
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Antibiot. , 09 May 2023
Sec. Antibiotic Resistance
Volume 2 - 2023 | https://doi.org/10.3389/frabi.2023.1095013
 Liam T. Martin1,2
Liam T. Martin1,2 Eleanor D. Lamming1
Eleanor D. Lamming1 Arundhati Maitra2†Parisa N. Mortazavi2†
Arundhati Maitra2†Parisa N. Mortazavi2† Rebecca Roddan1,2
Rebecca Roddan1,2 John M. Ward3
John M. Ward3 Sanjib Bhakta2*‡
Sanjib Bhakta2*‡ Helen C. Hailes1*‡
Helen C. Hailes1*‡Introduction: The emergence of extensively drug-resistant strains of Mycobacterium tuberculosis threatens decades of progress in the treatment of a disease which remains one of the leading infectious causes of death worldwide. The development of novel antimycobacterial compounds is therefore essential to reinforce the existing antitubercular drug discovery pipeline. There is also interest in new compounds which can synergize with existing antitubercular drugs and can be deployed as part of a combination therapy. This strategy could serve to delay the emergence of resistance to first-line anti-tuberculosis drugs and increase their efficacy against resistant strains of tuberculosis. Previous research has established that several C-1 substituted tetrahydroisoquinolines have antimycobacterial activity. Here we sought to expand our understanding of their antimycobacterial structure activity relationships and their potential to act as adjunct therapies alongside existing antitubercular drugs.
Methods: Three chemical series were synthesised and assayed for their antimycobacterial potency, mammalian cell toxicity, inhibition of whole-cell efflux and synergism with isoniazid, rifampicin, and ethambutol.
Results: Several compounds were found to inhibit the growth of mycobacteria. Potent inhibitors of whole-cell efflux were also identified, as well as compounds which exhibited synergism with rifampicin and ethambutol.
Conclusions: Structure-activity relationships were identified for antimycobacterial potency, improved selectivity, whole cell efflux inhibition and synergism. Potent whole-cell efflux inhibitors and synergistic compounds were identified, suggesting potential development as adjuncts to existing anti-tuberculosis chemotherapy.
Tuberculosis (TB) is one of the leading infectious causes of death worldwide, and the emergence of extensively drug-resistant (XDR) strains of Mycobacterium tuberculosis (Mtb) threatens decades of progress in the treatment of this disease. In 2021, the World Health Organization (WHO: http://www.who.int) estimated that 1.6 million people died of tuberculosis, and 10.6 million new cases were contracted (World Health Organisation, 2022). Of those who developed active TB in 2021, an estimated 450,000 have rifampicin-resistant or multidrug-resistant TB (RR- or MDR-TB) (World Health Organisation, 2022). Consequently, there is an urgent need for the identification of new drug candidates to strengthen the antitubercular drug development pipeline.
In recent years there has been a growing interest in antimycobacterial isoquinolines and tetrahydroisoquinolines (THIQs) (Iwasa et al., 2001; Magnet et al., 2010; Muthusaravanan et al., 2010; Matviiuk et al., 2014; Rotthier et al., 2016; Singh et al., 2017; Singh et al., 2019; Lu et al., 2020). In particular, Singh et al. (2017, 2019) have conducted structure-activity relationship (SAR) studies on a potent C-5-substituted isoquinoline which was initially identified through a high-throughput screening effort with the More Medicines for Tuberculosis (MM4TB) consortium (Magnet et al., 2010). This work led to the identification of several benzylurea derivatives which were potent inhibitors of both Mtb growth and the drug target inosine-5’-monophosphate dehydrogenase (IMPDH) (Singh et al., 2017; Singh et al., 2019). Lu et al. have also recently reported SAR studies of a series of N-5,8-trisubstituted THIQs with potent activity against Mtb, some of which show promising selectivity for toxicity to Mtb H37Rv over the VERO cell line (Lu et al., 2020). A prior SAR study from our research group found that C-1 substituted THIQs bearing a C-5-halogen and a C-8 hydroxy or methoxy group demonstrated good antimycobacterial potency, with some limited selectivity for toxicity to mycobacteria over mammalian cells (Guzman et al., 2015).
Efflux pumps are membrane-bound proteins which transport toxic substances across the bacterial cell wall and out of the cell. These proteins contribute to drug resistance by decreasing the critical concentration of drug inside the cell to a sub-inhibitory level (Machado et al., 2017; Du et al., 2018). This represents a particular problem as it increases the likelihood of resistance-conferring genetic mutations emerging (Machado et al., 2012). Combination therapy using efflux pump inhibitors (EPIs) alongside existing anti-TB drugs therefore represents a potential strategy to improve the efficacy of anti-TB drugs and limit the emergence of drug resistant TB (Rodrigues et al., 2020). Well-established EPIs include verapamil (Gupta et al., 2013; Adams et al., 2014), chlorpromazine (Rodrigues et al., 2008; Grimsey et al., 2020) and the plant alkaloid reserpine (Schmitz et al., 1998; Markham, 1999), while a range of novel efflux inhibitors of both natural (Nakamura de Vasconcelos et al., 2018; Solnier et al., 2020; Tran et al., 2020; Laws et al., 2022) and synthetic (Abate et al., 2016; Lentz et al., 2016; Scalacci et al., 2017; Lentz et al., 2018; Sen et al., 2018; Laws et al., 2022) origin have been identified in recent years. It has been reported that the use of verapamil in combination with rifampicin or bedaquiline resulted in reductions in their in vitro minimum inhibitory concentration (MIC) against Mtb of 4-fold or 8-fold, respectively (Gupta et al., 2014; Singh et al., 2014). Studies performed in in vivo mouse models have also demonstrated the efficacy of verapamil in combination with isoniazid, rifampicin and pyrazinamide (Gupta et al., 2013). Furthermore, co-administration of verapamil and bedaquiline has been shown to allow for lower dosing and while reducing the emergence of drug-resistance in mice (Gupta et al., 2014). These studies highlight the potential of EPIs to increase the efficacy of the existing anti-TB arsenal, but there are currently no inhibitors of mycobacterial efflux pumps in clinical use.
In this work, we aimed to further probe the antimycobacterial potential of the THIQ scaffold through the synthesis and SAR analysis of three groups (I-III) of analogues. The high-throughput spot-culture growth inhibition (HT-SPOTi) assay (Danquah et al., 2016) was used to determine the MIC of all compounds against the Mtb model organism M. aurum (Gupta and Bhakta, 2012), with a selection of compounds additionally screened against M. bovis BCG (Altaf et al., 2010). Compounds were also assayed for mammalian cell cytotoxicity against murine RAW 264.7 cells with some compounds also screened against the human THP-1 cell line. This allowed for a determination of the selectivity of these compounds for toxicity to mycobacteria over mammalian cells. To provide preliminary insights into the synergistic properties of some of the compounds, including brominated analogues which had previously been reported as having potentially interesting activities, selected compounds were evaluated for their ability to inhibit whole-cell efflux mechanisms of M. aurum using an ethidium bromide accumulation assay (Rodrigues et al., 2008; Paixão et al., 2009) and for their ability to synergize with isoniazid, rifampicin and ethambutol using a HT-SPOTi checkerboard method (Guzman et al., 2013).
Structure-activity relationships for the antimycobacterial activity of this compound class were established. THIQs which were able to inhibit multidrug efflux in M. aurum were also identified, with initial insights made into the structural features associated with efflux inhibition. In addition, four THIQs were found to synergize with either rifampicin or ethambutol at low concentrations, presenting the opportunity to further develop these compounds as adjunct therapies to the existing first-line antituberculosis drug regimen.
The bacterial species used in this study were Mycobacterium aurum (ATCC 23366) and M. bovis BCG Pasteur (ATCC 35734). The mammalian cell lines used for cytotoxicity assays were the murine macrophage cell line RAW 264.7 (ATCC-71) and human leukemia monocytic cell line THP-1 (ATCC TIB-202). Mycobacteria were cultured in Middlebrook 7H9 broth (BD Difco™) or Middlebrook 7H10 agar media (BD Difco™) supplemented with albumin/dextrose/catalase (ADC, Remel™) or oleic acid/albumin/dextrose/catalase (OADC, BD Difco™) enrichments, respectively. Mammalian cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 media with L-glutamine (Gibco®) supplemented with fetal bovine serum (FBS, Sigma Aldrich, Non-US origin, sterile filtered, heat-inactivated).
The procedures employed for chemical synthesis are reported in the Supplementary Information together with the relevant characterization data.
All compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 50 mg/ml. 2 µl aliquots from a 2-fold serial dilution of this stock were transferred into a 96-well microtiter plate. Isoniazid was included as a positive control and DMSO as a negative control. Middlebrook 7H10 agar medium containing 0.5% glycerol and 10% (v/v) OADC (200 µl) was dispensed into each well and allowed to solidify. A mid-logarithmic phase culture of M. aurum or M. bovis BCG was diluted in Middlebrook 7H9 broth to give a cell density of 1 x 106 cells/ml (Gupta and Bhakta, 2012) and then dispensed (2 µl, ~ 2000 cells) into each well aseptically. The plates were then incubated at 35 °C (for M. aurum) and 37 °C (for M. bovis BCG). The results were observed after 5 days for M. aurum or 14 days for M. bovis BCG. MICs were visually determined by observing the lowest concentration of each compound at which no bacterial growth could be observed. These assays were performed in biological triplicate.
All compounds were dissolved in DMSO to a concentration of 50 mg/ml. Aliquots (2 µl) from a 2-fold serial dilution of this stock were transferred into a 96-well microtiter plate, with a DMSO-only well included as a negative control. Cultures of RAW 264.7 or THP-1 cells (5 ml), were diluted to a cell density of 5 x 105 cells/ml in RPMI and aliquots (100 µl) were then transferred into each well of the 96-well plate. One row of the 96-well plate was left without cells, as a sterility control. The cells were then incubated without shaking (37 °C, 5% CO2) for 48 hours. The spent media was removed by pipetting and the media was replaced with complete RPMI (170 µl) and resazurin solution (0.01%, 30 µl). For non-adherent THP-1 cells, the cells were pelleted by centrifugation of the 96-well plates (270 x g, 2 min) before aspiration of the media. The plates for both cell lines were then incubated for a further 24 h (37 °C, 5% CO2). Growth inhibitory concentrations (GICs) were visually determined by observing the lowest concentration of each compound at which the resazurin dye was not reduced from blue to pink. Visual determinations were confirmed by fluorescence readings (Excitation wavelength: 560 nm, Emission wavelength: 590 nm) for each plate using a BioTek® Synergy 2™ Multi-Detection Plate Reader. These assays were performed in biological triplicate.
The assay was modified from a previously published protocol (Rodrigues et al., 2008). Early log phase cells of M. aurum (OD600 ∼ 0.8) were harvested by centrifugation and resuspended in 1 x PBS to an OD600 of 0.4. The test samples contained 4−6 × 107 CFU/ml in PBS, glucose (0.4%), ethidium bromide (0.5 mg/l), and the compounds of interest at ¼ × MIC. Blank samples contained all of the components mentioned above, except the bacterial suspension, which was replaced with 1 × PBS. Verapamil, a known efflux pump inhibitor, was used as the positive control at a concentration of 125 µg/ml. The experiment was performed in a fluorimeter (FLUOstar OPTIMA, BMG Labtech) programmed with the following parameters: wavelengths of 544 nm for excitation and 590 nm for detection of fluorescence, gain 2200, a temperature of 37°C, and a cycle of measurement every minute for a total period of 60 min. The accumulation or efflux of ethidium bromide was monitored on a real-time basis. The ability of compounds to inhibit ethidium bromide efflux was quantified by determining the relative final fluorescence (RFF) of each compound. This was calculated according to Formula 1:
RFtreated is the relative fluorescence (RF) at the last time point of the EtBr accumulation assay for the experimental sample, while RFcontrol is the RF of the untreated control sample at the last time point of the assay (Machado et al., 2011; Coelho et al., 2015).
The synergistic effect of THIQs were examined in combination with isoniazid, ethambutol and rifampicin against M. aurum. The assay was conducted in a 96-well microtiter plate using a SPOTi checkerboard distribution as reported previously (Guzman et al., 2013). The compounds were serially diluted in DMSO across a range of concentrations spanning both above and below the MIC of each compound/drug. Each row of the 96-well plate contained different concentrations of the THIQ derivatives, and each column contained different concentrations of the first-line anti-TB drug. The checkerboard was constructed by adding 1 µl of each of the stock concentrations to the corresponding well and then dispensing 200 µl of warm Middlebrook 7H10 agar medium. The plates were then spotted with M. aurum. The plates were incubated in sealed bags at 35° C for 5 days and Fractional Inhibitory Concentration Indices A FICI value were calculated using Formula 2:
An FICI value ≤ 0.5 indicates synergism; a value between 0.5 and 4.0 indicates no interaction; and a value higher than 4.0 indicates antagonism (Odds, 2003).
An initial group of C-1 substituted THIQs (Group I) was prepared via a phosphate-mediated, biomimetic Pictet-Spengler reaction between dopamine hydrochloride and a range of aldehydes (Table 1) (Pesnot et al., 2011). Both the major (6,7-, 1-10a) and minor (7,8-, 1-10b) regioisomers were isolated by preparative HPLC (Guzman et al., 2015). The C-1 alkyl chain length was systematically explored, as previous work had highlighted a longer alkyl chain at this position as having promising activities. C-1 cyclopentyl, 2-methyl benzyl and 3,4-methylenedioxybenzyl moieties were also explored, the latter of which had been present in several active analogues identified previously (Guzman et al., 2015). Moreover, a hydroxyl group at C-8 had also been associated with higher activities in some cases, so both 6,7- and 7,8-regioisomers were generated (Guzman et al., 2015). All compounds were prepared using commercially available aldehydes with the exception of 9a and 9b, for which (m-tolyl)acetaldehyde was prepared from the commercially available alcohol by a Parikh-Doering oxidation (Parikh and Doering, 1967), and 10a and 10b, for which 3,4-(methylenedioxy)phenylacetaldehyde was prepared as previously described (Guzman et al., 2015). Regioisomers 11a and 11b were prepared from a Pictet-Spengler reaction between m-tyramine and hexanal. THIQs 4a.HCl and 11a.HCl were prepared as the hydrochloride salts of their respective free-bases. The enantio-enriched THIQs (1S)-4a.HCl and (1S)-11a.HCl were synthesized using norcoclaurine synthase (TfNCSΔ33) in a biocatalytic Pictet-Spengler reaction between hexanal and dopamine or m-tyramine, respectively (Table 1) and were included in Group I to explore C-1 stereochemistry (Pesnot et al., 2012; Ruff et al., 2012; Lichman et al., 2015).
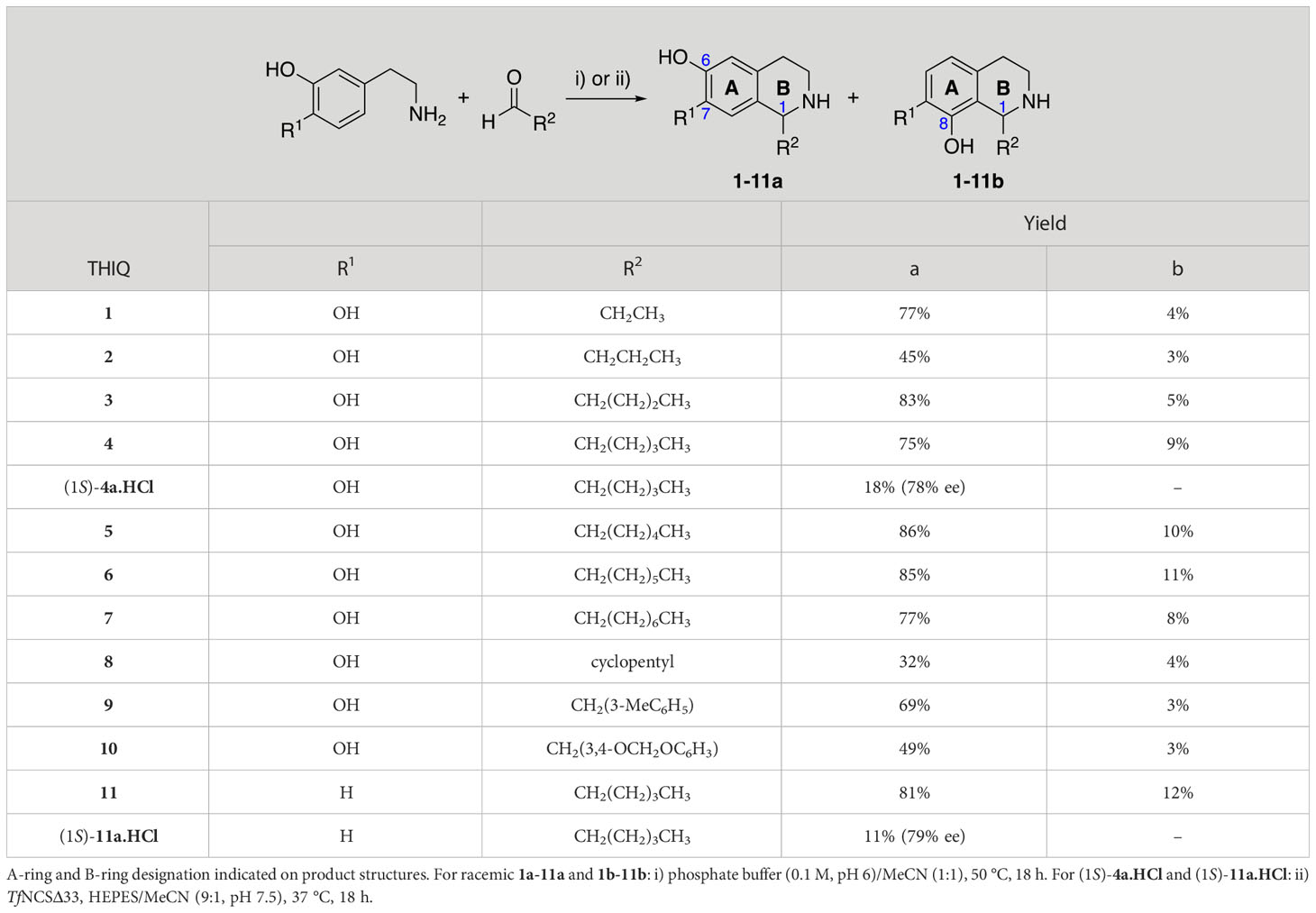
Table 1 Synthesis of Group I THIQs.
