
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Aging, 16 May 2023
Sec. Molecular Mechanisms of Aging
Volume 4 - 2023 | https://doi.org/10.3389/fragi.2023.1145198
This article is part of the Research TopicInsights in Molecular Mechanisms of Aging 2022View all 10 articles
 Annika Traa1,2,3
Annika Traa1,2,3 Hazel Shields1,2,3
Hazel Shields1,2,3 Abdelrahman AlOkda1,2,3Zenith D. Rudich1,2,3
Abdelrahman AlOkda1,2,3Zenith D. Rudich1,2,3 Bokang Ko1,2,3
Bokang Ko1,2,3 Jeremy M. Van Raamsdonk1,2,3,4*
Jeremy M. Van Raamsdonk1,2,3,4*Mutations that result in a mild impairment of mitochondrial function can extend longevity. Previous studies have shown that the increase in lifespan is dependent on stress responsive transcription factors, including DAF-16/FOXO, which exhibits increased nuclear localization in long-lived mitochondrial mutants. We recently found that the localization of DAF-16 within the cell is dependent on the endosomal trafficking protein TBC-2. Based on the important role of DAF-16 in both longevity and resistance to stress, we examined the effect of disrupting tbc-2 on lifespan and stress resistance in the long-lived mitochondrial mutants nuo-6 and isp-1 in Caenorhabditis elegans. Loss of tbc-2 markedly reduced the long lifespans of both mitochondrial mutants. Disruption of tbc-2 also decreased resistance to chronic oxidative stress in nuo-6 and isp-1 mutants but had little or no detrimental effect on resistance to other stressors. In contrast, tbc-2 inhibition had no effect on oxidative stress resistance or lifespan in isp-1 worms when DAF-16 is absent, suggesting that the effect of tbc-2 on mitochondrial mutant lifespan may be mediated by mislocalization of DAF-16. However, this result is complicated by the fact that deletion of daf-16 markedly decreases both phenotypes in isp-1 worms, which could result in a floor effect. In exploring the contribution of DAF-16 further, we found that disruption of tbc-2 did not affect the nuclear localization of DAF-16 in isp-1 worms or prevent the upregulation of DAF-16 target genes in the long-lived mitochondrial mutants. This suggests the possibility that the effect of tbc-2 on lifespan and stress resistance in the long-lived mitochondrial mutants is at least partially independent of its effects on DAF-16 localization. Overall, this work demonstrates the importance of endosomal trafficking for the extended longevity and enhanced stress resistance resulting from mild impairment of mitochondrial function.
Mitochondria are membrane-bound organelles that account for most of the energy production in the cell and have important roles in intracellular signaling and metabolism. Despite the importance of mitochondria to cellular function, mild impairment of mitochondrial function can extend lifespan and increase resistance to stress (Wong et al., 1995; Lakowski and Hekimi, 1996; Feng et al., 2001; Yang and Hekimi, 2010; Schaar et al., 2015; Dues et al., 2017; Wu et al., 2018). The ability of mutations affecting the mitochondria to extend lifespan has been demonstrated in multiple species including worms (Wong et al., 1995; Lakowski and Hekimi, 1996; Feng et al., 2001; Yang and Hekimi, 2010), flies (Copeland et al., 2009) and mice (Dell’agnello et al., 2007; Liu et al., 2005), thereby indicating conservation across species.
Among the most well studied mitochondrial mutants in Caenorhabditis elegans are nuo-6 and isp-1. The nuo-6 gene encodes a subunit of Complex I of the mitochondrial electron transport chain (Yang and Hekimi, 2010), while the isp-1 gene encodes a subunit of the Rieske iron sulfur protein in Complex III of the electron transport chain (Feng et al., 2001). Although a number of studies have identified factors that are important for the long lifespan of these mitochondrial mutants (Lee et al., 2010; Walter et al., 2011; Baruah et al., 2014; Hwang et al., 2014; Yee et al., 2014; Munkacsy et al., 2016; Senchuk et al., 2018; Wu et al., 2018; Campos et al., 2021; Jung et al., 2021; Harris-Gauthier et al., 2022), the mechanism by which mild mitochondrial impairment extends longevity remains incompletely understood.
Among the factors that are required for the longevity of the long-lived mitochondrial mutants is the FOXO transcription factor DAF-16 (Senchuk et al., 2018). nuo-6 and isp-1 mutants exhibit increased expression of DAF-16 target genes, which is dependent on DAF-16 and the DAF-16 deubiquitylase MATH-33. DAF-16 is part of the insulin/IGF-1 signaling pathway. Under conditions of decreased insulin/IGF-1 signaling or increased stress, DAF-16 translocates from the cytoplasm to the nucleus to modulate gene expression. Disrupting DAF-16 reverts nuo-6 and isp-1 lifespan to wild-type (Senchuk et al., 2018).
We recently showed that cytoplasmic DAF-16 can be localized to endosomes (Meras et al., 2022). Disruption of the insulin/IGF-1 signaling pathway decreases endosomal localization of DAF-16 while increasing its localization to the nucleus. Endosomal localization of DAF-16 is also affected by disruption of endosomal trafficking proteins. Specifically, loss of the Rab GTPase activating protein (GAP) TBC-2 increases the localization of DAF-16 to endosomes, while inhibition of the TBC-2 targets RAB-5 and RAB-7 has the opposite effect (Chotard et al., 2010; Law and Rocheleau, 2017; Meras et al., 2022). The increased endosomal localization of DAF-16 in tbc-2 mutants results in a decreased ability of DAF-16 to translocate to the nucleus in response to specific stresses (Traa et al., 2022). In addition, disruption of tbc-2 reduces the upregulation of DAF-16 target genes in long-lived daf-2 mutants and decreases their lifespan (Meras et al., 2022).
Under normal conditions, internalized vesicles containing cargo from the plasma membrane, such as ligand bound receptors, fuse together to form early endosomes, which contain RAB-5. From the early endosome, the internalized cargo can be sorted to the recycling endosome, the Trans-Golgi network or to late endosomes, which contain RAB-7. Cargo transported to late endosomes can then be degraded by the lysosome. When tbc-2 is disrupted, there is an accumulation of enlarged late endosomes resulting from increased activation of RAB-5 (Chotard et al., 2010).
In this work, we examined the role of TBC-2 in the extended longevity and enhanced stress resistance of long-lived C. elegans mitochondrial mutants. We found that disruption of tbc-2 decreases lifespan and resistance to specific exogenous stressors in nuo-6 and isp-1 worms. Surprisingly, the tbc-2 deletion did not affect the increased nuclear localization of DAF-16 in isp-1 mutants and failed to prevent upregulation of DAF-16 target genes in the long-lived mitochondrial mutants. Overall, this work demonstrates the importance of TBC-2 in stress resistance and longevity and suggests that tbc-2 disruption may be affecting these phenotypes in long-lived mitochondrial mutants at least partially through DAF-16-independent pathways.
