 Friederike I. L. Hucke*
Friederike I. L. Hucke* Joachim J. Bugert
Joachim J. Bugert- Department of Virology, Bundeswehr Institute of Microbiology, Munich, Germany
Chikungunya virus (CHIKV) is the causative agent of chikungunya fever (CHIKF) and is categorized as a(n) (re)emerging arbovirus. CHIKV has repeatedly been responsible for outbreaks that caused serious economic and public health problems in the affected countries. To date, no vaccine or specific antiviral therapies are available. This review gives a summary on current antivirals that have been investigated as potential therapeutics against CHIKF. The mode of action as well as possible compound targets (viral and host targets) are being addressed. This review hopes to provide critical information on the in vitro efficacies of various compounds and might help researchers in their considerations for future experiments.
Introduction Chikungunya Virus
Chikungunya virus (CHIKV) is a single-stranded RNA virus with a positive sense genome of about 11,800 nucleotides. CHIKV structure and genome organization follow those of all alphaviruses. The virion has a lipid-bilayer envelope that is tightly associated with an icosahedral nucleocapsid shell (240 capsid copies) which encapsidates genomic RNA (1). The genome contains two open reading frames (ORFs), which encode the non-structural (ns) or replicase polyprotein and the structural polyprotein.
CHIKV is primarily transmitted to humans by the bite of an infected mosquito, mainly of the Aedes species. CHIKV causes the so-called chikungunya fever (CHIKF) which is characterized by high fever, headache and the hallmarks of the disease, myalgia and polyarthralgia (1). The latter especially can last for months or even years after the acute phase of the illness has passed, causing a severely deteriorated quality of life for the patient. The resulting stooped bearing and rigid gait of infected individuals are described in the word origin of the disease “kungunyala,” which is Makonde for “that which bends up.” CHIKV was first described in 1955 by Robinson and Lumsden after an outbreak in present-day Tanzania in 1952 (2).
Until 2004, CHIKV was mainly distributed in tropical and subtropical regions of sub-Sahara Africa and Southeast Asia. It caused sporadic outbreaks mainly during the rainy season. In 2004, however, a massive outbreak in Kenya led to close to half a million infected people. This epidemic initiated the spread to more than 22 countries, including countries with a moderate climate such as France and Italy (Figure 1) (5).
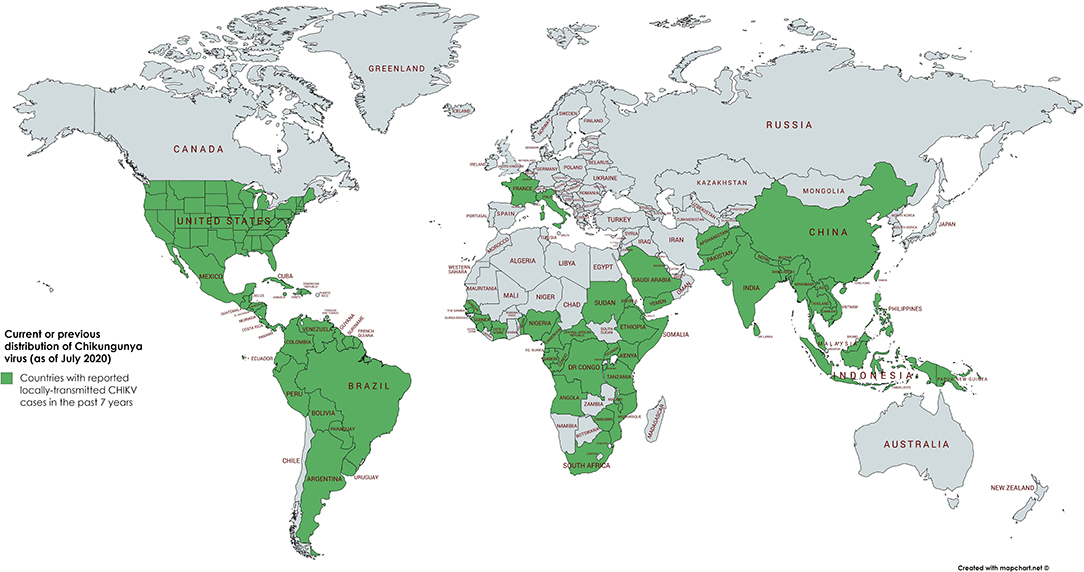
Figure 1. Geographical distribution of CHIKV. Word map showing the distribution of autochthonous CHIKV transmissions in the past 7 years (2013–2020) highlighted in green. In the continental United States of America, only travel associated cases have been reported in the past 3 years. As of July 2020, the latest autochthonous cases in Europe have been recorded in France and Italy in 2017 (3, 4).
Following the bite of a CHIKV infected mosquito, the virus is transported to the nearest lymph node and transferred to monocytes and macrophages which enter the bloodstream. At this point, viremia sets in by the active infection of human blood monocytes and other peripheral blood mononuclear cells. CHIKV then reaches the muscles and joints, where the infection causes the main symptoms of CHIKF—myalgia and arthralgia (6). Apart from muscles and joints, CHIKV may also target a range of secondary organs and thus cause severe complications in patients (i.e., renal, respiratory, hepatic, cardiac, and neural syndromes) (7). As neither specific antiviral drugs nor a licensed vaccine are available, the therapy of CHIKF is based on supportive measures and the treatment of symptoms [non-steroid anti-inflammatory drugs (NSAIDs) and fluid therapy (8)].
For detailed information on CHIKV epidemiology, replication, disease mechanism, and prophylaxis, we refer to the reviews of Silva and Dermody (1), Pietila et al. (9), and Hucke et al. (10).
Antivirals Against Chikungunya Virus
Direct-Acting Antivirals
The following chapter will deal with various compounds that are or have been in the focus of research and showed some promising results in vitro mainly against CHIKV and/or other relevant alphaviruses. Various compounds made it to in vivo studies but so far there is no licensed therapeutic drug acting directly against CHIKV or any other alphavirus. There are many compounds which are currently under investigation for their anti-CHIKV efficacy. However, as the scope of this review is limited, we will only discuss compounds that either showed efficacy in a variety of in vitro assays or were repeatedly investigated by different (independent) research groups. It must be noted that compound efficacy can vary considerably, depending on the cell line, virus strain, or assay method that is being used. Table 1 illustrates this fact and gives an overview of EC50/CC50 values of common substances used as experimental controls in in vitro trials. For the interested reader I refer to the reviews of Abdelnabi et al. (38), Subudhi et al. (39) and the review of da Silva-Junior et al. (40), focussing on the medicinal chemistry of synthetic and natural compounds against CHIKV. Furthermore, the review of Bugert et al. (41) inspects antivirals against alphaviruses and other viral agents relevant in medical biodefence.
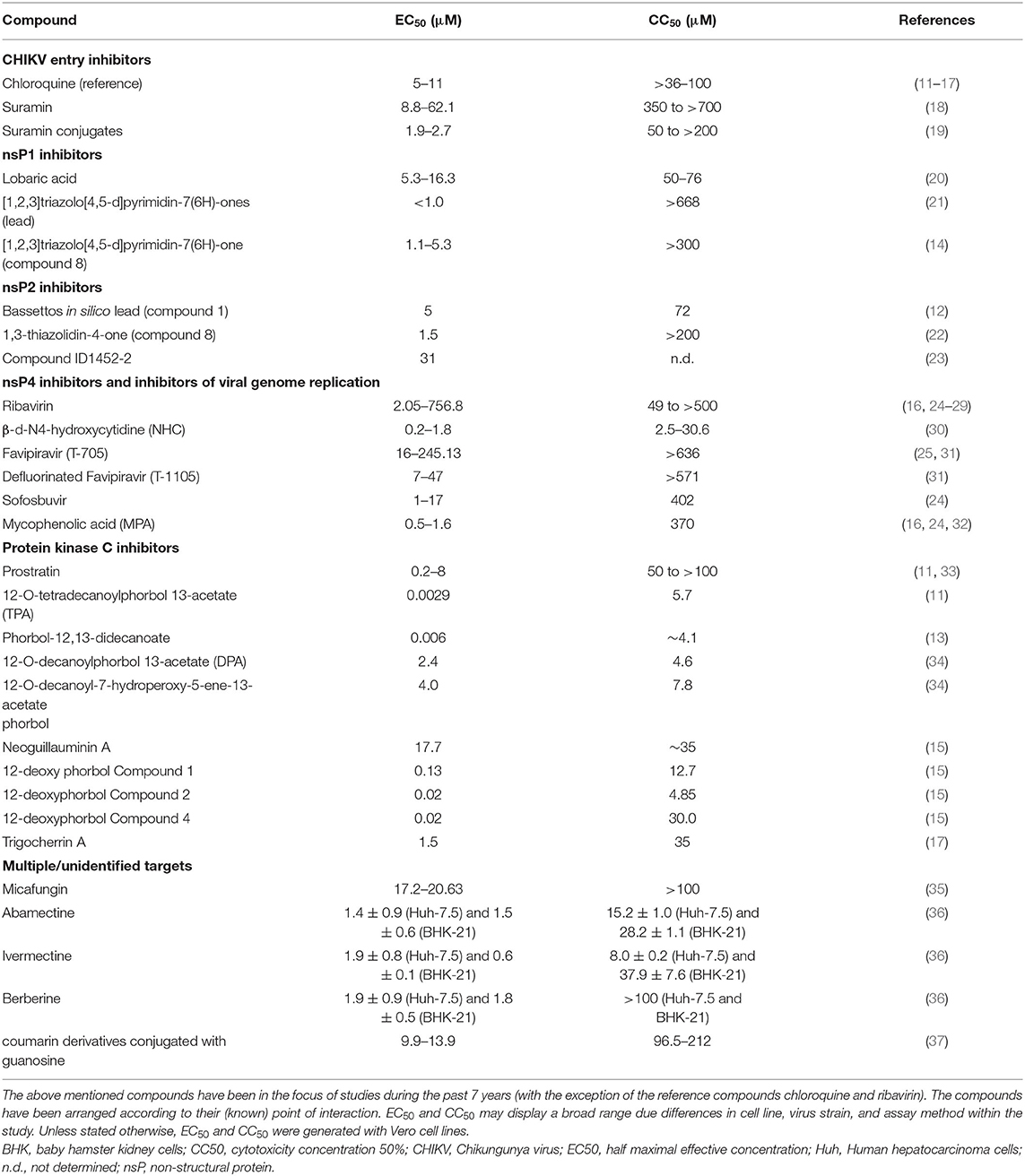
Table 1. Comparison of compounds with anti-CHIKV property.
