 MaKayla F. Cox
MaKayla F. Cox Erin R. Hascup
Erin R. Hascup Andrzej Bartke
Andrzej Bartke Kevin N. Hascup
Kevin N. Hascup- 1Dale and Deborah Smith Center for Alzheimer’s Research and Treatment, Department of Neurology, Neurosciences Institute, Southern Illinois University School of Medicine, Springfield, IL, United States
- 2Department of Pharmacology, Southern Illinois University School of Medicine, Springfield, IL, United States
- 3Department of Internal Medicine, Southern Illinois University School of Medicine, Springfield, IL, United States
- 4Department of Medical Microbiology, Immunology and Cell Biology, Southern Illinois University School of Medicine, Springfield, IL, United States
Aging is a naturally occurring decline of physiological processes and biological pathways that affects both the structural and functional integrity of the body and brain. These physiological changes reduce motor skills, executive function, memory recall, and processing speeds. Aging is also a major risk factor for multiple neurodegenerative disorders including Alzheimer’s disease (AD). Identifying a biomarker, or biomarkers, that signals the transition from physiological to pathological aging would aid in earlier therapeutic options or interventional strategies. Considering the importance of glutamate signaling in synaptic plasticity, motor movement, and cognition, this neurotransmitter serves as a juncture between cognitive health and disease. This article discusses glutamatergic signaling during physiological aging and the pathological changes observed in AD patients. Findings from studies in mouse models of successful aging and AD are reviewed and provide a biological context for this transition. Finally, current techniques to monitor brain glutamate are highlighted. These techniques may aid in elucidating time-point specific therapeutic windows to modify disease outcome.
GRAPHICAL ABSTRACT
Introduction
Aging is a complex biological process accompanied by declines in physiological function leading to increased susceptibility for disease and death (Carmona and Michan 2016). Aging is inevitable, occurring in all organisms with time. Over the past decades, innovative improvements in medicine have extended human life span and prolonged the aging process. But, a concomitant improvement to geriatric health has not followed. This unprecedented aging of the human population in developed nations has resulted in people living with multiple chronic health conditions (Barzilai et al., 2018). Maintaining their quality of life has created a significant socio-economic burden on both family members and governments. Accordingly, the geroscience hypothesis postulates that slowing the rate of biological aging can prevent, delay, or ameliorate the development of these chronic conditions leading to successful aging.
Research over the past decades has elucidated nutritional, genetic, and pharmacological interventions that can increase life span of numerous model organisms such as yeast, Caenorhabditis elegans, Drosophila melanogaster, and mammals. Some of these life span extension mechanisms have shown therapeutic potential in numerous model organisms of neurodegenerative disorders including Alzheimer’s disease (AD) (Soo et al., 2020). These beneficial effects have translated poorly into human clinical trials. By the time chronic illness becomes evident, the biological mechanisms governing healthy aging have succumbed to the chronic condition. Intervening years before the onset of chronic illness is ideal, but involves individual life style modifications such as diet and exercise. Despite their known health benefits long term adherence to these regimens is poor. Identifying the juncture when physiological aging transitions into a pathological disease state provides an opportune pharmacological window to modify disease outcome before onset of clinical symptoms.
Biomarkers are becoming increasingly common for use in understanding the physiological processes that occur with age and disease onset, (Crimmins et al., 2008). Biomarkers are molecules present in blood, body fluid, and tissues that can be quantitatively measured to directly assess disease onset or progression. The continuous progression of molecular diagnostics allows for accurate detection and quantification of biomarkers for various uses. Biomarkers have been separated into two types: biomarkers of exposure and biomarkers of disease. Our laboratory focuses on biomarkers of disease with a goal of earlier diagnosis and tighter monitoring of disease progression (Mayeux 2004). Within biomarkers of disease are functional biomarkers which provide a direct measurement of target engagement. In order to use a biomarker, there must be a distinguishable quantifiable change in the amount of the marker present in the healthy versus diseased population. For example, amyloid-β42, phosphorylated tau, and neurofilament light chain provide diagnostic utility when used in concert to evaluate AD progression (Jack et al., 2016). Although these proteins provide an indication of disease severity, they do not indicate the initial change from physiological aging to pathological disease. We propose examination of central nervous system (CNS) glutamate levels as a functional biomarker due to its role in procognitive changes before mild cognitive impairment (MCI) and eventual onset of AD. The focus of this article is to discuss changes in glutamatergic regulation as it relates to aging and AD. Based on our work and the research of others, we believe monitoring glutamate dynamics as a viable biomarker for identifying the conversion from healthy physiological aging to pathological dysfunctional aging.
Overview of Glutamatergic Neurotransmission
Glutamate is the most abundant neurotransmitter in the CNS with glutamate receptors present on more than 90% of neurons and 40% of synapses (Bukke et al., 2020; Conway, 2020; Gasiorowska et al., 2021). Currently, there are over 20 glutamate receptors identified within the CNS with each individual receptor having multiple subtypes (Pankevich et al., 2011). Glutamate homeostasis is critical for healthy aging and reducing the risk for various neurological diseases including epilepsy, addiction, amyotrophic lateral sclerosis, Parkinson’s disease, AD, and more (Scofield et al., 2016; Alcoreza et al., 2021). Physiologically, glutamate plays a critical role in synaptic stability and plasticity (Tapiero et al., 2002). The functional elements encompassing glutamate neurotransmission are the pre and postsynaptic neurons along with glial cells which are conjunctionally defined as the “tripartite synapse,” (Halassa et al., 2007; Lalo et al., 2021). The tripartite synapse functions through both metabotropic and ionotropic receptors. A comprehensive review of these receptors and their functions are detailed elsewhere (Findley et al., 2019) and briefly discussed below.
Glutamate is synthesized from glutamine by glutaminase in presynaptic neurons where it is then transported to synaptic terminals (Revett et al., 2013). While in the synaptic terminal, glutamate is stored into vesicles by vesicular glutamate transporters (VGLUT) 1–3 for immediate release upon neuronal depolarization (Takamori et al., 2000; Fremeau et al., 2004). Signaling is mediated through ionotropic and metabotropic glutamate receptors (iGluR and mGluR) located throughout the tripartite synapse. Signal transduction is terminated by uptake into high-efficiency excitatory amino acid transporters (EAATs) which are predominantly located on astrocytes. Once in the astrocyte, glutamine synthetase converts glutamate into glutamine where it can be released back into the extracellular space for return to the presynaptic terminal in a process referred to as the glutamate/glutamine cycle. Together, iGluR, mGluR, and EAATs act as critical regulators of the glutamatergic system through modulating signal strength and extracellular concentrations of glutamate. Meticulous regulation of extracellular glutamate is crucial to prevent persistent receptor activation that can result in excitotoxicity and neuronal loss.
iGluRs are formed by heterotetrametric and homotetrameric subunits and are voltage sensitive and fast acting due to the influx of ions across the plasma membrane (Pankevich et al., 2011). These receptors are expressed both pre- and post-synaptically (Wisden and Seeburg 1993). There are three subtypes of ionotropic receptors: N-methyl D-aspartate (NMDAr), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPAr), and kainate receptors. The NMDAr and AMPAr are colocalized on the postsynaptic membrane as they are functionally dependent on one another during membrane depolarization (Takumi et al., 1999). The quick activation of AMPAr facilitates the NMDAr by depolarizing the membrane and allowing for NMDAr to overcome the Mg2+ blockade. However, NMDAr and AMPAr interact with glutamate independently. NMDAr are comprised of subunits that provide different functions. The NR2A and NR2B subunits bind with glutamate and have been shown to mediate excitotoxicity in cultured cortical neurons (Engelhardt et al., 2007). The high density of NMDAr expressed on neurons and astrocytes throughout the brain, and most notably in the hippocampus, contribute to learning, memory, and synaptic plasticity (Barco et al., 2006; Lee et al., 2010), but overactivation drives neuronal loss. This dichotomy is due to the cellular localization of these receptors. Synaptic NMDAr activation drives calcium-mediated transcriptional changes to promote neuronal health and resistance to cellular insults. Extrasynaptic NMDAr activation is coupled to multiple signaling pathways that reduces stress resistance and drives apoptosis which may be an initiating factor for multiple neurodegenerative disorders (Hardingham and Bading, 2010).
AMPAr interact with glutamate through four subunits (GluA1–GluA4) that are highly homologous with conserved transmembrane and extracellular domains (Collingridge et al., 2004; Chater and Goda 2014). Presynaptically, AMPAr function to promote synapse and spine formation (Isaac et al., 2007). The functions of AMPAr in learning and memory are not well characterized, however, AMPAr are suspected to play a role in synaptic plasticity through augmentation of Ca2+ entry into NMDAr (Chater and Goda 2014). AMPAr are also important for potentiating other receptors in the cell signaling process. iGluRs are important for mediating rapid neuronal communication whereas metabotropic receptors gradually mediate changes through signal transduction pathways.
mGluRs are slow acting signal transducers present on both presynaptic and postsynaptic neurons (Revett et al., 2013). There are three subgroups of mGluRs distinguishable by their pharmacological and signaling properties. Group I is comprised of postsynaptically expressed Gq-coupled receptors while Groups II and III are Gi/o-coupled inhibitory auto receptors expressed both presynaptically and postsynaptically (Petralia and Wenthold 1996; Rudy et al., 2015). Dysregulated mGluR signaling contributes to anxiety, depression, learning impairments, cognitive decline, and choreas. mGluR1 and mGluR5 are identified as Group I receptors and facilitate the activity of various kinases (Ménard and Quirion 2012). Activation of mGluR1 and mGluR5 has also been shown to induce long term depression (LTD), a type of synaptic plasticity that decreases responsiveness to glutamate. Therefore, Group I receptors are important for memory formation and learning processes (Kang and Kaang 2016). Group II receptors include mGluR2 and mGluR3. These receptors are inhibitory autoreceptors and play a critical role in glutamate regulation as they are situated both presynaptically and postsynaptically (Hascup et al., 2012). Group III receptors are comprised of mGluR4, mGluR7, and mGluR8 in the cerebrum. These three receptors have been established as neuroprotectants with the capability to induce both long-lasting potentiation of glutamatergic transmission and inhibition of glutamatergic transmission (Neugebauer, et al., 1997; Schrader and Tasker 1997; Grueter and Winder 2005). The dual location of mGluRs facilitates both suppression and increased glutamate signaling (Swanson et al., 2005; Lesage and Steckler 2010). The extensive distribution of mGluRs can help to monitor extracellular glutamate levels and modulate the glutamatergic tone. Therefore, the conservation of iGluRs and mGluRs and their ability to modulate the glutamatergic system across aging are important for maintaining cognitive health.
