 Maria I. Lazaro-Pena1†
Maria I. Lazaro-Pena1† Zachary C. Ward1†
Zachary C. Ward1† Sifan Yang1,2†
Sifan Yang1,2† Alexandra Strohm1,3,4†
Alexandra Strohm1,3,4† Alyssa K. Merrill3,4
Alyssa K. Merrill3,4 Celia A. Soto5,6
Celia A. Soto5,6 Andrew V. Samuelson1*
Andrew V. Samuelson1*- 1Department of Biomedical Genetics, University of Rochester Medical Center, Rochester, NY, United States
- 2Department of Biology, University of Rochester, Rochester, NY, United States
- 3Department of Environmental Medicine, University of Rochester Medical Center, Rochester, NY, United States
- 4Toxicology Training Program, University of Rochester Medical Center, Rochester, NY, United States
- 5Department of Pathology, University of Rochester Medical Center, Rochester, NY, United States
- 6Cell Biology of Disease Graduate Program, University of Rochester Medical Center, Rochester, NY, United States
Discoveries made in the nematode Caenorhabditis elegans revealed that aging is under genetic control. Since these transformative initial studies, C. elegans has become a premier model system for aging research. Critically, the genes, pathways, and processes that have fundamental roles in organismal aging are deeply conserved throughout evolution. This conservation has led to a wealth of knowledge regarding both the processes that influence aging and the identification of molecular and cellular hallmarks that play a causative role in the physiological decline of organisms. One key feature of age-associated decline is the failure of mechanisms that maintain proper function of the proteome (proteostasis). Here we highlight components of the proteostatic network that act to maintain the proteome and how this network integrates into major longevity signaling pathways. We focus in depth on the heat shock transcription factor 1 (HSF1), the central regulator of gene expression for proteins that maintain the cytosolic and nuclear proteomes, and a key effector of longevity signals.
Overview of the Genetics of Longevity in C. elegans
Regulation of Insulin-Like Signaling Determines Metazoan Aging
Perhaps the most profound discovery in the last 30 years of aging research was the discovery in Caenorhabditis elegans that aging is under genetic control. Single mutations found in age-1 and daf-2 were the first genetic evidence that longevity is influenced by regulated processes (Friedman and Johnson 1988; Johnson 1990; Kenyon et al., 1993). Shortly after this discovery, genetic and molecular analysis revealed that age-1 and daf-2 function as a part of a conserved insulin-like signaling (ILS) pathway (Dorman et al., 1995). A seminal discovery in C. elegans was that the ILS promotes aging by repressing the FOXO transcription factor DAF-16 in wild-type animals (Lin et al., 1997; Ogg et al., 1997). This had major implications for the understanding of diabetes and cancer as it linked a specific transcription factor to mammalian insulin and insulin growth factor (IGF-1) for the first time [for a synopsis of this period of discovery see (Kenyon 2011)]. A large body of work has since demonstrated that this core signaling pathway influences aging across metazoan animals (Holzenberger et al., 2003; Kenyon 2005; Suh et al., 2008).
Early discoveries in the genetics of longevity using C. elegans were founded on prior genetic insight into dauer formation (Riddle et al., 1981). Developing C. elegans proceed through for larval stages (L1–L4), marked by molts before becoming reproductive adults (Cassada and Russell 1975; Felix and Duveau 2012; Frezal and Felix 2015; Lazetic and Fay 2017). At the L2 to L3 transition, C. elegans can enter an alternative L3 developmental stage called dauer, which is a genetic program of developmental diapause, allowing animals to survive food scarcity, overcrowding, or a number of harsh environmental conditions and survive for up to 4 months (Golden and Riddle 1984a; Butcher et al., 2007; Fielenbach and Antebi 2008; Felix and Duveau 2012). Genetic epistasis between mutations that favored constitutive dauer formation under normal conditions, with mutations that rendered animals unable to enter dauer (dauer defective), delineated not only the pathways that regulated dauer development but also informed early subsequent discoveries into the genetics of longevity, which occurred prior to knowing the molecular identity of pathway components (Vowels and Thomas 1992; Kenyon et al., 1993; Gottlieb and Ruvkun 1994; Dorman et al., 1995; Larsen et al., 1995; Gems et al., 1998; Fielenbach and Antebi 2008). It is worth noting that the mechanisms regulating dauer entry and longevity are separable; for example, C. elegans with temperature sensitive daf-2 mutations grown at the permissive temperature bypass the dauer checkpoint and develop into normal adulthood, shifting to higher non-permissive temperatures reduces ILS and extends longevity in these adult animals (Golden and Riddle 1984b; Ailion and Thomas 2000; Ailion and Thomas 2003). This genetic analysis revealed two parallel genetic pathways that converge to regulate dauer development, which we now know are the ILS and TGF-β signaling pathways (Ren et al., 1996; Kimura et al., 1997).
Early studies to identify the Daf genes revealed that organismal longevity was determined via canonical ILS signaling. DAF-2 (insulin/IGF1 receptor) activates AGE-1 (PI3K) to generate phosphatidylinositol-3,4,5-triphosphate (PIP3) (Morris et al., 1996; Kimura et al., 1997), which is opposed by DAF-18 (PTEN, Phosphatase And Tensin Homolog) (Ogg and Ruvkun 1998). Accumulation of PIP3 activates PDK-1 (PDPK1, 3-phosphoinositide-dependent protein kinase-1), which activates AKT-1/2 (AKT Serine/Threonine Kinase, protein kinase B) via phosphorylation (Paradis et al., 1999). In turn activated AKT-1/2 directly inhibits DAF-16 (FOXO) through phosphorylation and sequestration in the cytosol (Paradis and Ruvkun 1998; Lee et al., 2001; Lin et al., 2001). Mutations within this pathway that decreased ILS lead to activation of DAF-16 and increased longevity (Kenyon et al., 1993; Lin et al., 1997; Ogg et al., 1997). Conversely, mutations that promote ILS, such as loss of daf-18, decrease lifespan (Larsen et al., 1995; Ogg and Ruvkun 1998).
Early genetic evidence suggested that TGF-β signaling regulated dauer formation but not lifespan (Larsen et al., 1995). However, it was later discovered that the longevity-regulating activity of the TGF-β pathway was masked by an egg-laying (Egl) phenotype that caused mortality from internal hatching of progeny; suppressing the latter by preventing production of progeny revealed that mutations within the TGF-β pathway doubled lifespan and induced transcriptional changes that overlapped with many DAF-16 regulated genes (Shaw et al., 2007). Molecular genetic analysis of Daf genes revealed an endocrine network that converges within steroidogenic tissues to promote production of a cholesterol derived ligand (dafachronic acid) of DAF-12, which encodes a nuclear hormone receptor orthologous to vertebrate farnesoid-X, liver-X and vitamin D-receptors [reviewed in (Antebi 2013)]. Significantly, many of the endocrine pathways that regulate dauer and longevity are evolutionarily conserved (Mooijaart et al., 2007; Keisala et al., 2009; Kenyon 2010; Tennessen and Thummel 2011).
One unresolved question is the insulin paradox in humans. Defects in insulin receptor signaling causes insulin resistance and diabetes. Deficiencies in IGF-1 or upstream growth hormone (GH) are associated with increased incidence of cardiovascular disease and atherosclerosis. Yet, polymorphisms in many components of insulin or IGF-1 pathways that decrease signaling is associated with improved longevity and found in centenarian populations (IGF-1R, PI3K, INSR, FOXO3). Furthermore, centenarian populations are associated with improved insulin sensitivity, low-serum IGF-1, and a mutation in the insulin receptor has been found in semi-supercentenarians (>105 years) [reviewed in (Arai et al., 2009; Calvo-Ochoa and Arias 2015)]. Early characterization of C. elegans with difference alleles of daf-2, all of which increase lifespan, noted different degrees of an effect on motility, stress resistance, morphology, development, reproductive lifespan and brood size (Gems et al., 1998). Mutations within the ligand binding domain tended to have less pleiotropies in contrast to mutations within the kinase domain of DAF-2, which suggested DAF-2 kinase activity has at least separable outputs on organismal physiology (i.e. longevity and other pleotropic effects) (Gems et al., 1998). Whether different alleles of daf-2 differentially impact the molecular and cellular hallmarks of aging (discussed below) remain unexplored.
One possible explanation of the insulin paradox is that there is an optimal reduction in ILS to increase longevity, and reduction below this rate results in metabolic syndromes and premature aging (Cohen and Dillin 2008). Consistent with that possibility, null-mutations of daf-2 and age-1 in C. elegans result in lethal constitutive dauer formation (Malone and Thomas 1994; Dorman et al., 1995; Larsen et al., 1995; Gems et al., 1998) and several daf-2 mutations are temperature sensitive loss of function: small increases in lifespan are observed at lower permissive temperatures and greater increases when temperature is increased (Gems et al., 1998). However, age-1 null mutant animals raised at lower temperature can bypass dauer and eventually develop into adults that have near normal feeding rates, motility, and remarkably live up to 145–190 days, which is 10-times longer than wild-type animals (Ayyadevara et al., 2008). This suggests that the insulin paradox cannot be solved based simply on levels of ILS. Possible explanations to the insulin paradox that are not mutually exclusive include: differences between insulin and IGF-1 signaling, tissue- or cell-type specific effects, background mutations, the nature of mutation, and timing of alterations in ILS.
An Emerging Theme: Genes and Pathways Linked to Metabolic Control Determine Aging
Since the discovery that ILS regulates longevity, a common theme has emerged: the evolutionarily conserved genes and pathways that have the largest impact on lifespan often act in nutrient and energy sensing. For instance, a key controller of nutrient sensing is the target of Rapamycin (TOR) response to decreased levels of amino acids and carbohydrates (Kapahi et al., 2010). Under nutrient-rich conditions, TOR promotes cellular growth by simultaneously activating protein translation (e.g., transcription of translation components) while inhibiting protein turnover (e.g., transcription of chaperone and autophagy genes (McCormick et al., 2011; Seo et al., 2013; Lapierre et al., 2015)), and by inhibiting the initiation of autophagy. TOR inhibition, or activation of targets of TOR inhibition, results in extension of longevity (Jia et al., 2004; Hansen et al., 2008). Similarly, AMP-activated protein kinase (AMPK) acts as a conserved energy sensor of increased levels of AMP and ADP. Energy-stress activation of AMPK induces autophagy, the oxidative stress response (OSR), and extends longevity (Apfeld et al., 2004; Greer et al., 2007; Greer et al., 2009; Salminen and Kaarniranta 2012). Sirtuins (SIRT1-7 in mammals, sir-2.1 and sir-2.4 in C. elegans) also play a key role in nutrient sensing and extension of longevity (Jedrusik-Bode et al., 2013; Jedrusik-Bode 2014). Sirtuins are (NAD+)-dependent deacetylases, which sense levels of NAD+, an important metabolite linked to longevity (Verdin 2015). Sirtuin-mediated extension of longevity has been linked to ILS, AMPK, and TOR signaling, and sirtuins are essential for both dietary restriction (DR) and exercise to increase lifespan (Dai et al., 2018). From these and many additional studies two conclusions become self-evident: aging is under genetic control and these mechanisms have been deeply conserved throughout evolution.
Why would nutrient sensing, the abundance of key metabolites and energy currency, be causally linked to genetic programs that determine organismal longevity? It is tempting to speculate that very early in evolution organisms able to couple physiology to energy resources had a survival advantage. Under conditions of plentiful resources, organisms able to develop and reproduce quickly could dominate an ecological niche. However, when food is scarce, organisms able to conserve or recycle resources and delay energetically-costly physiological processes. such as development and reproduction, in favor of mechanisms that protect the Soma, would have a survival advantage. Delaying the production of offspring has the added benefit of limiting competition for limited resources. Consistent with this hypothesis, in C. elegans many long-lived mutant animals have one or more of the following characteristics: slower development, links to dauer formation (a form of developmental diapause), reduced numbers of overall progeny, and/or an extended period of progeny production (i.e., reproductive span) (Szewczyk et al., 2006; Mukhopadhyay and Tissenbaum 2007). In fact, loss of the C. elegans germline through mutation of glp-1, which encodes an ortholog to the Notch receptor, increases lifespan by preventing germ cell development in early adulthood, which also requires DAF-16, implying a connection to ILS (Arantes-Oliveira et al., 2002).
Refined genetic analysis has revealed that many of the phenotypes associated with reproduction or development are separable from longevity. For example, early discoveries in C. elegans aging research using temperature-sensitive alleles in the ILS pathway revealed that dauer formation and extended longevity were genetically separable (Kenyon et al., 1993), implying that strategies to improve healthy aging based on the genetics of longevity may not require a cost in developmental or reproductive fitness.
The aforementioned longevity signals converge on a limited number of transcription factors, which also respond to numerous additional stress signals. For example, skn-1 encodes the C. elegans ortholog of the nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the “Cap’n'Collar” basic leucine zipper family of transcription factors, which is best known for regulating the expression of the OSR (An and Blackwell 2003). However, specific splice isoforms of skn-1 play key roles in the endoplasmic reticulum (ER) unfolded protein response (ER-UPR), maintaining proteostasis in the cytosol, and the response to starvation (Glover-Cutter et al., 2013; Lehrbach and Ruvkun 2016; Denzel et al., 2019; Lehrbach and Ruvkun 2019). DR activates SKN-1 within two head neurons (ASI) and is essential for increased longevity and cell non-autonomous changes in metabolic activity within peripheral tissues (Bishop and Guarente 2007). Additionally, amino acid and carbohydrate starvation activate skn-1 through TOR signaling (Robida-Stubbs et al., 2012), and reduced ILS activates SKN-1 in conjunction with DAF-16 (Tullet et al., 2008). Additional evidence of signal convergence is AMPK phosphorylation and activation of DAF-16, after a distinct method of DR (Greer et al., 2007; Greer et al., 2009). Furthermore pha-4, which encodes the FOXA forkhead transcription factor is critical for lifespan extension phenotypes related to germline inhibition and DR, but not reduced ILS, through regulation of autophagy (Panowski et al., 2007; Lapierre et al., 2011). One of the key transcriptional effectors of longevity signaling is the heat shock transcription factor (HSF-1 in C. elegans, HSF1 in more complex metazoans), which we discuss in detail in the latter part of this review. Collectively, a growing number of C. elegans studies have begun to unravel the complex integrated networks that maintain organismal homeostasis from an extensive array of diverse extrinsic and intrinsic signals that converge on distinct but overlapping adaptive transcriptional responses (Greer and Brunet 2009; Denzel et al., 2019).
Longevity is Determined via Cell Non-Autonomous Signals
A strength of C. elegans as a model is the relative ease in achieving tissue- and cell-type specific genetic perturbation. Overexpression or rescue is easily achieved through the use of either tissue specific promoters (in wild-type or mutant backgrounds) or through mosaic analysis (Yochem et al., 2000; Yochem and Herman 2003; Meister et al., 2010; Prelich 2012). Spatial and temporal gene inactivation can be achieved classically through the use of tissue specific RNAi (e.g., tissue specific expression of rde-1 in RNAi-deficient rde-1 mutant animals) (Qadota et al., 2007; Miles and van Oosten-Hawle 2020; Watts et al., 2020) or more recently with the development of the Tir1-auxin system, which provides spatial and temporally controlled protein degradation (Zhang et al., 2015). Collectively, these approaches allowed early efforts in the emerging field of aging research identify the tissues where a longevity gene or pathway functioned. These studies raised two possibilities: 1) longevity functions restricted to a specific tissue are regions with a metazoan that decline cell-intrinsically during aging and ultimately result in death, or 2) the longevity functions within a specific tissue generate cell non-autonomous paracrine or endocrine signals that orchestrate cellular aging across tissues. The latter view has become widely accepted and is based on numerous studies ranging from early observations to ongoing discoveries. Distinct tissues which influence aging throughout the Soma through endocrine signals include neurons, the somatic gonad, the germ line/gonadal stem cells, and intestinal cells (Kleemann and Murphy 2009). Examples of some of the longevity signaling pathways that act cell non-autonomously through endocrine signaling include ILS, bile acid signaling, TGF-β signaling, serotonin signaling, pregnenolone signaling, TORC1 signaling, and AMPK signaling (Kleemann and Murphy 2009; Ulgherait et al., 2014; Zhang et al., 2019). Signaling is likely to occur through lipophilic hormones (Hansen et al., 2005). These signals are distinct, for example: alterations in gonadal stem cell signaling communicates cell non-autonomously with somatic intestinal cells through lipophilic hormone signaling and kri-1 (ortholog of human KRIT1) to activate DAF-16. This cell non-autonomous mechanism is specific to germline to Soma signaling, as these functions are not required for ILS-mediated longevity or DAF-16 activation (Berman and Kenyon 2006). Similarly, endocrine germline signals and DR differentially regulate protein quality control mechanisms (Shpigel et al., 2019). For a recent review on cell non-autonomous signaling in longevity see (Miller et al., 2020).
Hallmarks of Molecular and Cellular Aging
Many molecular and cellular hallmarks of aging have been discovered, which have been broadly classified as: altered intercellular communication, genomic instability, telomere attrition, epigenomic alterations, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and dysfunction, and decline of protein homeostasis (proteostasis) (Lopez-Otin et al., 2013). While many of the mechanisms that alter aging impact multiple hallmarks, and similarly these hallmarks impact one another; in this review we focus primarily on proteostasis through an in depth discussion of the mechanisms that preserve proper function and folding of the cytosolic and nuclear proteomes, how these mechanisms intersect with the aforementioned longevity signals, and give special emphasis to the key transcription factor that acts as the guardian of the nuclear and cytosolic proteome: HSF1. We provide a detailed analysis of discoveries in mammals and C. elegans, and highlight areas of future investigation where iterative analysis between systems would provide deeper mechanistic insight to how proteostasis preserves the functional integrity of complex metazoans.
Inherent Challenges to Maintaining Proteostasis
A challenge inherent to proteostasis is ensuring all proteins are properly folded de novo into a native conformation and maintained in a soluble state, despite overall protein concentrations approaching levels found in crystals (Powers et al., 2009). This is further amplified by the varied nature of protein size, amino acid composition, structural conformation, stability, turnover, and expression (Wolff et al., 2014). Thus, maintaining proteostasis is a generalized problem, not unique to a subset of proteins. Layered within this complexity is the need for proteins to be localized within the proper cellular compartment, to be maintained in correct stoichiometric ratios relative to other components of larger protein complexes, to undergo the correct modifications in response to a diverse array of internal and extrinsic cues, and to be maintained within a proteome with a composition unique to each cell type or tissue. To put it simply, the proper function and folding of the proteome is a complex and dynamic process, vital for maintaining cellular function.
Overview of the Proteostatic Network
Proteostasis is maintained through the coordinated action of a large proteostatic network (PN), consisting of approximately 2,000 unique proteins, which regulate the synthesis, folding, trafficking, and degradation within the proteome [reviewed in (Morimoto 2008; Wolff et al., 2014; Hipp et al., 2019; Morimoto 2019)]. This network regulates de novo protein folding from the emergence of the nascent polypeptide from the ribosome to the final subcellular localization of the mature protein. The PN responds to acute stress on the proteome, and fine-tunes the rates of translation and degradation in response to a myriad of cell intrinsic and extrinsic cues. The major components are several large families of molecular chaperones, co-chaperones, and protein clearance mechanisms, predominately degradation through the ubiquitin-proteasome system (UPS) and autophagy at the lysosome (Figure 1).
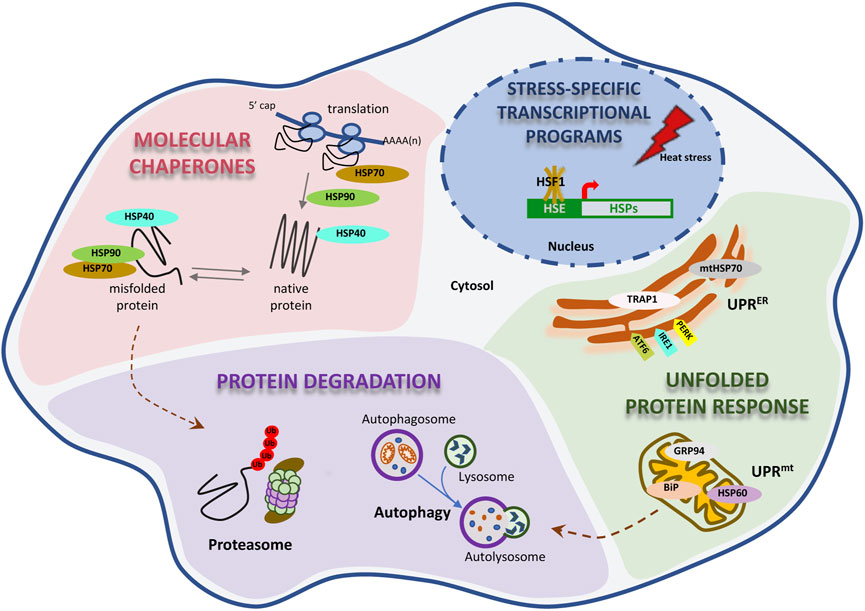
FIGURE 1. Proteostasis is maintain by a proteostasis network. Proteostasis has multiple levels of regulation. A great number of molecular chaperones are present in the cytosol, assisting on the appropriate folding and quality of the proteome. The proteosome and autophagy protein degradation pathways are essential for the clearance of unneeded proteins. Two distinct, but analogous, unfolded protein responses in the mitochondria and endoplasmic reticulum (mitoUPR and ER-UPR, respectively) are necessary to activate the expression of chaperones and to protect the function of these organelles from unfolding stress. Ultimately, the PN network is equipped with the adaptive activation of different transcriptional programs that get induced by different types of stress.
Declining Proteostasis During Aging
Decline of Cellular Proteostasis in C. elegans
Key discoveries in C. elegans first revealed that declining proteostasis is a hallmark of aging, and provided a generalized cellular explanation for the manifestation and progression of neurodegenerative disease. The decline of cellular proteostasis is a hallmark of aging across species [reviewed in (Rubinsztein et al., 2011; Taylor and Dillin 2011; Labbadia and Morimoto 2015a; Klaips et al., 2018; Hipp et al., 2019)]. For instance, even in the absence of overt disease, there is a growing body of evidence demonstrating that the loss of autophagy and declining levels of molecular chaperones are conserved hallmarks of aging (Ben-Zvi et al., 2009; Morimoto and Cuervo 2009; Vellai et al., 2009). The use of transgenic C. elegans expressing polyglutamine fused to a fluorescent reporter remains a powerful system to identify age-associated declines in proteostasis in vivo (Satyal et al., 2000; Brignull et al., 2006; Morimoto 2006; Morimoto 2008). Early studies demonstrated that co-expression of both a proteotoxic polyQ reporter and endogenous metastable proteins further exacerbates protein misfolding and degenerative phenotypes, consistent with the notion that prolonged proteotoxic stress overloads the limited buffering capacity of the PN (Gidalevitz et al., 2006). Widespread failures in protein folding have since been shown to occur in early adulthood across tissues and coincides with reduced activation of HSF-1 and chaperone protein expression (Ben-Zvi et al., 2009). In C. elegans, the transcriptional inducibility of multiple forms of stress response rapidly decline within a few hours after the onset of reproduction due to the formation of repressive chromatin marks at stress loci (Shemesh et al., 2013; Labbadia and Morimoto 2015a; Labbadia and Morimoto 2015b). During normal aging, solubility of the C. elegans proteome declines, resulting in an accumulation of aggregates (David et al., 2010; Reis-Rodrigues et al., 2012; Walther et al., 2015). Additionally, during aging, there is an increase in misfolded and oxidatively-damaged proteins, especially in neurons (Powers et al., 2009; Kundra et al., 2017; Sala et al., 2017).