N2 (WT)
QR15 tbc-2(tm2241) II
JVR171 isp-1(qm150) IV
MQ1333 nuo-6(qm200) I
JVR555 isp-1(qm150) IV;tbc-2(tm2241)
JVR556 nuo-6(qm200) I;tbc-2(tm2241)
JVR502 daf-16(mu86) I;isp-1(qm150) IV
JVR596 daf-16(mu86) I;tbc-2(tm2241) II
JVR619 daf-16(mu86) I;tbc-2(tm2241) II;isp-1(qm150) IV
JVR333 daf-16(mu86) I;zIs356 [daf-16p::daf-16a/b::GFP + rol-6(su1006)] IV
JVR640 daf-16(mu86) I;isp-1(qm150) IV;zIs356 [daf-16p::daf-16a/b::GFP + rol-6(su1006)] IV
JVR641 daf-16(mu86) I;isp-1(qm150 IV; tbc-2(tm2241) II; zIs356 [daf-16p::daf-16a/b::GFP + rol-6(su1006)] IV
Worms were maintained at 20°C on NGM plates seeded with OP50 bacteria.
Resistance to heat stress was measured by transferring 25 pre-fertile young adult worms to freshly seeded NGM plates, incubating at 37°C and monitoring survival every 2 h for a total of 10 h.
Resistance to chronic oxidative stress was measured by transferring 30 pre-fertile young adult worms to 60 mm NGM plates containing 4 mM paraquat (methyl viologen, Sigma Catalog No. 856177) and 100 μM FUdR and seeded with concentrated OP50 bacteria (see (Senchuk et al., 2017) for detailed protocol). Worms were kept at 20°C and survival was monitored daily until all worms died.
Resistance to acute oxidative stress was measured by transferring 30 pre-fertile young adult worms to 300 μM juglone plates seeded with OP50 bacteria and monitoring survival at 20°C every 2 h for a total of 8 h. Plates were made fresh on the day of the assay as juglone toxicity diminishes rapidly with time (see http://wbg.wormbook.org/2016/07/14/measuring-sensitivity-to-oxidative-stress-the-superoxide-generator-juglone-rapidly-loses-potency-with-time/).
To measure resistance to osmotic stress, 25 pre-fertile young adult worms were transferred to 450 or 550 mM NaCl plates seeded with OP50 bacteria and survival was scored after 24 h at 20°C.
To measure resistance to anoxic stress, 45 pre-fertile young adult worms were transferred to NGM plates freshly seeded with OP50 bacteria and sealed in low-oxygen Becton-Dickinson Bio-Bag Type A Environmental Chambers. After 48 h at 20°C, worms were allowed to recover outside of the chamber for 24 h before survival was measured.
Resistance to bacterial pathogen stress was performed as previously described (Campos et al., 2021). 45 L4 worms were transferred to 100 mg/L FUdR plates seeded with OP50 bacteria and were grown at 20°C until day 3 of adulthood. Day 3 adult worms were then transferred from these plates to 20 mg/L FudR plates seeded with Pseudomonas aeruginosa (PA14). Worms were kept at 20°C and survival was monitored daily until all worms died.
To measure lifespan, 50 pre-fertile young adult worms were transferred to 25 µM FUdR plates seeded with OP50 bacteria and kept at 20°C. Low concentrations of FUdR limit internal hatching and inhibit development of progeny without affecting longevity in wild-type worms (Van Raamsdonk and Hekimi, 2011). Survival was monitored daily until all worms died.
Quantitative RT-PCR was performed by collecting worms in M9 buffer. RNA was extracted using Trizol as we have described previously (Machiela et al., 2016) and converted to cDNA using a high-capacity cDNA Reverse Transcription kit (Applied Biosystems 4368814). Quantitative PCR was performed using a PowerUp SYBR Green Master Mix (Applied Biosystems A25742) in a MicroAmp Optical 96-well reaction plate (Applied Biosystems N8010560) and a Viia 7 applied biosystems qPCR machine. mRNA levels were calculated as the copy number of the gene of interest relative to the copy number of the endogenous control, act-3, and expressed as a percentage of WT. Primer sequences for each target gene are as follows:
sod-3 (AAAGGAGCTGATGGACACTATTAAGC, AAGTTATCCAGGGAACCGAAGTC), dod-3 (AAGTGCTCCGATTGTTACGC, ACATGAACACCGGCTCATTC), mtl-1 (ATGGCTTGCAAGTGTGACTG, GCTTCTGCTCTGCACAATGA), sodh-1 (GAAGGAGCTGGAAGTGTTGTTC, CTCCACGTATAGTGAGGTACTCCTG), ftn-1 (GAGTGGGGAACTGTCCTTGA, CGAATGTACCTGCTCTTCCA), icl-1 (TGTGAAGCCGAGGACTACCT, TCTCCGATCCAAGCTGATCT), act-3 (TGCGACATTGATATCCGTAAGG, GGTGGTTCCTCCGGAAAGAA).
DAF-16 localization to the nucleus was imaged in young adult hermaphrodite worms at the level of the whole body using the DAF-16::GFP reporter (zIs356[daf-16p::daf-16::GFP]) in isp-1;daf-16 worms and isp-1;daf-16;tbc-2 worms. We used worms with mutations in the endogenous daf-16 gene to ensure that the DAF-16 being localized to the nucleus has the GFP reporter. Worms were mounted onto 2% agarose pads and immobilized with 5–10 μL of 10 mM levamisole. Images were obtained used a ZEISS LSM780 confocal microscope using a ×40 objective lens. Images were then analyzed on Fiji (ImageJ) where the number of GFP positive foci was determined using the particle analysis tool. Approximately 20 worms per genotype were imaged over 3 replicates.
At least three biological replicates were completed in each experiment. For lifespan and stress assays, the experimenter was blinded to the genotype to ensure unbiased results. Worms were excluded from lifespan and stress assays if they crawled off the agar and died on the side of the plate, had internal hatching of progeny or expulsion of internal organs. Statistical significance of differences between genotypes was determined using a one-way ANOVA with Dunnet’s multiple comparison test, two-way ANOVA with Šidák’s multiple comparison test, a repeated measures ANOVA, or a log-rank test using Graphpad Prism. Complete statistical analysis can be found in Supplementary Table S1.
We previously showed that both daf-2 mutants and the long-lived mitochondrial mutants nuo-6 and isp-1 are dependent on DAF-16 for their enhanced longevity (Senchuk et al., 2018; Dues et al., 2019). We also showed that loss of tbc-2 decreases the lifespan of long-lived daf-2 insulin-IGF1 receptor mutants (Meras et al., 2022). Since deletion of tbc-2 affects the ability of DAF-16 to translocate to the nucleus (Traa et al., 2022), we wondered whether the disruption of tbc-2 would also decrease the lifespan of the long-lived mitochondrial mutants. Accordingly, we crossed tbc-2 deletion mutants to nuo-6 and isp-1 worms and quantified the resulting effect on lifespan. We found that nuo-6 and isp-1 worms both exhibit a marked decrease in lifespan when tbc-2 is disrupted (Figure 1).
