 Yi Guo
Yi Guo Ke Hu
Ke Hu Yuxuan Li
Yuxuan Li Chanjun Lu
Chanjun Lu Ken Ling
Ken Ling Chuanqi Cai
Chuanqi Cai Weici Wang
Weici Wang Dawei Ye
Dawei Ye
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Public Health, 11 February 2022
Sec. Infectious Diseases – Surveillance, Prevention and Treatment
Volume 10 - 2022 | https://doi.org/10.3389/fpubh.2022.833967
This article is part of the Research TopicT Cell Response in COVID-19View all 6 articles
Recent advances in the pathophysiologic understanding of coronavirus disease 2019 (COVID-19) suggests that cytokine release syndrome (CRS) has an association with the severity of disease, which is characterized by increased tumor necrosis factor α (TNF-α), interleukin (IL)-6, IL-2, IL-7, and IL-10. Hence, managing CRS has been recommended for rescuing severe COVID-19 patients. TNF-α, one of the pro-inflammatory cytokines commonly upregulated in acute lung injury, triggers CRS and facilitates SARS-CoV-2 interaction with angiotensin-converting enzyme 2 (ACE2). TNF-α inhibitors, therefore, may serve as an effective therapeutic strategy for attenuating disease progression in severe SARS-CoV-2 infection. Below, we review the possibilities and challenges of targeting the TNF-α pathway in COVID-19 treatment.
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), was declared a pandemic by the World Health Organization on 11th March 2020 and has become a major global health concern. According to recent statistical data among all COVID-19 patients, ~19% of them were severe or critical, most of which displayed remarkably increased serum levels of pro-inflammatory cytokines and subsets of immune cells (1–3). This kind of unprecedented spike in cytokines levels in critically ill COVID-19 patients is termed as cytokines release syndrome (CRS). Data obtained from COVID-19 patients have also shown that severe cases may be characterized by a CRS-induced irreversible progression to acute respiratory distress syndrome (ARDS) (4–6). To date, new therapeutic strategies for severe COVID-19 remain limited, and several antiviral drugs like lopinavir/ritonavir have shown no significant improvement in COVID-19 patients compared to those in standard care (7). It is noteworthy that tackling the immune response may be as important as controlling viral replication. Though the vaccines have developed a lot, the uncertainty around global uptake and vaccine safety remain, how to effectively treat patients with COVID-19 still attracts worldwide attention (8).
Recent advances in the pathophysiologic understanding of COVID-19 suggests that CRS has a great association with the severity of disease due to increased serum levels of tumor necrosis factor α (TNF-α), granulocyte-colony stimulating factor (G-CSF), interferon gamma-induced protein 10 (IP-10), monocyte chemoattractant protein 1 (MCP-1) and macrophage inflammatory protein 1α (MIP1A) (4, 6). A study of a longitudinal serum cytokine analysis of 207 COVID-19 patients showed that in very early inflammatory responses, IL-6, TNF-a, IL-10, and IL-1b rose in those with more severe disease (9). Additionally, based on a meta-analysis included about 23 studies, IL-6, IL-8, IL-10, IL-2R, and TNF-α cytokine levels were significantly higher and T-lymphocyte levels were significantly lower among serum of severe cases as compared to non-severe COVID-19 patients (10). Higher expressions of interleukin (IL)-2R, IL-6, IL-10, IL-18, Type-I interferon (IFN)-γ, and TNF-α cytokines in serum seemed to predict the severity and prognosis of SARS-CoV-2 infected patients (11, 12). These cytokines and chemokines were found to be positively associated with SARS-CoV-2 viral load (13). Peripheral blood flow cytometric analysis also indicated that overactivation of T cells accounted for the severe immune injury in this patient to some extent (14). Furthermore, interstitial mononuclear inflammatory infiltrates in both lungs, which are dominated by lymphocytes, were found in the pathological examination of a biopsy sample from a patient who died from COVID-19 (14). The large area of lung injury (≥50%) was closely correlated with the increased level of IL-6 as well as the subgroup of lymphocytes in the peripheral blood, according to the assessment of pulmonary infiltration in patients with ARDS (15). Thus, CRS may serve as an effective target in the treatment of COVID-19.
Initial reports have also determined that a multisystem inflammatory syndrome in children (MIS-C) associated with COVID-19 may cause public concern. It has been reported that patients with MIS-C tend to have variable symptoms such as unrelenting fever, exanthema, conjunctivitis, lymphadenopathy, peripheral oedema, and generalized extremity pain with significant gastrointestinal symptoms. Verdoni et al. (16) found that patients with mild also have a constellation of features that could be classified under the term CRS. It was found that levels of cytokines TNF-α and IL-10 discriminated between patients with MIS-C and severe COVID-19 (17). Another study in France found an elevation of inflammatory parameters including leukocytosis, levels of C-reactive protein (CRP), procalcitonin, and serum IL-6 (18). Moreover, Whittaker et al. found that a group of patients who presented well-respond to a variety of immunomodulatory treatments, including corticosteroids and intravenous immunoglobulin (IVIG), as well as some biologics such as infliximab (IFX), a TNF-α inhibitor, and anakinra, an IL-1 inhibitor (19).
Though Kawasaki disease (KD) and MIS-C share much common ground and similar clinical manifestations, patients with MIS-C were not only generally older than those with KD, but also had a higher white blood cell count, neutrophil count, and CRP (20). They were also inclined to suffer from profound lymphopenia and anemia, as well as lower platelet counts, higher fibrinogen levels, and greater elevation of troponin. This may be related to an overreaction of the body's natural defenses or immune system. It has been hypothesized that some children are predisposed to a more exaggerated inflammatory response to SARS-CoV-2, therefore inducing CRS, resulting in the clinical manifestation of MIS-C.
In summary, all of the aforementioned results suggested that SARS-CoV-2 triggers the aberrant production of cytokines and chemokines as well as alterations in the level of the subgroup of lymphocytes. This immune dysregulation may be responsible for CRS and further tissue damage.
TNF-α, a major pleiotropic mediator of acute and chronic systemic inflammatory responses, can simultaneously regulate cells apoptosis and proliferation while promoting the production of other chemokines and cytokines. It is also involved in a series of physiological processes such as anti-tumor responses, control inflammation, and immune system homeostasis (21, 22). TNF-α is also one of the most important pro-inflammatory cytokines of the innate immune response, dysregulated TNF-α signaling can trigger CRS (23).
