 Tongjing Xing
Tongjing Xing Xuequan Wang
Xuequan Wang Shanshan He
Shanshan He
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Public Health , 24 July 2024
Sec. Infectious Diseases: Epidemiology and Prevention
Volume 12 - 2024 | https://doi.org/10.3389/fpubh.2024.1398254
Objective: This study aimed to explore the potential causal relationship between the gut microbiota and/or its metabolites and the progression of chronic hepatitis B (CHB).
Method: The gut microbiota was used as the exposure factor. The training set exposure data were obtained from the China Nucleotide Sequence Archive (CNSA). Genome-wide association study (GWAS) data from Asia were used as the outcome variables. Outcome data for both the training and validation sets were sourced from the GWAS Catalog database. A dual-sample Mendelian randomization approach was used to analyze the causal relationships, with the inverse variance-weighted method serving as the main analytical strategy. Sensitivity analysis was conducted to assess the robustness of Mendelian randomization analysis results.
Result: In the training set database, analysis using the inverse variance-weighted method revealed a positive correlation between Fusobacterium varium and chronic hepatitis B [OR = 1.122, 95% CI (1.016, 1.240), p = 0.022]. Conversely, Veillonella parvula exhibited a negative correlation with chronic hepatitis B [OR = 0.917, 95% CI (0.852, 0.987), p = 0.021]. Sensitivity analysis revealed no evidence of pleiotropy and heterogeneity. No gut microbiota metabolites with a causal effect on chronic hepatitis B were identified. Additionally, no associations between the gut microbiota and the progression of chronic hepatitis B were found in the validation data from the European cohort.
Conclusion: This study suggests that F. varium may facilitate the progression of chronic hepatitis B, whereas V. parvula may impede it. No causal relationships between gut microbiota metabolites and chronic hepatitis B were established.
Chronic hepatitis B virus (HBV) infection is a serious public health issue that threatens human health. Currently, there are approximately 254 million people worldwide with chronic HBV infection. In China alone, there are an estimated 86 million people with chronic HBV infection and 26 million with active chronic hepatitis B. Between 20 and 40% of chronic hepatitis B patients may die from liver failure, cirrhosis, or hepatocellular carcinoma (1, 2). Chronic persistent HBV infection is primarily related to a weakened HBV-specific immune response. The recurrent occurrence of intrahepatic inflammation and necrosis can lead to the progression of chronic HBV infection to cirrhosis and liver cancer. Antivirus therapy is an effective treatment for chronic hepatitis B. Nucleos(t)ide analogs (NAs) are the most commonly used antivirus drugs, especially the application of highly efficient and low-level drug-resistant NAs, which has greatly reduced the incidence of cirrhosis and liver cancer, significantly improving patient prognoses (3, 4). However, disease progression still occurs in some patients despite long-term NA treatment (5), suggesting that there may be other mechanisms influencing disease progression.
Gut microbiota, the microbial community in the human gut, is closely related to the normal physiological functions of the host. Dysfunction of the gut microbiota plays an important role in the pathogenesis of various diseases, such as gastrointestinal diseases, chronic liver diseases, and tumors (6, 7). Studies have demonstrated significant alterations in the diversity and abundance of gut microbiota in patients with chronic hepatitis B, which may influence their peripheral immunity and disease progression (8, 9). Gut microbiota exhibits specificity at different stages of chronic HBV infection (10). Patients with chronic hepatitis B exhibited a significant reduction in intestinal bifidobacteria and lactobacilli, along with a higher abundance of Enterobacteriaceae and Bacteroidetes (11). Lactic acid bacteria are positively correlated with disease progression, whereas Clostridium is negatively correlated (10). Changes in microbial communities are highly correlated with alterations in host metabolism, which in turn may be associated with the progression of chronic hepatitis B. Metabolites derived from the gut microbiota could regulate the host’s immune system (12, 13). Comprehensive analyses of the microbiome and metabolomics have revealed significant changes in the gut microbiota and metabolites in HBV-related chronic liver disease (9, 14). However, most studies have only revealed correlations and have not provided direct causal evidence between these factors.
Mendelian randomization (MR) utilizes genome-wide association studies (GWASs) to obtain single-nucleotide polymorphisms (SNPs) that are strongly correlated with specific outcomes. MR serves as a tool to infer causal relationships between exposure factors and outcomes by using SNPs, which, on the basis of random Mendelian genetic variations, allow for the evaluation of such relationships while minimizing the influence of confounding factors (15). The annotation of SNPs in the gut microbiota, as validated by MR analysis, can reveal relevant genes. Zhang et al. (16) reported that several gut microbiota genera have a causal relationship with the progression of chronic hepatitis B, as determined by MR analysis. However, their study included both European and Asian populations and did not perform cluster analysis. Furthermore, no reports currently exist on MR analysis of gut microbiota metabolites in relation to chronic hepatitis B. In this study, double-sample Mendelian randomization analysis was performed to investigate the causal relationship between the types and abundance of gut microbiota or its metabolites and the progression of chronic hepatitis B, with the effectiveness of the findings validated using multiple methods.
We obtained the exposure data for the training set from the China Nucleotide Sequence Archive (CNSA), a convenient and rapid online submission database for biological research projects, samples, experiments, and data. We used the metabolites and gut microbiota data of Asians, as provided in the study with reference (17) from the CNSA database as the training set for exposure data (17).1 The exposure data for validation were from the GWAS Catalog database.2 As of September 2018, the GWAS Catalog database contains 5,687 GWA entries featuring 71,673 genetic variation phenotype trait associations from 3,567 publications. For our validation set exposure data, we used the European gut microbiota data presented in the publication associated with reference (18).
We sourced the outcome data for the training set from the GWAS Catalog database (see text footnote 2) and the UK BioBank database.3 The UK BioBank database was developed by the Medical Research Council Integrative Epidemiology Unit (MRC IEU) OpenGWAS project. The settings of the OpenGWAS database are designed to be scalable and open-source, allowing the importation and publication of complete GWAS summary and summary datasets for use by the scientific community. For the training set outcome data, we used the data with identifiers bbj-a-99 and GCST90018584 pertaining to Asian populations. We also obtained the outcome data for the validation set from the GWAS Catalog database with identifiers GCST90038627, GCST90041715, GCST90041716, and GCST90077701 pertaining to European populations.
To ensure the core assumptions of Mendelian randomization, we adhered to the following three criteria during this study. (1) The association hypothesis: genetic variation needs to be strongly associated with the exposure factors. (2) The independence hypothesis: genetic variation is not associated with any possible confounding factors. (3) The exclusivity hypothesis: genetic variation affects the outcomes solely through the exposure factors and not directly. We used a series of conditions to screen the instrumental variables to ensure sufficient instrumental variables. To screen instrumental variables and ensure their adequacy, we selected SNPs (F statistics = beta2/se2) with a genome-wide significance level (p < 5 × 10−6) and F statistics ≥10, which indicated a strong correlation with the exposure factors. For the independence test criterion, we tested the selected instrumental variables and set the linkage disequilibrium threshold at r2 < 0.001 and a genetic distance of 10,000 kb to mitigate the effects of linkage disequilibrium. Finally, to ensure consistency in the allelic directions of the SNP effects on exposure and outcomes, we excluded palindromic SNPs with ambiguous flips, such as those with A/T or G/C alleles.
We conducted a double-sample MR analysis using the inverse variance weighted (IVW), MR Egger, weighted media, simple mode, and weighted mode methods to estimate the effect of exposure factors on outcome data (19). Regarding algorithmic principles, the IVW method integrates the Wald ratio of causal effects for each SNP through meta-analysis to provide the most accurate estimate. Hence, when multiple instrumental variables are used, the IVW method is the primary approach, with the other four methods serving as supplementary analyses. The entire research process is depicted in Figure 1.