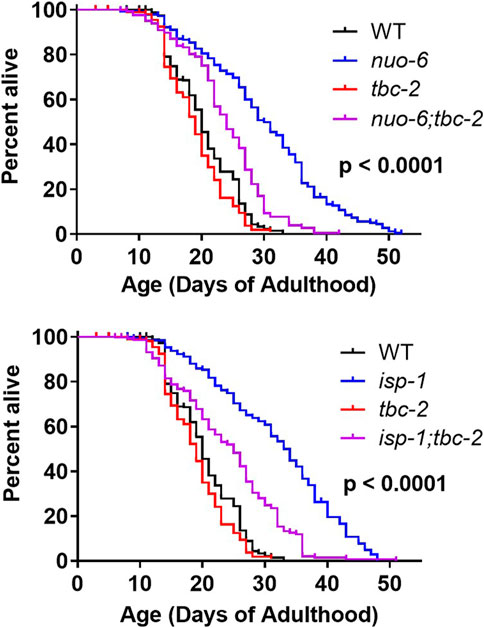
FIGURE 1. TBC-2 is required for the longevity of long-lived mitochondrial mutants. The long-lived mitochondrial mutants nuo-6 and isp-1 were crossed to tbc-2 deletion mutants. Disruption of tbc-2 markedly reduced the lifespan of both mutants, while only having a minor impact on wild-type lifespan. Significance indicates differences between blue and purple lifespan curves and was determined using the log-rank test. The Bonferroni-corrected threshold for significance is p = 0.00833. A minimum of three biological replicates per strain were performed. Raw lifespan data and full statistical comparisons can be found in Supplementary Table S1.
Next, we sought to determine the role of TBC-2 in the enhanced stress resistance of the long-lived mitochondrial mutants and whether this might be contributing to the effect of tbc-2 disruption on their lifespan. We quantified resistance to chronic oxidative stress (4 mM paraquat), acute oxidative stress (300 µM juglone), heat stress (37°C), bacterial pathogen stress (P. aeruginosa strain PA14), osmotic stress (450 mM and 550 mM NaCl) and anoxic stress.
In nuo-6 mutants, disruption of tbc-2 significantly decreased resistance to both chronic and acute oxidative stress (Figures 2A, B). In contrast, the loss of tbc-2 did not significantly decrease resistance to heat stress (Figure 2C), bacterial pathogen stress (Figure 2D), osmotic stress (Figures 2E, F) or anoxic stress (Figure 2G) in nuo-6 worms. In isp-1 mutants, deletion of tbc-2 decreased resistance to chronic oxidative stress (Figure 3A) but did not reduce resistance to acute oxidative stress (Figure 3B), heat stress (Figure 3C) or bacterial pathogens (Figure 3D). Disruption of tbc-2 decreased resistance to osmotic stress in isp-1 worms (Figures 3E, F), but did not decrease resistance to anoxia (Figure 3G). Combined, these results indicate that disruption of tbc-2 alters resistance to some, but not all, external stressors in long-lived mitochondrial mutants, and primarily affects resistance to chronic oxidative stress.
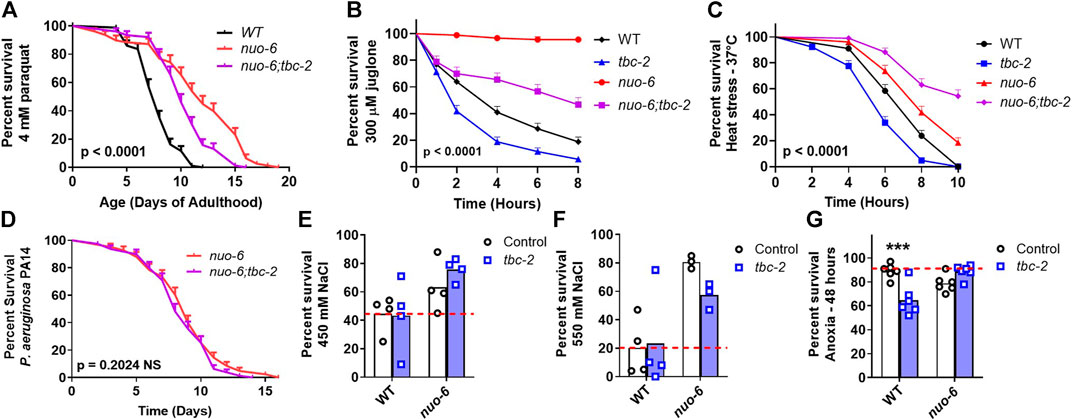
FIGURE 2. Disruption of tbc-2 decreases resistance to oxidative stress in long-lived nuo-6 mutants. Disruption of tbc-2 decreases resistance to both chronic (A); 4 mM paraquat) and acute (B); 300 µM juglone) oxidative stress in nuo-6 worms but has no significant effect on resistance heat stress (C); 37°C) in these worms. Disruption of tbc-2 did not affect resistance to bacterial pathogen stress (D); Pseudomonas aeruginosa strain PA14), osmotic stress (E, F); 450 mM NaCl, 550 mM NaCl) or anoxia (G); 48 h) in nuo-6 worms. A minimum of three biological replicates were performed. Statistical significance was assessed using the log-rank test in panels (A–D). In panels (A–D), p-values indicate the significance of difference between nuo-6 (red line) and nuo-6;tbc-2 (purple line). Significance was determined using a two-way ANOVA with Šidák’s multiple comparison test in panels (E–G). Full statistical analyses can be found in Supplementary Table S1. Error bars indicate standard error (SE). ***p < 0.001.
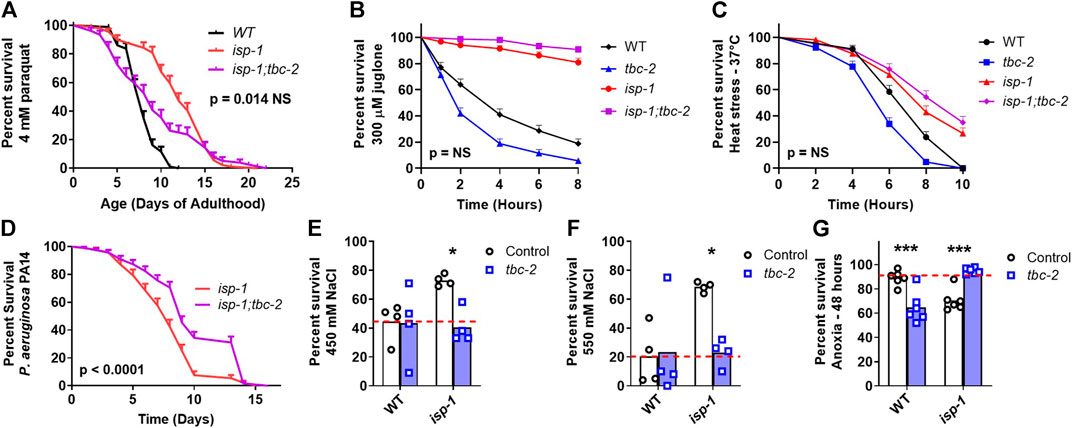
FIGURE 3. Disruption of tbc-2 decreases stress resistance in long-lived isp-1 mutants. Disruption of tbc-2 decreases resistance to chronic oxidative stress in isp-1 worms (A); 4 mM paraquat) but has no effect on resistance to acute oxidative stress (B); 300 µM juglone) or heat stress (C); 37°C) in these worms. The tbc-2 deletion increases resistance to bacterial pathogen stress in isp-1 worms (D); P. aeruginosa strain PA14). Disruption of tbc-2 decreased resistance to osmotic stress in isp-1 worms (E, F); 450 mM NaCl, 550 mM NaCl), but resulted in an increased resistance to anoxia in isp-1 mutants (G); 48 h). Control data on wild-type and tbc-2 mutants is repeated from Figure 2 for comparison. A minimum of three biological replicates were performed. Statistical significance was assessed using the log-rank test in panels (A–D), and a two-way ANOVA with Šidák’s multiple comparison test in panels (E–G). In panels (A–D), p-values indicate significance of difference between isp-1 (red line) and isp-1;tbc-2 (purple line). Full statistical analyses can be found in Supplementary Table S1. Error bars indicate standard error (SE). *p < 0.05, ***p < 0.001.