A team studied the immune responses of 54 COVID-19 patients and found that TNF-α and IL-6 production by circulating monocytes was sustained. All their patients with pneumonia caused by SARS-CoV-2 who develop SRF display hyper-inflammatory responses with two features: over-production of pro-inflammatory cytokines by monocytes and dysregulation of lymphocytes characterized by CD4 lymphopenia and subsequently B cell lymphopenia (24). A rise in TNF-α could result in the facilitation of viral infection and organ damage (25). It has been reported that anti-TNF therapy can significantly improve the severe respiratory syncytial virus and influenza in mice, thereby reducing the recruitment of inflammatory cells, disease duration, cytokine production induced by T cells, and the severity of illness (26). Blocking TNF-α can strongly modulate the balance between effector T cells and regulatory T cells; as a result, approved TNF-α inhibitors play a crucial role in some severe autoimmune inflammatory diseases as well as numerous chronic inflammatory diseases such as psoriasis, rheumatoid arthritis (RA), inflammatory bowel disease (IBD) and ankylosing spondylitis, refractory hemophagocytic lymphohistiocytosis, systemic juvenile idiopathic arthritis, Behcet's disease, dermatomyositis, and lupus (27–36). Although TNF coordinates the inflammatory response during the acute phase of inflammation, excessive TNF will suppress the immune system with the development of disease, which may lead to the adverse result (37).
However, inhibiting TNF has a direct effect in reducing the level of IL-6 and IL-1 along with adhesion molecules and vascular endothelial growth factor (VEGF) in RA patients (38). Notably, IL-6, IL-1, adhesion molecules, and VEGF have already been reported in many clinical cohort studies of COVID-19. The plasma level of IL-6, a significant cytokine contributing to macrophage activation syndrome (MAS), which is also known as a form of CRS, has been shown to increase in both mild and severe COVID-19 patients (1, 5, 6, 39). TNF inhibitors have already been successfully used to treat MAS. Significantly, adhesion molecules and VEGF also play a crucial role in pulmonary capillary leak (40–44). TNF can also reduce other inflammatory proteins, such as CRP and serum amyloid A protein (45). TNF-targeting aptamer treatment has also been reported to attenuate the poor prognosis of LPS-induced acute lung injury by improving oxygen saturation and lung injury scores, reducing protein-rich fluid leakage and neutrophil infiltration in alveolar spaces, and suppressing pro-inflammatory cytokines and chemokines expressions in lung tissues (46).
During SARS-CoV-2 infection, aggressive inflammatory response and cytokine storm contribute to severe systemic tissue damage and mortality. Recently, original research demonstrated that the synergism between TNF-α and IFN-γ is critical in triggering robust cell death during activating JAK/STAT1/IRF1 axis in the human monocytic cell line THP-1 and primary human umbilical vein endothelial cells (12). What's more, the kinetics of cell death induced by TNF-α and IFN-γ co-stimulation were proportional to the concentrations of TNF-α and IFN-γ. Collectively, the TNF-α signaling pathway plays a vital role in CRS (Figure 1). Blocking the cytokine-mediated inflammatory cell death signaling pathway may benefit patients with COVID-19 or other infectious diseases by limiting tissue damage (47).
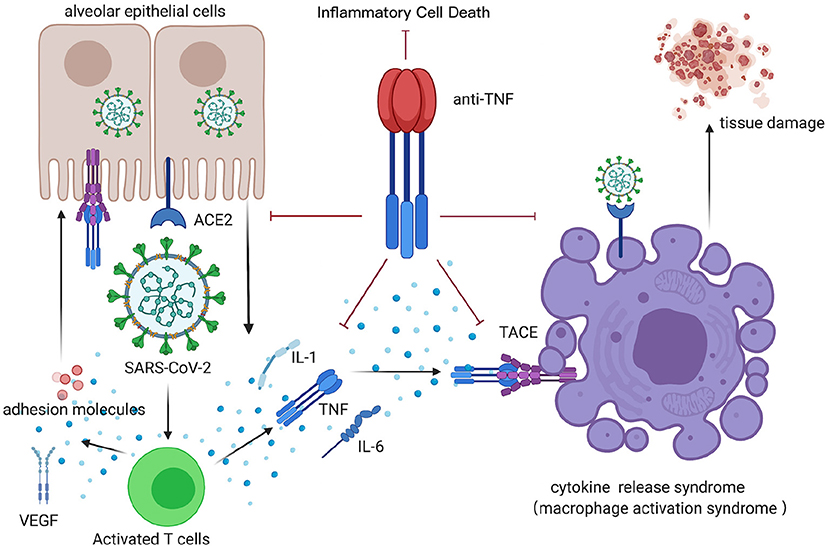
Figure 1. CRS after COVID-19 infection, inhibited by anti-TNF therapy. TNF, tumor necrosis factor; ACE, angiotensin-converting enzyme; TACE, TNF-α-converting enzyme; VEGF, vascular endothelial growth factor; IL-1, interleukin-1; IL-6, interleukin-6. This figure was generated in Biorender (https://Biorender.com).
According to in vitro studies, TNF-α facilitates the SARS-CoV interaction with angiotensin-converting enzyme 2 (ACE2) (48). ACE2, a type I membrane protein detected in many organs, especially lung AT2 alveolar epithelial cells that are particularly susceptible to viral infection, has been established as the host cell-surface receptor for both SARS-CoV and SARS-CoV-2 (49). Though a few studies provided implications that SARS-CoV-2 could accelerate its entrance into the infected cells by leveraging up-regulation of its receptor gene ACE2 via induction of cytokine storm involving IFNG, TNF-α as well as NFKβ (50, 51), physiologically ACE2 still has a significant anti-inflammatory role. The normal level of ACE2 is important to protect vital organs, SARS-CoV-2 infection causes the downregulation of ACE2 expression, leading to an overwhelming production of angiotensin II and resulting in increased pulmonary vascular permeability and lung damage. Loss of ACE2 expression may also increase neutrophil accumulation, lung edema, and diminish lung function (52). Reduced ACE2 activity can indirectly activate the kallikrein-bradykinin pathway and increase vascular permeability (53). Potdar et al. (54) reported that through their univariate analyses, small bowel ACE2 levels were restored after anti-TNF (infliximab) therapy and that it was statistically significant in anti-TNF responders. Moreover, some inflammatory cytokines like IL-1β and TNF-a can strengthen ACE2 shedding (48, 55, 56). The SARS-CoV spike protein can induce a TNF-α-converting enzyme (TACE)-dependent shedding of the ACE2 ectodomain, which may be the crux of the virus penetrating the cell (48). A close correlation between the SARS-CoV spike protein-induced ACE2 shedding and TNF-α production in cell culture conditions have been shown, which may suggest the significance of modulating TNF-α in SARS-CoV (48). SARS-CoV and SARS-CoV-2 share a highly similar mechanism of structures of spike proteins and semblable pathogenicity, and the surface of human ACE2 (hACE2) plays a critical role in both of them, which indicates that modulation of TNF-α may be the crux in SARS-COV-2 as well (57–60).