Physiological Aging
Aging is a naturally occurring decline of physiological processes modifying various biochemical pathways throughout the life span of all organisms (Bartke 2021). Physiological declines in motor movement, metabolism, inflammation, and executive function are prevalent and vary between individuals and sexes (Peters 2006). These declines are attributed to numerous factors including sarcopenia, adiposity, and arteriosclerosis. Aging also affects both structural and functional integrity of the brain contributing to these physiological declines.
Glutamate Regulation in Physiological Aging
The abundance of glutamate and glutamate receptors present in the brain contribute to the desire to understand how the glutamatergic system can contribute to the aging processes. Brain morphological changes such as cortical thinning and synaptic pruning are important aspects of brain development, but these become more pronounced with age leading to altered glutamatergic neurotransmission (Segovia et al., 2001; Anderton 2002; Brans et al., 2010). These age-related changes in glutamate occur in the hippocampus, prefrontal cortex, and motor and sensory areas (Segovia et al., 2001; Kaiser et al., 2005; Raininko and Mattsson 2010) that helps to explain the subsequent cognitive, motor, and sensory decline in healthy aging.
During healthy aging, there is general agreement that the total pool for neuronal glutamate signaling decreases (Gasiorowska et al., 2021). Examination of healthy individuals between the ages of 24 and 68 found that older subjects had lower glutamate concentrations in the motor cortex as compared to younger subjects (Kaiser et al., 2005). Similar age-related glutamate decline was seen in the striatum but not the pons or cerebellum (Zahr et al., 2013). Two meta-analyses of healthy aging humans using magnetic resonance spectroscopy (MRS) revealed that brain glutamate concentrations decrease in conjunction with brain volume shrinkage (Roalf et al., 2020; Sydnor and Roalf 2020). These physiological changes may start by midlife. Glutamate concentrations decreased 0.33 mM/year in the medial frontal cortex of 18–31 years-old study subjects (Marsman et al., 2013). This decline occurred in combination with grey matter thinning, but these anatomical changes may not solely explain decreased glutamate levels. Additionally, men more than women may be more affected by age-related brain glutamate decreases (Sailasuta et al., 2008).
Despite the importance of glutamate signaling in health and disease, this has yet to be evaluated in the oldest-old. Nonagenarians and centenarians have demonstrated a resiliency against the onset of age-related disease (Engberg et al., 2009) and have been proposed as the optimal study participants to identify genes related to successful aging (Marcos-Pérez et al., 2021). A majority of these individuals have normal cognitive and functional ability (Sachdev et al., 2013). This suggests glutamate levels decrease at a slower rate or other compensatory mechanisms occur to modulate signaling.
Glutamate Regulation in Animal Models of Normal Aging
Similar to what has been observed in humans, rats and mice show a subtle decrease in glutamatergic synapses and neurons during aging—particularly when compared to pathological conditions (Temido-Ferreira et al., 2019; Gasiorowska et al., 2021). This causes an age-related shift in synaptic plasticity that favors LTD over LTP (Kumar and Foster, 2019) that contributes to the natural cognitive decline consistently observed in multiple species across numerous behavioral tasks (Tanila et al., 1997). Over the past 40 years researchers have studied hippocampal glutamatergic changes to disentangle aging effects from disease progression. Unless specifically stated, the studies discussed below use rodents that were at least 24 months old. Hippocampal glutamate protein content is reduced in Fisher 344 rats (Banay-Schwartz et al., 1989) and in the temporal cortex of Sprague-Dawley rats (Jing et al., 2016). This lower protein content contributes to reduced tonic and stimulus-evoked glutamate release observed in the Fisher 344 rat model of aging (Zhang et al., 1991; Stephens et al., 2011). Similar findings are routinely reported in aging mouse models. C57BL/6 mice have reduced neurometabolic activity and hippocampal glutamatergic neurotransmitter cycling (Bahadur Patel et al., 2021) along with reduced VGLUT1 protein concentration (Rozycka et al., 2019). This longitudinally decreases cortical, striatal, and hippocampal glutamate in both sexes of C57BL/6 mice as measured by MRS (Duarte et al., 2014). Evoked hippocampal glutamate release is reduced in 17 months old C57BL/6J mice compared to younger (7 months) littermates (Minkeviciene et al., 2008). Changes to glutamatergic receptor density are also prominent in aged rats, mice, and nonhuman primates. 25–32 year old rhesus macques show subregion specific declines in NMDAr subtype expression that correlates with cognitive deficits (Hara et al., 2012). Hippocampal NMDAr and AMPAr density was decreased in Fisher 344 rats while EAATs remained consistent (Mitchell and Anderson, 1998; Segovia et al., 2001). Additionally, glutamate binding to NMDAr decreases with age in C57BL/6 and BALB/c mice (Peterson and Cotman, 1989). The decreased glutamatergic signaling across species can be interpreted as a biologically conserved mechanism of aging similar to sarcopenia or osteoporosis. Accordingly, this decline should be blunted or delayed in models of successful aging.
Glutamate Regulation in Animal Models of Longevity
Our laboratory is interested in the role of glutamatergic signaling in healthy aging and how changes can drive the transition to neurodegenerative disorders. Animal models of successful aging are important in biogerontology research for understanding mechanistic factors that contribute to the aging process. Suppression of the growth hormone/insulin-like growth factor 1/insulin axis is known to positively influence life span and health span through pleiotropic factors including protection against neurodegeneration. Mice with disruption of the growth hormone receptor (GHRKO) have reduced insulin-like growth factor 1 and elevated growth hormone that results in a diminutive phenotype with exceptional longevity. Disruption of this signaling pathway also increases neuronal differentiation and increases cortical neuronal density (Turnley et al., 2002; Ransome et al., 2004). Examination of CNS tissue by our laboratory revealed conserved or reduced expression of glutamatergic markers as GHRKO mice age (Hascup et al., 2015). In particular, hippocampal VGLUT1, GluN2B, and GluA1 expression levels decline with age that may provide GHRKO mice protection against excitotoxicity. In a follow up study, we examined hippocampal glutamatergic dynamics in 20–24 months old GHRKO and littermate control mice. To monitor in vivo glutamate our laboratory uses an enzyme-based microelectrode array with high spatial (micron) and temporal (millisecond) resolution that cause minimal damage to the surrounding parenchyma (Hascup et al., 2009; Hascup et al., 2010). We found that aged GHRKO mice maintained cognition and glutamate signaling throughout the hippocampus compared with age-matched littermate controls (Hascup et al., 2016). The GHRKO memory retention at advanced age was similar to previous studies (Kinney et al., 2001a) and may suggest the delayed cognitive aging in these mice is conferred through glutamatergic regulation.
Ames Dwarf mice are homozygous deficient in the Prophet of Pituitary Factor 1 gene an upstream regulator of Pituitary Factor 1 that confers deficiency in growth hormone, prolactin, and thyroid stimulating hormone. Similar to GHRKO mice, this mutation results in a diminutive size with an 1 year average lifespan extension compared to littermates (Brown-Borg et al., 1996). At 3 months of age, Ames Dwarf mice have higher hippocampal NMDAr expression (Sharma et al., 2012) and cAMP response element-binding protein (Sun et al., 2005) that may contribute to their enhanced learning and memory at older ages (Kinney et al., 2001b). Together, these animal models of successful aging may suggest that healthy cognitive aging requires maintenance of glutamatergic signaling throughout the aging process. When disruption occurs, the mechanisms of healthy aging are no longer conserved and may precipitate the onset of neurodegenerative disorders (Li et al., 2021).
Pathological Aging
Deviations from healthy aging to pathological aging are the driving force of age-related disease. The risk for cardiovascular diseases, cancer, neurodegenerative diseases, autoimmune diseases, and musculoskeletal issues increases significantly with age (Li et al., 2021). Therefore, understanding the mechanisms behind physiological aging could facilitate the direction of future studies on the biological drivers of pathological aging. For example, EAAT density and function decrease with age (Segovia et al., 2001) leading to extrasynaptic glutamate spillover. Activation of extrasynaptic glial NMDAr is a contributing factor to excitotoxicity during pathological aging. Our laboratory focuses on the glutamatergic system and its contributions to AD. We propose alterations in brain glutamate concentrations as a critical proponent for the pathological aging processes.
Alzheimer’s Disease
AD is an age-related neurodegenerative disorder characterized by progressive anterograde amnesia and the seventh leading cause of death in the US (Ahmad and Anderson 2021). Aging is the primary risk factor for developing AD, with 65 years of age designated as the cutoff that defines early-onset versus late-onset AD. Only 2%–10% of cases are categorized as early-onset (Cacace et al., 2016) with one in nine individuals aged 65 years or older diagnosed with AD (Rajan et al., 2021). Currently, an estimated 5.8 million Americans are diagnosed with AD and this number is projected to double by 2050 (Alzheimer’s Association, 2020) posing a significant socioeconomic burden. Despite onset differences, the structural pathologies are similar with hippocampal atrophy being the most prominent shared feature (Eckerström et al., 2018). The mechanisms behind development of cognitive deficits are numerous, but research has predominantly focused on accumulation of misfolded protein aggregates.
The deposition of amyloid beta (Aβ)42 that aggregates into extracellular plaques and hyperphosphorylated tau protein that misfolds into intracellular neurofibrillary tangles (NFT) are the hallmark pathological changes typically observed in postmortem tissue. These proteinopathies are hypothesized to begin abnormally accumulating in the brain 10–20 years prior to the onset of overt symptoms (Giannakopoulos et al., 1995; Bateman et al., 2012; Beason-Held et al., 2013). Various genetic and environmental components have been identified as risk factors contributing to these proteinopathies (Hascup and Hascup 2020). The complexity of AD has made early detection and treatment difficult. Current treatments are symptomatic with discordant results towards disease modification for next generation medications focused on plaque removal. This lack of efficacy is often attributed to poor biomarker identification for earlier interventional strategies. Current biomarkers do not adequately identify asymptomatic AD nor disease progression making it difficult to provide disease-stage specific treatments for optimal efficacy. The identification of proteins associated with changes occurring prior to the onset of symptoms could lead to the potential to develop timepoint dependent therapeutics for improved specificity.
