CD8+ T cells acquire an NK cell-like phenotype (Strauss-Albee et al., 2014), upregulating a range of NK receptors (NKRs) such as the activating receptor NKG2D, which is dependent on the expression of Sestrin 2 (Pereira et al., 2020). Senescent NKG2D+ T cells, displaying defective TCR-dependent signalling, are capable of an alternative NKG2D-mediated activation to elicit their cytotoxic functions (Pereira et al., 2020). This process could feasibly have evolved to compensate for the declining canonical T cell activation response in ageing. However, in the context of autoimmune disease, it has been suggested that NKR-activated T cells may drive the destruction of self-tissues in which cells upregulate NK ligands due to the high-stress inflammatory environment (Covre et al., 2020; Pereira et al., 2020). This has been hypothesised to drive immune-mediated tissue damage, implicating a causal role of senescent T cells in driving immunopathology. Hence DNA damage and ATM activation in senescent T cells can drive both loss-of-function (e.g., poor proliferation) and gain-of-function (e.g., NKR upregulation) phenotypes, both of which contribute to poor antigen-specific immunity and immunopathology during ageing.
Immunosenescence is also driven by the increasingly inflammatory niche of older tissues, due to the increased burden of senescent non-haematopoietic ‘stromal’ cells (Chambers et al., 2021a). Since persistent DNA damage is a major driver of senescence, as previously discussed, efforts to promote DNA repair may attenuate senescent cell burden and the inflammatory tissue environment, with the potential to rejuvenate immune responses during ageing.
Immunosenescence contributes to the accumulation of senescent stromal cell during ageing. In healthy young individuals, turnover of senescent cells is homeostatically regulated: senescent cells are generated (for example, acutely at sites of wounds (Demaria et al., 2014)) and then killed by immune cells such as CD8+ T cells and Natural Killer (NK) cells, and cleared by macrophages through phagocytosis (Oishi and Manabe, 2016; Pereira et al., 2019). However, senescent cells accumulate during ageing and cause age-related disease pathology. This accumulation is driven both by an increase in stressors that generate senescent cells, and by impaired immunosurveillance and reduced clearance of senescent cells during ageing, partly due to immunosenescence (Ovadya et al., 2018; Pereira et al., 2019). For example, senescent cells increase their expression of immune evasion molecules such as HLA-E, thus inhibiting their clearance by engaging the immunosuppressive T cell NKG2A receptor, an NKR which is upregulated upon T cell senescence (Pereira et al., 2019; Pereira et al., 2020). Similarly, senescent cell PDL-1 expression prevents clearance by engaging the PD-1 receptor on CD8+ T cells (Wang et al., 2022). Furthermore, the cytotoxicity of NK cells decreases in older people, with the cells acquiring defects in their ability to deliver cytotoxic granules to the immune synapse (Hazeldine et al., 2012), which may impair their ability to kill senescent cells. The ability of macrophages to undergo phagocytosis also becomes defective during ageing, at least in mice (Solana et al., 2012; Stranks et al., 2015). Indeed, strategies that remobilise the immune system to target senescent cell (or ‘seno’-) antigens using CAR T cells (Amor et al., 2020), seno-directed vaccines (Suda et al., 2021), and immune checkpoint blockade (Wang et al., 2022) have been successful in attenuating senescent cell burden and promoting health in ageing mice.
The aged immune system is also capable of inducing senescence of non-haematopoietic cells. In the Vav-iCre+/−;Ercc1-/fl immunosenescence model, DNA damage in haematopoietic cells alone led to accelerated ageing and increased senescent cell burden in non-haematopoietic tissues, indicating a potential role of bystander/paracrine senescence between the different cell types (Yousefzadeh et al., 2021b). Immune cells also perpetuate tissue senescence by inducing DNA damage. Senescent liver cells secrete IL-8/CXCL8 within their SASP, which recruits neutrophils that release ROS that in turn damage the telomeres of bystander hepatocytes in a process termed TelOxidation (Lagnado et al., 2021). Indeed, TelOxidation may be exacerbated during ageing through non-directional tissue infiltration by neutrophils with impaired chemotaxis, which can cause widespread collateral damage and bystander senescence (Sapey et al., 2014). Therefore, DNA damage of cells both within and outside of the immune system contributes to the increased senescent cell burden and subsequent inflammatory niche, which is capable of impairing immunity during ageing.
Senescent cells compromise immunity in an aged context. The primary mechanism by which this occurs is through the secretion of components of the SASP. For example, the cytokine CCL2/MCP-1, produced by senescent fibroblasts in culture and the skin of old people, is an inflammatory monocyte chemoattractant (Pereira et al., 2019; Chambers et al., 2021a). Monocytes are recruited to the skin at sites of cutaneous immune challenge in old people, and through their release of PGE2, suppress the adaptive proliferative responses of local resident T cells (Chambers et al., 2021a). SASP components also impact the differentiation of effector T cells recruited during an immune response and promote an excessively suppressive environment. This may account for the observation that old mice and people exhibit poor clearance of influenza infection compared to younger individuals (Thompson et al., 2004; Lorenzo et al., 2022). TGF-β, a component of the SASP, can promote the differentiation of FoxP3+ regulatory T cells (Tregs). There are more Tregs in the lungs of old mice than young mice after influenza infection, and this can be attenuated by mAb-mediated blockade of TGF-β, supporting the importance of TGF-β in immune suppression (Lorenzo et al., 2022). To determine the role of senescent cells in dampening this influenza-specific immunity, old mice were treated prior to infection with dasatinib and quercetin (D + Q), a senolytic combination that inhibits the pro-survival pathways that are hyperactivated in senescent cells. Clearance of senescent cells in this way reduced both TGF-β levels and FoxP3+ Treg accumulation, and improved the response to influenza infection, suggesting that senescent cells were responsible for suppressing adequate immune responses during lung infection in old mice (Lorenzo et al., 2022). In younger life, this pathway may be advantageous in restricting an overexuberant response in the lung. However, an over-suppressive immune environment generated by accumulation of Tregs may contribute to the impaired clearance of influenza infection in an aged context. Furthermore, senescent cells and their SASP drive mortality during coronavirus infection: indeed, in vivo senolysis of coronavirus-induced senescent cells, by the senolytics D + Q or navitoclax, reduced peripheral SASP levels, COVID-19-like immunopathology, and mortality in animal models of the infection (Camell et al., 2021; Lee et al., 2021; Pastor-Fernandez et al., 2023). It would be interesting to investigate whether the senolytic removal of certain cell types, such as those either of immune or non-immune origin, would be sufficient to limit the SASP and senescence-related pathology in these diseases.
Under our hypothesis that immunosenescence is caused by DNA damage, it follows that interventions that inhibit DNA damage and promote DNA repair should attenuate immunosenescence. However, in the absence of known DNA damage inhibitors, this has not been tested. Nevertheless, a variety of treatment interventions have been reported to restore immunity in older people. Interestingly, the efficacy of many of these can be partly explained by their ability to promote genome stability - such interventions therefore may be described as “genoprotective.” This serves as indirect evidence that inhibiting DNA damage and promoting DNA repair may improve immune resilience in old people. This section will consider some of these potential immunoprotective treatments (spermidine, metformin, mTOR inhibitors, p38 MAPK inhibitors, and vitamin D3) and offer a genoprotective mechanism of action that may contribute to their success. We further discuss the potential merit in exploring nicotinamide riboside (NR) or enoxacin supplementation as interventions to target immunosenescence, given the evidence for their genoprotective abilities in limiting DNA damage. These interventions, along with their relevant effective in vitro and in vivo doses, are summarised in Table 2.
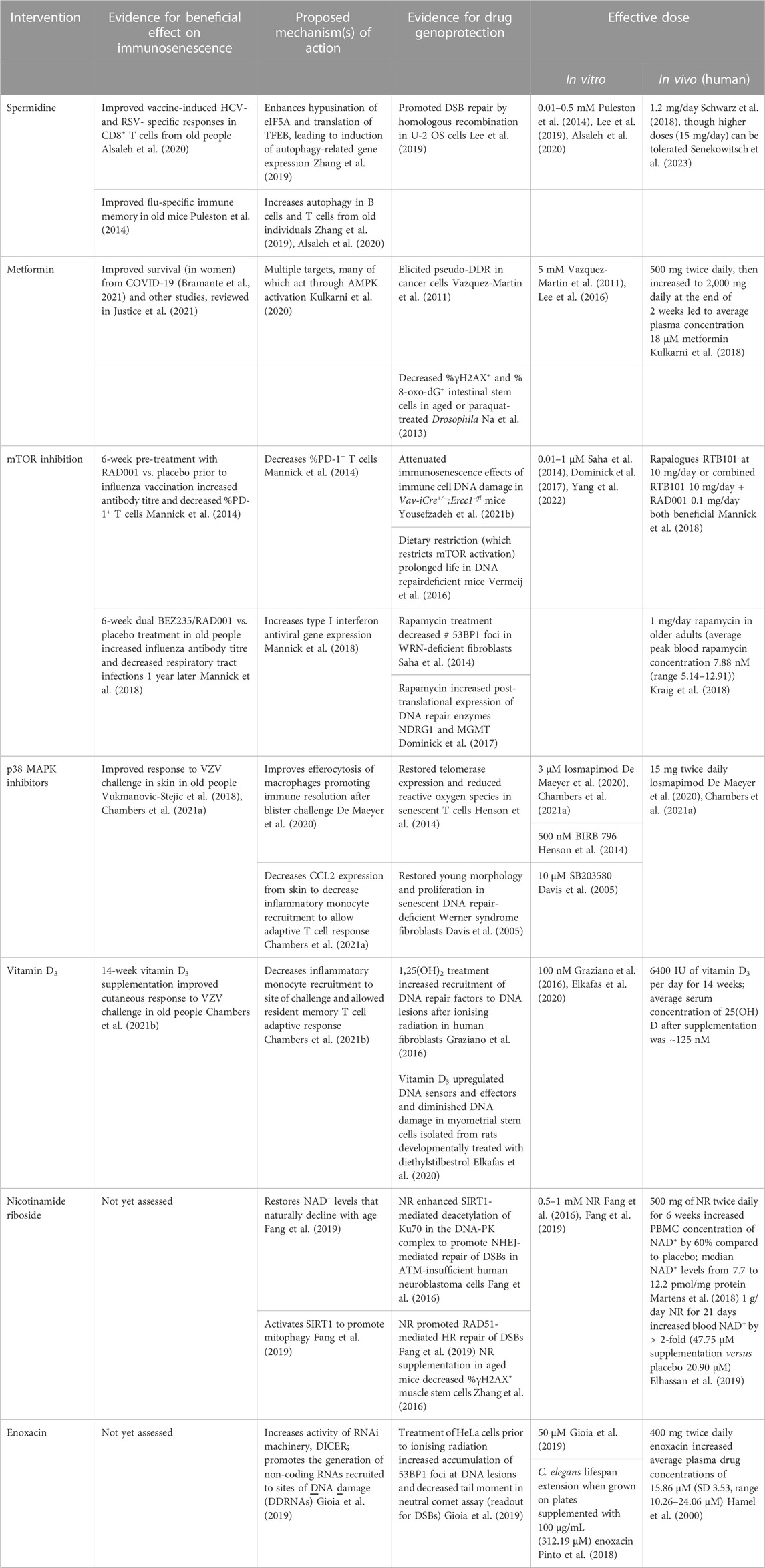
TABLE 2. Summary of immunosenescence interventions that may genoprotective.