Group II was designed primarily to further the exploration of A-ring substituents, including hydroxy substitutions unexplored in Group I; C-5 bromo substitution and A-ring methoxy substituents. A C-1 pentyl chain was retained for all compounds as the most potent and selective compounds in Group I featured this substituent (see below). The initial phenethylamines required for the synthesis of this series were either acquired commercially or prepared by reduction of the appropriate phenylacetonitrile (see supplementary information). Brominated phenethylamines were prepared by bromination and subsequent demethylation of the appropriate methoxy phenethylamines (Scheme S1). THIQs 12-15 were then prepared via a phosphate-mediated Pictet-Spengler reaction (Table 2) (Pesnot et al., 2011). THIQ 16 with a C-7 hydroxyl group was also prepared to compare to 11a which features a C-6 hydroxyl group. The synthesis of 16 (Scheme 1A) began with an initial amide coupling reaction between 4-methoxyphenyethylamine and hexanoic acid to give 17. This was followed by Bischler-Napieralski cyclisation (Heravi et al., 2014) and subsequent imine reduction to afford 18 and 19 as a mixture of regioisomers. These compounds were then demethylated and separated by preparative HPLC to isolate 16 (and 11a; Scheme 1A). Methoxy-substituted THIQs at C-6; C-6 and C-7; or C-6 and C-8 (18, 20 and 21) were synthesized in a similar manner (Scheme 1B) to compare to their hydroxy-substituted counterparts 4a, 11a and 12. First, amides 22-24 were prepared by a Sheppard amidation (Sabatini et al., 2017), before Bischler-Napieralski cyclisation to afford dihydroisoquinolines 25-27, which then underwent imine reduction to give the desired THIQs 18, 20 and 21.
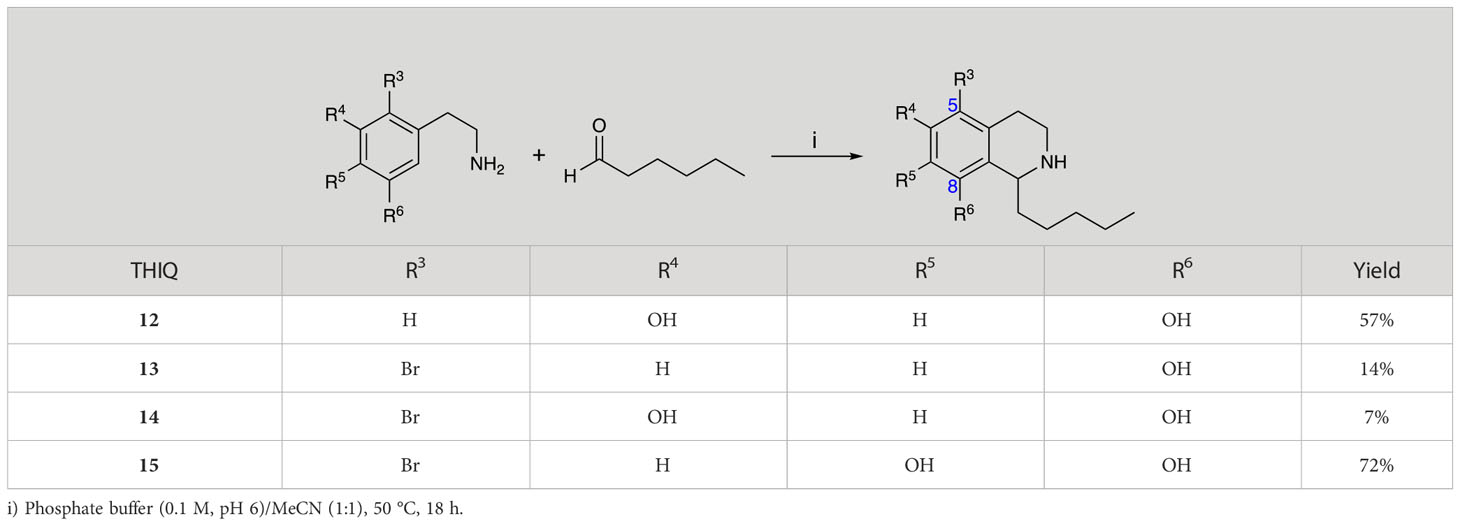
Table 2 Synthesis of Group II THIQs.
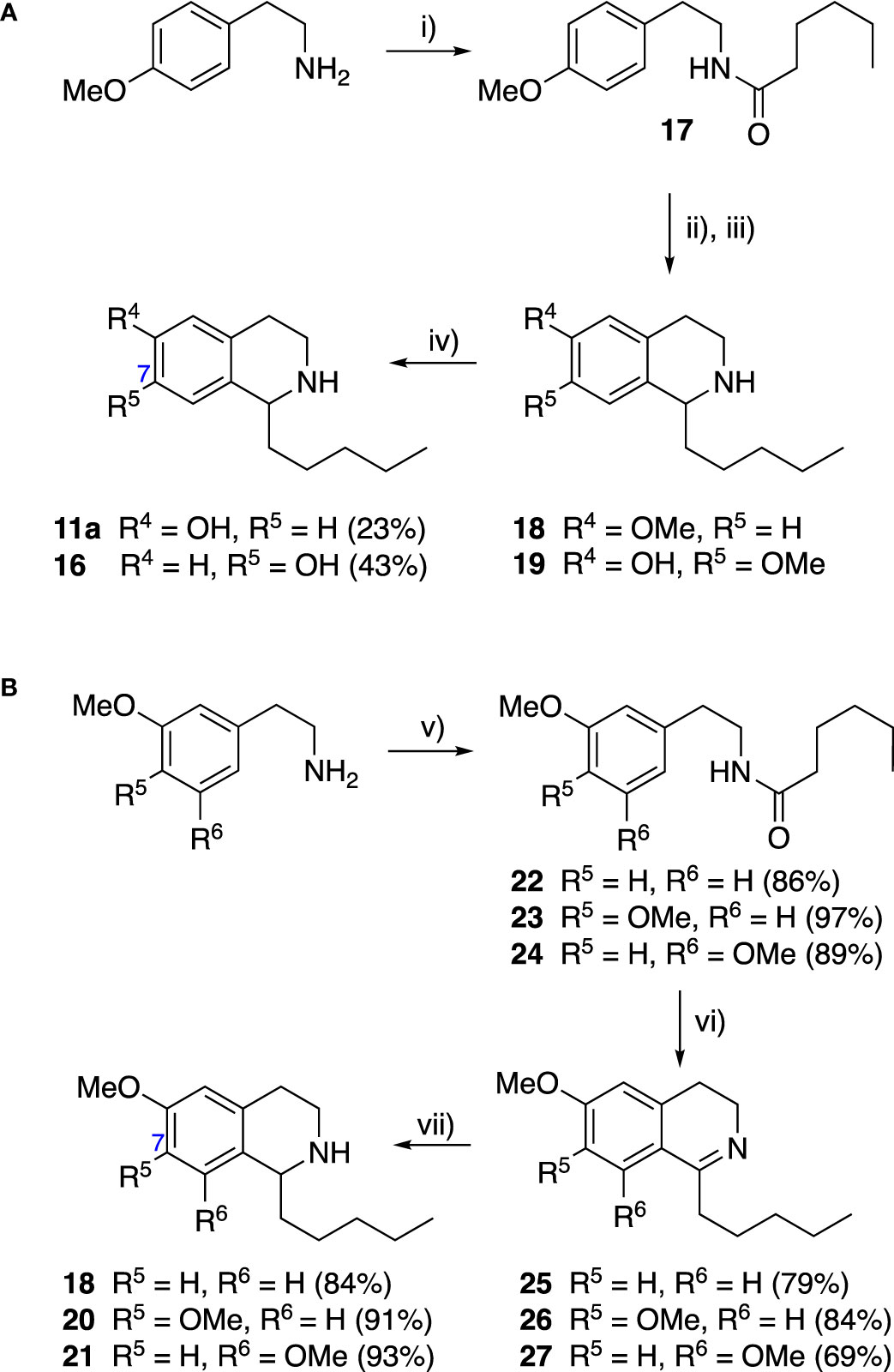
Scheme 1 (A) Synthesis of 7-hydroxy-substituted THIQ 16. Reaction conditions: i) hexanoyl chloride, Et3N, CH2Cl2, (90%), ii) P2O5, POCl3, xylene, 20 h, iii) NaBH4, MeOH (37% over two steps, 1:1.6 ratio of regioisomers 18 and 19), iv) BBr3, CH2Cl2, 78°C, rt, 24 h. (B) Synthesis of 6-methoxy-, 7-methoxy-, and 8-methoxy-substituted THIQs 18, 21 and 22. Reaction conditions: v) B(OCH2CF3), TAME, reflux, 18 h, vi) POCl3, xylene, reflux, 18 h, vii) NaBH4, MeOH, rt, 18 h.