Glutamate Dysregulation in Alzheimer’s Disease Patients
Various imaging techniques and postmortem tissue analyses have shown dysregulation of the glutamatergic system throughout the tripartite synapse. These changes vary across the continuum of cognitive impairment in AD and are largely affected by protein accumulation and aggregation status. Initially, either Aβ or hyperphosphorylated tau contributes to neuronal excitability that facilitates overexcitation and in turn leads to glutamatergic excitotoxicity (Huijbers et al., 2019; 2015). Hyperactivity along with morphological changes in neuronal structure are thought to be early contributors to AD pathology and cognitive dysfunction (Ghatak et al., 2019). The persistent overactivation of NMDAr perturbs recognition of physiological signals thereby affecting memory consolidation and recall (Danysz and Parsons, 2012). Later stages of AD are contraindicative of hyperactivity and demonstrate markedly decreased glutamate levels. This is attributed to neuronal loss caused by either hyperphosphorylated tau microtubule destabilization or the exposure to an excitotoxic environment.
Multiple receptors and transporters are responsible for altered glutamate signaling that show biphasic changes across the AD continuum. Presynaptically, soluble Aβ preferentially binds to VGLUT1 potentiating release characteristics in the early stages (Sokolow et al., 2012). While decreased VGLUT1 levels have been found in later stages of post-mortem brain tissue (Kirvell et al., 2006; Rodriguez-Perdigon et al., 2016). mGluR2 expression was increased in hippocampal pyramidal neurons and associated with hyperphosphorylated tau deposition in postmortem AD tissue (Lee et al., 2004). Considering the autoinhibitory nature of Group II receptors on synaptic signaling, this increase may be a compensatory mechanism to attenuate extracellular glutamate levels. Postsynaptically, AMPAr and NMDAr subunits change during the AD continuum with upregulation seen early in disease progression followed by downregulation in later disease stages (Findley et al., 2019). These changes may serve to modulate the strength of synaptic signaling to maintain cognitive processing during receptor over activation. mGLUR5 is ubiquitously expressed throughout the medial prefrontal cortex that potentiates NMDAr function and expression. Aβ42 complexes to mGluR5 along with cellular prion protein (Rammes et al., 2011; Piers et al., 2012). This binding disrupts multiple signal transduction pathways (Abd-Elrahman and Ferguson, 2022) that subsequently reduces receptor expression in later disease stages (Mecca et al., 2020). Glial EAATs are both downregulated (Jacob et al., 2007; Hoshi et al., 2018; Kobayashi et al., 2018) and have reduced transporter capacity due to altered splice variants (Scott et al., 2011). Finally reduced γ-aminobutyric acid neuronal density tips the excitatory/inhibitory scale to favor glutamate release (Palop et al., 2007; Lauterborn et al., 2021).
Glutamate Dysregulation in Alzheimer’s Disease Animal Models
Accumulation of Aβ42 is hypothesized to begin decades prior to cognitive decline (Sperling et al., 2014) These soluble isoforms are considered the neurotoxic species associated with AD progression and have been shown to bind to the alpha 7 nicotinic acetylcholine receptor (α7nAChR) (Wang et al., 2002). Our laboratory and others have shown that binding of soluble Aβ42 to this receptor elicits glutamate release (Mura et al., 2012) from presynaptic receptors (Hascup and Hascup 2016). Within the hippocampus, the dentate (DG) and CA1 had the largest responses to the lowest concentrations of Aβ42 applied. Considering our laboratory is focused on understanding glutamatergic dynamics during aging and its dysregulation in disease, we have strived to understand these changes throughout disease progression using an animal model of AD.
The APP/PS1 mouse is a transgenic amyloidogenic AD model that initially develop plaque pathology and subtle cognitive deficits at 6 months. By 12 months of age these mice have plaques prevalent throughout the hippocampus and observable cognitive deficits (Webster et al., 2014). While fast progressing AD models reduce study duration, the slower progression of both pathology and cognitive decline in APP/PS1 mice and other models is ideal for several reasons. First, the slower onset of amyloid deposition allows mice to naturally age. This is more conducive to identifying dysregulated aging processes that contribute to disease progression. Second, we can design early interventional strategies without potential confounds from developmental biology that have a greater potential to occur with faster progressing models. Third, we can identify windows of disease progression for improving the translational relevance of disease-stage specific interventional strategies. Based on the pathology and cognitive decline in the APP/PS1 mice compared to their littermate control C57BL/6 mice, 6 months of age corresponds with mild cognitie impairment while 12–15 and 18+ months correspond with mild-AD and AD, respectively.
We have shown that hippocampal basal and stimulus-evoked glutamate signaling in APP/PS1 mice becomes hyperactive in a subregion specific manner prior to onset of cognitive decline (Hascup and Hascup 2015). The first changes are seen in the CA1 where stimulus-evoked glutamate release is markedly elevated by 3 months of age. At 6 months, when hippocampal plaque accumulation begins, basal glutamate is elevated throughout the hippocampus and continues to increase with age. Glutamate release also increases in the DG at this age and stays elevated throughout disease progression. CA3 evoked glutamate release does not become elevated until 12 months of age when plaque accumulation is more prominent (Hascup et al., 2020a). We also observed that plaque deposition was anatomically aligned, but temporally delayed with hyperactive glutamate signaling (Hascup et al., 2020b), further supporting a role of the glutamatergic system as a potential early biomarker and therapeutic target. An increase in Aβ42 with age provides mechanistic insight into the temporal profile of hippocampal hyperglutamatergic signaling. In summary, dysregulation of glutamatergic signaling followed a subregion specific pattern with the CA1 > DG > CA3; not unlike the sensitivity of these regions to locally applied soluble Aβ42 (Hascup and Hascup 2016).
By 12 months, all regions of the APP/PS1 hippocampus routinely show hyperglutamatergic signaling. Figure 1 provides a grouped comparison of control APP/PS1 and age-matched C57BL/6J cohorts taken with multiple studies from our laboratory. The graphs depict the reproducibility of elevated hippocampal basal glutamate and stimulus-evoked release (Figures 1A–C) across multiple published datasets. We have also observed that APP/PS1 mice have a compensatory increase in glutamate clearance (Figures 1D,E) suggesting either an upregulation to transporter density or function. Despite the increased clearance, this still fails to modulate glutamate to nonpathological levels. Besides the aging associated changes to Aβ42, we have also shown VGLUT1 density is elevated at 12 months in APP/PS1 mice (Hascup E. R. et al., 2019). When taken together, the increases to both presynaptic stimulation and glutamatergic vesicles accounts for the elevated levels observed.
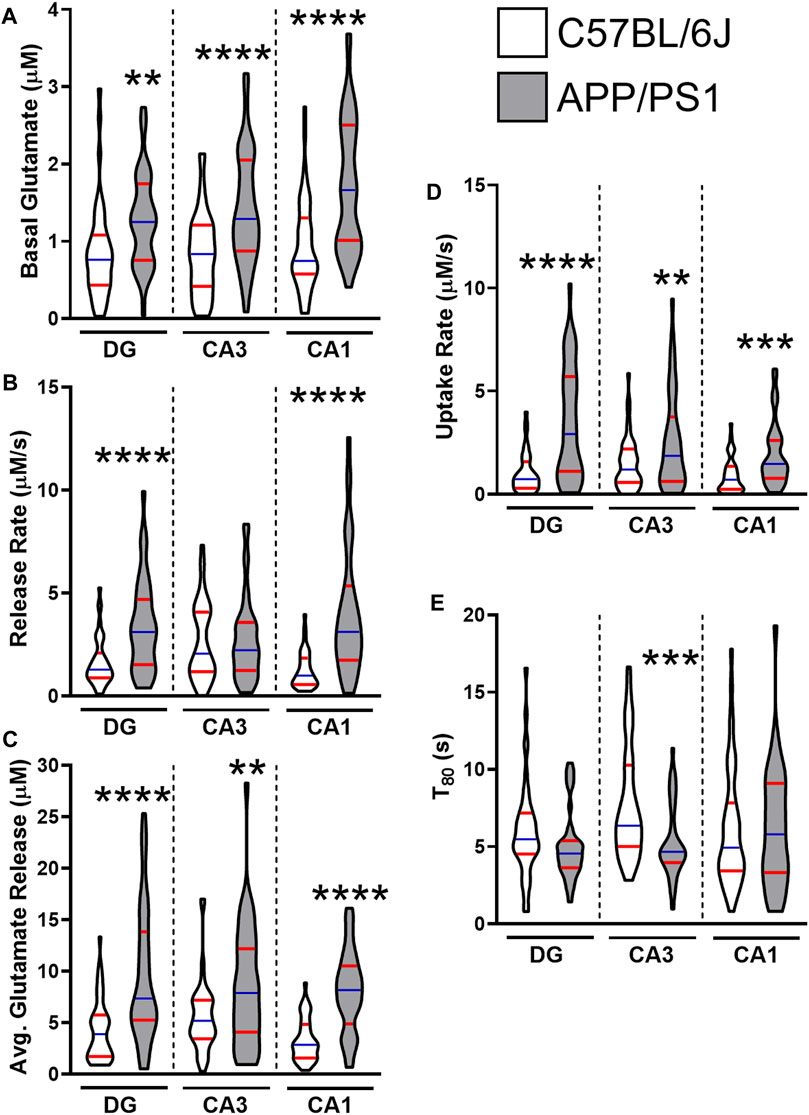
FIGURE 1. Glutamate dynamics in APP/PS1 and C57BL/6J mice at 12 months. The figures were created using control 12–15 months male C57BL/6J (white) and APP/PS1 (gray) mice across multiple datasets (Hascup E. R. et al., 2019; Hascup K. N. et al., 2019; Hascup et al., 2020a). Graphs depict violin plots with median (blue line) and quartiles (red line). (A) Basal glutamate levels were measured in the CA1, CA3, and the DG of the hippocampus using a microelectrode array (MEA). (B) Glutamate release rate was calculated using the change in amplitude between the maximal response and baseline over the duration (s) to reach maximal response after stimulation. (C) Average glutamate release was determined using the maximal change after stimulation from baseline. (D) Glutamate clearance followed first-order-rate kinetics. A logarithmic slope for glutamate concentration decay (k−1) versus time (s−1) is estimated using regression analysis (R2 ≥ 0.9) to determine the uptake rate. (E) T80 refers to the duration of time needed for 80% of the maximal glutamate signal to be cleared from the extracellular space. A two tailed t-test was used to compare genotypes in each hippocampal subregion. **p < 0.01, ***p < 0.001, ****p < 0.0001; n = 36–47.