Spermidine is a naturally-derived polyamine that has been shown to improve antigen-specific B and T cell responses in cells from old donors, through increasing autophagy (Zhang et al., 2019; Alsaleh et al., 2020). Spermidine is used as a substrate for the post-translational hypusination of eIF5A, a factor that enhances the translation of otherwise hard-to-translate poly-proline sequences. One such poly-proline-containing protein is TFEB, a transcription factor that drives the expression of autophagy genes. Thus, spermidine treatment increases autophagy in both old B and T cells through increased production of TFEB (Zhang et al., 2019; Alsaleh et al., 2020). As discussed, dysfunctional mitochondria accumulate in T cells from old people, and these produce ROS that damage the DNA and promote telomere attrition (Sanderson and Simon, 2017). Promoting autophagy by a different drug, metformin, has been shown to restore mitochondrial function in CD4+ T cells in old people (Bharath et al., 2020); by extension the autophagy-activating effect of spermidine may also restore mitophagy and mitochondrial health, thus protecting the DNA from ROS-mediated damage. Rejuvenating autophagy by spermidine may therefore be considered as an intervention that protects the genome, i.e., that is genoprotective.
In addition to their action through autophagy, polyamines such as spermidine may directly limit DNA damage and promote DNA repair. For example, spermine (spermidine’s derivative) reduces the presence of DNA oxidation products after damage by H2O2 (Muscari et al., 1995). Spermidine was also found to promote homologous recombination repair (HRR) mediated by RAD51 after DNA damage (Lee et al., 2019). Consistent with a core role for polyamines in DNA repair, inhibition of the polyamine biosynthesis pathway using DFMO (which inhibits ornithine decarboxylase) prolonged the presence of DNA damage after ionising radiation or PARP inhibition by olaparib (Lee et al., 2019). Though several polyamines improved HRR after DNA damage, spermine was found to be most efficient (Lee et al., 2019). Spermidine and other related polyamines may therefore elicit genoprotective effects and alleviate immunosenescence by acting as antioxidants, promoting repair, and reducing ROS burden through increased autophagy. Spermidine supplementation in humans is tolerated at doses tested up to 15 mg/day, though we note that supplementation does not detectably change blood spermidine levels, despite eliciting its beneficial anti-ageing effects (Schwarz et al., 2018; Wirth et al., 2018; Senekowitsch et al., 2023). Current clinical trials investigating whether spermidine supplementation (e.g., NCT05421546, spermidine at 6 mg/day) improves vaccine responses in old people will be greatly informative in translating in vitro studies to population interventions.
The anti-diabetic drug metformin is a potential geroprotector affecting a number of the hallmarks of ageing (Kulkarni et al., 2020); it may also improve immune resilience in older people (Justice et al., 2021). Metformin has polypharmacological effects, one of which we propose is genoprotective. Metformin is an AMPK activator and thus induces autophagy via activating phosphorylation of ULK1 (Kulkarni et al., 2020). In this way it can induce the removal of dysfunctional mitochondria (sources of genotoxic ROS) to prevent DNA damage. Indeed, metformin treatment of T cells from older people restored their autophagy and mitochondrial bioenergetics.
Metformin can affect the DNA damage response by driving a pseudo-DDR that protects genome stability. Epidermoid carcinoma cells treated for 2 days with metformin accumulated γH2AX foci and high levels of the phosphorylated DDR orchestrator, p-ATM (Ser 1981), in the absence of any detectable DNA lesions (Vazquez-Martin et al., 2011). Furthermore, metformin treatment of A549 lung cancer cells modulated the expression of p-Chk2, p53 and p53R2 (a p53 target gene) after UVC-induced DNA damage (Lee et al., 2016). Importantly, supplementation of metformin in Drosophila diminished the proportion of γH2AX+ and 8-oxo-dG+ intestinal stem cells undergoing acute paraquat-induced oxidative stress or chronological ageing, indicating the genoprotective effects of metformin (Na et al., 2013). Though some in vitro studies have used high metformin concentrations (5 mM) that would not be attainable in vivo, it is encouraging that a well-tolerated treatment regimen in older people was associated with upregulated expression of several DNA repair pathways in muscle biopsies (base excision repair, mismatch repair, and BRCA-mediated repair) (Kulkarni et al., 2018). Metformin may therefore ensure DNA stability by limiting damage by oxidative stress and further kickstarting a DDR and DNA repair.
Mammalian Target of Rapamycin (mTOR) is a master regulator kinase involved in nutrient sensing and cellular anabolic activity. mTOR inhibition is a potent life extension strategy in mammals (Harrison et al., 2009) and may have therapeutic advantages in many age-related disease contexts, including immunosenescence (Walters and Cox, 2018; Cox et al., 2020). At high doses, mTOR inhibitors such as rapamycin are immunosuppressive. However, low-dose rapamycin is immunoprotective; it promoted survival against lethal influenza infection in mice (Keating et al., 2013), and other acute pathogenic insults as reported in a recent meta-analysis of 23 studies in mice (Phillips and Simons, 2022). Low-dose mTOR inhibition strongly attenuates immunosenescence, as demonstrated by clinical trials in old people: mTOR inhibition by either RAD001 monotherapy, or dual RAD001/BEZ235 therapy, improved influenza vaccination responses and decreased subsequent respiratory tract infection rates 1 year after treatment (Mannick et al., 2014; Mannick et al., 2018), while treatment with BEZ235 (also named RTB101) showed trends towards immune improvement (Mannick et al., 2021). Moreover, the low nanomolar doses used in in vitro studies are achieved in vivo [e.g., in one study, 1 mg/day of rapamycin raised the average blood concentrations of the drug to 7.88 nM (range 5.14–12.91) (Kraig et al., 2018)]. The mechanism for immunoprotection by mTOR inhibition is not fully understood, but it has been observed to decrease %PD-1+ T cells, increase B cell antibody titres, and increase type I interferon gene expression in peripheral blood (Mannick et al., 2014; Mannick et al., 2018; Mannick et al., 2021).
mTOR inhibition modulates senescence and may promote genome stability. In the Vav-iCre+/−;Ercc1-/fl DNA damage immunosenescence model, rapamycin improved the immune response to a model antigen, and reduced the expression of the cell cycle inhibitors, CDKN2A and CDKN1A in T cells and several other tissues, though the effect of rapamycin treatment on markers of DNA damage in the immune cells was not assessed (Yousefzadeh et al., 2021b). Direct evidence that mTOR inhibition is genoprotective comes from the observation that long-term culture with rapamycin reduces the number of 53BP1+ DNA damage foci in fibroblasts deficient in the DNA repair factor, WRN (Saha et al., 2014). In addition to this, dietary restriction (DR), a well-known life extension strategy that converges on the inhibition of the mTOR signalling pathway (Cox and Mattison, 2009), elicited a 3-fold increase in median lifespan in both DNA repair-deficient Ercc1Δ/− and Xpg−/− progeroid mice compared to ad libitum-fed mutant mice (Vermeij et al., 2016). Importantly, genome damage, as measured by the fraction of neuronal cells showing γH2AX staining, was reduced in DR-fed mice, suggesting that DR could alleviate genome instability in the context of DNA repair deficiency (Vermeij et al., 2016). Since the mTOR pathway is inhibited during nutrient deprivation (particularly via reduced phosphorylation of one of its downstream targets, p-Akt (Ser473), this indirectly suggests that DR-induced genome stability may be through mTOR inhibition (Vermeij et al., 2016). However, since neither rapamycin treatment nor genetic inhibition of mTOR could fully recapitulate the DR-induced life extension in Ercc1Δ/− mice, the pro-longevity effects of DR may act through additional mechanisms independent of mTOR (Birkisdóttir et al., 2021).