The age-associated collapse of proteostasis is also intrinsic to human senescence. During aging of higher metazoans, cells can enter a permanent form of G1 arrest commonly referred to as cellular senescence [reviewed in (McHugh and Gil 2018; Prata et al., 2018; Song et al., 2020)]. Cellular senescence is a genetic program (Serrano et al., 1997), and a key hallmark of aging, which can be induced by various age-associated damage drivers including: telomere damage, epigenetic dysregulation, DNA damage, and mitochondrial dysfunction. Accumulating senescence cells play a key role in age-associated diseases by promoting stem cell exhaustion, chronic inflammation, disruption of nutrient signaling, and proteostatic dysfunction (McHugh and Gil 2018; Prata et al., 2018; Borghesan et al., 2020; Song et al., 2020). The age-associated collapse of proteostatic networks observed in C. elegans also occurs in human senescent cells. For example, within senescent cells the transcriptional activation of the heat shock response deteriorates, activation of HSF1 is impaired, UPR-related transcriptional responses are impaired, and the proteosome is dysfunctional (Sabath et al., 2020).
Components of the Proteostatic Network and Links to Longevity
Molecular Chaperones
The “workhorse” components of the PN are the molecular chaperones, which assist in all aspects of proteostasis both under normal conditions and in response to challenge from intrinsic and extrinsic factors. Chaperones are tightly regulated; their relative abundance is closely linked to the rest of the proteome, with a limited buffering capacity (Gidalevitz et al., 2011). Thus, the consequence of proteotoxic stress is overload upon the chaperone system, resulting in protein misfolding. In the context of proteostatic disease, aggregate formation and toxic gain-of-function disruption of normal cellular physiology occurs and ultimately results in cell death if unresolved (Balch et al., 2008).
As key components of the PN, there are numerous molecular chaperones with diverse regulatory roles in maintaining proper function and folding of the proteome [for excellent reviews see (Richter et al., 2010; Balchin et al., 2016; Hipp et al., 2019)]. An informatic analysis identified 332 human genes encoding chaperones and co-chaperones (Brehme et al., 2014), which fall into nine families: HSP90, HSP70, HSP60, HSP40, prefoldin, small heat shock protein (sHSP), TPR-domain containing (Hartl and Hayer-Hartl 2002), and organellar-specific chaperones of the endoplasmic reticulum (Kleizen and Braakman 2004) and mitochondria (Tatsuta et al., 2005). The number of paralogous genes within a family can be extensive: members may be essential or dispensable for viability, act in specific contexts (e.g., de novo protein folding or refolding misfolded proteins), be expressed constitutively or regulated via specific signals, be expressed in specific cell types, or be localized in specific subcellular regions (Richter et al., 2010; Balchin et al., 2016). For example, cytosolic HSP70 paralogs differ in that heat shock cognate protein HSC70 is constitutively expressed, while in contrast expression HSP70 expression is induced after heat shock. The HSP90s and HSP70s are highly abundant chaperones in the cytosol and nucleus (Taipale et al., 2010; Labbadia and Morimoto 2015a). Paralogs, such as BiP and GRP94 are essential for ER function, while the paralogs mortalin and TRAP1 function in the mitochondria. HSP90s, HSP70s, HSP60s, and sHSPs mediate de novo protein folding of nascent polypeptides, a process directly coupled to translation, acting alone or in conjunction with co-chaperones. In response to stress, other HSP70 and HSP90 family members are induced to resolve protein misfolding. Small molecular chaperones are somewhat distinct, as they interact reversibly with a broad range of unfolded substrates independent of ATP. sHSPs do not primarily refold proteins, rather they prevent the formation of highly stable, proteotoxic aggregates, acting as a storage depot for unfolded proteins until they can be refolded or degraded [for detailed reviews on the chaperone system see (Bar-Lavan et al., 2016; Biebl and Buchner 2019; Rosenzweig et al., 2019; Jayaraj et al., 2020; Reinle et al., 2022)].
Molecular chaperones are central to the proper regulation of protein homeostasis in aging cells (Labbadia and Morimoto 2015a; Margulis et al., 2020). Induction of molecular chaperones in response to stress is essential for normal development, and is significantly reduced during aging (Lopez-Otin et al., 2013; Dues et al., 2016; Sabath et al., 2020). In addition to regulating proteostasis, molecular chaperones and co-chaperones have been implicated in C. elegans longevity (Hsu et al., 2003; Morley and Morimoto 2004); direct manipulation of chaperone levels can alter C. elegans lifespan. For example, loss of HSP90 shortens lifespan (Somogyvari et al., 2018), while overexpression of HSP16 or HSP70 increases lifespan (Lithgow et al., 1995; Tatar et al., 1997; Yokoyama et al., 2002; Walker and Lithgow 2003). Molecular chaperone levels of expression are also regulated by longevity pathways. For instance, ILS regulates molecular chaperone expression, as the long-lived daf-2 and age-1 mutant animals have increased expression levels (Hsu et al., 2003; McElwee et al., 2003; Murphy et al., 2003; Walker and Lithgow 2003; Morley and Morimoto 2004; Halaschek-Wiener et al., 2005; Lamitina and Strange 2005; Wentz et al., 2018). Similarly, inactivation of TORC1 (e.g., daf-15 encodes the C. elegans ortholog of Raptor) or downstream S6 kinase (rsks-1) is sufficient to induce chaperone expression and interestingly sHSPs, but hsp70s are not essential for the increased lifespan of rsks-1 mutant animals (Seo et al., 2013). Chaperone functions also feedback to longevity signaling pathways. For example, the HSP90 family member DAF-21 directly regulates nuclear localization and transcriptional activity of the DAF-16A isoform (Somogyvari et al., 2018). Chaperones also coordinate adaptive transcriptional responses to changes in longevity signals. For instance, prefoldin 6 (pfd-6) encodes a chaperone, which under conditions of decreased ILS integrates HSF-1 and DAF-16 transcriptional activity (Son et al., 2018). These studies highlight a few examples of increasing evidence that molecular chaperones are not merely effectors of mechanisms to maintain the proteome through changes in protein folding, degradation and aggregation. Rather, molecular chaperones play key roles in connecting the PN with longevity signals and pathways.
Ubiquitin Proteasome System and Autophagy
Protein degradation is a fundamental mechanism for maintaining proteostasis. Turnover of unfolded polypeptides via proteasome- and autophagy-mediated degradation pathways are additional components of the PN. These effector mechanisms not only safeguard de novo protein quality control but are continually adjusted in response to stress from both internal cues and endocrine signals through transcriptional regulators of the PN.
The UPS is the main protein degradation system within the cell and is an integral part of the PN, assisting by clearing misfolded or toxic proteins. The proteasome is composed by a 19S regulatory cap and a 20S proteolytic core (Finley 2009). The ubiquitinated substrate attaches to the 19S regulatory cap via ubiquitin receptors to be translocated to the 20S core where it is hydrolyzed, effectively degrading it. Substrates targeted to the proteasome are tagged by polyubiquitin chains by a series of steps of E1, E2, and E3 ligases. Targets for degradation are redirected to the proteasome through interaction with co-chaperones, including the C-terminus of HSC70-interacting protein (CHIP) and Bcl2-associated athanogene 1 (BAG1), in conjunction with HSP70 and HSP90 complexes (Connell et al., 2001). When proteome stability cannot be maintained by protein degradation through the UPS, such as after heat shock, the accumulation of misfolded proteins is alleviated through increased autophagy. Thus, the major degradation mechanisms also function as a part of an integrated system.
A second major degradation system is mediated via the lysosome. The process of macroautophagy (hereafter referred to as autophagy), chaperone-mediated autophagy, and microautophagy are clearance mechanisms for a growing number of substrates, including proteins, aggregates, damaged organelles, nucleic acids, and pathogens (Rabinowitz and White 2010; Tooze and Yoshimori 2010; Workman and van Montfort 2010; Mizushima 2011; Benbrook and Long 2012; Pyo et al., 2012; Khaminets et al., 2015; Khaminets et al., 2016). Like the ubiquitin-dependent proteasome pathway, autophagy is tightly regulated (He and Klionsky 2009; Kroemer et al., 2010). Autophagosomes, which are double-membrane vesicles, form during autophagy to sequester substrates for degradation. These loaded autophagosomes then fuse to lysosomes to form autolysosomes, and substrates are degraded by lysosomal hydrolytic activity. The activation of autophagy is important to protect cells against multiple stressors such as heat shock and nutrient deprivation (Kroemer et al., 2010), protecting the organism from diseases associated with degeneration, infections, and inflammation, among others (Levine and Kroemer 2008; Mizushima et al., 2008; Aman et al., 2021; Kaushik et al., 2021; Nieto-Torres and Hansen 2021).
The UPS is essential for normal aging in C. elegans (Papaevgeniou and Chondrogianni 2016; Margulis et al., 2020; Ottens et al., 2021). The ubiquitin E3 ligase CHIP promotes longevity through ILS by regulating insulin receptor turnover (Tawo et al., 2017). Additionally, the CUL-1 E3 ligase complex regulates DAF-16 transcriptional activity (Ghazi et al., 2007). Another ubiquitin E3 ligase, RLE-1, targets DAF-16 for polyubiquitination-mediated degradation (Li et al., 2007). The proteasome itself also plays a key role in aging. Multiple studies have shown that loss of proteasome subunits lead to premature aging (Yun et al., 2008; Chondrogianni et al., 2015). Upregulated proteasomal activity is observed in the long-lived glp-1 mutant animals and in DR models (Vilchez et al., 2012; Depuydt et al., 2013). The role of deubiquitination enzymes (DUBs) has also been implied in proteasome activity and aging in C. elegans (Papaevgeniou and Chondrogianni 2016). Repression of ubh-4 (C. elegans DUB gene) by DAF-16 induces proteasome activity (Matilainen et al., 2013).
Lysosomal proteolytic activity deteriorates with aging (Sarkis et al., 1988; Kaushik et al., 2021). In C. elegans, many studies have reported the direct link between autophagy and longevity [e.g. (Melendez et al., 2003; Hansen et al., 2008; Chang et al., 2017), and many more]. Mutational inactivation of autophagy genes (unc-51, bec-1, atg-18, atg-9, lgg-1) shortens C. elegans lifespan (Toth et al., 2008). Many autophagy genes contribute to longevity paradigms; Bec-1, unc-51, lgg-1, and atg-18 are crucial for lifespan extension through the ILS, TOR signaling, or under conditions of DR (Melendez et al., 2003; Jia and Levine 2007; Hansen et al., 2008; Kenyon 2010). Activation of the AMPK pathway to promote longevity is also autophagy-dependent (Egan et al., 2011; Mihaylova and Shaw 2011). Furthermore, some autophagy receptors promote autophagy-dependent proteostasis and longevity in a tissue specific manner (Kumsta et al., 2017).
The transcriptional regulation of autophagy genes affects longevity and some transcription factors extend lifespan or delay aging in an autophagy-dependent manner in C. elegans (Lapierre et al., 2015; Minnerly et al., 2017; Liu et al., 2020; Kaushik et al., 2021). For example, HLH-30/TFEB regulates multiple autophagy genes (atg-18, vha-16, lmp-1, lipl-1, lipl-3) and is required for lifespan extension in glp-1, eat-2, daf-2, clk-1, rsks-1, and TOR mutants, indicating roles in multiple longevity paradigms (Lapierre et al., 2013). HLH-30 is also implicated in lipid metabolism to promote longevity (Lapierre et al., 2011). PHA-4 regulates autophagy gene expression and is required to extend lifespan (Panowski et al., 2007; Sheaffer et al., 2008; Zhong et al., 2010; Lapierre et al., 2011). DAF-16 also regulates autophagy gene expression to extend longevity (Wang et al., 2008; McColl et al., 2010; Lapierre et al., 2015; Kaushik et al., 2021). The Myc-Mondo:Mlx transcriptional activation complex and the Mad:Max transcriptional repression complex links autophagy to longevity through ILS, DR, and germline signaling (Johnson et al., 2014; Nakamura et al., 2016). The homeodomain interacting protein kinase (hpk-1) is a transcriptional co-factor and nuclear kinase that regulates longevity and preserves proteostasis, at least in part, through an essential role in the induction of autophagy in response to inhibition of TOR or under conditions of DR (Das et al., 2017). How these transcription factors regulate autophagy under stress conditions (i.e., diverse metabolic or non-metabolic stressors), across cell types (either cell-intrinsically or cell non-autonomously), or compensate and coordinate specific types of autophagy, are all areas for future investigation.
Transcriptional Regulation of the Proteostatic Network
Vital components of the PN are adaptive transcriptional responses activated in response to acute or chronic damage to the proteome, as well as in response to metabolic and mitogenic signals. The most well-characterized adaptive transcriptional responses to proteotoxic stress are the HSR, OSR, mitochondrial unfolded protein response (mitoUPR), and ER-UPR. The HSR, ER-UPR, and mitoUPR are induced in response to proteotoxic stress within the cytosol/nucleus, ER, and mitochondria, respectively. Regulation of these adaptive transcriptional programs are critical aspects of the larger PN that act in concordance with each other and additional protein quality control components. There is growing evidence for crosstalk and compensatory mechanisms among these adaptive responses. Furthermore, each has cell-intrinsic and non-autonomous components [reviewed in (Dillin et al., 2014; Taylor et al., 2014; van Oosten-Hawle and Morimoto 2014; Morimoto 2019)]. Below, we focus on HSF1, which maintains proper function of the cytoplasmic and nuclear proteomes. Breakdown of the OSR, mitoUPR, and ER-UPR components of the PN have direct ties to cancer, neurodegenerative disease, and aging, but are beyond the scope of this review [see (Ryan and Hoogenraad 2007; Sykiotis et al., 2011; Wolff and Dillin 2013; Dufey et al., 2014; Jovaisaite et al., 2014; Mottis et al., 2014; Wolff et al., 2014; Karagoz et al., 2019a; Karagoz et al., 2019b; Preissler and Ron 2019) for detailed reviews on the OSR, mitoUPR and ER-UPR]. For a comprehensive review of transcriptional and epigenetic regulation of stress response in C. elegans longevity, see (Denzel et al., 2019).
Tissue-Specific Utilization of the PN
While core components of the chaperone system are uniformly expressed across tissues, each cell type preferentially utilizes specific subsets of molecular chaperones, presumably in alignment with the demands of a proteome that is unique to each particular cell type [(Shemesh et al., 2021), and reviewed in (Labbadia and Morimoto 2015a; Sala et al., 2017)]. For example, the proteomes of cells within the pancreas and muscle significantly differ; the former have elevated expression of secreted proteins, while the latter are enriched in mitochondrial localized proteins (compared to the mean expression in all tissues) (Sala et al., 2017). Thus, it is not surprising that ER-specific chaperones are the major class of chaperones expressed in the secretory tissues of the pancreas, small intestine, and liver. In contrast, sHSPs are overrepresented in skeletal and cardiac muscle, consistent with their role in maintaining the folding of filament components. In accordance with their key role in proteome maintenance, the proportion of HSP70, HSP40, and HSP90 chaperones are relatively constant across tissues (Taipale et al., 2010; Guisbert et al., 2013; Sala et al., 2017; Nisaa and Ben-Zvi 2021). Nevertheless, specific members within these families, as well as other PN components, can be enriched to support specialized functions within a given tissue (Hageman and Kampinga 2009). Consistently, the regulatory pathways that control the heat shock response (HSR) comprise a heat shock regulatory network with tissue-selective effects: in total 59 regulators of the HSR were identified through a genome-wide functional genomic screen and include both molecular chaperones and additional components of the PN (Guisbert et al., 2013). Recent analysis of the transcriptional landscape of molecular chaperones have delineated core chaperones expressed across human tissues from variable chaperones differentially expressed to match tissue specific requirements, which collectively form conserved tissue-specific functional networks (Brehme et al., 2014; Shemesh et al., 2021). Interestingly, these networks are formed during development and differentiation to rewire the cell chaperoning capacities and alter usage of mechanisms to maintain protein quality control (Nisaa and Ben-Zvi 2021; Shemesh et al., 2021). Thus, when considering targeting the PN for the effective treatment of disease, one must account for the tissue of origin, the unique nature of the proteome within that tissue, cell fate/differentiation status, and specialized components of the PN acting within a tissue or cell type.
Cell Non-Autonomous Regulation of Proteostasis and Longevity are Linked
In addition to cell-intrinsic mechanisms, organisms maintain proteostasis through cell non-autonomous mechanisms. To date, the majority of these discoveries have come from studies in C. elegans. For instance, a regulatory component of the HSR has a cell non-autonomous component; the maintenance of proteostasis throughout the organism is controlled by thermosensory neurons in a serotonin-dependent manner (Prahlad et al., 2008; Tatum et al., 2015). In contrast, a second regulatory component from the GABAergic and cholinergic system normally limits muscle cell proteostasis (Garcia et al., 2007; Silva et al., 2013). Furthermore, a moderate increase in cholinergic signaling in the neuromuscular junction triggers calcium influx to the cytosol of muscle cells, activating a downstream signaling cascade leading to the transcriptional activation of HSF-1, and thereby the expression of molecular chaperones (Silva et al., 2013). These findings suggest that upstream neuronal signals regulate proteostatic mechanisms in distal tissues. The GATA transcription factor: PQM-1, functions as a mediator of transcellular chaperone signaling, acting in either a neuron or intestinal-specific route to trigger hsp-90 in remote tissues to preserve proteostasis and metastable proteins in muscle cells induces systemic stress response across multiple tissues through transcellular chaperone signaling (van Oosten-Hawle and Morimoto 2014; O’Brien et al., 2018; Morimoto 2019). Moreover, the expression of HSP90 within intestinal or neuronal cells is sufficient to suppress protein misfolding in muscle cells (van Oosten-Hawle et al., 2013). Proteostasis and stress resilience in reproductive adult C. elegans are also regulated by communication from internally fertilized embryos (Sala et al., 2020).
Cell non-autonomous signals regulates components of the PN and extends longevity in C. elegans. For example, specific downregulation of electron transport chain components in the nervous system causes an increase in the mitoUPR in non-neuronal tissues (Durieux et al., 2011). Surprisingly, neuronal downregulation of the respiratory chain complex IV promotes longevity to a similar level as observed when it is downregulated throughout the Soma, and induces mitochondrial chaperones in the intestine. Furthermore, neuronal proteotoxic stress targeting mitochondria elicits a global induction of the mitoUPR through serotonin signaling (Berendzen et al., 2016). Interestingly, cell non-autonomous regulation of mitochondrial stress has also been observed in mammals, where the fibroblast growth factor 21 (FGF21) mediates a signaling event from muscle to peripheral tissue triggered by mitochondrial dysfunction in mice (Kim et al., 2013). Two lines of research indicate that the ER-UPR is cell non-autonomously regulated in C. elegans. First, neuronal expression of the spliced and activated form of the ER-UPR transcription factor X box-binding protein 1 (XBP-1) is sufficient to induce the activation of ER-UPR in intestinal cells, which in turn increases stress resistance and extends longevity (Taylor and Dillin 2013). Second, expression of octopamine receptor 1 (OCTR-1) in ASH, ASI, AIY or ADE chemosensory neurons is necessary to inhibit the activation of XBP-1 and the expression of non-canonical ER-UPR genes in distal cells (Urano et al., 2002; Sun et al., 2011). Interestingly, activation of the ER-UPR within distinct neuronal cell types activate unique responses in peripheral tissues (Higuchi-Sanabria et al., 2020). In mammals, ER-UPR interaction between tissues has been proposed as the mechanism of induction of ER stress in tumor cell lines that promotes the activation of the ER-UPR in macrophages, resulting in the production of pro-inflammatory cytokines leading to tumor growth (Mahadevan et al., 2011). Lastly, gonadal stem cell non-autonomous signaling also links proteotoxic stress resistance and longevity: loss of C. elegans gonadal stem cells results in increased somatic maintenance through increased proteosomal activity: overexpression of the 19S proteasome subunit rpn-6 is sufficient to fortify proteostasis and increase lifespan (Vilchez et al., 2012).
HSF1: Guardian of the Cytosolic and Nuclear Proteome
Heat shock transcription factors are conserved throughout eukaryotes. The Drosophila melanogaster, C. elegans, and Saccharomyces cerevisiae genomes each encode one heat shock transcription factor, whereas HSF has expanded in vertebrates to encode four paralogs (HSF 1 through 4). Here we focus primarily focus on HSF1 [for more on HSF family members and their discovery, see (Sorger and Pelham 1988; Wiederrecht et al., 1988; Clos et al., 1990; Rabindran et al., 1991; Liu et al., 1997; Nakai 1999; Gomez-Pastor et al., 2018; Roos-Mattjus and Sistonen 2021)].
Regulation of HSF1
As a transcription factor, HSF1 is regulated extensively by post-translational modifications including acetylation, sumoylation, and phosphorylation (summarized in Table 1). These modifications regulate HSF1 activity at multiple stages, including release from inhibitors, nuclear translocation, homotrimerization, promoter binding, and recruitment of RNA Polymerase II [reviewed in (Gomez-Pastor et al., 2018; Roos-Mattjus and Sistonen 2021)].