CHIKV Entry Inhibitors
Chloroquine
Chloroquine is a licensed drug for the prophylaxis and treatment of malaria. Furthermore, it is prescribed for the treatment of systemic lupus erythematosus and rheumatoid arthritis (42). Chloroquine also shows in vitro antiviral activity against several viruses, such as human immunodeficiency virus (HIV), severe acute respiratory syndrome (SARS) coronavirus and alphaviruses (43). Khan demonstrated that chloroquine is able to inhibit CHIKV replication in VeroA cells in a dose-dependent manner. Apart from this mode of action, it is also assumed that the drug interferes with the endosome-mediated CHIKV internalization. Bernard et al. (44) showed that chloroquine raises the endosomal pH by interfering with the protonation of the endocytic vesicles and thereby prevents the E1 fusion step needed for the release of CHIKV RNA into the cell cytoplasm. Various research groups used chloroquine as a reference compound in their in vitro studies (Table 2) (11–15, 45).
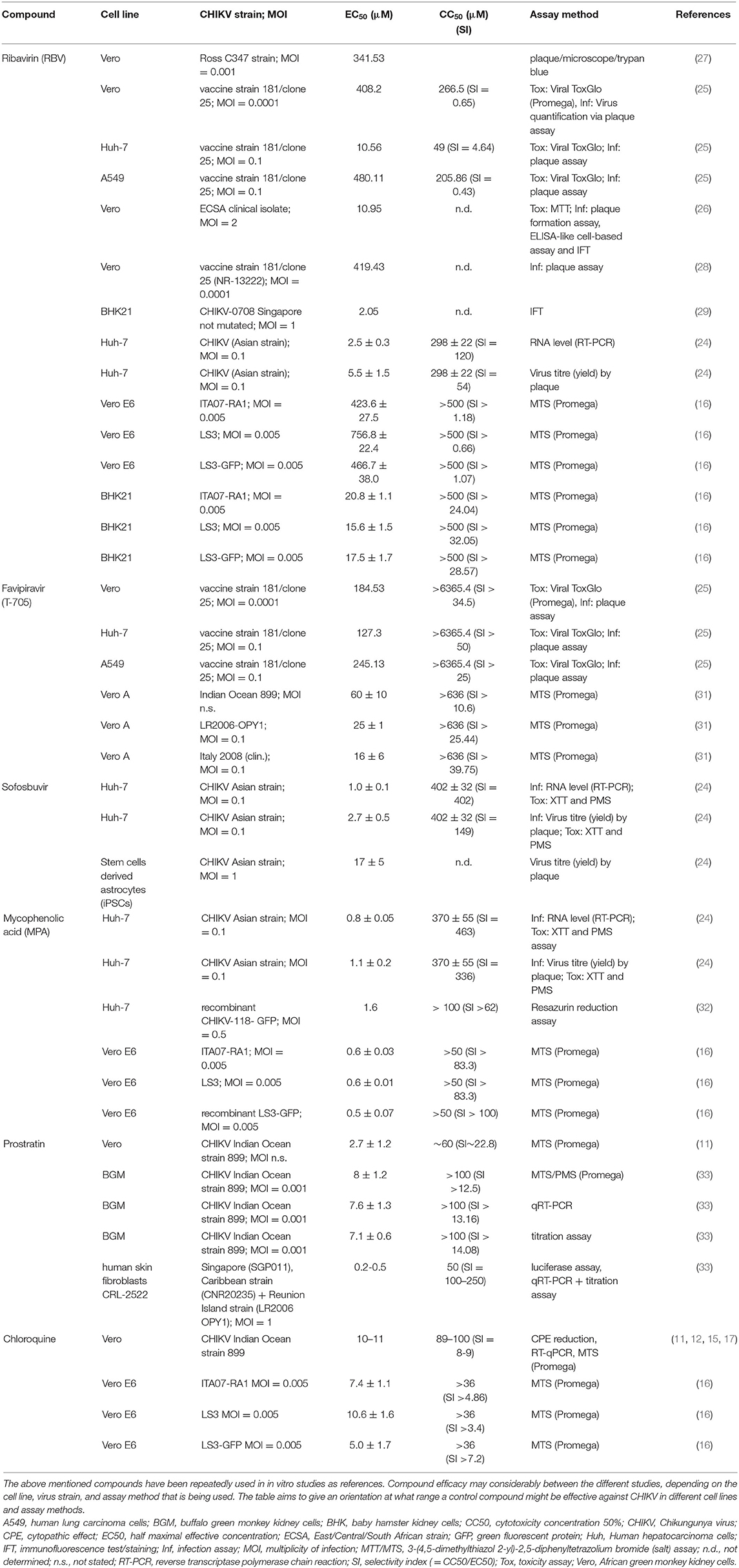
Table 2. Efficacy of selected compounds against CHIKV according to different studies.
Despite the promising results chloroquine displays in in vitro studies, clinical trials with the drug failed to prove any benefit for the patient. Trials for prophylaxis or treatment of CHIKV infection either in macaque models or human patients could not demonstrate advantage of chloroquine over meloxicam (an NSAID) administration (46, 47). The discrepancy between in vitro and in vivo effectiveness of chloroquine has been described before.
Epigallocatechin Gallate (Green Tea Component)
Epigallocatechin gallate (EGCG) is an active polyphenolic catechin and the essential element of green tea (Camellia sinensis) extract. Various independent research groups discovered the antiviral properties of EGCG against a number of viruses and recent studies revealed that EGCG also inhibits CHIKV replication in vitro. Weber et al. (48) demonstrated that EGCG inhibits CHIKV replication in HEK 293T cells by blocking the entry of CHIKV pseudo-particles that carried the CHIKV envelope proteins. Thus, EGCG prevented the attachment of CHIKV to the target cells.
More recently, Lu et al. (49) showed the benefits of synergism in the combination treatment of CHIKV infected U2OS cells (human bone osteosarcoma cells) with EGCG and suramin. Lu tested EGCG combined with suramin against the CHIKV strain S27 and two clinical isolates. Besides the synergistic effect of the two compounds, Lu could confirm that the EGCG inhibits virus entry, replication, progeny yield as well as CPE of CHIKV in vitro.
Suramin
Suramin, also known as germanin or Bayer-205, is a symmetrical hexasulfonated naphthylurea compound that has been market-authorized by the U.S. Food and Drug Administration (FDA) for the treatment of trypanosomiasis (trypanosome-caused river blindness, onchocerciasis). The drug acts as a competitive inhibitor of sulphated glycosaminoglycans (GAGs) and heparin. As a number of viruses attach to cells via GAGs, suramin may consequently have anti-viral activity by inhibiting virus entry. The drug proved effective against a number of viruses, including DENV and Venezuelan equine encephalitis virus (VEEV) (50, 51). Against CHIKV, suramin proved effective in various in vitro studies (18, 52, 53). Suramin diminished CPE, virus replication and yield in a dose-dependent manner. Ho et al. (18) demonstrated that suramin was broadly effective in vitro against various CHIKV strains (Table 1). Ho used BHK-21, U2OS and MRC-5 cells. His group was the first to prove that the compound inhibits entry and transmission of CHIKV through binding onto E1/E2 glycoproteins. Furthermore, they showed that CHIKV infection was hampered in early stages. Virus binding and fusion was disrupted by the binding of suramin with viral glycoproteins. The compound also interfered with virus release. According to their research the EC50 of suramin for the inhibition of CHIKV in vitro (EC50 of 8.8–62.1 μM) is well within the range of non-toxic serum concentrations in humans (70 μM) when treated for river blindness (54).
Henß et al. (53) were also able to verify that suramin blocks CHIKV at early stages of the infection. Furthermore, her group tested the compound successfully against Ebola virus. All her tests were done in vitro (HEK 293T, MCF7, and Huh-7 cells). According to Henß however, the drug's side effects on the patient (nausea, vomiting, reversible urticarial rash, kidney damage, and exfoliative dermatitis; furthermore, suramin is connected to hepatic and bone marrow toxicity) might make suramin inappropriate for the treatment of CHIKV infections, a rather mild disease compared to Ebola. To avoid these side effects, Hwu et al. (19) chemically modified suramin and used 20 new conjugated compounds in a CPE screening assay against CHIKV. He identified six compounds with promising activity against CHIKV.
Inhibitors of Viral Genome Replication and Translation
RNA Interference (RNAi) Targeting CHIKV Genes
Small interfering RNA (siRNA) is able to regulate gene expression by the cleavage of the corresponding messenger RNA (mRNA) (55). The most commonly understood effect of this mechanism is the inhibition of the protein synthesis of certain genes because the mRNA is no longer available. This is referred to as “gene silencing.” The discovery that siRNA is able to inhibit specific genes has led to a vast interest in this particular field. SiRNA was hoped to be used as a potential therapy for the treatment of genetic disorders, cancer, viruses, and other diseases. Bitko and Barik (56) showed that RNA interference (RNAi) was able to inhibit a negative-strand RNA virus.
Since RNAi is an endogenous biological process, potentially every gene can be supressed. In addition to that, siRNAs are easier to identify, synthesize and produce on a large scale than traditional drugs (57). Multiple studies have been conducted to test the possible efficacy of siRNA against viruses in vitro and in vivo (mice, guinea pigs, macaques and humans) (58). There are two approaches for recruiting RNA interference as antivirals: (1) targeting specific viral sequences; (2) targeting the host cell.
(1) Targeting specific viral sequences with synthetic siRNA:
SiRNA can be created in the laboratory and preferably targets conserved regions. Theoretically any specific viral gene can be disabled. This is an advantage over classical small drug molecules that have to be fitted to a target protein which usually is only present at certain sites in the cell (59).