The mechanism by which mTOR inhibition may promote genome stability is not fully understood. As mTOR inhibits ULK1 and autophagy, mTOR suppression and subsequent induction of autophagy may be genoprotective by removing intracellular sources of genotoxicity, such as the damaged mitochondria that produce high levels of ROS. mTOR may also influence DDR pathways. Several strains of long-lived mice show decreased mTOR activity in a number of tissues (Dominick et al., 2015), associated with high levels of the DNA repair factors MGMT and NDRG1 (Dominick et al., 2017). This is recapitulated by mTOR inhibition both in vitro and in vivo: mTOR inhibition by rapamycin treatment led to increased protein expression of MGMT and NDRG1 via a post-transcriptional mechanism through modulation of the CCR4-NOT complex (Dominick et al., 2017). Increased protein expression of these DNA repair factors may therefore promote DNA repair of existing damage, thus attenuating senescent phenotypes. Interestingly, in the case of CD8+ T cells where mTOR activity is low, rapamycin treatment may have beneficial effects that are independent of mTOR (Henson, 2015). These mTOR-independent mechanisms remain to be explored.
MAPK signalling occurs in response to growth factors, leading to cell proliferation; alternatively, members of the MAPK family are implicated in stress responses, including following DNA damage (Yacoub et al., 2001). Consistent with this, and the high levels of DNA damage in senescence, p38 MAPK is more active in both senescent T cells and non-haematopoietic cells compared to non-senescent counterparts (Lanna et al., 2014; Davis et al., 2016; Callender et al., 2018). p38 MAPK inhibitors also successfully dampen the inflammatory SASP of senescent cells, partly through inhibiting p38-DDR signalling (Alimbetov et al., 2016). p38 MAPK inhibitors, such as losmapimod and BIRB796, improve both senescent phenotypes and responses to immune challenge in older people (Vukmanovic-Stejic et al., 2018; Chambers et al., 2021a). p38 MAPK inhibition increases autophagy and telomerase expression, and reduces levels of intracellular ROS in senescent T cells (Henson et al., 2014). Thus, by restoring T cell telomere length and removing genotoxic ROS, p38 MAPK inhibition is a genoprotective strategy. Further evidence for the genoprotective effects of p38 MAPK inhibition is evidenced by the effects of SB203580 treatment, which rejuvenates both the senescent morphology and proliferative potential of DNA repair-deficient Werner Syndrome fibroblasts (Davis et al., 2005), although this treatment may have off-target effects (Lali et al., 2000). Furthermore, long-term (>3 months) treatment with current p38 MAPK inhibitors (such as losmapimod) is unfavourable due to hepatotoxicity, though short-term treatment prior to immune challenge may be beneficial. Although current p38 MAPK inhibitors are limited by their low therapeutic index and specificity, drugs with higher specificity and selectivity for MAPK isoforms are in development (Davis et al., 2016).
Vitamin D3 supplementation has been shown to improve cutaneous immune responses in old people, in part due to the effects of reducing inflammatory monocyte recruitment to the site of antigen challenge (Chambers et al., 2021b). Interestingly, vitamin D3 modulates DNA repair factor activity and cellular senescence (Graziano et al., 2016). For example, fibroblasts that have undergone oncogene-induced senescence (OIS) fail to repair DNA damage due to insufficient expression of BRCA1 and the vitamin D receptor (VDR); VDR knockdown recapitulated both BRCA1 insufficiency and the senescence phenotypes observed in OIS (Graziano et al., 2016). Intriguingly, treatment of OIS fibroblasts with active vitamin D3 (1,25(OH)2-D3) enhanced the recruitment of DNA repair factors to DNA lesions after ionising radiation (Graziano et al., 2016). This reveals an axis connecting vitamin D3, DNA repair factor activity, and senescence. Furthermore, vitamin D3 treatment reduced DNA damage foci in myometrial stem cells isolated from rats developmentally exposed to diethylstilbestrol (DES) (Elkafas et al., 2020). Indeed, in vitro treatment with vitamin D3 enhanced expression of both DNA damage sensors (in the MRN complex) and DDR effector proteins (RAD51 and BRCA2) after DES treatment (Elkafas et al., 2020). Additionally, vitamin D3 antagonised the effects of DNA-damaging treatment by upregulating MRE11 and other DDR genes (Elkafas et al., 2020). These data suggest that vitamin D3 is genoprotective and its supplementation in humans could improve immune responses in old people by limiting DNA damage. This vitamin D3-DNA repair axis deficiency may be particularly pertinent in older adults, where vitamin D deficiency is common. Importantly, supplementation in humans elevates vitamin D3 blood to concentrations similar to those used in in vitro studies that demonstrate improved DNA repair (Table 2); however, the effect of supplementation on DNA damage readouts in either haematopoietic or non-haematopoietic cells in people remains to be explored.
Energy homeostasis is critical for correct immune function, yet immune energetics have remained largely ignored within the extensive body of research on NAD+. NAD+ levels decline during normal ageing and this contributes both to increased DNA damage and mitochondrial dysfunction, both processes discussed in this review to be drivers of immunosenescence (Yoshino et al., 2018). The age-related decline in NAD+ is due, in part, to the increase in its consumption by NADases, such as CD38, which are upregulated in tissue-resident macrophages in response to the SASP (Chini et al., 2020; Covarrubias et al., 2020). Additionally, NADases, such as DNA damage-induced poly-ADP ribosyl polymerases (PARPs), become hyperactivated in senescence due to the increase in DNA lesion burden, leading to NAD+ decline (Fang et al., 2014; Yoshino et al., 2018). The overactivation of these NAD-consuming enzymes restricts the activity of other pro-longevity NAD-dependent enzymes, the sirtuins: e.g., SIRT1, an NAD-dependent deacetylase that regulates mitochondrial biogenesis by deacetylating PGC-1α (Fang et al., 2016). Restoring SIRT1 activity by NR supplementation increases mitophagy, removal of ROS-producing dysfunctional mitochondria, and lifespan in progeroid worms mutant for the DNA repair factor, wrn (wrn-1(gk99)), and in progeroid mice that are deficient in the DDR master regulator, Atm (Fang et al., 2016; Fang et al., 2019). Indeed, in progeroid Tfamfl/fl Cd4Cre mice that accumulate senescent T cells with dysfunctional mitochondria, NR supplementation reversed tissue inflammation and senescence, although the explicit effect of this treatment on immune responses was not reported (Desdin-Mico et al., 2020). NR supplementation also attenuated inflammageing in Atm−/− progeroid and wildtype old mice, and restored their lymphoid vs. myeloid haematopoietic output (Zong et al., 2021). Restoring sirtuin activity in this way may be beneficial in immunosenescence, since antigen-experienced senescent CD8+CD28− T cells, for example, have decreased SIRT1 expression compared to naïve T cells, which partly drives their glycolytic and cytotoxic aged phenotype (Jeng et al., 2018). It would be interesting to investigate whether NR supplementation may alleviate immunosenescence in other DNA repair-defective models, e.g., the Vav-iCre+/−;Ercc1-fl/ mouse model (Yousefzadeh et al., 2021b).
The impact of NR supplementation on mitophagy is also accompanied by better DNA repair. In ATM-knockdown primary neurons treated with DNA-damaging etoposide, NR reduced 53BP1 staining (Fang et al., 2016). Mechanistically this was due to an increase in SIRT1-mediated deacetylation of Ku70 in the DNA-PK complex during DSB repair by NHEJ (Fang et al., 2016). Similarly, in wrn-1(gk99) mutant worms, NR treatment decreased the RAD51 signal 24 h after ionising radiation, this time suggesting an improvement in HR-mediated repair of DSBs (Fang et al., 2019). Accordingly, NR supplementation starting at 2 years-old in wildtype mice reduced the accumulation of γH2AX+ DNA-damaged muscle stem cells (Zhang et al., 2016). Taken together, these data suggest that NR may be genoprotective by improving both mitochondrial quality and DNA repair. We note that in vitro studies use relatively high concentrations of NR (0.5–1 mM) for improvement of DNA repair (Fang et al., 2016; Fang et al., 2019). Though current supplementation regimens in older adults robustly increase the NAD+ levels of peripheral immune cells, investigations into the effective doses of NR for in vivo genoprotection are needed (Martens et al., 2018).
We have discussed the evidence that persistent DNA damage is a primary driver of immunosenescence during ageing, and how existing interventions that ameliorate immunosenescence may be genoprotective by acting to promote DNA repair. Pharmacological interventions that target DNA repair pathways are common in the field of cancer chemotherapy, and often work to elicit cancer cell death by inhibiting DNA repair. However, drugs that act to promote DNA repair to elicit genoprotection have received less attention.
One exception is enoxacin, a fluoroquinolone antibiotic that boosts DNA repair by enhancing the RNAi machinery component, DICER, which increases the generation of DNA damage response non-coding RNAs (DDRNAs) (Gioia et al., 2019). Deficiency of the RNAi machinery impairs the DDR and increases genome instability (Swahari et al., 2016). However, treatment of cells with 50 µM enoxacin prior to ionising radiation caused a rapid accumulation of 53BP1 at DSBs which corresponded with a subsequent decrease in DNA lesions (as measured by comet assay and γH2AX levels) after the acute DDR, indicating a boost in overall DNA repair (Gioia et al., 2019). Thus, enoxacin appears to be genoprotective by enhancing DSB repair. In the context of ageing, enoxacin has been reported to increase C. elegans lifespan (Pinto et al., 2018), but little is known on its immune effects. It is of note that the SARS-CoV2 N-protein binds to DDRNAs, and by doing so outcompetes the 53BP1-DDRNA binding that is necessary for 53BP1 recruitment to DSBs (Gioia et al., 2023). In this way, SARS-CoV2 infection was recently demonstrated to increase DNA damage and induce cellular senescence, driving disease pathology (Gioia et al., 2023). It may be that enoxacin treatment, by increasing DDRNA expression, could promote 53BP1-mediated repair of DNA lesions, thus preventing senescence and potentially limiting morbidity and even mortality in COVID-19 patients. Most importantly, enoxacin is already well tolerated in older adults, and current treatment regimens increase enoxacin blood concentrations to a level relatively comparable to those used in in vitro studies for eliciting genoprotection (Table 2) (Hamel et al., 2000). Given the urgent call for novel drugs attenuating immunosenescence, future studies investigating enoxacin’s effect on DNA damage/repair in immune and non-immune cells, and on immune system function in older people, may be beneficial. More generally, we propose that genoprotective drug discovery campaigns may be a valuable approach towards attenuating immunosenescence.