Nuclear factor kappa B (NF-κB), a transcription factor modulating both innate and adaptive immunity, is also crucial in the progress of COVID-19 (61). Relevant changes that occur in innate and adaptive immunity in COVID-19 have been highlighted by several studies. Specifically, NF-κB can affect pro-inflammatory cytokines and chemokines, anti-apoptotic proteins, and stress-response proteins by inducing the transcription of a wide spectrum of their encoding genes. NF-κB is also crucial for survival and activation and plays a significant role in initiating and propagating optimal immune responses (62). A study is reported that an exacerbated inflammatory response in severe COVID-19 patients associated with increased TNF-a and IL-6 potentially driven by NF-κB (63). Notably, respiratory viruses including SARS-CoV can induce the exacerbated activation of NF-κB, leading to lung inflammatory immunopathology (64, 65). Additionally, blocking NF-κB can protect against pulmonary pathology and enhance mice survival after SARS-CoV infection (64). All of the aforementioned evidence suggests that blocking rate NF-κB may be a consequential choice in counteracting disease progress in COVID-19 patients. However, blocking NF-κB may bring about undesirable side effects, such as the broad suppression of innate immunity due to the lack of specificity of the relevant treatment and the significance of NF-κB in cellular homeostasis (66). The interaction of TNF receptor-associated factor (TRAF) 2 with the I-kB kinase (IKK) complex, as well as the support of receptor-interacting protein (RIP), can finally activate NF-κB (67). TRAF 2, a vital constituent of the TNF receptor signaling complexes, participates in the TNF-R1-mediated activation of NF-kB (68). As a result, blocking TNF-α may be a more effective strategy, which have been clinically and effectually used in several pathological conditions (Figure 2).
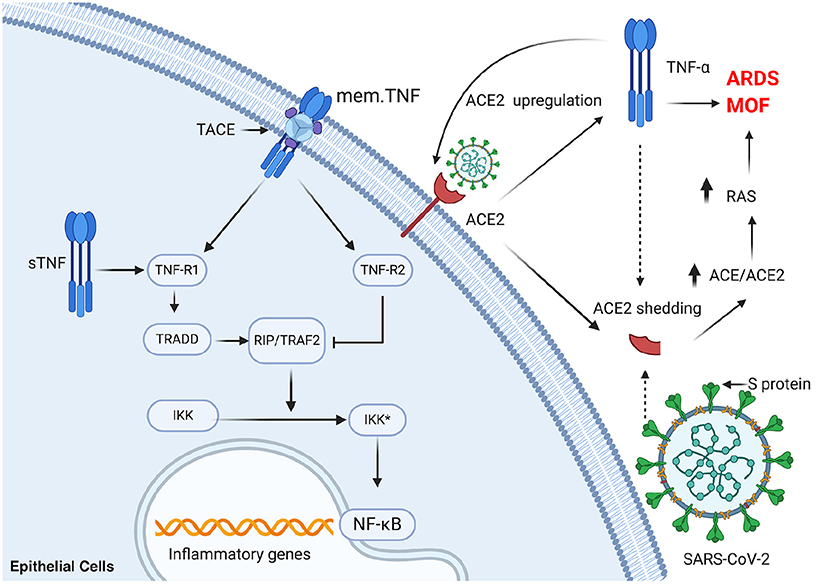
Figure 2. TNF-a plays a significant role in activation of NF-κB and ACE2 shedding in SARS-CoV-2 infection. mem.TNF, membrane-bound TNF; sTNF, soluble TNF; TACE, TNF-α-converting enzyme; TNF-R1, TNF-receptor 1; TNF-R2, TNF-receptor 2; TRADD, TNF-receptor-associated protein; RIP, receptor-interacting protein; TRAF2, TNF receptor-associated factor 2; IKK, I-κB kinase; NF-κB, nuclear factor kappa B; ACE, angiotensin-converting enzyme; ARDS, acute respiratory distress syndrome; MOF, multi-organ failure; RAS, renin-angiotensin system. This figure was generated in Biorender (https://Biorender.com).
Since TNF-α production plays a vital role in these processes, inhibiting TNF-α may be an effective strategy in attenuating both SARS-CoV2 infection and the resulting organ damage (69). Feldmann et al. suggested to evaluate anti-TNF therapy as soon as COVID-19 patients are admitted to the hospital to prevent their condition from persisting or worsening, which would then require intensive care support. It may be reasonable to initiate anti-TNFα therapy as early as possible (70). Existing anti-TNF therapies include Humira (adalimumab, AbbVie), Remicade (infliximab, Janssen), Simponi (golimumab, Janssen), Cimzia (certolizumab pegol, UCB), and Entyvio (vedolizumab, Takeda). Currently, a clinical trial comprised of COVID-19 patients evaluating adalimumab, an anti-TNF-α drug, has been registered in China (ChiCTR2000030089). A phase II Trial of IFX in COVID-19 registered in the US (NCT04425538) has been completed and reported that IFX may abrogate pathological inflammatory signaling rapidly to facilitate clinical recovery in severe and critical COVID-19 (71). Additionally, a prospective multicenter cohort of patients with IBD Treated by IFX or Vedolizumab during the COVID-19 pandemic was registered in the US (NCT04344249). A few studies have reported recovery in a patient with COVID-19 treated with TNF-α inhibitor etanercept successfully (32, 34, 72, 73). Several trials with different TNF antagonists will provide information on the effectiveness of these therapeutic interventions along with the risks of these measures in COVID-19 therapy, are shown in Table 1.