Our previous work suggests that the effect of TBC-2 disruption on stress resistance in wild-type and daf-2 worms is primarily dependent on DAF-16, while its effect on wild-type and daf-2 lifespan is also mediated by TBC-2’s impact on DAF-16-independent pathways (Traa et al., 2022). To gain insight into the extent to which deletion of tbc-2 decreases oxidative stress resistance and lifespan through DAF-16-dependent pathways, we examined the effect of disrupting tbc-2 in isp-1 worms lacking DAF-16 (isp-1;daf-16 mutants). We did not examine the DAF-16 dependency of the effect of tbc-2 disruption on nuo-6 worms because we are unable to generate nuo-6;daf-16 or nuo-6;daf-16;tbc-2 worms because of the close proximity of nuo-6 and daf-16 on the same chromosome.
We found that isp-1;daf-16 mutants have markedly decreased resistance to oxidative stress (4 mM paraquat) compared to isp-1 worms and that this deficit was not significantly exacerbated by disruption of tbc-2 (Figures 4A, B). Similarly, isp-1;daf-16 worms have a greatly reduced lifespan compared to isp-1 worms and their lifespan is not further decreased by loss of tbc-2 (Figures 4C, D). In both cases, however, there was a trend towards decrease in the absence of tbc-2.
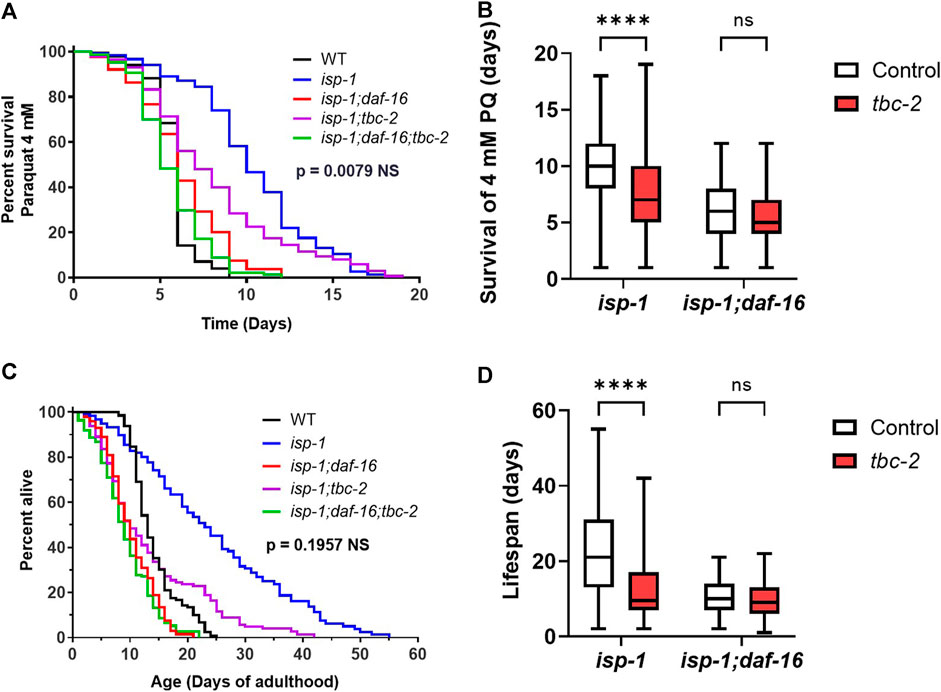
FIGURE 4. Loss of TBC-2 does not exacerbate the effects of disrupting DAF-16 on oxidative stress resistance or lifespan in isp-1 mutants. To determine the role of DAF-16 in tbc-2’s detrimental impact on oxidative stress resistance and longevity, tbc-2 was disrupted in isp-1 worms with a deletion in daf-16. In isp-1;daf-16 worms, deletion of tbc-2 did not significantly decrease resistance to oxidative stress when exposed to 4 mM paraquat (A, B). Similarly, disruption of tbc-2 did not significantly decrease isp-1;daf-16 lifespan (C, D). In both cases, there was a trend towards decrease with the loss of tbc-2. Four biological replicates were performed. Statistical significance was assessed using a log-rank test in panels (A, C), or a two-way ANOVA with Šidák’s multiple comparison test in panels (B, D). In panels (A, C), the p-value indicated is the significance of the difference between the isp-1;daf-16(blue line) and isp-1;daf = 16;tbc-2 (purple line). Full statistical analysis can be found in Supplementary Table S1. Box plot error bars show minimum and maximum values. Box plot line indicates average value. NS = not significant, ****p < 0.0001.
In daf-2 mutants, the nuclear localization of DAF-16 and the upregulation of DAF-16 target genes is inhibited by the loss of tbc-2 (Meras et al., 2022). Similarly, TBC-2 is required for nuclear localization of DAF-16 in response to some stresses and for the full upregulation of DAF-16 target genes in wild-type worms exposed to exogenous stressors (Traa et al., 2022). Since the long-lived mitochondrial mutants also exhibit increased nuclear localization of DAF-16 and upregulation of DAF-16 target genes (Senchuk et al., 2018), we wondered whether TBC-2 is required for the increased expression of DAF-16 targets in these mutants and if the prevention of this gene upregulation might be contributing to the decrease in stress resistance and lifespan resulting from tbc-2 deletion.
To examine the effect of tbc-2 disruption on the nuclear localization of DAF-16 in the long-lived mitochondrial mutants, we used a strain expressing DAF-16 linked to GFP under its endogenous promoter (zIs356[daf-16p::daf-16::GFP]). These experiments were performed in a daf-16 mutant background, as previous studies have shown that it is important to remove the endogenous daf-16 allele in order to properly visualize the nuclear localization of DAF-16::GFP likely because DAF-16 moves to the nucleus more readily than DAF-16::GFP (Putker et al., 2013; Senchuk et al., 2018). As we have observed previously, we found that isp-1 worms have increased nuclear localization of DAF-16::GFP compared to wild-type worms (Senchuk et al., 2018). However, the increased nuclear localization of DAF-16 in isp-1 worms was not affected by the disruption of TBC-2 (Figure 5A).