To explore the influence of B-ring aromaticity, Group III was prepared. This group contained fully aromatic isoquinolines rather than THIQs. The A-ring-unsubstituted isoquinoline 28 was prepared by first synthesizing β-hydroxy amide 29 via a Sheppard amidation (Sabatini et al., 2017) between 2-amino-1-phenylethanol and hexanoic acid (Scheme 2A). This was followed by a Pictet-Gams cyclisation (Wang, 2010) to directly afford the fully aromatic isoquinoline. A-ring substituted isoquinolines were prepared by oxidation of previously prepared dihydroisoquinolines 25-27 with Pd/C under air to afford the methoxy-substituted compounds 30-32 (Scheme 2B) (Awuah and Capretta, 2010). These were then demethylated to afford their hydroxy-substituted counterparts 33-35. Compound 36 (Scheme 2C) was synthesized as previously reported, for use in synergism studies (Guzman et al., 2015).
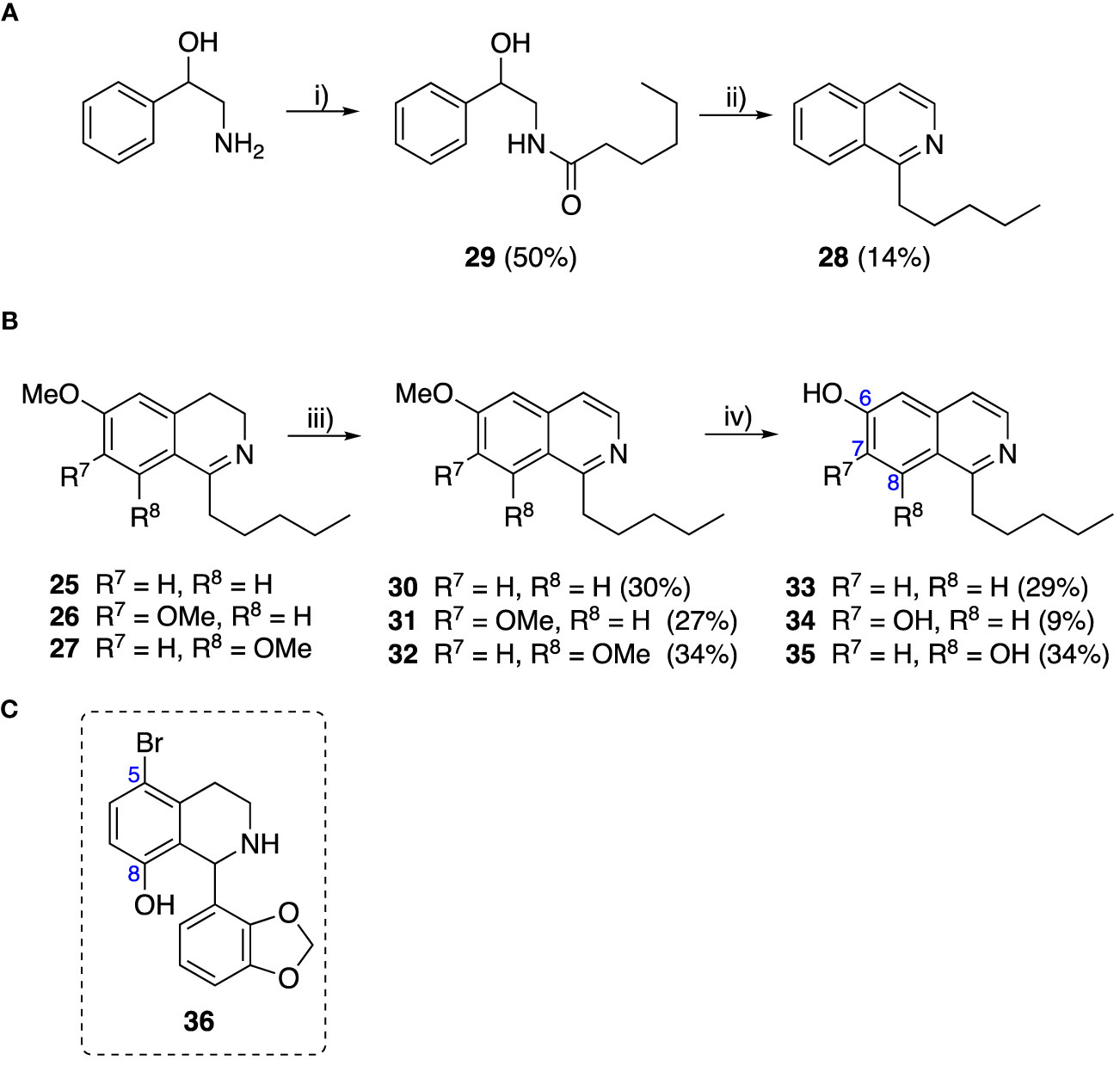
Scheme 2 (A) Synthesis of A-ring unsubstituted isoquinoline. Reaction conditions: i) B(OCH2CF3)3, TAME, reflux, 18 h (50%), ii) POCl3, xylene, reflux, 18 h. (B) Synthesis of A-ring substituted isoquinolines. Reaction conditions: iii) Pd/C, 150°C, iv) BBr3, CH2Cl2, 18 h. (C) Structure of 36.
Generally, for the 6,7-dihydroxy-substituted THIQs, 1a-7a, potency against M. aurum was found to increase with increasing chain length (Table 3). Toxicity to RAW 264.7 cells was high for THIQs 1a, 2a, 4a, 6a and 7a. However, 3a and 5a displayed lower cytotoxicity, with GIC values of ≥ 62.5 µg/ml, indicating a non-linear relationship between carbon chain length and cytotoxicity. The same trends were not observed for the 7,8-dihydroxy-substituted THIQs, 1b-7b, all of which had MIC values of ≥ 125 µg/ml, with no clear relationship between chain length and activity (Table 3). All of these compounds had high cytotoxicity against the RAW 264.7 cell line with GIC values of 15.6 µg/ml, suggesting no relationship between chain length and cytotoxicity. Of the alkyl chain-bearing THIQs, the C-1 pentyl THIQs, 4a and 4b, appeared to confer the best balance of antimycobacterial potency and mammalian cell cytotoxicity. For the THIQs with cyclic C-1 substituents, it was observed that the 7,8-hydroxy-substituted examples had greater potency against M. aurum than their 6,7-substituted counterparts (Table 3). However, these compounds were also more toxic to RAW 264.7 cells and did not show improved selectivity. Only the 3,4-methylenedioxybenzyl moiety (10a & 10b) was found to confer comparable potency and selectivity to the C-1 pentyl group (4a & 4b). The 6- and 8-mono-hydroxy-substituted THIQs (11a & 11b) were observed to have similar activity to 4a and 4b (Table 3). Both 4a.HCl and 11a.HCl were found to have comparable activity to their respective free-bases, 4a and 11a, however the hydrochloride salts were found to exhibit reduced cytotoxicity against the RAW 264.7 cell line. Many THIQs and isoquinolines in Group II and all of Group III were therefore formulated as their HCl salts to reduce cytotoxicity and to improve the solubility of the more lipophilic compounds. Interestingly, (1S)-4a.HCI and (1S)-11a.HCI, were found to have low potency and cytotoxicity. When compared to the equivalent racemic THIQs, these results suggest that the (1R)-THIQs may be more potent and cytotoxic than the corresponding (1S)-THIQs. All compounds in this series displayed selectivity for toxicity to the mammalian RAW 264.7 cells over M. aurum; an issue which would require optimization to overcome.

Table 3 Antimycobacterial and cytotoxicity data for Group I.