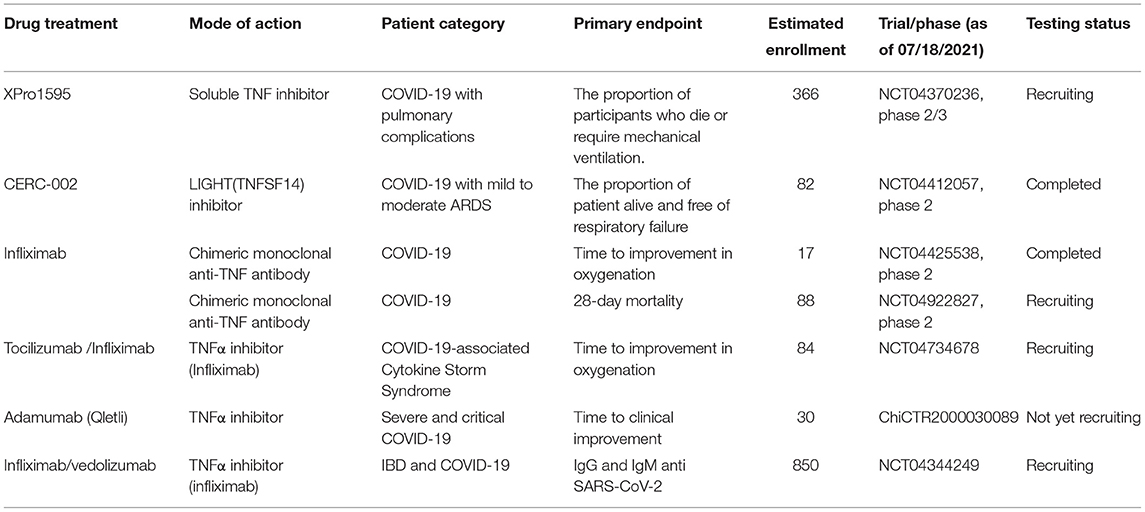
Table 1. Drugs targeting TNF or its receptor in clinical studies in patients with COVID-19.
IFX, a recombinant humanized monoclonal antibody against TNF-α, is now approved for use in several immune-mediated disorders, including psoriasis, RA, IBD, ankylosing spondylitis, and IVIG resistant KD (74–77). Numerous studies have reported that combined treatment with IFX was related to clinical and biochemical improvement in KD complicated with MAS (78). Considering the potential similar pathophysiology between KD complicated with MAS and MIS-C, which is also potentially related to CRS to a certain extent, IFX may play a vital role in CRS with COVID-19. Additionally, IFX has already been reported in successfully treating patients with MIS-C when treating the COVID-19 inflammatory cascade (79). IFX has also demonstrated collateral benefits for an adult patient diagnosed with severe ulcerative colitis and COVID-19 as both diseases successfully improved (80).
A coronavirus and IBD reporting database on Surveillance Epidemiology of Coronavirus Under Research Exclusion (SECURE-IBD) has shown a cluster of outcomes for IBD patients diagnosed with COVID-19. As of December 7, 2021, 2216 patients have been reported to use anti-TNF therapy alone, of which 2004 patients were rehabilitated without hospitalization, while 10 died with a mortality rate of <1%. Meanwhile, 25 required transfer to the ICU, of which 18 required a ventilator. Together, the rate of patients with poor prognosis involving ICU transfer, ventilation or death was 1%. However, only 1,593 of 2,035 patients who used sulfasalazine/mesalamine recovered without hospitalization, while 53 died with a mortality rate of 3%. Here, 82 were transferred to the ICU, of which 70 needed ventilation. The rate of those using sulfasalazine/mesalamine with a poor prognosis was about 6%. Moreover, anti-TNF therapy maintained the highest outpatient rate of 90% among all kinds of IBD medications in the database, even compared to the IL 12/23 inhibitor and JAK inhibitor. Research based on the SECURE-IBD database also suggests that TNF antagonist treatment was not an independent risk factor for severe COVID-19 and plays a significant protective role (81, 82). Anti-TNF therapy was found to be related to reduced probability of hospitalization while prednisone use ≥10 mg/day was linked with a higher probability of hospitalization (83). Evidently, thiopurine monotherapy and the combination thiopurines with TNF antagonists are associated with significantly increased risk of severe COVID-19 compared with TNF monotherapy. Therefore, consideration of discontinuing the thiopurine during the COVID-19 pandemic may be warranted in those high-risk patients (e.g., older age or multiple comorbidities) in stable remission on TNF antagonist combination therapy (84).
Furthermore, there have been many case reports of patients using TNF inhibitors as treatment for COVID-19, demonstrating no respiratory complications or death (72, 85, 86). More significantly, Bezzio and colleagues found no association between the use of biological therapies and negative outcomes of COVID-19 in IBD patients (87). In a study of 70 rheumatic disease patients, patients on anti-TNF alpha medications were hospitalized less frequently (88). Another study reported that COVID-19 prevalence in patients with IBD was comparable with that in the general population and anti-TNF agents seem to mitigate the course of COVID-19 (89). According to a case-control study which investigated the frequency of COVID-19 incidence in 254 eligible patients with rheumatoid arthritis (RA) or seronegative spondyloarthropathies (SpA), TNF-α blockers including adalimumab, infliximab, and etanercept decreased significantly the risk of developing COVID-19 in patients with RA and SpA. And the highest percentage of impact on the prevention of COVID-19 was related to adalimumab (96.8%). Their result could also confirm that TNF-α inhibitors may mitigate or ameliorate the COVID-19 disease course (90). Overall, patients with COVID-19 using anti-TNF therapy do not fare worse than those treated with other drugs.
TNF-α inhibitors may trigger or worsen disease progression (33). TNF-α inhibitors can increase bacterial and mycobacterial infections in patients as well as reactivate latent infections, including hepatitis B and tuberculosis, with its immunosuppressive activity (91). A systematic review and meta-analysis of published trials found a higher incidence rate of any infection (20%), serious infection (40%), and tuberculosis (250%) related to anti-TNF therapy (92). Moreover, patients already taking immunosuppressive drugs such as TNF inhibitors are potentially an at-risk group for COVID-19 infection and its complications (93, 94). Meanwhile, paradoxical effects such as sarcoidosis and other granulomatous disorders like rheumatoid nodules and vasculitis, paradoxical psoriasis and psoriasiform lesions, uveitis and other eye diseases, and miscellaneous auto-immune diseases like central and peripheral nervous system demyelinating disorders, have also been reported to be associated with anti-TNF drug use (95). However, most symptoms could resolve after the discontinuation of anti-TNF therapy or switching to another anti-TNF agent.
