 Hisham N. Altayb1,2*
Hisham N. Altayb1,2* Salman Hosawi1
Salman Hosawi1 Othman Baothman1
Othman Baothman1 Imran Kazmi1Kamel Chaieb1Isam M. Abu Zeid3
Imran Kazmi1Kamel Chaieb1Isam M. Abu Zeid3 Hana S. Elbadawi4
Hana S. Elbadawi4 Bruno Silvester Lopes5,6*Ehssan Moglad7
Bruno Silvester Lopes5,6*Ehssan Moglad7- 1Department of Biochemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
- 2Center of Artificial Intelligence in Precision Medicines, King Abdulaziz University, Jeddah, Saudi Arabia
- 3Department of Biological Sciences, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
- 4Microbiology and Parasitology Department, Soba University Hospital, University of Khartoum, Khartoum, Sudan
- 5School of Health and Life Sciences, Teesside University, Middlesbrough, United Kingdom
- 6National Horizons Centre, Teesside University, Darlington, United Kingdom
- 7Department of Pharmaceutics, College of Pharmacy, Prince Sattam bin Abdulaziz University, Al-Kharj, Saudi Arabia
Introduction: The emergence of bacterial pathogens in environmental hosts represents a major risk to public health. This study aimed at characterizing seven novel environmental strains of K. quasipneumoniae using a genomic approach which was misidentified by phenotypic methods in a previous batch of 27 species thought to be K. pneumoniae.
Methods: Whole-genome sequencing was performed using the Illumina platform, and the generated raw reads were de novo assembled. Comparative genomic, resistome, virulome, mobilome, and phylogeny were then investigated using dierent bioinformatics tools.
Results: Six strains were identified as K. quasipneumoniae subsp similipneumoniae and one as K. quasipneumoniae subsp. quasipneumoniae. All isolates were resistant to ampicillin, cephalexin, and amoxicillin-clavulanic acid and harbored the fosA, blaOKP types, oqxB, and oqxA genes. One isolate additionally harbored a gene cassettes consisting of blaSHV−1, blaOXA−1, aac(6′)-Ib-cr, catB genes. The aminoglycoside-modifying enzyme gene aph(3”)-Ia was bracketed by two insertion elements. Plasmid analyses showed that IncFIBK was the most prevalent plasmid, circulating in six isolates, while one isolate exhibited seven different plasmids. The isolates have virulence genes responsible for capsule formation, lipopolysaccharide, iron uptake aerobactin (iutA), salmochelins (iroE, iroN), enterobactin siderophore, adherence, and biofilm formation (mrkA, mrkB, mrkC, mrkD, mrkF, and mrkH).
Conclusion: Our study highlights the ecology and transmission of K. quasipneumoniae (which have the ability to disseminate to other environmental sources including animals) outside the clinical setting and the contribution of water, vegetables, and table surfaces as potential reservoirs of farm-to-fork transmission of disease via local markets in Khartoum, Sudan.
Introduction
Klebsiella pneumoniae represents an ongoing and growing challenge for humans and is classified as a critical priority pathogen listed by the WHO. Hypervirulent community-acquired invasive strains of Klebsiella represent a major problem in developing countries with the growing threat of convergence of multidrug-resistant (MDR) and hypervirulent phenotypes (1). K. pneumoniae is not only capable of causing serious clinical infections but can acquire significant levels of antibiotic resistance genes leading to failure in the treatment of patients with urinary tract and respiratory tract infections, liver abscess, endophthalmitis, and meningitis (2). K. pneumoniae is a familiar MDR nosocomial pathogen and has the capacity to adapt to various environmental and clinical settings (3). Recent taxonomic classification has delineated at least seven phylogroups of K. pneumoniae (Kp1–Kp7) which contribute to the K. pneumoniae species complex (KpSC). The KpSC consists of five species, these include K. pneumoniae sensu stricto (Kp1), K. quasipneumoniae [subsp. quasipneumoniae (Kp2) and subsp. similipneumoniae (Kp4)], K. variicola [subsp. variicola (Kp3) and subsp. tropica (Kp5)] and K. quasivariicola (Kp6, which is not formally defined yet), and K. africana (Kp7) (4–6). Originally, K. quasipneumoniae was found to be associated with the environment. However, there are now increasing reports of it being a human pathogen associated which underlying hospital-acquired infections (7, 8). Identification of K. quasipneumoniae is challenging when using conventional laboratory methods due to the similarity in their biochemical tests with other Klebsiella spp. leading to misidentification and false reporting (5).
Multidrug-resistant (MDR) hypervirulent strains of K. quasipneumoniae subsp. quasipneumoniae are becoming an issue for public health worldwide. Emerging antimicrobial resistance genes (ARGs) conferring resistance to last-resort antibiotics including cephalosporins, carbapenems, and tigecycline, have been reported in clinical and environmental isolates. Such genes include blaCTX−M−2, blaDHA−1, blaNDM, blaIMP, blaKHM−1, blaOXA−48, blaOXA−162, blaOXA−10, blaKPC, tet(A), and tet(K) genes (9–12). Environmental settings, especially water surfaces, soil, animal waste, and sewage treatment plants, are implicated in promoting the distribution of ARGs and as a result act as a reservoir of multidrug-resistant (MDR) K. quasipneumoniae (13).
Furthermore, K. quasipneumoniae has the ability to uptake ARGs and plasmids from other Enterobacteria, including plasmids belonging to different incompatibility groups such as IncU/IncX5 which harbors blaKPC, IncHI2 which harbors mcr-9, and IncFII/IncFIB which harbors mcr-8.2 (7).
The emergence of MDR pathogens in the environment, particularly from a one-health perspective is hence becoming a global threat to public health (14). K. quasipneumoniae has been identified as an opportunistic pathogen capable of harboring different ARGs that render clinically important antibiotics ineffective. Hence, this study aimed at characterizing the genomes of seven K. quasipneumoniae isolated from environmental sources such as water, vegetables, and table surfaces from the local market in Khartoum Bahre, Sudan.
Materials and methods
Isolate selection
A total of 27 bacterial isolates were collected from clinical (n = 10) and environmental samples (n = 17) between March and July 2021, the isolates were initially identified as K. pneumoniae using standard biochemical tests (15). Among the K. pneumoniae (n = 27) 7 were reidentified as K. quasipneumoniae using genomic tools. More details on the rest of K. pneumoniae (n = 20) isolates, sample collection, and processing have been published elsewhere (16). The environmental samples were obtained via swabs collected from vegetables and table surfaces in the vegetable market in the Khartoum locality. In addition, water samples were collected from water stations in the Khartoum locality. The samples were collected and processed according to published protocols (17, 18). Chromogenic UTI agar media (bioMérieux, France) and MacConkey agar (HiMedia, India) were used for the differentiation of the isolates according to their phenotypic characteristics. Standard biochemical tests were used for the identification of the isolates (15).
Antimicrobial sensitivity testing and minimum inhibitory concentration
Disk-diffusion method was used for the determination of antimicrobial susceptibility testing. The antibiotics tested were, amoxicillin-clavulanate (30 μg), cefuroxime (30 μg), ceftriaxone (30 μg), ceftazidime (30 μg), cephalexin (30 μg), meropenem (10 μg), imipenem (10 μg), amikacin (30 μg), gentamicin (10 μg), ciprofloxacin (5 μg), trimethoprim-sulfamethoxazole (25 μg), and chloramphenicol (10 μg). Minimum inhibitory concentration (MIC) was performed for ampicillin, chloramphenicol and ciprofloxacin using the broth dilution method in a serial dilution ranging from 0.5 to 256 μg/ml (19). K. pneumoniae ATCC 700603 and E. coli ATCC 25922 were used as quality controls and the results were interpreted according to the CLSI guidelines (20).
Whole-genome sequencing and molecular analysis
DNA was extracted from overnight bacterial growth of pure colonies on Nutrient Agar (HiMedia, India), by the quinidine chloride protocol as described by Sabeel et al. (21). The DNA quality was checked by gel electrophoresis where clear crisp bands indicate DNA of high quality and on the contrary, a smear indicates DNA degradation or low quality. It was also quantitatively checked using nanodrop and Qubit (Thermo Scientific, USA). The extracted DNA was subjected to next-generation sequencing with 100X coverage. Paired-end (2 × 150 bp) whole-genome sequencing was achieved using Illumina HiSeq 2500 platform (Illumina, USA), by Novogene Company (China). PATRIC server assembly was used to obtain de-novo assemblies. Identification of species and strains was performed by MLST 2.0 and PubMLST (22). The assignment of the new sequence types and subspecies identification were achieved by the Pasteur MLST database curators. Genomes were annotated by PATRIC server and NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (23). The Pathogenwatch platform was used for capsule (K) and O serotype identification (24).
The resistome profile was analyzed by Resistance Gene Identifier (RGI) and ResFinder (25). Mobile genetic elements, insertion sequences and plasmids were identified by Mobile Element Finder (26), IS Finder, and Plasmid Finder 2.1, respectively. Geneious Prime was used for the visualization of resistant genes and transposon cassettes. Virulence factors were investigated using VirulenceFinder 2.0 and the Virulence Factor Database (VFDB) (27) where genomes of K. quasipneumoniae subsp. quasipneumoniae (18A069 and MGH96) and K. quasipneumoniae subsp. similipneumoniae (HKUOPA4, HKUOPL4, and ATCC700603) were used as reference.
Core-genome and average nucleotide identity analysis
Core-genome multi-locus sequence typing (cgMLST) classification and core genome MLST profile comparison were achieved using Pathogenwatch, which includes tools that infer Klebsiella lineage codes based on references from the Pasteur/PubMLST resource.