Dash et al. (60) designed and evaluated siRNA sequences targeting CHIKV nsP3 and E1 genes in Vero cells. They could demonstrate that these siRNAs curbed CHIKV titres by 99.6% in siRNA transfected cells 24 h after infection. However, this reduction could not be sustained at 72 h, possibly because of the intracellular degradation of the siRNA. In 2013, Parashar et al. conducted in vitro studies in Vero-E6 cells, where he used siRNAs targeting nsP1 and/or E2 mRNA. He succeeded in downregulation of CHIKV replication for more than 90%. In vivo studies in CHIKV-infected Swiss albino and C57 BL/6 mice showed a complete inhibition of CHIKV replication when these siRNAs were administered 72 h post-infection (61). Lam et al. (62) could also demonstrate that CHIKV infection could effectively be supressed in the mouse model when pre-treating the animals with (small hairpin) shRNA (a precursor form of siRNA) against CHIKV E1 and nsP1 (62).
More recently, due to its advantages over siRNA and shRNA as far as stability, effectiveness, and toxicity are concerned, the artificial miRNA (amiRNA) based approach is in the focus of research. Bhomia et al. (63) showed the effectiveness of amiRNA for inhibition of Venezuelan equine encephalitis virus (VEEV). Saha et al. (64) successfully tested vector-delivered amiRNA against CHIKV infected Vero cells and efficiently inhibited CHIKV replication. One problem arising from this approach is the development of resistant mutants. A possible solution might be a combination therapy with a cocktail of various siRNAs.
(2) Targeting the host cell with siRNA:
It is also possible to target mRNAs for cellular accessory or entry proteins so that they can no longer be used by the virus during infection. Researchers tried to use the mutationally more stable host proteins as targets instead of the rapidly mutating viral proteins (58).
Rathore et al. were able to show in 2014 that by silencing the heat shock protein 90 (Hsp90) transcripts with siRNA, CHIKV replication is interrupted in cultured cells. Heat shock protein 90 (Hsp90) is known to play a key role in the replication of CHIKV and other viruses and is a highly abundant molecular chaperone (65). Rathore found out that Hsp90 interacts with the nsP3 and nsP4 proteins of CHIKV to promote virus replication (66). For further “Host-targeting Antivirals” (see section Antivirals Against Chikungunya Virus).
Both siRNA approaches (viral or host target approach) share the same issues in bioavailability, delivery, and specificity. siRNA is not very stable. It is rapidly degraded in the cell/organism. Furthermore, when systemically applied, siRNA has to reach the target cells. Effective pharmacological use of siRNA requires “carriers” that deliver the siRNA to its intended site of action. siRNA displays poor cellular uptake and is not able to pass through the blood-brain-barrier (67). Small hairpin RNAs (shRNAs) present a solution to some of these flaws. shRNAs are ~70 nt long precursor siRNAs that are introduced into the cell by viral or bacterial vectors (e.g., plasmids). After expression in the nucleus, the shRNA is being transported to the cytoplasm where it is further processed by Dicer proteins. It is subsequently loaded into the RISC for specific gene silencing activity in the same manner as synthetic siRNAs (68).
siRNA often turns out to be unspecific. The suppression of other genes (the so-called “off target effects”) may lead to unknown consequences due to dangerous mutations and unwanted gene expression (69). SiRNA may also interfere with the host immune response (70). Consequently, the long-term safety of si/shRNA treatment is yet unclear as there are only few in vivo RNAi long-term studies (58).
Inhibitors of CHIKV nsP1
The non-structural protein 1 (nsP1) is a palmitoylated protein with methytransferase (MTase) and guanylyl transferase (GTase) activity. The protein consists of 535 amino acid residues and is responsible for the capping and the methylation of the newly synthesized viral and genomic RNAs (39). The added cap structure on the viral mRNA ensures the translation of the RNA and prevents its degradation from cellular 5′-endonucleases. On its N-terminal domain, the nsP1 has a α-helical amphipathic loop as well as a palmitoylation, which both act as anchors to attach the nsP1 and the nsP1-containing polyproteins/replication complex (RC) to the host's cellular membrane (71). Various studies could show that the palmitoylation of nsP1 is an important feature for the replication of some alphaviruses (72, 73). Depalmoylated Semliki Forest virus (SFV) mutants displayed a diminished pathogenesis in mice (72). Likewise, Zhang and colleagues (74) demonstrated in vitro that by inhibiting the enzyme responsible for the palmitoylation of proteins during CHIKV infection, CHIKV replication could be suppressed. There is evidence suggesting that nsP1 has additional functions during alphavirus infections like the development of cell filopodia and the rearrangement of actin filaments (73). Especially the MTase and GTase-like activities of nsP1 present a viable target for antiviral compounds since both enzymatic properties are essential for virus replication. The GT activity of nsP1 is dependent on successful MTase activity (75). Interestingly, unlike cellular MTase and GTase enzymes, the nsP1 does not contain canonical signature motifs and the mechanism of the enzymatic action differs from the cellular cap formation. Thus, there is the possibility of identifying molecules that selectively inhibit viral nsP1 without affecting the host cell capping enzymes' activity (76). Compared to the other nsPs, the research on antivirals that target nsP1 has been poor. Lampio et al. tested 50 guanosine/cap analogs for their activity of inhibiting SFV nsP1 20 years ago (77). Recently, Bullard-Feibelman developed an assay to screen and identify possible CHIKV nsP1 inhibitors (78). Two years later, the same research group presented their results on a high throughput screening (HTS) of 3,051 compounds and their successful identification of promising hit compounds like the naturally derived compound “lobaric acid” (Table 1) (20). Gigante et al. found a strong inhibitor of CHIKV replication among a new family of compounds named [1,2,3]triazolo[4,5-d]pyrimidin-7(6H)-ones (Table 1) (21). It was not until 2016 when reverse genetics carried out by Delang et al. could identify the CHIKV nsP1 as the target for this potent compound (79). New derivatives of these compounds also inhibited the GTase activity of CHIKV and VEEV nsP1 (14). A report from Jones et al. (80) postulated that nsP1 was an antagonist of tetherin (an antiviral host factor that helps to retain the viruses at the surface of the infected cells). These findings gave rise to hope that nsP1 could be considered as a target for developing tetherin-mediated therapeutics against CHIKV (80). However, a more recent study on the subject could not confirm Jones' report since no evidence for tetherin-antagonists in alphaviruses was found (81).
Inhibitors of CHIKV nsP2
The CHIKV nsP2 has multiple enzymatic activities and thus plays a central role in CHIKV replication. nsP2 has auto-protease activity at its C-terminal end for cleaving the non-structural viral polyprotein (nsP1234) into the individual nsPs. There is a methyltransferase-like region of unknown function. The N-terminal half has terminal helicase, nucleoside triphosphatase (NTPase), and RNA triphosphatase activities (82). The triphosphatases are involved in RNA capping and also fuel the RNA helicase domain with energy. Additionally, CHIKV nsP2 is a virulence factor as it is able to stop the host cells mRNA transcription and translation, thus tampering with the hosts immune response. This is referred to as “transcriptional shut-off” (83). In fact, a recent study was able to show that nsP2 (as well as nsP3) exhibit RNA interference (RNAi) suppressor activity (84). Viral suppressors of the RNAi pathway (VSR) have been found encoded in various viruses (including flaviviruses) before. Yet, the report of Mathur et al. was the first to show VSR in alphaviruses. Moreover, Fros and colleagues found out that CHIKV nsP2 suppresses the type I/II interferon-stimulated JAK/STAT signaling pathway, which consequently inhibits the hosts antiviral response and defense mechanisms (82). It has previously been shown in other viruses that especially the protease function poses an interesting target for antiviral drugs (85).
Compounds designed in silico. Marcella Bassetto and colleagues applied a structure-based virtual screening strategy to find possible CHIKV nsP2 inhibitors. The molecules in question have been modeled to potentially fit and thus block the nsP2 protease binding site.
Based on this model, Bassetto performed a virtual screening of ~5 million compounds and investigated the structure-activity relationship of the identified hits. After a final visual inspection, 15 derivates were selected to be potential CHIKV nsP2 inhibitors. As only 9 were commercially available, those were evaluated in a virus-cell-based CPE reduction assay. Compound 1 performed best and was predicted to fit the central portion of the nsP2 protease active site (Table 1). The compounds' ability to act as a selective CHIKV replication inhibitor was then further investigated by performing a virus yield assay on Vero cells. The assay confirmed the findings of the CPE reduction assay.
Furthermore, Bassetto created structural analogs of Compound 1 and tried to chemically optimize the properties of the compounds. She designed and synthesized two new derivates with one showing a slightly better antiviral activity profile than compound 1. With her work Bassetto proved that a combination of molecular modeling with different in silico techniques and classical medical chemistry methods can lead to the discovery of novel and selective antiviral compounds.
Jadav et al. (86) tested a series of derivates of 1,3-thiazolidin-4-ones for their antiviral activity in a CPE reduction assay on Vero cells. Five compounds showed promising CHIKV inhibition properties. The authors assumed the mode of action may be that of protease inhibition, after they carried out molecular docking simulation with the available X-ray crystal structure of the CHIKV nsP2 protease (86). Here, the computer-aided binding model was used to explain possible mechanism of action, while Bassetto used the docking simulation to model compounds accordingly. Still, neither of these studies actually tested the ability of the predicted compounds to inhibit the protease activity of CHIKV nsP2.
It was the group of Das that actually designed and tested 12 compounds specifically on their ability to block the nsP2 (22). The researchers managed to create a test to validate whether the compounds actually inhibit nsP2. Das designed the compounds specifically to fit the nsP2 active site, using the same method as Bassetto and employing Compound 1 of Bassetto as a template for his products.
The group then systematically analyzed the ability of the compounds to inhibit the protease activity of the purified enzyme in cell-free assays. Two different cell free assays were employed, one being an end-point assay, the second one being continuous. In the end point assay, Das used full-length recombinant CHIKV nsP2 as the protease and a recombinant protein substrate containing the nsP2 cleavage site that was located between enhanced green fluorescent protein (EGFP) and thioredoxin. If the nsP2 was fully functional, the protein substrate was being processed, making it possible to detect the products by separating them by SDS-PAGE and visualizing the results with a Coomassie blue staining. The method on how to express and purify the recombinant proteins has been described earlier by the same group (87).