As a result, it is of great importance to differentiate patient cohorts with a bleak prognosis and amendable clinical outcomes who require anti-TNF therapy from those who can be rehabilitated on their own to minimize potential risks linked with immunosuppression. Prophylactic treatment and screening for latent tuberculosis while assessing the risk of venous thromboembolism prior to commencing anti-TNF therapy are still highly recommended.
IFX exposure was found to be independent of increased risk of hemophagocytic lymphohistiocytosis in pediatric patients with IBD who were followed for 20 years in a prospective cohort study (96). A study conducted comprised of 434 pediatric patients with KD received IFX in Japan, which also showed no MAS cases have been reported. In this study, the median days of IFX treatment were 9, and single dose IFX 5 mg/kg was administered to 412 patients (94.9%) (76). As an approved drug for pediatric patients with low risk of MAS occurrence, IFX may serve as a proper choice among all TNF inhibitors. However, although using IFX (5 mg/kg) against CRS with organ failure in COVID-19 patients 3 days has shown temporary reductions in pro-inflammatory cytokines, it may increase the risk of concomitant infection according to an analysis of a cautionary case series (97).
All of the corresponding results suggest the security and potential significance of TNF inhibitors to a certain degree. Thus far, anti-TNF therapy seems to have had a protective effect on the progression of COVID-19, especially in severe forms. In addition, anti-TNF therapy has a well-demonstrated ability to reduce inflammation and can prevent CRS associated with the immune pathogenesis of infection. Research based on the SECURE-IBD database has shown that TNF antagonist treatment reduces the ratio of COVID-19 patients with a poor prognosis and plays a significant protective role. However, there are still some limitations, such as the data from the SECURE-IBD database, anti-TNF drugs are only used in COVID-19 patients with IBD, not including non-IBD COVID-19 patients. Nevertheless, some other clinical trial results suggest that anti-TNF therapy is meaningful in COVID-19 patients. The currently completed clinical trials are mainly aimed at IFX, which has been proved to facilitate clinical recovery in severe and critical COVID-19. The clinical trial on adalimumab also has been recruited, and the role of the TNF-inhibitor family in covid-19 patients is appreciable. Notably, the adoption of anti-TNF therapy may be limited by its potential side effects, which underlines the significance of differentiating patient cohorts with a bleak prognosis and amendable clinical outcomes from those who can be rehabilitated on their own, as discussed earlier. Finding the timing of the therapy in COVID-19 patients also appears to be crucial. Therefore, we're looking forward to the results of clinical trials to bring us more guiding advice. In a word, the role of TNF inhibitors in the treatment of COVID-19 warrants further study.
YG, KH, and YL: served as a guarantor, contributed to literature search, manuscript editing, manuscript preparation, interpretation of data, and study design. CL: contributed to design, interpretation of data, literature search, and manuscript preparation. KL and CC: contributed to manuscript preparation, interpretation of data, and manuscript review. WW and DY: served as a guarantor, contributed to literature search, manuscript review, manuscript editing, manuscript preparation, interpretation of data, and study design. All authors read and approved the final version to be published.
This review was supported by funding from the National Natural Science Foundation of China (Grant Numbers 82000729 and 81873529) and Shanxi Province 136 Revitalization Medical Project Construction Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. (2020) 55:102763. doi: 10.1016/j.ebiom.2020.102763
2. Wang F, Nie J, Wang H, Zhao Q, Xiong Y, Deng L, et al. Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J Infect Dis. (2020) 221:1762–9. doi: 10.1093/infdis/jiaa150
3. Dong YM, Sun J, Li YX, Chen Q, Liu QQ, Sun Z, et al. Development and validation of a nomogram for assessing survival in patients with COVID-19 pneumonia. Clin Infect Dis. (2021) 72:652–60. doi: 10.1093/cid/ciaa963
4. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20):30183-5
5. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. (2020) 71:762–8. doi: 10.1093/cid/ciaa248
6. Chen L, Liu H, Liu W, Liu J, Liu K, Shang J, et al. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Zhonghua jie he he hu xi za zhi. (2020) 43:203–8. doi: 10.3760/cma.j.issn.1001-0939.2020.03.013
7. Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. New Engl J Med. (2020) 382:1787–99. doi: 10.1056/NEJMc2008043
8. Ronald RN, Briggs MB, Porter AL, Spandidos DA, Tsatsakis A. [Comment] COVID-19 vaccine safety. Int J Mol Med. (2020) 46:1599–602. doi: 10.3892/ijmm.2020.4733
9. Bergamaschi L, Mescia F, Turner L, Hanson AL, Kotagiri P, Dunmore BJ, et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity. (2021) 54:1257–75.e8. doi: 10.1016/j.immuni.2021.05.010
10. Mulchandani R, Lyngdoh T, Kakkar AK. Deciphering the COVID-19 cytokine storm: systematic review and meta-analysis. Eur J Clin Invest. (2021) 51:e13429. doi: 10.1111/eci.13429
11. Li X, Xu S, Yu M, Wang K, Tao Y, Zhou Y, et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol. (2020) 146:110–8. doi: 10.1016/j.jaci.2020.04.006
12. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. (2021) 184:149–68.e17. doi: 10.1016/j.cell.2020.11.025
13. Yang Y, Shen C, Li J, Yuan J, Yang M, Wang F, et al. Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. medRxiv [preprint]. (2020). doi: 10.1101/2020.03.02.20029975. Available online at: https://www.medrxiv.org/content/10.1101/2020.03.02.20029975v1 (accessed March 6, 2020).
14. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
15. Zinkernagel RM. Immunology taught by viruses. Science. (1996) 271:173–8. doi: 10.1126/science.271.5246.173
16. Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. (2020) 395:1771–8. doi: 10.1016/S0140-6736(20)31103-X
17. Diorio C, Henrickson SE, Vella LA, McNerney KO, Chase J, Burudpakdee C, et al. Multisystem inflammatory syndrome in children and COVID-19 are distinct presentations of SARS-CoV-2. J Clin Invest. (2020) 130:5967–75. doi: 10.1172/JCI140970
18. Grimaud M, Starck J, Levy M, Marais C, Chareyre J, Khraiche D, et al. Acute myocarditis and multisystem inflammatory emerging disease following SARS-CoV-2 infection in critically ill children. Ann Intensive Care. (2020) 10:1–5. doi: 10.1186/s13613-020-00690-8
19. Whittaker E, Bamford A, Kenny J, Kaforou M, Jones CE, Shah P, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA. (2020) 324:259–69. doi: 10.1001/jama.2020.10369
20. Belhadjer Z, Méot M, Bajolle F, Khraiche D, Legendre A, Abakka S, et al. Acute heart failure in multisystem inflammatory syndrome in children (MIS-C) in the context of global SARS-CoV-2 pandemic. Circulation. (2020) 142:429–36. doi: 10.1161/CIRCULATIONAHA.120.048360
21. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nature Rev Immunol. (2009) 9:271–85. doi: 10.1038/nri2526
22. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nature Rev Immunol. (2003) 3:745–56. doi: 10.1038/nri1184
23. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol. (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
24. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. (2020) 27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009
25. Deng X, Yu X, Pei J. Regulation of interferon production as a potential strategy for COVID-19 treatment. arXiv. preprint arXiv. (2020) 200300751. Available online at: https://arxiv.org/abs/2003.00751 (Accessed March 2, 2020).
26. Hussell T, Pennycook A, Openshaw PJ. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol. (2001) doi: 10.1002/1521-414131:9<2566::AID–IMMU2566>3.0.CO;2-L
27. Henzan T, Nagafuji K, Tsukamoto H, Miyamoto T, Gondo H, Imashuku S, et al. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol. (2006) 81:59–61. doi: 10.1002/ajh.20462
28. Ideguchi H, Ohno S, Takase K, Hattori H, Kirino Y, Takeno M, et al. Successful treatment of refractory lupus-associated haemophagocytic lymphohistiocytosis with infliximab. Rheumatology. (2007) 46:1621–2. doi: 10.1093/rheumatology/kem205
29. Komiya Y, Saito T, Mizoguchi F, Kohsaka H. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. (2017) 56:3237–41. doi: 10.2169/internalmedicine.7966-16
30. Kikuchi H, Yamamoto T, Asako K, Takayama M, Shirasaki R, Ono Y. Etanercept for the treatment of intractable hemophagocytic syndrome with systemic lupus erythematosus. Modern Rheumatol. (2012) 22:308–11. doi: 10.3109/s10165-011-0500-1
31. Sellmer A, Stausbøl-Grøn B, Krag-Olsen B, Herlin T. Successful use of infliximab in macrophage activation syndrome with severe CNS involvement. Scand J Rheumatol. (2011) 40:156. doi: 10.3109/03009742.2010.508468
32. Makay B, Yilmaz S, Türkyilmaz Z, Ünal N, Ören H, Ünsal E. Etanercept for therapy-resistant macrophage activation syndrome. Pediatr Blood Cancer. (2008) 50:419–21. doi: 10.1002/pbc.21019
33. Buonuomo PS, Campana A, Insalaco A, Bracaglia C, Pardeo M, Cortis E. Necrotizing fasciitis in a pediatric patient treated with etanercept and cyclosporine for macrophage activation syndrome. Rheumatol Int. (2013) 33:1097. doi: 10.1007/s00296-011-2319-7
34. Prahalad S, Bove KE, Dickens D, Lovell DJ, Grom AA. Etanercept in the treatment of macrophage activation syndrome. J Rheumatol. (2001) 28:2120–4. Available online at: https://www.jrheum.org/content/28/9/2120.long
35. Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine. (2018) 101:14–8. doi: 10.1016/j.cyto.2016.08.003
36. Mylonas A, Conrad C. Psoriasis: classical vs. Paradoxical The Yin-Yang of TNF and type I interferon. Front Immunol. (2018) 9:2746. doi: 10.3389/fimmu.2018.02746
37. Clark J, Vagenas P, Panesar M, Cope A. What does tumour necrosis factor excess do to the immune system long term? Ann Rheum Dis. (2005) 64(Suppl. 4)iv70–iv6. doi: 10.1136/ard.2005.042523
38. Macías I, García-Pérez S, Ruiz-Tudela M, Medina F, Chozas N, Girón-González JA. Modification of pro-and antiinflammatory cytokines and vascular-related molecules by tumor necrosis factor-a blockade in patients with rheumatoid arthritis. J Rheumatol. (2005) 32:2102–8. Available online at: https://www.jrheum.org/content/32/11/2102.long
39. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol. (2019) 10:e00119. doi: 10.3389/fimmu.2019.00119
40. Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN. Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor α and interleukin-1 in rheumatoid arthritis. Arthritis Rheum. (1998) 41:1258–65. doi: 10.1002/1529-013141:7<1258::AID–ART17>3.0.CO;2-1
41. Majewska E, Paleolog E, Baj Z, Kralisz U, Feldmann M, Tchorzewski H. Role of tyrosine kinase enzymes in TNF-α and IL-1 induced expression of ICAM-1 and VCAM-1 on human umbilical vein endothelial cells. Scand J Immunol. (1997) 45:385–92. doi: 10.1046/j.1365-3083.1997.d01-412.x
42. Feldmann M, Maini RN. Anti-TNFα therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. (2001) 19:163–96. doi: 10.1146/annurev.immunol.19.1.163
43. Charles P, Elliott MJ, Davis D, Potter A, Kalden JR, Antoni C, et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-α therapy in rheumatoid arthritis. J Immunol. (1999) 163:1521–8.