Average nucleotide identity analysis was conducted using fastANI v. 1.33 (28). For ANI analysis of our isolates, five reference genomes were used for comparison, which includes K. quasipneumoniae subsp. quasipneumoniae (18A069 and MGH96) and K. quasipneumoniae subsp. similipneumoniae (HKUOPA4, HKUOPL4, and ATCC700603). Heat map and SNP matrix were built by FastANI_heatmap using R code for building a heat map and histogram with the output of FastANI. ANI with more than 95% identity was considered suitable to identify species and ≥ 98% to identify subspecies (29–31).
Comparative genomics and phylogenomics analysis
A comparative genomics study was performed using the Gview tools (32) and PATRIC Proteome Comparison tool (33). The phylogenetic tree was generated by PATRIC phylogenetic tree builder, using the assembled contigs as input. In PATRIC the phylogenetic tree was created by using Codon Tree approach, which utilized PATRIC global protein families (PGFams) as homology groups. A group of PGFams were discovered among these chosen genomes using the Codon Tree analysis, and the aligned proteins and coding DNA from single-copy genes were used for RAxML analysis (34). The tree was built for the seven isolates of K. quasipneumoniae and the most similar genomes of K. quasipneumoniae (n = 27) from NCBI database. Figtree (35) was used for the modification and visualization of the generated tree.
Nucleotide sequence accession numbers
The Bioproject for isolates is PRJNA767482, and the complete chromosomal sequences were submitted to GenBank under accessions JAJOZL000000000, JAJOZM000000000, JAJOZN000000000, JAJOZO000000000, JAJHNR000000000, JAJOZP000000000, and JAJONG000000000 for isolates 3KE, 4KE, 5KE, 6KE, 8KE, 10KE, and 14KE respectively.
Results
Isolates identification
From a batch of 27 Klebsiella spp. identified phenotypically as K. pneumoniae, 7 (26%) were primarily misidentified as K. pneumoniae which after WGS were reidentified as K. quasipneumoniae by MLST 2.0, and PubMLST databases.
Antimicrobial susceptibility testing and MIC
All isolates were resistant to ampicillin, amoxicillin-clavulanic acid and cephalexin, while they were sensitive to meropenem, imipenem, amikacin, gentamicin, ciprofloxacin, ceftazidime, ceftriaxone, cefuroxime, chloramphenicol, and trimethoprim-sulfamethoxazole (Table 1).
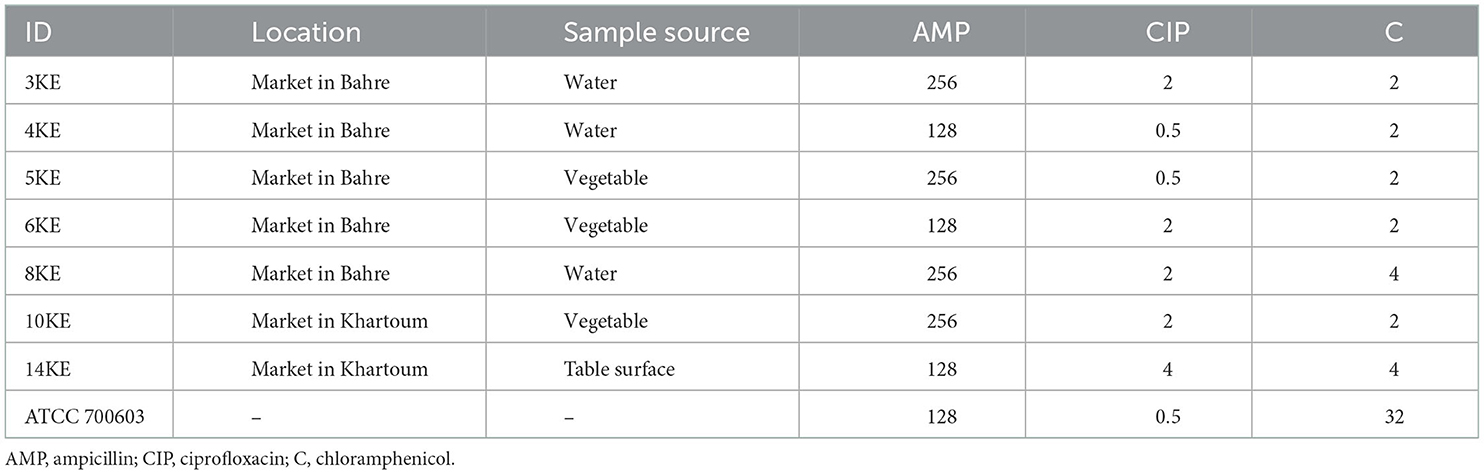
Table 1. The minimum inhibitory concentration (μg/ml) of antibiotics against K. quasipneumoniae isolates.
Genomic features and sequence types of the isolate
All assembled reads had coverage in a range of 171–198, an average genomic length of 5,320,537 bp, a number of contigs in a range of 213–386, GC content of 57%, and an average number of coding sequences (CDS) of 5,322 (Table 2).
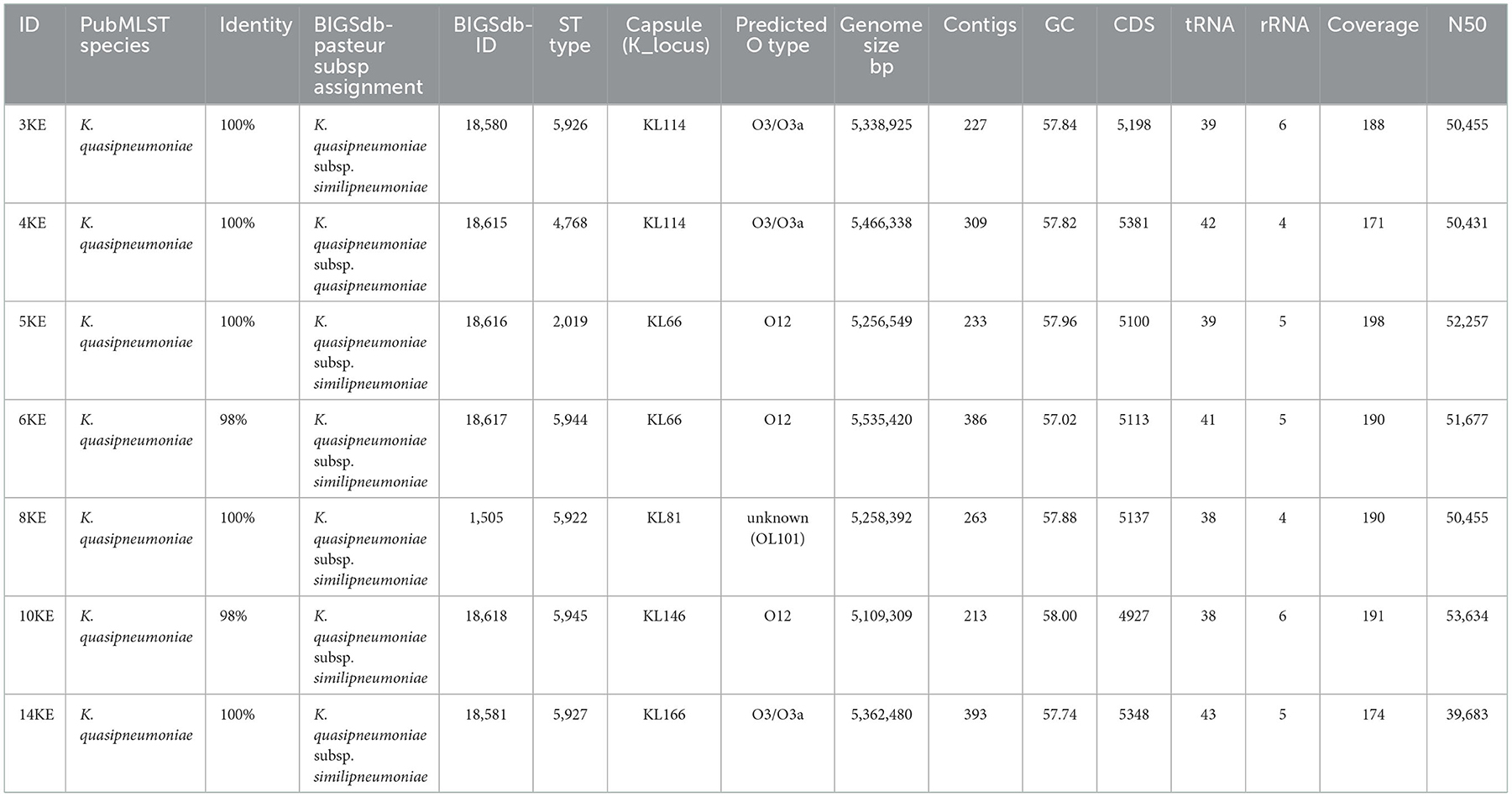
Table 2. Genomic features of different strains of K. quasipneumoniae.
When queried with the rMLST database sited at PubMLST, five isolates showed 100% identity, and two showed 98% identity to K. quasipneumoniae (https://pubmlst.org/bigsdb?db=pubmlst_rmlst_seqdef_kiosk, last accessed, on 21st September 2022). The isolates were subsequently identified as K. quasipneumoniae subsp. similipneumoniae (n = 6) and K. quasipneumoniae subsp. quasipneumoniae (n = 1). Six isolates were identified at the subspecies level and assigned the novel sequence types and IDs by the Institut Pasteur team (Table 2). The isolate 5KE was not defined as a novel ST due to partial sequence alignment of tonB gene alleles. Isolate 4KE was identified with unknown sublineage, clonal group and core genome sequence type (cgST), while isolates 5KE, 6KE, and 8KE were identified with known clonal group, and cgST as shown in Supplementary material 1 and Supplementary Table 1.