To verify his finding, Das additionally used a fluorescence resonance energy transfer (FRET)-based assay to compare the efficiencies of different inhibitors. This kind of assay had originally been described for the HIV protease by Matayoshi et al. (88). It is a continuous assay that makes it possible to collect information on the initial period of the reaction. In Das' assay, the nsP2 protease processed a peptide substrate with the nsP3/nsP4 cleavage site of CHIKV P1234 polyprotein (89). The substrate had a quencher at the N terminus and a fluorescent molecule at the C terminus. Cleavage of the substrate by nsP2 protease results in fluorescence that can be detected at an emission wavelength of 490 nm.
With these assays, Das managed to show that the majority of his compounds inhibited the nsP2s ability to process recombinant protein and synthetic peptide substrates. He also discovered that the original template molecule from Bassetto performed very poorly as a specific nsP2 inhibitor in these cell free assays, despite the fact that it had an EC50 of ~5 μM in cell-based assays against CHIKV (12). Das then tested his compounds successfully in cell-based assays against CHIKV. The fact that some compounds did not inhibit the CHIKV nsP2 protease function in the cell free assays and yet managed to curb CHIKV infection in cell-based assays suggests that the antiviral activity of these compounds may be at least in part due to other mechanisms than the inhibition of protease activity of nsP2 (Table 1) (22).
Compounds inhibiting the nsP2 mediated “transcriptional shut-off”. Lucas-Hourani et al. (23) developed a phenotypic cell-based functional assay to detect CHIKV nsP2 protease inhibitors. In particular, compounds that inhibited the nsP2 mediated “transcriptional shut-off” mechanism were to be detected. As mentioned before, the nsP2 protease is able to bind to cellular transcription factors and thus induce downregulation of the cell's immune response. In Lucas-Houranis' assay luciferase expression is induced when the cellular functions are working at a normal level. If nsP2 protease is blocked by antivirals, the cells mRNA transcription is properly restored and thus a replication of luciferase takes place, resulting in an increased signal.
The assay is thus based on a recombinant human cell line (HEK-293T) that expresses CHIKV nsP2 together with various reporter gene constructs (on three plasmids). Lucas-Hourani used this transfected cell line to establish an assay suitable for screening compounds for their nsP2 inhibition activity. From a pool of 3,040 molecules, he detected one with no toxicity that particularly blocked nsP2 activity in vitro (Table 1) (23).
Inhibitors of CHIKV nsP4 and Viral Genome Replication
The nsP4 is the sole protein with a polymerase function and is responsible for the RNA synthesis of the (replication complexes) RCs. The ~100 residues at the N-terminal region are specific to alphaviruses. The nsP4 has ~70 kDa and contains the core RNA-dependent RNA polymerase (RdRp) domain at its C-terminal end. The structure of the RdRp is typical and encompasses fingers, palm containing the GDD motif at the active site and thumb domains (90). The RdRp is able to copy the genome into a complementary minus-strand which is in turn copied into genomic and subgenomic RNAs by the polymerase with the help of the other viral nsPs in the RC. Mutation studies revealed a TATase (tyrosine aminotransferase) activity in the RdRp domain. Thus, the nsP4 may be generating the poly(A) tail at the 3'terminal of the genome (91). For more details on the nsP4s role during genome replication and its fundamental function I refer to the review of Pietila et al. (9).
Research has recently focussed on finding antiviral compounds against viruses of the Flaviviridae family [hepatitis C virus (HCV), Zika, Dengue, Yellow Fever virus (YFV), tick borne encephalitis virus (TBEV)], most of which are arboviruses. Especially Zika and Dengue can cause coinfections with CHIKV and the initial symptoms of the three diseases look very similar. Since the diagnosis is costly and time consuming, it is crucial to find a pan-antiviral that works against all of them. All three viruses are +ssRNA viruses and there is a reasonable chance that they share conserved motifs in the orthologous RdRp enzyme (24, 91). The remarkable homology of the nsP4 among the alphaviruses makes it possible that antivirals blocking the nsP4 may exhibit their activity over a broad spectrum of viruses. With human cells lacking this specific polymerase the chances of adverse side effects of RdRp inhibitors are minimized (92).
Nucleoside analogs and proTides. Nucleoside analogs (NAs) are synthetic, chemically modified nucleosides consisting of a sugar and a nucleic acid analog. Nucleotide analogs additionally have one to three phosphate groups attached to the 5′-site. In the cell, they are processed the same way as the natural (endogenous) nucleosides. After their uptake into the cell and their metabolization, the NAs can act on cellular functions. They mimic their physiological counterparts and block cellular division or viral replication by impairing DNA/RNA synthesis (they usually cause termination of the nascent DNA/RNA chain) or by inhibition of cellular or viral enzymes involved in the nucleoside/tide metabolism (93, 94). The FDA has approved more than 25 nucleoside analog drugs used for the therapy of viral infections such as HIV/AIDS (tenofovir), hepatitis B (lamivudine/entecavir), and C (sofosbuvir) or herpes (acyclovir) (93, 95). Besides being antiviral agents, NA drugs are also applied in the therapy of cancer, rheumatologic diseases and even bacterial infections (96).
Before NAs can actually work as antivirals, they have to be phosphorylated in the host organism. Three consecutive phosphorylation reactions are necessary to activate the prodrug. The first reaction to the 5′-monophosphate is usually a rate-limiting step, which also means that if this first phosphorylation does not take place, the drugs remains inactive (97). This might happen either because the virus does not induce a specific kinase or has acquired a mutation in this particular enzyme resulting in resistance to the compound because the host cell is not able to phosphorylate the NA.
Monophosphate NAs have come into focus in order to avoid this problem and improve the therapeutic properties. However, these phosphate analogs (possessing a CO–P bond) proved to be prone to esterase and phosphatase hydrolysis. As an alternative, chemists investigated replacing the phosphate group by an isosteric and isoelectronic phosphonate moiety (CH2-P bond). This led to the discovery of nucleoside phosphonate analogs (NPs), which are chemically and enzymatically more stable than the phosphate analogs (98).
Toxicity and side effects of nucleoside/-tide analog drugs often result from their off-target use by host polymerases and their incorporation into RNA or DNA. The observed toxicities tend to be highly unpredictable and even closely related analogs may prove toxic for different organs (95). Various mechanisms for NAs toxicity have been discovered, the most characteristic is due to their affinity to host mitochondrial gamma polymerase (99). The NAs enter the mitochondria and are either incorporated into the mitochondrial DNA or block its synthesis.
Since NAs, nucleoside 5′-monophosphates or 5′-phosphonates are charged molecules and penetrate the cell membrane very poorly, they are not suited for oral administration. Research tried to improve the pharmacological properties and bioavailability of this class of compounds. This led to the discovery of the ProTides approach by McGuigan in 1998 (100, 101). The researchers designed a novel prodrug in which the phosphate was chemically protected or masked. This group of prodrugs became known as “ProTides” (pronucleotide) and as a result from the masked phosphate, this construct is able to pass the cell membrane via facilitated passive diffusion (94).
In the cell, the ProTide is enzymatically cleaved, thus releasing the masking groups from the nucleoside monophosphate/phosphonate which can be further transformed into the active 5′-triphosphate form of the NA. Various natural and unnatural amino acids can serve as the masking amino acid motif. All ProTide drugs that have reached the clinic, feature l-alanine (94). With the prodrug strategy, medical chemists were able to solve the main pharmacological problems associated with NAs, namely poor cellular uptake and poor metabolism into their phosphorylated forms.
Ribavirin. Ever since its discovery in 1972, ribavirin (1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide, also known as Virazole), a synthetic guanosine nucleoside analog, has been used as a compound against various viruses (102).
Ribavirin (RBV) is one of few FDA approved antiviral drugs in clinical use that is effective against respiratory syncytial virus in infants and chronic hepatitis C virus infections in combination with pegylated interferon (IFN)-α (103, 104). Apart from the FDA approved indications, RBV has shown efficacy against a variety of virus infections including haemorrhagic fever and measles (105, 106). Huggins and colleagues could also prove RBV's effectiveness against viruses of the alphavirus family in vitro (107). Multiple studies confirmed his findings by testing RBV in vitro against CHIKV either as a monotherapy (25) or in combination with doxycycline (26) or IFN-α (27, 28). Especially, Franco et al. (25) demonstrate that the effectiveness of antiviral agents against CHIKV differs considerably between host cell lines (Table 2).
Various different mechanisms of action have been attributed to RBV which might explain its broad-spectrum antiviral activity. The major mechanism, by which the replication of RNA viruses is being inhibited, is curbing the cellular guanosine triphosphate (GTP) pools by blocking the inosine monophosphate dehydrogenase (IMPDH) (108). Another indirect mechanism is the immunomodulation of the host's adaptive immune response: RBV triggers a suppression of the T-helper type 2 response and an induction of the T-helper type 1 response (109). The type 1 response is responsible for an increased clearance of infected cells. Additionally, RBV is believed to directly inhibit RNA capping. Other findings suggested that RBV interferes with the guanylyl transferase and/or methyltransferase activity of the nsP1, leading to a production of mRNAs that are not fit for translation (110). RBV is said to directly inhibit the viral polymerases, thus hampering the virus' genome replication (111). This has also been proposed by other studies that suggested RBV to directly inhibits nsP4 RdRp by interacting with its Cys483 residue, resulting in a decrease in replication fidelity (112). This would confirm the theory that RBV leads to error catastrophe via increased mutation frequency (nucleotide transitions) because of the incorporation of ribavirin triphosphate (RTP) into the newly synthesized viral genomes (113). Others found indications that RBV promotes IFN signaling by modulating specific genes and thus potentiating IFN action (114).