In THIQ Group II (Table 2 and Scheme 1), it was observed that the transition away from catechol-bearing THIQs generally resulted in increased potency and selectivity (Table 4). Dimethoxy-substituted THIQ 21 was the most promising compound in this series, with the highest potency observed against both M. aurum and M. bovis BCG. The high cytotoxicity of this compound against RAW 264.7 resulted in SI values of 1 or 2 (M. aurum or M. bovis BCG, respectively). These SI values are in line with those observed for the most selective THIQs reported in our previous work (Guzman et al., 2015). The two dimethoxy-substituted THIQs, 20 and 21, were also among the most selective compounds in the series, suggesting that A-ring methoxy-substitution may improve selectivity.

Table 4 Antimycobacterial and cytotoxicity activity for Group II.
The isoquinolines in Group III (Schemes 2A, B) were found to have similar activity to their THIQ counterparts (Table 5), suggesting that B-ring aromaticity had a limited influence on the activity of these compounds. A notable exception to this trend is 33, which was observed to be significantly more antimycobacterial and selective than the corresponding THIQ, 11a. This compound is also notable for having an SI of 2 for both M. aurum and M. bovis BCG over RAW 264.7 cells and is the only compound in this study with any selectivity for toxicity to M. aurum over the RAW 264.7 cell line. Compound 32 was the most potent compound in this series against both mycobacterial species. It was not found to be selective for toxicity to M. aurum but had an SI of 2 for M. bovis BCG over RAW 264.7.
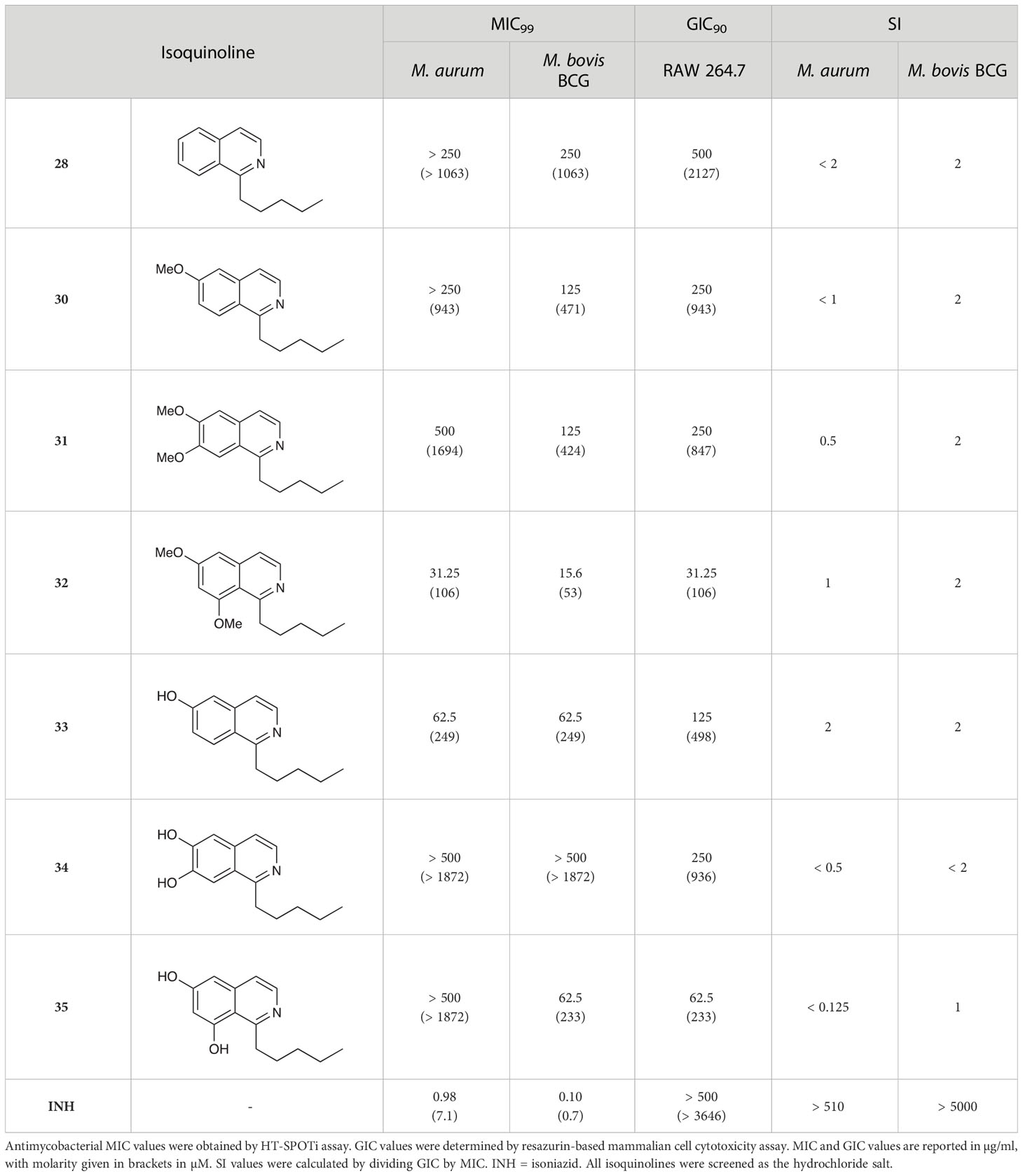
Table 5 Antimycobacterial and cytotoxicity data for Group III isoquinolines.
The compounds in Group III were also screened for cytotoxicity against the human THP-1 cell line, alongside selected compounds from Groups I and II (Table S1). In general, these compounds showed similar cytotoxicity against this cell line, however, some displayed lower cytotoxicity, resulting in increased SI values. Notably, Compound 11a had an SI value of 2 for activity against M. bovis BCG over THP-1 cells, while 21 and 30 had SI values of 4.
The 6,7-dihydroxy-THIQs (2a, 5a & 6a) displayed little to no inhibition of ethidium bromide efflux (Table 6). However, all 7,8-dihydroxy-THIQs (2b-5b), with the exception of 6b, displayed efflux inhibition. 5b resulted in the greatest accumulation of ethidium bromide of the THIQs tested, with an RFF of 1.42, demonstrating some potency as an efflux inhibitor, but still significantly less than the RFF of 2.09 observed for the positive control Verapamil. The activity of the 7,8-dihydroxy substituted THIQs alongside the inactivity of the 6,7-dihydroxy-subsituted analogues, suggested that either a C-8 hydroxy substituent was a requirement for efflux pump inhibition, or that a C-6 hydroxy substituent abolishes this activity. The activity of the 5-bromo-substituted THIQ 13 suggested that a bromide atom at this position may confer efflux pump inhibition, while the low activity of 36 demonstrated that the C-1 substituent is also an important factor in efflux pump inhibition. Interestingly, no correlation was observed between efflux pump activity and compound MIC.
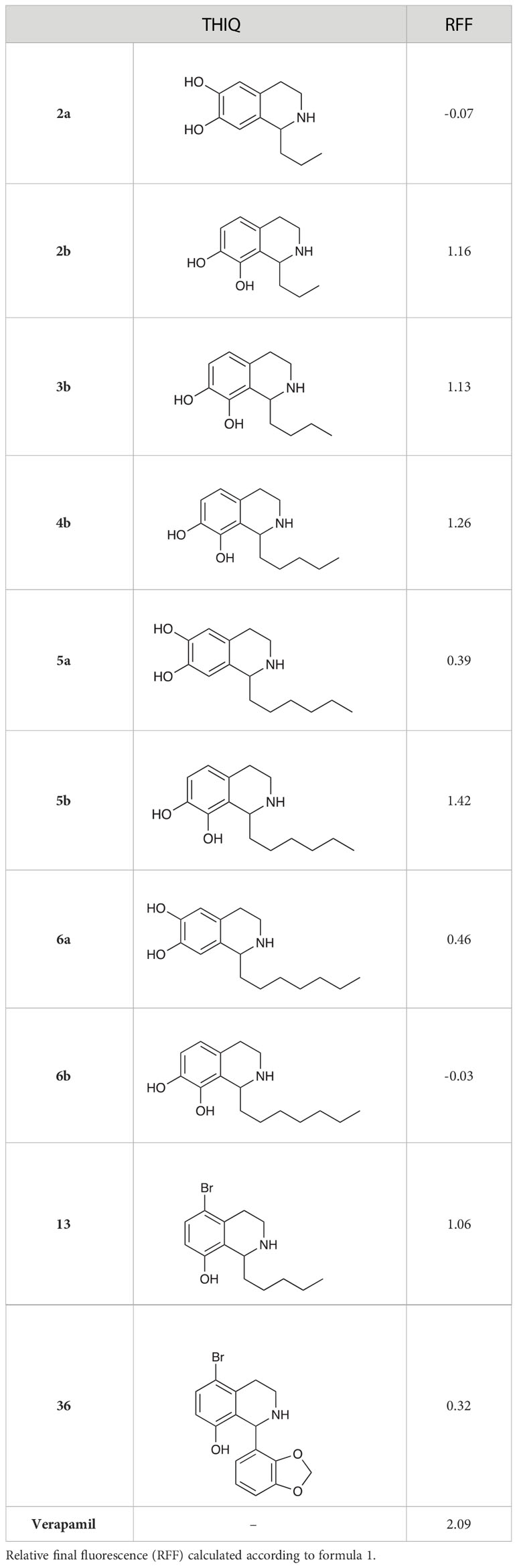
Table 6 Efflux pump inhibition by selected THIQs.