Prediction of the capsule (K) and O serotypes revealed that isolates 3KE and 4KE possessed KL114 capsule-type whereas isolates 5KE, 6KE, and 14KE harbored the KL66 K-loci. The KL81 and KL146 capsule (K) types were present in 8KE and 10KE respectively. Isolates 3KE, 4KE, and 14KE belonged to O3/O3a serotype and isolates 5KE, 6KE, and 19KE had the O12 serotype. In isolate 8KE the serotype was unknown (Table 2).
ANI analysis was performed on the whole genome sequences for species and subspecies identification; the 95% identity criteria were considered for species identification, and 98% for subspecies identification. The K. quasipneumoniae subsp. quasipneumoniae (4KE) strain showed an average of 96.5% ANI compared to other K. quasipneumoniae subsp. similipneumoniae strains, while the 4KE showed 99% identity with reference strains of the same subspecies K. quasipneumoniae subsp. quasipneumoniae reference strains (18A069 and MGH96). At the same time, the comparison of K. quasipneumoniae subsp. similipneumoniae strains (3KE, 5KE, 6KE, 8KE, 10KE, and 14KE) showed high ANI (ranging from 98.84 to 99.96%) similarity with K. quasipneumoniae subsp. Similipneumoniae reference strains (HKUOPA4, HKUOPL4 and ATCC700603). 5KE and 6KE were the closest strains showing 99.96% identity, while 10KE showed the lowest (with an average of 98.87%) identity among subspecies of K. quasipneumoniae subsp. similipneumoniae (Table 3, Figure 1). Alleles differences generated by Pathogenwatch are described using distance matrices as shown in Supplementary Table 2.
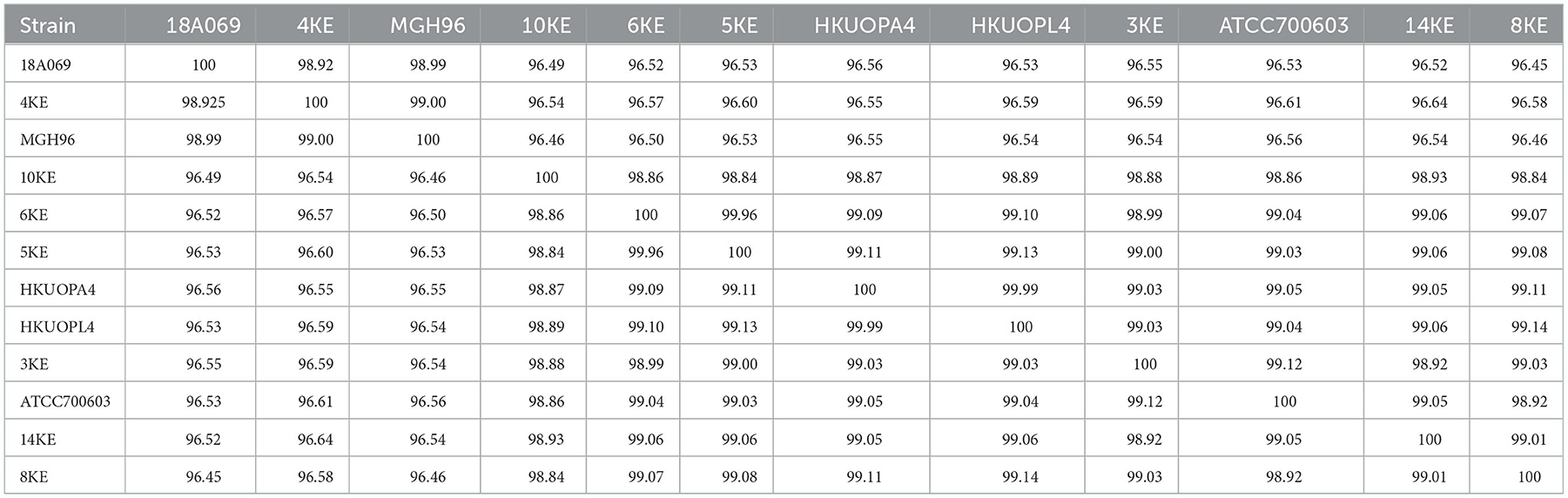
Table 3. ANIs comparison among subspecies of K. quasipneumoniae subsp. quasipneumoniae and K. quasipneumoniae subsp. similipneumoniae.
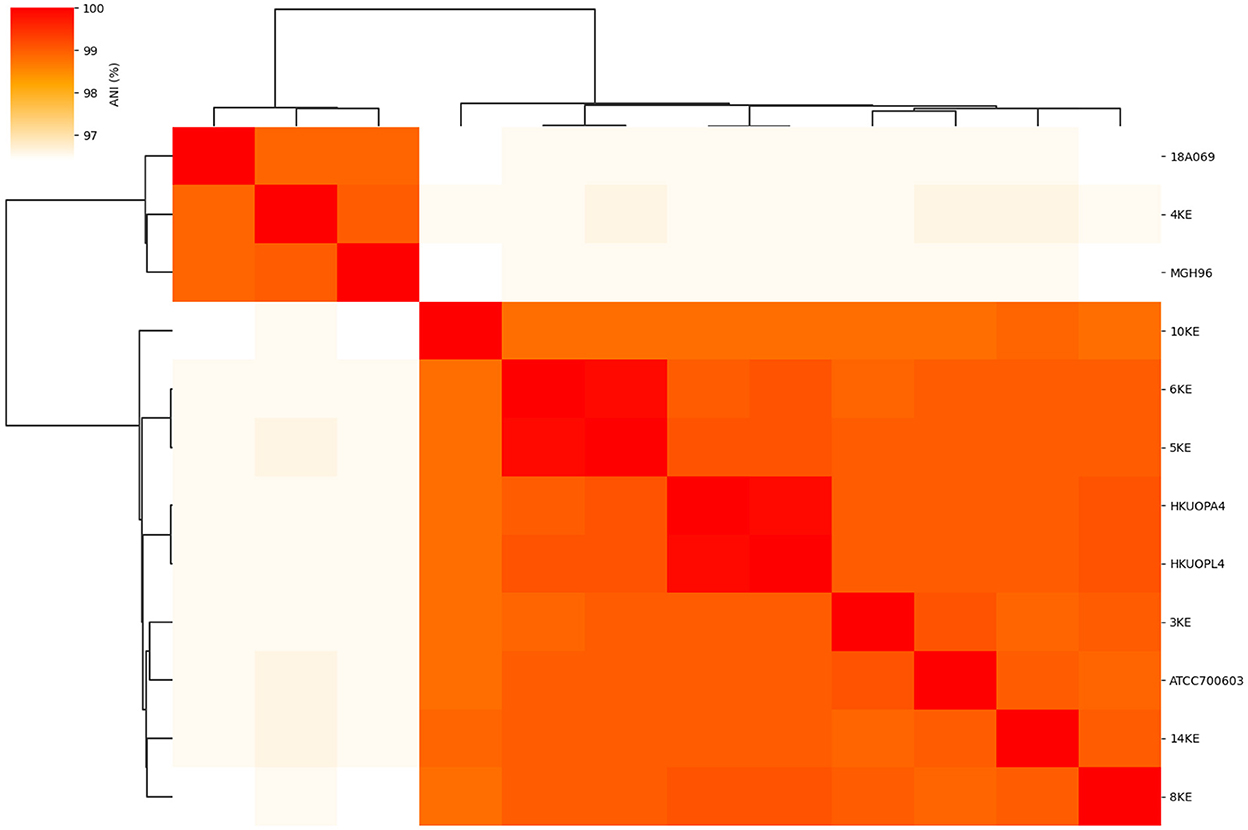
Figure 1. Heat map of the average nucleotide identity (ANI) and phylogenetic tree of subspecies of K. quasipneumoniae strains.
Predicted antimicrobial resistance mechanism, virulence genes, and plasmids
All the isolates harbored the fosfomycin resistance gene (fosA), the chromosomally expressed beta-lactamases (blaOKP types), and olaquindox/quinolone AB (oqxAB) efflux pump genes. The ompK36 and ompK37 genes involved in reduced permeability to beta-lactams were present in all the isolates except 5KE. The fosA6 gene was present only in isolate 4KE, and the fosA7 gene was present only in isolate 3KE. Isolate 6KE harbored a cluster of blaSHV−1, blaOXA−1, aac(6′)-Ib-cr, and catB (Table 4, Figure 2). The aminoglycoside-modifying enzyme gene [aph(3”)-Ia] was harbored in isolate 6KE and was bracketed by two transposable insertion elements IS5075 at 5′ and IS91 transposase at the 3′ ends (Figure 3).