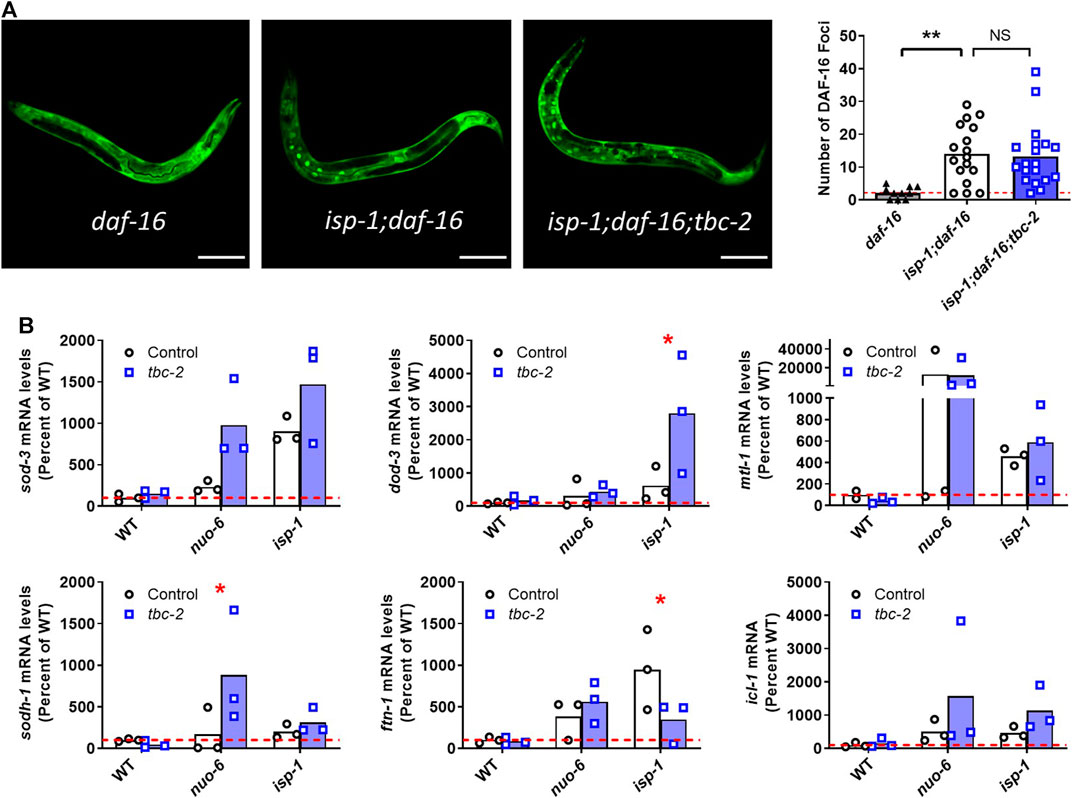
FIGURE 5. Disruption of TBC-2 does not affect nuclear localization of DAF-16 or prevent upregulation of DAF-16 target genes in long-lived mitochondrial mutants. (A) The nuclear localization of DAF-16 was assessed using zIs356[daf-16p::daf-16::GFP] worms. This experiment was performed in a daf-16 mutant background because DAF-16 moves to the nucleus more readily than DAF-16::GFP. isp-1 mutants have increased nuclear localization of DAF-16::GFP compared to wild-type worms. The nuclear localization of DAF-16 in isp-1 worms was not affected by the disruption of tbc-2. Statistical significance was assessed using a one-way ANOVA with Dunnett’s multiple comparison test. (B) Quantitative RT-PCR was used to quantify the effect of tbc-2 deletion on the upregulation of DAF-16 target genes (sod-3, dod-3, mtl-1, sodh-1, ftn-1, and icl-1) in the long-lived mitochondrial mutants, nuo-6 and isp-1. All of the DAF-16 target genes were upregulated in nuo-6 and isp-1 mutants. In all but one instance, the disruption of tbc-2 did not prevent the upregulation of DAF-16 target genes in the long-lived mitochondrial mutants. This suggests that TBC-2 is not required for the upregulation of DAF-16 target genes in the long-lived mitochondrial mutants. Three biological replicates were performed. Statistical significance was assessed using a two-way ANOVA with Šidák’s multiple comparison test. Full statistical analyses can be found in Supplementary Table S1. Error bars indicates SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we used quantitative RT-PCR to compare the expression of DAF-16 target genes between long-lived mitochondrial mutants in a wild-type and tbc-2 mutant background. For this purpose, we chose six DAF-16 target genes (sod-3, dod-3, mtl-1, sodh-1, ftn-1, icl-1) that were among the top fifty DAF-16 upregulated genes identified by a meta-analysis of DAF-16 gene expression studies (Tepper et al., 2013), and which we have previously used to examine DAF-16 activity (Senchuk et al., 2018; Campos et al., 2021; Meras et al., 2022; Traa et al., 2022).
Consistent with our past results (Senchuk et al., 2018), we found that all six DAF-16 target genes are upregulated in both nuo-6 and isp-1 mutants (Figure 5B). Disruption of tbc-2 did not result in a strong or consistent effect on the expression of DAF-16 target genes in the long-lived mitochondrial mutants. There was one example in which loss of tbc-2 significantly decreased DAF-16 target gene expression (ftn-1 levels in isp-1 mutants) and two examples in which the loss of tbc-2 resulted in a significant increase in the expression of DAF-16 target genes (dod-3 in isp-1 worms and sodh-1 in nuo-6 mutants). Disruption of tbc-2 did not significantly alter the expression of most of the DAF-16 target genes examined in wild-type, nuo-6 or isp-1 worms (Figure 5B). Combined, this suggests that disruption of tbc-2 does not markedly inhibit the ability of nuo-6 and isp-1 worms to upregulate DAF-16 target genes. However, it is possible that the upregulation of other DAF-16 target genes in nuo-6 and isp-1 worms is affected by tbc-2 disruption.
What is the role of TBC-2 is stress resistance and lifespan? The results from our current and previous studies indicate that stress responsive factors such as the FOXO transcription factor DAF-16 can be localized to endosomes and that this localization is affected by TBC-2. Under normal conditions a small amount of DAF-16 is present on endosomes, while disruption of tbc-2 results in increased levels of endosomal DAF-16 (Meras et al., 2022). When we originally observed DAF-16 on endosomes, we hypothesized that the increase in endosomal DAF-16 might allow for a stronger DAF-16 response to stress because of the larger stockpile of DAF-16 at the endosomes. However, it was also possible that disruption of TBC-2 would cause a weaker DAF-16 response because DAF-16 was stuck to endosomes. It turns out that both may be true depending on the stress. For heat stress, bacterial pathogen stress and anoxia, the nuclear localization of DAF-16 was slower in tbc-2 mutants than wild-type worms, suggesting that endosomal DAF-16 is less able to move to the nucleus under these stresses (Traa et al., 2022). In contrast, DAF-16 moved to the nucleus more rapidly under chronic oxidative stress and osmotic stress in tbc-2 mutants, suggesting that having a stockpile of DAF-16 at the endosomes enhances DAF-16 nuclear localization under these stresses.