The most notable trend from the synergism studies (Table 7) is that the 5-bromo-substituted THIQs, 15 and 36, were both able to affect a 2-fold increase in the potency of rifampicin at a concentration of 0.1 µg/ml, with FICIs of 0.5, indicating synergism. Compound 7b was also found to have the same activity alongside rifampicin. None of the compounds assayed were found to have synergistic activity with isoniazid, and only compound 5b was found to have any synergistic activity with ethambutol, causing a 2-fold increase in potency, at a concentration of 0.1 µg/ml.
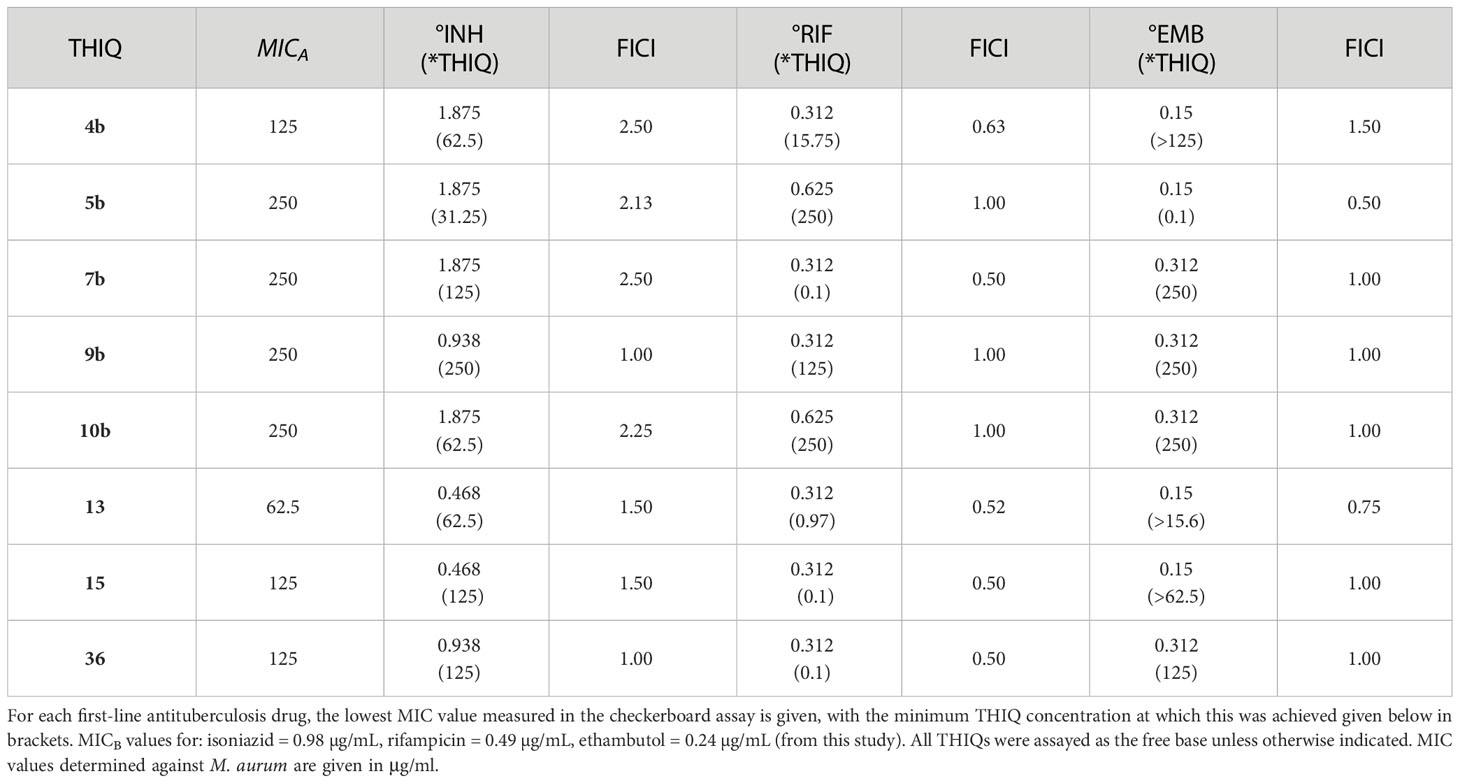
Table 7 Synergism Data.
Structure activity relationships were established for C-1 substituted THIQs and isoquinolines. An n-pentyl chain was found to be the optimal length of C-1 alkyl substituent, while the 3,4-methylenedioxyphenyl group was also found to perform well in this position. A comparison of the (1S)-THIQs, (1S)-4a.HCl and (1S)-11a.HCl, with their racemic counterparts suggested that (1R)-THIQs may be more potent and cytotoxic than the corresponding (1S)-THIQs. This suggests that the (1R)-THIQs may confer no advantage in selectivity, as increased cytotoxicity would negate any gains in potency. However, only two pair-wise matches were compared in this study, and it may be yet possible to leverage the stereochemistry of this scaffold to improve the selectivity of other THIQs with further research.
It was also observed that the replacement of A-ring hydroxy groups with methoxy groups typically increased selectivity. The presence of a C-8 methoxy substituent was typically found to confer greater antimycobacterial potency to compounds in this study. Notably, the 6,8-dimethoxy-substituted THIQ 21 was found to be a potent inhibitor of M. bovis BCG growth (MIC = 15.6 µg/ml), while also having an SI of 4 over the human THP-1 cell line. This suggests that the careful selection of A-ring substituents can be an effective approach to engineering the activity of this compound class. A-ring substitution was found to have a much greater influence on the activity of these compounds than B-ring aromaticity, for which no clear impact on compound activity could be determined.
For those compounds which were screened against both M. aurum and M. bovis BCG, it was notable that greater potency was typically observed against the latter. While both are valid model organisms for Mtb, M. bovis BCG is the more closely related of the two (Garnier et al., 2003; Altaf et al., 2010; Gupta and Bhakta, 2012; Phelan et al., 2015), and so it may be expected that the antitubercular activity of these compounds more closely reflects that observed against M. bovis BCG. Small differences in cytotoxicity were also observed when screening against THP-1 over RAW 264.7, however these were minor and both mammalian cell lines generally gave similar results.
Overall, despite the identification of key structure activity relationships, no compound in this study was sufficiently potent or selective to take forward for hit-to-lead optimisation (Katsuno et al., 2015). In order to conduct further optimization of the most promising compounds in from this study it would be prudent to engage in further exploration of A and B-ring substituents. Potent antitubercular isoquinolines and THIQs with large substituents in the C-5 and C-8 positions have been reported in the literature and it may be possible to take inspiration from these studies to build upon the most promising C-5 and C-8 substituted compounds reported in this work (Magnet et al., 2010; Singh et al., 2017; Singh et al., 2019; Lu et al., 2020). N-substituted THIQs could also be explored. This would increase the lipophilicity of the THIQs in this series, potentially improving their permeability through the mycobacterial membrane. Several potent antitubercular N-substituted THIQs have also been reported previously (Guzman et al., 2010; Matviiuk et al., 2014; Rotthier et al., 2016).
An area in which these compounds may be developed to add value in the fight against DR-TB is as EPIs for use in combination with existing antitubercular drugs. Several compounds, at their sub-MICs, were found to be inhibitors of whole-cell efflux of ethidium bromide, and some early structure activity relationships have been identified. As only hydroxyl and bromo substituents have so far been explored in these assays, there is still space to expand on these to identify more potent efflux inhibitors with improved drug-like properties.
Four THIQs (5b, 7b, 15 and 36) were found to synergize with either rifampicin or ethambutol and it was observed that a 5-bromo substituent appeared important in conferring synergism with rifampicin, though other substituents in this position are yet to be explored. These compounds therefore merit further study and optimization for their synergistic activity. That synergism was observed at a concentration significantly below their GIC values suggests that these compounds may have a broad therapeutic window when applied as an adjunct therapy. Further studies will therefore be required to unpick the relationship between efflux inhibition and synergism in these cases, and to confirm the method through which synergism was occurring.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
LM and EL synthesized compounds. LM, AM, and PM performed assays and evaluated biological data. RR and JW purified enzymes for biocatalytic reactions. HCH and SB contributed conceptualization and supervision. LM wrote the first draft of the article. All authors contributed to designing the article. All authors contributed to the article and approved the submitted version.