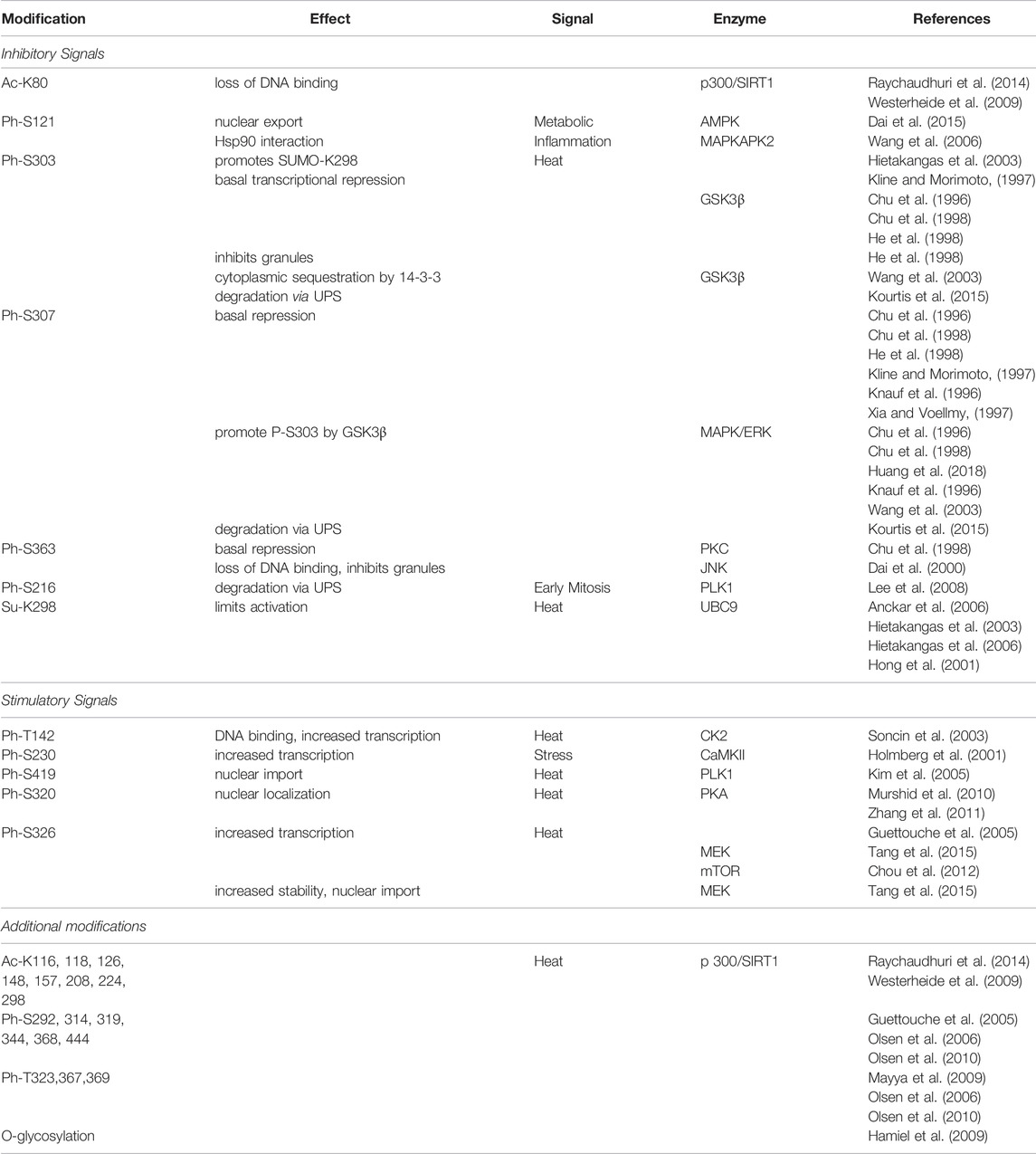
TABLE 1. HSF1 post-translational modifications.
Under normal conditions, HSF1 exists as an inactive monomer, stabilized by hydrophobic interactions between the heptad repeats in the N- and C-terminal regions (Sorger 1990; Sarge et al., 1993; Orosz et al., 1996; Farkas et al., 1998). One model for HSF1 activation is intrinsic activation; HSF1 effectively acts as a sensor responsive to changing thermodynamic conditions and thereby converts from an inactive monomer to an active trimer. This idea that HSF1 functions as an intrinsic “thermosensor” is consistent with in vitro data demonstrating that HSF1 can be activated in response to increasing temperature alone (Hentze et al., 2016). A second “chaperone titration model” posits that under basal conditions HSF1 is sequestered within the cytoplasm by molecular chaperones including HSP70, HSP90, and the chaperonin tailless complex polypeptide 1 (TCP1) ring complex (TRiC) (Abravaya et al., 1992; Baler et al., 1992; Baler et al., 1996; Duina et al., 1998; Shi et al., 1998; Zou et al., 1998; Guo et al., 2001; Akerfelt et al., 2010; Neef et al., 2010; Neef et al., 2011; Neef et al., 2014; Zheng et al., 2016). Upon acute proteotoxic stress, such as heat shock, protein misfolding titrates away chaperones that normally sequester HSF1 as a monomer. Released HSF1 monomers trimerize, translocate to the nucleus, bind to DNA promoters, and upregulate transcription of multiple genes. HSF1 activation induces expression of molecular chaperones, which initiates a negative feedback loop to inactivate HSF1 once stress upon the proteome is resolved (Zheng et al., 2016; Krakowiak et al., 2018). HSF1 has been shown to be regulated by additional means, including intrinsic refolding mechanisms (Mosser et al., 1990; Goodson and Sarge 1995; Xia and Voellmy 1997; Farkas et al., 1998; Zhong et al., 1998; Ahn and Thiele 2003), non-coding RNA (Shamovsky et al., 2006) and in vivo through cell non-autonomous signals from thermosensory neurons in C. elegans (Clark et al., 2007; Prahlad et al., 2008).
Conserved HSF-1 Regulation in C. elegans
There is a general conservation of mechanisms of HSF-1 regulation between C. elegans and mammals, yet the precise molecular mechanisms of control and how, for instance, post-translational modifications and direct interactors of mammalian HSF1, intersect with the wealth of genetic information linking C. elegans HSF-1 to longevity and stress signaling remains poorly understood. In both mammalian cell culture and C. elegans, under basal conditions HSF-1 is transcriptionally inactive due to cytoplasmic sequestration. Upon stress in either system, HSF-1 nuclear localization is governed by phosphorylation (Chiang et al., 2012; Dai et al., 2015; Tang et al., 2015). Similarly, nuclear HSF-1 forms stress granules upon heat stress in both C. elegans and mammals (Alastalo et al., 2003; Morton and Lamitina 2013). As in mammals, C. elegans HSF-1 transcriptional activation is dependent on the regulation of trimerization (Chiang et al., 2012). At least one direct regulator of HSF1 is conserved between mammals and C. elegans: the heat shock binding factor 1 (HSB-1) negatively regulates HSF-1 transcriptional activity (Satyal et al., 1998). Other direct regulators in mammals, such as TOR and AMPK, also show genetic interactions in C. elegans (discussed below), but whether direct interactions occur in C. elegans has yet to be determined. Knockdown of hsp-70 also activates HSF-1, implying that the chaperone titration model regulation of HSF1 is conserved (Guisbert et al., 2013).
Secondary Structure of HSF1
Structurally, HSF1 is highly conserved across metazoan animals (Figure 2). The N-terminal region of approximately the first 100 amino acids is the most well-conserved region of the HSF protein family and encodes a helix-turn-helix loop DNA-binding domain (DBD), which recognizes DNA heat shock elements (HSE) (identified as nGAAn DNA repeats) (Wu 1995). Two regions of leucine zippers (HR-A/B and HR-C) allow oligomerization of HSF1 monomers (Bjork and Sistonen 2010; Anckar and Sistonen 2011; Dayalan Naidu and Dinkova-Kostova 2017). HR-A/B immediately follows the DBD and flanks a regulatory domain (RD) on one side, while HR-C is downstream of the RD. Intrinsic interactions of HR-A/B with HR-C prevents spontaneous HSF1 trimerization and activation under basal conditions. Approximately the last 100 amino acids of HSF1 contains the trans-activation domain (TAD), which is the region through which HSP70 interacts with HSF1.
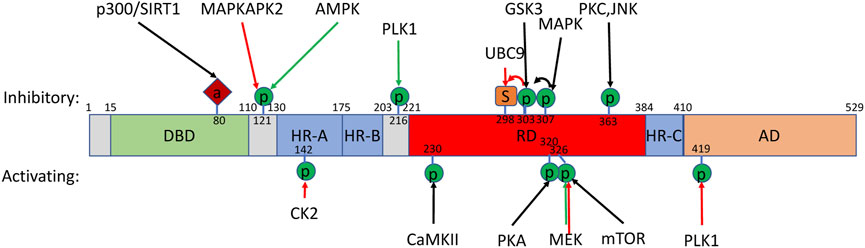
FIGURE 2. HSF1 post-translational modifications. Schematic of mammalian HSF1 secondary structure with post-translational modifications with known regulators and overall effect on activity. Additional modifications without known regulators are listed in Table 1. Additional regulators where a specific post-translational modification has not been identified have been omitted. “DBD” indicates the helix-turn-helix loop DNA-binding domain. “HR-A/B” and “HR-C″ identify two regions of leucine zippers. “RD” indicates the regulatory domain. “TAD” indicates the transcriptional transactivation domain. Modifications in response to heat (red arrows) or metabolic/mitogenic signals (green) are shown. Amino acid numbers are indicated. “P” indicates phosphorylation. “S” indicates sumoylation. “A” indicates acetylation.
HSF1 Regulation Through Phosphorylation
Phosphorylation of mammalian HSF1 is one of the most well studied mechanisms of HSF1 regulation; mass spectrometry and site-directed mutagenesis have identified phosphorylation on multiple serine or threonine residues (S121, S127, T142, S195, S216, S230, S292, S303, S307, S314, S319, S320, T323, S326, S338, S344, S363, T367, S368, T369, S419, and S444) [e.g. (Guettouche et al., 2005; Anckar and Sistonen 2011; Xu et al., 2012; Gomez-Pastor et al., 2018; Roos-Mattjus and Sistonen 2021), summary in Table 1]. Some residues appear to be phosphorylated under basal conditions (Chu et al., 1996; Knauf et al., 1996; Kline and Morimoto 1997; Chu et al., 1998; He et al., 1998) whereas other sites undergo inducible phosphorylation (Holmberg et al., 2002). Stress induces phosphorylation at multiple residues (i.e., hyperphosphorylation) and is concurrent with transactivation (Cotto et al., 1996; Xia and Voellmy 1997; Holmberg et al., 2001; Holmberg et al., 2002; Guettouche et al., 2005).
Most phosphorylation events repress transcriptional activity. For example, sequential phosphorylation of S307 by mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) and S303 by glycogen synthase kinase 3 (GSK3) (Chu et al., 1996; Chu et al., 1998; He et al., 1998) recruits 14-3-3 proteins to export HSF1 from the nucleus and sequester it in the cytoplasm (Wang et al., 2003). Several serine residues are targeted by multiple kinases to repress HSF1 activity: acute inflammatory signals activate mitogen-activated protein kinase 2 (MAPK2) to phosphorylate HSF1 at S121, which promotes direct interaction with HSP90, subsequently reducing binding to DNA at heat shock elements and therefore lessening the activation of corresponding gene expression (Wang et al., 2006). This same residue is also phosphorylated by AMPK under conditions of metabolic stress to impair HSF1 activity by impeding nuclear translocation, thereby rendering cells sensitive to proteotoxic stress (Dai et al., 2015).
HSF1 (S363) phosphorylation also inhibits activity and is the target of both protein kinase C (PKC) and c-Jun N-terminal kinase (JNK). PKC was shown to inhibit HSF1 without affecting trimerization or binding to heat shock elements, and it may limit HSF1 activity during increases in PKC activity, such as after activation by growth receptors (Chu et al., 1998). JNK, a stress-responsive member of the MAPK pathway, also inactivates HSF1 through S363 phosphorylation, which rapidly clears HSF1 from the sites of transcription (Dai et al., 2000). Thus, the outcome of HSF1 (S363) phosphorylation on DNA binding is context-dependent. JNK has also been shown to phosphorylate the TAD of HSF1 after severe heat shock, and JNK inhibitors prevent the activation of HSF1 target genes, but whether this is occurring at S363 is unclear (Park and Liu 2001). Collectively, these findings indicate that diverse negative regulatory signals can converge on a common HSF1 residues to limit activity.
HSF1 phosphorylation can also activate transcription, which has been best-studied in the context of hyperphosphorylation due to acute heat stress. By mass spectrometry, twelve serine residues were identified that are phosphorylated after heat stress (Guettouche et al., 2005). During heat stress, Polo-like kinase (PLK1), one of the major protein kinases involved in cell division, and specifically in APC/C regulation, directly phosphorylates HSF1 on S419, and a S419A mutation inhibits HSF1 translocation to the nucleus, suggesting this is an early step in HSF1 activation (Kim et al., 2005). However, PLK1 also phosphorylates HSF1 (S216) in early mitosis, facilitating degradation, which is essential for proper mitotic progression (Lee et al., 2008). Thus, PLK1 can regulate HSF1 in opposing manners through phosphorylation of different residues in response to distinct signals. Similar to PLK1 action at HSF1 (S419), protein kinase A (PKA) phosphorylates HSF1 (S320) in response to heat shock, and this modification is required for translocation to the nucleus, DNA binding at HSE, and to activate expression of molecular chaperones (Murshid et al., 2010; Zhang et al., 2011). Casein kinase 2 phosphorylates HSF1 on T142 and mutation to alanine (T142A) inhibits trans-activation of HSP70 by HSF1 and blocks binding to HSE, without affecting translocation to the nucleus (Soncin et al., 2003).
Both S230 and S326 have also been shown to be essential for the increased HSF1 transcriptional activity in response to acute heat shock (Holmberg et al., 2001; Guettouche et al., 2005; Tang et al., 2015). S230 lies within a consensus site for calcium/calmodulin-dependent protein kinase II (CaMKII), and CaMKII overexpression enhances both the level of in vivo S230 phosphorylation and transactivation of HSF1 (Holmberg et al., 2001). Furthermore, S230 is not needed for either the heat-induced DNA-binding activity or granule formation but is essential for the transcriptional activity of HSF1 (Holmberg et al., 2001). HSF1 (S326) has been shown to be directly phosphorylated by mTOR, and inhibition of mTOR limits induction of molecular chaperones (Chou et al., 2012), yet it is unclear whether mTOR is essential for HSF1 (S326) phosphorylation after heat stress. mTOR is a major regulator of cellular growth and translation, which suggests that mTOR regulation of HSF1 activity may directly balance the total protein abundance within the proteome and molecular chaperone abundance in response to cell size. Interestingly, MAPK/ERK Kinase, or mitogen-activated protein kinase (MEK), phosphorylates S326 both in vitro and in vivo after heat stress to activate and stabilize HSF1 to preserve proteostasis (Tang et al., 2015). Furthermore, under heat stress, ERK, MEK, and HSF1 assemble into a ternary protein complex wherein ERK suppresses HSF1 (S326) phosphorylation through inhibitory phosphorylation of MEK.
The emerging picture of regulation of mammalian HSF1 through phosphorylation is one of an integrated, combinatorial process. Indeed, in contrast to the aforementioned studies where HSF1 function was compromised after mutating a single phosphorylation site, an HSF1 isoform with 15 phosphorylation sites mutated, including residues targeted by both inhibitory and stimulatory signals, was still able to translocate to the nucleus, bind DNA, and activate transcription (Budzynski et al., 2015). Thus, HSF1 activity is carefully balanced between positive and negative regulators, which is likely dependent on cell type and context. While in C. elegans HSF-1 has been shown to undergo phosphorylation (Chiang et al., 2012), only a fraction of modified amino acids in mammalian HSF1 are directly conserved (Supplementary File S1). Furthermore, the molecular and cellular details of regulation, and subsequent consequences on stress response, proteostasis, and longevity in an intact multicellular organism remains underexplored.
Ubiquitination of HSF1 and Degradation via the UPS
HSF1 protein levels are also regulated via degradation by the UPS. HSF1 can be ubiquitinated and degraded by the Skp1–Cul1–F box (SCF) ubiquitin ligase complex (Skaar et al., 2013). Ubiquitination of HSF1 by the SCF complex occurs during mitosis upon phosphorylation of HSF1 (S216) by PLK1, which releases HSF1 from the Cdc20 complex (Lee et al., 2008). In another study, the SCF complex was shown to target HSF1 for degradation via F-box and WD repeat domain containing protein 7 alpha (FBXW7α), a substrate-targeting subunit of the SCF complex. Interestingly, interaction occurs through a conserved degron motif phosphorylated by GSK3β and ERK1; FBXW7α ubiquitylates HSF1, and loss of FBXW7α results in impaired degradation of nuclear HSF1 and defective HSR attenuation (Kourtis et al., 2015). This suggests the possibility that distinct stimuli converge through phospho-regulation of HSF1 to recruit the SCF complex and target HSF1 for degradation. HSF1 may be transported to the proteasome through the Filamin A interacting protein 1-like (FILIP-1L) protein, which has been found in a complex with HSF1, Hsp72, and the ubiquitin-binding domain of hHR23A, a receptor that transports polyubiquitinated proteins to the proteasome for degradation; cells co-expressing HSF1 and FILIP-1L exhibit reduction in the HSF1 protein levels and inhibition of stress granule formation following exposure to heat shock (Hu and Mivechi 2011).
Sumoylation of HSF1
HSF1 is negatively regulated through sumoylation of HSF1 in both C. elegans and mammalian cell culture (Hong et al., 2001; Hietakangas et al., 2003; Anckar et al., 2006; Hietakangas et al., 2006; Das et al., 2017). SUMO protein catalyzes a small ubiquitin like modification, which frequently targets transcription factors to limit activity (Gill 2005; Cubenas-Potts and Matunis 2013; Deyrieux and Wilson 2017; Wotton et al., 2017). Mammals have four genes encoding SUMO, while C. elegans possess a single gene, smo-1 (Kamitani et al., 1998; Jones et al., 2002). In mammals, mild heat stress results in HSF1 (S303) phosphorylation, which promotes subsequent SUMO-1 addition to HSF1 (K298) and limits HSF1 transcriptional activation. More severe and prolonged heat stress results in desumoylation and increased expression of heat shock genes (Hietakangas et al., 2003; Anckar et al., 2006). Thus, sumoylation has been proposed to act as a mechanism to fine-tune HSF1 activity to levels of acute proteotoxic stress (i.e., protein misfolding). Mapping of the human SUMO proteome has identified many additional lysine residues of HSF1 that putatively undergo sumoylation, but the biological significance of these modifications remains unknown (Hendriks et al., 2017; Gomez-Pastor et al., 2018; Roos-Mattjus and Sistonen 2021). In C. elegans, sumoylation also limits the inducibility of the HSR (Das et al., 2017), indicating that this mechanism regulates HSF-1 activity in vivo, as well as in mammalian cell culture. Consistently, loss of the SUMO isopeptidase ulp-1 shortens lifespan under conditions of mild heat stress (Samuelson et al., 2007). Additionally, the transcriptional cofactor HPK-1 prevents HSF1 sumoylation under basal conditions (Das et al., 2017). Interestingly the yeast ortholog of HPK-1, Yak1, which directly phosphorylates HSF1 in response to altered metabolic conditions, is induced by heat stress and is required for thermal stress survival (Garrett et al., 1991; Hartley et al., 1994), suggesting that a regulatory role of HPK-1 upon HSF1 is evolutionarily conserved.
Acetylation of HSF1
HSF1 is also regulated through acetylation. At least nine lysine residues of HSF1 have been shown to be acetylated (Table 1). Perhaps the most well-studied is K80 acetylation of HSF1; the transcriptional co-activator p300/CBP (CREB-binding protein) acetylates HSF1 (K80) to attenuate the HSR by inhibiting DNA binding. This activity is opposed by the deacetylase activity of SIRT1: downregulation of SIRT1 resulting in the weaker induction of molecular chaperones due to greater acetylation of HSF1, which prevents binding to HSE (Westerheide et al., 2009; Raychaudhuri et al., 2014). Interestingly, acetylation of HSF1 has been found to both increase and decrease stability (Westerheide et al., 2009; Kim et al., 2016); acetylation at different positions alters the HSF1 stability. Specifically, acetylation of HSF1 at position K118 and K80 lead to attenuation of the HSR due to HSF1 degradation by the ubiquitin-proteasome. In contrast, acetylation at position K208 and K298 stabilizes HSF1 (Raychaudhuri et al., 2014). As SIRT1 is a critical deacetylase of HSF1, it is tempting to speculate that SIRT1 may promote HSF1 stability or degradation depending on the position of acetylation.
Additional Direct Regulators of Mammalian HSF1
A number of additional proteins have been identified that regulate HSF1 activity through direct interactions. For example, heat shock factor binding protein 1 (Hsbp1) is a negative regulator of HSF1 activity (Satyal et al., 1998). CHIP, Ral-binding protein 1 (RalBP1), and Death-associated protein 6 (Daxx) function in the activation of HSF1 (Dai et al., 2003; Hu and Mivechi 2003; Boellmann et al., 2004). Metastasis-associated protein 1 (MTA1) and Cdc20 are additional HSF1 regulators (Khaleque et al., 2008; Lee et al., 2008).
Control of Gene Expression by HSF1
HSF1 preserves proteome vitality by commanding a transcriptional program whose physiological purpose is to maintain proper folding and function of the proteome in the face of both global and localized forms of protein misfolding stress. This transcriptional program marshals multiple chaperone systems when protein homeostasis is compromised, either in response to intrinsic or extrinsic cues. In addition to regulating the HSR, HSF1 activity is also responsive to metabolic and mitogenic signals and plays an important role in development and organismal longevity [reviewed in (Li et al., 2017)]. Additionally, HSF1 participates in physiological and pathological processes including: differentiation, immune response, multidrug resistance, longevity, neurodegeneration, and cancer. Interestingly, recent study comprehensively cataloged all known HSF1 target genes and preformed an enrichment analysis of HSF1 targets across tissues, cell types, and organisms (hsf1base.org) (Kovacs et al., 2019) and found that HSF1 targets, expressed in all tissues and cell types, are generally related to maintaining proteostasis. Furthermore, HSF1 targets that are conserved across various animal taxa operate mostly in cellular stress pathways (e.g., autophagy), chromatin remodeling, ribosome biogenesis, and aging; highlighting the diverse roles for HSF1 in regulating gene expression.
The Heat Shock Response (Unfolded Protein Response in the Cytosol and Nucleus)
The “heat shock response,” defined as the rapid induction of heat shock proteins, was initially described in Drosophila 60 years ago (Ritossa 1962). We now know that the HSR is an ancient genetic program shared across all organisms and constitutes one key component of a larger network that responses to stress on the proteome. The HSR could more accurately be described as the Unfolded Protein Response to proteotoxic stress within the cytosol and nucleus, analogous to the mitoUPR and ER-UPR.
Protein stability, and therefore normal cellular function, are highly sensitive to changes in temperature. Acute heat stress not only denatures and aggregates proteins, but also damages the cytoskeleton, breaks down organelles such as Golgi apparatus and the ER, diminished the numbers of functional mitochondria and lysosomes, and produces cytoplasmic stress granules. The cellular consequences of this damage are: a collapse of actin and microtubule networks, disruption of intracellular transport, decreased availability of ATP, a global decrease in translation, a drop in cytosolic pH, and cell cycle arrest [reviewed in (Richter et al., 2010)]. Due to the inherent danger heat has on cellular function, it is not surprising that transcriptional programs evolved early in evolution to respond to heat. Life exists in a wide range of temperatures, for example Pyrodictium abyssi grows in hot vents over 100°C, and species of the Thermoproteus genus live in boiling mud. Yet, shifting Pyrodictium occultum from 102°C to 108°C induces transcriptional changes in response to heat stress (Stetter 2006). Thus, organisms thrive in only a narrow temperature range, and shifts of only a few degrees induces a universal and ancient transcriptional response to heat: the “heat shock response” (Brown and Lupas 1998; Takai et al., 1998; D'Amico et al., 2006; Richter et al., 2010).