A couple of limitations should be considered when interpreting these results. First, these studies were conducted only in male mice. Others have shown that stimulus-evoked hippocampal glutamate was decreased at 17 months of age in female APP/PS1 mice (Minkeviciene et al., 2008). While this was similar to our observations in the CA1 of male APP/PS1 mice, additional research is required to determine if the glutamatergic signaling profile differs during disease progression between sexes of APP/PS1 mice. Second, we have only assayed glutamate dynamics in a single model of cerebral amyloidosis. Multiple laboratories have reported on these changes in other amyloid and tau AD models, which are discussed below. To further address these limitations, our laboratory is actively probing sex as a biological variable to understand hippocampal glutamate changes throughout disease progression of additional AD models such as the APPNL-F/NL-F mice.
Similar glutamate signaling changes during disease progression have been reported in other AD animal models and summarized in Table 1. The rTg4510 mice express human tau containing the P301L mutation. This model develops neurofibrillary tangle, neuronal loss, and age-dependent cognitive deficits. By 6 months, these mice also have elevated hippocampal glutamate that correlated with acquisition errors on the Barnes maze behavioral task (Hunsberger et al., 2014; Hunsberger et al., 2015). Hippocampal glutamate levels are also higher at 3 months of age in the PS19 tauopathy AD model. As these mice continue to age, the number of synapses, and neurons decrease causing a corresponding reduction in glutamate levels. The elevated glutamate early in life coupled with neuronal loss at later stages suggests an excitotoxic mechanism (Crescenzi et al., 2017). However, it should be noted that the human P301L transgene insertion disrupts an endogenous mouse gene that contributes to the molecular changes and cognitive decline observed in this model (Gamache et al., 2019). The 3xTg AD model contains familial mutations to the amyloid precursor protein, presenilin 1, and microtubule associated protein tau genes resulting in dual amyloid and tau pathology by 12 months of age. Persistent hyperactive glutamatergic synapses were detected using electrophysiological techniques in the entorhinal cortex at this age (Arsenault et al., 2011). 5xFAD mice have multiple transgene insertions for human amyloid precursor protein and presenilin 1 that causes rapidly progressing amyloid accumulation starting at 6 weeks of age. Whole-cell patch clamp recordings of CA1 pyramidal neurons show higher spontaneous excitatory post synaptic currents at 2 and 4 months of age (Li et al., 2021). All together these studies show that hyperactive glutamatergic signaling either precedes or coincides with amyloid and/or tau accumulation.
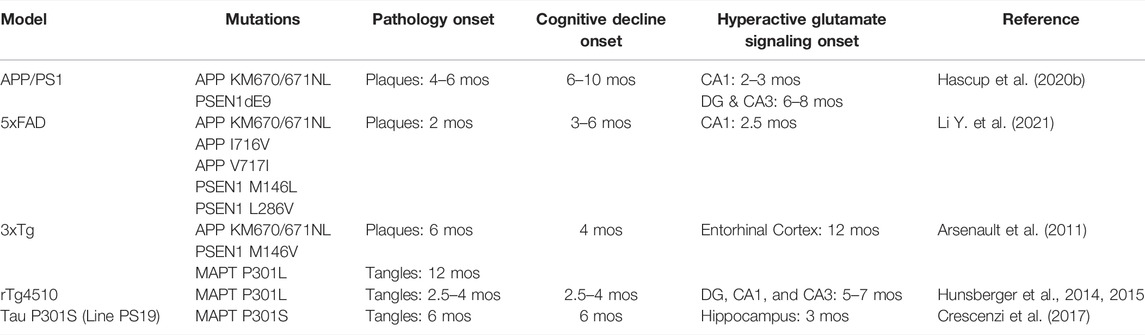
TABLE 1. The onset of pathological, cognitive and glutamatergic changes in AD mouse models.
The build-up of pathological proteins during disease progression may initiate the transition from low glutamate levels that are observed in healthy aging. These temporal hippocampal glutamate changes could serve as a biomarker for identifying a switch between physiological and pathological aging. Understanding this progression would help provide disease-stage specific interventions. For example, we treated APP/PS1 mice with riluzole, a glutamate modulator, starting when we first observed changes in the CA1. This treatment not only attenuated both basal and evoked glutamate release, but also provided procognitive effects (Hascup et al., 2020a). Other laboratories have shown these effects are modulated through EAAT2 as well as NMDAr subunit expression levels (Pereira et al., 2017; Okamoto et al., 2018).
Diagnostic Methods for In Vivo Glutamate Monitoring
Increasing evidence supports the importance of modulating glutamatergic signaling as a therapeutic treatment for AD. Therefore, methods to monitor glutamate as it transitions from physiological to pathological signaling are needed. The following sections discusses minimally invasive and non-invasive techniques that can be utilized in both humans and preclinical AD models.
Cerebrospinal fluid (CSF) provides an indication of the brain parenchyma and is currently used to measure deposition of amyloid and tau while determining neuronal loss by neurofilament light chain. Samples are collected and analyzed after a lumbar puncture commonly referred to as a spinal tap. During this 30–40 min procedure, the patient is given a local anesthetic before a needle is inserted between two lumbar vertebrae to collect CSF. The lumbar puncture can be difficult to perform and leave post-procedural issues including a “spinal headache.” Several studies have analyzed glutamate in the CSF of MCI and AD patients indicating increases or decreases compared to healthy aging (Madeira et al., 2018). Rather these discordant results may reflect progression through the AD continuum. While lumbar punctures are minimally invasive, they have the benefit of being 3–4 times less expensive than the imaging techniques discussed below (Tariciotti et al., 2018).
Currently, advanced neuroimaging methods are being explored to detect changes in glutamate receptors and glutamate excitation in healthy aging, MCI, and AD patients. Imaging offers the potential to detect changes in metabolic functions within the brain and monitor structural transformations throughout the aging process. Positron emission tomography (PET) imaging can be used with multiple radiotracers to quantitatively assess synaptic density. Radiotracers are molecules where one or more atoms are replaced with a radioactive isotope and their in vivo decay is monitored. Radiotracers can be created to target specific receptors in the brain that can be visualized using gamma ray emissions (Schaffer et al., 2015). Currently, PET imaging is used to assess multiple AD pathophysiological targets including amyloid, tau, neuronal density, and neurotransmitter signaling—including glutamate (Bao et al., 2021). Using radiotracers targeting mGluR5 researchers found decreased hippocampal expression in early AD patients that correlated with lower episodic memory scores and decreased global function (Mecca et al., 2020; Treyer et al., 2020). The small number of glutamate signaling radiotracers, short decay times, and large financial costs all limit the clinical utility of PET imaging.
Magnetic resonance (MR) uses a magnetic field to create a three-dimensional image of the brain without the need for ionizing radiation or radiotracers. Unlike PET imaging, MR is unable to directly measure neural activity (Osborne et al., 2015). MRS is capable of detecting the chemical composition of the tissue by examining the magnetic moment of nuclei such as protons (1H) and carbons (13C). MRS has allowed for the noninvasive quantification of regional neurotransmitters including glutamate in numerous diseases and preclinical disease models. Multiple MRS studies have shown decreased hippocampal glutamate concentrations in AD patients (Lin et al., 2003; Fayed et al., 2011; Graff-Radford and Kantarci, 2013).
In the last decade MR has been used in conjunction with chemical exchange saturation transfer (CEST) allowing for the indirect detection of brain metabolite changes using endogenous proteins. CEST uses exchangeable amide protons to monitor glycogen, glycosaminoglycans, γ-aminobutyric acid, and glutamate (GluCEST) (Kogan et al., 2013). GluCEST has been validated in humans (Nanga et al., 2018) and is capable of sub-hippocampal measurements. The majority of GluCEST studies, however, have focused on preclinical aging and disease models. Aging reduced GluCEST contrast in multiple networks associated with cognition in mouse lemur primates (Garin et al., 2022). Decreased hippocampal glutamate has also been observed in 18+ month old amyloid (Haris et al., 2013) and tau (Crescenzi et al., 2014) mouse models of AD similar to observations using implantable biosensors. Considering GluCEST is noninvasive, longitudinal analysis in mouse models would provide initial clinical utility for examination across the AD continuum in humans.
Conclusion
The extensive amount of knowledge accumulated through decades of research on the biology of glutamate leaves this system poised to make major advancements in the realm of neurological disorders. Our laboratory has shown the potential for using glutamate as a biomarker for determining the juxtaposition between physiological and pathological aging. Advances using minimally and noninvasive techniques allows for longitudinally monitoring regional changes in glutamate levels. These methods have the potential to provide disease state dependent therapeutic interventions for improving patient outcome.
Author Contributions
All authors wrote and edited the manuscript. KH analyzed the pooled data and prepared the figures.
Funding
This work was supported by the National Institutes of Health (NIA R01-AG057767 and NIA R01-AG061937), Dale and Deborah Smith Center for Alzheimer’s Research and Treatment, Kenneth Stark Endowment (MC, EH, and KH), and NIA R21-AG062985 (AB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
α7nAChR, α7 nicotinic acetylcholine receptor; AMPAr, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors; AD, Alzheimer’s disease; Aβ, amyloid-β; CR, caloric restriction; CNS, central nervous system; DG, dentate gyrus; EAAT, excitatory amino acid transporters; GHRKO, growth hormone receptor knockout; GluCEST, glutamate chemical exchange saturation transfer; iGluR, ionotropic glutamate receptors; MR, magnetic resonance; MRS, magnetic resonance spectroscopy; mGluR, metabotropic glutamate receptors; MCI, mild cognitive impairment; NMDAr, N-methyl-D-aspartate receptors; PET, positron emission tomography; VGLUT, vesicular glutamate transporter.