Improvements in global health have resulted in a steady increase in population lifespan over the past century. However, the widening gap between healthspan and lifespan has accelerated the need to uncover ageing mechanisms and interventions for promoting healthy ageing. DNA damage and repair play important roles in immunity and ageing: intentional induction of DNA lesions is crucial in the generation of a diverse immune repertoire, but poor resolution of DNA damage can induce a persistent DNA damage response that drives permanent exit from the cell cycle and senescence. Ageing is accompanied by an accumulation of senescent immune cells, exemplified by T cells, with persistent DNA damage that may directly cause ageing.
In this review we draw together evidence from the fields of DNA repair, ageing, and immunology, to explore why genome instability increases in old T cells and non-immune cells during ageing, and how this drives their maladaptive senescent phenotypes and promotes immunosenescence. We propose that existing geroprotective therapeutic approaches can limit DNA damage and promote DNA repair, and so we term these “genoprotective” interventions. We discuss potential genoprotective interventions, including dietary supplementation (vitamin D3 and NR), as well as treatments with repurposed drugs (metformin, mTOR inhibitors, p38 MAPK inhibitors), and finally a novel drug, enoxacin, that may boost DNA repair and which remains largely unexplored in longevity science. Overall, we suggest that investigations into improving DNA repair processes and genome stability, both for immune and non-immune cell types, are valuable strategies for targeting immunosenescence.
LK conceived of and wrote the first draft of the manuscript. GA, AS, and LC all contributed to manuscript critical feedback and revision. All authors contributed to the article and approved the submitted version.
LK is funded by the Mellon Longevity Graduate Programme at the University of Oxford, and a UK SPINE (Research England) proof-of-concept translational fund (2021–22). AS is funded by the Wellcome Trust, UK SPINE (Research England), and the Helmholtz Society. GA is funded by Versus Arthritis grants 22617. LC reports funding from BBSRC (the Biotechnology and Biological Sciences Research Council) [BB/W01825X/1], MRC (Medical Research Council); Public Health England (now UK Health Security Agency); UK SPINE (Research England); Diabetes UK/BIRAX; and the Mellon Longevity Science Programme at Oxford.
LK would like to thank James Cai for his helpful comments and discussion when conceptualising the manuscript. Figure 1 and 2 were made using BioRender.
AS consults for Oxford Healthspan, The Longevity Labs, and Calico. LC is Program Director (Dynamic Resilience) for non-profit Wellcome Leap and is co-director of UK Ageing Research Networks and BLAST ageing network (UKRI funded). LC holds voluntary roles with the All Party Parliamentary Group for Longevity (UK); European Geriatric Medicine Society special interest group in Ageing Biology; Clinical and Translational Theme panel, Biochemical Society (UK); and Medical Research Council Ageing Research Steering Group. GA and LK report no conflicts of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agius, E., Lacy, K. E., Vukmanovic-Stejic, M., Jagger, A. L., Papageorgiou, A. P., Hall, S., et al. (2009). Decreased TNF-alpha synthesis by macrophages restricts cutaneous immunosurveillance by memory CD4+ T cells during aging. J. Exp. Med. 206, 1929–1940. doi:10.1084/jem.20090896
Aiello, A., Farzaneh, F., Candore, G., Caruso, C., Davinelli, S., Gambino, C. M., et al. (2019). Immunosenescence and its hallmarks: How to oppose aging strategically? A review of potential options for therapeutic intervention. Front. Immunol. 10, 2247. doi:10.3389/fimmu.2019.02247
Akbar, A. N., Henson, S. M., and Lanna, A. (2016). Senescence of T Lymphocytes: Implications for enhancing human immunity. Trends Immunol. 37, 866–876. doi:10.1016/j.it.2016.09.002
Alimbetov, D., Davis, T., Brook, A. J., Cox, L. S., Faragher, R. G., Nurgozhin, T., et al. (2016). Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 17, 305–315. doi:10.1007/s10522-015-9610-z
Alonso-Arias, R., Moro-García, M. A., López-Vázquez, A., Rodrigo, L., Baltar, J., Suárez García, F. M., et al. (2011). NKG2D expression in CD4+ T lymphocytes as a marker of senescence in the aged immune system. Age (Dordr) 33 (4), 591–605. doi:10.1007/s11357-010-9200-6
Alsaleh, G., Panse, I., Swadling, L., Zhang, H., Richter, F. C., Meyer, A., et al. (2020). Autophagy in T cells from aged donors is maintained by spermidine and correlates with function and vaccine responses. Elife 9, e57950. doi:10.7554/eLife.57950
Alsaleh, G., Richter, F. C., and Simon, A. K. (2022). Age-related mechanisms in the context of rheumatic disease. Nat. Rev. Rheumatol. 18, 694–710. doi:10.1038/s41584-022-00863-8
Amor, C., Feucht, J., Leibold, J., Ho, Y. J., Zhu, C., Alonso-Curbelo, D., et al. (2020). Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132. doi:10.1038/s41586-020-2403-9
Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Sieben, C. J., Zhong, J., et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189. doi:10.1038/nature16932
Barnett, K., Mercer, S. W., Norbury, M., Watt, G., Wyke, S., and Guthrie, B. (2012). Epidemiology of multimorbidity and implications for health care, research, and medical education: A cross-sectional study. Lancet 380, 37–43. doi:10.1016/S0140-6736(12)60240-2
Bektas, A., Schurman, S. H., Gonzalez-Freire, M., Dunn, C. A., Singh, A. K., Macian, F., et al. (2019). Age-associated changes in human CD4(+) T cells point to mitochondrial dysfunction consequent to impaired autophagy. Aging (Albany NY) 11, 9234–9263. doi:10.18632/aging.102438
Bharath, L. P., Agrawal, M., Mccambridge, G., Nicholas, D. A., Hasturk, H., Liu, J., et al. (2020). Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32, 44–55 e6. doi:10.1016/j.cmet.2020.04.015
Birkisdóttir, M. B., Jaarsma, D., Brandt, R. M. C., Barnhoorn, S., van Vliet, N., Imholz, S., et al. (2021). Unlike dietary restriction, rapamycin fails to extend lifespan and reduce transcription stress in progeroid DNA repair-deficient mice. Aging Cell 20, e13302. doi:10.1111/acel.13302
Bonassi, S., Ceppi, M., Moller, P., Azqueta, A., Milic, M., Neri, M., et al. (2021). DNA damage in circulating leukocytes measured with the comet assay may predict the risk of death. Sci. Rep. 11, 16793. doi:10.1038/s41598-021-95976-7
Bramante, C. T., Ingraham, N. E., Murray, T. A., Marmor, S., Hovertsen, S., Gronski, J., et al. (2021). Metformin and risk of mortality in patients hospitalised with COVID-19: a retrospective cohort analysis. Lancet Healthy Longev. 2 (1), e34–e41. doi:10.1016/S2666-7568(20)30033-7
Buchwalter, A., and Hetzer, M. W. (2017). Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 8, 328. doi:10.1038/s41467-017-00322-z
Callender, L. A., Carroll, E. C., Bober, E. A., Akbar, A. N., Solito, E., and Henson, S. M. (2020). Mitochondrial mass governs the extent of human T cell senescence. Aging Cell 19, e13067. doi:10.1111/acel.13067
Callender, L. A., Carroll, E. C., Beal, R. W. J., Chambers, E. S., Nourshargh, S., Akbar, A. N., et al. (2018). Human CD8(+) EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 17, e12675. doi:10.1111/acel.12675
Callender, L. A., Schroth, J., Carroll, E. C., Garrod-Ketchley, C., Romano, L. E. L., Hendy, E., et al. (2021). GATA3 induces mitochondrial biogenesis in primary human CD4(+) T cells during DNA damage. Nat. Commun. 12, 3379. doi:10.1038/s41467-021-23715-7
Camell, C. D., Yousefzadeh, M. J., Zhu, Y., Langhi Prata, L. G. P., Huggins, M. A., Pierson, M., et al. (2021). Senolytics reduce coronavirus-related mortality in old mice. Science 373, eabe4832. doi:10.1126/science.abe4832
Chambers, E. S., Vukmanovic-Stejic, M., Shih, B. B., Trahair, H., Subramanian, P., Devine, O. P., et al. (2021a). Recruitment of inflammatory monocytes by senescent fibroblasts inhibits antigen-specific tissue immunity during human aging. Nat. Aging 1, 101–113. doi:10.1038/s43587-020-00010-6
Chambers, E. S., Vukmanovic-Stejic, M., Turner, C. T., Shih, B. B., Trahair, H., Pollara, G., et al. (2021b). Vitamin D3 replacement enhances antigen-specific immunity in older adults. Immunother. Adv. 1, ltaa008. doi:10.1093/immadv/ltaa008
Chini, C. C. S., Peclat, T. R., Warner, G. M., Kashyap, S., Espindola-Netto, J. M., de Oliveira, G. C., et al. (2020). CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2, 1284–1304. doi:10.1038/s42255-020-00298-z
Coppe, J. P., Patil, C. K., Rodier, F., Sun, Y., Munoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868. doi:10.1371/journal.pbio.0060301
Covarrubias, A. J., Kale, A., Perrone, R., Lopez-Dominguez, J. A., Pisco, A. O., Kasler, H. G., et al. (2020). Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2, 1265–1283. doi:10.1038/s42255-020-00305-3
Covre, L. P., de Maeyer, R. P. H., Gomes, D. C. O., and Akbar, A. N. (2020). The role of senescent T cells in immunopathology. Aging Cell 19, e13272. doi:10.1111/acel.13272