LM received funding from the Wellcome Trust (grant code: 108877/Z/15/Z). EL received funding from GlaxoSmithKline and the EPSRC (Engineering and Physical Sciences Research Council). AM and RR were funded by a Birkbeck Anniversary PhD Scholarship. The EPSRC (EP/P020410/1) provided 700 MHz NMR (Nuclear Magnetic Resonance) equipment support. The funders were not involved in the study design, collection, analysis or interpretation of data, the writing of this article or the decision to submit it for publication. All authors declare no other competing interests.
We thank Dr Abil Aliev for NMR support, and Dr Kersti Karu and Dr Malgorzata Puchnarewicz for mass spectrometry assistance. We thank Dr Joanna Redmond and Dr Deborah Needham at GlaxoSmithKline for helpful discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frabi.2023.1095013/full#supplementary-material
Abate G., Ruminiski P. G., Kumar M., Singh K., Hamzabegovic F., Hoft D. F., et al. (2016). New verapamil analogs inhibit intracellular mycobacteria without affecting the functions of mycobacterium-specific T cells. Antimicrob. Agents Chemother. 60, 1216–1225. doi: 10.1128/AAC.01567-15
Adams K. N., Szumowski J. D., Ramakrishnan L. (2014). Verapamil, and its metabolite norverapamil, inhibit macrophage-induced, bacterial efflux pump-mediated tolerance to multiple anti-tubercular drugs. J. Infect. Dis. 210, 456–466. doi: 10.1093/infdis/jiu095
Altaf M., Miller C. H., Bellows D. S., O’Toole R. (2010). Evaluation of the mycobacterium smegmatis and BCG models for the discovery of mycobacterium tuberculosis inhibitors. Tuberculosis 90, 333–337. doi: 10.1016/j.tube.2010.09.002
Awuah E., Capretta A. (2010). Strategies and synthetic methods directed toward the preparation of libraries of substituted isoquinolines. J. Org. Chem. 75, 5627–5634. doi: 10.1021/jo100980p
Coelho T., Machado D., Couto I., Maschmann R., Ramos D., von Groll A., et al. (2015). Enhancement of antibiotic activity by efflux inhibitors against multidrug resistant mycobacterium tuberculosis clinical isolates from Brazil. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00330
Danquah C. A., Maitra A., Gibbons S., Faull J., Bhakta S. (2016). HT-SPOTi: a rapid drug susceptibility test (DST) to evaluate antibiotic resistance profiles and novel chemicals for anti-infective drug discovery. Curr. Protoc. Microbiol. 40, 17.8.1–17.8.12. doi: 10.1002/9780471729259.mc1708s40
Du D., Wang-Kan X., Neuberger A., van Veen H. W., Pos K. M., Piddock L. J. V., et al. (2018). Multidrug efflux pumps: structure, function and regulation. Nat. Rev. Microbiol. 16, 523–539. doi: 10.1038/s41579-018-0048-6
Garnier T., Eiglmeier K., Camus J. C., Medina N., Mansoor H., Pryor M., et al. (2003). The complete genome sequence of mycobacterium bovis. Proc. Natl. Acad. Sci. 100, 7877–7882. doi: 10.1073/pnas.1130426100
Grimsey E. M., Fais C., Marshall R. L., Ricci V., Ciusa M. L., Stone J. W., et al. (2020). Chlorpromazine and amitriptyline are substrates and inhibitors of the AcrB multidrug efflux pump. MBio 11, e00465–20. doi: 10.1128/mBio.00465-20
Gupta A., Bhakta S. (2012). An integrated surrogate model for screening of drugs against mycobacterium tuberculosis. J. Antimicrob. Chemother. 67, 1380–1391. doi: 10.1093/jac/dks056
Gupta S., Cohen K. A., Winglee K., Maiga M., Diarra B., Bishai W. R. (2014). Efflux inhibition with verapamil potentiates bedaquiline in mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 574–576. doi: 10.1128/AAC.01462-13
Gupta S., Tyagi S., Almeida D. V., Maiga M. C., Ammerman N. C., Bishai W. R. (2013). Acceleration of tuberculosis treatment by adjunctive therapy with verapamil as an efflux inhibitor. Am. J. Respir. Crit. Care Med. 188, 600–607. doi: 10.1164/rccm.201304-0650OC
Guzman J. D., Evangelopoulos D., Gupta A., Birchall K., Mwaigwisya S., Saxty B., et al. (2013). Antitubercular specific activity of ibuprofen and the other 2-arylpropanoic acids using the HT-SPOTi whole-cell phenotypic assay. BMJ Open 3, e002672. doi: 10.1136/bmjopen-2013-002672
Guzman J. D., Gupta A., Evangelopoulos D., Basavannacharya C., Pabon L. C., Plazas E. A., et al. (2010). Anti-tubercular screening of natural products from Colombian plants: 3-methoxynordomesticine, an inhibitor of MurE ligase of mycobacterium tuberculosis. J. Antimicrob. Chemother. 65, 2101–2107. doi: 10.1093/jac/dkq313
Guzman J. D., Pesnot T., Barrera D. A., Davies H. M., McMahon E., Evangelopoulos D., et al. (2015). Tetrahydroisoquinolines affect the whole-cell phenotype of mycobacterium tuberculosis by inhibiting the ATP-dependent MurE ligase. J. Antimicrob. Chemother. 70, 1691–1703. doi: 10.1093/jac/dkv010
Heravi M. M., Khaghaninejad S., Nazari N. (2014). Bischler–napieralski reaction in the syntheses of isoquinolines. Adv. Heterocyclic Chem. vol 112, 183–234. doi: 10.1016/B978-0-12-800171-4.00005-6
Iwasa K., Moriyasu M., Tachibana Y., Kim H.-S., Wataya Y., Wiegrebe W., et al. (2001). Simple isoquinoline and benzylisoquinoline alkaloids as potential antimicrobial, antimalarial, cytotoxic, and anti-HIV agents. Bioorg. Med. Chem. 9, 2871–2884. doi: 10.1016/S0968-0896(01)00154-7
Katsuno K., Burrows J. N., Duncan K., van Huijsduijnen R. H., Kaneko T., Kita K., et al. (2015). Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discovery 14, 751–758. doi: 10.1038/nrd4683
Laws M., Jin P., Rahman K. M. (2022). Efflux pumps in mycobacterium tuberculosis and their inhibition to tackle antimicrobial resistance. Trends Microbiol. 30, 57–68. doi: 10.1016/j.tim.2021.05.001
Lentz F., Hemmer M., Reiling N., Hilgeroth A. (2016). Discovery of novel n- phenyl 1,4-dihydropyridines with a dual mode of antimycobacterial activity. Bioorg. Med. Chem. Lett. 26, 5896–5898. doi: 10.1016/j.bmcl.2016.11.010
Lentz F., Reiling N., Martins A., Molnár J., Hilgeroth A. (2018). Discovery of novel enhancers of isoniazid toxicity in mycobacterium tuberculosis. Molecules 23, 825. doi: 10.3390/molecules23040825
Lichman B. R., Gershater M. C., Lamming E. D., Pesnot T., Sula A., Keep N. H., et al. (2015). ‘Dopamine-first’ mechanism enables the rational engineering of the norcoclaurine synthase aldehyde activity profile. FEBS J. 282, 1137–1151. doi: 10.1111/febs.13208
Lu G.-L., Tong A. S. T., Conole D., Sutherland H. S., Choi P. J., Franzblau S. G., et al. (2020). Synthesis and structure-activity relationships for tetrahydroisoquinoline-based inhibitors of mycobacterium tuberculosis. Bioorg. Med. Chem. 28, 115784. doi: 10.1016/j.bmc.2020.115784
Machado D., Coelho T. S., Perdigão J., Pereira C., Couto I., Portugal I., et al. (2017). Interplay between mutations and efflux in drug resistant clinical isolates of mycobacterium tuberculosis. Front. Microbiol. 8, e34538. doi: 10.3389/fmicb.2017.00711
Machado D., Couto I., Perdigão J., Rodrigues L., Portugal I., Baptista P., et al. (2012). Contribution of efflux to the emergence of isoniazid and multidrug resistance in mycobacterium tuberculosis. PloS One 7. doi: 10.1371/journal.pone.0034538
Machado L., Spengler G., Evaristo M., Handzlik J., Molnár J., Viveiros M., et al. (2011). Biological activity of twenty-three hydantoin derivatives on intrinsic efflux pump system of salmonella enterica serovar enteritidis NCTC 13349. In Vivo (Brooklyn) 25, 769–772.