Previous transcriptional and proteomic studies have identified a vast number of heat-inducible genes, which are involved in diverse cellular processes. The HSR not only induces the expression of molecular chaperones, but also: increases protein degradation via autophagy and expression of proteasome subunits, promotes stabilization of cellular energetics via altered expression of metabolic enzymes, inhibits unnecessary processes through the activation of additional regulatory proteins, induces repair of DNA/RNA and changes in gene expression to sustain cellular structures, and repairs membranes to restore transport and detoxification within the cell [reviewed in (Richter et al., 2010)].
HSF1 also restores proteostasis after stress by increased expression of genes involved in autophagy. As previously mentioned, autophagy is a crucial protagonist of the proteostasis network that functions to recycle cytosolic components after stress, including: toxic protein aggregates, nutrient deprivation, hypoxia, and damaged organelles, among others (Kroemer et al., 2010). In C. elegans, heat shock and hsf-1 overexpression induce autophagy in multiple tissues (Kumsta et al., 2017). In mammals, HSF1 regulates the phosphorylation and activity of the SQSTM1/p62 autophagy receptor, suggesting that the HSF1 stress response pathway is involved in autophagic clearance of protein aggregates (Watanabe et al., 2017). Moreover, HSF1 controls autophagy activity induced by chemotherapeutic agents by regulating the transcription of autophagy-related protein 7 (ATG7) (Desai et al., 2013).
The Role of HSF1 in Development is Distinct From the HSR
Periods of rapid growth during development require proteome expansion, in turn demanding an expansion of the PN to regulate developmental processes. Certain windows during development demand an excess amount of energy and nutritional resources. Organisms consequently experience stress during stages where meeting these needs requires divergence from optimal developmental trajectory (Puscheck et al., 2015). In response to stress, HSF1, along with mammalian paralogs HSF2 and HSF3, acts to compensate and ensure survival of the developing organism (Akerfelt et al., 2010; Puscheck et al., 2015). The role of HSF1 in development with and without canonical heat shock stressors is still under ongoing investigation in other model systems.
In Drosophila, mutations in a single base of the HSF coding sequence causes arrest at the first or second larval instar stage of development. However, HSF mutations induced past these larval stages do not affect cell growth or viability under normal conditions, suggesting that HSF is only required in the early stages of development in Drosophila (Table 2). Additionally, the expression of canonical heat shock genes did not change in these mutants, suggesting that the developmental role of HSF1 could be distinct from the HSR (Jedlicka et al., 1997).

TABLE 2. HSF1 loss and organismal development.
The developmental transcriptional program of HSF-1 in C. elegans is distinct from the canonical HSR (Li et al., 2016; Li et al., 2017). hsf-1 null mutants arrest at the L2-L3 larval stage of development (Table 2). Activation of HSF-1 during development depends on a GC-rich E2F/DP transcription factor binding to a motif that allows HSF-1 to bind to a heat shock element distinct from the classical HSR. Through this, E2F and HSF-1 facilitate regulation of biogenesis and anabolic metabolism during development. However, loss of hsf-1 also results in lower basal levels of molecular chaperones (Chiang et al., 2012). Additionally, knockdown of components of the mTOR pathway can rescue these defective developmental phenotypes. Specifically, rescue utilizing either knockdown of the TORC1 component daf-15 (Raptor), a positive regulator of mTORC1 ragc-1 (orthologous to RAG GTPase), or loss of the downstream effector that regulates rates of translation; rsks-1 (ortholog of S6 Kinase), prevented developmental arrest (Chisnell et al., 2018). This implies that decreased rates of protein synthesis resulting from mTORC1 inactivation mitigates damage to the proteome associated with loss of HSF function. Alternatively, decreased mTORC1 activity may rescue hsf-1 developmental defects resulting from diminished basal chaperone expression, at least in part by reducing either the total concentration or specific components of the cellular proteome.
HSF-1 Integrates Diverse Metabolic and Stress Signals to Preserve Proteostasis and Longevity
HSF-1 Preserves Longevity
HSF-1 has emerged as a key regulator of organismal longevity through the integration of signals of cellular energy metabolism and diverse forms of stress. This has been well studied in C. elegans; loss of hsf-1 shortens lifespan, impairs survival to a diverse array of cellular stresses, and compromises proteostasis. Conversely, hsf-1 overexpression increases lifespan, stress resistance, and delays age-associated proteostatic decline (Garigan et al., 2002; Hsu et al., 2003; Morley and Morimoto 2004; Kourtis et al., 2012; Das et al., 2017). Recently, it has been shown that HSF-1 requires the transcriptional cofactor hpk-1 to extend longevity, to induce molecular chaperones after thermal stress, to enhance hormetic extension of longevity, and is required in conjunction with HSF-1 for maintenance of proteostasis (Das et al., 2017). HPK-1 antagonizes sumoylation of HSF-1 and inhibiting sumoylation increases the induction of molecular chaperones after heat shock (Das et al., 2017). While persistent heat stress is detrimental to nematode survival, either intermittent heat shock or mild hormetic heat shock also extends longevity via HSF-1 activation (Das et al., 2017; Kumsta et al., 2017). It is generally believed that hormesis extends longevity by bolstering organismal and cellular stress response pathways, which subsequently offsets aging-related decline in these pathways (Epel and Lithgow 2014).
How HSF-1 extends longevity remains an active area of investigation. Early work suggested that HSF-1 delays aging through expression of molecular chaperones, as overexpression of molecular chaperones can suppress polyglutamine aggregation in body wall muscle and increase C. elegans lifespan (Satyal et al., 2000; Hsu et al., 2003; Walker and Lithgow 2003). However, emerging evidence suggesting a more complex picture. First, there are conflicting reports as to whether a hypomorphic hsf-1 allele (premature stop codon removing the transactivation domain) sensitizes C. elegans to heat stress, despite having an impaired HSR (Prahlad et al., 2008; McColl et al., 2010). Interestingly, overexpression of a mutant HSF-1 lacking the transactivation domain is able to increase thermotolerance and lifespan through maintaining cytoskeletal integrity, despite being impaired in the ability to induce molecular chaperones after heat shock (Baird et al., 2014), but it is possible that the truncated hsf-1 isoform is a neomorph. Another possibility is that HSF-1 may extend longevity through links to other components of the PN, such as the regulation of autophagy (Kumsta et al., 2017). As previously mentioned, efforts to directly link the genetic interactions and cell biological activity of HSF-1 in C. elegans to specific HSF1 post-translational modifications that occur in mammalian cells is still lacking, yet it is widely postulated that HSF-1 functions that extend longevity will be the same as those that preserve proteostasis, and by extension possibly delay aging and prevent the manifestation of neurodegenerative age-associated proteotoxic diseases in humans.
HSF-1 Functions Cell Non-Autonomously to Regulate Proteostasis, Stress Response and Longevity
One transformative discovery in C. elegans was the finding that HSF-1 functions cell non-autonomously within neurons to increase longevity and maintain proteostasis in distal tissues (Morley and Morimoto 2004; Prahlad et al., 2008; Prahlad and Morimoto 2011; Douglas et al., 2015), and that this occurs through serotonin signaling (Tatum et al., 2015). Increased neuronal expression of HSF-1 is sufficient to extend longevity and improve stress resistance. Interestingly, signals that increase the HSR in peripheral tissues through thermosensory neuronal circuits are separable from those that increase longevity (Douglas et al., 2015). In accordance to these findings, another study identifies that integrin-linked kinase (ILK) inhibition activates HSF-1 cell non-autonomous effect on stress resistance and lifespan in a thermosensory-dependent manner (Kumsta et al., 2014). Of note, HSF-1 acts in multiple tissues to regulate longevity (Morley and Morimoto 2004) and also acts cell non-autonomously outside of the nervous system. For example, intestinal HSF-1 activity upregulates the mir-83/miR-29 secreted microRNA to disrupt macroautophagy both within intestinal and body wall muscle (Zhou et al., 2019). However somewhat paradoxically, hormetic heat shock to activate HSF-1 or HSF-1 overexpression induces autophagy in multiple tissues (Kumsta et al., 2017), yet whether this occurs cell intrinsically or non-autonomously was not explored. Altogether, this implies that strategies to target HSF-1 in the treatment of disease should consider both cell intrinsic changes and effects in distal tissues. It will be interesting to learn whether pro-longevity functions of HSF-1 that are independent of molecular chaperone induction, can improve neuronal proteostasis but not protect cancer cells from chronic proteotoxic stress.
HSF-1 Integrates Metabolic Signals to Extend C. elegans Longevity
The most potent influencers of C. elegans longevity sense changes in metabolic status, which in turn leads to the activation of cytoprotective stress response and adaptive transcriptional programs, including the PN. HSF-1 is essential for many of these metabolic pathways or signals to extend longevity: including decreased ILS (Hsu et al., 2003; Morley and Morimoto 2004), germline deficiency (Hansen et al., 2005), reduced TORC1 signaling or inhibition of rsks-1 (Seo et al., 2013), and dietary deprivation (a distinct method of C. elegans DR) (Steinkraus et al., 2008). Furthermore, HSF-1 is subjected to complex regulation at times of simultaneously applying thermal stress and DR, through the integrin-linked kinase PAT-4 (human integrin linked kinase) and the deacetylase SIR-2.1 (Raynes et al., 2012; Kumsta et al., 2014).
Not all longevity signals are dependent on HSF-1, or at least the interrelationship between longevity signaling and HSF-1 are complex. For instance, early work on disruption of the electron transport chain showed increased lifespan independently of hsf-1 (Hsu et al., 2003). However, more recent work showed lifespan extension of HSB-1/HSF-1 signaling could be in part through modulation of mitochondrial function via mediating histone H4-dependent regulation of mtDNA gene expression (Sural et al., 2020), indicating that the complex signaling interactions between the mitochondrial and nuclear genomes is not fully understood. Another example is AMPK and HSF1. In mammals, AMPK activation directly phosphorylates HSF1 to suppress proteotoxic stress response and conversely, either proteotoxic stress or HSF1 itself inactivates AMPK (Dai et al., 2015; Su et al., 2019). In C. elegans as AMPK lifespan extension appears to be independent of HSF-1 (Greer and Brunet 2009; Hwang et al., 2014; Kuo et al., 2020). It is unclear whether this regulatory mechanism is simply not conserved, restricted to specific cell types, or compensated for when examined in a multicellular organism.
As discussed above, HSF-1 is required for a specific type of DR (referred to as dietary deprivation) to increase lifespan in C. elegans (Hsu et al., 2003; Greer and Brunet 2009). HSF-1 is integral in the extension of longevity under this specific form of DR, but surprisingly not the other methods of DR, indicating that HSF-1 plays a specific role. For example, eat-2 mutant animals are a genetic model of DR: animals have reduced acetylcholine channel activity, which results in decreased pharyngeal pumping, increased lifespan, and stress resistance. The increased lifespan of eat-2 animals is hsf-1 independent (Hsu et al., 2003). In contrast, the increased stress resistance of eat-2 mutant animals requires hsf-1 (Shpigel et al., 2019). However, “dietary restriction” is a vague term that attempts to capture all forms of nutritional stress, and multiple methods for inducing DR have been described. Since empirical findings indicate that distinct types of DR have many different genetic requirements, it suggests one of the possible explanations: 1) different feeding regiments or nutrient uptake either titrate a similar signal, and differing genetic requirements are observed when threshold effects trigger a specific response, 2) different types of DR trigger distinct forms of nutrient stress, or 3) some types of DR may trigger multiple forms of nutrient stress, which enact combinatorial responses.
HSF-1 is an essential transcriptional effector of the increased longevity in ILS mutant animals (Hsu et al., 2003; Morley and Morimoto 2004). In wild type animals, DAF-2 inhibits HSF-1 activity through DDL-1 and DDL-2 (Chiang et al., 2012). DDL-1 is homologous to the human coiled-coil domain-containing protein 53 (CCDC53), and DDL-2 is the homolog of human Wiskott-Aldrich syndrome protein and SCAR homolog (WASH2) protein. WASH2 and CCDC53 are both components of the Arp2/3 complex involved in actin polymerization, and intracellular motility of endosomes (Derivery et al., 2009). CCDC53 has also been reported to potentially interact with heat shock factor binding protein 1 (HSBP1) (Rual et al., 2005). Consistently, the Arp2/3 complex as well as multiple components involved in endocytic trafficking to the lysosome are essential for decreased ILS to extend longevity (Samuelson et al., 2007). As noted above, HSF-1 cooperates with DAF-16 through PFD-6, a component of the molecular chaperone prefoldin-like complex, which relays longevity signals from HSF-1 to FOXO under conditions of reduced ILS (Son et al., 2018).
As a key nutrient sensor for NAD + levels, SIRT1 deacetylates HSF1 during nutrient stress promoting stress survival, thus making the acetylation of HSF1 an integral part of sirtuin signaling (Westerheide et al., 2009; Raynes et al., 2013). Deacetylation of mammalian HSF1 frees up its DNA binding domain, allowing it to bind to a HSE, activate the HSR; and inhibition of SIRT1 accelerates the release from the HSE, decreases HSF1 protein expression, and activation of the HSR (Westerheide et al., 2009; Kim et al., 2016). In mammals SIRT1 is also required for maintenance of the proteome as SIRT1 deficiency results in defective protein quality control (Tomita et al., 2015), reinforcing the notion that SIRT1 regulation of HSF1 connects longevity signaling to the maintenance of proteostasis. HSF1 regulation by Sirtuins is conserved in Saccharomyces cerevisiae, and C. elegans (Anderson et al., 2003; Brunquell et al., 2018). In mammals, AROS (a positive regulator of SIRT1) increases deacetylation of HSF1, while CCAR2 and DBC1 negatively regulate SIRT1 dependent HSF1 deacetylation (Zhao et al., 2008; Raynes et al., 2013). In C. elegans, as CCAR-1 negatively regulates SIR-2.1, increasing HSF-1 acetylation and decreasing HSF-1 ability to regulate the HSR (Brunquell et al., 2018), demonstrating a conserved regulatory mechanism for HSF-1 activation. Furthermore, DR induces heat shock gene expression in a sir-2.1 dependent manner (Raynes et al., 2012). This suggests that signals that modulate Sirtuin function can affect HSF-1. However, increasing NAD + levels promotes longevity in C. elegans via sir-2.1, DAF-16 activation, and the mito-UPR, without changing hsf-1 expression (Mouchiroud et al., 2013). We could not find direct experimental evidence in C. elegans to demonstrate that the extended longevity conferred by sir-2.1 overexpression is hsf-1 dependent.
Mammalian HSF1 is directly activated by TOR [see above and reviewed in (Antikainen et al., 2017; Saxton and Sabatini 2017; Blackwell et al., 2019)]. Furthermore, experiments in yeast suggest that activated HSF1 might inhibit rapamycin resistance and TOR signaling (Bandhakavi et al., 2008). While a bulk of HSF1 and TOR interactions have been studied in cell culture and yeast, studies in C. elegans reveal that inactivation of TOR signaling increases lifespan in a manner dependent on hsf-1. Consistently, HSF-1 can also be activated with rapamycin treatment (Seo et al., 2013).
Loss of C. elegans germ cells cause a metabolic shift fat mobilization and lipolysis (Wang et al., 2008). This shift in results in aged animals assuming a younger metabolic transcriptional state (Wan et al., 2017). Gonadal stem cells are depleted in glp-1 mutant animals, which encodes the C. elegans ortholog of the NOTCH receptor, and glp-1 mutations increase lifespan dependent on hsf-1 (Hansen et al., 2005). Interestingly, shortly after the onset of reproduction the inducibility of the HRS drops dramatically (Labbadia and Morimoto 2015b), and HSF-1 forms nuclear stress bodies at the initiation of reproduction throughout the germline and upon transition to adulthood HSF-1 stress bodies form in most somatic cells (Deonarine et al., 2021). However, this repression of the HSR does not occur in glp-1 mutant animals (Shemesh et al., 2013) and genetic loss of the germline suppressed nuclear stress body formation with age (Deonarine et al., 2021), which suggests alterations in gonadal stem cell signaling cell non-autonomously reprograms somatic maintenance, at least in part through HSF-1 activity. Interestingly, genetic perturbation of the extracellular vitelline layer of an internally fertilized embryo initiates a transcellular signal to improve proteostasis and stress resistance in an HSF-1 dependent manner (Sala et al., 2020). Thus, HSF-1 links metabolic reprograming and stress signals from the germline to improved proteostasis in the Soma and extend longevity.
Future Perspectives
Science is an iterative process, where discoveries in one model system informs and generates new hypotheses best tested in others. Emerging technologies, such as clustered regularly interspaced short palindromic repeats (CRISPR) and single-cell RNA sequencing, along with the development of large datasets (e.g., human sequence data), and the field of systems biology hold the promise of translating discoveries made in model organisms to ultimately improve human healthspan. Conversely, model organisms provide a powerful platform to segregate causative mutations from correlations identified within human sequence variation; and can quickly elucidate gene function within an intact metazoan. The proteostasis field is currently undergoing a transformation thanks to the development of AlphaFold, an artificial intelligence program that can accurately predict protein structure (Jumper et al., 2021), which will ultimately provide a predictive iterative framework to merge findings in genetics, structural biology, and biochemistry. These are just a few examples of how advancing technical innovation in the biological sciences will be applied to further unify our understanding of how aging negatively impacts biological systems.
Work in C. elegans has been instrumental in understanding the basis of aging, and is well positioned to remain a premiere system of discovery for years to come. For example, in the specific area of transcriptional control of proteostasis, HSF1 regulation through multiple post-translational modifications and direct interactors, reveals the underlying complexity required to maintain proteome function, yet surprisingly little is known about the specific HSF-1 residues that modified within an intact multicellular organism, such as C. elegans. Conversely, whether the longevity signals that converge on HSF-1 to delay C. elegans aging have analogous levels of regulation in mammals remains an area of exploration. Discovering how HSF-1 is regulated in a cell-type specific manner within an intact multicellular organism would provide insight into how extrinsic and intrinsic signals are integrated via HSF-1 to regulate the PN, and how this system breaks down during normal aging. As an integration point for diverse signals of metabolic, mitogenic, and proteotoxic stress; distinguishing between modifications acting as molecular switches, versus providing rheostat regulation to fine-tune gene expression levels, those responsible for selective gene expression and tissue specific regulation, could be paramount in guiding the development of strategies to extend human healthspan.
The past 30 years of aging research has led to a wealth of discovery into the major regulatory mechanisms that have a deterministic role on organismal aging, the molecular and cellular hallmarks of aging, and how organisms respond to myriad forms of stress. Major challenges will be to elucidate how organisms, as biological systems, integrate signals and coordinate adaptive responses to maintain homeostasis, how these processes breakdown during aging, and whether greater understanding will facilitate the development of strategies to maximize human health span and minimize the onset and progression of age-associated diseases.
Author Contributions
AVS conceived and wrote the manuscript. MIP, ZCW, SY, and AS made significant intellectual contributions and major contributions to writing specific sections of the manuscript. AM and CS made significant intellectual contributions and minor contributions to writing.
Funding
Development of this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Numbers RF1AG062593 and R21AG064519 granted to AVS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in design, analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The concept for this review was formulated by AVS as a workshop within his module of a Foundations Course (IND432) for incoming graduate students at the University of Rochester Medical Center. AnS would like to thank Dr. Scott Butler (URMC), the IND432 course director, for encouragement and support. We would also like to thank current and past members of the Samuelson laboratory for fruitful scientific discussions. We would like to thank the reviewers for their thoughtful comments, which greatly improved the quality of the manuscript. We sincerely apologize to authors of many relevant discoveries not cited in this review for reasons of space.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fragi.2022.861686/full#supplementary-material
References
Abravaya, K., Myers, M. P., Murphy, S. P., and Morimoto, R. I. (1992). The Human Heat Shock Protein Hsp70 Interacts with HSF, the Transcription Factor that Regulates Heat Shock Gene Expression. Genes Dev. 6, 1153–1164. doi:10.1101/gad.6.7.1153
Ahn, S.-G., and Thiele, D. J. (2003). Redox Regulation of Mammalian Heat Shock Factor 1 Is Essential for Hsp Gene Activation and Protection from Stress. Genes Dev. 17, 516–528. doi:10.1101/gad.1044503
Ailion, M., and Thomas, J. H. (2000). Dauer Formation Induced by High Temperatures in Caenorhabditis elegans. Genetics 156, 1047–1067. doi:10.1093/genetics/156.3.1047
Ailion, M., and Thomas, J. H. (2003). Isolation and Characterization of High-Temperature-Induced Dauer Formation Mutants in Caenorhabditis elegans. Genetics 165, 127–144. doi:10.1093/genetics/165.1.127
Åkerfelt, M., Morimoto, R. I., and Sistonen, L. (2010). Heat Shock Factors: Integrators of Cell Stress, Development and Lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555. doi:10.1038/nrm2938
Alastalo, T.-P., Hellesuo, M., Sandqvist, A., Hietakangas, V., Kallio, M., and Sistonen, L. (2003). Formation of Nuclear Stress Granules Involves HSF2 and Coincides with the Nucleolar Localization of Hsp70. J. Cell Sci. 116, 3557–3570. doi:10.1242/jcs.00671
Aman, Y., Schmauck-Medina, T., Hansen, M., Morimoto, R. I., Simon, A. K., Bjedov, I., et al. (2021). Autophagy in Healthy Aging and Disease. Nat. Aging 1, 634–650. doi:10.1038/s43587-021-00098-4
An, J. H., and Blackwell, T. K. (2003). SKN-1 Links C. elegans Mesendodermal Specification to a Conserved Oxidative Stress Response. Genes Dev. 17, 1882–1893. doi:10.1101/gad.1107803
Anckar, J., Hietakangas, V., Denessiouk, K., Thiele, D. J., Johnson, M. S., and Sistonen, L. (2006). Inhibition of DNA Binding by Differential Sumoylation of Heat Shock Factors. Mol. Cell Biol. 26, 955–964. doi:10.1128/mcb.26.3.955-964.2006
Anckar, J., and Sistonen, L. (2011). Regulation of HSF1 Function in the Heat Stress Response: Implications in Aging and Disease. Annu. Rev. Biochem. 80, 1089–1115. doi:10.1146/annurev-biochem-060809-095203
Anderson, R. M., Bitterman, K. J., Wood, J. G., Medvedik, O., and Sinclair, D. A. (2003). Nicotinamide and PNC1 Govern Lifespan Extension by Calorie Restriction in Saccharomyces cerevisiae. Nature 423, 181–185. doi:10.1038/nature01578
Antebi, A. (2013). Steroid Regulation of C. elegans Diapause, Developmental Timing, and Longevity. Curr. Top. Dev. Biol. 105, 181–212. doi:10.1016/b978-0-12-396968-2.00007-5
Antikainen, H., Driscoll, M., Haspel, G., and Dobrowolski, R. (2017). TOR-mediated Regulation of Metabolism in Aging. Aging Cell 16, 1219–1233. doi:10.1111/acel.12689
Apfeld, J., O'Connor, G., McDonagh, T., DiStefano, P. S., and Curtis, R. (2004). The AMP-Activated Protein Kinase AAK-2 Links Energy Levels and Insulin-like Signals to Lifespan in C. elegans. Genes Dev. 18, 3004–3009. doi:10.1101/gad.1255404
Arai, Y., Kojima, T., Takayama, M., and Hirose, N. (2009). The Metabolic Syndrome, IGF-1, and Insulin Action. Mol. Cell Endocrinol. 299, 124–128. doi:10.1016/j.mce.2008.07.002
Arantes-Oliveira, N., Apfeld, J., Dillin, A., and Kenyon, C. (2002). Regulation of Life-Span by Germ-Line Stem Cells in Caenorhabditis elegans. Science 295, 502–505. doi:10.1126/science.1065768
Ayyadevara, S., Alla, R., Thaden, J. J., and Shmookler Reis, R. J. (2008). Remarkable Longevity and Stress Resistance of Nematode PI3K-Null Mutants. Aging Cell 7, 13–22. doi:10.1111/j.1474-9726.2007.00348.x
Baird, N. A., Douglas, P. M., Simic, M. S., Grant, A. R., Moresco, J. J., Wolff, S. C., et al. (2014). HSF-1-mediated Cytoskeletal Integrity Determines Thermotolerance and Life Span. Science 346, 360–363. doi:10.1126/science.1253168
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting Proteostasis for Disease Intervention. Science 319, 916–919. doi:10.1126/science.1141448
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In Vivo aspects of Protein Folding and Quality Control. Science 353, aac4354. doi:10.1126/science.aac4354
Baler, R., Welch, W. J., and Voellmy, R. (1992). Heat Shock Gene Regulation by Nascent Polypeptides and Denatured Proteins: Hsp70 as a Potential Autoregulatory Factor. J. Cell Biol. 117, 1151–1159. doi:10.1083/jcb.117.6.1151
Baler, R., Zou, J., and Voellmy, R. (1996). Evidence for a Role of Hsp70 in the Regulation of the Heat Shock Response in Mammalian Cells. Cell stress chaperones 1, 33–39. doi:10.1379/1466-1268(1996)001<0033:efaroh>2.3.co;2
Bandhakavi, S., Xie, H., O'Callaghan, B., Sakurai, H., Kim, D. H., and Griffin, T. J. (2008). Hsf1 Activation Inhibits Rapamycin Resistance and TOR Signaling in Yeast Revealed by Combined Proteomic and Genetic Analysis. PLoS One 3, e1598. doi:10.1371/journal.pone.0001598
Bar-Lavan, Y., Shemesh, N., and Ben-Zvi, A. (2016). Chaperone Families and Interactions in Metazoa. Essays Biochem. 60, 237–253. doi:10.1042/ebc20160004
Ben-Zvi, A., Miller, E. A., and Morimoto, R. I. (2009). Collapse of Proteostasis Represents an Early Molecular Event in Caenorhabditis elegans Aging. Proc. Natl. Acad. Sci. U. S. A. 106, 14914–14919. doi:10.1073/pnas.0902882106
Benbrook, D. M., and Long, A. (2012). Integration of Autophagy, Proteasomal Degradation, Unfolded Protein Response and Apoptosis. Exp. Oncol. 34, 286–297.