RBV, albeit a success as a broad-spectrum antiviral in vitro, has rarely been reported to be the subject of in vivo trials against CHIKV in humans. Ravichandran and Manian (115) treated 10 patients with confirmed CHIKV infection. Before treatment the infection had not been resolved after 2 weeks and resulted in crippling lower limb pains and arthritis. The patients were treated with 200 mg RBV twice daily for 7 days. A control group of 10 similar patients was only given analgesics when required. According to Ravichandran and Manian the patients of the RBV group showed a significant improvement in the joint pains and 8 patients out of 10 had a reduction in tissue swelling. Ravichandran concluded that RBV may indeed have a direct antiviral property against CHIKV infection and might lead to a faster recovery of the patients. However, the study had some flaws: (1) only a small number of patients were considered; (2) the study was not a randomized controlled study (a so-called double-blind study) where the RBV group was compared with a group receiving placebo; (3) the drug was administered in the subacute phase of the disease, thus some of the improvement could be attributed to a normal course of healing. A recent in vitro study of Mishra et al. (116) suggested that RBV is only effective in the earlier stages of the CHIKV lifecycle; the benefit of giving the drug in a subacute or chronic phase might therefore be questioned.
The doses at which RBV would have to be administered in order to reach its full potential as an antiviral in vivo are associated with severe side effects such as haemolytic anemia, pulmonary, dermatologic, and teratogenic effects and can thus only be justified if the infection is life-threatening (117).
RBV's success as an antiviral is probably attributed to its ability to act simultaneously via multiple mechanisms. Usually, when an antiviral interacts at various cellular and viral processes, the chances for drug resistant mutants are diminished. But, in case of RBV, various resistant viruses have been reported, such as Sindbis virus, Hepatitis C Virus and CHIKV, showing yet again, how quickly viruses are able to adapt (52, 110, 118). Taking these developments into account, RBV might still be interesting as a component in an antiviral “cocktail” consisting of multiple drugs with various modes of action, where the dosages of the drugs themselves could be reduced due to synergism and the risk of adverse effects could thus be minimized.
β-d-N4-hydroxycytidine (NHC). A report from Ehteshami et al. (30) stated the outcome of experiments dealing with β-d-N4-hydroxycytidine (NHC), another modified NA. NHC was identified to successfully inhibit CHIKV replication in different replicon cell lines as well as in infectious models in vitro (Table 1). One year later, another group published that NHC was able to curb the release of genome RNA-containing VEE virions and their infectivity in in vitro test with Vero cells (119). This discovery supports the idea that the polymerase activity of the nsP4 is quite conserved and that drugs targeting this particular activity might show efficacy against various alphaviruses. The antiviral activities of NHC are probably due to the compound acting as a pyrimidine analog that may directly target the viral polymerase and cause chain-termination. Alternatively, the compound might induce accumulation of mutations in virus-specific RNAs which are either lethal or lead to viral genomes that are incapable of replication (30). Urakova suspects a dual effect of NHC on VEEV by causing a modest decrease in virion release and a strong decrease in virion infectivity. This idea supports the theory that mutations caused during the replication process lead to “error catastrophe” or “lethal defection” (119, 120).
Urakova reported that NHC only triggered the development of a low-level resistance in VEEV against NHC, which makes it a very promising compound that might substitute RBV. These findings are very encouraging. Nevertheless, further studies with more relevant human cell lines, animal models as well as other viruses are needed to confirm whether this compound has a future as a broad-spectrum antiviral.
Favipiravir (T-705) and its defluorinated analog (T-1105). Favipiravir (T-705, 6-fluoro-3-hydroxy-2-pyrazinecarboxamide) is an approved drug in Japan for the treatment of influenza virus infections (121, 122). The drug is a purine analog and functions as a broad-spectrum antiviral agent which has also been reported to inhibit (in vitro and in vivo) the replication of a number of RNA viruses such as arenaviruses, bunyaviruses (123) and alphaviruses (124–126). During the 2014/2015 Ebola epidemic in western Africa, T-705 proved beneficial for infected patients (127).
Favipiravir is a prodrug, which is phosphoribosylated in the cell into its active form, a ribofuranosyl 5′-triphosphate metabolite (favipiravir-RTP). It acts as a pseudo purine and inhibits the viral replication of influenza. Two modes of action have been suggested: There is evidence that favirpiravir-RTP specifically blocks the influenza virus RNA-dependent RNA polymerase (RdRp) by binding at certain domains of the enzyme (122). Others suggested that favipiravir-RTP is incorporated into the nascent viral RNA, thus leading to lethal mutagenesis or preventing further extension of the RNA strand entirely by chain termination (128, 129).
As favipiravir is relatively novel, the information on its in vitro efficacy is limited. Values vary depending on the assay, cell line and virus strain used (Tables 1, 2). Apart from favipiravir itself, the defluorinated analog T-1105 has worked as an antiviral drug against CHIKV in in vivo experiments with mice (31). The drug prevented mice from developing severe neurological disease and reduced the mortality rate of the CHIKV infected animals. A dosage of 300 mg/kg T-705 daily and orally proved especially beneficial for CHIKV infected mice during the acute phase of the disease (125). Delang also identified T-705 resistant CHIKV variants in vitro. The mutant had acquired a mutation in the motif F1 of the RdRp, which seems to be important in the nucleoside triphosphate binding during and in the initiation of the viral RNA synthesis of +ssRNA viruses (130). Yet, Abdelnabi et al. (126) suggest that favipiravir has a high barrier of resistance. Abdelnabi made experiments in which he tried to create T-705-resistant coxsackievirus B3 (CVB3) (another +ssRNA virus), by point-mutating the same F1 motif. These efforts resulted in either low-fidelity RdRp or unviable virus. Since NTP binding is a major fidelity checkpoint, point mutations in this F1 motif could destroy the activity of the polymerase or reduce catalysis (131).
The fact that resistant mutants develop, demonstrates how quickly RNA viruses can adapt to selective pressure via mutations. Understanding the role of conserved motifs like F1 is of great importance in order to understand the mode of action of certain drugs and possibly design more potent compounds.
Sofosbuvir. Sofosbuvir (β-D-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine, formerly known as PS-7977 or GS-7977) is a RdRp inhibitor approved by the FDA for the treatment of HCV infections (132). The drug is a nucleotide analog that is orally available and functions as a prodrug. In hepatocytes, sofosbuvir is metabolized to 2′-F-2′-C-methyluridine monophosphate (UMP) and further phosphorylated into its active triphosphate form (UTP). During the viral genome synthesis, UTP functions as a chain terminator, thus inhibiting HCV replication and production at the site of infection, in this case the liver (133). Sofosbuvir has recently been reported to inhibit YFV and ZIKV replication in vitro and in vivo (134–136).
Sofosbuvir has been tested against CHIKV in vitro (Huh-7 cells and astrocytes) and in vivo (mice) (24). The drug inhibited CHIKV replication and was three times more potent in inhibiting CHIKV in human hepatoma cells than RBV (Table 2). In human induced pluripotent stem cell-derived astrocytes, sofosbuvir did impair virus production and cell death in a MOI-dependent manner, yet not to such a degree as in the Huh-7 cells. This may be due to the fact that hepatocytes have the most effective system of turning the prodrug sofosbuvir into its active form (UTP), whereas astrocytes show less metabolic activity in this respect and thus have less of the active UTP form of the drug available (133). Furthermore, sofosbuvir prevented CHIKV-induced arthralgia-related paw oedema in adult mice as well as mortality in neonate mice (24). Since CHIKF can lead to chronic arthralgia, further studies are needed to evaluate if sofosbuvir in a combination therapy alongside anti-inflammatory drugs is beneficial to patients suffering from chronic CHIKV associated arthritis.
Interestingly, humans tolerate the drug better than mice. A 400 mg daily dose over a period of 12–24 weeks is the standard therapy for HCV patients (133), while doses of >33 mg/kg/day in a 7 day regime proved to be toxic to mice (136). The reason for this observation might be the decreased stability of sofosbuvir in rodent serum. This raises the question of how significant rodent models are for the evaluation of sofosbuvir or whether other (animal) models might be more representative.
Similar to favipiravir, sofosbuvir resistant HCV strains have been reported (137). Yet, the development of sofosbuvir resistant mutants seems to be slower compared to HCV inhibitors targeting other proteins. Researchers hold the high degree of amino acid conservation within the RdRp domain as well as the lack of fitness in mutated viruses responsible for this phenomenon (136).
Nevertheless, the fact that sofosbuvir blocks the viral replication of CHIKV as well as several flaviviruses is strong evidence for the presence of conserved motifs among RNA polymerases from +ssRNA viruses. The recent advances in elucidating the nsP4 structure and core domain function of CHIKV highlight these observations and may confirm that the RdRp is a feasible target for pan-antiviral molecules (91).
Other Viral Genome Replication Inhibitors
Mycophenolic acid (MPA). Mycophenolic acid (MPA) had already been discovered in 1893 and was isolated in 1896 as an antibacterial molecule produced by Penicillium brevicompactum (138). MPA is licensed by the FDA as a drug for transplantation rejection (139). The drug inhibits cellular inosine monophosphate dehydrogenase (IMPDH) and thus decreases the intracellular pools of guanosine triphosphate (GTP) and 2′-deoxyguanosine triphosphate (dGTP). This causes a disruption of viral and cellular RNA, DNA, and protein synthesis (140). Two derivatives of MPA are available for clinical use: mycophenolate mofetil (MMF, CellCept) and mycophenolate sodium (MPS, Myfortic). Mycophenolate mofetil is the orally bioavailable prodrug form of MPA. MPA has shown antiviral activity against DENV and Orthopoxvirus (141, 142).
Although MPA was reported to inhibit CHIKV in vitro in 2011, tests done in 2018 could not confirm these findings (143, 144). However, Ferreira tested MPA as a control alongside his compounds and indeed received good EC50 values, with MPA even performing slightly better than sofosbuvir and with a much better selectivity index (SI = CC50/IC50) than RBV (24). Likewise, other research groups used MPA as a reference against CHIKV and evaluated the efficacy against CHIKV (Table 2) (16, 32).
There are various studies confirming the antiviral, antibacterial, antifungal, immunosuppressive, and anticancer properties of MPA or its derivates (145). Yet it is important to deliberate whether the benefits of MPA as an antiviral outweigh its adverse effects as an immunosuppressant.
NsP3 and Possible Inhibitors
The nsP3 consists of three domains. The N-terminus has a macrodomain, while the C-terminus holds a hypervariable domain (HVD). The central part of the protein contains a zinc-binding domain which is sometimes referred to as the alphavirus-unique domain (AUD), a region that shares a strong sequence homology across the alphaviruses. The role of the AUD is so far undefined but the domain seems to be important in RNA replication and in the synthesis of negative sense and sub-genomic RNA (146).