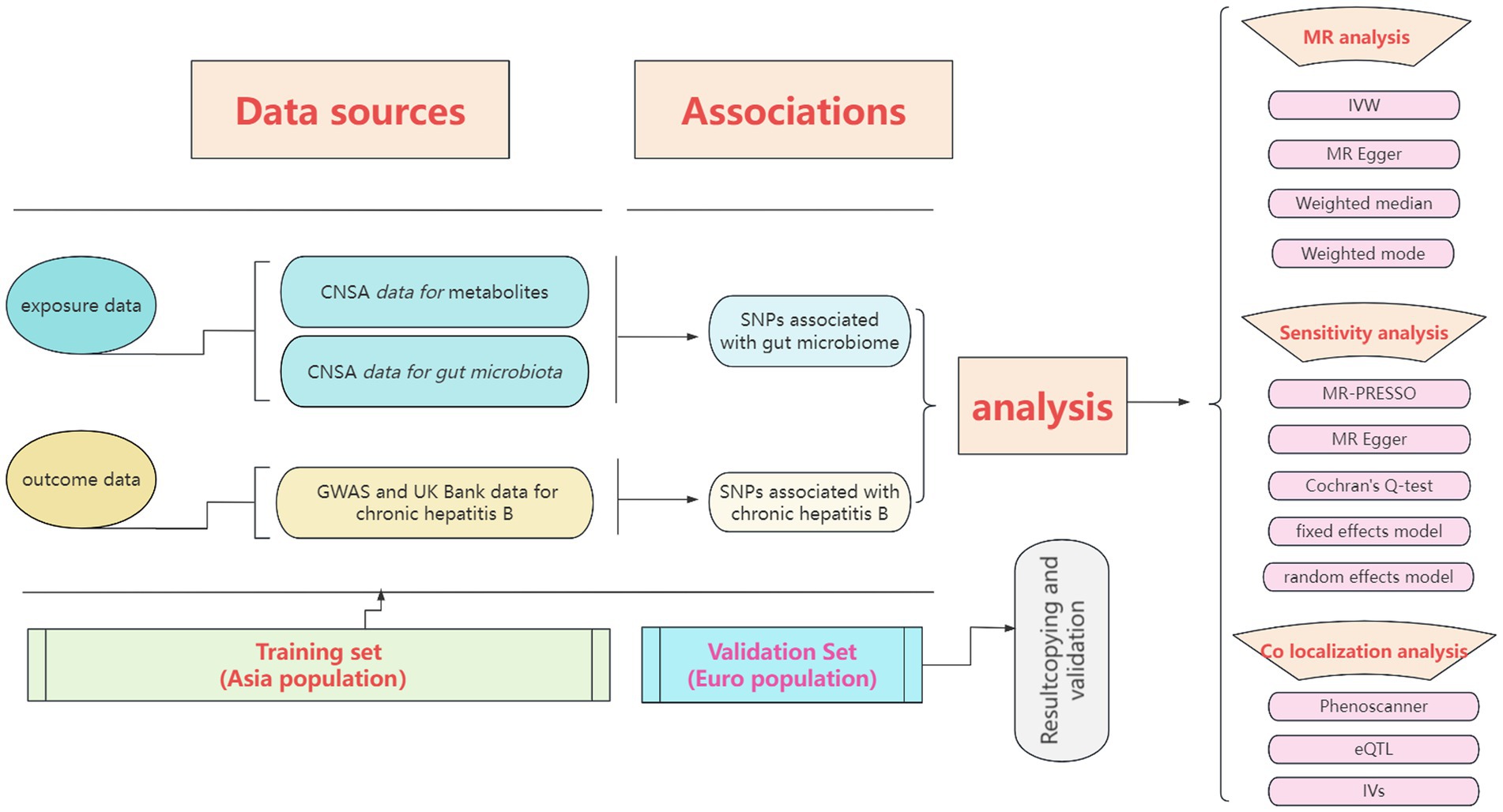
Figure 1. The entire research process of Mendelian randomization analysis.
Multiple statistical methods were used for sensitivity analysis. Initially, MR pleiotropy residual sum and outlier tests were conducted to detect horizontal pleiotropy (p < 0.05) and remove outlier SNPs (20). Simultaneously, the MR Egger intercept was used to evaluate the level of pleiotropy (21). Subsequently, Cochran’s Q test (a heterogeneity test) was used to evaluate the heterogeneity among the instrumental variables. Depending on the degree of heterogeneity, a fixed-effects model was used for IVW calculations when Q > 0.05. Conversely, a random-effects model was used when Q < 0.05. Further analysis was then conducted using either the fixed-effects model or the random-effects model, as appropriate. Additionally, a one-way analysis was performed to ascertain whether the significant association between exposure factors and outcomes is driven by any individual SNP. This involved sequentially removing different SNPs for MR analysis in each iteration. All the aforementioned analyses, including sensitivity analysis and MR analysis, were performed in the R package (R version 4.2.2) (22).
Colocalization analysis is commonly used to identify whether two phenotypes are influenced by the same causal variant within a certain region, thereby strengthening the evidence of an association between the two phenotypes. After establishing the causal relationship between exposure factors and outcomes, the following methods were used for gene colocalization calculation. (1) Based on PhenoScanner’s exploration of the correlation between expression quantitative trait loci and instrumental variables (IVs) in GTEx data, the IVs related to gene expression were screened with a threshold of p < 1e−05. (2) Each IV identified through this screening was treated as lead SNP, and SNPs within the 50 kb range upstream and downstream were examined for colocalization. Gene-level pleiotropy was assessed using a posterior probability threshold of 0.75. This analysis was conducted using the “coloc” package in R.
After controlling for linkage disequilibrium effects and F-statistic values, instrumental variables were extracted from the summary GWAS data of exposure factors. In the positive result, Veillonella parvula had the highest number of SNPs, totaling eight, as presented in Table 1. For Fusobacterium varium, four SNPs were identified as instrumental variables, which are presented in Table 2.
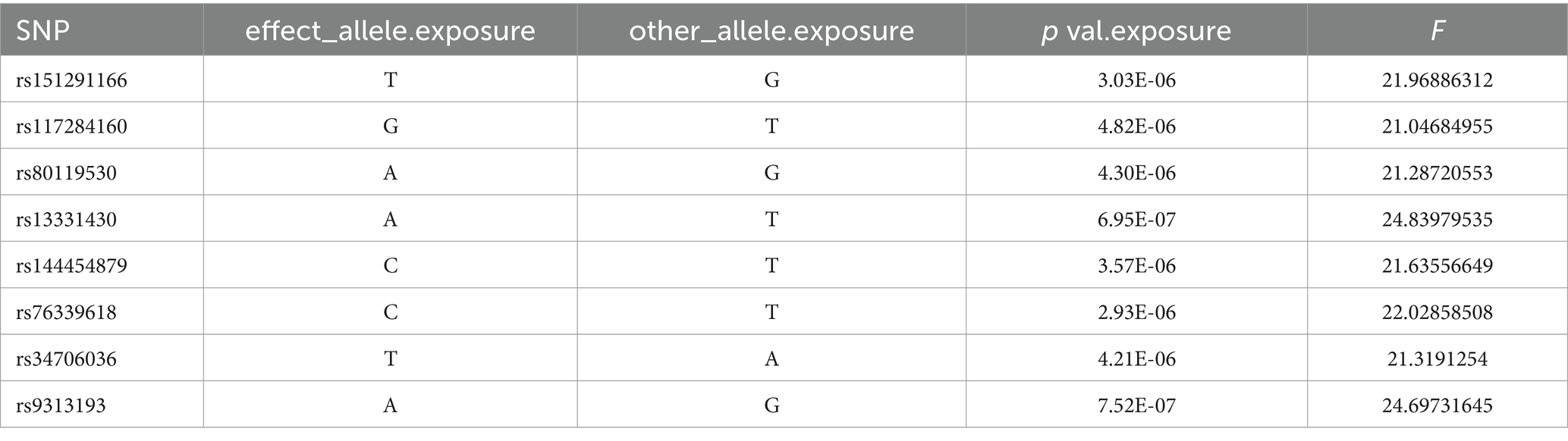
Table 1. SNP of Veillonella_parvula.