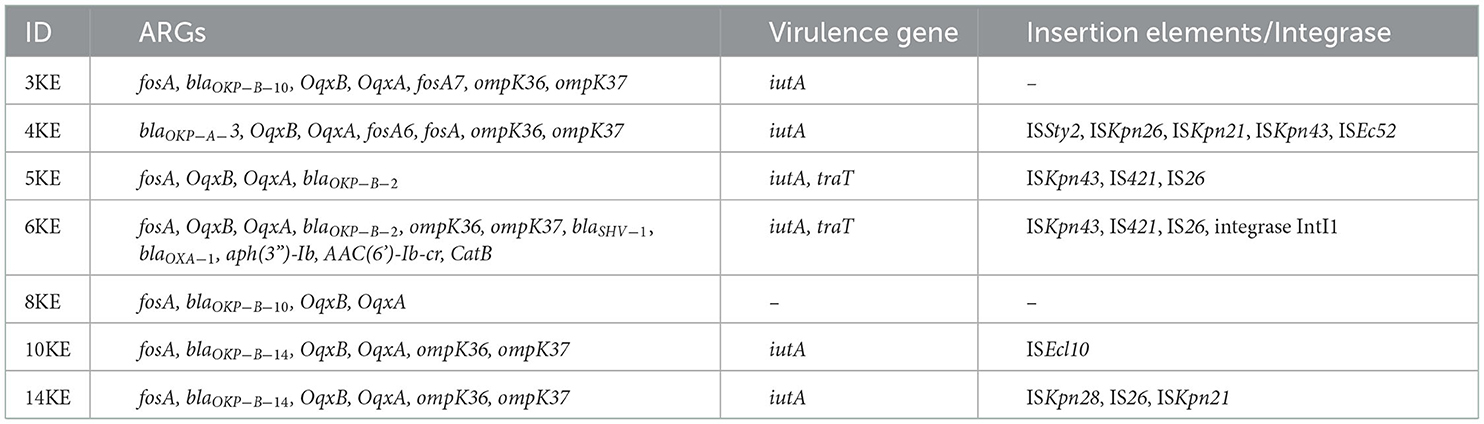
Table 4. Antibiotic resistance genes and mobile genetic elements present in K. quasipneumoniae isolates.
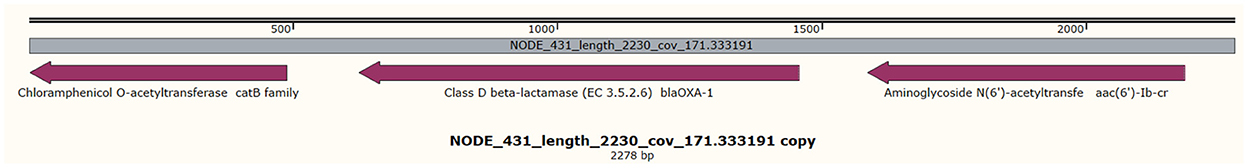
Figure 2. Clustering of blaOXA−1, aac(6')-Ib-cr, and catB genes in contig 431 in isolate 6KE, the gray middle line indicates the length, and coverage of the contig, and the purple arrows indicate the reported genes and their orientations.

Figure 3. Showing the presence of aph(3′)-Ib, sul2 genes harbored in isolate 6KE and is found in brackets of two mobile elements. The gray middle line indicates the length, and coverage of the contig, and the purple arrows indicate the reported genes and their orientations.
The most common efflux pumps identified in the isolates were KpnG, LptD, CRP, eptB, ArnT, KpnF. Gene point mutations associated with drug resistance were also investigated and the acrR: Y114F, V165I, and EF-Tu: R234F point mutations were identified commonly in the isolates (Supplementary material 1, Supplementary Table 3).
Upon investigation of virulence factors, the type 3 fimbriae adherence-related genes were more common in the isolates. These were the major fimbrial subunit (mrkA), chaperone (mrkB), usher (mrkC), adhesins (mrkD, mrkH), and minor fimbrial subunit (mrkF). All type I fimbriae adherence-related genes (fim A-H) were identified in isolates 3KE, 6KE, 10KE, 14KE and the reference strains, while isolates 4KE lacked fimA, and fimF genes. Isolate 5KE lacked the fimB, while isolate 8KE possessed only fimD. The iron uptake aerobactin (iutA), salmochelins (iroE, iroN), and enterobactin siderophores (ent A-F, feb A-G), entS, and fes were identified in all the isolates, with notable exceptions: the iutA gene which was absent in 8KE, the entD gene was absent in 6KE and fes gene was absent in 4KE. Isolate 8KE was characterized by the presence of 16 types of secretion systems [T6SS-I (15) and (T6SS-II)], while 4KE was characterized by the presence of 14 types of T6SS-III. Six lipopolysaccharides biosynthetic (rfb) loci were identified in isolates 3KE, 4KE, 14KE, and the reference ATCC 35657, while only one locus was identified in the rest of the isolates except the reference MGH96 which is lacked all the lipopolysaccharide genes (Supplementary material 1, Supplementary Table 4).
Plasmid prediction showed that IncFIBK was the most prevalent plasmid, circulating in six isolates; one isolate carried two plasmids IncFIBK Col440I, while other strains carried five plasmids ColpVC, IncN, IncFIBK, IncFIIpKPX1, and IncR. Isolate 4KE contained 7 plasmids: ColpHAD28, ColpHAD28, ColpVC, IncFIBK, IncFIIpKPX1, IncN, and IncR (Table 5).
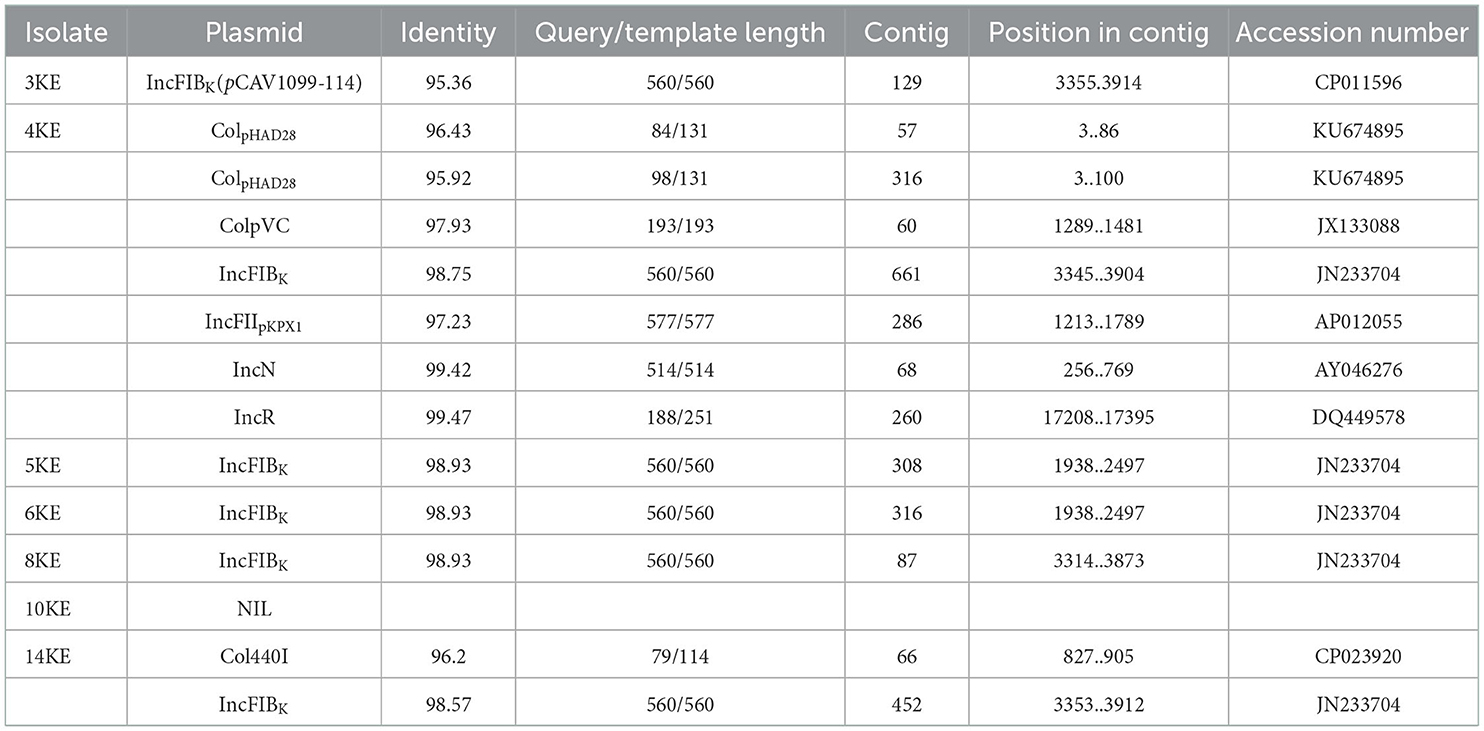
Table 5. Predicted plasmids in K. quasipneumoniae.
Antimicrobial susceptibility testing was interpreted according to CLSI guidelines (20).
Comparative genomics
The comparison of the genomes of K. quasipneumoniae isolates (3KE, 4KE, 5KE, 6KE, 8KE, 10KE, and 14KE) with K. quasipneumoniae strain ATCC 700603 revealed 9 unique regions among our isolates (Figure 4). These regions contain different types of unique proteins; the R1 region contains mobile elements (plasmids, AMR genes, and transposases), proteins associated with phage integration, anti-restriction protein klcA, and hypothetical proteins. Isolate 4KE contains, exclusively in region 1, a unique set of IncI1 plasmid conjugative transfer proteins and they include: IncI1 plasmid conjugative transfer protein TraU, IncI1 plasmid conjugative transfer protein TraW, IncI1 plasmid conjugative transfer protein TraX, IncI1 plasmid conjugative transfer integral membrane protein TraY, and IncI1 plasmid conjugative transfer protein TraQ. Region 2 contains transposases and phage-associated proteins, while region 3 contains proteins that regulate the length and adhesion of type 1 fimbriae and contains several genes involved in capsule production, fimbrial elements, and putative transcriptional regulatory proteins. Isolates 4KE, 5KE, 8KE, and 10KE are harbored in regions 4, 5, and 9 phages and phages-associated proteins in addition to hypothetical proteins (Supplementary material 2). Phylogenetic analysis revealed that isolates 3KE, 5KE, 8KE, 10KE, and 14KE were clustered at the same branch, indicating their relatedness, in contrast with 6KE and 4KE, which formed two separate branches (Figure 5).