Covre, L. P., Martins, R. F., Devine, O. P., Chambers, E. S., Vukmanovic-Stejic, M., Silva, J. A., et al. (2018). Circulating senescent T cells are linked to systemic inflammation and lesion size during human cutaneous leishmaniasis. Front. Immunol. 9, 3001. doi:10.3389/fimmu.2018.03001
Cox, L. S., Bellantuono, I., Lord, J. M., Sapey, E., Mannick, J. B., Partridge, L., et al. (2020). Tackling immunosenescence to improve COVID-19 outcomes and vaccine response in older adults. Lancet Healthy Longev. 1, e55–e57. doi:10.1016/S2666-7568(20)30011-8
Cox, L. S., and Faragher, R. G. (2007). From old organisms to new molecules: Integrative biology and therapeutic targets in accelerated human ageing. Cell Mol. Life Sci. 64, 2620–2641. doi:10.1007/s00018-007-7123-x
Cox, L. S., and Lord, J. M. (2021). Targeting aging cells improves survival. Science 373, 281–282. doi:10.1126/science.abi4474
Cox, L. S., and Mattison, J. A. (2009). Increasing longevity through caloric restriction or rapamycin feeding in mammals: Common mechanisms for common outcomes? Aging Cell 8, 607–613. doi:10.1111/j.1474-9726.2009.00509.x
Czesnikiewicz-Guzik, M., Lee, W. W., Cui, D., Hiruma, Y., Lamar, D. L., Yang, Z. Z., et al. (2008). T cell subset-specific susceptibility to aging. Clin. Immunol. 127, 107–118. doi:10.1016/j.clim.2007.12.002
Davis, T., Baird, D. M., Haughton, M. F., Jones, C. J., and Kipling, D. (2005). Prevention of accelerated cell aging in Werner syndrome using a p38 mitogen-activated protein kinase inhibitor. J. Gerontol. A Biol. Sci. Med. Sci. 60, 1386–1393. doi:10.1093/gerona/60.11.1386
Davis, T., Brook, A. J., Rokicki, M. J., Bagley, M. C., and Kipling, D. (2016). Evaluating the role of p38 MAPK in the accelerated cell senescence of werner syndrome fibroblasts. Pharm. (Basel) 9, 23. doi:10.3390/ph9020023
de Cecco, M., Ito, T., Petrashen, A. P., Elias, A. E., Skvir, N. J., Criscione, S. W., et al. (2019). L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78. doi:10.1038/s41586-018-0784-9
de Maeyer, R. P. H., van de Merwe, R. C., Louie, R., Bracken, O. V., Devine, O. P., Goldstein, D. R., et al. (2020). Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat. Immunol. 21, 615–625. doi:10.1038/s41590-020-0646-0
Demaria, M., Ohtani, N., Youssef, S. A., Rodier, F., Toussaint, W., Mitchell, J. R., et al. (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733. doi:10.1016/j.devcel.2014.11.012
Desdin-Mico, G., Soto-Heredero, G., Aranda, J. F., Oller, J., Carrasco, E., Gabande-Rodriguez, E., et al. (2020). T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376. doi:10.1126/science.aax0860
di Mitri, D., Azevedo, R. I., Henson, S. M., Libri, V., Riddell, N. E., Macaulay, R., et al. (2011). Reversible senescence in human CD4+CD45RA+CD27-memory T cells. J. Immunol. 187, 2093–2100. doi:10.4049/jimmunol.1100978
Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. 92, 9363–9367. doi:10.1073/pnas.92.20.9363
Dominick, G., Berryman, D. E., List, E. O., Kopchick, J. J., Li, X., Miller, R. A., et al. (2015). Regulation of mTOR activity in Snell dwarf and GH receptor gene-disrupted mice. Endocrinology 156, 565–575. doi:10.1210/en.2014-1690
Dominick, G., Bowman, J., Li, X., Miller, R. A., and Garcia, G. G. (2017). mTOR regulates the expression of DNA damage response enzymes in long-lived Snell dwarf, GHRKO, and PAPPA-KO mice. Aging Cell 16, 52–60. doi:10.1111/acel.12525
Dong, X., Milholland, B., and Vijg, J. (2016). Evidence for a limit to human lifespan. Nature 538, 257–259. doi:10.1038/nature19793
Elhassan, Y. S., Kluckova, K., Fletcher, R. S., Schmidt, M. S., Garten, A., Doig, C. L., et al. (2019). Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 28, 1717–1728 e6. doi:10.1016/j.celrep.2019.07.043
Elkafas, H., Ali, M., Elmorsy, E., Kamel, R., Thompson, W. E., Badary, O., et al. (2020). Vitamin D3 ameliorates DNA damage caused by developmental exposure to endocrine disruptors in the uterine myometrial stem cells of eker rats. Cells 9, 1459. doi:10.3390/cells9061459
Fang, E. F., Hou, Y., Lautrup, S., Jensen, M. B., Yang, B., Sengupta, T., et al. (2019). NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 10, 5284. doi:10.1038/s41467-019-13172-8
Fang, E. F., Kassahun, H., Croteau, D. L., Scheibye-Knudsen, M., Marosi, K., Lu, H., et al. (2016). NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581. doi:10.1016/j.cmet.2016.09.004
Fang, E. F., Scheibye-Knudsen, M., Brace, L. E., Kassahun, H., Sengupta, T., Nilsen, H., et al. (2014). Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157, 882–896. doi:10.1016/j.cell.2014.03.026
Fletcher, J. M., Vukmanovic-Stejic, M., Dunne, P. J., Birch, K. E., Cook, J. E., Jackson, S. E., et al. (2005). Cytomegalovirus-specific CD4+ T cells in healthy carriers are continuously driven to replicative exhaustion. J. Immunol. 175, 8218–8225. doi:10.4049/jimmunol.175.12.8218
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F., de Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254. doi:10.1111/j.1749-6632.2000.tb06651.x
Fumagalli, M., Rossiello, F., Clerici, M., Barozzi, S., Cittaro, D., Kaplunov, J. M., et al. (2012). Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365. doi:10.1038/ncb2466
Fujii, H., Shao, L., Colmegna, I., Goronzy, J. J., and Weyand, C. M. (2009). Telomerase insufficiency in rheumatoid arthritis. Proc. Natl. Acad. Sci. U. S. A. 106, 4360–4365. doi:10.1073/pnas.0811332106
Fumagalli, M., Rossiello, F., Mondello, C., and D'Adda di Fagagna, F. (2014). Stable cellular senescence is associated with persistent DDR activation. PLoS One 9, e110969. doi:10.1371/journal.pone.0110969
Gellert, M. (2002). V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 71, 101–132. doi:10.1146/annurev.biochem.71.090501.150203
Gioia, U., Francia, S., Cabrini, M., Brambillasca, S., Michelini, F., Jones-Weinert, C. W., et al. (2019). Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci. Rep. 9, 6460. doi:10.1038/s41598-019-42892-6
Gioia, U., Tavella, S., Martinez-Orellana, P., Cicio, G., Colliva, A., Ceccon, M., et al. (2023). SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nat. Cell Biol. 25, 550–564. doi:10.1038/s41556-023-01096-x
Goronzy, J. J., and Weyand, C. M. (2012). Immune aging and autoimmunity. Cell Mol. Life Sci. 69, 1615–1623. doi:10.1007/s00018-012-0970-0
Graziano, S., Johnston, R., Deng, O., Zhang, J., and Gonzalo, S. (2016). Vitamin D/vitamin D receptor axis regulates DNA repair during oncogene-induced senescence. Oncogene 35, 5362–5376. doi:10.1038/onc.2016.77
Gullickson, P., Xu, Y. W., Niedernhofer, L. J., Thompson, E. L., and Yousefzadeh, M. J. (2022). The role of DNA repair in immunological diversity: From molecular mechanisms to clinical ramifications. Front. Immunol. 13, 834889. doi:10.3389/fimmu.2022.834889
Hamel, B., Mottet, N., Audran, M., Costa, P., and Bressolle, F. (2000). Pharmacokinetics of enoxacin and its oxometabolite after multiple oral dosing and penetration into prostatic tissue. J. Antimicrob. Chemother. 46, 993–996. doi:10.1093/jac/46.6.993
Harley, C. B., Futcher, A. B., and Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460. doi:10.1038/345458a0
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. doi:10.1038/nature08221
Hayflick, L., and Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. doi:10.1016/0014-4827(61)90192-6
Hazeldine, J., Hampson, P., and Lord, J. M. (2012). Reduced release and binding of perforin at the immunological synapse underlies the age-related decline in natural killer cell cytotoxicity. Aging Cell 11, 751–759. doi:10.1111/j.1474-9726.2012.00839.x
Henson, S. M. (2015). CD8+ T-cell senescence: No role for mTOR. Biochem. Soc. Trans. 43, 734–739. doi:10.1042/BST20150092
Henson, S. M., Franzese, O., Macaulay, R., Libri, V., Azevedo, R. I., Kiani-Alikhan, S., et al. (2009). KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood 113 (26), 6619–6628. doi:10.1182/blood-2009-01-199588
Henson, S. M., Lanna, A., Riddell, N. E., Franzese, O., Macaulay, R., Griffiths, S. J., et al. (2014). p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8⁺ T cells. J. Clin. Invest. 124, 4004–4016. doi:10.1172/JCI75051
Henson, S. M., Macaulay, R., Riddell, N. E., Nunn, C. J., and Akbar, A. N. (2015). Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur. J. Immunol. 45, 1441–1451. doi:10.1002/eji.201445312
Hewitt, G., Jurk, D., Marques, F. D., Correia-Melo, C., Hardy, T., Gackowska, A., et al. (2012). Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 708. doi:10.1038/ncomms1708
Hoare, M., Gelson, W. T., Das, A., Fletcher, J. M., Davies, S. E., Curran, M. D., et al. (2010). CD4+ T-lymphocyte telomere length is related to fibrosis stage, clinical outcome and treatment response in chronic hepatitis C virus infection. J. Hepatol. 53, 252–260. doi:10.1016/j.jhep.2010.03.005
Hohensinner, P. J., Goronzy, J. J., and Weyand, C. M. (2011). Telomere dysfunction, autoimmunity and aging. Aging Dis. 2, 524–537.