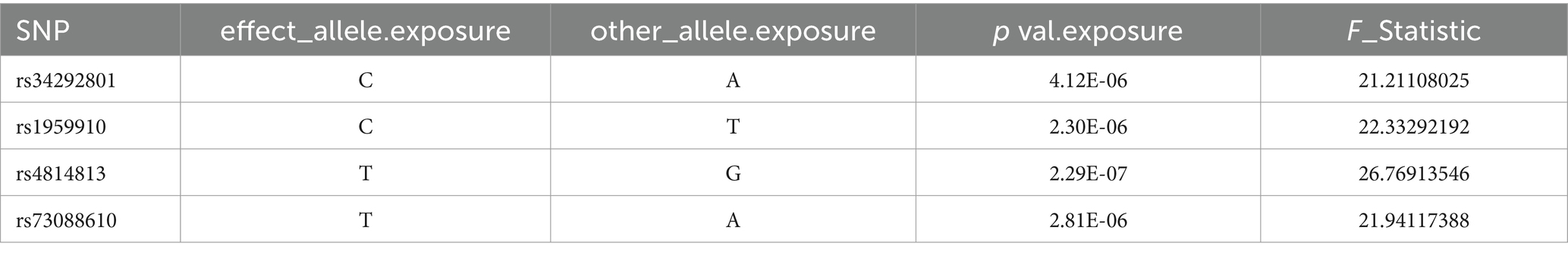
Table 2. SNP of Fusobacterium_varium.
For the training set exposure data and training set outcome data, one-on-one MR analyses were conducted. Within the training set, IVW analysis indicated causal relationships between two types of gut microbiota and chronic hepatitis B at a significance threshold of 5 × 10−6 as presented in Table 3 (Figure 2). F. varium exhibited a promotive effect in both datasets analyzed, whereas V. parvula demonstrated an inhibitory effect. Results from the MR Egger intercept and MR pleiotropy residual sum and outlier pleiotropy tests indicated no pleiotropy (Supplementary Table S1). The results of the MR analysis are presented in Figures 2–4.
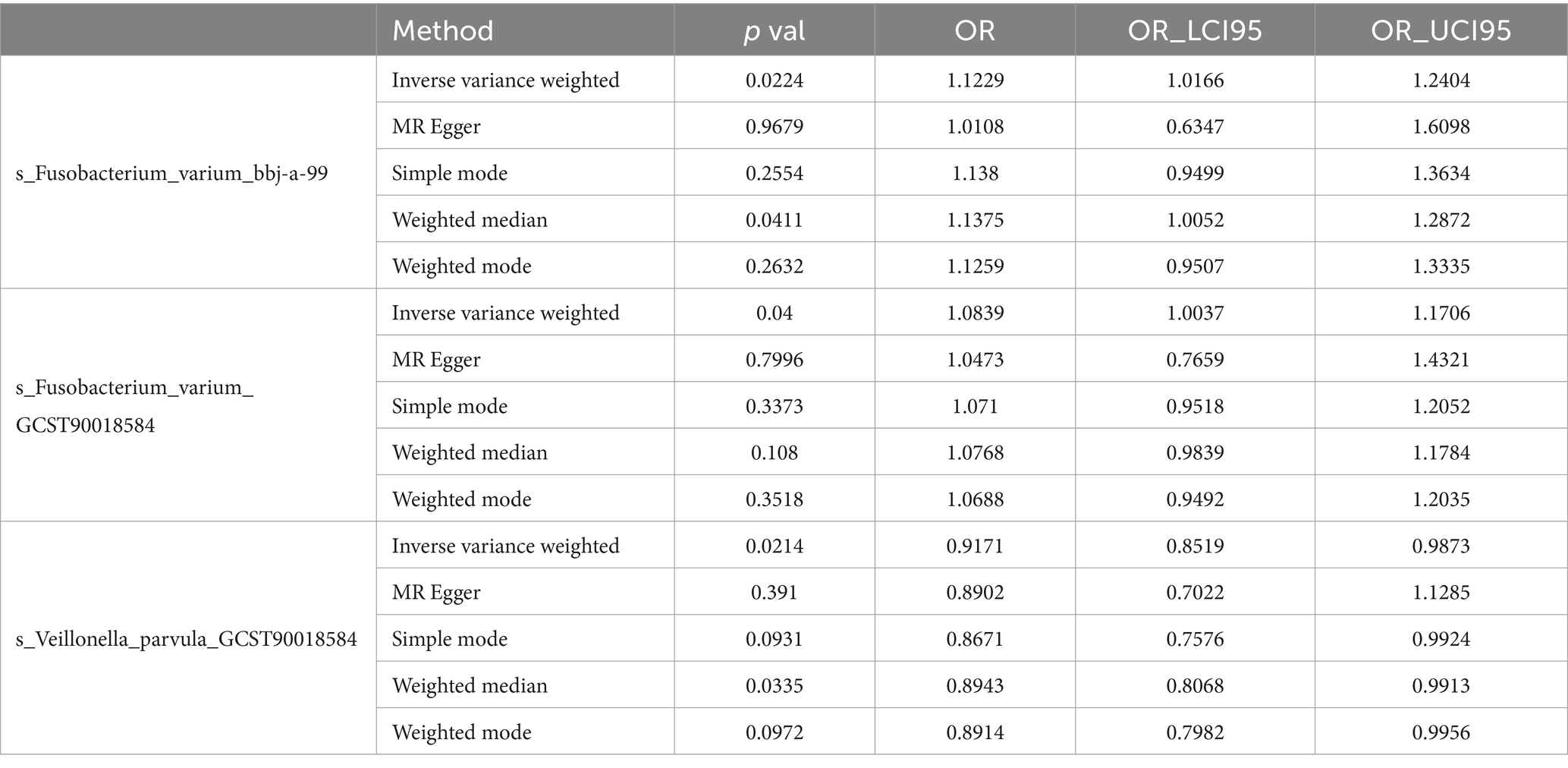
Table 3. Intestinal microbiota with significant causal effects on CHB.