Berendzen, K. M., Durieux, J., Shao, L. W., Tian, Y., Kim, H. E., Wolff, S., et al. (2016). Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 166, 1553–1563 e1510. doi:10.1016/j.cell.2016.08.042
Berman, J. R., and Kenyon, C. (2006). Germ-cell Loss Extends C. elegans Life Span through Regulation of DAF-16 by Kri-1 and Lipophilic-Hormone Signaling. Cell 124, 1055–1068. doi:10.1016/j.cell.2006.01.039
Biebl, M. M., and Buchner, J. (2019). Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 11. doi:10.1101/cshperspect.a034017
Bishop, N. A., and Guarente, L. (2007). Two Neurons Mediate Diet-Restriction-Induced Longevity in C. elegans. Nature 447, 545–549. doi:10.1038/nature05904
Bjork, J. K., and Sistonen, L. (2010). Regulation of the Members of the Mammalian Heat Shock Factor Family. FEBS J. 277, 4126–4139. doi:10.1111/j.1742-4658.2010.07828.x
Blackwell, T. K., Sewell, A. K., Wu, Z., and Han, M. (2019). TOR Signaling in Caenorhabditis elegans Development, Metabolism, and Aging. Genetics 213, 329–360. doi:10.1534/genetics.119.302504
Boellmann, F., Guettouche, T., Guo, Y., Fenna, M., Mnayer, L., and Voellmy, R. (2004). DAXX Interacts with Heat Shock Factor 1 during Stress Activation and Enhances its Transcriptional Activity. Proc. Natl. Acad. Sci. U. S. A. 101, 4100–4105. doi:10.1073/pnas.0304768101
Borghesan, M., Hoogaars, W. M. H., Varela-Eirin, M., Talma, N., and Demaria, M. (2020). A Senescence-Centric View of Aging: Implications for Longevity and Disease. Trends Cell Biol. 30, 777–791. doi:10.1016/j.tcb.2020.07.002
Brehme, M., Voisine, C., Rolland, T., Wachi, S., Soper, J. H., Zhu, Y., et al. (2014). A Chaperome Subnetwork Safeguards Proteostasis in Aging and Neurodegenerative Disease. Cell Rep. 9, 1135–1150. doi:10.1016/j.celrep.2014.09.042
Brignull, H. R., Moore, F. E., Tang, S. J., and Morimoto, R. I. (2006). Polyglutamine Proteins at the Pathogenic Threshold Display Neuron-specific Aggregation in a Pan-Neuronal Caenorhabditis elegans Model. J. Neurosci. 26, 7597–7606. doi:10.1523/jneurosci.0990-06.2006
Brown, J. R., and Lupas, A. N. (1998). What Makes a Thermophile? Trends Microbiol. 6, 349–350. doi:10.1016/s0966-842x(98)01351-1
Brunquell, J., Raynes, R., Bowers, P., Morris, S., Snyder, A., Lugano, D., et al. (2018). CCAR-1 Is a Negative Regulator of the Heat-Shock Response in Caenorhabditis elegans. Aging Cell 17, e12813. doi:10.1111/acel.12813
Budzynski, M. A., Puustinen, M. C., Joutsen, J., and Sistonen, L. (2015). Uncoupling Stress-Inducible Phosphorylation of Heat Shock Factor 1 from its Activation. Mol. Cell Biol. 35, 2530–2540. doi:10.1128/MCB.00816-14
Butcher, R. A., Fujita, M., Schroeder, F. C., and Clardy, J. (2007). Small-molecule Pheromones that Control Dauer Development in Caenorhabditis elegans. Nat. Chem. Biol. 3, 420–422. doi:10.1038/nchembio.2007.3
Calvo-Ochoa, E., and Arias, C. (2015). Cellular and Metabolic Alterations in the hippocampus Caused by Insulin Signalling Dysfunction and its Association with Cognitive Impairment during Aging and Alzheimer's Disease: Studies in Animal Models. Diabetes Metab. Res. Rev. 31, 1–13. doi:10.1002/dmrr.2531
Cassada, R. C., and Russell, R. L. (1975). The Dauerlarva, a Post-embryonic Developmental Variant of the Nematode Caenorhabditis elegans. Dev. Biol. 46, 326–342. doi:10.1016/0012-1606(75)90109-8
Chang, J. T., Kumsta, C., Hellman, A. B., Adams, L. M., and Hansen, M. (2017). Spatiotemporal Regulation of Autophagy during Caenorhabditis elegans Aging. Elife 6. doi:10.7554/eLife.18459
Chiang, W. C., Ching, T. T., Lee, H. C., Mousigian, C., and Hsu, A. L. (2012). HSF-1 Regulators DDL-1/2 Link Insulin-like Signaling to Heat-Shock Responses and Modulation of Longevity. Cell 148, 322–334. doi:10.1016/j.cell.2011.12.019
Chisnell, P., Parenteau, T. R., Tank, E., Ashrafi, K., and Kenyon, C. (2018). The mTOR Target S6 Kinase Arrests Development in Caenorhabditis elegans when the Heat-Shock Transcription Factor Is Impaired. Genetics 210, 999–1009. doi:10.1534/genetics.118.301533
Chondrogianni, N., Georgila, K., Kourtis, N., Tavernarakis, N., and Gonos, E. S. (2015). 20S Proteasome Activation Promotes Life Span Extension and Resistance to Proteotoxicity in Caenorhabditis elegans. FASEB J. 29, 611–622. doi:10.1096/fj.14-252189
Chou, S. D., Prince, T., Gong, J., and Calderwood, S. K. (2012). mTOR Is Essential for the Proteotoxic Stress Response, HSF1 Activation and Heat Shock Protein Synthesis. PLoS One 7, e39679. doi:10.1371/journal.pone.0039679
Chu, B., Soncin, F., Price, B. D., Stevenson, M. A., and Calderwood, S. K. (1996). Sequential Phosphorylation by Mitogen-Activated Protein Kinase and Glycogen Synthase Kinase 3 Represses Transcriptional Activation by Heat Shock Factor-1. J. Biol. Chem. 271, 30847–30857. doi:10.1074/jbc.271.48.30847
Chu, B., Zhong, R., Soncin, F., Stevenson, M. A., and Calderwood, S. K. (1998). Transcriptional Activity of Heat Shock Factor 1 at 37 Degrees C Is Repressed through Phosphorylation on Two Distinct Serine Residues by Glycogen Synthase Kinase 3 and Protein Kinases Calpha and Czeta. J. Biol. Chem. 273, 18640–18646. doi:10.1074/jbc.273.29.18640
Clark, D. A., Gabel, C. V., Gabel, H., and Samuel, A. D. (2007). Temporal Activity Patterns in Thermosensory Neurons of Freely Moving Caenorhabditis elegans Encode Spatial Thermal Gradients. J. Neurosci. 27, 6083–6090. doi:10.1523/jneurosci.1032-07.2007
Clos, J., Westwood, J. T., Becker, P. B., Wilson, S., Lambert, K., and Wu, C. (1990). Molecular Cloning and Expression of a Hexameric Drosophila Heat Shock Factor Subject to Negative Regulation. Cell 63, 1085–1097. doi:10.1016/0092-8674(90)90511-c
Cohen, E., and Dillin, A. (2008). The Insulin Paradox: Aging, Proteotoxicity and Neurodegeneration. Nat. Rev. 9, 759–767. doi:10.1038/nrn2474
Connell, P., Ballinger, C. A., Jiang, J., Wu, Y., Thompson, L. J., Hohfeld, J., et al. (2001). The Co-chaperone CHIP Regulates Protein Triage Decisions Mediated by Heat-Shock Proteins. Nat. Cell Biol. 3, 93–96. doi:10.1038/35050618
Cotto, J. J., Kline, M., and Morimoto, R. I. (1996). Activation of Heat Shock Factor 1 DNA Binding Precedes Stress-Induced Serine Phosphorylation. Evidence for a Multistep Pathway of Regulation. J. Biol. Chem. 271, 3355–3358. doi:10.1074/jbc.271.7.3355
Cubenas-Potts, C., and Matunis, M. J. (2013). SUMO: a Multifaceted Modifier of Chromatin Structure and Function. Dev. Cell 24, 1–12. doi:10.1016/j.devcel.2012.11.020
D'Amico, S., Collins, T., Marx, J. C., Feller, G., and Gerday, C. (2006). Psychrophilic Microorganisms: Challenges for Life. EMBO Rep. 7, 385–389. doi:10.1038/sj.embor.7400662
Dai, H., Sinclair, D. A., Ellis, J. L., and Steegborn, C. (2018). Sirtuin Activators and Inhibitors: Promises, Achievements, and Challenges. Pharmacol. Ther. 188, 140–154. doi:10.1016/j.pharmthera.2018.03.004
Dai, Q., Zhang, C., Wu, Y., McDonough, H., Whaley, R. A., Godfrey, V., et al. (2003). CHIP Activates HSF1 and Confers Protection against Apoptosis and Cellular Stress. EMBO J. 22, 5446–5458. doi:10.1093/emboj/cdg529
Dai, R., Frejtag, W., He, B., Zhang, Y., and Mivechi, N. F. (2000). c-Jun NH2-terminal Kinase Targeting and Phosphorylation of Heat Shock Factor-1 Suppress its Transcriptional Activity. J. Biol. Chem. 275, 18210–18218. doi:10.1074/jbc.m000958200
Dai, S., Tang, Z., Cao, J., Zhou, W., Li, H., Sampson, S., et al. (2015). Suppression of the HSF1-Mediated Proteotoxic Stress Response by the Metabolic Stress Sensor AMPK. EMBO J. 34, 275–293. doi:10.15252/embj.201489062
Das, R., Melo, J. A., Thondamal, M., Morton, E. A., Cornwell, A. B., Crick, B., et al. (2017). The Homeodomain-Interacting Protein Kinase HPK-1 Preserves Protein Homeostasis and Longevity through Master Regulatory Control of the HSF-1 Chaperone Network and TORC1-Restricted Autophagy in Caenorhabditis elegans. PLoS Genet. 13, e1007038. doi:10.1371/journal.pgen.1007038
David, D. C., Ollikainen, N., Trinidad, J. C., Cary, M. P., Burlingame, A. L., and Kenyon, C. (2010). Widespread Protein Aggregation as an Inherent Part of Aging in C. elegans. PLoS Biol. 8, e1000450. doi:10.1371/journal.pbio.1000450
Dayalan Naidu, S., and Dinkova-Kostova, A. T. (2017). Regulation of the Mammalian Heat Shock Factor 1. FEBS J. 284, 1606–1627. doi:10.1111/febs.13999
Denzel, M. S., Lapierre, L. R., and Mack, H. I. D. (2019). Emerging Topics in C. elegans Aging Research: Transcriptional Regulation, Stress Response and Epigenetics. Mech. Ageing Dev. 177, 4–21. doi:10.1016/j.mad.2018.08.001
Deonarine, A., Walker, M. W. G., and Westerheide, S. D. (2021). HSF-1 Displays Nuclear Stress Body Formation in Multiple Tissues in Caenorhabditis elegans upon Stress and Following the Transition to Adulthood. Cell stress chaperones 26, 417–431. doi:10.1007/s12192-020-01188-9
Depuydt, G., Xie, F., Petyuk, V. A., Shanmugam, N., Smolders, A., Dhondt, I., et al. (2013). Reduced Insulin/insulin-like Growth Factor-1 Signaling and Dietary Restriction Inhibit Translation but Preserve Muscle Mass in Caenorhabditis elegans. Mol. Cell Proteomics 12, 3624–3639. doi:10.1074/mcp.m113.027383
Derivery, E., Sousa, C., Gautier, J. J., Lombard, B., Loew, D., and Gautreau, A. (2009). The Arp2/3 Activator WASH Controls the Fission of Endosomes through a Large Multiprotein Complex. Dev. Cell 17, 712–723. doi:10.1016/j.devcel.2009.09.010
Desai, S., Liu, Z., Yao, J., Patel, N., Chen, J., Wu, Y., et al. (2013). Heat Shock Factor 1 (HSF1) Controls Chemoresistance and Autophagy through Transcriptional Regulation of Autophagy-Related Protein 7 (ATG7). J. Biol. Chem. 288, 9165–9176. doi:10.1074/jbc.m112.422071
Deyrieux, A. F., and Wilson, V. G. (2017). Sumoylation in Development and Differentiation. Adv. Exp. Med. Biol. 963, 197–214. doi:10.1007/978-3-319-50044-7_12
Dillin, A., Gottschling, D. E., and Nystrom, T. (2014). The Good and the Bad of Being Connected: the Integrons of Aging. Curr. Opin. Cell Biol. 26, 107–112. doi:10.1016/j.ceb.2013.12.003
Dorman, J. B., Albinder, B., Shroyer, T., and Kenyon, C. (1995). The Age-1 and Daf-2 Genes Function in a Common Pathway to Control the Lifespan of Caenorhabditis elegans. Genetics 141, 1399–1406. doi:10.1093/genetics/141.4.1399
Douglas, P. M., Baird, N. A., Simic, M. S., Uhlein, S., McCormick, M. A., Wolff, S. C., et al. (2015). Heterotypic Signals from Neural HSF-1 Separate Thermotolerance from Longevity. Cell Rep. 12, 1196–1204. doi:10.1016/j.celrep.2015.07.026
Dues, D. J., Andrews, E. K., Schaar, C. E., Bergsma, A. L., Senchuk, M. M., and Van Raamsdonk, J. M. (2016). Aging Causes Decreased Resistance to Multiple Stresses and a Failure to Activate Specific Stress Response Pathways. Aging (Albany NY) 8, 777–795. doi:10.18632/aging.100939
Dufey, E., Sepulveda, D., Rojas-Rivera, D., and Hetz, C. (2014). Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. 1. An Overview. Am. J. Physiol. Cell Physiol. 307, C582–C594. doi:10.1152/ajpcell.00258.2014
Duina, A. A., Kalton, H. M., and Gaber, R. F. (1998). Requirement for Hsp90 and a CyP-40-type Cyclophilin in Negative Regulation of the Heat Shock Response. J. Biol. Chem. 273, 18974–18978. doi:10.1074/jbc.273.30.18974
Durieux, J., Wolff, S., and Dillin, A. (2011). The Cell-Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell 144, 79–91. doi:10.1016/j.cell.2010.12.016
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 331, 456–461. doi:10.1126/science.1196371
Epel, E. S., and Lithgow, G. J. (2014). Stress Biology and Aging Mechanisms: toward Understanding the Deep Connection between Adaptation to Stress and Longevity. J. Gerontol. A Biol. Sci. Med. Sci. 69 (Suppl. 1), S10–S16. doi:10.1093/gerona/glu055
Farkas, T., Kutskova, Y. A., and Zimarino, V. (1998). Intramolecular Repression of Mouse Heat Shock Factor 1. Mol. Cell Biol. 18, 906–918. doi:10.1128/mcb.18.2.906
Felix, M. A., and Duveau, F. (2012). Population Dynamics and Habitat Sharing of Natural Populations of Caenorhabditis elegans and C. briggsae. BMC Biol. 10, 59. doi:10.1186/1741-7007-10-59
Fielenbach, N., and Antebi, A. (2008). C. elegans Dauer Formation and the Molecular Basis of Plasticity. Genes Dev. 22, 2149–2165. doi:10.1101/gad.1701508
Finley, D. (2009). Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 78, 477–513. doi:10.1146/annurev.biochem.78.081507.101607
Frezal, L., and Felix, M. A. (2015). C. elegans outside the Petri Dish. Elife 4. doi:10.7554/eLife.05849
Friedman, D. B., and Johnson, T. E. (1988). A Mutation in the Age-1 Gene in Caenorhabditis elegans Lengthens Life and Reduces Hermaphrodite Fertility. Genetics 118, 75–86. doi:10.1093/genetics/118.1.75
Garcia, S. M., Casanueva, M. O., Silva, M. C., Amaral, M. D., and Morimoto, R. I. (2007). Neuronal Signaling Modulates Protein Homeostasis in Caenorhabditis elegans Post-synaptic Muscle Cells. Genes Dev. 21, 3006–3016. doi:10.1101/gad.1575307
Garigan, D., Hsu, A. L., Fraser, A. G., Kamath, R. S., Ahringer, J., and Kenyon, C. (2002). Genetic Analysis of Tissue Aging in Caenorhabditis elegans: a Role for Heat-Shock Factor and Bacterial Proliferation. Genetics 161, 1101–1112. doi:10.1093/genetics/161.3.1101
Garrett, S., Menold, M. M., and Broach, J. R. (1991). The Saccharomyces cerevisiae YAK1 Gene Encodes a Protein Kinase that Is Induced by Arrest Early in the Cell Cycle. Mol. Cell Biol. 11, 4045–4052. doi:10.1128/mcb.11.8.4045-4052.1991
Gems, D., Sutton, A. J., Sundermeyer, M. L., Albert, P. S., King, K. V., Edgley, M. L., et al. (1998). Two Pleiotropic Classes of Daf-2 Mutation Affect Larval Arrest, Adult Behavior, Reproduction and Longevity in Caenorhabditis elegans. Genetics 150, 129–155. doi:10.1093/genetics/150.1.129
Ghazi, A., Henis-Korenblit, S., and Kenyon, C. (2007). Regulation of Caenorhabditis elegans Lifespan by a Proteasomal E3 Ligase Complex. Proc. Natl. Acad. Sci. U. S. A. 104, 5947–5952. doi:10.1073/pnas.0700638104
Gidalevitz, T., Ben-Zvi, A., Ho, K. H., Brignull, H. R., and Morimoto, R. I. (2006). Progressive Disruption of Cellular Protein Folding in Models of Polyglutamine Diseases. Science 311, 1471–1474. doi:10.1126/science.1124514
Gidalevitz, T., Prahlad, V., and Morimoto, R. I. (2011). The Stress of Protein Misfolding: from Single Cells to Multicellular Organisms. Cold Spring Harb. Perspect. Biol. 3. doi:10.1101/cshperspect.a009704
Gill, G. (2005). Something about SUMO Inhibits Transcription. Curr. Opin. Genet. Dev. 15, 536–541. doi:10.1016/j.gde.2005.07.004
Glover-Cutter, K. M., Lin, S., and Blackwell, T. K. (2013). Integration of the Unfolded Protein and Oxidative Stress Responses through SKN-1/Nrf. PLoS Genet. 9, e1003701. doi:10.1371/journal.pgen.1003701
Golden, J. W., and Riddle, D. L. (1984b). A Pheromone-Induced Developmental Switch in Caenorhabditis elegans: Temperature-Sensitive Mutants Reveal a Wild-type Temperature-dependent Process. Proc. Natl. Acad. Sci. U. S. A. 81, 819–823. doi:10.1073/pnas.81.3.819
Golden, J. W., and Riddle, D. L. (1984a). The Caenorhabditis elegans Dauer Larva: Developmental Effects of Pheromone, Food, and Temperature. Dev. Biol. 102, 368–378. doi:10.1016/0012-1606(84)90201-x
Gomez-Pastor, R., Burchfiel, E. T., and Thiele, D. J. (2018). Regulation of Heat Shock Transcription Factors and Their Roles in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 19, 4–19. doi:10.1038/nrm.2017.73
Goodson, M. L., and Sarge, K. D. (1995). Heat-inducible DNA Binding of Purified Heat Shock Transcription Factor 1. J. Biol. Chem. 270, 2447–2450. doi:10.1074/jbc.270.6.2447
Gottlieb, S., and Ruvkun, G. (1994). daf-2, Daf-16 and Daf-23: Genetically Interacting Genes Controlling Dauer Formation in Caenorhabditis elegans. Genetics 137, 107–120. doi:10.1093/genetics/137.1.107
Greer, E. L., Banko, M. R., and Brunet, A. (2009). AMP-activated Protein Kinase and FoxO Transcription Factors in Dietary Restriction-Induced Longevity. Ann. N. Y. Acad. Sci. 1170, 688–692. doi:10.1111/j.1749-6632.2009.04019.x
Greer, E. L., and Brunet, A. (2009). Different Dietary Restriction Regimens Extend Lifespan by Both Independent and Overlapping Genetic Pathways in C. elegans. Aging Cell 8, 113–127. doi:10.1111/j.1474-9726.2009.00459.x
Greer, E. L., Dowlatshahi, D., Banko, M. R., Villen, J., Hoang, K., Blanchard, D., et al. (2007). An AMPK-FOXO Pathway Mediates Longevity Induced by a Novel Method of Dietary Restriction in C. elegans. Curr. Biol. 17, 1646–1656. doi:10.1016/j.cub.2007.08.047
Guettouche, T., Boellmann, F., Lane, W. S., and Voellmy, R. (2005). Analysis of Phosphorylation of Human Heat Shock Factor 1 in Cells Experiencing a Stress. BMC Biochem. 6, 4. doi:10.1186/1471-2091-6-4
Guisbert, E., Czyz, D. M., Richter, K., McMullen, P. D., and Morimoto, R. I. (2013). Identification of a Tissue-Selective Heat Shock Response Regulatory Network. PLoS Genet. 9, e1003466. doi:10.1371/journal.pgen.1003466
Guo, Y., Guettouche, T., Fenna, M., Boellmann, F., Pratt, W. B., Toft, D. O., et al. (2001). Evidence for a Mechanism of Repression of Heat Shock Factor 1 Transcriptional Activity by a Multichaperone Complex. J. Biol. Chem. 276, 45791–45799. doi:10.1074/jbc.m105931200
Hageman, J., and Kampinga, H. H. (2009). Computational Analysis of the Human HSPH/HSPA/DNAJ Family and Cloning of a Human HSPH/HSPA/DNAJ Expression Library. Cell stress chaperones 14, 1–21. doi:10.1007/s12192-008-0060-2
Hajdu-Cronin, Y. M., Chen, W. J., and Sternberg, P. W. (2004). The L-Type Cyclin CYL-1 And The Heat-Shock-Factor HSF-1 Are Required For Heat-Shockinduced Protein Expression In Caenorhabditis Elegans. Genetics 168, 1937–1949. doi:10.1534/genetics.104.028423