44. Dvorak HF, Nagy J, Feng D, Brown L, Dvorak A. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Vasc Growth Fact Angiog. (1999) 237:97–132. doi: 10.1007/978-3-642-59953-8_6
45. Kuroda T, Wada Y, Kobayashi D, Murakami S, Sakai T, Hirose S, et al. Effective anti-TNF-α therapy can induce rapid resolution and sustained decrease of gastroduodenal mucosal amyloid deposits in reactive amyloidosis associated with rheumatoid arthritis. J Rheumatol. (2009) 36:2409–15. doi: 10.3899/jrheum.090101
46. Lai W-Y, Wang J-W, Huang B-T, Lin EP-Y, Yang P-C, A. novel TNF-α-targeting aptamer for TNF-α-mediated acute lung injury and acute liver failure. Theranostics. (2019) 9:1741. doi: 10.7150/thno.30972
47. Luo W, Li YX, Jiang LJ, Chen Q, Wang T, Ye DW. Targeting JAK-STAT signaling to control cytokine release syndrome in COVID-19. Trends Pharmacol Sci. (2020) 41:531–43. doi: 10.1016/j.tips.2020.06.007
48. Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, et al. Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc Nat Acad Sci. (2008) 105:7809–14. doi: 10.1073/pnas.0711241105
49. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
50. Cheng J, Zhang J, Wu Z, Sun X. Inferring microenvironmental regulation of gene expression from single-cell RNA sequencing data using scMLnet with an application to COVID-19. Brief Bioinform. (2021) 22:988–1005. doi: 10.1093/bib/bbaa327
51. Collin J, Queen R, Zerti D, Dorgau B, Georgiou M, Djidrovski I, et al. Co-expression of SARS-CoV-2 entry genes in the superficial adult human conjunctival, limbal and corneal epithelium suggests an additional route of entry via the ocular surface. Ocul Surf. (2021) 19:190–200. doi: 10.1016/j.jtos.2020.05.013
52. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. (2005) 436:112–6. doi: 10.1038/nature03712
53. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nature Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
54. Potdar AA, Dube S, Naito T, Botwin G, Haritunians T, Li D, et al. Reduced expression of COVID-19 host receptor, ACE2 is associated with small bowel inflammation, more severe disease, and response to anti-TNF therapy in Crohn's disease. medRxiv [preprint]. (2020). doi: 10.1101/2020.04.19.20070995. Available online at: www.medrxiv.org/content/10.1101/2020.04.19.20070995v1 (accessed April 23, 2020).
55. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. (2005) 280:30113–9. doi: 10.1074/jbc.M505111200
56. Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L84–L96. doi: 10.1152/ajplung.00071.2009
57. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. (2005) 309:1864–8. doi: 10.1126/science.1116480
58. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. (2003) 426:450–4. doi: 10.1038/nature02145
59. Li F. Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J Virol. (2008) 82:6984–91. doi: 10.1128/JVI.00442-08
60. Wu K, Peng G, Wilken M, Geraghty RJ Li F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J Biol Chem. (2012) 287:8904–11. doi: 10.1074/jbc.M111.325803
61. Li Q, Verma IM. NF-κB regulation in the immune system. Nature Rev Immunol. (2002) 2:725–34. doi: 10.1038/nri910
62. Hayden M, West A, Ghosh S. NF-κ B and the immune response. Oncogene. (2006) 25:6758–80. doi: 10.1038/sj.onc.1209943
63. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. (2020) 369:718–24. doi: 10.1126/science.abc6027
64. DeDiego ML, Nieto-Torres JL, Regla-Nava JA, Jimenez-Guardeño JM, Fernandez-Delgado R, Fett C, et al. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J Virol. (2014) 88:913–24. doi: 10.1128/JVI.02576-13
65. Smits SL, de Lang A, van den Brand JM, Leijten LM, van IJcken WF, Eijkemans MJ, et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathogens. (2010) 6:e1000756. doi: 10.1371/journal.ppat.1000756
66. Hiscott J, Nguyen TA, Arguello M, Nakhaei P, Paz S. Manipulation of the nuclear factor-κ B pathway and the innate immune response by viruses. Oncogene. (2006) 25:6844–67. doi: 10.1038/sj.onc.1209941
67. Kalliolias GD, Ivashkiv LB, TNF. biology, pathogenic mechanisms and emerging therapeutic strategies. Nature Rev Rheumatol. (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
68. Wajant H, Scheurich P. Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int J Biochem Cell Biol. (2001) 33:19–32. doi: 10.1016/S1357-2725(00)00064-9
69. Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. (2020) 35:266–71. doi: 10.1007/s12250-020-00207-4
70. Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M, et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet. (2020) 395:1407–9. doi: 10.1016/S0140-6736(20)30858-8
71. Hachem H, Godara A, Schroeder C, Fein D, Mann H, Lawlor C, et al. Rapid and sustained decline in CXCL-10 (IP-10) annotates clinical outcomes following TNFα-antagonist therapy in hospitalized patients with severe and critical COVID-19 respiratory failure. J Clin Transl Sci. (2021) 5:e146. doi: 10.1017/cts.2021.805
72. Duret P-M, Sebbag E, Mallick A, Gravier S, Spielmann L, Messer L. Recovery from COVID-19 in a patient with spondyloarthritis treated with TNF-alpha inhibitor etanercept. Ann Rheum Dis. (2020) 79:1251–2. doi: 10.1136/annrheumdis-2020-217362
73. Flammiger A, Fiedler W, Bacher U, Bokemeyer C, Schneider M, Binder M. Critical imbalance of TNF-α and soluble TNF receptor 1 in a patient with macrophage activation syndrome: potential implications for diagnostics and treatment. Acta Haematol. (2012) 128:69–72. doi: 10.1159/000338179
74. Son MB, Gauvreau K, Burns JC, Corinaldesi E, Tremoulet AH, Watson VE, et al. Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study. J Pediatr. (2011) 158:644–9.e1. doi: 10.1016/j.jpeds.2010.10.012
75. Mori M, Hara T, Kikuchi M, Shimizu H, Miyamoto T, Iwashima S, et al. Infliximab versus intravenous immunoglobulin for refractory Kawasaki disease: a phase 3, randomized, open-label, active-controlled, parallel-group, multicenter trial. Sci Rep. (2018) 8:1–10. doi: 10.1038/s41598-017-18387-7