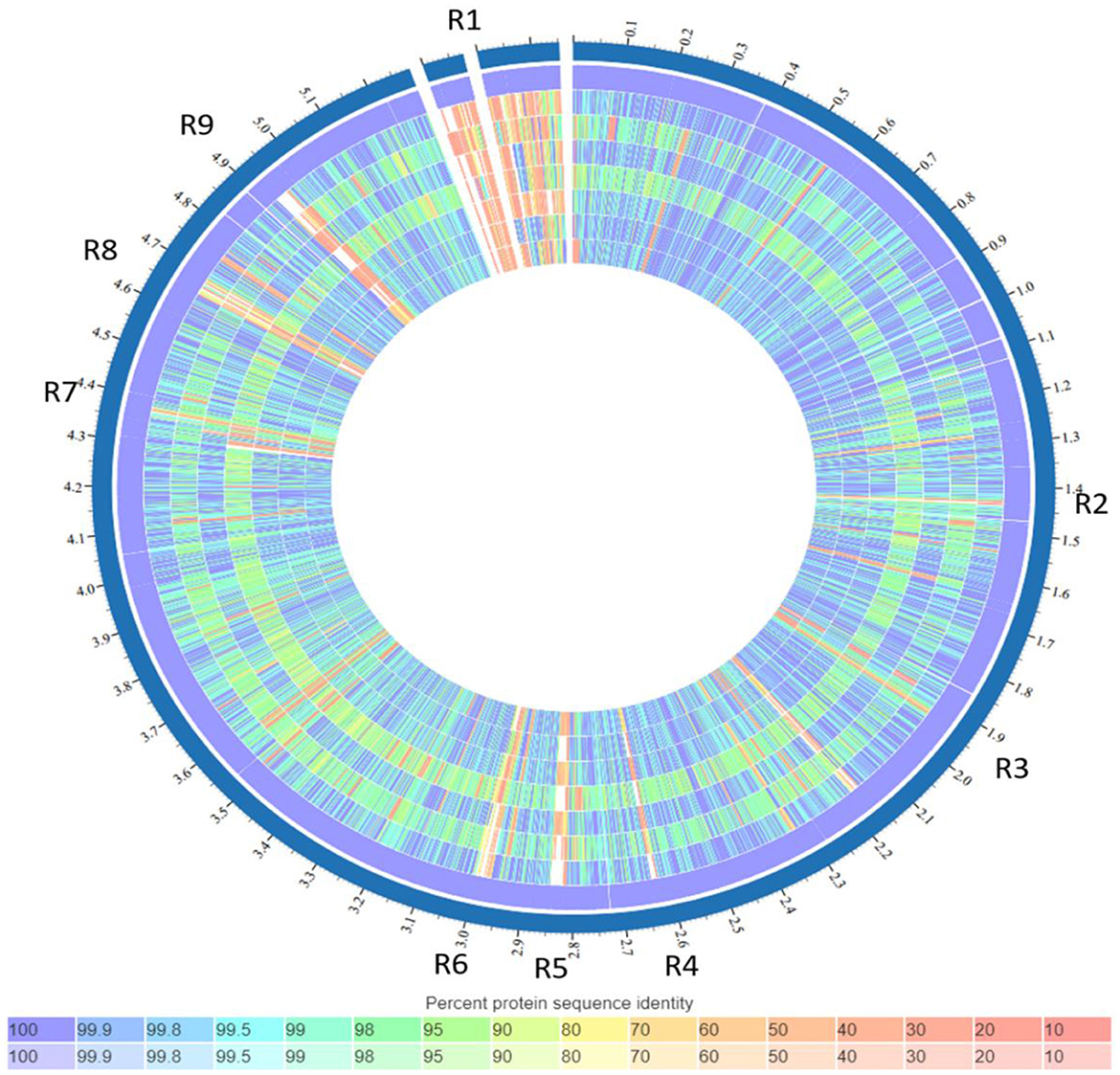
Figure 4. List of tracks, from outside to inside: K. quasipneumoniae strain ATCC 700603, isolate 3KE, isolate 4KE, isolate 5KE, isolate 6KE, isolate 10KE, isolate 14KE, isolate 8KE. Regions containing unique genes or features (named R1–R9). Colors indicate the percent protein identity of sequences to the reference, blue indicates high similarity (100%) while red indicates low similarity (0%).
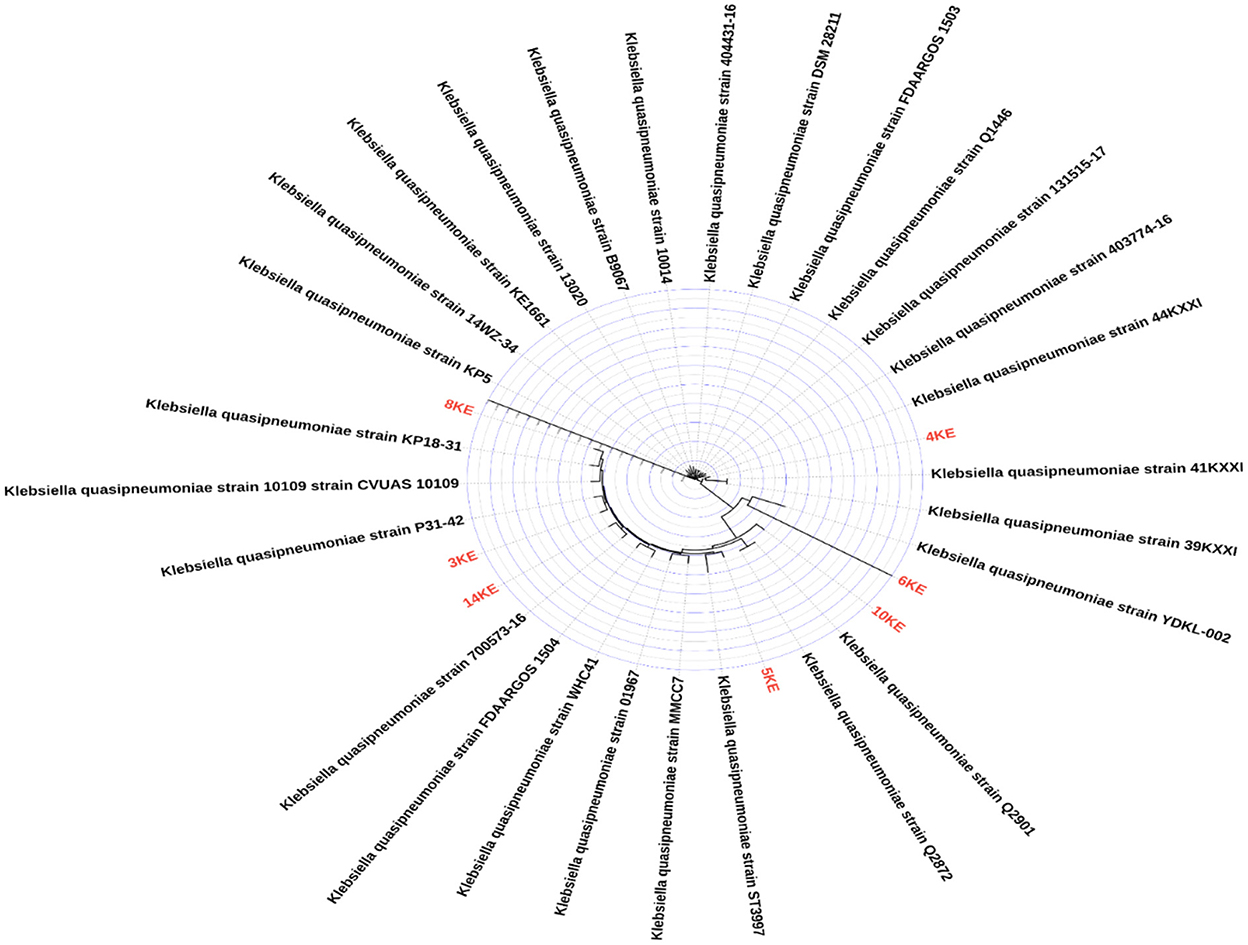
Figure 5. Phylogenetic tree of our isolates colored red and other K. quasipneumoniae isolates from Genbank database (n = 27).
Resistome comparison among our isolates and reference strains, showed the unique presence of antimicrobial resistance genes blaSHV−1, blaOXA−1, aph(3”)-Ib, AAC(6′)-Ib-cr, and CatB in 6KE. The fosA, blaOKP, OqxB, OqxA, and fosA7 were commonly identified in all isolates including references (Supplementary material 1, Supplementary Table 1).
The virulome comparison revealed the presence of type I secretory system (T6SS-I) associated genes (clpV/tssH, dotU/tssL, hcp/tssD, icmF/tssM, ompA, sciN/tssJ, tssF, tssG, vasE/tssK, vgrG/tssI, vipA/tssB, and vipB/tssC) only in isolate 8KE and three reference strains (ATCC35657, MGH96, and 18A069), while the clpV gene was found to be dominant in all isolates. Type III secretory system (T6SS-III) virulence genes (dotU, icmF, impA, impF, impG, impH, impJ, ompA, sciN, and vgrG) have been identified in isolate 4KE and three reference strains (ATCC35657, MGH96, and 18A069). Five LPS rfb loci have been identified in isolates 3KE and 4KE and two reference strains (ATCC35657 and 18A069). Fimbrial adherence determinants of Salmonella species (stcB) have been identified in 3KE and 10KE genes (Supplementary material 1, Supplementary Table 4).