Magnet S., Hartkoorn R. C., Székely R., Pató J., Triccas J. A., Schneider P., et al. (2010). Leads for antitubercular compounds from kinase inhibitor library screens. Tuberculosis 90, 354–360. doi: 10.1016/j.tube.2010.09.001
Markham P. N. (1999). Inhibition of the emergence of ciprofloxacin resistance in streptococcus pneumoniae by the multidrug efflux inhibitor reserpine. Antimicrob. Agents Chemother. 43, 988–989. doi: 10.1128/AAC.43.4.988
Matviiuk T., Mori G., Lherbet C., Rodriguez F., Pasca M. R., Gorichko M., et al. (2014). Synthesis of 3-heteryl substituted pyrrolidine-2,5-diones via catalytic Michael reaction and evaluation of their inhibitory activity against InhA and mycobacterium tuberculosis. Eur. J. Med. Chem. 71, 46–52. doi: 10.1016/j.ejmech.2013.10.069
Muthusaravanan S., Perumal S., Yogeeswari P., Sriram D. (2010). Facile three-component domino reactions in the regioselective synthesis and antimycobacterial evaluation of novel indolizines and pyrrolo[2,1-a]isoquinolines. Tetrahedron Lett. 51, 6439–6443. doi: 10.1016/j.tetlet.2010.09.128
Nakamura de Vasconcelos S. S., Caleffi-Ferracioli K. R., Hegeto L. A., Baldin V. P., Nakamura C. V., Stefanello T. F., et al. (2018). & cardoso, r. f. carvacrol activity & morphological changes in mycobacterium tuberculosis. Future Microbiol. 13, 877–888. doi: 10.2217/fmb-2017-0232
Odds F. C. (2003). Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52, 1. doi: 10.1093/jac/dkg301
Paixão L., Rodrigues L., Couto I., Martins M., Fernandes P., de Carvalho C. C. C. R., et al. (2009). Fluorometric determination of ethidium bromide efflux kinetics in escherichia coli. J. Biol. Eng. 3, 18. doi: 10.1186/1754-1611-3-18
Parikh J. R., Doering W. (1967). Von e. sulfur trioxide in the oxidation of alcohols by dimethyl sulfoxide. J. Am. Chem. Soc 89, 5505–5507. doi: 10.1021/ja00997a067
Pesnot T., Gershater M. C., Ward J. M., Hailes H. C. (2011). Phosphate mediated biomimetic synthesis of tetrahydroisoquinoline alkaloids. Chem. Commun. 47, 3242. doi: 10.1039/c0cc05282e
Pesnot T., Gershater M. C., Ward J. M., Hailes H. C. (2012). The catalytic potential of coptis japonica NCS2 revealed - development and utilisation of a fluorescamine-based assay. Adv. Synth. Catal. 354, 2997–3008. doi: 10.1002/adsc.201200641
Phelan J., Maitra A., McNerney R., Nair M., Gupta A., Coll F., et al. (2015). The draft genome of mycobacterium aurum, a potential model organism for investigating drugs against mycobacterium tuberculosis and mycobacterium leprae. Int. J. Mycobacteriol. 4, 207–216. doi: 10.1016/j.ijmyco.2015.05.001
Rodrigues L., Cravo P., Viveiros M. (2020). Efflux pump inhibitors as a promising adjunct therapy against drug resistant tuberculosis: a new strategy to revisit mycobacterial targets and repurpose old drugs. Expert Rev. Anti Infect. Ther. 18, 741–757. doi: 10.1080/14787210.2020.1760845
Rodrigues L., Wagner D., Viveiros M., Sampaio D., Couto I., Vavra M., et al. (2008). Thioridazine and chlorpromazine inhibition of ethidium bromide efflux in mycobacterium avium and mycobacterium smegmatis. J. Antimicrob. Chemother. 61, 1076–1082. doi: 10.1093/jac/dkn070
Rotthier G., Cappoen D., Nguyen Q. T., Dang Thi T. A., Mathys V., Nguyen V. T., et al. (2016). Synthesis and anti-tubercular activity of n 2 -arylbenzo[g]isoquinoline-5,10-dione-3-iminium bromides. Org. Biomol. Chem. 14, 2041–2051. doi: 10.1039/C5OB02138C
Ruff B. M., Bräse S., O’Connor S. E. (2012). Biocatalytic production of tetrahydroisoquinolines. Tetrahedron Lett. 53, 1071–1074. doi: 10.1016/j.tetlet.2011.12.089
Sabatini M. T., Boulton L. T., Sheppard T. D. (2017). Borate esters: simple catalysts for the sustainable synthesis of complex amides. Sci. Adv. 3, e1701028. doi: 10.1126/sciadv.1701028
Scalacci N., Brown A. K., Pavan F. R., Ribeiro C. M., Manetti F., Bhakta S., et al. (2017). Synthesis and SAR evaluation of novel thioridazine derivatives active against drug-resistant tuberculosis. Eur. J. Med. Chem. 127, 147–158. doi: 10.1016/j.ejmech.2016.12.042
Schmitz F. J., Fluit A. C., Luckefahr M., Engler B., Hofmann B., Verhoef J., et al. (1998). The effect of reserpine, an inhibitor of multidrug efflux pumps, on the in-vitro activities of ciprofloxacin, sparfloxacin and moxifloxacin against clinical isolates of staphylococcus aureus. J. Antimicrob. Chemother. 42, 807–810. doi: 10.1093/jac/42.6.807
Sen T., Neog K., Sarma S., Manna P., Deka Boruah H. P., Gogoi P., et al. (2018). Efflux pump inhibition by 11H-pyrido[2,1-b]quinazolin-11-one analogues in mycobacteria. Bioorg. Med. Chem. 26, 4942–4951. doi: 10.1016/j.bmc.2018.08.034
Singh V., Donini S., Pacitto A., Sala C., Hartkoorn R. C., Dhar N., et al. (2017). The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect. Dis. 3, 5–17. doi: 10.1021/acsinfecdis.6b00102
Singh K., Kumar M., Pavadai E., Naran K., Warner D. F., Ruminski P. G., et al. (2014). Synthesis of new verapamil analogues and their evaluation in combination with rifampicin against mycobacterium tuberculosis and molecular docking studies in the binding site of efflux protein Rv1258c. Bioorg. Med. Chem. Lett. 24, 2985–2990. doi: 10.1016/j.bmcl.2014.05.022
Singh V., Pacitto A., Donini S., Ferraris D. M., Boros S., Illyés E., et al. (2019). Synthesis and structure–activity relationship of 1-(5-isoquinolinesulfonyl)piperazine analogues as inhibitors of mycobacterium tuberculosis IMPDH. Eur. J. Med. Chem. 174, 309–329. doi: 10.1016/j.ejmech.2019.04.027
Solnier J., Martin L., Bhakta S., Bucar F. (2020). Flavonoids as novel efflux pump inhibitors and antimicrobials against both environmental and pathogenic intracellular mycobacterial species. Molecules 25, 734. doi: 10.3390/molecules25030734
Tran H. T., Solnier J., Pferschy-Wenzig E.-M., Kunert O., Martin L., Bhakta S., et al. (2020). Antimicrobial and efflux pump inhibitory activity of carvotacetones from sphaeranthus africanus against mycobacteria. Antibiotics 9, 390. doi: 10.3390/antibiotics9070390
Wang Z. (2010). “Pictet-gams synthesis,” in Comprehensive organic name reactions and reagents (John Wiley & Sons, Inc.), 2206–2209.
Keywords: tuberculosis, mycobacteria, antimicrobial resistance, isoquinolines, synergism, efflux pump inhibition, antibiotic
Citation: Martin LT, Lamming ED, Maitra A, Mortazavi PN, Roddan R, Ward JM, Bhakta S and Hailes HC (2023) C-1 Substituted isoquinolines potentiate the antimycobacterial activity of rifampicin and ethambutol. Front. Antibiot. 2:1095013. doi: 10.3389/frabi.2023.1095013
Received: 10 November 2022; Accepted: 31 March 2023;
Published: 09 May 2023.
Edited by:
Tejas M. Dhameliya, Nirma University, IndiaReviewed by:
Saeed Khoshnood, Ilam University of Medical Sciences, IranCopyright © 2023 Martin, Lamming, Maitra, Mortazavi, Roddan, Ward, Bhakta and Hailes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanjib Bhakta, cy5iaGFrdGFAYmJrLmFjLnVr; Helen C. Hailes, aC5jLmhhaWxlc0B1Y2wuYWMudWs=
†Present address: Arundhati Maitra, Institute for Global Health, UCL, Royal Free Hospital, London, United Kingdom Parisa N. Mortazavi, Pathology Laboratory, Microbiology Department, Wexham Park Hospital, Slough, United Kingdom
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.