Halaschek-Wiener, J., Khattra, J. S., McKay, S., Pouzyrev, A., Stott, J. M., Yang, G. S., et al. (2005). Analysis of Long-Lived C. elegans Daf-2 Mutants Using Serial Analysis of Gene Expression. Genome Res. 15, 603–615. Epub 2005 Apr 2018. doi:10.1101/gr.3274805
Hamiel, C. R., Pinto, S., Hau, A., and Wischmeyer, P. E. (2009). Glutamine Enhances Heat Shock Protein 70 Expression Via Increased Hexosamine Biosynthetic Pathway Activity. Am. J. Physiol. Cell Physiol. 297, C1509–C1519. doi:10.1152/ajpcell.00240.2009
Hansen, M., Chandra, A., Mitic, L. L., Onken, B., Driscoll, M., and Kenyon, C. (2008). A Role for Autophagy in the Extension of Lifespan by Dietary Restriction in C. elegans. PLoS Genet. 4, e24. doi:10.1371/journal.pgen.0040024
Hansen, M., Hsu, A. L., Dillin, A., and Kenyon, C. (2005). New Genes Tied to Endocrine, Metabolic, and Dietary Regulation of Lifespan from a Caenorhabditis elegans Genomic RNAi Screen. PLoS Genet. 1, 119–128. doi:10.1371/journal.pgen.0010017
Hartl, F. U., and Hayer-Hartl, M. (2002). Molecular Chaperones in the Cytosol: from Nascent Chain to Folded Protein. Science 295, 1852–1858. doi:10.1126/science.1068408
Hartley, A. D., Ward, M. P., and Garrett, S. (1994). The Yak1 Protein Kinase of Saccharomyces cerevisiae Moderates Thermotolerance and Inhibits Growth by an Sch9 Protein Kinase-independent Mechanism. Genetics 136, 465–474. doi:10.1093/genetics/136.2.465
He, B., Meng, Y. H., and Mivechi, N. F. (1998). Glycogen Synthase Kinase 3beta and Extracellular Signal-Regulated Kinase Inactivate Heat Shock Transcription Factor 1 by Facilitating the Disappearance of Transcriptionally Active Granules after Heat Shock. Mol. Cell Biol. 18, 6624–6633. doi:10.1128/mcb.18.11.6624
He, C., and Klionsky, D. J. (2009). Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 43, 67–93. doi:10.1146/annurev-genet-102808-114910
Hendriks, I. A., Lyon, D., Young, C., Jensen, L. J., Vertegaal, A. C., and Nielsen, M. L. (2017). Site-specific Mapping of the Human SUMO Proteome Reveals Co-modification with Phosphorylation. Nat. Struct. Mol. Biol. 24, 325–336. doi:10.1038/nsmb.3366
Hentze, N., Le Breton, L., Wiesner, J., Kempf, G., and Mayer, M. P. (2016). Molecular Mechanism of Thermosensory Function of Human Heat Shock Transcription Factor Hsf1. Elife 5. doi:10.7554/eLife.11576
Hietakangas, V., Ahlskog, J. K., Jakobsson, A. M., Hellesuo, M., Sahlberg, N. M., Holmberg, C. I., et al. (2003). Phosphorylation of Serine 303 Is a Prerequisite for the Stress-Inducible SUMO Modification of Heat Shock Factor 1. Mol. Cell Biol. 23, 2953–2968. doi:10.1128/mcb.23.8.2953-2968.2003
Hietakangas, V., Anckar, J., Blomster, H. A., Fujimoto, M., Palvimo, J. J., Nakai, A., et al. (2006). PDSM, a Motif for Phosphorylation-dependent SUMO Modification. Proc. Natl. Acad. Sci. U. S. A. 103, 45–50. doi:10.1073/pnas.0503698102
Higuchi-Sanabria, R., Durieux, J., Kelet, N., Homentcovschi, S., de Los Rios Rogers, M., Monshietehadi, S., et al. (2020). Divergent Nodes of Non-autonomous UPR(ER) Signaling through Serotonergic and Dopaminergic Neurons. Cell Rep. 33, 108489. doi:10.1016/j.celrep.2020.108489
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019). The Proteostasis Network and its Decline in Ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435. doi:10.1038/s41580-019-0101-y
Holmberg, C. I., Hietakangas, V., Mikhailov, A., Rantanen, J. O., Kallio, M., Meinander, A., et al. (2001). Phosphorylation of Serine 230 Promotes Inducible Transcriptional Activity of Heat Shock Factor 1. EMBO J. 20, 3800–3810. doi:10.1093/emboj/20.14.3800
Holmberg, C. I., Tran, S. E., Eriksson, J. E., and Sistonen, L. (2002). Multisite Phosphorylation Provides Sophisticated Regulation of Transcription Factors. Trends Biochem. Sci. 27, 619–627. doi:10.1016/s0968-0004(02)02207-7
Holzenberger, M., Dupont, J., Ducos, B., Leneuve, P., Geloen, A., Even, P. C., et al. (2003). IGF-1 Receptor Regulates Lifespan and Resistance to Oxidative Stress in Mice. Nature 421, 182–187. doi:10.1038/nature01298
Hong, Y., Rogers, R., Matunis, M. J., Mayhew, C. N., Goodson, M. L., Park-Sarge, O. K., et al. (2001). Regulation of Heat Shock Transcription Factor 1 by Stress-Induced SUMO-1 Modification. J. Biol. Chem. 276, 40263–40267. doi:10.1074/jbc.m104714200
Hsu, A. L., Murphy, C. T., and Kenyon, C. (2003). Regulation of Aging and Age-Related Disease by DAF-16 and Heat-Shock Factor. Science 300, 1142–1145. doi:10.1126/science.1083701
Hu, Y., and Mivechi, N. F. (2003). HSF-1 Interacts with Ral-Binding Protein 1 in a Stress-Responsive, Multiprotein Complex with HSP90 In Vivo. J. Biol. Chem. 278, 17299–17306. doi:10.1074/jbc.m300788200
Hu, Y., and Mivechi, N. F. (2011). Promotion of Heat Shock Factor Hsf1 Degradation via Adaptor Protein Filamin A-Interacting Protein 1-like (FILIP-1L). J. Biol. Chem. 286, 31397–31408. doi:10.1074/jbc.m111.255851
Huang, C.-Y., Lee, F.-L., Peng, S.-F., Lin, K.-H., Chen, R.-J., Ho, T.-J., et al. (2018). HSF1 Phosphorylation By ERK/GSK3 Suppressesrnf126 To Sustain IGF-IIR Expression For Hypertension-Inducedcardiomyocyte Hypertrophy. J. Cell Physiol. 233, 979–989. doi:10.1002/jcp.25945
Hwang, A. B., Ryu, E. A., Artan, M., Chang, H. W., Kabir, M. H., Nam, H. J., et al. (2014). Feedback Regulation via AMPK and HIF-1 Mediates ROS-dependent Longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 111, E4458–E4467. doi:10.1073/pnas.1411199111
Jayaraj, G. G., Hipp, M. S., and Hartl, F. U. (2020). Functional Modules of the Proteostasis Network. Cold Spring Harb. Perspect. Biol. 12. doi:10.1101/cshperspect.a033951
Jedlicka, P., Mortin, M. A., and Wu, C. (1997). Multiple Functions of Drosophila Heat Shock Transcription Factor In Vivo. EMBO J. 16, 2452–2462. doi:10.1093/emboj/16.9.2452
Jedrusik-Bode, M. (2014). C. elegans Sirtuin SIR-2.4 and its Mammalian Homolog SIRT6 in Stress Response. Worm 3, e29102. doi:10.4161/worm.29102
Jedrusik-Bode, M., Studencka, M., Smolka, C., Baumann, T., Schmidt, H., Kampf, J., et al. (2013). The Sirtuin SIRT6 Regulates Stress Granule Formation in C. elegans and Mammals. J. Cell Sci. 126, 5166–5177. doi:10.1242/jcs.130708
Jia, K., Chen, D., and Riddle, D. L. (2004). The TOR Pathway Interacts with the Insulin Signaling Pathway to Regulate C. elegans Larval Development, Metabolism and Life Span. Development 131, 3897–3906. doi:10.1242/dev.01255
Jia, K., and Levine, B. (2007). Autophagy Is Required for Dietary Restriction-Mediated Life Span Extension in C. elegans. Autophagy 3, 597–599. doi:10.4161/auto.4989
Johnson, D. W., Llop, J. R., Farrell, S. F., Yuan, J., Stolzenburg, L. R., and Samuelson, A. V. (2014). The Caenorhabditis elegans Myc-Mondo/Mad Complexes Integrate Diverse Longevity Signals. PLoS Genet. 10, e1004278. doi:10.1371/journal.pgen.1004278
Johnson, T. E. (1990). Increased Life-Span of Age-1 Mutants in Caenorhabditis elegans and Lower Gompertz Rate of Aging. Science 249, 908–912. doi:10.1126/science.2392681
Jones, D., Crowe, E., Stevens, T. A., and Candido, E. P. (2002). Functional and Phylogenetic Analysis of the Ubiquitylation System in Caenorhabditis elegans: Ubiquitin-Conjugating Enzymes, Ubiquitin-Activating Enzymes, and Ubiquitin-like Proteins. Genome Biol. 3, RESEARCH0002. doi:10.1186/gb-2001-3-1-research0002
Jovaisaite, V., Mouchiroud, L., and Auwerx, J. (2014). The Mitochondrial Unfolded Protein Response, a Conserved Stress Response Pathway with Implications in Health and Disease. J. Exp. Biol. 217, 137–143. doi:10.1242/jeb.090738
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly Accurate Protein Structure Prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kamitani, T., Kito, K., Nguyen, H. P., Fukuda-Kamitani, T., and Yeh, E. T. (1998). Characterization of a Second Member of the Sentrin Family of Ubiquitin-like Proteins. J. Biol. Chem. 273, 11349–11353. doi:10.1074/jbc.273.18.11349
Kapahi, P., Chen, D., Rogers, A. N., Katewa, S. D., Li, P. W., Thomas, E. L., et al. (2010). With TOR, Less Is More: a Key Role for the Conserved Nutrient-Sensing TOR Pathway in Aging. Cell metab. 11, 453–465. doi:10.1016/j.cmet.2010.05.001
Karagoz, G. E., Acosta-Alvear, D., and Walter, P. (2019a). The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 11. doi:10.1101/cshperspect.a033886
Karagoz, G. E., Aragon, T., and Acosta-Alvear, D. (2019b). Recent Advances in Signal Integration Mechanisms in the Unfolded Protein Response. F1000Res 8. doi:10.12688/f1000research.19848.1
Kaushik, S., Tasset, I., Arias, E., Pampliega, O., Wong, E., Martinez-Vicente, M., et al. (2021). Autophagy and the Hallmarks of Aging. Ageing Res. Rev. 72, 101468. doi:10.1016/j.arr.2021.101468
Keisala, T., Minasyan, A., Lou, Y. R., Zou, J., Kalueff, A. V., Pyykko, I., et al. (2009). Premature Aging in Vitamin D Receptor Mutant Mice. J. Steroid Biochem. Mol. Biol. 115, 91–97. doi:10.1016/j.jsbmb.2009.03.007
Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. (1993). A C. elegans Mutant that Lives Twice as Long as Wild Type. Nature 366, 461–464. doi:10.1038/366461a0
Kenyon, C. (2011). The First Long-Lived Mutants: Discovery of the insulin/IGF-1 Pathway for Ageing. Philos. Trans. R. Soc. Lond B Biol. Sci. 366, 9–16. doi:10.1098/rstb.2010.0276
Kenyon, C. (2005). The Plasticity of Aging: Insights from Long-Lived Mutants. Cell 120, 449–460. doi:10.1016/j.cell.2005.02.002
Khaleque, M. A., Bharti, A., Gong, J., Gray, P. J., Sachdev, V., Ciocca, D. R., et al. (2008). Heat Shock Factor 1 Represses Estrogen-dependent Transcription through Association with MTA1. Oncogene 27, 1886–1893. doi:10.1038/sj.onc.1210834
Khaminets, A., Behl, C., and Dikic, I. (2016). Ubiquitin-Dependent and Independent Signals in Selective Autophagy. Trends Cell Biol. 26, 6–16. doi:10.1016/j.tcb.2015.08.010
Khaminets, A., Heinrich, T., Mari, M., Grumati, P., Huebner, A. K., Akutsu, M., et al. (2015). Regulation of Endoplasmic Reticulum Turnover by Selective Autophagy. Nature 522, 354–358. doi:10.1038/nature14498
Kim, H. W., Kim, S. A., and Ahn, S. G. (2016). Sirtuin Inhibitors, EX527 and AGK2, Suppress Cell Migration by Inhibiting HSF1 Protein Stability. Oncol. Rep. 35, 235–242. doi:10.3892/or.2015.4381
Kim, K. H., Jeong, Y. T., Oh, H., Kim, S. H., Cho, J. M., Kim, Y. N., et al. (2013). Autophagy Deficiency Leads to Protection from Obesity and Insulin Resistance by Inducing Fgf21 as a Mitokine. Nat. Med. 19, 83–92. doi:10.1038/nm.3014
Kim, S. A., Yoon, J. H., Lee, S. H., and Ahn, S. G. (2005). Polo-like Kinase 1 Phosphorylates Heat Shock Transcription Factor 1 and Mediates its Nuclear Translocation during Heat Stress. J. Biol. Chem. 280, 12653–12657. doi:10.1074/jbc.m411908200
Kimura, K. D., Tissenbaum, H. A., Liu, Y., and Ruvkun, G. (1997). daf-2, an Insulin Receptor-like Gene that Regulates Longevity and Diapause in Caenorhabditis elegans. Science 277, 942–946. doi:10.1126/science.277.5328.942
Klaips, C. L., Jayaraj, G. G., and Hartl, F. U. (2018). Pathways of Cellular Proteostasis in Aging and Disease. J. Cell Biol. 217, 51–63. doi:10.1083/jcb.201709072
Kleemann, G. A., and Murphy, C. T. (2009). The Endocrine Regulation of Aging in Caenorhabditis elegans. Mol. Cell Endocrinol. 299, 51–57. doi:10.1016/j.mce.2008.10.048
Kleizen, B., and Braakman, I. (2004). Protein Folding and Quality Control in the Endoplasmic Reticulum. Curr. Opin. Cell Biol. 16, 343–349. doi:10.1016/j.ceb.2004.06.012
Kline, M. P., and Morimoto, R. I. (1997). Repression of the Heat Shock Factor 1 Transcriptional Activation Domain Is Modulated by Constitutive Phosphorylation. Mol. Cell Biol. 17, 2107–2115. doi:10.1128/mcb.17.4.2107
Knauf, U., Newton, E. M., Kyriakis, J., and Kingston, R. E. (1996). Repression of Human Heat Shock Factor 1 Activity at Control Temperature by Phosphorylation. Genes Dev. 10, 2782–2793. doi:10.1101/gad.10.21.2782
Kourtis, N., Moubarak, R. S., Aranda-Orgilles, B., Lui, K., Aydin, I. T., Trimarchi, T., et al. (2015). FBXW7 Modulates Cellular Stress Response and Metastatic Potential through HSF1 Post-translational Modification. Nat. Cell Biol. 17, 322–332. doi:10.1038/ncb3121
Kourtis, N., Nikoletopoulou, V., and Tavernarakis, N. (2012). Small Heat-Shock Proteins Protect from Heat-Stroke-Associated Neurodegeneration. Nature 490, 213–218. doi:10.1038/nature11417
Kovacs, D., Sigmond, T., Hotzi, B., Bohar, B., Fazekas, D., Deak, V., et al. (2019). HSF1Base: A Comprehensive Database of HSF1 (Heat Shock Factor 1) Target Genes. Int. J. Mol. Sci. 20. doi:10.3390/ijms20225815
Krakowiak, J., Zheng, X., Patel, N., Feder, Z. A., Anandhakumar, J., Valerius, K., et al. (2018). Hsf1 and Hsp70 Constitute a Two-Component Feedback Loop that Regulates the Yeast Heat Shock Response. Elife 7. doi:10.7554/eLife.31668
Kroemer, G., Marino, G., and Levine, B. (2010). Autophagy and the Integrated Stress Response. Mol. Cell 40, 280–293. doi:10.1016/j.molcel.2010.09.023
Kumsta, C., Chang, J. T., Schmalz, J., and Hansen, M. (2017). Hormetic Heat Stress and HSF-1 Induce Autophagy to Improve Survival and Proteostasis in C. elegans. Nat. Commun. 8, 14337. doi:10.1038/ncomms14337
Kumsta, C., Ching, T. T., Nishimura, M., Davis, A. E., Gelino, S., Catan, H. H., et al. (2014). Integrin-linked Kinase Modulates Longevity and Thermotolerance in C. elegans through Neuronal Control of HSF-1. Aging Cell 13, 419–430. doi:10.1111/acel.12189
Kundra, R., Ciryam, P., Morimoto, R. I., Dobson, C. M., and Vendruscolo, M. (2017). Protein Homeostasis of a Metastable Subproteome Associated with Alzheimer's Disease. Proc. Natl. Acad. Sci. U. S. A. 114, E5703–E5711. doi:10.1073/pnas.1618417114
Kuo, C. T., You, G. T., Jian, Y. J., Chen, T. S., Siao, Y. C., Hsu, A. L., et al. (2020). AMPK-mediated Formation of Stress Granules Is Required for Dietary Restriction-Induced Longevity in Caenorhabditis elegans. Aging Cell 19, e13157. doi:10.1111/acel.13157
Labbadia, J., and Morimoto, R. I. (2015b). Repression of the Heat Shock Response Is a Programmed Event at the Onset of Reproduction. Mol. Cell 59, 639–650. doi:10.1016/j.molcel.2015.06.027
Labbadia, J., and Morimoto, R. I. (2015a). The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 84, 435–464. doi:10.1146/annurev-biochem-060614-033955
Lamitina, S. T., and Strange, K. (2005). Transcriptional Targets of DAF-16 Insulin Signaling Pathway Protect C. elegans from Extreme Hypertonic Stress. Am. J. Physiol. Cell Physiol. 288, C467–C474. Epub 2004 Oct 2020. doi:10.1152/ajpcell.00451.2004
Lapierre, L. R., De Magalhaes Filho, C. D., McQuary, P. R., Chu, C. C., Visvikis, O., Chang, J. T., et al. (2013). The TFEB Orthologue HLH-30 Regulates Autophagy and Modulates Longevity in Caenorhabditis elegans. Nat. Commun. 4, 2267. doi:10.1038/ncomms3267
Lapierre, L. R., Gelino, S., Melendez, A., and Hansen, M. (2011). Autophagy and Lipid Metabolism Coordinately Modulate Life Span in Germline-Less C. elegans. Curr. Biol. 21, 1507–1514. doi:10.1016/j.cub.2011.07.042
Lapierre, L. R., Kumsta, C., Sandri, M., Ballabio, A., and Hansen, M. (2015). Transcriptional and Epigenetic Regulation of Autophagy in Aging. Autophagy 11, 867–880. doi:10.1080/15548627.2015.1034410
Larsen, P. L., Albert, P. S., and Riddle, D. L. (1995). Genes that Regulate Both Development and Longevity in Caenorhabditis elegans. Genetics 139, 1567–1583. doi:10.1093/genetics/139.4.1567
Lee, R. Y., Hench, J., and Ruvkun, G. (2001). Regulation of C. elegans DAF-16 and its Human Ortholog FKHRL1 by the Daf-2 Insulin-like Signaling Pathway. Curr. Biol. 11, 1950–1957. doi:10.1016/s0960-9822(01)00595-4
Lee, Y. J., Kim, E. H., Lee, J. S., Jeoung, D., Bae, S., Kwon, S. H., et al. (2008). HSF1 as a Mitotic Regulator: Phosphorylation of HSF1 by Plk1 Is Essential for Mitotic Progression. Cancer Res. 68, 7550–7560. doi:10.1158/0008-5472.can-08-0129
Lehrbach, N. J., and Ruvkun, G. (2019). Endoplasmic Reticulum-Associated SKN-1A/Nrf1 Mediates a Cytoplasmic Unfolded Protein Response and Promotes Longevity. Elife 8. doi:10.7554/eLife.44425
Lehrbach, N. J., and Ruvkun, G. (2016). Proteasome Dysfunction Triggers Activation of SKN-1A/Nrf1 by the Aspartic Protease DDI-1. Elife 5. doi:10.7554/eLife.17721
Levine, B., and Kroemer, G. (2008). Autophagy in the Pathogenesis of Disease. Cell 132, 27–42. doi:10.1016/j.cell.2007.12.018
Li, J., Chauve, L., Phelps, G., Brielmann, R. M., and Morimoto, R. I. (2016). E2F Coregulates an Essential HSF Developmental Program that Is Distinct from the Heat-Shock Response. Genes Dev. 30, 2062–2075. doi:10.1101/gad.283317.116
Li, J., Labbadia, J., and Morimoto, R. I. (2017). Rethinking HSF1 in Stress, Development, and Organismal Health. Trends Cell Biol. 27, 895–905. doi:10.1016/j.tcb.2017.08.002
Li, W., Gao, B., Lee, S. M., Bennett, K., and Fang, D. (2007). RLE-1, an E3 Ubiquitin Ligase, Regulates C. elegans Aging by Catalyzing DAF-16 Polyubiquitination. Dev. Cell 12, 235–246. doi:10.1016/j.devcel.2006.12.002
Lin, K., Dorman, J. B., Rodan, A., and Kenyon, C. (1997). daf-16: An HNF-3/forkhead Family Member that Can Function to Double the Life-Span of Caenorhabditis elegans. Science 278, 1319–1322. doi:10.1126/science.278.5341.1319
Lin, K., Hsin, H., Libina, N., and Kenyon, C. (2001). Regulation of the Caenorhabditis elegans Longevity Protein DAF-16 by insulin/IGF-1 and Germline Signaling. Nat. Genet. 28, 139–145. doi:10.1038/88850
Lithgow, G. J., White, T. M., Melov, S., and Johnson, T. E. (1995). Thermotolerance and Extended Life-Span Conferred by Single-Gene Mutations and Induced by Thermal Stress. Proc. Natl. Acad. Sci. U. S. A. 92, 7540–7544. doi:10.1073/pnas.92.16.7540
Liu, W., Lin, H., Mao, Z., Zhang, L., Bao, K., Jiang, B., et al. (2020). Verapamil Extends Lifespan in Caenorhabditis elegans by Inhibiting Calcineurin Activity and Promoting Autophagy. Aging (Albany NY) 12, 5300–5317. doi:10.18632/aging.102951