There are hints indicating that the nsP3 is involved in inhibiting the assembly of the host cells stress granules (SG) which are essential for the degradation of viral mRNA (147). NsP3 is usually found in complex with other nsPs during infection. It also interacts with host factors. Saul et al. (148) discovered that the amount of nsP4 increased in a recombinant SFV with a duplicated nsP3-encoding sequence. Saul concluded that nsP3 is involved in the stabilization of nsP4. He could furthermore back other studies' findings that nsP3 is important for the (neuro-) virulence of old-world alphaviruses (148). In New-World alphaviruses, neurovirulence is mainly determined by structural proteins, particularly E2 (149).
So far, the complete function of the nsP3 macrodomain has not been fully unraveled although its crystal structure has been known since 2009 (PDB id: 3GPG and 3GPO) (150). The N-terminal macrodomain is highly conserved among alphaviruses but also occurs in other positive-strand RNA viruses such as coronaviruses and hepatitis E virus (151). There is evidence that the viral macrodomains bind ADP-ribose, dephosphorylate ADP-ribose-1”-phosphate and act as de-ADP-ribosylating enzymes thus counteracting antiviral ADP-ribosylation (152). Other studies indicated that the most likely biochemical function of viral macrodomains is de-ADP-ribosylation. By enzymatically removing mono- and poly-ADP-ribose from proteins, macrodomains might oppose the host cells' antiviral response (153). Furthermore, the mono(ADP-ribosyl)hydrolase activity of the nsP3 is critical for CHIKV replication in vertebrate hosts and insect vectors, and determines virulence in mice (154). These findings suggest that the macrodomain plays an important part in the host-pathogen conflict.
Nguyen et al. virtually screened a database of 1,541 compounds for possible hits that might block the nsP3 macrodomain of CHIKV (155). The group combined molecular docking, virtual screening, and molecular dynamics simulations to identify potential inhibitors. They ended up with three ligands that might have potential as nsP3 inhibitors. However, these findings were achieved in silico and still need to be verified by experimental studies in vivo.
Until Varjak et al. discovered a degradation signal at its C-terminus, nsP3 was thought to be a rather stable protein. Varjak could demonstrate that the nsP3 of SFV and Sinbis Virus (SINV) was degraded rapidly when the protein was expressed individually. On the other hand, nsP3 was significantly stabilized when it was expressed in the nsP123 polyprotein form (156). The role of this C-terminal degradation signal is still unknown but there are various hints that it may contribute to granting the optimal stoichiometry of the nsPs.
Especially the HVD at the C-terminal region of the nsP3 seems to be a center for interactions with host cell proteins, including stress granule (SG) components which might help the virus adapt to distinct cellular environments. Data suggests that the HVD interacts with several host factors through a conserved proline (P)-rich and duplicate FGDF motif. The letters of the motif correspond to the according amino acids, two phenylalanine residues which are separated from each other by a glycine and an aspartate residue (157). These interactions are needed for the assembly of virus genome replication complexes (158, 159). The FGDF motif seems particularly important for the successful replication of alphaviruses in mammalian cells. Experiments with CHIKV revealed that the virus' nsP3 has two FGDF motifs that bind to certain domains of the SG components in mammalian cells (160). SGs usually block host and viral translation. The interactions between the CHIKV nsP3 and the SG domains impede the organization of the SGs and thus may allow virus replication (147, 161, 162). When the alphavirus nsP3 HVD is mutated in a way that both FGDF motifs are disrupted, CHIKV is inactivated and SFV as well as SINV are attenuated in mammalian cells. If only one FGDF motif is present in CHIKV or SFV nsP3, the affinity for the SG domains is reduced and the virus is attenuated as well. This leads to the conclusion that alphaviruses need two FGDF motifs for a successful viral replication in mammalian cells (146, 160, 161).
The HVD seems also to be a determinant for virulence in some viruses. There is evidence that the conserved FGDF motifs in the HVD of chikungunya virus nsP3 are required for the effective transmission of the virus from Aedes aegypti mosquito saliva to a vertebrate host (163).
The nsP3 seems to be an important protein in determining vector specificity. ONNV, which is closely related to CHIKV, is the only alphavirus known to be transmitted by Anopheline mosquito species. CHIKV on the other hand, is mainly transmitted by Aedes mosquitoes. Experiments with chimeric CHIKV expressing ONNV nsP3 revealed that Anopheles gambiae mosquitoes become susceptible for CHIKV although being naturally immune to WT CHIKV (164). This observation is in line with previous findings suggesting that nsP3 might be involved in specific protein-protein interactions and thus carries out host cell-dependent functions (165). A recent study revealed that nsP3 suppresses RNAi alongside nsP2 in CHIKV infected insect cells (84). As RNAi is an antiviral defense mechanism in various organisms that leads to a degradation of viral RNA, the suppression of RNAi by viral proteins enhances infection.
The impact of these interactions on biological and biochemical processes of the host cell at early stages of the infection are still under heavy investigation. There is hope that the interacting regions might prove valuable targets for intervention and opens new possibilities for vaccine development and antiviral drug discovery.
Kaur et al. (29) reported the discovery of the anti-CHIKV properties of harringtonine, a cephalotoxin alkaloid from the Cephalotaxus harrintonica trees. It was suggested that the compound inhibits the early stages of CHIKV infection after cellular endocytosis (29). Harringtonine was proposed to interfere with the protein translation of CHIKV since it seemed to inhibit the production of nsP3, E2 proteins, and CHIKV RNA (29, 166). Harringtonine was approved in 2012 by the FDA as a drug for the treatment of chronic myeloid leukemia (167). Homoharringtonine, an analog of harringtonine with an additional methyl group, was reported to have anti-CHIKV properties as well. According to Kaur, both compounds display minimal cytotoxicity on BHK-21 cells and primary human skeletal myoblasts at the dosage needed for inhibiting CHIKV. However, the drug itself is labeled as a cytotoxic agent and according to the Globally Harmonized System (GHS) harringtonine is fatal if swallowed (H300), in contact with skin (H310) or if inhaled (330) (168). This may be the reason that although Kaur's original article has been cited repeatedly, no studies on the anti-alphavirus properties of harringtonine have been published in the past 7 years.
Host-Targeting Antivirals
Many viruses depend on host factors to ensure their replication or are inhibited by such. Host factors present a valuable target for drugs to interfere in the virus' life cycle either by inhibiting host factors on which the virus relies on or by promoting host factors that curb virus infection. Since host factors also play vital roles in normal physiology, their inhibition or promotion can lead to abnormal physiological function and toxicity. The impact such interference may have on the host organism must thus be critically elucidated. Ideally therapeutics would target interactions between host and viral factors without disrupting essential cellular processes. For the interested reader we refer to the review of Wong and Chu (169) that summarizes the current knowledge on the interplay of viral and host factors in CHIKV infection as well as potential targets for antivirals.
Viperin, Hsp90 Inhibitors, and Interferons
Viperin
Viperin (virus inhibitory protein, endoplasmic reticulum-associated, interferon-inducible) is an interferon (IFN)-induced host cell protein that has come into focus because it is responsible for inhibiting viral replication via multiple pathways. It thus represents an interesting target for antiviral drugs (170). Viperin has been reported to inhibit a broad spectrum of DNA and RNA viruses, including members of the herpesvirus, flavivirus, alphavirus, orthomyxovirus, paramyxovirus, rhabdovirus, and retrovirus family (170). CHIKV infection is also curbed via IFN-induction of viperin and compounds leading to the up-regulation of viperin may present a strategy to manage CHIKV infections. Studies could demonstrate that CHIKV infection is controlled via type I IFNs that induce the interferon-stimulated gene (ISG) RSAD2 (radical SAM domain-containing 2) which encodes viperin (171). Teng et al. showed that mice lacking RSAD2/viperin had a higher rate of CHIKV replication and more severe inflammatory symptoms in the joints. A recent study tried to elucidate the role of viperin in shaping the pathogenic CHIKV-specific CD4 T-cell adaptive immune response during late acute disease phase (172). The group used viperin deficient mice in which CD4 T-cell had been depleted. They could demonstrate that increased late acute joint inflammation was exclusively mediated by CD4 T cells and that Th1-IFNγ-producing T cells played a pivotal role in the joint pathology. Further experiments showed that viperin expression contributes to reducing disease severity in both haematopoietic and non-haematopoietic cells (172).
Hsp90 Inhibitors
Chaperones help in the folding, assembly and maturation of host- and viral proteins. Almost all viruses depend on the chaperone Hsp90 (heat shock protein 90) especially during replication to ensure their life cycle (173). This causes viruses to be hypersensitive to Hsp90 inhibition and provides a way to curb virus replication. Compounds interfering in Hsp90 function have a potential as broad-spectrum antiviral drugs, especially since experiments with picornaviruses demonstrated that Hsp90 inhibitors are refractory to the development of drug resistance (174). As mentioned before, Hsp90 also plays an important role during CHIKV replication due to its interaction with the nsP3 and nsP4 of CHIKV. The chaperone furthermore stabilizes CHIKV nsP2 and thus promotes virus replication (65). Studies demonstrated that the Hsp90 inhibitor geldanamycin (GA) reduce CHIKV replication, particle formation and infection in vitro (65, 66). Yet, inhibiting Hsp90 very often results in toxicity, especially for the liver, presumably because Hsp90 is very abundant in liver cells and interacts with multiple proteins at crucial points in the cellular function. A lot of clinical trials with anti-Hsp90 drugs have been abandoned due to the in vivo toxicity (175). This also holds true for GA which is hepatotoxic as well as structurally instable, and thus has so far not been approved for clinical usage (176). Research is currently focussing on developing Hsp90 inhibitors with better pharmacological profile, such as ganetespib, which is relatively hydrophobic and less toxic (177). Ganetespib is currently under investigation in phase 1-3 clinical trials for the treatment of breast cancer, small cell lung cancer, acute myeloid leukemia, and myelodysplastic syndrome. However, its potential as an antiviral is not known but might be worth investigating once the drug is approved by the FDA.