76. Masuda H, Kobayashi T, Hachiya A, Nakashima Y, Shimizu H, Nozawa T, et al. Infliximab for the treatment of refractory Kawasaki disease: a nationwide survey in Japan. J Pediatr. (2018) 195:115–20.e3. doi: 10.1016/j.jpeds.2017.10.013
77. Tremoulet AH. Adjunctive therapies in Kawasaki disease. Int J Rheum Dis. (2018) 21:76–9. doi: 10.1111/1756-185X.13208
78. Rivera-Rodriguez L, Pardo-Díaz E, Moreno-Espinosa S, Scheffler-Mendoza S, Ruiz-Ontiveros MA, Garrido-García LM, et al. Use of infliximab in the treatment of macrophage activation syndrome complicating Kawasaki disease. J Pediatr Hematol Oncol. (2021) 43:e448–e51. doi: 10.1097/MPH.0000000000001756
79. Dolinger MT, Person H, Smith R, Jarchin L, Pittman N, Dubinsky MC, et al. Pediatric Crohn's disease and multisystem inflammatory syndrome in children (MIS-C) and COVID-19 treated with infliximab. J Pediatr Gastroenterol Nutr. (2020) 71:153–5. doi: 10.1097/MPG.0000000000002809
80. Bezzio C, Manes G, Bini F, Pellegrini L, Saibeni S. Infliximab for severe ulcerative colitis and subsequent SARS-CoV-2 pneumonia: a stone for two birds. Gut. (2021) 70:623–4. doi: 10.1136/gutjnl-2020-321760
81. Brenner EJ, Ungaro RC, Gearry RB, Kaplan GG, Kissous-Hunt M, Lewis JD, et al. Corticosteroids, but not TNF antagonists, are associated with adverse COVID-19 outcomes in patients with inflammatory Bowel diseases: results from an international registry. Gastroenterology. (2020) 159:481–91.e3. doi: 10.1053/j.gastro.2020.05.032
82. Cappello M, Busacca A, Guida L. The course of COVID-19 in inflammatory Bowel disease: protective role of TNF antagonists. Gastroenterology. (2021) 160:1885–6. doi: 10.1053/j.gastro.2020.06.087
83. Gianfrancesco M, Hyrich KL, Al-Adely S, Carmona L, Danila MI, Gossec L, et al. Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease: data from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann Rheum Dis. (2020) 79:859–66. doi: 10.1136/annrheumdis-2020-217871
84. Ungaro RC, Brenner EJ, Gearry RB, Kaplan GG, Kissous-Hunt M, Lewis JD, et al. Effect of IBD medications on COVID-19 outcomes: results from an international registry. Gut. (2021) 70:725–32. doi: 10.1136/gutjnl-2020-322539
85. Monti S, Balduzzi S, Delvino P, Bellis E, Quadrelli VS, Montecucco C. Clinical course of COVID-19 in a series of patients with chronic arthritis treated with immunosuppressive targeted therapies. Ann Rheum Dis. (2020) 79:667–8. doi: 10.1136/annrheumdis-2020-217424
86. Tomelleri A, Sartorelli S, Campochiaro C, Baldissera EM, Dagna L. Impact of COVID-19 pandemic on patients with large-vessel vasculitis in Italy: a monocentric survey. Ann Rheum Dis. (2020) 79:1252–3. doi: 10.1136/annrheumdis-2020-219414
87. Bezzio C, Saibeni S, Variola A, Allocca M, Massari A, Gerardi V, et al. Outcomes of COVID-19 in 79 patients with IBD in Italy: an IG-IBD study. Gut. (2020) 69:1213–7. doi: 10.1136/gutjnl-2020-321411
88. Arleo T, Tong D, Shabto J, O'Keefe G, Khosroshahi A. Clinical course and outcomes of COVID-19 in rheumatic disease patients: a case cohort study with a diverse population. Clin Rheumatol. (2021) 40:2633–42. doi: 10.1007/s10067-021-05578-x
89. Papa A, Gasbarrini A, Tursi A. Epidemiology and the impact of therapies on the outcome of COVID-19 in patients with inflammatory Bowel disease. Am J Gastroenterol. (2020) 115:1722–4. doi: 10.14309/ajg.0000000000000830
90. Salesi M, Shojaie B, Farajzadegan Z, Salesi N, Mohammadi E. TNF-α blockers showed prophylactic effects in preventing COVID-19 in patients with rheumatoid arthritis and seronegative spondyloarthropathies: a case-control study. Rheumatol Ther. (2021) 23:1–16. doi: 10.1007/s40744-021-00342-8
91. Chiu Y-M, Chen D-Y. Infection risk in patients undergoing treatment for inflammatory arthritis: non-biologics versus biologics. Expert Rev Clin Immunol. (2020) 207–28. doi: 10.1080/1744666X.2019.1705785
92. Minozzi S, Bonovas S, Lytras T, Pecoraro V, González-Lorenzo M, Bastiampillai AJ, et al. Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin Drug Saf. (2016) 15:11–34. doi: 10.1080/14740338.2016.1240783
93. Brito CA, Paiva JG, Pimentel FN, Guimarães RS, Moreira MR. COVID-19 in patients with rheumatological diseases treated with anti-TNF. Ann Rheum Dis. (2021) 80:e62. doi: 10.1136/annrheumdis-2020-218171
94. Mikuls TR, Johnson SR, Fraenkel L, Arasaratnam RJ, Baden LR, Bermas BL, et al. American College of rheumatology guidance for the management of adult patients with rheumatic disease during the COVID-19 pandemic. Arthritis Rheumatol. (2020) 72:1241–51. doi: 10.1002/art.41437
95. Wendling D, Prati C. Paradoxical effects of anti-TNF-α agents in inflammatory diseases. Expert Rev Clin Immunol. (2014) 10:159–69. doi: 10.1586/1744666X.2014.866038
96. Hyams JS, Dubinsky MC, Baldassano RN, Colletti RB, Cucchiara S, Escher J, et al. Infliximab is not associated with increased risk of malignancy or hemophagocytic lymphohistiocytosis in pediatric patients with inflammatory bowel disease. Gastroenterology. (2017) 152:1901–14.e3. doi: 10.1053/j.gastro.2017.02.004
Keywords: COVID-19, cytokine release syndrome, MIS-C, TNF-α inhibitor, infliximab
Citation: Guo Y, Hu K, Li Y, Lu C, Ling K, Cai C, Wang W and Ye D (2022) Targeting TNF-α for COVID-19: Recent Advanced and Controversies. Front. Public Health 10:833967. doi: 10.3389/fpubh.2022.833967
Received: 12 December 2021; Accepted: 17 January 2022;
Published: 11 February 2022.
Edited by:
Emma Grant, La Trobe University, AustraliaReviewed by:
Xueqin Chen, Ningbo First Hospital, ChinaCopyright © 2022 Guo, Hu, Li, Lu, Ling, Cai, Wang and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weici Wang, d2VpY2l3YW5nQGdtYWlsLmNvbQ==; Dawei Ye, ZHkwNzExQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.