Liu, X. D., Liu, P. C., Santoro, N., and Thiele, D. J. (1997). Conservation of a Stress Response: Human Heat Shock Transcription Factors Functionally Substitute for Yeast HSF. EMBO J. 16, 6466–6477. doi:10.1093/emboj/16.21.6466
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The Hallmarks of Aging. Cell 153, 1194–1217. doi:10.1016/j.cell.2013.05.039
Mahadevan, N. R., Rodvold, J., Sepulveda, H., Rossi, S., Drew, A. F., and Zanetti, M. (2011). Transmission of Endoplasmic Reticulum Stress and Pro-inflammation from Tumor Cells to Myeloid Cells. Proc. Natl. Acad. Sci. U. S. A. 108, 6561–6566. doi:10.1073/pnas.1008942108
Malone, E. A., and Thomas, J. H. (1994). A Screen for Nonconditional Dauer-Constitutive Mutations in Caenorhabditis elegans. Genetics 136, 879–886. doi:10.1093/genetics/136.3.879
Margulis, B., Tsimokha, A., Zubova, S., and Guzhova, I. (2020). Molecular Chaperones and Proteolytic Machineries Regulate Protein Homeostasis in Aging Cells. Cells 9. doi:10.3390/cells9051308
Matilainen, O., Arpalahti, L., Rantanen, V., Hautaniemi, S., and Holmberg, C. I. (2013). Insulin/IGF-1 Signaling Regulates Proteasome Activity through the Deubiquitinating Enzyme UBH-4. Cell Rep. 3, 1980–1995. doi:10.1016/j.celrep.2013.05.012
Mayya, V., Lundgren, D. H., Hwang, S. I., Rezaul, K., Wu, L., Eng, J. K., et al. (2009). Quantitative Phosphoproteomic Analysis of T Cell Receptor Signaling Reveals System-Wide Modulation of Protein-Protein Interactions. Sci. Signal. 2 (84), ra46. doi:10.1126/scisignal.2000007
McColl, G., Rogers, A. N., Alavez, S., Hubbard, A. E., Melov, S., Link, C. D., et al. (2010). Insulin-like Signaling Determines Survival during Stress via Posttranscriptional Mechanisms in C. elegans. Cell metab. 12, 260–272. doi:10.1016/j.cmet.2010.08.004
McCormick, M. A., Tsai, S. Y., and Kennedy, B. K. (2011). TOR and Ageing: a Complex Pathway for a Complex Process. Philos. Trans. R. Soc. Lond B Biol. Sci. 366, 17–27. doi:10.1098/rstb.2010.0198
McElwee, J., Bubb, K., and Thomas, J. H. (2003). Transcriptional Outputs of the Caenorhabditis elegans Forkhead Protein DAF-16. Aging Cell 2, 111–121. doi:10.1046/j.1474-9728.2003.00043.x
McHugh, D., and Gil, J. (2018). Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol. 217, 65–77. doi:10.1083/jcb.201708092
Meister, P., Towbin, B. D., Pike, B. L., Ponti, A., and Gasser, S. M. (2010). The Spatial Dynamics of Tissue-specific Promoters during C. elegans Development. Genes Dev. 24, 766–782. doi:10.1101/gad.559610
Melendez, A., Talloczy, Z., Seaman, M., Eskelinen, E. L., Hall, D. H., and Levine, B. (2003). Autophagy Genes Are Essential for Dauer Development and Life-Span Extension in C. elegans. Science 301, 1387–1391. doi:10.1126/science.1087782
Mihaylova, M. M., and Shaw, R. J. (2011). The AMPK Signalling Pathway Coordinates Cell Growth, Autophagy and Metabolism. Nat. Cell Biol. 13, 1016–1023. doi:10.1038/ncb2329
Miles, J., and van Oosten-Hawle, P. (2020). Tissue-Specific RNAi Tools to Identify Components for Systemic Stress Signaling. J. Vis. Exp. 159, 1. doi:10.3791/61357
Miller, H. A., Dean, E. S., Pletcher, S. D., and Leiser, S. F. (2020). Cell Non-autonomous Regulation of Health and Longevity. Elife 9. doi:10.7554/eLife.62659
Minnerly, J., Zhang, J., Parker, T., Kaul, T., and Jia, K. (2017). The Cell Non-autonomous Function of ATG-18 Is Essential for Neuroendocrine Regulation of Caenorhabditis elegans Lifespan. PLoS Genet. 13, e1006764. doi:10.1371/journal.pgen.1006764
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy Fights Disease through Cellular Self-Digestion. Nature 451, 1069–1075. doi:10.1038/nature06639
Mizushima, N. (2011). Autophagy in Protein and Organelle Turnover. Cold Spring Harb. Symp. Quant. Biol. 76, 397–402. doi:10.1101/sqb.2011.76.011023
Mooijaart, S. P., Kuningas, M., Westendorp, R. G., Houwing-Duistermaat, J. J., Slagboom, P. E., Rensen, P. C., et al. (2007). Liver X Receptor Alpha Associates with Human Life Span. J. Gerontol. A Biol. Sci. Med. Sci. 62, 343–349. doi:10.1093/gerona/62.4.343
Morimoto, R. I. (2019). Cell-Nonautonomous Regulation of Proteostasis in Aging and Disease. Cold Spring Harb. Perspect. Biol. 12 (4), a034074. doi:10.1101/cshperspect.a034074
Morimoto, R. I., and Cuervo, A. M. (2009). Protein Homeostasis and Aging: Taking Care of Proteins from the Cradle to the Grave. J. Gerontol. A Biol. Sci. Med. Sci. 64, 167–170. doi:10.1093/gerona/gln071
Morimoto, R. I. (2008). Proteotoxic Stress and Inducible Chaperone Networks in Neurodegenerative Disease and Aging. Genes Dev. 22, 1427–1438. doi:10.1101/gad.1657108
Morimoto, R. I. (2006). Stress, Aging, and Neurodegenerative Disease. N. Engl. J. Med. 355, 2254–2255. doi:10.1056/nejmcibr065573
Morley, J. F., and Morimoto, R. I. (2004). Regulation of Longevity in Caenorhabditis elegans by Heat Shock Factor and Molecular Chaperones. Mol. Biol. Cell 15, 657–664. doi:10.1091/mbc.e03-07-0532
Morris, J. Z., Tissenbaum, H. A., and Ruvkun, G. (1996). A Phosphatidylinositol-3-OH Kinase Family Member Regulating Longevity and Diapause in Caenorhabditis elegans. Nature 382, 536–539. doi:10.1038/382536a0
Morton, E. A., and Lamitina, T. (2013). Caenorhabditis elegans HSF-1 Is an Essential Nuclear Protein that Forms Stress Granule-like Structures Following Heat Shock. Aging Cell 12, 112–120. doi:10.1111/acel.12024
Mosser, D. D., Kotzbauer, P. T., Sarge, K. D., and Morimoto, R. I. (1990). In Vitro activation of Heat Shock Transcription Factor DNA-Binding by Calcium and Biochemical Conditions that Affect Protein Conformation. Proc. Natl. Acad. Sci. U. S. A. 87, 3748–3752. doi:10.1073/pnas.87.10.3748
Mottis, A., Jovaisaite, V., and Auwerx, J. (2014). The Mitochondrial Unfolded Protein Response in Mammalian Physiology. Mamm. Genome 25, 424–433. doi:10.1007/s00335-014-9525-z
Mouchiroud, L., Houtkooper, R. H., Moullan, N., Katsyuba, E., Ryu, D., Canto, C., et al. (2013). The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154, 430–441. doi:10.1016/j.cell.2013.06.016
Mukhopadhyay, A., and Tissenbaum, H. A. (2007). Reproduction and Longevity: Secrets Revealed by C. elegans. Trends Cell Biol. 17, 65–71. doi:10.1016/j.tcb.2006.12.004
Murphy, C. T., McCarroll, S. A., Bargmann, C. I., Fraser, A., Kamath, R. S., Ahringer, J., et al. (2003). Genes that Act Downstream of DAF-16 to Influence the Lifespan of Caenorhabditis elegans. Nature 424, 277–283. doi:10.1038/nature01789
Murshid, A., Chou, S. D., Prince, T., Zhang, Y., Bharti, A., and Calderwood, S. K. (2010). Protein Kinase A Binds and Activates Heat Shock Factor 1. PLoS One 5, e13830. doi:10.1371/journal.pone.0013830
Nakai, A. (1999). New Aspects in the Vertebrate Heat Shock Factor System: Hsf3 and Hsf4. Cell stress chaperones 4, 86–93. doi:10.1379/1466-1268(1999)004<0086:naitvh>2.3.co;2
Nakamura, S., Karalay, O., Jager, P. S., Horikawa, M., Klein, C., Nakamura, K., et al. (2016). Mondo Complexes Regulate TFEB via TOR Inhibition to Promote Longevity in Response to Gonadal Signals. Nat. Commun. 7, 10944. doi:10.1038/ncomms10944
Neef, D. W., Jaeger, A. M., Gomez-Pastor, R., Willmund, F., Frydman, J., and Thiele, D. J. (2014). A Direct Regulatory Interaction between Chaperonin TRiC and Stress-Responsive Transcription Factor HSF1. Cell Rep. 9, 955–966. doi:10.1016/j.celrep.2014.09.056
Neef, D. W., Jaeger, A. M., and Thiele, D. J. (2011). Heat Shock Transcription Factor 1 as a Therapeutic Target in Neurodegenerative Diseases. Nat. Rev. Drug Discov. 10, 930–944. doi:10.1038/nrd3453
Neef, D. W., Turski, M. L., and Thiele, D. J. (2010). Modulation of Heat Shock Transcription Factor 1 as a Therapeutic Target for Small Molecule Intervention in Neurodegenerative Disease. PLoS Biol. 8, e1000291. doi:10.1371/journal.pbio.1000291
Nieto-Torres, J. L., and Hansen, M. (2021). Macroautophagy and Aging: The Impact of Cellular Recycling on Health and Longevity. Mol. Asp. Med. 82, 101020. doi:10.1016/j.mam.2021.101020
Nisaa, K., and Ben-Zvi, A. (2021). Chaperone Networks Are Shaped by Cellular Differentiation and Identity. Trends Cell Biol. 32 (6), 470–474. doi:10.1016/j.tcb.2021.11.001
O'Brien, D., Jones, L. M., Good, S., Miles, J., Vijayabaskar, M. S., Aston, R., et al. (2018). A PQM-1-Mediated Response Triggers Transcellular Chaperone Signaling and Regulates Organismal Proteostasis. Cell Rep. 23, 3905–3919. doi:10.1016/j.celrep.2018.05.093
Ogg, S., Paradis, S., Gottlieb, S., Patterson, G. I., Lee, L., Tissenbaum, H. A., et al. (1997). The Fork Head Transcription Factor DAF-16 Transduces Insulin-like Metabolic and Longevity Signals in C. elegans. Nature 389, 994–999. doi:10.1038/40194
Ogg, S., and Ruvkun, G. (1998). The C. elegans PTEN Homolog, DAF-18, Acts in the Insulin Receptor-like Metabolic Signaling Pathway. Mol. Cell 2, 887–893. doi:10.1016/s1097-2765(00)80303-2
Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., et al. (2006). Global, In Vivo, And Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 127, 635–648. doi:10.1016/j.cell.2006.09.026
Olsen, J. V., Vermeulen, M., Santamaria, A., Kumar, C., Miller, M. L., Jensen, L. J., et al. (2010). Quantitative Phosphoproteomics Reveals Widespread Full Phosphorylation Site Occupancy During Mitosis. Science signaling 3, ra3. doi:10.1126/scisignal.2000475
Orosz, A., Wisniewski, J., and Wu, C. (1996). Regulation of Drosophila Heat Shock Factor Trimerization: Global Sequence Requirements and Independence of Nuclear Localization. Mol. Cell Biol. 16, 7018–7030. doi:10.1128/mcb.16.12.7018
Ottens, F., Franz, A., and Hoppe, T. (2021). Build-UPS and Break-Downs: Metabolism Impacts on Proteostasis and Aging. Cell Death Differ. 28, 505–521. doi:10.1038/s41418-020-00682-y
Panowski, S. H., Wolff, S., Aguilaniu, H., Durieux, J., and Dillin, A. (2007). PHA-4/Foxa Mediates Diet-Restriction-Induced Longevity of C. elegans. Nature 447, 550–555. doi:10.1038/nature05837
Papaevgeniou, N., and Chondrogianni, N. (2016). UPS Activation in the Battle against Aging and Aggregation-Related Diseases: An Extended Review. Methods Mol. Biol. Clift. N. J. 1449, 1–70. doi:10.1007/978-1-4939-3756-1_1
Paradis, S., Ailion, M., Toker, A., Thomas, J. H., and Ruvkun, G. (1999). A PDK1 Homolog Is Necessary and Sufficient to Transduce AGE-1 PI3 Kinase Signals that Regulate Diapause in Caenorhabditis elegans. Genes Dev. 13, 1438–1452. doi:10.1101/gad.13.11.1438
Paradis, S., and Ruvkun, G. (1998). Caenorhabditis elegans Akt/PKB Transduces Insulin Receptor-like Signals from AGE-1 PI3 Kinase to the DAF-16 Transcription Factor. Genes Dev. 12, 2488–2498. doi:10.1101/gad.12.16.2488
Park, J., and Liu, A. Y. (2001). JNK Phosphorylates the HSF1 Transcriptional Activation Domain: Role of JNK in the Regulation of the Heat Shock Response. J. Cell Biochem. 82, 326–338. doi:10.1002/jcb.1163
Powers, E. T., Morimoto, R. I., Dillin, A., Kelly, J. W., and Balch, W. E. (2009). Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Annu. Rev. Biochem. 78, 959–991. doi:10.1146/annurev.biochem.052308.114844
Prahlad, V., Cornelius, T., and Morimoto, R. I. (2008). Regulation of the Cellular Heat Shock Response in Caenorhabditis elegans by Thermosensory Neurons. Science 320, 811–814. doi:10.1126/science.1156093
Prahlad, V., and Morimoto, R. I. (2011). Neuronal Circuitry Regulates the Response of Caenorhabditis elegans to Misfolded Proteins. Proc. Natl. Acad. Sci. U. S. A. 108, 14204–14209. doi:10.1073/pnas.1106557108
Prata, L., Ovsyannikova, I. G., Tchkonia, T., and Kirkland, J. L. (2018). Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin. Immunol. 40, 101275. doi:10.1016/j.smim.2019.04.003
Preissler, S., and Ron, D. (2019). Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 11. doi:10.1101/cshperspect.a033894
Prelich, G. (2012). Gene Overexpression: Uses, Mechanisms, and Interpretation. Genetics 190, 841–854. doi:10.1534/genetics.111.136911
Puscheck, E. E., Awonuga, A. O., Yang, Y., Jiang, Z., and Rappolee, D. A. (2015). Molecular Biology of the Stress Response in the Early Embryo and its Stem Cells. Adv. Exp. Med. Biol. 843, 77–128. doi:10.1007/978-1-4939-2480-6_4
Pyo, J. O., Nah, J., and Jung, Y. K. (2012). Molecules and Their Functions in Autophagy. Exp. Mol. Med. 44, 73–80. doi:10.3858/emm.2012.44.2.029
Qadota, H., Inoue, M., Hikita, T., Koppen, M., Hardin, J. D., Amano, M., et al. (2007). Establishment of a Tissue-specific RNAi System in C. elegans. Gene 400, 166–173. doi:10.1016/j.gene.2007.06.020
Rabindran, S. K., Giorgi, G., Clos, J., and Wu, C. (1991). Molecular Cloning and Expression of a Human Heat Shock Factor, HSF1. Proc. Natl. Acad. Sci. U. S. A. 88, 6906–6910. doi:10.1073/pnas.88.16.6906
Rabinowitz, J. D., and White, E. (2010). Autophagy and Metabolism. Science 330, 1344–1348. doi:10.1126/science.1193497
Raychaudhuri, S., Loew, C., Korner, R., Pinkert, S., Theis, M., Hayer-Hartl, M., et al. (2014). Interplay of Acetyltransferase EP300 and the Proteasome System in Regulating Heat Shock Transcription Factor 1. Cell 156, 975–985. doi:10.1016/j.cell.2014.01.055
Raynes, R., Leckey, B. D., Nguyen, K., and Westerheide, S. D. (2012). Heat Shock and Caloric Restriction Have a Synergistic Effect on the Heat Shock Response in a sir2.1-dependent Manner in Caenorhabditis elegans. J. Biol. Chem. 287, 29045–29053. doi:10.1074/jbc.m112.353714
Raynes, R., Pombier, K. M., Nguyen, K., Brunquell, J., Mendez, J. E., and Westerheide, S. D. (2013). The SIRT1 Modulators AROS and DBC1 Regulate HSF1 Activity and the Heat Shock Response. PLoS One 8, e54364. doi:10.1371/journal.pone.0054364
Reinle, K., Mogk, A., and Bukau, B. (2022). The Diverse Functions of Small Heat Shock Proteins in the Proteostasis Network. J. Mol. Biol. 434, 167157. doi:10.1016/j.jmb.2021.167157
Reis-Rodrigues, P., Czerwieniec, G., Peters, T. W., Evani, U. S., Alavez, S., Gaman, E. A., et al. (2012). Proteomic Analysis of Age-dependent Changes in Protein Solubility Identifies Genes that Modulate Lifespan. Aging Cell 11, 120–127. doi:10.1111/j.1474-9726.2011.00765.x
Ren, P., Lim, C. S., Johnsen, R., Albert, P. S., Pilgrim, D., and Riddle, D. L. (1996). Control of C. elegans Larval Development by Neuronal Expression of a TGF-Beta Homolog. Science 274, 1389–1391. doi:10.1126/science.274.5291.1389
Richter, K., Haslbeck, M., and Buchner, J. (2010). The Heat Shock Response: Life on the Verge of Death. Mol. Cell 40, 253–266. doi:10.1016/j.molcel.2010.10.006
Riddle, D. L., Swanson, M. M., and Albert, P. S. (1981). Interacting Genes in Nematode Dauer Larva Formation. Nature 290, 668–671. doi:10.1038/290668a0
Ritossa, F. (1962). A New Puffing Pattern Induced by Temperature Shock and DNP in drosophila. Experientia 18, 571–573. doi:10.1007/bf02172188
Robida-Stubbs, S., Glover-Cutter, K., Lamming, D. W., Mizunuma, M., Narasimhan, S. D., Neumann-Haefelin, E., et al. (2012). TOR Signaling and Rapamycin Influence Longevity by Regulating SKN-1/Nrf and DAF-16/FoxO. Cell metab. 15, 713–724. doi:10.1016/j.cmet.2012.04.007
Roos-Mattjus, P., and Sistonen, L. (2021). Interplay between Mammalian Heat Shock Factors 1 and 2 in Physiology and Pathology. FEBS J. 2021, 1. doi:10.1111/febs.16178
Rosenzweig, R., Nillegoda, N. B., Mayer, M. P., and Bukau, B. (2019). The Hsp70 Chaperone Network. Nat. Rev. Mol. Cell Biol. 20, 665–680. doi:10.1038/s41580-019-0133-3
Rual, J. F., Venkatesan, K., Hao, T., Hirozane-Kishikawa, T., Dricot, A., Li, N., et al. (2005). Towards a Proteome-Scale Map of the Human Protein-Protein Interaction Network. Nature 437, 1173–1178. doi:10.1038/nature04209
Rubinsztein, D. C., Marino, G., and Kroemer, G. (2011). Autophagy and Aging. Cell 146, 682–695. doi:10.1016/j.cell.2011.07.030
Ryan, M. T., and Hoogenraad, N. J. (2007). Mitochondrial-nuclear Communications. Annu. Rev. Biochem. 76, 701–722. doi:10.1146/annurev.biochem.76.052305.091720
Sabath, N., Levy-Adam, F., Younis, A., Rozales, K., Meller, A., Hadar, S., et al. (2020). Cellular Proteostasis Decline in Human Senescence. Proc. Natl. Acad. Sci. U. S. A. 117, 31902–31913. doi:10.1073/pnas.2018138117
Sala, A. J., Bott, L. C., Brielmann, R. M., and Morimoto, R. I. (2020). Embryo Integrity Regulates Maternal Proteostasis and Stress Resilience. Genes Dev. 34, 678–687. doi:10.1101/gad.335422.119
Sala, A. J., Bott, L. C., and Morimoto, R. I. (2017). Shaping Proteostasis at the Cellular, Tissue, and Organismal Level. J. Cell Biol. 216, 1231–1241. doi:10.1083/jcb.201612111
Salminen, A., and Kaarniranta, K. (2012). AMP-activated Protein Kinase (AMPK) Controls the Aging Process via an Integrated Signaling Network. Ageing Res. Rev. 11, 230–241. doi:10.1016/j.arr.2011.12.005
Samuelson, A. V., Carr, C. E., and Ruvkun, G. (2007). Gene Activities that Mediate Increased Life Span of C. elegans Insulin-like Signaling Mutants. Genes Dev. 21, 2976–2994. doi:10.1101/gad.1588907
Sarge, K. D., Murphy, S. P., and Morimoto, R. I. (1993). Activation of Heat Shock Gene Transcription by Heat Shock Factor 1 Involves Oligomerization, Acquisition of DNA-Binding Activity, and Nuclear Localization and Can Occur in the Absence of Stress. Mol. Cell Biol. 13, 1392–1407. doi:10.1128/mcb.13.3.1392-1407.1993
Sarkis, G. J., Ashcom, J. D., Hawdon, J. M., and Jacobson, L. A. (1988). Decline in Protease Activities with Age in the Nematode Caenorhabditis elegans. Mech. Ageing Dev. 45, 191–201. doi:10.1016/0047-6374(88)90001-2
Satyal, S. H., Chen, D., Fox, S. G., Kramer, J. M., and Morimoto, R. I. (1998). Negative Regulation of the Heat Shock Transcriptional Response by HSBP1. Genes Dev. 12, 1962–1974. doi:10.1101/gad.12.13.1962