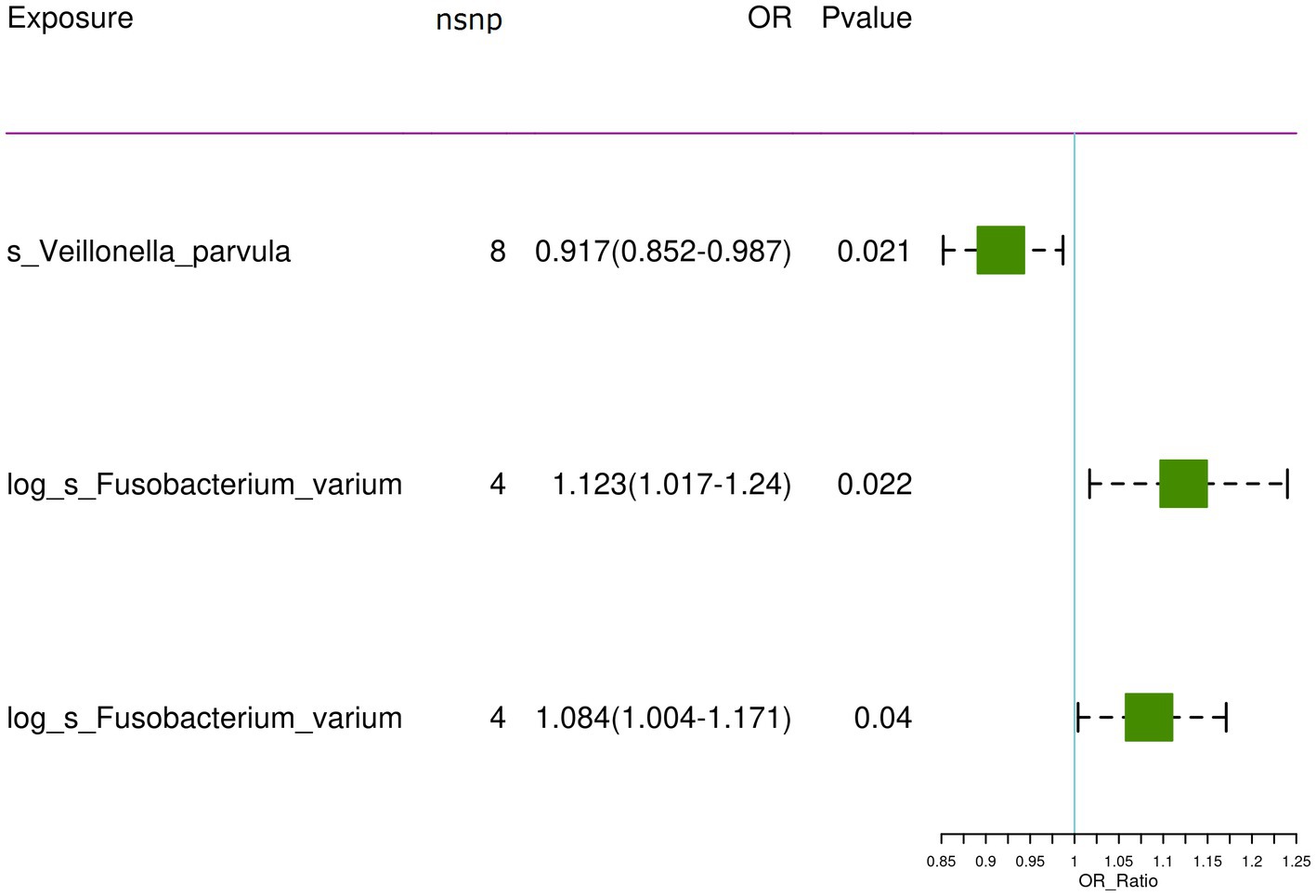
Figure 2. The significant causal relationship between intestinal microbiota and CHB.
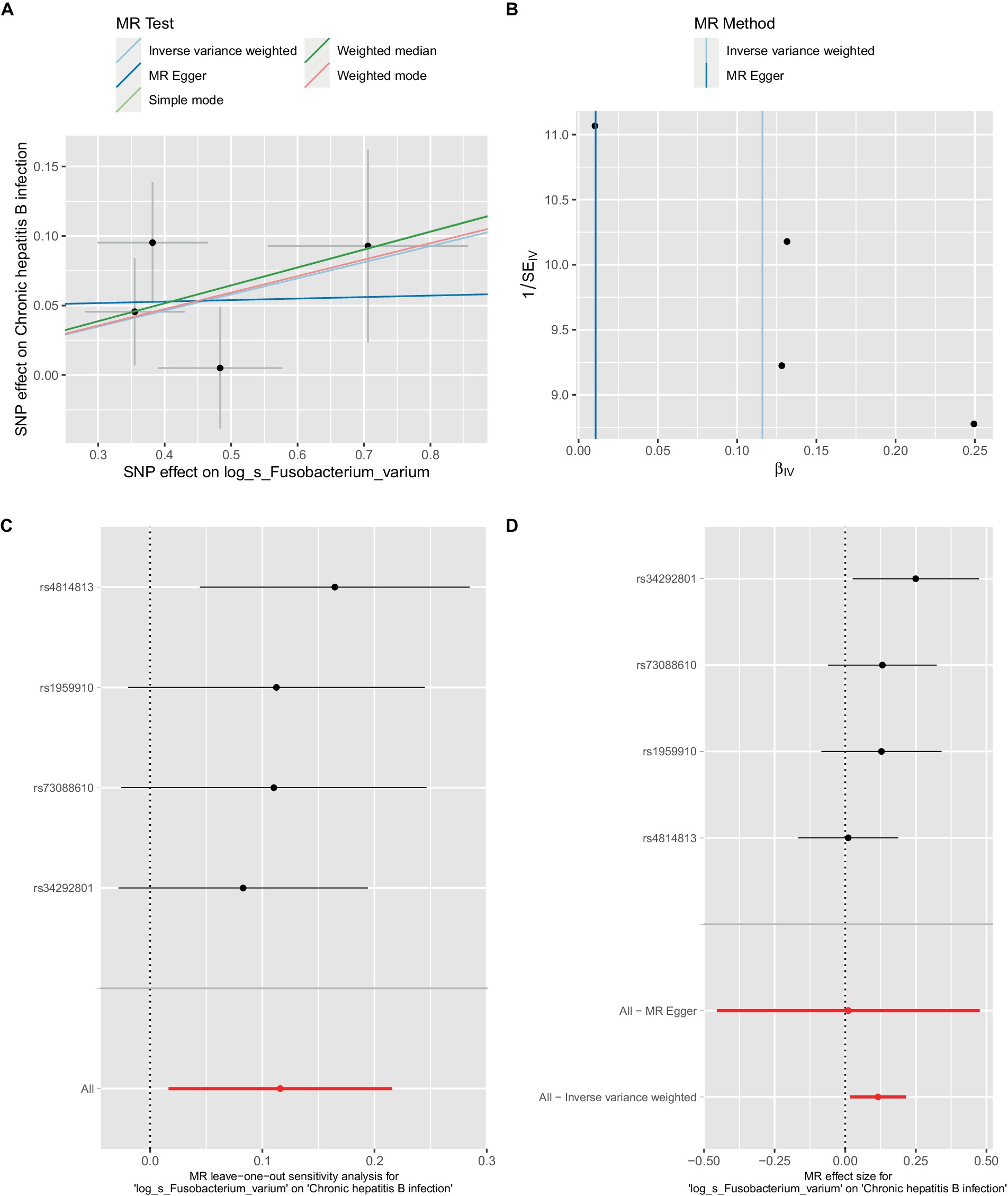
Figure 3. Mendelian randomization analysis between Fusobacterium_varium and CHB in bbj-a-99. (A) IVW model analysis between F. varium and CHB. (B) Cochran’s Q-test for heterogeneity. (C) Analysis of the retention method of single SNP affected on the overall relationship estimation. (D) Analysis of effect of individual SNPs on CHB.