In our current work, we find that the loss of TBC-2 decreases both stress resistance and lifespan in nuo-6 mutants and isp-1 mutants. We previously showed that disruption of tbc-2 also decreases stress resistance and lifespan in long-lived daf-2 mutants (Traa et al., 2022). Interestingly, the pattern of decreased stress resistance resulting from deletion of tbc-2 differs between these three strains (Supplementary Table S2). While disruption of tbc-2 decreases resistance to oxidative stress, heat stress, bacterial pathogen stress, osmotic stress and anoxia in daf-2 mutants, resistance to heat stress, bacterial pathogen stress and anoxic stress is not decreased in either of the long-lived mitochondrial mutants when tbc-2 is deleted. As our previous work suggests that the effect of tbc-2 deletion on resistance to stress is primarily mediated by DAF-16 (Traa et al., 2022), it is plausible that stress resistance is more affected in daf-2 mutants than nuo-6 or isp-1 mutants because the enhanced stress resistance of daf-2 worms is entirely dependent on DAF-16 (Dues et al., 2019), while the long-lived mitochondrial mutants also rely on other pathways such as the mitochondrial unfolded protein response (Wu et al., 2018), the p38-mediated innate immune signaling pathway (Campos et al., 2021) and the mitochondrial thioredoxin system (Harris-Gauthier et al., 2022).
The only type of stress resistance that is significantly affected in all three strains is resistance to chronic oxidative stress. While this result is consistent with the possibility that disruption of tbc-2 is decreasing lifespan by diminishing resistance to oxidative stress, the deletion of tbc-2 also decreases lifespan in wild-type worms without affecting their ability to survive chronic oxidative stress (Traa et al., 2022). In addition, there are multiple examples in which resistance to oxidative stress can be experimentally dissociated from longevity (Van Raamsdonk and Hekimi, 2009; Van Raamsdonk et al., 2010; Shields et al., 2021).
To determine the extent to which deletion of tbc-2 is decreasing oxidative stress resistance and lifespan through its effect on DAF-16, we examined the effect of disrupting tbc-2 when DAF-16 is absent. We reasoned that if the tbc-2 deletion is acting entirely through DAF-16 then disruption of tbc-2 should not further decrease stress resistance or lifespan when DAF-16 is absent. We found that disruption of tbc-2 resulted in a trend towards decreased resistance to chronic oxidative stress and lifespan in isp-1;daf-16 mutants, which did not reach significance. This result is consistent with the effects of tbc-2 deletion being mediated by DAF-16 but could also be explained by a floor effect since disruption of daf-16 markedly reduces the lifespan and stress resistance of isp-1 worms.
In exploring further, we found that disruption of tbc-2 did not affect the nuclear localization of DAF-16 or prevent the upregulation of DAF-16 target genes in the long-lived mitochondrial mutants. These findings are consistent with the effects of tbc-2 deletion being mediated by DAF-16-independent pathways (Figure 6). These differing conclusions may stem from the daf-16 deletion having such a severe impact on isp-1 stress resistance and longevity that the effects of tbc-2 disruption on DAF-16-independent pathways are being masked. This possibility is supported by the fact that we did observe a trend towards decrease in isp-1;daf-16;tbc-2 mutants compared to isp-1;daf-16 mutants.
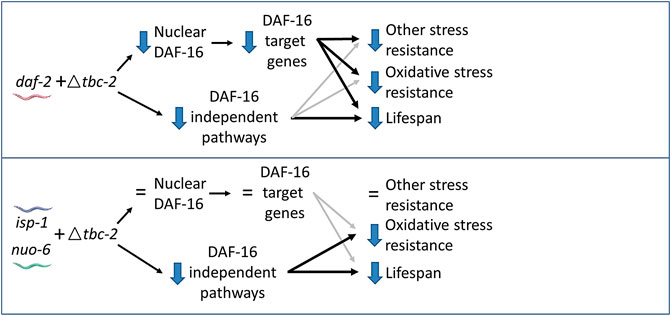
FIGURE 6. Model for contribution of DAF-16-dependent and DAF-16-independent pathways to effect of tbc-2 deletion on stress resistance and longevity. In daf-2 mutants, deletion of tbc-2 decreases the nuclear localization of DAF-16 and decreases the expression of DAF-16 target genes. Decreased activation of DAF-16 is primarily responsible for the decreased stress resistance in daf-2;tbc-2 mutants and partially contributes to their decrease in lifespan compared to daf-2 worms. Decreased activation of DAF-16-independent pathways also contributes to the detrimental effect of tbc-2 deletion on daf-2 lifespan. In the long-lived mitochondrial mutants, isp-1 and nuo-6, deletion of tbc-2 does not affect the nuclear localization of DAF-16 or decrease the expression of DAF-16-target genes. Deletion of tbc-2 also has little effect on resistance to most types of stress in isp-1 and nuo-6 worms. Disruption of tbc-2 decreases chronic oxidative stress resistance and lifespan in isp-1 and nuo-6 worms primarily through DAF-16-independent pathways.
In contrast to the long-lived mitochondrial mutants, TBC-2 is required for the full upregulation of DAF-16 target genes in daf-2 worms (Meras et al., 2022) and also important for the nuclear localization of DAF-16 in daf-2 mutants (Traa et al., 2022). In these worms, the disruption of TBC-2 appears to be affecting stress resistance primarily through DAF-16 but affecting lifespan through DAF-16 and DAF-16-independent pathways (Traa et al., 2022). These differences likely stem from daf-2 mutants being more dependent on DAF-16 for their stress resistance and longevity than the long-lived mitochondrial mutants, which rely more on DAF-16-independent pathways compared to daf-2 worms. These DAF-16-independent pathways may include signaling through the nuclear hormone receptor NHR-49, epidermal growth factor signaling and notch signaling, all of which have been shown to be present on endosomes and influence longevity (Arantes-Oliveira et al., 2002; Wang et al., 2002; Curran and Ruvkun, 2007; Fortini and Bilder, 2009; Iwasa et al., 2010; Walter et al., 2011; Yu and Driscoll, 2011; Khan et al., 2013; Zheng et al., 2013; Dresen et al., 2015; Watterson et al., 2022). In the future, it will be important to assess the contribution of these and other signaling pathways that are present on endosomes in the long-lived mitochondrial mutants.
Two other endosome related proteins were recently shown to be required for the longevity of isp-1 worms. After using proteomics to identify differentially expressed proteins in long-lived clk-1 and isp-1 mutants, a targeted RNAi screen was performed to assess the contribution of these proteins to their longevity. It was found that RNAi knockdown or genetic deletion of mon-2 decreases isp-1 lifespan (Jung et al., 2021). MON-2 regulates trafficking between endosomes and the Golgi apparatus (Mahajan et al., 2013). Similarly, TBC-3 is also involved in transport between endosomes and Golgi (Kanamori et al., 2008) and is required for isp-1 lifespan (Jung et al., 2021). While MON-2 appears to be promoting longevity by upregulating autophagy, it is unclear how elevated endosomal MON-2 increases autophagy (Jung et al., 2021; Artan et al., 2022). Future studies will be needed to determine whether MON-2 and TBC-3 are required for endosomal signaling pathways, similar to TBC-2, or whether these endosome-related proteins are all acting through independent mechanisms.