Lillsunde et al. (178) investigated the antiviral activity of a number of marine alkaloid-oroidin analogs that are synthetic compounds and target the Hsp90. Lillsunde tested the compounds in replicon models against HCV and CHIKV. While 4 compounds selectively inhibited the HCV replicon, the compounds exhibited only moderate selectivity and efficacy against the CHIKV replicon in dose-response and cytotoxicity studies.
Interferons
Interferons (IFNs) play a vital role in the innate immune response to counter virus infections and thus have been the subjects of multiple studies. IFNs have been tested widely for their potential use as antivirals against a variety of viruses including HIV, Hepatitis C and B, and Influenza A (179). Type I IFNs [alpha/beta interferon (IFN-α/β)] are produced by the host cell upon sensing virus invasion. IFNs upregulate a variety of interferon-stimulated genes (ISGs). The protein products of the ISGs contribute to countering viral infections by suppressing viral spread and supporting the initiation of adaptive immunity [reviewed in (180)]. IFNs Type I are considered a “standard of care” in suppressing chronic HCV and HBV infections, while Type III IFNs have generated encouraging results as a treatment for HCV infection in phase III clinical trials (181). Various studies have confirmed that alphaviruses are also highly sensitive to the antiviral activity of Type-I IFNs (IFN-α/β) (182, 183).
Briolant et al. (27) compared the antiviral efficacy of IFN-α, glycyrrhizin, 6-azauridine, and RBV of inhibiting CHIKV and SFV infection in vitro. When combined with RBV, IFN-α2b had a sub-synergistic antiviral effect on both alphaviruses (27). A more recent study by Gallegos et al. (28) confirmed the highly synergistic effect of RBV and IFN α when administered as combination therapy in vitro.
In vivo studies with IFN-α/β receptor-deficient mice also demonstrated the importance of IFNs against CHIKV infection. The deficient mice lacked adequate IFN-α/β responses to the viral infection and CHIKV caused haemorrhagic fever, shock, and finally resulted in death (184).
Brehin et al. (185) investigated the role of IFN-induced 2′,5′-Oligoadenylate Synthetase (OAS) protein family in innate immunity to CHIKV. OAS proteins are critical components of innate immunity and the group was able to show that the antiviral actions of IFN-α/β in HeLa cells are mediated due to the induction of these proteins. Various ISGs that affect alphavirus replication have been identified, including ISG15, ISG20, P56, ZAP, and Viperin (185).
Tetherin
Tetherin [also known as bone marrow stromal antigen 2 (BST-2)] is a host transmembrane protein with antiviral activity that is induced by IFN. Tetherin binds budded viral particles directly to the plasma membrane (PM) and thus restricts the release of enveloped viruses. The virus particles which are thus bound to the PM can then be endocytosed and degraded (186). Two isoforms of tetherin that differ in length are known. They are referred to as L-(long) and S-(short) tetherin and each has distinct biological properties (187). Tetherin showed antiviral activity against alphavirus release and studies demonstrated that tetherin does not affect viral entry or protein expression. L-tetherin is significantly more efficient in inhibiting the SFV release than the short isoform (186).
In response to this antiviral countermeasure, many viruses have evolved tetherin antagonists. Jones (80) postulated that CHIKV nsP1 is such a BST-2/tetherin antagonist. However, Wan et al. (81) could not confirm Jones' findings and suggested that the sole physical tethering of virus particles to the PM is not sufficient to restrict alphaviruses and that the subsequent virus endocytosis is a requirement for efficient inhibition of alphavirus release.
Silvestrol
The natural compound silvestrol (a cyclopenta[b]benzofuran flavagline) is an isolate from plants of the genus Aglaia and has been the focus of various antiviral studies over the past 5 years. Flavaglines have been the interest of anticancer research for more than two decades because they display antitumor activity (188). Silvestrol is a highly efficient, non-toxic and specific inhibitor of the host RNA helicase eIF4A (eukaryotic initiation factor-4A), which is part of the heterotrimeric translation initiation complex in eukaryotes (189). The host cell needs the RNA helicase eIF4A to unwind structured 5′-untranslated regions (UTRs) of mRNAs to allow translation. Since 5′-capped viral mRNAs often contain structured 5′-UTRs as well, it has been suggested that RNA viruses which have these structures might depend on eIF4A for their translation. Silvestrol proved to be a successful antiviral in multiple in vitro studies against a variety of RNA viruses, such as Ebola, Corona-, Picornaviruses and CHIKV (189–191).
Henß et al. (191) demonstrated that by delaying the protein synthesis of CHIKV nsPs and structural proteins, silvestrol also retarded the innate response to CHIKV infection. By curbing the amount of nsPs, silvestrol reduced CHIKV RNA replication. The compound also decreased the host protein shut-off which was induced by CHIKV infection, probably because of the lower total amount of nsP2. In accordance with this, silvestrol seemed not to impair the IFN-induced STAT1 phosphorylation and eIF2 did not become phosphorylated. All these in vitro findings suggest that inhibition of the host helicase eIF4A with silvestrol might be a therapeutic strategy to treat CHIKV infections. Further research is needed to find out how and if silvestrol can actually be of benefit against CHIKV infection in vivo.
Protein Kinase C Modulators and Plant Extracts
Plants have always been an important source of active substances and to date about 50% of the licensed drugs are natural products or were inspired by them (192). Natural compounds quite frequently have striking differences compared to chemical molecules, which often result in better pharmacological properties (193). The introduction of today's modern drug discovery process has led to a certain neglect of considering plants as a resource for bioactive compounds. But with the technological improvement in the field of natural product isolation, synthesis and screening, the interest in plants as a source for anti-infective natural compounds has been renewed (194).
After the massive CHIKV outbreak in the Indian Ocean region in 2005–2006, a large-scale quest for novel and selective antiviral compounds was initiated. A project called “Biodiversity and emerging viruses in the Indian Ocean: selection of drug candidates targeting the Chikungunya virus” was financially supported by the Center for Research and Monitoring of Emerging Diseases in the Indian Ocean (CRVOI) and carried out from March 2009 to December 2011 (195). Its goal was to find new selective antiviral compounds derived from plants from the Indian Ocean Region, an area with a vast botanical biodiversity. Soon after the program started, virologists, and natural product chemists discovered that the plant family with the most promising components was the Euphorbiaceae.
Especially polycyclic and macrocyclic diterpenoids as well as molecules derived from them came into focus of antiviral research. Within the family of Eurphorbia more than several hundred different macrocyclic diterpenoids of interest have been discovered. These molecules possess various types of carbon skeletons (e.g., jatrophane, lathyrane, myrsinane, ingenane, tigliane, daphnane, etc.). More than 20 skeletal types can only be found in this particular plant family (196). These molecules possess a broad structural diversity due to their different macrocyclic skeletons and the various aliphatic and aromatic ester groups.
Macrocyclic diterpenoids have the ability to modulate protein kinase C (PKC) activity (196). Particularly the phorbol esters or phorboids have a tendency to bind to phospholipid membrane receptors and activate the PKC (197). PKCs are a multigene family of related serine/threonine kinases that are involved in many signal transduction pathways and cellular responses. PKCs play a role in a multitude of cellular functions such as cell mitogenesis, differentiation and apoptosis, smooth muscle contraction, platelet aggregation, tumor-modulation, and anti-HIV activity (198). PKCs are classified into three sub-families with different isoforms depending on the way of their activation. The classical PKC (cPKC) isoforms (α, β, and γ) require calcium (Ca2+) and the membrane-embedded ligand diacylglycerol (DAG) for activation, while the novel PKC (nPKC δ, ε, θ, η) are activated by DAG alone. The atypical PKC (aPKC Mζ- ι/λ) are not dependent on either ligand, but on proteins for activation (199).
All PKCs have an N-terminal regulatory moiety with a C1A domain and a C-terminal catalytic moiety for phosphorylation. Conventional and novel PKC isozymes have a second C1 domain (C1B) to which DAG binds (199). Phorbol esters have a two-order higher affinity to the C1B domain of conventional and novel PKC isoforms than DAG. This leads to the activation of the PKCs.
Recently a study reviewed the anti-CHIKV activity of about 80 naturally occurring macrocyclic diterpenes originating from the Euphorbiaceae plant family and about 30 commercially available natural diterpenoids (198) (Table 1). Some of these compounds have also been tested against other alphaviruses, like SFV or SINV. Other studies evaluated the antiviral properties of different plant compounds in vitro and found out that the phorbol esters prostratin (12-deoxyphorbol 13-acetate) and 12-O-tetradecanoylphorbol 13-acetate (TPA) are potent inhibitors of CHIKV (11, 200). Allard et al. published on the anti-CHIKV properties of trigocherrierin A, an unusual chlorinated daphnane diterpenoid orthoester (DDO) from the plant Trigonostemon cherrieri (Euphorbiaceae), and analog compounds from the same plant (17, 45). Likewise, Nothias-Scaglia et al. found Phorbol-12,13-didecanoate to be the most potent inhibitor of CHIKV replication among 29 commercially available natural diterpenoids (201). Phorbol-12,13-didecanoate is structurally related to TPA. Corlay et al. tested 12-O-decanoylphorbol 13-acetate (DPA), a molecule that differs from TPA only by the length of the side chain that is attached at C-12 (10 carbons for DPA vs. 14 carbons for TPA) (34). DPA had anti-CHIKV properties but a small SI of 2.0 reflecting a narrow therapeutic window making this compound a poor choice as a future antiviral drug. A novel DDO called neoguillauminin A and four 12-deoxyphorbols from Euphorbiaceae plants were recently found to have significant in vitro anti-CHIKV properties, three with an SI above 50 (Table 1) (15).