References
Abd-Elrahman, K. S., and Ferguson, S. S. G. (2022). Noncanonical Metabotropic Glutamate Receptor 5 Signaling in Alzheimer's Disease. Annu. Rev. Pharmacol. Toxicol. 62, 235–254. doi:10.1146/annurev-pharmtox-021821-091747
Ahmad, F. B., and Anderson, R. N. (2021). The Leading Causes of Death in the US for 2020. JAMA 325 (18), 1829–1830. doi:10.1001/JAMA.2021.5469
Alcoreza, O. B., Patel, D. C., Tewari, B. P., and Sontheimer, H. (2021). Dysregulation of Ambient Glutamate and Glutamate Receptors in Epilepsy: An Astrocytic Perspective. Front. Neurology 1, 652159. doi:10.3389/fneur.2021.652159
Alzheimer's Association (2020). 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dementia 16 (3), 391–460. doi:10.1002/ALZ.12068
Anderton, B. H. (2002). Ageing of the Brain. Mech. Ageing Dev. 123 (7), 811–817. doi:10.1016/S0047-6374(01)00426-2
Arsenault, D., Julien, C., Tremblay, C., and Calon, F. (2011). DHA Improves Cognition and Prevents Dysfunction of Entorhinal Cortex Neurons in 3xTg-AD Mice. PLoS ONE 6 (2), e17397. doi:10.1371/journal.pone.0017397
Bahadur Patel, A., Veeraiah, P., Shameem, M., Mahesh Kumar, J., and Saba, K. (2021). Impaired GABAergic and Glutamatergic Neurometabolic Activity in Aged Mice Brain as Measured by 1 H‐[ 13 C]‐NMR Spectroscopy. FASEB J. 35, e21321. doi:10.1096/fj.202001704rr
Banay-Schwartz, M., Lajtha, A., and Palkovits, M. (1989). Changes with Aging in the Levels of Amino Acids in Rat CNS Structural Elements I. Glutamate and Related Amino Acids. Neurochem. Res. 14, 555–562. doi:10.1007/bf00964918
Bao, W., Xie, F., Zuo, C., Guan, Y., and Huang, Y. H. (2021). PET Neuroimaging of Alzheimer’s Disease: Radiotracers and Their Utility in Clinical Research. Front. Aging Neurosci. 13, 114. doi:10.3389/fnagi.2021.624330
Barco, A., Bailey, C. H., and Kandel, E. R. (2006). Common Molecular Mechanisms in Explicit and Implicit Memory. J. Neurochem. 97 (6), 1520–1533. doi:10.1111/J.1471-4159.2006.03870.X
Bartke, A. (2021). , 1, 1–7. doi:10.1007/s12015-021-10305-9New Directions in Research on AgingStem Cell Rev. Rep. 2021
Barzilai, N., Cuervo, A. M., and Austad, S. (2018). Aging as a Biological Target for Prevention and Therapy. JAMA 320, 1321. doi:10.1001/jama.2018.9562
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and Biomarker Changes in Dominantly Inherited Alzheimer's Disease. N. Engl. J. Med. 367 (9), 795–804. doi:10.1056/NEJMOA1202753
Beason-Held, L. L., Goh, J. O., An, Y., Kraut, M. A., O'Brien, R. J., Ferrucci, L., et al. (2013). Changes in Brain Function Occur Years before the Onset of Cognitive Impairment. J. Neurosci. 33 (46), 18008–18014. doi:10.1523/JNEUROSCI.1402-13.2013
Brans, R. G. H., Kahn, R. S., Schnack, M., van Baal, G. C. M., Posthuma, D., van Haren, N. E. M., et al. (2010). Brain Plasticity and Intellectual Ability Are Influenced by Shared Genes. J. Neurosci. 30 (16), 5519–5524. doi:10.1523/jneurosci.5841-09.2010
Brown-Borg, H. M., Borg, K. E., Meliska, C. J., and Bartke, A. (1996). Dwarf Mice and the Ageing Process. Nature 384, 33. doi:10.1038/384033a0
Bukke, V. N., Archana, M., Villani, R., Romano, A. D., Wawrzyniak, A., Balawender, K., et al. (2020). The Dual Role of Glutamatergic Neurotransmission in Alzheimer's Disease: From Pathophysiology to Pharmacotherapy. Ijms 21, 7452. doi:10.3390/ijms21207452
Cacace, R., Sleegers, K., and Van Broeckhoven, C. (2016). Molecular Genetics of Early‐onset Alzheimer's Disease Revisited. Alzheimer's & Dement. 12 (6), 733–748. doi:10.1016/J.JALZ.2016.01.012
Carmona, J. J., and Michan, S. (2016). Biology of Healthy Aging and Longevity. Rev. Invest Clin. 68 (1), 7–16.
Chater, T. E., and Goda, Y. (2014). The Role of AMPA Receptors in Postsynaptic Mechanisms of Synaptic Plasticity. Front. Cell. Neurosci. 8, 401. doi:10.3389/FNCEL.2014.00401/BIBTEX
Collingridge, G. L., Isaac, J. T., and Wang, Yu. (2004). Receptor Trafficking and Synaptic Plasticity. Nat. Rev. Neurosci. 5 (12), 952–962. doi:10.1038/NRN1556
Conway, M. E. (2020). Alzheimer's Disease: Targeting the Glutamatergic System. Biogerontology 21, 257–274. doi:10.1007/s10522-020-09860-4
Crescenzi, R., DeBrosse, C., Nanga, R. P. R., Byrne, M. D., Krishnamoorthy, G., D'Aquilla, K., et al. (2017). Longitudinal Imaging Reveals Subhippocampal Dynamics in Glutamate Levels Associated with Histopathologic Events in a Mouse Model of Tauopathy and Healthy Mice. Hippocampus 27 (3), 285–302. doi:10.1002/hipo.22693
Crescenzi, R., DeBrosse, C., Nanga, R. P. R., Reddy, S., Haris, M., Hariharan, H., et al. (2014). In Vivo measurement of Glutamate Loss Is Associated with Synapse Loss in a Mouse Model of Tauopathy. Neuroimage 101, 185–192. doi:10.1016/j.neuroimage.2014.06.067
Crimmins, E., Vasunilashorn, S., Kim, J. K., and Alley, D. (2008). Chapter 5 Biomarkers Related to Aging in Human Populations. Adv. Clin. Chem. 46 (January), 161–216. doi:10.1016/s0065-2423(08)00405-8
Danysz, W., and Parsons, C. G. (2012). Alzheimer's Disease, β-amyloid, Glutamate, NMDA Receptors and Memantine - Searching for the Connections. Br. J. Pharmacol. 167, 324–352. doi:10.1111/j.1476-5381.2012.02057.x
Duarte, J. M. N., Do, K. Q., and Gruetter, R. (2014). Longitudinal Neurochemical Modifications in the Aging Mouse Brain Measured In Vivo by 1H Magnetic Resonance Spectroscopy. Neurobiol. Aging 35, 1660–1668. doi:10.1016/j.neurobiolaging.2014.01.135
Eckerström, C., Klasson, N., Olsson, E., Selnes, P., Rolstad, S., and Wallin, A. (2018). Similar Pattern of Atrophy in Early‐ and Late‐onset Alzheimer's Disease. Alzheimer's & Dement. Diagn. Assess. & Dis. Monit. 10, 253–259. doi:10.1016/j.dadm.2018.02.001
Engberg, H., Oksuzyan, A., Jeune, B., Vaupel, J. W., and Christensen., K. (2009). Centenarians - a Useful Model for Healthy Aging? A 29-year Follow-Up of Hospitalizations Among 40 000 Danes Born in 1905. Aging Cell 8 (3), 270–276. doi:10.1111/J.1474-9726.2009.00474.X
Fayed, N., Modrego, P. J., Rojas-Salinas, G., and Aguilar, K. (2011). Brain Glutamate Levels Are Decreased in Alzheimer's Disease. Am. J. Alzheimers Dis. Other Demen 26, 450–456. doi:10.1177/1533317511421780
Findley, C. A., Bartke, A., Hascup, K. N., and Hascup, E. R. (2019). Amyloid Beta-Related Alterations to Glutamate Signaling Dynamics during Alzheimer's Disease Progression. ASN Neuro 11, 175909141985554. doi:10.1177/1759091419855541
Fremeau, R. T., Kam, K., Qureshi, T., Johnson, J., Copenhagen, D. R., Storm-Mathisen, J., et al. (2004). Vesicular Glutamate Transporters 1 and 2 Target to Functionally Distinct Synaptic Release Sites. Science 304 (5678), 1815–1819. doi:10.1126/SCIENCE.1097468
Gamache, Julia., Benzow, Kellie., Forster, Colleen., Kemper, Lisa., Hlynialuk, Chris., Furrow, Eva., et al. (2019). Factors Other Than HTau Overexpression that Contribute to Tauopathy-like Phenotype in RTg4510 Mice. Nature 10 (1), 2479. doi:10.1038/s41467-019-10428-1
Garin, C. M., Nadkarni, N. A., Pépin, J., Flament, J., and Dhenain, M. (2022). Whole Brain Mapping of Glutamate Distribution in Adult and Old Primates at 11.7T. Neuroimage 251, 118984. doi:10.1016/j.neuroimage.2022.118984
Gasiorowska, A., Wydrych, M., Drapich, P., Zadrozny, M., Steczkowska, M., Niewiadomski, W., et al. (2021). The Biology and Pathobiology of Glutamatergic, Cholinergic, and Dopaminergic Signaling in the Aging Brain. Front. Aging Neurosci. 13. doi:10.3389/fnagi.2021.654931
Ghatak, S., Dolatabadi, N., Trudler, D., Zhang, X., Wu, Y., Mohata, M., et al. (2019). Mechanisms of Hyperexcitability in Alzheimer's Disease hiPSC-Derived Neurons and Cerebral Organoids vs Isogenic Controls. ELife 8 (November). doi:10.7554/eLife.50333
Giannakopoulos, P., Hof, P. R., Vallet, P. G., Giannakopoulos, A. S., Charnay, Y., and Bouras, C. (1995). Quantitative Analysis of Neuropathologic Changes in the Cerebral Cortex of Centenarians. Prog. Neuropsychopharmacol. Biol. Psychiatry 19 (4), 577–592. doi:10.1016/0278-5846(95)00103-3
Graff-Radford, J., and Kantarci, K. (2013). Magnetic Resonance Spectroscopy in Alzheimer's Disease. Neuropsychiatr. Dis. Treat. 9, 687–696. doi:10.2147/NDT.S35440
Grueter, B. A., and Winder, D. G. (2005). Group II and III Metabotropic Glutamate Receptors Suppress Excitatory Synaptic Transmission in the Dorsolateral Bed Nucleus of the Stria Terminalis. Neuropsychopharmacol 30, 1302–1311. doi:10.1038/sj.npp.1300672