We find that disruption of the endosomal trafficking protein TBC-2 decreases oxidative stress resistance and lifespan in the long-lived mitochondrial mutants nuo-6 and isp-1. Interestingly, the loss of TBC-2 does not prevent the upregulation of DAF-16 target genes in these mutants suggesting that the disruption of tbc-2 may be affecting lifespan and resistance to stress in these worms by affecting other DAF-16-independent signaling pathways localized to endosomes. This work demonstrates an important role of endosomal signaling in stress resistance and lifespan.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Conceptualization: JV. methodology: AT, HS, AA, ZR, BK, and JV. investigation: AT, HS, AA, ZR, and BK. Analysis: AT, HS, AA, ZR, BK, and JV. visualization AT, HS, AA, ZR, BK, and JV. writing—original draft: AT and JV. writing—review and editing: AT, HS, AA, ZR, BK, and JV. supervision: JV. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication. All authors contributed to the article and approved the submitted version.
This work was supported by the Canadian Institutes of Health Research (CIHR; http://www.cihr-irsc.gc.ca/; JV) and the Natural Sciences and Engineering Research Council of Canada (NSERC; https://www.nserc-crsng.gc.ca/index_eng.asp; JV). JV received a salary award from Fonds de Recherche du Quebec Santé (FRQS) and Parkinson Quebec. AT received scholarships from NSERC, FRQS and CIHR. HS received an undergraduate student research award (URSA) from NSERC. AA received a scholarship from Healthy Brains Healthy Lives (HBHL). ZR received a postdoctoral award from FRQS.
We would like to thank Christian Rocheleau for discussions about this work and for reviewing the manuscript prior to submission. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P30 OD010440). We would also like to acknowledge the C. elegans knockout consortium and the National Bioresource Project of Japan for providing strains used in this research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fragi.2023.1145198/full#supplementary-material
Arantes-Oliveira, N., Apfeld, J., Dillin, A., and Kenyon, C. (2002). Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 295, 502–505. doi:10.1126/science.1065768
Artan, M., Sohn, J., Lee, C., Park, S. Y., and Lee, S. V. (2022). Mon-2, a Golgi protein, promotes longevity by upregulating autophagy through mediating inter-organelle communications. Autophagy 18, 1208–1210. doi:10.1080/15548627.2022.2039523
Baruah, A., Chang, H., Hall, M., Yuan, J., Gordon, S., Johnson, E., et al. (2014). CEP-1, the Caenorhabditis elegans p53 homolog, mediates opposing longevity outcomes in mitochondrial electron transport chain mutants. PLoS Genet. 10, e1004097. doi:10.1371/journal.pgen.1004097
Campos, J. C., Wu, Z., Rudich, P. D., Soo, S. K., Mistry, M., Ferreira, J. C., et al. (2021). Mild mitochondrial impairment enhances innate immunity and longevity through ATFS-1 and p38 signaling. EMBO reports, e52964.
Chotard, L., Mishra, A. K., Sylvain, M. A., Tuck, S., Lambright, D. G., and Rocheleau, C. E. (2010). TBC-2 regulates RAB-5/RAB-7-mediated endosomal trafficking in Caenorhabditis elegans. Mol. Biol. Cell 21, 2285–2296. doi:10.1091/mbc.e09-11-0947
Copeland, J. M., Cho, J., Lo, T., Hur, J. H., Bahadorani, S., Arabyan, T., et al. (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598. doi:10.1016/j.cub.2009.08.016
Curran, S. P., and Ruvkun, G. (2007). Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 3, e56. doi:10.1371/journal.pgen.0030056
Dell'agnello, C., Leo, S., Agostino, A., Szabadkai, G., Tiveron, C., Zulian, A., et al. (2007). Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431–444. doi:10.1093/hmg/ddl477
Dresen, A., Finkbeiner, S., Dottermusch, M., Beume, J. S., Li, Y., Walz, G., et al. (2015). Caenorhabditis elegans OSM-11 signaling regulates SKN-1/Nrf during embryonic development and adult longevity and stress response. Dev. Biol. 400, 118–131. doi:10.1016/j.ydbio.2015.01.021
Dues, D. J., Andrews, E. K., Senchuk, M. M., and Van Raamsdonk, J. M. (2019). Resistance to stress can Be experimentally dissociated from longevity. journals gerontology Ser. A, Biol. Sci. Med. Sci. 74, 1206–1214. doi:10.1093/gerona/gly213
Dues, D. J., Schaar, C. E., Johnson, B. K., Bowman, M. J., Winn, M. E., Senchuk, M. M., et al. (2017). Uncoupling of oxidative stress resistance and lifespan in long-lived isp-1 mitochondrial mutants in Caenorhabditis elegans. Free Radic. Biol. Med. 108, 362–373. doi:10.1016/j.freeradbiomed.2017.04.004
Feng, J., Bussiere, F., and Hekimi, S. (2001). Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644. doi:10.1016/s1534-5807(01)00071-5
Fortini, M. E., and Bilder, D. (2009). Endocytic regulation of Notch signaling. Curr. Opin. Genet. Dev. 19, 323–328. doi:10.1016/j.gde.2009.04.005
Harris-Gauthier, N., Traa, A., AlOkda, A., Moldakozhayev, A., Anglas, U., Soo, S. K., et al. (2022). Mitochondrial thioredoxin system is required for enhanced stress resistance and extended longevity in long-lived mitochondrial mutants. Redox Biol. 53, 102335. doi:10.1016/j.redox.2022.102335
Hwang, A. B., Ryu, E. A., Artan, M., Chang, H. W., Kabir, M. H., Nam, H. J., et al. (2014). Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 111, E4458–E4467. doi:10.1073/pnas.1411199111
Iwasa, H., Yu, S., Xue, J., and Driscoll, M. (2010). Novel EGF pathway regulators modulate C. elegans healthspan and lifespan via EGF receptor, PLC-gamma, and IP3R activation. Aging Cell 9, 490–505. doi:10.1111/j.1474-9726.2010.00575.x
Jung, Y., Artan, M., Kim, N., Yeom, J., Hwang, A. B., Jeong, D. E., et al. (2021). Mon-2, a Golgi protein, mediates autophagy-dependent longevity in Caenorhabditis elegans. Sci. Adv. 7, eabj8156. doi:10.1126/sciadv.abj8156
Kanamori, T., Inoue, T., Sakamoto, T., Gengyo-Ando, K., Tsujimoto, M., Mitani, S., et al. (2008). Beta-catenin asymmetry is regulated by PLA1 and retrograde traffic in C. elegans stem cell divisions. EMBO J. 27, 1647–1657. doi:10.1038/emboj.2008.102
Khan, M. H., Ligon, M., Hussey, L. R., Hufnal, B., Farber, R., Munkacsy, E., et al. (2013). TAF-4 is required for the life extension of isp-1, clk-1 and tpk-1 Mit mutants. Albany NY): Aging.