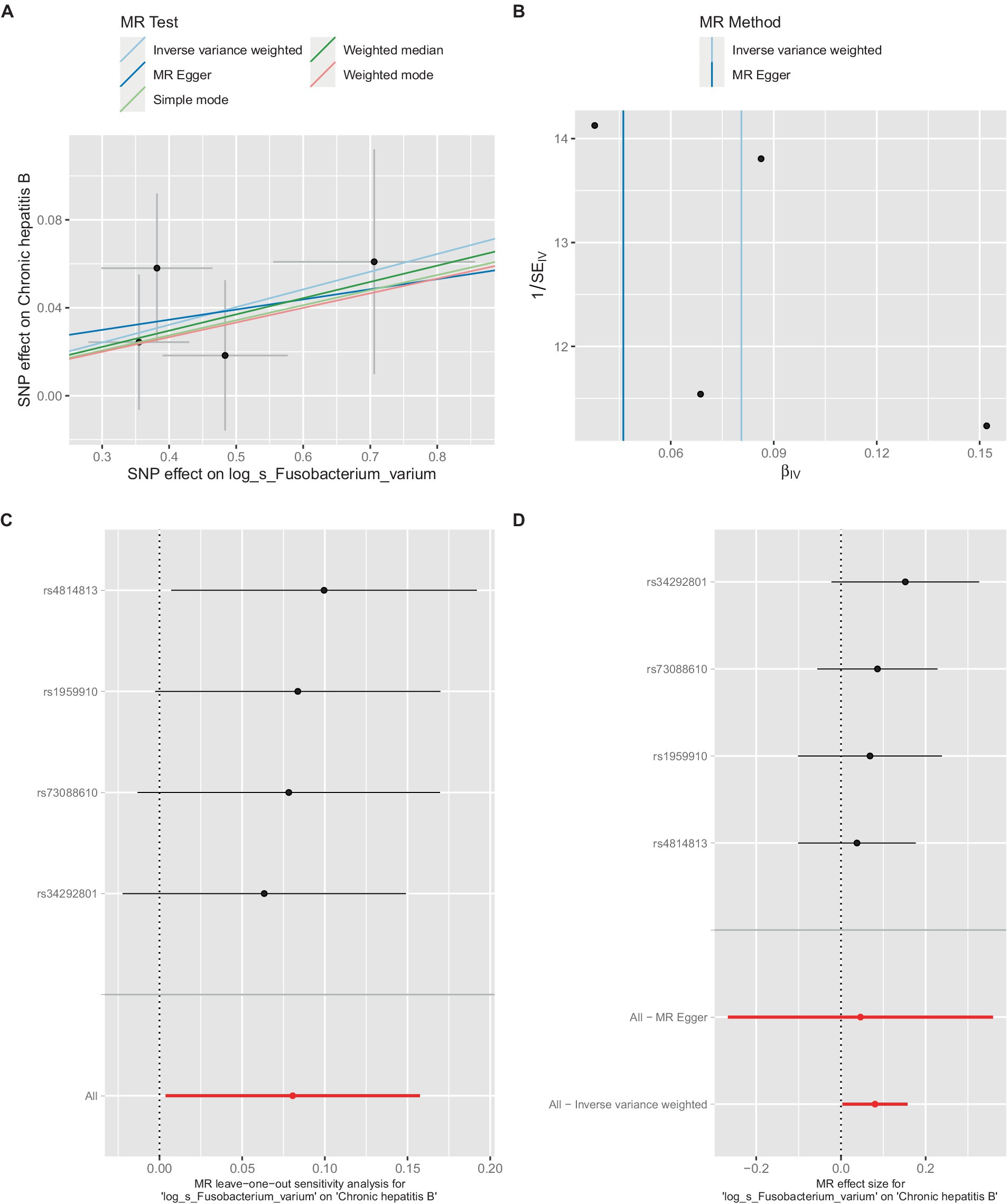
Figure 4. Mendelian randomization analysis between Fusobacterium_varium and CHB in GCST90018584. (A) IVW model analysis between Fusobacterium_varium and CHB. (B) Cochran’s Q-test for heterogeneity. (C) Analysis of the retention method of single SNP affected on the overall relationship estimation. (D) Analysis of effect of individual SNPs on CHB.
In the bbi-a-99 dataset, the IVW model analysis revealed a significant causal relationship between F. varium and chronic hepatitis B [OR = 1.1229, 95% CI (1.0166, 1.2404), p = 0.0224] (Figure 3A). Cochran’s Q-test results indicated no heterogeneity (Figure 3B). The analysis results of the retention method suggested that the overall relationship estimation is not significantly affected by any single SNP (Figure 3C). Analysis of individual SNPs revealed that some SNPs may confer a protective effect against chronic hepatitis B (Figure 3D).
Similarly, in the GCST90018584 dataset, IVW model results confirmed a significant causal relationship between F. varium and chronic hepatitis B [OR = 1.0839, 95% CI (1.0037, 1.1706), p = 0.04] (Figure 4A), with Cochran’s Q-test results indicating no heterogeneity (Figure 4B). The results of the retention method analysis indicated that the overall relationship estimate was not significantly affected by any single SNP (Figure 4C), whereas single-SNP analysis suggested a certain protective trend against chronic hepatitis B (Figure 4D).
Additionally, within the GCST90018584 dataset, IVW model results indicated a significant causal relationship between V. parvula and chronic hepatitis B [OR = 0.9171, 95% CI (0.8519, 0.9873), p = 0.0214] (Figure 5A), with Cochran’s Q-test results indicating no heterogeneity (Figure 5B). The results of the retention method analysis indicated that the overall relationship estimation is not significantly affected by any single SNP (Figure 5C). Single-SNP analysis indicated a certain protective effect against chronic hepatitis B (Figure 5D).
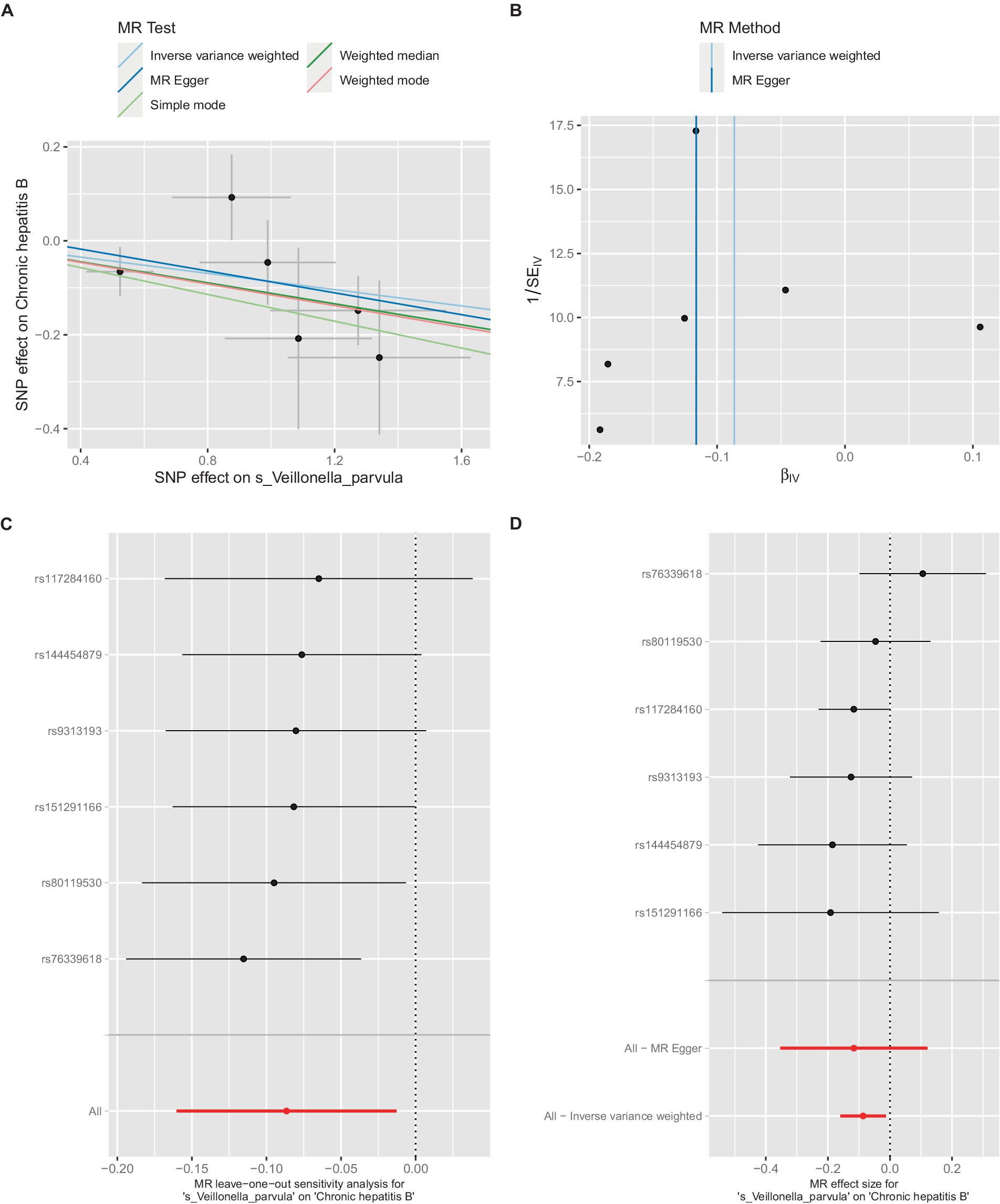
Figure 5. Mendelian randomization analysis between Veillonella_parvula and CHB in GCST90018584. (A) IVW model analysis between Veillonella_parvul and CHB. (B) Cochran’s Q-test for heterogeneity. (C) Analysis of the retention method of single SNP affected on the overall relationship estimation. (D) Analysis of effect of individual SNPs on CHB.