Halassa, M. M., Fellin, T., and Haydon, P. G. (2007). The Tripartite Synapse: Roles for Gliotransmission in Health and Disease. Trends Mol. Med. 13 (2), 54–63. doi:10.1016/j.molmed.2006.12.005
Hara, Y., Rapp, P. R., and Morrison, J. H. (2012). Neuronal and Morphological Bases of Cognitive Decline in Aged Rhesus Monkeys. Age 34, 1051–1073. doi:10.1007/s11357-011-9278-5
Hardingham, G. E., and Bading, H. (2010). Synaptic versus Extrasynaptic NMDA Receptor Signalling: Implications for Neurodegenerative Disorders. Nat. Rev. Neurosci. 11, 682–696. doi:10.1038/nrn2911
Haris, M., Nath, K., Cai, K., Singh, A., Crescenzi, R., Kogan, F., et al. (2013). Imaging of Glutamate Neurotransmitter Alterations in Alzheimer's Disease. NMR Biomed. 26, 386–391. doi:10.1002/nbm.2875
Hascup, E. R., Hascup, K. N., Pomerleau, F., Huettl, P., Hajos-Korcsok, E., Kehr, J., et al. (2012). An Allosteric Modulator of Metabotropic Glutamate Receptors (mGluR₂), (+)-TFMPIP, Inhibits Restraint Stress-Induced Phasic Glutamate Release in Rat Prefrontal Cortex. J. Neurochem. 122, 619–627. doi:10.1111/j.1471-4159.2012.07784.x
Hascup, E. R., af Bjerkén, S., Hascup, K. N., Pomerleau, F., Huettl, P., Strömberg, I., et al. (2009). Histological Studies of the Effects of Chronic Implantation of Ceramic-Based Microelectrode Arrays and Microdialysis Probes in Rat Prefrontal Cortex. Brain Res. 1291, 12–20. doi:10.1016/j.brainres.2009.06.084
Hascup, E. R., Broderick, S. O., Russell, M. K., Fang, Y., Bartke, A., Boger, H. A., et al. (2019). Diet-induced Insulin Resistance Elevates Hippocampal Glutamate as Well as VGLUT1 and GFAP Expression in AβPP/PS1 Mice. J. Neurochem. 148 (2), 219–237. doi:10.1111/jnc.14634
Hascup, E. R., Hascup, K. N., Stephens, M., Pomerleau, F., Huettl, P., Gratton, A., et al. (2010). Rapid Microelectrode Measurements and the Origin and Regulation of Extracellular Glutamate in Rat Prefrontal Cortex. J. Neurochem. 115 (6), 1608–1620. doi:10.1111/j.1471-4159.2010.07066.x
Hascup, E. R., and Hascup, K. N. (2020). Toward Refining Alzheimer's Disease into Overlapping Subgroups. A&D Transl Res Clin Interv 6 (1). doi:10.1002/TRC2.12070
Hascup, E. R., Wang, F., Kopchick, J. J., and Bartke, A. (2015). Inflammatory and Glutamatergic Homeostasis Are Involved in Successful Aging. Gerona 71, 281–289. doi:10.1093/gerona/glv010
Hascup, K. N., Lynn, M. K., Fitzgerald, P. J., Randall, S., Kopchick, J. J., Boger, H. A., et al. (2016). Enhanced Cognition and Hypoglutamatergic Signaling in a Growth Hormone Receptor Knockout Mouse Model of Successful Aging. J. Gerontol. A Biol. Sci. Med. Sci. 72, 329–337. doi:10.1093/gerona/glw088
Hascup, K. N., Britz, J., Findley, C. A., Tischkau, S., and Hascup, E. R. (2019). LY379268 Does Not Have Long-Term Procognitive Effects Nor Attenuate Glutamatergic Signaling in AβPP/PS1 Mice. Jad 68 (3), 1193–1209. doi:10.3233/JAD-181231
Hascup, K. N., Findley, C. A., Britz, J., Esperant‐Hilaire, N., Broderick, S. O., Delfino, K., et al. (2020a). Riluzole Attenuates Glutamatergic Tone and Cognitive Decline in AβPP/PS1 Mice. J. Neurochem. 156, 513–523. doi:10.1111/jnc.15224
Hascup, K. N., Findley, C. A., Sime, L. N., and Hascup, E. R. (2020b). Hippocampal Alterations in Glutamatergic Signaling during Amyloid Progression in AβPP/PS1 Mice. Sci. Rep. 10 (1), 14503. doi:10.1038/s41598-020-71587-6
Hascup, K. N., and Hascup, E. R. (2015). Altered Neurotransmission Prior to Cognitive Decline in AβPP/PS1 Mice, a Model of Alzheimer's Disease. Jad 44 (3), 771–776. doi:10.3233/JAD-142160
Hascup, K. N., and Hascup, E. R. (2016). Soluble Amyloid-Β42 Stimulates Glutamate Release through Activation of the α7 Nicotinic Acetylcholine Receptor. J. Alzheimer’s Dis. 53 (1). doi:10.3233/JAD-160041
Hoshi, A., Tsunoda, A., Yamamoto, T., Tada, M., Kakita, A., and Ugawa, Y. (2018). Altered Expression of Glutamate Transporter-1 and Water Channel Protein Aquaporin-4 in Human Temporal Cortex with Alzheimer's Disease. Neuropathol. Appl. Neurobiol. 44, 628–638. doi:10.1111/nan.12475
Huijbers, W., Mormino, E. C., Schultz, A. P., Wigman, S., Ward, A. M., Larvie, M., et al. (2015). Amyloid-β Deposition in Mild Cognitive Impairment Is Associated with Increased Hippocampal Activity, Atrophy and Clinical Progression. Brain 138 (4), 1023–1035. doi:10.1093/brain/awv007
Huijbers, W., Schultz, A. P., Papp, K. V., LaPoint, M. R., Hanseeuw, B., Chhatwal, J. P., et al. (2019). Tau Accumulation in Clinically Normal Older Adults Is Associated with Hippocampal Hyperactivity. J. Neurosci. 39 (3), 548–556. doi:10.1523/JNEUROSCI.1397-18.2018
Hunsberger, H. C., Rudy, C. C., Seth, R. B., Gerhardt, G. A., and Reed, M. N. (2014). P301L Tau Expression Affects Glutamate Release and Clearance in the Hippocampal Trisynaptic Pathway. J. Neurochem. 132 (2), 169–182. doi:10.1111/jnc.12967
Hunsberger, H. C., Weitzner, D. S., Rudy, C. C., Hickman, J. E., Libell, E. M., Speer, R. R., et al. (2015). Riluzole Rescues Glutamate Alterations, Cognitive Deficits, and Tau Pathology Associated with P301L Tau Expression. J. Neurochem. 135 (2), 381–394. doi:10.1111/jnc.13230
Isaac, J. T. R., Ashby, M., and McBain, C. J. (2007). The Role of the GluR2 Subunit in AMPA Receptor Function and Synaptic Plasticity. Neuron 54 (6), 859–871. doi:10.1016/j.neuron.2007.06.001
Jack, C. R., Bennett, D. A., Blennow, K., Carrillo, M. C., Feldman, H. H., Frisoni, G. B., et al. (2016). A/T/N: An Unbiased Descriptive Classification Scheme for Alzheimer Disease Biomarkers. Neurology 87 (5), 539–547. doi:10.1212/WNL.0000000000002923
Jacob, C. P., Koutsilieri, E., Bartl, J., Neuen-Jacob, E., Arzberger, T., Zander, N., et al. (2007). Alterations in Expression of Glutamatergic Transporters and Receptors in Sporadic Alzheimer's Disease. J. Alzheimers Dis. 11, 97–116. doi:10.3233/jad-2007-11113
Jing, Y., Liu, P., and Leitch, B. (2016). Region-specific Changes in Presynaptic Agmatine and Glutamate Levels in the Aged Rat Brain. Neuroscience 312, 10–18. doi:10.1016/j.neuroscience.2015.11.002
Kaiser, L. G., Schuff, N., Cashdollar, N., and Weiner, M. W. (2005). Age-related Glutamate and Glutamine Concentration Changes in Normal Human Brain: 1H MR Spectroscopy Study at 4 T. Neurobiol. Aging 26 (5), 665–672. doi:10.1016/J.NEUROBIOLAGING.2004.07.001
Kang, S. J., and Kaang, B. K. (2016). Metabotropic Glutamate Receptor Dependent Long-Term Depression in the Cortex. Korean J. Physiol. Pharmacol. 20 (6), 557–564. doi:10.4196/kjpp.2016.20.6.557
Kinney, B. A., Coschigano, K. T., Kopchick, J. J., Steger, R. W., and Bartke, A. (2001). Evidence that Age-Induced Decline in Memory Retention Is Delayed in Growth Hormone Resistant GH-R-KO (Laron) Mice. Physiol. Behav. 72, 653–660. doi:10.1016/s0031-9384(01)00423-1
Kinney, B. A., Meliska, C. J., Steger, R. W., and Bartke, A. (2001). Evidence that Ames Dwarf Mice Age Differently from Their Normal Siblings in Behavioral and Learning and Memory Parameters. Horm. Behav. 39, 277–284. doi:10.1006/hbeh.2001.1654
Kirvell, S. L., Esiri, M., and Francis, P. T. (2006). Down-regulation of Vesicular Glutamate Transporters Precedes Cell Loss and Pathology in Alzheimer's Disease. J. Neurochem. 98, 939–950. (Accessed June 18, 2015).doi:10.1111/j.1471-4159.2006.03935.x
Kobayashi, E., Nakano, M., Kubota, K., Himuro, N., Mizoguchi, S., Chikenji, T., et al. (2018). Activated Forms of Astrocytes with Higher GLT-1 Expression Are Associated with Cognitive Normal Subjects with Alzheimer Pathology in Human Brain. Sci. Rep. 8, 1712–12. doi:10.1038/s41598-018-19442-7
Kogan, F., Hariharan, H., and Reddy, R. (2013). Chemical Exchange Saturation Transfer (CEST) Imaging: Description of Technique and Potential Clinical Applications. Curr. Radiol. Rep. 1 (2), 102–114. doi:10.1007/s40134-013-0010-3
Kumar, A., and Foster, T. C. (2019). Alteration in NMDA Receptor Mediated Glutamatergic Neurotransmission in the Hippocampus during Senescence. Neurochem. Res. 44, 38–48. doi:10.1007/s11064-018-2634-4
Lalo, U., Koh, W., Lee, C. J., and Pankratov, Y. (2021). The Tripartite Glutamatergic Synapse. Neuropharmacology 199, 108758. doi:10.1016/j.neuropharm.2021.108758
Lauterborn, J. C., Scaduto, P., Cox, C. D., Schulmann, A., Lynch, G., Gall, C. M., et al. (2021). Increased Excitatory to Inhibitory Synaptic Ratio in Parietal Cortex Samples from Individuals with Alzheimer’s Disease. Nat. Commun. 12, 1–15.