Different plasmids were identified in our isolates: seven plasmids (ColpHAD28, ColpHAD28, ColpVC, IncFIBK, IncFIIpKPX1, IncN, and IncR) were identified in 4KE isolate; IncFIBK plasmid was identified in 3KE, 5KE, 6KE, 8KE, and 14KE, while there was no plasmid identified in reference isolates.
Discussion
The emergence of bacterial pathogens in environmental niches represents a continuous risk to human health (36). K. quasipneumoniae is a newly identified bacterial species discovered in 2014, which has not been fully understood until now (7). Due to the large similarity between K. variicola, K. pneumoniae, and K. quasipneumoniae isolates, there is difficulty in their identification and classification using routine biochemical tests (5, 37, 38). Additionally, identification based on public databases represents a source of misidentification due to the wrong submission to these databases (39). This may lead to misclassification even by using WGS. In this study, 26% (7/27) of K. quasipneumoniae species were misidentified as K. pneumoniae as a part of a previous study intended to study the genomics of K. pneumoniae and E. coli (16). This finding is consistent with Long et al. who showed that 30% of K. pneumoniae are misidentified by conventional biochemical tests (40).
cgMLST, core-genome SNP analysis, and ANI schemes demonstrated their ability to subtype species with the same MLST denominations (28, 31, 41). ANI with more than 95% identity was considered suitable to identify species and ≥98% to identify subspecies (29–31). In this study, the comparison of K. quasipneumoniae subsp. quasipneumoniae and K. quasipneumoniae subsp. similipneumoniae showed an average ANI of 96.5%, while the comparison of the same subspecies showed ANI ranging from 98.84 to 99.96%, supporting their subspecies relativeness. These findings are in line with Nicolás et al. (37) who found the same difference (96.52%) between subspecies of K. quasipneumoniae subsp. quasipneumoniae and K. quasipneumoniae subsp. Similipneumoniae; they also found 99% similarity among the same subspecies.
Our results showed that one strain (6KE) possessed a variety of genes such as aph(3”)-Ib, aac(6′)-Ib-cr, and catB, which conferred resistance to aminoglycosides and chloramphenicol, but it was phenotypically sensitive to these antibiotics. Although gene sequences were intact, the activity was not observed, which may be attributed to their low expression level (42). Isolates expressed catB and sensitive to chloramphenicol were previously reported (43), which was attributed to the decreased levels of acetyl coenzyme A in the isolates.
Furthermore, the presence of plasmids, insertion sequences (ISKpn43, IS421, and IS26), and integron integrase (intI1) can aid in the mobility of AMR genes which could increase the risk of transmission and dissemination of resistance in other susceptible strains (42, 44, 45).
Although Klebsiella species are intrinsically resistant to ampicillin (46), in this study we noted a high resistance rate to ampicillin (MIC ≥ 128 μg/ml). This could be attributed to the presence of different variants of class A beta-lactamases (blaOKP) that are associated with intrinsic ampicillin resistance in Klebsiella species (47, 48). This finding is higher than what was reported recently in Saudi Arabia for ampicillin-resistant K. quasipneumoniae (MIC = 32 μg/ml) (38). In this study, the isolates were resistant to cephalexin and ampicillin, while being susceptible to other beta-lactam antibiotics (meropenem, imipenem, ceftazidime, ceftriaxone, and cefuroxime). Although Klebsiella species have no intrinsic resistance to cephalosporins, the chromosomally expressed blaOKP beta-lactamases can cause a low level of resistance to cephalexin (47).
Plasmid screening showed that IncFIBK was the most prevalent plasmid, circulating in six isolates except 10KE. The IncFIBK plasmid is known to be a vehicle for the transmission of AMR genes in Enterobacterales (49). This plasmid is more common in environmental isolates of K. quasipneumoniae and has also been documented commonly in clinical isolates of K. pneumoniae (50), which supports our finding.
Several key features are common in our isolates when compared to K. quasipneumoniae strain ATCC 700603, such as putative transcriptional regulatory protein flanking Lysine 2,3-aminomutase (EC 5.4.3.2) which is involved in Lysine degradation (51). This was uniquely detected in all isolates. Genes encoding methyl-directed repair DNA adenine methylase, and twitching motility protein (PilT) which is associated with the surface-associated bacterial movement, were observed as mutations in PilT and pilU in Pseudomonas aeruginosa and are shown to be defective twitching motility (52). Antirestriction protein klcA was reported in all isolates and is located near incF plasmid proteins; the presence of anti-restriction protein klcAHS has been reported to increase plasmid transformation by 3–6 folds thereby increasing competence (53).
Genes associated with drug resistance and virulence mechanism are used for differentiating the commensals from pathogenic bacteria (54). The virulence factors investigated in our isolates showed aerobactin siderophore receptor gene (iutA) and salmochelin (iroE, iroN) genes commonly reported in the isolates, and are considered as clear markers of hypervirulent strains of K. pneumoniae (55). These genes were reported in K. pneumoniae strains isolated recently from the same location in Sudan (Khartoum locality) (56). Additionally, the type 3 fimbrial proteins (mrkA, mrkB, mrkC, mrkD, mrkF, and mrkH) were detected in all the isolates, which could increase the chance of biofilm production (57); isolates producing biofilm are associated with hospital-acquired infections and chronic infections (58). In addition to the presence of type 3 fimbrial (mrkD) gene, the isolates possessed type III secretory system (T6SS-III). Isolates positive for mrkD and T6SS-III fimbriae can establish biofilm formation in harsh environments (59).
The key virulence factors of pathogenic Klebsiella spp. are two cell surface-associated glycoproteins called capsular polysaccharides (CPS) and lipopolysaccharides (LPS) (60). In this study, the analysis of the capsule structure of the isolates revealed that isolates 3KE and 4KE possessed KL114 capsule type, which is a rare capsule type in K. pneumoniae (61) that has been reported recently by Long et al. in drug-resistant human pathogenic K. quasipneumoniae strains (40). Isolates 5KE, 6KE, and 14KE harboring the KL66 K-loci have been identified in K. oxytoca, which is closely associated with K. quasipneumoniae (62). The sharing of these genes among Klebsiella species suggests the horizontal gene transfer among these species (63). Moreover, the KL81 and KL146 capsule types were found in 8KE and 10KE respectively. Regarding the prediction of LPS O-antigen gene, three isolates (3KE, 4KE and 14KE) belonged to O3/O3a serotype. Although O3/O3a serotypes pathogenic strains of K. quasipneumoniae have been reported, and with the strong adjuvant effect of O3/O3a serotypes, the clinical impact of this serotype is still unknown (59). Isolates 5KE, 6KE and 10KE belonged O12 serotype. In earlier studies, analyses of 573 pathogenic strains of K. pneumoniae revealed that 9.2% belonged to O12 serotype (63).
Recently discovered T6SS in K. pneumoniae strains plays a role in bacterial warfare and long-term gastrointestinal colonization (64). Although, the presence of T6SS in K. quasipneumoniae is low, in this study isolate 8KE was characterized by the presence of 16 types of secretion systems (T6SS-I) and 15 different clustered T6SS-II genes, while 4KE was characterized by the presence of 14 types of T6SS-III, suggesting their virulence and pathogenic activity.
Conclusion
In summary, we characterized genomes of novel strains of K. quasipneumoniae subsp. similipneumoniae (n = 6) and K. quasipneumoniae subsp. quasipneumoniae (n = 1), harboring different ARGs circulating in drinking water, table surfaces and vegetables in Khartoum markets. The aminoglycoside-modifying enzyme gene [aph(3”)-Ia] was harbored in isolate 6KE and was bracketed by two transposable insertion elements IS5075 and IS91 transposase. The isolates were identified with key virulence factors occurring in pathogenic Klebsiella spp. (CPS and LPS), and possessed a group of other virulence genes such as type 3 fimbriae (mrkA, mrkB, mrkC, mrkD, mrkF, mrkH) associated with adherence and biofilm formation. Additionally, the iron uptake aerobactin (iutA), salmochelins (iroE, iroN), and enterobactin siderophores (ent A-F, feb-G), entS, and fes were identified in all of the isolates except the iutA gene which was absent in one isolate (8KE). Such isolates represent a potential risk of being transmitted to humans and can cause hospital or community-acquired infections. We also demonstrated that a large percentage (23%) of K. pneumoniae isolates were misidentified; this implies that routine biochemical tests are not enough for species identification and more robust molecular detection methods need to be used in order to improve our understanding which will have a direct impact on improving public health.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
This study was approved by the Ethics Committee of the Khartoum State Ministry of Health (Ref: 2/2021).
Author contributions
Conceptualization: HA and HE. Methodology: HA, EM, and HE. Validation: BL and EM. Formal analysis: BL, IK, and HA. Writing—original draft preparation: IK, OB, SH, IA, HE, and BL. Writing—review and editing: BL and HA. Supervision: SH, HA, and IA. Analysis of fastANI and genome comparison: EM. All authors have read and agreed to the published version of the manuscript.
Funding
This research work was funded by Institutional Fund Projects under grant no. (IFPIP: 263-130-1443). The authors gratefully acknowledge the technical and financial support provided by the Ministry of Education and King Abdulaziz University, DSR, Jeddah, Saudi Arabia.