Lakowski, B., and Hekimi, S. (1996). Determination of life-span in Caenorhabditis elegans by four clock genes. Science 272, 1010–1013. doi:10.1126/science.272.5264.1010
Law, F., and Rocheleau, C. E. (2017). Vps34 and the armus/TBC-2 Rab GAPs: Putting the brakes on the endosomal Rab5 and Rab7 GTPases. Cell Logist. 7, e1403530. doi:10.1080/21592799.2017.1403530
Lee, S. J., Hwang, A. B., and Kenyon, C. (2010). Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 20, 2131–2136. doi:10.1016/j.cub.2010.10.057
Liu, X., Jiang, N., Hughes, B., Bigras, E., Shoubridge, E., and Hekimi, S. (2005). Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes & Dev. 19, 2424–2434. doi:10.1101/gad.1352905
Machiela, E., Dues, D. J., Senchuk, M. M., and Van Raamsdonk, J. M. (2016). Oxidative stress is increased in C. elegans models of Huntington's disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol. Dis. 96, 1–11. doi:10.1016/j.nbd.2016.08.008
Mahajan, D., Boh, B. K., Zhou, Y., Chen, L., Cornvik, T. C., Hong, W., et al. (2013). Mammalian Mon2/Ysl2 regulates endosome-to-Golgi trafficking but possesses no guanine nucleotide exchange activity toward Arl1 GTPase. Sci. Rep. 3, 3362. doi:10.1038/srep03362
Meras, I., Chotard, L., Liontis, T., Ratemi, Z., Wiles, B., Seo, J. H., et al. (2022). The Rab GTPase activating protein TBC-2 regulates endosomal localization of DAF-16 FOXO and lifespan. PLoS Genet. 18, e1010328. doi:10.1371/journal.pgen.1010328
Munkacsy, E., Khan, M. H., Lane, R. K., Borror, M. B., Park, J. H., Bokov, A. F., et al. (2016). DLK-1, SEK-3 and PMK-3 are required for the life extension induced by mitochondrial bioenergetic disruption in C. elegans. PLoS Genet. 12, e1006133. doi:10.1371/journal.pgen.1006133
Putker, M., Madl, T., Vos, H. R., de Ruiter, H., Visscher, M., van den Berg, M. C., et al. (2013). Redox-dependent control of FOXO/DAF-16 by transportin-1. Mol. Cell 49, 730–742. doi:10.1016/j.molcel.2012.12.014
Schaar, C. E., Dues, D. J., Spielbauer, K. K., Machiela, E., Cooper, J. F., Senchuk, M., et al. (2015). Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 11, e1004972. doi:10.1371/journal.pgen.1004972
Senchuk, M. M., Dues, D. J., Schaar, C. E., Johnson, B. K., Madaj, Z. B., Bowman, M. J., et al. (2018). Activation of DAF-16/FOXO by reactive oxygen species contributes to longevity in long-lived mitochondrial mutants in Caenorhabditis elegans. PLoS Genet. 14, e1007268. doi:10.1371/journal.pgen.1007268
Senchuk, M. M., Dues, D. J., and Van Raamsdonk, J. M. (2017). Measuring oxidative stress in Caenorhabditis elegans: Paraquat and juglone sensitivity assays. Bio-protocol 7, e2086. doi:10.21769/BioProtoc.2086
Shields, H. J., Traa, A., and Van Raamsdonk, J. M. (2021). Beneficial and detrimental effects of reactive oxygen species on lifespan: A comprehensive review of comparative and experimental studies. Front. Cell Dev. Biol. 9, 628157. doi:10.3389/fcell.2021.628157
Tepper, R. G., Ashraf, J., Kaletsky, R., Kleemann, G., Murphy, C. T., and Bussemaker, H. J. (2013). PQM-1 complements DAF-16 as a key transcriptional regulator of DAF-2-mediated development and longevity. Cell 154, 676–690. doi:10.1016/j.cell.2013.07.006
Traa, A., Soo, S. K., AlOkda, A., Ko, B., Rocheleau, C., and Van Raamsdonk, J. M. (2022). Endosomal trafficking protein TBC-2 modulates stress resistance and lifespan through DAF-16-dependent and independent mechanisms. Aging cell In Press.
Van Raamsdonk, J. M., and Hekimi, S. (2009). Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000361. doi:10.1371/journal.pgen.1000361
Van Raamsdonk, J. M., and Hekimi, S. (2011). FUdR causes a twofold increase in the lifespan of the mitochondrial mutant gas-1. Mech. ageing Dev. 132, 519–521. doi:10.1016/j.mad.2011.08.006
Van Raamsdonk, J. M., Meng, Y., Camp, D., Yang, W., Jia, X., Benard, C., et al. (2010). Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics 185, 559–571. doi:10.1534/genetics.110.115378
Walter, L., Baruah, A., Chang, H. W., Pace, H. M., and Lee, S. S. (2011). The homeobox protein CEH-23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans. PLoS Biol. 9, e1001084. doi:10.1371/journal.pbio.1001084
Wang, Y., Pennock, S., Chen, X., and Wang, Z. (2002). Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol. Cell. Biol. 22, 7279–7290. doi:10.1128/mcb.22.20.7279-7290.2002
Watterson, A., Tatge, L., Wajahat, N., Arneaud, S. L. B., Solano Fonseca, R., Beheshti, S. T., et al. (2022). Intracellular lipid surveillance by small G protein geranylgeranylation. Nature 605, 736–740. doi:10.1038/s41586-022-04729-7
Wong, A., Boutis, P., and Hekimi, S. (1995). Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139, 1247–1259. doi:10.1093/genetics/139.3.1247
Wu, Z., Senchuk, M. M., Dues, D. J., Johnson, B. K., Cooper, J. F., Lew, L., et al. (2018). Mitochondrial unfolded protein response transcription factor ATFS-1 promotes longevity in a long-lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 16, 147. doi:10.1186/s12915-018-0615-3
Yang, W., and Hekimi, S. (2010). Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9, 433–447. doi:10.1111/j.1474-9726.2010.00571.x
Yee, C., Yang, W., and Hekimi, S. (2014). The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 157, 897–909. doi:10.1016/j.cell.2014.02.055
Yu, S., and Driscoll, M. (2011). EGF signaling comes of age: Promotion of healthy aging in C. elegans. Exp. Gerontol. 46, 129–134. doi:10.1016/j.exger.2010.10.010
Keywords: aging, lifespan, stress resistance, endosomal trafficking, C. elegans, genetics, TBC-2, DAF-16 transcription factor
Citation: Traa A, Shields H, AlOkda A, Rudich ZD, Ko B and Van Raamsdonk JM (2023) Endosomal trafficking protein TBC-2 is required for the longevity of long-lived mitochondrial mutants. Front. Aging 4:1145198. doi: 10.3389/fragi.2023.1145198
Received: 15 January 2023; Accepted: 04 May 2023;
Published: 16 May 2023.
Edited by:
Marco P. Brotto, University of Texas at Arlington, United StatesReviewed by:
Anders Olsen, Aalborg University, DenmarkCopyright © 2023 Traa, Shields, AlOkda, Rudich, Ko and Van Raamsdonk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy M. Van Raamsdonk, amVyZW15LnZhbnJhYW1zZG9ua0BtY2dpbGwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.