Lee, H. G., Ogawa, O., Zhu, X., O'Neill, M. J., Petersen, R. B., Castellani, R. J., et al. (2004). Aberrant Expression of Metabotropic Glutamate Receptor 2 in the Vulnerable Neurons of Alzheimer's Disease. Acta Neuropathol. 107, 365–371. doi:10.1007/s00401-004-0820-8
Lee, M. C., Ting, K. K., Adams, S., Brew, B. J., Chung, R., and Guillemin, G. J. (2010). Characterisation of the Expression of NMDA Receptors in Human Astrocytes. PLoS One 5 (11), e14123. doi:10.1371/JOURNAL.PONE.0014123
Lesage, A., and Steckler, T. (2010). Metabotropic Glutamate mGlu1 Receptor Stimulation and Blockade: Therapeutic Opportunities in Psychiatric Illness. Eur. J. Pharmacol. 639 (1–3), 2–16. doi:10.1016/J.EJPHAR.2009.12.043
Li, Y., Zhu, K., Li, N., Wang, X., Xiao, X., Li, L., et al. (2021). Reversible GABAergic Dysfunction Involved in Hippocampal Hyperactivity Predicts Early-Stage Alzheimer Disease in a Mouse Model. Alzheimer’s Res. Ther. 13, 1–14.
Li, Z., Zhang, Z., Ren, Y., Wang, Y., Fang, J., Han, Y., et al. (2021). Aging and Age‐related Diseases: From Mechanisms to Therapeutic Strategies. Biogerontology 22 (2), 1. doi:10.1007/S10522-021-09910-5
Lin, A. P., Shic, F., Enriquez, C., and Ross, B. D. (2003). Reduced Glutamate Neurotransmission in Patients with Alzheimer's Disease -- an In Vivo (13)C Magnetic Resonance Spectroscopy Study. MAGMA 16, 29–42. doi:10.1007/s10334-003-0004-x
Madeira, C., Vargas-Lopes, C., Brandão, C. O., Reis, T., Laks, J., Panizzutti, R., et al. (2018). Elevated Glutamate and Glutamine Levels in the Cerebrospinal Fluid of Patients with Probable Alzheimer's Disease and Depression. Front. Psychiatry 9, 561. doi:10.3389/FPSYT.2018.00561/BIBTEX
Marcos-Pérez, D., Saenz-Antoñanzas, A., and Matheu, A. (2021). Centenarians as Models of Healthy Aging: Example of REST. Ageing Res. Rev. 70, 101392. doi:10.1016/J.ARR.2021.101392
Marsman, A., Mandl, R. C., van den Heuvel, M. P., Boer, V. O., Wijnen, J. P., Klomp, D. W., et al. (2013). Glutamate Changes in Healthy Young Adulthood. Eur. Neuropsychopharmacol. 23 (11), 1484–1490. doi:10.1016/J.EURONEURO.2012.11.003
Mayeux, R. (2004). Biomarkers: Potential Uses and Limitations. NeuroRx 1 (2), 182. doi:10.1602/NEURORX.1.2.182
Mecca, A. P., McDonald, J. W., Michalak, H. R., Godek, T. A., Harris, J. E., Pugh, E. A, et al. (2020). PET Imaging of MGluR5 in Alzheimer’s Disease. Alzheimer’s Res. Ther. 12 (1), 15. doi:10.1186/s13195-020-0582-0
Ménard, C., and Quirion, R. (2012). Group 1 Metabotropic Glutamate Receptor Function and its Regulation of Learning and Memory in the Aging Brain. Front. Pharmacol. 3, 182. doi:10.3389/FPHAR.2012.00182
Minkeviciene, R., Ihalainen, J., Malm, T., Matilainen, O., Keksa-Goldsteine, V., Goldsteins, G., et al. (2008). Age-related Decrease in Stimulated Glutamate Release and Vesicular Glutamate Transporters in APP/PS1 Transgenic and Wild-type Mice. J. Neurochem. 105, 584–594. doi:10.1111/j.1471-4159.2007.05147.x
Mitchell, J. J., and Anderson, K. J. (1998). Age-related Changes in [3H]MK-801 Binding in the Fischer 344 Rat Brain. Neurobiol. Aging 19, 259–265. doi:10.1016/s0197-4580(98)00058-x
Mura, E., Zappettini, S., Preda, S., Biundo, F., Lanni, C., Grilli, M., et al. (2012). Dual Effect of Beta-Amyloid on Α7 and Α4β2 Nicotinic Receptors Controlling the Release of Glutamate, Aspartate and GABA in Rat Hippocampus. PloS One 7 (1), e29661. doi:10.1371/journal.pone.0029661
Nanga, R. P. R., DeBrosse, C., Kumar, D., Roalf, D., McGeehan, B., D'Aquilla, K., et al. (2018). Reproducibility of 2D GluCEST in Healthy Human Volunteers at 7 T. Magn. Reson Med. 80, 2033–2039. doi:10.1002/mrm.27362
Neugebauer, V., Bradley Keele, N., and Shinnick-Gallagher, P. (1997). Epileptogenesis In Vivo Enhances the Sensitivity of Inhibitory Presynaptic Metabotropic Glutamate Receptors in Basolateral Amygdala Neurons In Vitro. J. Neurosci. 17 (3), 983–995. doi:10.1523/JNEUROSCI.17-03-00983.1997
Okamoto, M., Gray, J. D., Larson, C. S., Kazim, S. F., Soya, H., McEwen, B. S., et al. (2018). Riluzole Reduces Amyloid Beta Pathology, Improves Memory, and Restores Gene Expression Changes in a Transgenic Mouse Model of Early-Onset Alzheimer's Disease. Transl. Psychiatry 8 (1), 153. doi:10.1038/s41398-018-0201-z
Osborne, J., Cole, S., Sarad, N., Ashton, D., Goswami, D., Herrmann, S., et al. (2015). Biomarkers in the Diagnosis and Prognosis of Alzheimer’s Disease. J. Laboratory Automation 20 (5), 589–600. doi:10.1177/2211068214559979
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer's Disease. Neuron 55, 697–711. doi:10.1016/j.neuron.2007.07.025
Pankevich, D. E., Davis, M., and Altevogt, B. M. (2011). GLUTAMATE-RELATED BIOMARKERS IN DRUG DEVELOPMENT for DISORDERS of the NERVOUS SYSTEM WORKSHOP SUMMARY Forum on Neuroscience and Nervous System Disorders Board on Health Sciences Policy. National Academies Press. Available at: http://www.nap.edu.
Pereira, A. C., Gray, J. D., Kogan, J. F., Davidson, R. L., Rubin, T. G., Okamoto, M., et al. (2017). Age and Alzheimer's Disease Gene Expression Profiles Reversed by the Glutamate Modulator Riluzole. Mol. Psychiatry 22 (2), 296–305. doi:10.1038/mp.2016.33
Peters, R. (2006). Ageing and the Brain. Postgrad. Med. J. 82 (964), 84–88. doi:10.1136/pgmj.2005.036665
Peterson, C., and Cotman, C. W. (1989). Strain-dependent Decrease in Glutamate Binding to the N-Methyl-D-Aspartic Acid Receptor during Aging. Neurosci. Lett. 104, 309–313. doi:10.1016/0304-3940(89)90594-6
Petralia, R. S., and Wenthold, R. J. (1996). “Types of Excitatory Amino Acid Receptors and Their Localization in the Nervous System and Hypothalamus,” in Excitatory Amino Acids: Their Role in Neuroendocrine Function. 1st ed. (New York: CRC Press).