Acknowledgments
The authors extend their appreciation to the institutional Fund Projects grant no. (IFPIP: 263-130-1443). We thank the Institut Pasteur teams for the curation and maintenance of BIGSdb-Pasteur databases at http://bigsdb.pasteur.fr, and for their role in strain identification and assigning numbers for the novel strains.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2022.1068888/full#supplementary-material
References
1. Wyres KL, Wick RR, Judd LM, Froumine R, Tokolyi A, Gorrie CL, et al. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. (2019) 15:e1008114. doi: 10.1371/journal.pgen.1008114
2. Catalán-Nájera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: two different but complementary Klebsiella spp. phenotypes? Virulence. (2017) 8:1111–23. doi: 10.1080/21505594.2017.1317412
3. Martínez-Romero E, Rodríguez-Medina N, Beltrán-Rojel M, Toribio-Jiménez J, Garza-Ramos U. Klebsiella variicola and Klebsiella quasipneumoniae with capacity to adapt to clinical and plant settings. Salud Publica Mex. (2018) 60:29–40. doi: 10.21149/8156
4. Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci USA. (2015) 112:E3574–81. doi: 10.1073/pnas.1501049112
5. Rodrigues C, Passet V, Rakotondrasoa A, Brisse S. Identification of Klebsiella pneumoniae, Klebsiella quasipneumoniae, Klebsiella variicola and related phylogroups by MALDI-TOF mass spectrometry. Front Microbiol. (2018) 9:3000. doi: 10.3389/fmicb.2018.03000
6. Blin C, Passet V, Touchon M, Rocha EP, Brisse S. Metabolic diversity of the emerging pathogenic lineages of Klebsiella pneumoniae. Environ Microbiol. (2017) 19:1881–98. doi: 10.1111/1462-2920.13689
7. Mathers AJ, Crook D, Vaughan A, Barry KE, Vegesana K, Stoesser N, et al. Klebsiella quasipneumoniae provides a window into carbapenemase gene transfer, plasmid rearrangements, and patient interactions with the hospital environment. Antimicrobiol Agents Chemother. (2019) 63:e02513–18. doi: 10.1128/AAC.02513-18
8. Venkitapathi S, Sharon BM, Ratna TA, Arute AP, Zimmern PE, De Nisco NJ. Complete genome sequences of three uropathogenic Klebsiella quasipneumoniae strains isolated from postmenopausal women with recurrent urinary tract infection. Microbiol Resourc Announce. (2021) 10:e00073–21. doi: 10.1128/MRA.00073-21
9. Suzuki Y, Ida M, Kubota H, Ariyoshi T, Murakami K, Kobayashi M, et al. Multiple β-lactam resistance gene-carrying plasmid harbored by Klebsiella quasipneumoniae isolated from urban sewage in Japan. Msphere. (2019) 4:e00391–19. doi: 10.1128/mSphere.00391-19
10. Hu X, Yu X, Shang Y, Xu H, Guo L, Liang Y, et al. Emergence and characterization of a novel IncP-6 plasmid harboring blaKPC−2 and qnrS2 genes in Aeromonas taiwanensis isolates. Front Microbiol. (2019) 10:2132. doi: 10.3389/fmicb.2019.02132
11. Ghiglione B, Haim MS, Penzotti P, Brunetti F, Di Conza J, Figueroa-Espinosa R, et al. Characterization of emerging pathogens carrying blaKPC-2 gene in IncP-6 plasmids isolated from urban sewage in Argentina. Front Cell Infect Microbiol. (2021) 11:722536. doi: 10.3389/fcimb.2021.722536
12. Wang Y, Zhu B, Liu M, Dong X, Ma J, Li X, et al. Characterization of IncHI1B plasmids encoding efflux pump TmexCD2-ToprJ2 in carbapenem-resistant Klebsiella variicola, Klebsiella quasipneumoniae, and Klebsiella michiganensis strains. Front Microbiol. (2021) 2933. doi: 10.3389/fmicb.2021.759208
13. Sekizuka T, Yatsu K, Inamine Y, Segawa T, Nishio M, Kishi N, et al. Complete genome sequence of a bla KPC-2-positive Klebsiella pneumoniae strain isolated from the effluent of an urban sewage treatment plant in Japan. msphere. (2018) 3:e00314–18. doi: 10.1128/mSphere.00314-18
14. Aslam B, Khurshid M, Arshad MI, Muzammil S, Rasool M, Yasmeen N, et al. Antibiotic resistance: one health one world outlook. Front Cell Infect Microbiol. (2021) 11:771510. doi: 10.3389/fcimb.2021.771510
15. Wauters G, Vaneechoutte M. Approaches to the identification of aerobic gram-negative bacteria. Man Clin Microbiol. (2015) 613–34. doi: 10.1128/9781555817381.ch33
16. Altayb HN, Elbadawi HS, Alzahrani FA, Baothman O, Kazmi I, Nadeem MS, et al. Co-occurrence of β-lactam and aminoglycoside resistance determinants among clinical and environmental isolates of Klebsiella pneumoniae and Escherichia coli: a genomic approach. Pharmaceuticals. (2022) 15:1011. doi: 10.3390/ph15081011
17. Mahmoud NE, Altayb HN, Gurashi RM. Detection of carbapenem-resistant genes in Escherichia coli isolated from drinking water in Khartoum, Sudan. J Environ Public Health. (2020) 2020:2571293. doi: 10.1155/2020/2571293
18. Biswas B, Azad MAK, Absar N, Islam S, Amin S. Isolation and Identification of Pathogenic Bacteria from Fresh Fruits and Vegetables in Chittagong, Bangladesh (2020) 10:55–8.
19. Kouidhi B, Zmantar T, Jrah H, Souiden Y, Chaieb K, Mahdouani K, et al. Antibacterial and resistance-modifying activities of thymoquinone against oral pathogens. Ann Clin Microbiol Antimicrobiol. (2011) 10:1–7. doi: 10.1186/1476-0711-10-29
20. Wayne P. Clinical and laboratory standards institute: Performance standards for antimicrobial susceptibility testing. Inform Suppl. (2020) 31:100–21.
21. Sabeel S, Salih MA, Ali M, El-Zaki S-E, Abuzeid N, Elgadi ZAM, et al. Phenotypic and genotypic analysis of multidrug-resistant Mycobacterium tuberculosis isolates from Sudanese patients. Tuberculosis Res Treat. (2017) 2017:8340746. doi: 10.1155/2017/8340746
22. Jolley KA, Bray JE, Maiden MC. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. (2018) 3:124. doi: 10.12688/wellcomeopenres.14826.1
23. Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. (2016) 44:6614–24. doi: 10.1093/nar/gkw569
24. Argimón S, Yeats CA, Goater RJ, Abudahab K, Taylor B, Underwood A, et al. A global resource for genomic predictions of antimicrobial resistance and surveillance of Salmonella typhi at pathogenwatch. Nat Commun. (2021) 12:2879. doi: 10.1038/s41467-021-23091-2
25. Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrobiol Chemother. (2012) 67:2640–4. doi: 10.1093/jac/dks261
26. Johansson MH, Bortolaia V, Tansirichaiya S, Aarestrup FM, Roberts AP, Petersen TN. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J Antimicrobiol Chemother. (2021) 76:101–9. doi: 10.1093/jac/dkaa390
27. Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. (2019) 47:D687–92. doi: 10.1093/nar/gky1080
28. Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. (2018) 9:5114. doi: 10.1038/s41467-018-07641-9
29. Jensen CS, Iversen KH, Dargis R, Shewmaker P, Rasmussen S, Christensen JJ, et al. Streptococcus pseudopneumoniae: use of whole-genome sequences to validate species identification methods. J Clin Microbiol. (2021) 59:e02503–20. doi: 10.1128/JCM.02503-20
30. Gan HM, Eng WWH, Dhanoa A. First genomic insights into carbapenem-resistant Klebsiella pneumoniae from Malaysia. J Glob Antimicrobiol Resist. (2020) 20:153–9. doi: 10.1016/j.jgar.2019.07.008
31. Pearce ME, Langridge GC, Lauer A, Grant K, Maiden MC, Chattaway MA. An evaluation of the species and subspecies of the genus Salmonella with whole genome sequence data: proposal of type strains and epithets for novel S. enterica subspecies VII, VIII, IX, X and XI. Genomics. (2021) 113:3152–62. doi: 10.1016/j.ygeno.2021.07.003
32. Petkau A, Stuart-Edwards M, Stothard P, Van Domselaar G. Interactive microbial genome visualization with GView. Bioinformatics. (2010) 26:3125–6. doi: 10.1093/bioinformatics/btq588
33. Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, et al. The PATRIC bioinformatics resource center: expanding data and analysis capabilities. Nucleic acids Res. (2020) 48:D606–12. doi: 10.1093/nar/gkz943
34. Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. (2008) 57:758–71. doi: 10.1080/10635150802429642
36. Buelow E, Ploy M-C, Dagot C. Role of pollution on the selection of antibiotic resistance and bacterial pathogens in the environment. Curr Opin Microbiol. (2021) 64:117–24. doi: 10.1016/j.mib.2021.10.005