Hou, Y., Zhou, Y., Jehi, L., Luo, Y., Gack, M. U., Chan, T. A., et al. (2022). Aging-related cell type-specific pathophysiologic immune responses that exacerbate disease severity in aged COVID-19 patients. Aging Cell 21, e13544. doi:10.1111/acel.13544
Huang, R., and Zhou, P. K. (2021). DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target Ther. 6, 254. doi:10.1038/s41392-021-00648-7
Ibler, A. E. M., Elghazaly, M., Naylor, K. L., Bulgakova, N. A., S, F. E.-K., and Humphreys, D. (2019). Typhoid toxin exhausts the RPA response to DNA replication stress driving senescence and Salmonella infection. Nat. Commun. 10, 4040. doi:10.1038/s41467-019-12064-1
Ivanov, A., Pawlikowski, J., Manoharan, I., van Tuyn, J., Nelson, D. M., Rai, T. S., et al. (2013). Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 202, 129–143. doi:10.1083/jcb.201212110
Jaskelioff, M., Muller, F. L., Paik, J. H., Thomas, E., Jiang, S., Adams, A. C., et al. (2011). Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469, 102–106. doi:10.1038/nature09603
Jeng, M. Y., Hull, P. A., Fei, M., Kwon, H. S., Tsou, C. L., Kasler, H., et al. (2018). Metabolic reprogramming of human CD8(+) memory T cells through loss of SIRT1. J. Exp. Med. 215, 51–62. doi:10.1084/jem.20161066
Ji, Y., Dang, X., Nguyen, L. N. T., Nguyen, L. N., Zhao, J., Cao, D., et al. (2019). Topological DNA damage, telomere attrition and T cell senescence during chronic viral infections. Immun. Ageing 16, 12. doi:10.1186/s12979-019-0153-z
Justice, J. N., Gubbi, S., Kulkarni, A. S., Bartley, J. M., Kuchel, G. A., and Barzilai, N. (2021). A geroscience perspective on immune resilience and infectious diseases: A potential case for metformin. Geroscience 43, 1093–1112. doi:10.1007/s11357-020-00261-6
Keating, R., Hertz, T., Wehenkel, M., Harris, T. L., Edwards, B. A., Mcclaren, J. L., et al. (2013). The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat. Immunol. 14, 1266–1276. doi:10.1038/ni.2741
Kraig, E., Linehan, L. A., Liang, H., Romo, T. Q., Liu, Q., Wu, Y., et al. (2018). A randomized control trial to establish the feasibility and safety of rapamycin treatment in an older human cohort: Immunological, physical performance, and cognitive effects. Exp. Gerontol. 105, 53–69. doi:10.1016/j.exger.2017.12.026
Kulkarni, A. S., Gubbi, S., and Barzilai, N. (2020). Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 32, 15–30. doi:10.1016/j.cmet.2020.04.001
Kulkarni, A. S., Brutsaert, E. F., Anghel, V., Zhang, K., Bloomgarden, N., Pollak, M., et al. (2018). Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell 17, e12723. doi:10.1111/acel.12723
Lanna, A., Henson, S. M., Escors, D., and Akbar, A. N. (2014). The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 15, 965–972. doi:10.1038/ni.2981
Lagnado, A., Leslie, J., Ruchaud-Sparagano, M. H., Victorelli, S., Hirsova, P., Ogrodnik, M., et al. (2021). Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 40, e106048. doi:10.15252/embj.2020106048
Lali, F. V., Hunt, A. E., Turner, S. J., and Foxwell, B. M. (2000). The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J. Biol. Chem. 275, 7395–7402. doi:10.1074/jbc.275.10.7395
Lanna, A., Coutavas, E., Levati, L., Seidel, J., Rustin, M. H., Henson, S. M., et al. (2013). IFN-α inhibits telomerase in human CD8⁺ T cells by both hTERT downregulation and induction of p38 MAPK signaling. J. Immunol. 191, 3744–3752. doi:10.4049/jimmunol.1301409
Lanna, A., Gomes, D. C., Muller-Durovic, B., Mcdonnell, T., Escors, D., Gilroy, D. W., et al. (2017). A sestrin-dependent Erk-Junk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 18, 354–363. doi:10.1038/ni.3665
Lanna, A., Vaz, B., D'Ambra, C., Valvo, S., Vuotto, C., Chiurchiu, V., et al. (2022). An intercellular transfer of telomeres rescues T cells from senescence and promotes long-term immunological memory. Nat. Cell Biol. 24, 1461–1474. doi:10.1038/s41556-022-00991-z
Lee, C. Y., Su, G. C., Huang, W. Y., Ko, M. Y., Yeh, H. Y., Chang, G. D., et al. (2019). Promotion of homology-directed DNA repair by polyamines. Nat. Commun. 10, 65. doi:10.1038/s41467-018-08011-1
Lee, S., Yu, Y., Trimpert, J., Benthani, F., Mairhofer, M., Richter-Pechanska, P., et al. (2021). Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599, 283–289. doi:10.1038/s41586-021-03995-1
Lee, Y. S., Doonan, B. B., Wu, J. M., and Hsieh, T. C. (2016). Combined metformin and resveratrol confers protection against UVC-induced DNA damage in A549 lung cancer cells via modulation of cell cycle checkpoints and DNA repair. Oncol. Rep. 35, 3735–3741. doi:10.3892/or.2016.4740
Li, Y., Shen, Y., Hohensinner, P., Ju, J., Wen, Z., Goodman, S. B., et al. (2016). Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity 45, 903–916. doi:10.1016/j.immuni.2016.09.013
Libri, V., Azevedo, R. I., Jackson, S. E., Di Mitri, D., Lachmann, R., Fuhrmann, S., et al. (2011). Cytomegalovirus infection induces the accumulation of short-lived, multifunctional CD4+CD45RA+CD27+ T cells: the potential involvement of interleukin-7 in this process. Immunology 132 (3), 326–339. doi:10.1111/j.1365-2567.2010.03386.x
Lin, A. W., Barradas, M., Stone, J. C., van Aelst, L., Serrano, M., and Lowe, S. W. (1998). Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 12, 3008–3019. doi:10.1101/gad.12.19.3008
Lin, Y., Damjanovic, A., Metter, E. J., Nguyen, H., Truong, T., Najarro, K., et al. (2015). Age-associated telomere attrition of lymphocytes in vivo is co-ordinated with changes in telomerase activity, composition of lymphocyte subsets and health conditions. Clin. Sci. (Lond) 128, 367–377. doi:10.1042/CS20140481
Liu, B., Wang, J., Chan, K. M., Tjia, W. M., Deng, W., Guan, X., et al. (2005). Genomic instability in laminopathy-based premature aging. Nat. Med. 11, 780–785. doi:10.1038/nm1266
Liu, X., Mo, W., Ye, J., Li, L., Zhang, Y., Hsueh, E. C., et al. (2018). Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 9, 249. doi:10.1038/s41467-017-02689-5
Liu, Y., Sanoff, H. K., Cho, H., Burd, C. E., Torrice, C., Ibrahim, J. G., et al. (2009). Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 8, 439–448. doi:10.1111/j.1474-9726.2009.00489.x
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi:10.1016/j.cell.2013.05.039
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell 186, 243–278. doi:10.1016/j.cell.2022.11.001
Lord, J. M. (2013). The effect of ageing of the immune system on vaccination responses. Hum. Vaccin Immunother. 9, 1364–1367. doi:10.4161/hv.24696
Lorenzo, E. C., Torrance, B. L., Keilich, S. R., Al-Naggar, I., Harrison, A., Xu, M., et al. (2022). Senescence-induced changes in CD4 T cell differentiation can be alleviated by treatment with senolytics. Aging Cell 21, e13525. doi:10.1111/acel.13525
Mannick, J. B., Del Giudice, G., Lattanzi, M., Valiante, N. M., Praestgaard, J., Huang, B., et al. (2014). mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 6, 268ra179. doi:10.1126/scitranslmed.3009892
Mannick, J. B., Morris, M., Hockey, H. P., Roma, G., Beibel, M., Kulmatycki, K., et al. (2018). TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 10, eaaq1564. doi:10.1126/scitranslmed.aaq1564
Mannick, J. B., Teo, G., Bernardo, P., Quinn, D., Russell, K., Klickstein, L., et al. (2021). Targeting the biology of ageing with mTOR inhibitors to improve immune function in older adults: Phase 2b and phase 3 randomised trials. Lancet Healthy Longev. 2, e250–e262. doi:10.1016/S2666-7568(21)00062-3
Marquez, E. J., Chung, C. H., Marches, R., Rossi, R. J., Nehar-Belaid, D., Eroglu, A., et al. (2020). Sexual-dimorphism in human immune system aging. Nat. Commun. 11, 751. doi:10.1038/s41467-020-14396-9
Martens, C. R., Denman, B. A., Mazzo, M. R., Armstrong, M. L., Reisdorph, N., Mcqueen, M. B., et al. (2018). Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 9, 1286. doi:10.1038/s41467-018-03421-7
Martinez-Zamudio, R. I., Dewald, H. K., Vasilopoulos, T., Gittens-Williams, L., Fitzgerald-Bocarsly, P., and Herbig, U. (2021). Senescence-associated beta-galactosidase reveals the abundance of senescent CD8+ T cells in aging humans. Aging Cell 20, e13344. doi:10.1111/acel.13344
Mason, P. A., and Cox, L. S. (2012). The role of DNA exonucleases in protecting genome stability and their impact on ageing. Age (Dordr) 34, 1317–1340. doi:10.1007/s11357-011-9306-5
Mathiasen, S. L., Gall-Mas, L., Pateras, I. S., Theodorou, S. D. P., Namini, M. R. J., Hansen, M. B., et al. (2021). Bacterial genotoxins induce T cell senescence. Cell Rep. 35, 109220. doi:10.1016/j.celrep.2021.109220
Mittelbrunn, M., and Kroemer, G. (2021). Hallmarks of T cell aging. Nat. Immunol. 22, 687–698. doi:10.1038/s41590-021-00927-z
Moro-García, M. A., Alonso-Arias, R., and López-Larrea, C. (2013). When aging reaches CD4+ T-cells: phenotypic and functional changes. Front. Immunol. 4, 107. doi:10.3389/fimmu.2013.00107
Muscari, C., Guarnieri, C., Stefanelli, C., Giaccari, A., and Caldarera, C. M. (1995). Protective effect of spermine on DNA exposed to oxidative stress. Mol. Cell Biochem. 144, 125–129. doi:10.1007/BF00944391