Piers, T. M., Kim, D. H., Kim, B. C., Regan, P., Whitcomb, D. J., and Cho, K. (2012). Translational Concepts of mGluR5 in Synaptic Diseases of the Brain. Front. Pharmacol. 3, 199. doi:10.3389/FPHAR.2012.00199/BIBTEX
Raininko, R., and Mattsson, P. (2010). Metabolite Concentrations in Supraventricular White Matter from Teenage to Early Old Age: A Short Echo Time 1H Magnetic Resonance Spectroscopy (MRS) Study. Acta Radiol. 51 (3), 309–315. doi:10.3109/02841850903476564
Rajan, K. B., Weuve, J., Barnes, L. L., McAninch, E. A., Wilson, R. S., and Evans, D. A. (2021). Population Estimate of People with Clinical Alzheimer’s Disease and Mild Cognitive Impairment in the United States (2020-2060). Alzheimer’s Dementia J. Alzheimer’s Assoc. 17 (12), 1966–1975. doi:10.1002/ALZ.12362
Rammes, G., Hasenjäger, A., Sroka-Saidi, K., Deussing, J. M., and Parsons, C. G. (2011). Therapeutic Significance of NR2B-Containing NMDA Receptors and mGluR5 Metabotropic Glutamate Receptors in Mediating the Synaptotoxic Effects of β-amyloid Oligomers on Long-Term Potentiation (LTP) in Murine Hippocampal Slices. Neuropharmacology 60 (6), 982–990. doi:10.1016/J.NEUROPHARM.2011.01.051
Ransome, M. I., Goldshmit, Y., Bartlett, P. F., Waters, M. J., and Turnley, A. M. (2004). Comparative Analysis of CNS Populations in Knockout Mice with Altered Growth Hormone Responsiveness. Eur. J. Neurosci. 19, 2069–2079. doi:10.1111/j.0953-816X.2004.03308.x
Revett, T. J., Glen, B. B., Jack, J., and Satyabrata, K. (2013). Glutamate System, Amyloid β Peptides and Tau Protein: Functional Interrelationships and Relevance to Alzheimer Disease Pathology. J. Psychiatry & Neurosci. JPN 38 (1), 6. doi:10.1503/JPN.110190
Roalf, D. R., Sydnor, V. J., Woods, M., Wolk, D. A., Scott, J. C., Reddy, R., et al. (2020). A Quantitative Meta-Analysis of Brain Glutamate Metabolites in Aging. Neurobiol. Aging 95, 240–249. doi:10.1016/j.neurobiolaging.2020.07.015
Rodriguez-Perdigon, M., Tordera, R. M., Gil-Bea, F. J., Gerenu, G., Ramirez, M. J., and Solas, M. (2016). Down-regulation of Glutamatergic Terminals (VGLUT1) Driven by Aβ in Alzheimer’s Disease. Hippocampus 26, 1303–1312. (Accessed April 11, 2022).doi:10.1002/hipo.22607
Rozycka, A., Charzynska, A., Misiewicz, Z., Maciej Stepniewski, T., Sobolewska, A., Kossut, M., et al. (2019). Glutamate, GABA, and Presynaptic Markers Involved in Neurotransmission Are Differently Affected by Age in Distinct Mouse Brain Regions. ACS Chem. Neurosci. 10, 4449–4461. doi:10.1021/acschemneuro.9b00220
Rudy, C. C., Hunsberger, H. C., Weitzner, D. S., and Reed, M. N. (2015). The Role of the Tripartite Glutamatergic Synapse in the Pathophysiology of Alzheimer's Disease. Aging Dis. 6 (2), 131–148. doi:10.14336/AD.2014.0423
Sachdev, P. S., Lipnicki, D. M., Crawford, J., Reppermund, S., Kochan, N. A., Trollor, J. N, et al. (2013). Factors Predicting Reversion from Mild Cognitive Impairment to Normal Cognitive Functioning: A Population-Based Study. PloS One 8 (3), e59649. doi:10.1371/JOURNAL.PONE.0059649
Sailasuta, N., Ernst, T., and Chang, L. (2008). Regional Variations and the Effects of Age and Gender on Glutamate Concentrations in the Human Brain. Magn. Reson Imaging 26, 667–675. doi:10.1016/j.mri.2007.06.007
Schaffer, C., Sarad, N., DeCrumpe, A., Goswami, D., Herrmann, S., Morales, J., et al. (2015). Biomarkers in the Diagnosis and Prognosis of Alzheimer’s Disease. J. Laboratory Automation 20 (5), 589–600. doi:10.1177/2211068214559979
Schrader, L. A., and Tasker, J. G. (1997). Modulation of Multiple Potassium Currents by Metabotropic Glutamate Receptors in Neurons of the Hypothalamic Supraoptic Nucleus. J. Neurophysiology 78 (6), 3428–3437. doi:10.1152/JN.1997.78.6.3428/ASSET/IMAGES/LARGE/JNP.DE27F6.JPEG
{kind=link}
Scofield, M. D., Heinsbroek, J. A., Gipson, C. D., Kupchik, Y. M., Spencer, S., Smith, A. C. W., et al. (2016). The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol. Rev. 68 (3), 816–871. doi:10.1124/pr.116.012484
Scott, H. A., Gebhardt, F. M., Mitrovic, A. D., Vandenberg, R. J., and Dodd, P. R. (2011). Glutamate Transporter Variants Reduce Glutamate Uptake in Alzheimer's Disease. Neurobiol. Aging 32, 553.e1–553.e11. doi:10.1016/j.neurobiolaging.2010.03.008
Segovia, G., Porras, A., Del Arco, A., and Mora, F. (2001). Glutamatergic Neurotransmission in Aging: a Critical Perspective. Mech. Ageing Dev. 122 (1), 1–29. doi:10.1016/s0047-6374(00)00225-6
Sharma, S., Darland, D., Lei, S., Rakoczy, S., and Brown-Borg, H. M. (2012). NMDA and Kainate Receptor Expression, Long-Term Potentiation, and Neurogenesis in the hippocampus of Long-Lived Ames Dwarf Mice. Age (Dordr) 34, 609–620. doi:10.1007/s11357-011-9253-1
Sokolow, S., Luu, S. H., Nandy, K., Miller, C. A., Vinters, H. V., Poon, W. W., et al. (2012). Preferential Accumulation of Amyloid-Beta in Presynaptic Glutamatergic Terminals (VGluT1 and VGluT2) in Alzheimer's Disease Cortex. Neurobiol. Dis. 45, 381–387. (Accessed July 7, 2017).doi:10.1016/j.nbd.2011.08.027
Soo, S. K., Rudich, P. D., Traa, A., Harris-Gauthier, N., Shields, H. J., and Van Raamsdonk, J. M. (2020). Compounds that Extend Longevity Are Protective in Neurodegenerative Diseases and Provide a Novel Treatment Strategy for These Devastating Disorders. Mech. Ageing Dev. 190, 111297. doi:10.1016/j.mad.2020.111297
Sperling, R. A., Rentz, D. M., Johnson, K. A., Karlawish, J., Donohue, M., Salmon, D. P., et al. (2014). The A4 Study: Stopping AD before Symptoms Begin? Sci. Transl. Med. 6 (228), 228fs13. doi:10.1126/scitranslmed.3007941
Stephens, M. L., Quintero, J. E., Pomerleau, F., Huettl, P., and Gerhardt, G. A. (2011). Age-related Changes in Glutamate Release in the CA3 and Dentate Gyrus of the Rat hippocampus. Neurobiol. Aging 32, 811–820. doi:10.1016/j.neurobiolaging.2009.05.009
Sun, L. Y., Al-Regaiey, K., Masternak, M. M., Wang, J., and Bartke, A. (2005). Local Expression of GH and IGF-1 in the hippocampus of GH-Deficient Long-Lived Mice. Neurobiol. Aging 26, 929–937. doi:10.1016/j.neurobiolaging.2004.07.010
Swanson, C. J., Bures, M., Johnson, M. P., Linden, A. M., Monn, J. A., and Schoepp, D. D. (2005). Metabotropic Glutamate Receptors as Novel Targets for Anxiety and Stress Disorders. Nat. Rev. Drug Discov. 4 (2), 131–144. doi:10.1038/NRD1630
Sydnor, V. J., and Roalf, D. R. (2020). A Meta-Analysis of Ultra-high Field Glutamate, Glutamine, GABA and Glutathione 1HMRS in Psychosis: Implications for Studies of Psychosis Risk. Schizophr. Res. 226, 61–69. doi:10.1016/J.SCHRES.2020.06.028
Takamori, S., Rhee, J. S., Rosenmund, C., and Jahn, R. (2000). Identification of a Vesicular Glutamate Transporter that Defines a Glutamatergic Phenotype in Neurons. Nature 407 (6801), 189–194. doi:10.1038/35025070
Takumi, Y., Ramírez-León, V., Laake, P., Rinvik, E., and Ottersen, O. P. (1999). Different Modes of Expression of AMPA and NMDA Receptors in Hippocampal Synapses. Nat. Neurosci. 2 (7), 618–624. doi:10.1038/10172
Tanila, H., Shapiro, M., Gallagher, M., and Eichenbaum, H. (1997). Brain Aging: Changes in the Nature of Information Coding by the hippocampus. J. Neurosci. 17, 5155–5166.
Tapiero, H., Mathé, G., Couvreur, P., and Tew, K. D. (2002). II. Glutamine and Glutamate. Biomed. Pharmacother. 56 (9), 446–457. doi:10.1016/s0753-3322(02)00285-8
Tariciotti, L., Casadei, M., Honig, L. S., Teich, A. F., McKhann Ii, G. M., Tosto, G., et al. (2018). Clinical Experience with Cerebrospinal Fluid Aβ42, Total and Phosphorylated Tau in the Evaluation of 1,016 Individuals for Suspected Dementia. J. Alzheimers Dis. 65 (4), 1417–1425. doi:10.3233/JAD-180548
Temido-Ferreira, M., Coelho, J. E., Pousinha, P. A., and Lopes, L. V. (2019). Novel Players in the Aging Synapse: Impact on Cognition. J. Caffeine Adenosine Res. 9, 104–127. doi:10.1089/caff.2019.0013
Treyer, V., Gietl, A. F., Suliman, H., Gruber, E., Meyer, R., Buchmann, A., et al. (2020). Reduced Uptake of [11C]-Abp688, a PET Tracer for Metabolic Glutamate Receptor 5 in Hippocampus and Amygdala in Alzheimer’s Dementia. Brain Behav. 10 (6), e01632. doi:10.1002/brb3.1632
Turnley, A. M., Faux, C. H., Rietze, R. L., Coonan, J. R., and Bartlett, P. F. (2002). Suppressor of Cytokine Signaling 2 Regulates Neuronal Differentiation by Inhibiting Growth Hormone Signaling. Nat. Neurosci. 5 (11), 1155–1162. doi:10.1038/nn954
von Engelhardt, J., Eliava, M., Meyer, A. H., Rozov, A., and Monyer, H. (2007). Functional Characterization of Intrinsic Cholinergic Interneurons in the Cortex. J. Neurosci. 27 (21), 5633–5642. doi:10.1523/jneurosci.4647-06.2007
Wang, H. Y., Lee, D. H., Davis, C. B., and Shank, R. P. (2002). Amyloid Peptide Aβ1-42 Binds Selectively and with Picomolar Affinity to Α7 Nicotinic Acetylcholine Receptors. J. Neurochem. 75 (3), 1155–1161. doi:10.1046/j.1471-4159.2000.0751155.x
Webster, S. J., Bachstetter, A. D., Nelson, P. T., Schmitt, F. A., and Van Eldik, L. J. (2014). Using Mice to Model Alzheimer’s Dementia: An Overview of the Clinical Disease and the Preclinical Behavioral Changes in 10 Mouse Models. Front. Genet. 5, 1–14. doi:10.3389/fgene.2014.00088
Wisden, W., and Seeburg, P. H. (1993). A Complex Mosaic of High-Affinity Kainate Receptors in Rat Brain. J. Neurosci. 13 (8), 3582–3598. doi:10.1523/JNEUROSCI.13-08-03582.1993
Zahr, N. M., Mayer, D., Rohlfing, T., Chanraud, S., Gu, M., Sullivan, E. V., et al. (2013). In vivo Glutamate Measured with Magnetic Resonance Spectroscopy: Behavioral Correlates in Aging. Neurobiol. Aging 34, 1265–1276. doi:10.1016/j.neurobiolaging.2012.09.014
Keywords: excitotoxcity, neurodegenerative disease, amyloid—beta, geroscience, biomarker, hippocampus, neuroimaging, growth hormone receptor knockout
Citation: Cox MF, Hascup ER, Bartke A and Hascup KN (2022) Friend or Foe? Defining the Role of Glutamate in Aging and Alzheimer’s Disease. Front. Aging 3:929474. doi: 10.3389/fragi.2022.929474
Received: 26 April 2022; Accepted: 17 May 2022;
Published: 16 June 2022.
Edited by:
Ana Margarida Ledo, University of Coimbra, PortugalReviewed by:
Miranda N. Reed, Auburn University, United StatesXinxing Wang, Stony Brook University, United States
Copyright © 2022 Cox, Hascup, Bartke and Hascup. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin N. Hascup, a2hhc2N1cDQ5QHNpdW1lZC5lZHU=