Colocalization analysis was performed on two types of bacteria, one of which was analyzed in two datasets (bbj-a-99 and GCST90018584). The correlation between Fusobacterium expression level and four SNPs was explored using PhenoScanner within GTEx data. No significant correlation was found between them in the bbj-a-99 datasets. Colocalization analysis within the 50-kb range upstream and downstream of these SNP sites indicated that the causal relationships do not exhibit gene-level pleiotropy, with a negative threshold of 0.75 (Table 4). Similarly, in the GCST90018584 dataset, no significant correlation was observed between Fusobacterium expression levels and the four SNPs (Table 5). The analysis of Veillonella expression level and six SNPs using PhenoScanner within GTEx data also indicated no significant correlation in the GCST90018584 dataset. Colocalization analysis of these SNP sites indicated a lack of gene-level pleiotropy with a negative threshold of 0.75 (Table 6).
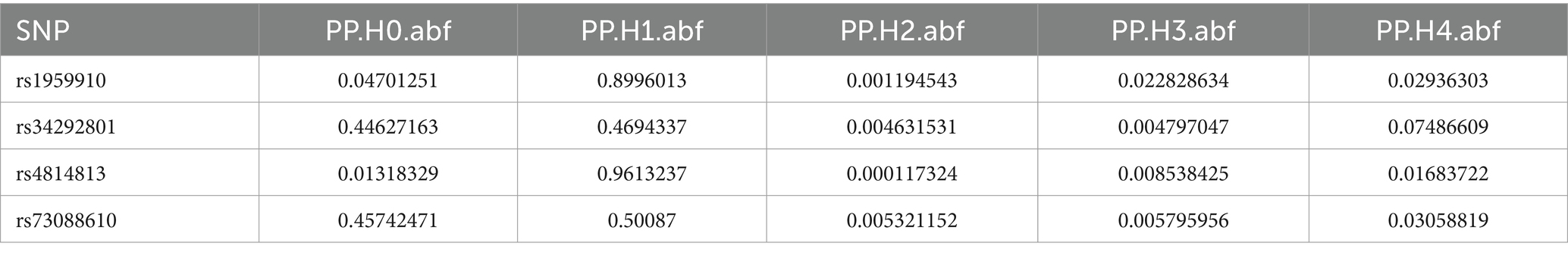
Table 4. Co localization results of Fusobacterium_varium in bbj-a-99.
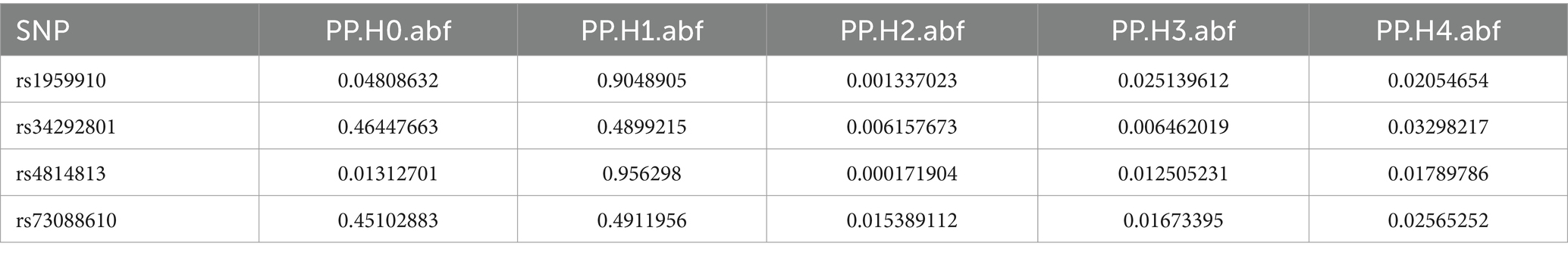
Table 5. Co localization results of Fusobacterium_varium in GCST90018584.
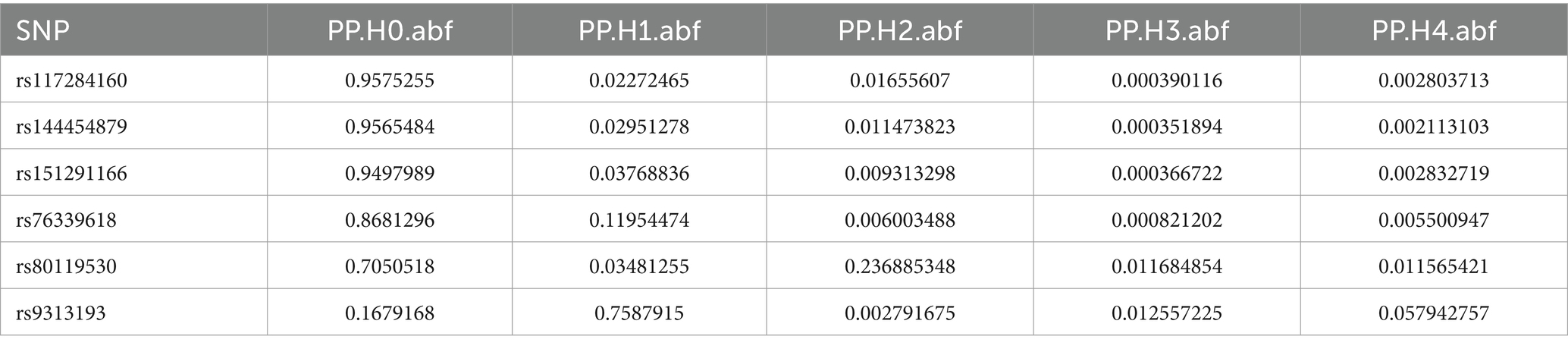
Table 6. Co localization results of Veillonella_parvula in GCST90018584.