Na, H. J., Park, J. S., Pyo, J. H., Lee, S. H., Jeon, H. J., Kim, Y. S., et al. (2013). Mechanism of metformin: Inhibition of DNA damage and proliferative activity in Drosophila midgut stem cell. Mech. Ageing Dev. 134, 381–390. doi:10.1016/j.mad.2013.07.003
Narita, M., Nunez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., et al. (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716. doi:10.1016/s0092-8674(03)00401-x
Nguyen, L. N., Zhao, J., Cao, D., Dang, X., Wang, L., Lian, J., et al. (2018). Inhibition of TRF2 accelerates telomere attrition and DNA damage in naive CD4 T cells during HCV infection. Cell Death Dis. 9, 900. doi:10.1038/s41419-018-0897-y
O'Driscoll, M., Ribeiro Dos Santos, G., Wang, L., Cummings, D. A. T., Azman, A. S., Paireau, J., et al. (2021). Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 590, 140–145. doi:10.1038/s41586-020-2918-0
Oeppen, J., and Vaupel, J. W. (2002). Demography, Broken limits to life expectancy. Science 296, 1029–1031. doi:10.1126/science.1069675
Oishi, Y., and Manabe, I. (2016). Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech. Dis. 2, 16018. doi:10.1038/npjamd.2016.18
Ovadya, Y., Landsberger, T., Leins, H., Vadai, E., Gal, H., Biran, A., et al. (2018). Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 9, 5435. doi:10.1038/s41467-018-07825-3
Passos, J. F., Nelson, G., Wang, C., Richter, T., Simillion, C., Proctor, C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, 347. doi:10.1038/msb.2010.5
Pastor-Fernandez, A., Bertos, A. R., Sierra-Ramirez, A., Del Moral-Salmoral, J., Merino, J., de Avila, A. I., et al. (2023). Treatment with the senolytics dasatinib/quercetin reduces SARS-CoV-2-related mortality in mice. Aging Cell 22, e13771. doi:10.1111/acel.13771
Patrick, M. S., Cheng, N. L., Kim, J., An, J., Dong, F., Yang, Q., et al. (2019). Human T cell differentiation negatively regulates telomerase expression resulting in reduced activation-induced proliferation and survival. Front. Immunol. 10, 1993. doi:10.3389/fimmu.2019.01993
Pereira, B. I., and Akbar, A. N. (2016). Convergence of innate and adaptive immunity during human aging. Front. Immunol. 7, 445. doi:10.3389/fimmu.2016.00445
Pereira, B. I., de Maeyer, R. P. H., Covre, L. P., Nehar-Belaid, D., Lanna, A., Ward, S., et al. (2020). Sestrins induce natural killer function in senescent-like CD8(+) T cells. Nat. Immunol. 21, 684–694. doi:10.1038/s41590-020-0643-3
Pereira, B. I., Devine, O. P., Vukmanovic-Stejic, M., Chambers, E. S., Subramanian, P., Patel, N., et al. (2019). Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat. Commun. 10, 2387. doi:10.1038/s41467-019-10335-5
Phadwal, K., Alegre-Abarrategui, J., Watson, A. S., Pike, L., Anbalagan, S., Hammond, E. M., et al. (2012). A novel method for autophagy detection in primary cells: Impaired levels of macroautophagy in immunosenescent T cells. Autophagy 8, 677–689. doi:10.4161/auto.18935
Phillips, E. J., and Simons, M. J. P. (2022). Rapamycin not dietary restriction improves resilience against pathogens: A meta-analysis. Geroscience 45, 1263–1270. doi:10.1007/s11357-022-00691-4
Pieren, D. K. J., Smits, N. A. M., Imholz, S., Nagarajah, B., van Oostrom, C. T., Brandt, R. M. C., et al. (2021). Compromised DNA repair promotes the accumulation of regulatory T cells with an aging-related phenotype and responsiveness. Front. Aging 2, 667193. doi:10.3389/fragi.2021.667193
Pinto, S., Sato, V. N., de-Souza, E. A., Ferraz, R. C., Camara, H., Pinca, A. P. F., et al. (2018). Enoxacin extends lifespan of C. elegans by inhibiting miR-34-5p and promoting mitohormesis. Redox Biol. 18, 84–92. doi:10.1016/j.redox.2018.06.006
Plunkett, F. J., Franzese, O., Finney, H. M., Fletcher, J. M., Belaramani, L. L., Salmon, M., et al. (2007). The loss of telomerase activity in highly differentiated CD8+CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation. J. Immunol. 178, 7710–7719. doi:10.4049/jimmunol.178.12.7710
Puleston, D. J., Zhang, H., Powell, T. J., Lipina, E., Sims, S., Panse, I., et al. (2014). Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 3, e03706. doi:10.7554/eLife.03706
Reed, J. R., Vukmanovic-Stejic, M., Fletcher, J. M., Soares, M. V., Cook, J. E., Orteu, C. H., et al. (2004). Telomere erosion in memory T cells induced by telomerase inhibition at the site of antigenic challenge in vivo. J. Exp. Med. 199, 1433–1443. doi:10.1084/jem.20040178
Riddell, N. E., Griffiths, S. J., Rivino, L., King, D. C. B., Teo, G. H., Henson, S. M., et al. (2015). Multifunctional cytomegalovirus (CMV)-specific CD8(+) T cells are not restricted by telomere-related senescence in young or old adults. Immunology 144 (4), 549–560. doi:10.1111/imm.12409
Rodier, F., Coppe, J. P., Patil, C. K., Hoeijmakers, W. A., Munoz, D. P., Raza, S. R., et al. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979. doi:10.1038/ncb1909
Rodier, F., Munoz, D. P., Teachenor, R., Chu, V., Le, O., Bhaumik, D., et al. (2011). DNA-SCARS: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 124, 68–81. doi:10.1242/jcs.071340
Rong, Z., Tu, P., Xu, P., Sun, Y., Yu, F., Tu, N., et al. (2021). The mitochondrial response to DNA damage. Front. Cell Dev. Biol. 9, 669379. doi:10.3389/fcell.2021.669379
Rossiello, F., Herbig, U., Longhese, M. P., Fumagalli, M., and D'Adda di Fagagna, F. (2014). Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 26, 89–95. doi:10.1016/j.gde.2014.06.009
Saha, B., Cypro, A., Martin, G. M., and Oshima, J. (2014). Rapamycin decreases DNA damage accumulation and enhances cell growth of WRN-deficient human fibroblasts. Aging Cell 13, 573–575. doi:10.1111/acel.12190
Sanderson, S. L., and Simon, A. K. (2017). In aged primary T cells, mitochondrial stress contributes to telomere attrition measured by a novel imaging flow cytometry assay. Aging Cell 16, 1234–1243. doi:10.1111/acel.12640
Sapey, E., Greenwood, H., Walton, G., Mann, E., Love, A., Aaronson, N., et al. (2014). Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: Toward targeted treatments for immunosenescence. Blood 123, 239–248. doi:10.1182/blood-2013-08-519520
Schmauck-Medina, T., Moliere, A., Lautrup, S., Zhang, J., Chlopicki, S., Madsen, H. B., et al. (2022). New hallmarks of ageing: A 2022 copenhagen ageing meeting summary. Aging (Albany NY) 14, 6829–6839. doi:10.18632/aging.204248
Schumacher, B., Pothof, J., Vijg, J., and Hoeijmakers, J. H. J. (2021). The central role of DNA damage in the ageing process. Nature 592, 695–703. doi:10.1038/s41586-021-03307-7
Schwarz, C., Stekovic, S., Wirth, M., Benson, G., Royer, P., Sigrist, S. J., et al. (2018). Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging (Albany NY) 10, 19–33. doi:10.18632/aging.101354
Senekowitsch, S., Wietkamp, E., Grimm, M., Schmelter, F., Schick, P., Kordowski, A., et al. (2023). High-dose spermidine supplementation does not increase spermidine levels in blood plasma and saliva of healthy adults: A randomized placebo-controlled pharmacokinetic and metabolomic study. Nutrients 15, 1852. doi:10.3390/nu15081852
Shao, L., Fujii, H., Colmegna, I., Oishi, H., Goronzy, J. J., and Weyand, C. M. (2009). Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J. Exp. Med. 206, 1435–1449. doi:10.1084/jem.20082251
Simon, M., van Meter, M., Ablaeva, J., Ke, Z., Gonzalez, R. S., Taguchi, T., et al. (2019). LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab. 29, 871–885 e5. doi:10.1016/j.cmet.2019.02.014
Soerens, A. G., Kunzli, M., Quarnstrom, C. F., Scott, M. C., Swanson, L., Locquiao, J. J., et al. (2023). Functional T cells are capable of supernumerary cell division and longevity. Nature 614, 762–766. doi:10.1038/s41586-022-05626-9
Solana, R., Tarazona, R., Gayoso, I., Lesur, O., Dupuis, G., and Fulop, T. (2012). Innate immunosenescence: Effect of aging on cells and receptors of the innate immune system in humans. Semin. Immunol. 24, 331–341. doi:10.1016/j.smim.2012.04.008
Stranks, A. J., Hansen, A. L., Panse, I., Mortensen, M., Ferguson, D. J., Puleston, D. J., et al. (2015). Autophagy controls acquisition of aging features in macrophages. J. Innate Immun. 7, 375–391. doi:10.1159/000370112
Strauss-Albee, D. M., Horowitz, A., Parham, P., and Blish, C. A. (2014). Coordinated regulation of NK receptor expression in the maturing human immune system. J. Immunol. 193, 4871–4879. doi:10.4049/jimmunol.1401821
Suda, M., Shimizu, I., Katsuumi, G., Yoshida, Y., Hayashi, Y., Ikegami, R., et al. (2021). Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat. Aging 1, 1117–1126. doi:10.1038/s43587-021-00151-2
Swahari, V., Nakamura, A., Baran-Gale, J., Garcia, I., Crowther, A. J., Sons, R., et al. (2016). Essential function of dicer in resolving DNA damage in the rapidly dividing cells of the developing and malignant cerebellum. Cell Rep. 14, 216–224. doi:10.1016/j.celrep.2015.12.037
Swift, L. P., Castle, L., and Mchugh, P. J. (2020). “Analysis of DNA interstrand cross-links and their repair by modified comet assay,” in DNA electrophoresis: Methods and protocols. Editor K. HANADA (New York, NY: Springer US).