37. Nicolás MF, Ramos PIP, Marques de Carvalho F, Camargo DR, de Fátima Morais Alves C, Loss de Morais G, et al. Comparative genomic analysis of a clinical isolate of Klebsiella quasipneumoniae subsp. similipneumoniae, a KPC-2 and OKP-B-6 beta-lactamases producer harboring two drug-resistance plasmids from southeast Brazil. Front Microbiol. (2018) 9:220. doi: 10.3389/fmicb.2018.00220
38. Hala S, Antony CP, Alshehri M, Althaqafi AO, Alsaedi A, Mufti A, et al. First report of Klebsiella quasipneumoniae harboring bla KPC-2 in Saudi Arabia. Antimicrobiol Resist Infect Control. (2019) 8:1–8. doi: 10.1186/s13756-019-0653-9
39. Martínez-Romero E, Rodríguez-Medina N, Beltrán-Rojel M, Silva-Sánchez J, Barrios-Camacho H, Pérez-Rueda E, et al. Genome misclassification of Klebsiella variicola and Klebsiella quasipneumoniae isolated from plants, animals and humans. Salud Públic México. (2018) 60:56–62. doi: 10.21149/8149
40. Long SW, Linson SE, Ojeda Saavedra M, Cantu C, Davis JJ, Brettin T, et al. Whole-genome sequencing of human clinical Klebsiella pneumoniae isolates reveals misidentification and misunderstandings of Klebsiella pneumoniae, Klebsiella variicola, Klebsiella quasipneumoniae. msphere. (2017) 2:e00290–17. doi: 10.1128/mSphereDirect.00290-17
41. Miro E, Rossen JW, Chlebowicz MA, Harmsen D, Brisse S, Passet V, et al. Core/whole genome multilocus sequence typing and core genome SNP-based typing of OXA-48-producing Klebsiella pneumoniae clinical isolates from Spain. Front Microbiol. (2020) 10:2961. doi: 10.3389/fmicb.2019.02961
42. Vk D, Srikumar S, Shetty S, van Nguyen S, Karunasagar I, Fanning S. Silent antibiotic resistance genes: a threat to antimicrobial therapy. Int J Infect Dis. (2019) 79:20. doi: 10.1016/j.ijid.2018.11.063
43. Potrykus J, Wegrzyn G. Chloramphenicol-sensitive Escherichia coli strain expressing the chloramphenicol acetyltransferase (cat) gene. Antimicrobiol Agents Chemother. (2001) 45:3610–2. doi: 10.1128/AAC.45.12.3610-3612.2001
44. Nagachinta S, Chen J. Transfer of class 1 integron-mediated antibiotic resistance genes from shiga toxin-producing Escherichia coli to a susceptible E. coli K-12 strain in storm water and bovine feces. Appl Environ Microbiol. (2008) 74:5063–7. doi: 10.1128/AEM.00517-08
45. Mbelle NM, Feldman C, Sekyere JO, Maningi NE, Modipane L, Essack SY. The resistome, mobilome, virulome and phylogenomics of multidrug-resistant Escherichia coli clinical isolates from Pretoria, South Africa. Sci Rep. (2019) 9:16457. doi: 10.1038/s41598-019-52859-2
46. Bouza E, Cercenado E. Klebsiella and enterobacter: antibiotic resistance and treatment implications. Semin Respirat Infect. (2002) 17:215–30. doi: 10.1053/srin.2002.34693
47. Brisse S, van Duijkeren E. Identification and antimicrobial susceptibility of 100 Klebsiella animal clinical isolates. Vet Microbiol. (2005) 105:307–12. doi: 10.1016/j.vetmic.2004.11.010
48. Henson SP, Boinett CJ, Ellington MJ, Kagia N, Mwarumba S, Nyongesa S, et al. Molecular epidemiology of Klebsiella pneumoniae invasive infections over a decade at Kilifi County Hospital in Kenya. Int J Med Microbiol. (2017) 307:422–9. doi: 10.1016/j.ijmm.2017.07.006
49. Muraya A, Kyany'a C, Kiyaga S, Smith HJ, Kibet C, Martin MJ, et al. Antimicrobial resistance and virulence characteristics of Klebsiella pneumoniae isolates in Kenya by whole-genome sequencing. Pathogens. (2022) 11:545. doi: 10.3390/pathogens11050545
50. Ramsamy Y, Mlisana KP, Amoako DG, Abia ALK, Ismail A, Allam M, et al. Mobile genetic elements-mediated Enterobacterales-associated carbapenemase antibiotic resistance genes propagation between the environment and humans: a one health South African study. Sci Total Environ. (2022) 806:150641. doi: 10.1016/j.scitotenv.2021.150641
51. Wu W, Booker S, Lieder KW, Bandarian V, Reed GH, Frey PA. Lysine 2, 3-aminomutase and trans-4, 5-dehydrolysine: characterization of an allylic analogue of a substrate-based radical in the catalytic mechanism. Biochemistry. (2000) 39:9561–70. doi: 10.1021/bi000658p
52. Zolfaghar I, Evans DJ, Fleiszig SM. Twitching motility contributes to the role of pili in corneal infection caused by Pseudomonas aeruginosa. Infect Immunity. (2003) 71:5389–93. doi: 10.1128/IAI.71.9.5389-5393.2003
53. Liang W, Xie Y, Xiong W, Tang Y, Li G, Jiang X, et al. Anti-restriction protein, KlcAHS, promotes dissemination of carbapenem resistance. Front Cell Infect Microbiol. (2017) 7:150. doi: 10.3389/fcimb.2017.00150
54. Amaretti A, Righini L, Candeliere F, Musmeci E, Bonvicini F, Gentilomi GA, et al. Antibiotic resistance, virulence factors, phenotyping, and genotyping of non-Escherichia coli Enterobacterales from the gut microbiota of healthy subjects. Int J Mol Sci. (2020) 21:1847. doi: 10.3390/ijms21051847
55. Li Q, Zhu J, Kang J, Song Y, Yin D, Guo Q, et al. Emergence of NDM-5-producing carbapenem-resistant Klebsiella pneumoniae and SIM-producing hypervirulent Klebsiella pneumoniae Isolated from aseptic body fluid in a large tertiary hospital, 2017–2018: genetic traits of blaNDM-like and blaSIM-like genes as determined by NGS. Infect Drug Resist. (2020) 13:3075. doi: 10.2147/IDR.S261117
56. Altayb HN, Elbadawi HS, Baothman O, Kazmi I, Alzahrani FA, Nadeem MS, et al. genomic analysis of multidrug-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae strain lacking the hypermucoviscous regulators (rmpA/rmpA2). Antibiotics. (2022) 11:596. doi: 10.3390/antibiotics11050596
57. Schroll C, Barken KB, Krogfelt KA, Struve C. Role of type 1 and type 3 fimbriae in Klebsiella pneumoniae biofilm formation. BMC Microbiol. (2010) 10:179. doi: 10.1186/1471-2180-10-179
58. Jamal M, Ahmad W, Andleeb S, Jalil F, Imran M, Nawaz MA, et al. Bacterial biofilm and associated infections. J Chin Med Assoc. (2018) 81:7–11. doi: 10.1016/j.jcma.2017.07.012
59. Perlaza-Jiménez L, Wu Q, Von Vergel LT, Zhang X, Li J, Rocker A, et al. Forensic genomics of a novel Klebsiella quasipneumoniae type from a neonatal intensive care unit in China reveals patterns of colonization, evolution and epidemiology. Microb Genom. (2020) 6:mgen000433. doi: 10.1099/mgen.0.000433
60. Patro L, Sudhakar KU, Rathinavelan T. K-PAM: a unified platform to distinguish Klebsiella species K-and O-antigen types, model antigen structures and identify hypervirulent strains. Sci Rep. (2020) 10:16732. doi: 10.1038/s41598-020-73360-1
61. Rodríguez-Medina N, Martínez-Romero E, De la Cruz MA, Ares MA, Valdovinos-Torres H, Silva-Sánchez J, et al. A Klebsiella variicola plasmid confers hypermucoviscosity-like phenotype and alters capsule production and virulence. Front Microbiol. (2020) 11:579612. doi: 10.3389/fmicb.2020.579612
62. Wyres KL, Wick RR, Gorrie C, Jenney A, Follador R, Thomson NR, et al. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb Genom. (2016) 2:e000102. doi: 10.1099/mgen.0.000102
63. Follador R, Heinz E, Wyres KL, Ellington MJ, Kowarik M, Holt KE, et al. The diversity of Klebsiella pneumoniae surface polysaccharides. Microb Genom. (2016) 2:e000073. doi: 10.1099/mgen.0.000073
Keywords: K. quasipneumoniae subsp. similipneumoniae, K. quasipneumoniae subsp. quasipneumoniae, misidentification, whole-genome sequencing, antimicrobial resistance
Citation: Altayb HN, Hosawi S, Baothman O, Kazmi I, Chaieb K, Abu Zeid IM, Elbadawi HS, Lopes BS and Moglad E (2023) Molecular insights into novel environmental strains of Klebsiella quasipneumoniae harboring different antimicrobial-resistance genes. Front. Public Health 10:1068888. doi: 10.3389/fpubh.2022.1068888
Received: 13 October 2022; Accepted: 21 December 2022;
Published: 12 January 2023.
Edited by:
Aleksandra Barac, Faculty of Medicine, University of Belgrade, SerbiaReviewed by:
Herrison Fontana, University of São Paulo, BrazilMohamed H. Al-Agamy, King Saud University, Saudi Arabia
Copyright © 2023 Altayb, Hosawi, Baothman, Kazmi, Chaieb, Abu Zeid, Elbadawi, Lopes and Moglad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hisham N. Altayb,  aGRlbW1haG9tQGthdS5lZHUuc2E=; Bruno Silvester Lopes, YnJ1bm9sZGxvcGV6QGdtYWlsLmNvbQ==
aGRlbW1haG9tQGthdS5lZHUuc2E=; Bruno Silvester Lopes, YnJ1bm9sZGxvcGV6QGdtYWlsLmNvbQ==