Based on the positive phenotype of the training set, data related to relevant phenotypes in the European population were obtained from the literature (PMID: 35115689) to serve as exposure data for the validation set of gut microbiota. Datasets GCST90038627, GCST90041715, GCST90041716, and GCST90077701 were used as outcome data for the validation set. No positive results were observed in the European population dataset (Supplementary Table S2).
The liver, as the principal immune organ, processes a large amount of gut-derived components and toxins to maintain immune homeostasis. Disruptions in the equilibrium of the gut microbiota are implicated in a spectrum of liver diseases (23). Gut microbiota imbalance is characterized by an increased proportions of Bacillus and Proteus, and a decreased proportions of Clostridium associated with the onset of bacterial translocation. These microorganisms infiltrate the bloodstream via the portal vein and then enter the liver tissue, where they activate Kupffer cells and hepatic stellate cells through Toll-like receptor 4, thereby promoting disease progression (24). Investigating the causal relationship between the gut microbiota, its metabolites, and the development of chronic hepatitis B may provide further insight into the pathogenesis of the disease and personalized approaches to microbiome modulation (25). In this study, we evaluated the causal relationship between the gut microbiota, its metabolites, and the advancement of chronic hepatitis B through MR analysis using publicly available GWAS summary data. Our findings indicate that F. varium exerts a promoting effect, whereas V. parvula exhibits an inhibitory effect. These outcomes diverge from those reported by Zhang et al. (16). Possible reasons may be related to factors such as the selected population and the diversity of gut microbiota. No metabolites were identified as having a causal relationship with the progression of chronic hepatitis B.
Fusobacterium is one of the symbiotic bacteria in humans and animals primarily mainly colonizing the oral and colonic mucosa. Some species, which are opportunistic pathogens, can cause bacteremia and various rapidly progressing infections. These microorganisms are more toxic than most normal anaerobic bacterial communities, as they produce endotoxins and hemolysin. Fusobacterium can trigger pro-inflammatory responses in the host and possesses virulence characteristics that promote adhesion to host epithelial cells and invasion of epithelial cells (26). Research has indicated that Fusobacterium may be associated with intestinal diseases. The concentration of Clostridium in colon cancer tissue is significantly higher than that in normal tissue, which may contribute to tumor development. Fusobacterium is also associated with inflammatory bowel disease and intestinal infections (27, 28). In the oral cavity, Fusobacterium is associated with oral diseases such as dental caries and periodontitis. However, Fusobacterium may also play a role in maintaining the homeostasis and metabolic function of the gut microbiota and can produce beneficial metabolites for the human body, such as pyruvate and butyric acid. The study by Liu et al. reported alterations in the microbial composition and function in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy by multi-omics studies (29). The abundance of mutated F. varium positively correlated with IL-1 β and IL-6 levels in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. This study indicates that F. varium may have a deleterious effect on the progression of chronic hepatitis B disease, suggesting that F. varium may promote the progression of chronic hepatitis B disease, potentially due to its role in elevating levels of inflammatory factors.
Veillonella belongs to the genus Gram-negative anaerobic Micrococcus, with more than 10 known within the genus. Recent research has revealed that Veillonella significantly affects the human microbiome, infection processes, and immune system development (30). Qin et al. found an increase in Veillonella abundance among the gut microbiota of patients with liver cirrhosis (31). Wei et al. reported that an increase in Veillonella abundance is strongly associated with autoimmune hepatitis and is positively correlated with serum AST and liver inflammation (32). V. parvula, isolated from patients with appendiceal abscess, has been found in multiple ecological niches within the human body, including the mouth, lungs, gastrointestinal tract, and vagina. Rojas et al. (33) found that V. parvula colonizes the intestine by using alterations in carbon metabolism and ATP production pathways during inflammation. The presence of V. parvula is associated with disease remission and stability. These specific bacterial genera may be biomarkers in response to immune checkpoint inhibitor therapy (34). Jin et al. (35) demonstrated that an increase in intestinal V. parvula induced liver cell damage, hepatic stellate cell activation, and subsequent progression of cirrhosis by interacting with the TLR4/NLRP3 signaling pathway in a mouse model. This study indicates that V. parvula has an inhibitory effect on chronic hepatitis B, suggesting that V. parvula may inhibit the progression of chronic hepatitis B.
The metabolites produced by the gut microbiota serve as signaling molecules and substrates, affecting both pathological and physiological processes. Many beneficial microorganisms, such as rumen bacteria, fecal bacteria, Clostridium, and Prevotella, are known to play important roles in enhancing short-chain fatty acid activity and increasing butyric acid abundance. The study of Li et al. identified 134 named metabolites (57 upregulated and 77 downregulated metabolites) in patients with chronic hepatitis B. This study predicted a total of 101 different metabolic functions with six metabolic pathways having the highest enrichments (14). The study of Zeng et al. (36) reported that certain fecal metabolites such as essential amino acids and several dipeptides may be associated with HBeAg seroconversion of chronic hepatitis B patients. These findings provided evidence for changes in gut microbiota–related metabolites in patients with chronic HBV infection. However, the causal relationship between gut metabolites and chronic hepatitis B has not yet been established. Our results indicated that there is no causal relationship between serum metabolites related to gut microbiota and the progression of chronic hepatitis B. These results indicate that although gut microbiota–related metabolites exhibit significant differences between patients with chronic hepatitis B and healthy individuals, their effect on the progression of chronic hepatitis B may be minimal or inconsequential. The presence of a specific bacterial genus or metabolite alone may not be sufficient to trigger disease progression. This could also relate to the typically prolonged natural course of chronic liver disease in humans, which often spans years or decades. Nevertheless, the potential synergistic effect of multiple metabolites or lower abundance of certain fecal metabolites on the progression of chronic hepatitis B cannot be completely discounted, warranting further in-depth investigation.
This study has several limitations. First, our MR analysis restricted by GWAS data was limited to the genus level and not the species level. Second, training group and validation group populations from different races may be affected by genetic variation. Additionally, longitudinal research was not performed in this study. Further longitudinal studies are required to validate our results in the population with chronic hepatitis B in the future.
In summary, this study indicates that F. varium may have a promoting effect, whereas V. parvula (Vibrio genus) appears to have an inhibitory effect. However, no gut microbiota metabolites have been definitively found to have a causal relationship with the progression of chronic hepatitis B. This provides new evidence for elucidating the potential relationships between the gut microbiota, its metabolites, and chronic hepatitis B.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
TX: Writing – original draft. XW: Writing – review & editing. SH: Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2024.1398254/full#supplementary-material
1. WHO Global hepatitis report 2024: action for access in low- and middle-income countries. Available at: https://www.who.int/publications/i/item/9789240091672 (Accessed April 9, 2024).
2. Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol. (2018) 3:383–403. doi: 10.1016/S2468-1253(18)30056-6
3. Iannacone, M, and Guidotti, LG. Immunobiology and pathogenesis of hepatitis B virus infection. Nat Rev Immunol. (2022) 22:19–32. doi: 10.1038/s41577-021-00549-4
4. Park, JJ, Wong, DK, Wahed, AS, Lee, WM, Feld, JJ, Terrault, N, et al. Hepatitis B virus-specific and global T-cell dysfunction in chronic hepatitis B[J]. Gastroenterology. (2016) 150:684–695.e5. doi: 10.1053/j.gastro.2015.11.050
5. Boni, C, Laccabue, D, Lampertico, P, Giuberti, T, Viganò, M, Schivazappa, S, et al. Restored function of HBV-specific T cells after long-term effective therapy with nucleos(t)ide analogues. Gastroenterology. (2012) 143:963–973.e9. doi: 10.1053/j.gastro.2012.07.014
6. Schmidt, TSB, Raes, J, and Bork, P. The human gut microbiome: from association to modulation. Cell. (2018) 172:1198–215. doi: 10.1016/j.cell.2018.02.044
7. Llorente, C, and Schnabl, B. The gut microbiota and liver disease. Cell Mol Gastroenterol Hepatol. (2015) 1:275–84. doi: 10.1016/j.jcmgh.2015.04.003
8. Chou, HH, Chien, WH, Wu, LL, Cheng, CH, Chung, CH, Horng, JH, et al. Age related immune clearance of hepatitis B virus infection requires the establishment of gut microbiota. Proc Natl Acad Sci U S A. (2015) 112:2175–80. doi: 10.1073/pnas.1424775112
9. Shen, Y, Wu, SD, Chen, Y, Li, XY, Zhu, Q, Nakayama, K, et al. Alterations in gut microbiome and metabolomics in chronic hepatitis B infection-associated liver disease and their impact on peripheral immune response. Gut Microbes. (2023) 15:2155018. doi: 10.1080/19490976.2022.2155018
10. Yang, XA, Lv, F, Wang, R, Chang, Y, Zhao, Y, Cui, X, et al. Potential role of intestinal microflora in disease progression among patients with different stages of hepatitis B. Gut Pathog. (2020) 12:50. doi: 10.1186/s13099-020-00391-4
11. Lu, H, Wu, Z, Xu, W, Yang, J, Chen, Y, and Li, L. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic patients. Microb Ecol. (2011) 61:693–703. doi: 10.1007/s00248-010-9801-8
12. Sun, M, Wu, W, Chen, L, Yang, W, Huang, X, Ma, C, et al. Microbiota derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat Commun. (2018) 9:3555. doi: 10.1038/s41467-018-05901-2
13. Guo, W, Zhou, X, Li, X, Zhu, Q, Peng, J, Zhu, B, et al. Depletion of gut microbiota impairs gut barrier function and antiviral immune defense in the liver. Front Immunol. (2021) 12:636803. doi: 10.3389/fimmu.2021.636803
14. Li, YN, Kang, NL, Jiang, JJ, Zhu, YY, Liu, YR, Zeng, DW, et al. Gut microbiota of hepatitis B virus-infected patients in the immune-tolerant and immune-active phases and their implications in metabolite changes. World J Gastroenterol. (2022) 28:5188–202. doi: 10.3748/wjg.v28.i35.5188
15. Birney, E. Mendelian randomization. Cold Spring Harb Perspect Med. (2022) 12:a041302. doi: 10.1101/cshperspect.a041302
16. Zhang, Q, Zhou, J, Zhang, X, Mao, R, and Zhang, C. Mendelian randomization supports causality between gut microbiota and chronic hepatitis B. Front Microbiol. (2023) 14:1243811. doi: 10.3389/fmicb.2023.1243811
17. Liu, X, Tong, X, Zou, Y, Lin, X, Zhao, H, Tian, L, et al. Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat Genet. (2022) 54:52–61. doi: 10.1038/s41588-021-00968-y
18. Qin, Y, Havulinna, AS, Liu, Y, Jousilahti, P, Ritchie, SC, Tokolyi, A, et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat Genet. (2022) 54:134–42. doi: 10.1038/s41588-021-00991-z
19. Burgess, S, Dudbridge, F, and Thompson, SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. (2016) 35:1880–906. doi: 10.1002/sim.6835
20. Bowden, J, Davey Smith, G, and Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol. (2015) 44:512–25. doi: 10.1093/ije/dyv080
21. Verbanck, M, Chen, CY, and NealeB, DR. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. (2018) 50:693–8. doi: 10.1038/s41588-018-0099-7
22. Hemani, G, Tilling, K, and Davey, SG. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. (2017) 13:e1007081. doi: 10.1371/journal.pgen.1007081
23. Li, SQ, Shen, Y, Zhang, J, Weng, CZ, Wu, SD, and Jiang, W. Immune modulation of gut microbiota and its metabolites in chronic hepatitis B. Front Microbiol. (2023) 14:1285556. doi: 10.3389/fmicb.2023.1285556
24. Maslennikov, R, Poluektova, E, Zolnikova, O, Sedova, A, Kurbatova, A, Shulpekova, Y, et al. Gut microbiota and bacterial translocation in the pathogenesis of liver fibrosis. Int J Mol Sci. (2023) 24:16502. doi: 10.3390/ijms242216502
25. Jain, N. The need for personalized approaches to microbiome modulation. Front Public Health. (2020) 8:144. doi: 10.3389/fpubh.2020.00144
26. Brennan, CA, and Garrett, WS. Fusobacterium nucleatum – symbiont, opportunist and oncobacterium. Nat Rev Microbiol. (2019) 17:156–66. doi: 10.1038/s41579-018-0129-6
27. Liu, S, Zhao, W, Lan, P, and Mou, X. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell. (2021) 12:331–45. doi: 10.1007/s13238-020-00745-3
28. Wang, J, Zhang, P, Chen, S, Duan, H, and Xie, L. Microbiota and gut health: promising prospects for clinical trials from bench to bedside. Advan Gut Microbiome Res. (2022) 2022:1–17. doi: 10.1155/2022/2290052
29. Liu, S, Men, X, Guo, Y, Cai, W, Wu, R, Gao, R, et al. Gut microbes exacerbate systemic inflammation and behavior disorders in neurologic disease CADASIL. Microbiome. (2023) 11:202. doi: 10.1186/s40168-023-01638-3
30. Bhatti, MA, and Frank, MO. Veillonella parvula meningitis: case report and review of Veillonella infections. Clin Infect Dis. (2000) 31:839–40. doi: 10.1086/314046
31. Qin, N, Yang, F, Li, A, Prifti, E, Chen, Y, Shao, L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. (2014) 513:59–64. doi: 10.1038/nature13568
32. Wei, Y, Li, Y, Yan, L, Sun, C, Miao, Q, Wang, Q, et al. Alterations of gut microbiome in autoimmune hepatitis. Gut. (2020) 69:569–77. doi: 10.1136/gutjnl-2018-317836
33. Rojas-Tapias, DF, Brown, EM, Temple, ER, Onyekaba, MA, Mohamed, AMT, Duncan, K, et al. Inflammation-associated nitrate facilitates ectopic colonization of oral bacterium Veillonella parvula in the intestine. Nat Microbiol. (2022) 7:1673–85. doi: 10.1038/s41564-022-01224-7
34. Sunakawa, Y, Matoba, R, Inoue, E, Sakamoto, Y, Kawabata, R, Ishiguro, A, et al. Genomic pathway of gut microbiome to predict efficacy of nivolumab in advanced gastric cancer: DELIVER trial (JACCRO GC-08). ASCO GI. (2021) 39:161. doi: 10.1200/JCO.2021.39.3_suppl.161
35. Jin, Y, Shi, M, Feng, J, Zhang, Z, Zhao, B, Li, Q, et al. Splenectomy ameliorates liver cirrhosis by restoring the gut microbiota balance. Cell Mol Life Sci. (2024) 81:32. doi: 10.1007/s00018-023-05055-5
Keywords: chronic hepatitis B, gut microbiota, metabolites, Mendelian randomization analysis, causal links
Citation: Xing T, Wang X and He S (2024) A Mendelian randomization study investigating causal links between gut microbiota or metabolites and chronic hepatitis B. Front. Public Health. 12:1398254. doi: 10.3389/fpubh.2024.1398254
Edited by:
Zongxin Ling, Zhejiang University, ChinaReviewed by:
Congwei Gu, Southwest Medical University, ChinaCopyright © 2024 Xing, Wang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tongjing Xing, eGluZ3RqNTE4QHNpbmEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.