Teissier, T., Boulanger, E., and Cox, L. S. (2022). Interconnections between inflammageing and immunosenescence during ageing. Cells 11, 359. doi:10.3390/cells11030359
Thompson, W. W., Shay, D. K., Weintraub, E., Brammer, L., Bridges, C. B., Cox, N. J., et al. (2004). Influenza-associated hospitalizations in the United States. JAMA 292, 1333–1340. doi:10.1001/jama.292.11.1333
Tuttle, C. S. L., Waaijer, M. E. C., Slee-Valentijn, M. S., Stijnen, T., Westendorp, R., and Maier, A. B. (2020). Cellular senescence and chronological age in various human tissues: A systematic review and meta-analysis. Aging Cell 19, e13083. doi:10.1111/acel.13083
van Deursen, J. M. (2014). The role of senescent cells in ageing. Nature 509, 439–446. doi:10.1038/nature13193
Vazquez-Martin, A., Oliveras-Ferraros, C., Cufi, S., Martin-Castillo, B., and Menendez, J. A. (2011). Metformin activates an ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle 10, 1499–1501. doi:10.4161/cc.10.9.15423
Vermeij, W. P., Dolle, M. E., Reiling, E., Jaarsma, D., Payan-Gomez, C., Bombardieri, C. R., et al. (2016). Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 537, 427–431. doi:10.1038/nature19329
von Zglinicki, T. (2002). Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344. doi:10.1016/s0968-0004(02)02110-2
von Zglinicki, T. (2000). Role of oxidative stress in telomere length regulation and replicative senescence. Ann. N. Y. Acad. Sci. 908, 99–110. doi:10.1111/j.1749-6632.2000.tb06639.x
Vukmanovic-Stejic, M., Chambers, E. S., Suarez-Farinas, M., Sandhu, D., Fuentes-Duculan, J., Patel, N., et al. (2018). Enhancement of cutaneous immunity during aging by blocking p38 mitogen-activated protein (MAP) kinase-induced inflammation. J. Allergy Clin. Immunol. 142, 844–856. doi:10.1016/j.jaci.2017.10.032
Wagner, A., Garner-Spitzer, E., Jasinska, J., Kollaritsch, H., Stiasny, K., Kundi, M., et al. (2018). Age-related differences in humoral and cellular immune responses after primary immunisation: Indications for stratified vaccination schedules. Sci. Rep. 8, 9825. doi:10.1038/s41598-018-28111-8
Walters, H. E., and Cox, L. S. (2018). mTORC inhibitors as broad-spectrum therapeutics for age-related diseases. Int. J. Mol. Sci. 19, 2325. doi:10.3390/ijms19082325
Walters, H. E., Deneka-Hannemann, S., and Cox, L. S. (2016). Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY) 8, 231–244. doi:10.18632/aging.100872
Wang, T. W., Johmura, Y., Suzuki, N., Omori, S., Migita, T., Yamaguchi, K., et al. (2022). Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 611, 358–364. doi:10.1038/s41586-022-05388-4
Wirth, M., Benson, G., Schwarz, C., Kobe, T., Grittner, U., Schmitz, D., et al. (2018). The effect of spermidine on memory performance in older adults at risk for dementia: A randomized controlled trial. Cortex 109, 181–188. doi:10.1016/j.cortex.2018.09.014
Wyllie, F. S., Jones, C. J., Skinner, J. W., Haughton, M. F., Wallis, C., Wynford-Thomas, D., et al. (2000). Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat. Genet. 24, 16–17. doi:10.1038/71630
Xu, M., Pirtskhalava, T., Farr, J. N., Weigand, B. M., Palmer, A. K., Weivoda, M. M., et al. (2018). Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256. doi:10.1038/s41591-018-0092-9
Yacoub, A., Park, J. S., Qiao, L., Dent, P., and Hagan, M. P. (2001). MAPK dependence of DNA damage repair: Ionizing radiation and the induction of expression of the DNA repair genes XRCC1 and ERCC1 in DU145 human prostate carcinoma cells in a MEK1/2 dependent fashion. Int. J. Radiat. Biol. 77, 1067–1078. doi:10.1080/09553000110069317
Yang, Q., XI, Q., Wang, M., Liu, J., Li, Z., Hu, J., et al. (2022). Rapamycin improves the developmental competence of human oocytes by alleviating DNA damage during IVM. Hum. Reprod. Open 2022, hoac050. doi:10.1093/hropen/hoac050
Ye, J., Huang, X., Hsueh, E. C., Zhang, Q., Ma, C., Zhang, Y., et al. (2012). Human regulatory T cells induce T-lymphocyte senescence. Blood 120, 2021–2031. doi:10.1182/blood-2012-03-416040
Yoshino, J., Baur, J. A., and Imai, S. I. (2018). NAD(+) intermediates: The biology and therapeutic potential of NMN and NR. Cell Metab. 27, 513–528. doi:10.1016/j.cmet.2017.11.002
Yousefzadeh, M. J., Flores, R. R., Zhu, Y., Schmiechen, Z. C., Brooks, R. W., Trussoni, C. E., et al. (2021b). An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105. doi:10.1038/s41586-021-03547-7
Yousefzadeh, M. J., Henpita, C., Vyas, R., Soto-Palma, C., Robbins, P., and Niedernhofer, L. (2021a). DNA damage-how and why we age? Elife 10, e62852. doi:10.7554/eLife.62852
Yousefzadeh, M. J., Zhu, Y., Mcgowan, S. J., Angelini, L., Fuhrmann-Stroissnigg, H., Xu, M., et al. (2018). Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28. doi:10.1016/j.ebiom.2018.09.015
Zhang, H., Alsaleh, G., Feltham, J., Sun, Y., Napolitano, G., Riffelmacher, T., et al. (2019). Polyamines control eIF5A hypusination, TFEB translation, and autophagy to reverse B cell senescence. Mol. Cell 76, 110–125 e9. doi:10.1016/j.molcel.2019.08.005
Zhang, H., Ryu, D., Wu, Y., Gariani, K., Wang, X., Luan, P., et al. (2016). NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. doi:10.1126/science.aaf2693
Zhao, J., Dang, X., Zhang, P., Nguyen, L. N., Cao, D., Wang, L., et al. (2018). Insufficiency of DNA repair enzyme ATM promotes naive CD4 T-cell loss in chronic hepatitis C virus infection. Cell Discov. 4, 16. doi:10.1038/s41421-018-0015-4
Zhou, D., Borsa, M., Puleston, D. J., Zellner, S., Capera, J., Sanderson, S., et al. (2022). Mapping autophagosome contents identifies interleukin-7 receptor-α as a key cargo modulating CD4+ T cell proliferation. Nat. Commun. 13, 5174. doi:10.1038/s41467-022-32718-x
Zhou, D., Borsa, M., and Simon, A. K. (2021). Hallmarks and detection techniques of cellular senescence and cellular ageing in immune cells. Aging Cell 20, e13316. doi:10.1111/acel.13316
Keywords: ageing, immunosenescence, senescence, immunology, DNA repair, DNA damage
Citation: Kell L, Simon AK, Alsaleh G and Cox LS (2023) The central role of DNA damage in immunosenescence. Front. Aging 4:1202152. doi: 10.3389/fragi.2023.1202152
Received: 07 April 2023; Accepted: 22 June 2023;
Published: 03 July 2023.
Edited by:
Jenna M. Bartley, University of Connecticut Health Center, United StatesReviewed by:
Satomi Miwa, Newcastle University, United KingdomCopyright © 2023 Kell, Simon, Alsaleh and Cox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lynne S. Cox, bHlubmUuY294QGJpb2NoLm94LmFjLnVr; Ghada Alsaleh, Z2hhZGEuYWxzYWxlaEBuZG9ybXMub3guYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.