Satyal, S. H., Schmidt, E., Kitagawa, K., Sondheimer, N., Lindquist, S., Kramer, J. M., et al. (2000). Polyglutamine Aggregates Alter Protein Folding Homeostasis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 97, 5750–5755. doi:10.1073/pnas.100107297
Saxton, R. A., and Sabatini, D. M. (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. doi:10.1016/j.cell.2017.02.004
Seo, K., Choi, E., Lee, D., Jeong, D. E., Jang, S. K., and Lee, S. J. (2013). Heat Shock Factor 1 Mediates the Longevity Conferred by Inhibition of TOR and insulin/IGF-1 Signaling Pathways in C. elegans. Aging Cell 12, 1073–1081. doi:10.1111/acel.12140
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D., and Lowe, S. W. (1997). Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and p16INK4a. Cell 88, 593–602. doi:10.1016/s0092-8674(00)81902-9
Shamovsky, I., Ivannikov, M., Kandel, E. S., Gershon, D., and Nudler, E. (2006). RNA-mediated Response to Heat Shock in Mammalian Cells. Nature 440, 556–560. doi:10.1038/nature04518
Shaw, W. M., Luo, S., Landis, J., Ashraf, J., and Murphy, C. T. (2007). The C. elegans TGF-Beta Dauer Pathway Regulates Longevity via Insulin Signaling. Curr. Biol. 17, 1635–1645. doi:10.1016/j.cub.2007.08.058
Sheaffer, K. L., Updike, D. L., and Mango, S. E. (2008). The Target of Rapamycin Pathway Antagonizes pha-4/FoxA to Control Development and Aging. Curr. Biol. 18, 1355–1364. doi:10.1016/j.cub.2008.07.097
Shemesh, N., Jubran, J., Dror, S., Simonovsky, E., Basha, O., Argov, C., et al. (2021). The Landscape of Molecular Chaperones across Human Tissues Reveals a Layered Architecture of Core and Variable Chaperones. Nat. Commun. 12, 2180. doi:10.1038/s41467-021-22369-9
Shemesh, N., Shai, N., and Ben-Zvi, A. (2013). Germline Stem Cell Arrest Inhibits the Collapse of Somatic Proteostasis Early in Caenorhabditis elegans Adulthood. Aging Cell 12, 814–822. doi:10.1111/acel.12110
Shi, Y., Mosser, D. D., and Morimoto, R. I. (1998). Molecular Chaperones as HSF1-specific Transcriptional Repressors. Genes Dev. 12, 654–666. doi:10.1101/gad.12.5.654
Shpigel, N., Shemesh, N., Kishner, M., and Ben-Zvi, A. (2019). Dietary Restriction and Gonadal Signaling Differentially Regulate Post-development Quality Control Functions in Caenorhabditis elegans. Aging Cell 18, e12891. doi:10.1111/acel.12891
Silva, M. C., Amaral, M. D., and Morimoto, R. I. (2013). Neuronal Reprograming of Protein Homeostasis by Calcium-dependent Regulation of the Heat Shock Response. PLoS Genet. 9, e1003711. doi:10.1371/journal.pgen.1003711
Skaar, J. R., Pagan, J. K., and Pagano, M. (2013). Mechanisms and Function of Substrate Recruitment by F-Box Proteins. Nat. Rev. Mol. Cell Biol. 14, 369–381. doi:10.1038/nrm3582
Somogyvari, M., Gecse, E., and Soti, C. (2018). DAF-21/Hsp90 Is Required for C. elegans Longevity by Ensuring DAF-16/FOXO Isoform A Function. Sci. Rep. 8, 12048. doi:10.1038/s41598-018-30592-6
Son, H. G., Seo, K., Seo, M., Park, S., Ham, S., An, S. W. A., et al. (2018). Prefoldin 6 Mediates Longevity Response from Heat Shock Factor 1 to FOXO in C. elegans. Genes Dev. 32, 1562–1575. doi:10.1101/gad.317362.118
Soncin, F., Zhang, X., Chu, B., Wang, X., Asea, A., Ann Stevenson, M., et al. (2003). Transcriptional Activity and DNA Binding of Heat Shock Factor-1 Involve Phosphorylation on Threonine 142 by CK2. Biochem. Biophys. Res. Commun. 303, 700–706. doi:10.1016/s0006-291x(03)00398-x
Song, S., Lam, E. W., Tchkonia, T., Kirkland, J. L., and Sun, Y. (2020). Senescent Cells: Emerging Targets for Human Aging and Age-Related Diseases. Trends Biochem. Sci. 45, 578–592. doi:10.1016/j.tibs.2020.03.008
Sorger, P. K., and Pelham, H. R. (1988). Yeast Heat Shock Factor Is an Essential DNA-Binding Protein that Exhibits Temperature-dependent Phosphorylation. Cell 54, 855–864. doi:10.1016/s0092-8674(88)91219-6
Sorger, P. K. (1990). Yeast Heat Shock Factor Contains Separable Transient and Sustained Response Transcriptional Activators. Cell 62, 793–805. doi:10.1016/0092-8674(90)90123-v
Steinkraus, K. A., Smith, E. D., Davis, C., Carr, D., Pendergrass, W. R., Sutphin, G. L., et al. (2008). Dietary Restriction Suppresses Proteotoxicity and Enhances Longevity by an Hsf-1-dependent Mechanism in Caenorhabditis elegans. Aging Cell 7, 394–404. doi:10.1111/j.1474-9726.2008.00385.x
Stetter, K. O. (2006). Hyperthermophiles in the History of Life. Philos. Trans. R. Soc. Lond B Biol. Sci. 361, 1837–1842. doi:10.1098/rstb.2006.1907
Su, K. H., Dai, S., Tang, Z., Xu, M., and Dai, C. (2019). Heat Shock Factor 1 Is a Direct Antagonist of AMP-Activated Protein Kinase. Mol. Cell 76, 546–561. doi:10.1016/j.molcel.2019.08.021
Suh, Y., Atzmon, G., Cho, M. O., Hwang, D., Liu, B., Leahy, D. J., et al. (2008). Functionally Significant Insulin-like Growth Factor I Receptor Mutations in Centenarians. Proc. Natl. Acad. Sci. U. S. A. 105, 3438–3442. doi:10.1073/pnas.0705467105
Sun, J., Singh, V., Kajino-Sakamoto, R., and Aballay, A. (2011). Neuronal GPCR Controls Innate Immunity by Regulating Noncanonical Unfolded Protein Response Genes. Science 332, 729–732. doi:10.1126/science.1203411
Sural, S., Liang, C. Y., Wang, F. Y., Ching, T. T., and Hsu, A. L. (2020). HSB-1/HSF-1 Pathway Modulates Histone H4 in Mitochondria to Control mtDNA Transcription and Longevity. Sci. Adv. 6. doi:10.1126/sciadv.aaz4452
Sykiotis, G. P., Habeos, I. G., Samuelson, A. V., and Bohmann, D. (2011). The Role of the Antioxidant and Longevity-Promoting Nrf2 Pathway in Metabolic Regulation. Curr. Opin. Clin. Nutr. Metab. Care 14, 41–48. doi:10.1097/mco.0b013e32834136f2
Szewczyk, N. J., Udranszky, I. A., Kozak, E., Sunga, J., Kim, S. K., Jacobson, L. A., et al. (2006). Delayed Development and Lifespan Extension as Features of Metabolic Lifestyle Alteration in C. elegans under Dietary Restriction. J. Exp. Biol. 209, 4129–4139. doi:10.1242/jeb.02492
Taipale, M., Jarosz, D. F., and Lindquist, S. (2010). HSP90 at the Hub of Protein Homeostasis: Emerging Mechanistic Insights. Nat. Rev. Mol. Cell Biol. 11, 515–528. doi:10.1038/nrm2918
Takai, K., Nunoura, T., Sako, Y., and Uchida, A. (1998). Acquired Thermotolerance and Temperature-Induced Protein Accumulation in the Extremely Thermophilic Bacterium Rhodothermus Obamensis. J. Bacteriol. 180, 2770–2774. doi:10.1128/jb.180.10.2770-2774.1998
Tang, Z., Dai, S., He, Y., Doty, R. A., Shultz, L. D., Sampson, S. B., et al. (2015). MEK Guards Proteome Stability and Inhibits Tumor-Suppressive Amyloidogenesis via HSF1. Cell 160, 729–744. doi:10.1016/j.cell.2015.01.028
Tatar, M., Khazaeli, A. A., and Curtsinger, J. W. (1997). Chaperoning Extended Life. Nature 390, 30. doi:10.1038/36237
Tatsuta, T., Model, K., and Langer, T. (2005). Formation of Membrane-Bound Ring Complexes by Prohibitins in Mitochondria. Mol. Biol. Cell 16, 248–259. doi:10.1091/mbc.e04-09-0807
Tatum, M. C., Ooi, F. K., Chikka, M. R., Chauve, L., Martinez-Velazquez, L. A., Steinbusch, H. W., et al. (2015). Neuronal Serotonin Release Triggers the Heat Shock Response in C. elegans in the Absence of Temperature Increase. Curr. Biol. 25, 163–174. doi:10.1016/j.cub.2014.11.040
Tawo, R., Pokrzywa, W., Kevei, E., Akyuz, M. E., Balaji, V., Adrian, S., et al. (2017). The Ubiquitin Ligase CHIP Integrates Proteostasis and Aging by Regulation of Insulin Receptor Turnover. Cell 169, 470–482. e413. doi:10.1016/j.cell.2017.04.003
Taylor, R. C., Berendzen, K. M., and Dillin, A. (2014). Systemic Stress Signalling: Understanding the Cell Non-autonomous Control of Proteostasis. Nat. Rev. Mol. Cell Biol. 15, 211–217. doi:10.1038/nrm3752
Taylor, R. C., and Dillin, A. (2011). Aging as an Event of Proteostasis Collapse. Cold Spring Harb. Perspect. Biol. 3. doi:10.1101/cshperspect.a004440
Taylor, R. C., and Dillin, A. (2013). XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell 153, 1435–1447. doi:10.1016/j.cell.2013.05.042
Tennessen, J. M., and Thummel, C. S. (2011). Coordinating Growth and Maturation - Insights from Drosophila. Curr. Biol. 21, R750–R757. doi:10.1016/j.cub.2011.06.033
Tomita, T., Hamazaki, J., Hirayama, S., McBurney, M. W., Yashiroda, H., and Murata, S. (2015). Sirt1-deficiency Causes Defective Protein Quality Control. Sci. Rep. 5, 12613. doi:10.1038/srep12613
Tooze, S. A., and Yoshimori, T. (2010). The Origin of the Autophagosomal Membrane. Nat. Cell Biol. 12, 831–835. doi:10.1038/ncb0910-831
Toth, M. L., Sigmond, T., Borsos, E., Barna, J., Erdelyi, P., Takacs-Vellai, K., et al. (2008). Longevity Pathways Converge on Autophagy Genes to Regulate Life Span in Caenorhabditis elegans. Autophagy 4, 330–338. doi:10.4161/auto.5618
Tullet, J. M., Hertweck, M., An, J. H., Baker, J., Hwang, J. Y., Liu, S., et al. (2008). Direct Inhibition of the Longevity-Promoting Factor SKN-1 by Insulin-like Signaling in C. elegans. Cell 132, 1025–1038. doi:10.1016/j.cell.2008.01.030
Ulgherait, M., Rana, A., Rera, M., Graniel, J., and Walker, D. W. (2014). AMPK Modulates Tissue and Organismal Aging in a Non-cell-autonomous Manner. Cell Rep. 8, 1767–1780. doi:10.1016/j.celrep.2014.08.006
Urano, F., Calfon, M., Yoneda, T., Yun, C., Kiraly, M., Clark, S. G., et al. (2002). A Survival Pathway for Caenorhabditis elegans with a Blocked Unfolded Protein Response. J. Cell Biol. 158, 639–646. doi:10.1083/jcb.200203086
van Oosten-Hawle, P., and Morimoto, R. I. (2014). Organismal Proteostasis: Role of Cell-Nonautonomous Regulation and Transcellular Chaperone Signaling. Genes Dev. 28, 1533–1543. doi:10.1101/gad.241125.114
van Oosten-Hawle, P., Porter, R. S., and Morimoto, R. I. (2013). Regulation of Organismal Proteostasis by Transcellular Chaperone Signaling. Cell 153, 1366–1378. doi:10.1016/j.cell.2013.05.015
Vellai, T., Takacs-Vellai, K., Sass, M., and Klionsky, D. J. (2009). The Regulation of Aging: Does Autophagy Underlie Longevity? Trends Cell Biol. 19, 487–494. doi:10.1016/j.tcb.2009.07.007
Verdin, E. (2015). NAD(+) in Aging, Metabolism, and Neurodegeneration. Science 350, 1208–1213. doi:10.1126/science.aac4854
Vilchez, D., Morantte, I., Liu, Z., Douglas, P. M., Merkwirth, C., Rodrigues, A. P., et al. (2012). RPN-6 Determines C. elegans Longevity under Proteotoxic Stress Conditions. Nature 489, 263–268. doi:10.1038/nature11315
Vowels, J. J., and Thomas, J. H. (1992). Genetic Analysis of Chemosensory Control of Dauer Formation in Caenorhabditis elegans. Genetics 130, 105–123. doi:10.1093/genetics/130.1.105
Walker, G. A., and Lithgow, G. J. (2003). Lifespan Extension in C. elegans by a Molecular Chaperone Dependent upon Insulin-like Signals. Aging Cell 2, 131–139. doi:10.1046/j.1474-9728.2003.00045.x
Walther, D. M., Kasturi, P., Zheng, M., Pinkert, S., Vecchi, G., Ciryam, P., et al. (2015). Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell 161, 919–932. doi:10.1016/j.cell.2015.03.032
Wan, Q. L., Shi, X., Liu, J., Ding, A. J., Pu, Y. Z., Li, Z., et al. (2017). Metabolomic Signature Associated with Reproduction-Regulated Aging in Caenorhabditis elegans. Aging (Albany NY) 9, 447–474. doi:10.18632/aging.101170
Wang, M. C., O'Rourke, E. J., and Ruvkun, G. (2008). Fat Metabolism Links Germline Stem Cells and Longevity in C. elegans. Science 322, 957–960. doi:10.1126/science.1162011
Wang, X., Grammatikakis, N., Siganou, A., and Calderwood, S. K. (2003). Regulation of Molecular Chaperone Gene Transcription Involves the Serine Phosphorylation, 14-3-3 Epsilon Binding, and Cytoplasmic Sequestration of Heat Shock Factor 1. Mol. Cell Biol. 23, 6013–6026. doi:10.1128/mcb.23.17.6013-6026.2003
Wang, X., Khaleque, M. A., Zhao, M. J., Zhong, R., Gaestel, M., and Calderwood, S. K. (2006). Phosphorylation of HSF1 by MAPK-Activated Protein Kinase 2 on Serine 121, Inhibits Transcriptional Activity and Promotes HSP90 Binding. J. Biol. Chem. 281, 782–791. doi:10.1074/jbc.m505822200
Watanabe, Y., Tsujimura, A., Taguchi, K., and Tanaka, M. (2017). HSF1 Stress Response Pathway Regulates Autophagy Receptor SQSTM1/p62-Associated Proteostasis. Autophagy 13, 133–148. doi:10.1080/15548627.2016.1248018
Watts, J. S., Harrison, H. F., Omi, S., Guenthers, Q., Dalelio, J., Pujol, N., et al. (2020). New Strains for Tissue-specific RNAi Studies in Caenorhabditis elegans. G3 10, 4167–4176. doi:10.1534/g3.120.401749
Wentz, J. M., Mendenhall, A. R., and Bortz, D. M. (2018). Pattern Formation in the Longevity-Related Expression of Heat Shock Protein-16.2 in Caenorhabditis elegans. Bull. Math. Biol. 80, 2669–2697. doi:10.1007/s11538-018-0482-7
Westerheide, S. D., Anckar, J., Stevens, S. M., Sistonen, L., and Morimoto, R. I. (2009). Stress-inducible Regulation of Heat Shock Factor 1 by the Deacetylase SIRT1. Science 323, 1063–1066. doi:10.1126/science.1165946
Wiederrecht, G., Seto, D., and Parker, C. S. (1988). Isolation of the Gene Encoding the S. cerevisiae Heat Shock Transcription Factor. Cell 54, 841–853. doi:10.1016/s0092-8674(88)91197-x
Wolff, S., and Dillin, A. (2013). Ageing: Beneficial Miscommunication. Nature 497, 442–443. doi:10.1038/497442a
Wolff, S., Weissman, J. S., and Dillin, A. (2014). Differential Scales of Protein Quality Control. Cell 157, 52–64. doi:10.1016/j.cell.2014.03.007
Workman, P., and van Montfort, R. L. (2010). Unveiling the Secrets of the Ancestral PI3 Kinase Vps34. Cancer Cell 17, 421–423. doi:10.1016/j.ccr.2010.04.016
Wotton, D., Pemberton, L. F., and Merrill-Schools, J. (2017). SUMO and Chromatin Remodeling. Adv. Exp. Med. Biol. 963, 35–50. doi:10.1007/978-3-319-50044-7_3
Wu, C. (1995). Heat Shock Transcription Factors: Structure and Regulation. Annu. Rev. Cell Dev. Biol. 11, 441–469. doi:10.1146/annurev.cb.11.110195.002301
Xia, W., and Voellmy, R. (1997). Hyperphosphorylation of Heat Shock Transcription Factor 1 Is Correlated with Transcriptional Competence and Slow Dissociation of Active Factor Trimers. J. Biol. Chem. 272, 4094–4102. doi:10.1074/jbc.272.7.4094
Xu, Y. M., Huang, D. Y., Chiu, J. F., and Lau, A. T. (2012). Post-translational Modification of Human Heat Shock Factors and Their Functions: a Recent Update by Proteomic Approach. J. Proteome Res. 11, 2625–2634. doi:10.1021/pr201151a
Yochem, J., and Herman, R. K. (2003). Investigating C. elegans Development through Mosaic Analysis. Development 130, 4761–4768. doi:10.1242/dev.00701
Yochem, J., Sundaram, M., and Bucher, E. A. (2000). Mosaic Analysis in Caenorhabditis elegans. Methods Mol. Biol. Clift. N. J. 135, 447–462. doi:10.1385/1-59259-685-1:447
Yokoyama, K., Fukumoto, K., Murakami, T., Harada, S., Hosono, R., Wadhwa, R., et al. (2002). Extended Longevity of Caenorhabditis elegans by Knocking in Extra Copies of hsp70F, a Homolog of Mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 516, 53–57. doi:10.1016/s0014-5793(02)02470-5
Yun, C., Stanhill, A., Yang, Y., Zhang, Y., Haynes, C. M., Xu, C. F., et al. (2008). Proteasomal Adaptation to Environmental Stress Links Resistance to Proteotoxicity with Longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 105, 7094–7099. doi:10.1073/pnas.0707025105
Zhang, L., Ward, J. D., Cheng, Z., and Dernburg, A. F. (2015). The Auxin-Inducible Degradation (AID) System Enables Versatile Conditional Protein Depletion in C. elegans. Development 142, 4374–4384. doi:10.1242/dev.129635
Zhang, Y., Lanjuin, A., Chowdhury, S. R., Mistry, M., Silva-Garcia, C. G., Weir, H. J., et al. (2019). Neuronal TORC1 Modulates Longevity via AMPK and Cell Nonautonomous Regulation of Mitochondrial Dynamics in C. elegans. Elife 8. doi:10.7554/eLife.49158
Zhang, Y., Murshid, A., Prince, T., and Calderwood, S. K. (2011). Protein Kinase A Regulates Molecular Chaperone Transcription and Protein Aggregation. PLoS One 6, e28950. doi:10.1371/journal.pone.0028950
Zhao, W., Kruse, J. P., Tang, Y., Jung, S. Y., Qin, J., and Gu, W. (2008). Negative Regulation of the Deacetylase SIRT1 by DBC1. Nature 451, 587–590. doi:10.1038/nature06515
Zheng, X., Krakowiak, J., Patel, N., Beyzavi, A., Ezike, J., Khalil, A. S., et al. (2016). Dynamic Control of Hsf1 during Heat Shock by a Chaperone Switch and Phosphorylation. Elife 5. doi:10.7554/eLife.18638
Zhong, M., Niu, W., Lu, Z. J., Sarov, M., Murray, J. I., Janette, J., et al. (2010). Genome-wide Identification of Binding Sites Defines Distinct Functions for Caenorhabditis elegans PHA-4/FOXA in Development and Environmental Response. PLoS Genet. 6, e1000848. doi:10.1371/journal.pgen.1000848
Zhong, M., Orosz, A., and Wu, C. (1998). Direct Sensing of Heat and Oxidation by Drosophila Heat Shock Transcription Factor. Mol. Cell 2, 101–108. doi:10.1016/s1097-2765(00)80118-5
Zhou, Y., Wang, X., Song, M., He, Z., Cui, G., Peng, G., et al. (2019). A Secreted microRNA Disrupts Autophagy in Distinct Tissues of Caenorhabditis elegans upon Ageing. Nat. Commun. 10, 4827. doi:10.1038/s41467-019-12821-2
Keywords: aging, proteostasis, cell stress and aging, HSF-1 = heat-shock factor-1, longevity, Caenorhabditis elegans (C. elegans), genetics
Citation: Lazaro-Pena MI, Ward ZC, Yang S, Strohm A, Merrill AK, Soto CA and Samuelson AV (2022) HSF-1: Guardian of the Proteome Through Integration of Longevity Signals to the Proteostatic Network. Front. Aging 3:861686. doi: 10.3389/fragi.2022.861686
Received: 25 January 2022; Accepted: 13 June 2022;
Published: 08 July 2022.
Edited by:
Cindy Voisine, Northeastern Illinois University, United StatesReviewed by:
Anat Ben-Zvi, Ben-Gurion University of the Negev, IsraelSrinivas Ayyadevara, Veterans Health Administration, United States
Daniel Czyz, University of Florida, United States
Copyright © 2022 Lazaro-Pena, Ward, Yang, Strohm, Merrill, Soto and Samuelson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew V. Samuelson, YW5kcmV3X3NhbXVlbHNvbkBVUk1DLnJvY2hlc3Rlci5lZHU=
†These authors have contributed equally to this work