Yet despite the promising results of resent studies, the question of how said compounds manage to curb CHIKV replication has not been fully answered. Most studies assume that PKCs modulation is the key mechanism, but specifics are still outstanding. At the same time, the manner of how PKCs isoforms regulate intracellular signal transduction pathways and influence biological responses is still under heavy investigation and not completely understood. There are hints indicating that different translocation patters of the PKCs might lead to different intracellular signal transduction and cellular functions (202, 203). The cell type in which the PKCs are activated play a role as to how the response affects the organism. Additionally, the chemical properties (e.g., hydrophobicity) of different phorbol esters seem to play a critical role as well, since they induce different translocation patterns of PKCs in the cell. As conventional PKCs depend on plasma membrane bound Ca2+ and DAG as ligands, phorbol esters translocate them primarily to the PM, while the novel PKCs only depend on DAG and translocate to the more abundant and diacylglycerol-rich Golgi membrane (199). Studies showed that the stimulation of PKC δ by different phorbol esters induced distinct patterns of enzyme translocation. This indicates that lipophilicity of phorbol esters may contribute to differential PKC δ localization and thus to potentially different biological activities (203). Nothias-Scaglia et al. demonstrated that the HIV-1 and HIV-2 inhibitory effects of phorbol esters were strongly correlated with those of CHIKV (13). This observation is even more interesting since CHIKV and HIV belong to two different virus genera (alphavirus and lentivirus). Thus, the most probable explanation would be a common PKC-based mechanism of action. Yet a broad and potent PKC modulator with very good anti-HIV activity showed no anti-CHIKV activity, which might indicate that different PKC isoforms are involved in the two different virus life cycles. Abdelnabi et al. (33) tried to shed light on the role of PKCs in the cellular antiviral response to CHIKV infection by studying the mechanism of how prostratin works as an antiviral against CHIKV. The group found out that different cell lines express varied levels of diverse PKC isoform. Abdelnabi used four different cell lines [buffalo green monkey kidney (BGM) cells, African green monkey kidney cells (Vero cells), human embryonic lung fibroblasts (HEL), and human skin fibroblast cells] and four different CHIKV strains. Prostratin curbed CHIKV RNA synthesis and the production of infectious virus progeny at a post-entry step during virus replication. The antiviral action of the compound was dose- and cell- dependent. The most potent antiviral effect was observed in human skin fibroblast cells which also showed the highest gene expression levels of the classical PKC isoforms (Table 1). The antiviral activity of prostratin was significantly reduced when PKC inhibitors were present. These results suggest that the activation of mostly classical PKCs is the reason for the antiviral effect of prostratin (33).
Multiple or Unidentified Targets
Many other molecules have been tested against CHIKV and other alphaviruses in the past 5 years, with a special focus on plant extracts or drugs originally licensed for other diseases. Some seemed promising at first but then, upon closer investigation and with different assay methods, turned out to have a narrow SI or bad chemical properties. For some, the mode of action is still unknown. Here only the most recent or promising will be mentioned if they have been subject to repeated studies. For details on their efficacy (see Table 1).
Micafungin
Various researchers successfully tested the antifungal drug micafungin against viruses such as CHIKV, SFV, and SINV in vitro (35, 159). Micafungin has been licensed for the treatment of invasive candidiasis in 2005 (204, 205). According to Ho et al., micafungin significantly reduced CHIKV infection, cytopathic effects, and progeny yield (35). The question of how micafungin inhibits viral infection is still not answered. It was observed that the drug proved to be more effective in inhibiting CHIKV progeny yield than in reducing RNA replication (35, 159). The researchers thus deducted that micafungin might have a major influence on the later stages of CHIKV infection. On the other hand, the inhibitory effects of micafungin were stronger in the full-time treatment group than in the post-treatment group. This finding allows the speculation that micafungin might target different intracellular events during virus infection, such as viral replication, intracellular and extracellular transmission, and virus stability. The antifungal action of micafungin comes from the non-reversible inhibition of the β-1,3-D-glucan synthase of fungi, thus blocking the cell wall synthesis (206). Since neither mammalian cells nor viruses contain 1,3-beta-D-glucan polymers, the mechanism of action of micafungin still has to be elucidated. On the other hand, the absence of these polymers in mammal cells indicates a lack of mechanism-based toxicity of the drug that might partially account for the good tolerability in patients.
Abamectin, Ivermectin, and Berberine
Varghese et al. (36) conducted HTS of about 3000 compounds for their ability to inhibit CHIKV infection. Some of the substances were already licensed drugs or under investigation in clinical trials. With the help of a Renilla reniformis luciferase (Rluc) reporter system in baby hamster kidney (BHK-21) cells, Varghese could evaluate the compounds' impact on viral replication. After a second validation with WT and reporter CHIKV infection essays of 25 initial hits, Varghese identified five compounds with the capacity to curb CHIKV replication (36). Among these secondary hit compounds, abamectin, ivermectin, and berberine performed best with an inhibition activity against CHIKV of over 85%. Toxicity evaluations of these three compounds were done in BHK-21 and human hepatocellular (Huh-7.5) cells (Table 1). All three compounds also exhibited antiviral action against other alphaviruses, including SFV and SINV (39).
Abamectin and ivermectin are macrocyclic lactones which originate from the fungus Streptomyces avemitilis and are the most commonly used compounds of the avermectin family. Both drugs are potent endo- and ectoparasitic agents with a broad spectrum of activity. Especially ivermectin has been used as an insecticide for vector control and it seems that apart from its insecticide properties against Aedes and Anopheles species, it also displays antiviral activity against some arboviruses (207). The fact that ivermectin has both mosquitocidal and antiviral action may come in handy for vector control and limiting virus spread as well as infection at the same time. The drug is currently under investigation in a phase 2 clinical trial as a therapeutic for Dengue haemorrhagic fever (ClinicalTrials.gov identifier: NCT03432442). In flaviviruses (DENV, YFV, TBEV) ivermectin inhibits the NS3 helicase activity and thus curbs viral replication (208). The mode of action of abamectin and ivermectin against CHIKV is not clear, but it is being speculated that the drugs inhibit the RNA synthesis and down-regulate the viral protein expression of the nsP1 and nsP3 (36).
Berberine is a plant-derived isoquinoline alkaloid that is also able to inhibit CHIKV replication in a dose-dependent manner. It is believed to curb RNA synthesis and interfere with the viral protein expression (39). However, berberine has a wide range of bioactivities and it is also possible that the alkaloid interferes with host factors which promote CHIKV replication (209). Berberine reduced the virus-induced activation of cellular mitogen-activated protein kinase signaling, a pathway which is relevant for maintaining the viral life cycle. Inhibiting this kinase cascade with specific drugs resulted in a decreased production of CHIKV progeny virions. Varghese tested berberine in vivo in a mouse model where it significantly reduced CHIKV-induced inflammatory disease (210). Berberine is currently under clinical investigation in a variety of trials; however, none of them test its use as an antiviral.
Coumarin Conjugates
Coumarins can be found in plants as well as certain microorganisms and animals. The (natural and/or synthetic) coumarins have a wide range of biological activities and they are in focus for the therapy of various conditions. A number of coumarins have been found to display antiviral, anticoagulant, anti-inflammatory, antimutagenic, antitumor, antitubercular, central nervous system stimulant, fungicidal or vasodilator activities (211).
Hwu designed and developed 22 compounds that were made up of uracil, arene, and coumarin derivatives (212). He tried to combine the antiviral properties previously described for uracil derivatives and coumarin compounds. Hwu tested the newly designed compounds against CHIKV in vitro. Five molecules displayed significant potency against CHIKV (212). In 2019, the same research group published a study after testing 21 new coumarin derivatives against CHIKV in vitro. This time coumarin derivatives had been conjugated with guanosine. Hwu had modified the design of the molecules and after HTS, three of these new conjugates were found to inhibit CHIKV in Vero cells with significant potency but with a better SI than the ones tested before (Table 1) (37). From the structure-activity relationship Hwu deduced that the coumarin moiety was essential and the presence of a –OMe group enhanced the antiviral activity. Still, Hwu did not try to elucidate the work mechanism of the antiviral activity of his compounds.
Discussion
As CHIKV transmission depends on arthropod vectors in a complex interaction between virus host and the environment, a thorough understanding of these interactions is essential for the development of strategies to curb infections and the geographical spread of vectors. Especially climate change is one factor that may help arboviruses manifest in new areas that were formerly unsuitable for their vectors. International travel might further contribute to importing newly emerging arboviral diseases (like CHIK, Zika, or Dengue Fever virus). With autochthonous infections of CHIKV in France and Italy and established populations of Aedes albopictus in southern Germany, it is only a question of time until CHIKV manifests in moderate regions (3).
Thus, antiviral research remains of utmost importance to counter CHIKV infection. The different antiviral modes of action (MoAs), direct (by inhibiting the virus themselves), and indirect (by inhibiting host factors), have different merits, but both need to be considered and possibly combined for synergic effects of different MoAs.
A number of directly inhibiting antivirals against CHIKV that were tested in vitro were either discovered via in silico approach, high throughput screening of libraries or classical pharmacology. Especially plants have been rediscovered as a source for possible antivirals and yielded promising compounds like prostratin. Other drug candidates have been repurposed and are already licensed for the treatment of different viral diseases, e.g., sofosbuvir, ribavirin, and favipiravir. As these molecules have already been intensely evaluated in patients, trials for them against CHIKV in humans may possibly be fast-tracked. Unfortunately, some failed to maintain their efficacy in in vivo experiments (e.g., chloroquine and ribavirin), while others (like favipiravir and sofosbuvir) look more promising in animal experiments but still have to be tested against CHIKV in humans.
Despite multiple efforts in antiviral research, there is no standardized protocol for determining efficacy and toxicity. This makes comparison of the different hits impossible. As demonstrated in Tables 1, 2, efficacy and toxicity values vary considerably depending on the assay method, virus strain, and cell line. Some cell lines are refractory to the toxic effects of the molecules, possibly whitewashing the SI of the potential hit. The same applies for the assay methods, where each has its merits and its flaws. The lack of standardization as well as polypharmacology in vivo might be reasons why multiple drugs, although having achieved promising results in vitro, failed to be of benefit in vivo. Standardized efficacy and toxicity assays would help in calculating the SI which in turn is important for selecting molecules to test in vivo. So far, there is no defined cut-off for the SI, but a value of ≥10 is usually considered for animal models (39). A more thorough validation of potential hits in pre-clinical studies (e.g., multiple assay methods of selected hits in vitro) might help to avoid disappointment in in vivo assays.
Furthermore, as CHIKV infection often go hand in hand with other arboviral infections that are transmitted by the same Aedes species (e.g., DENV and ZIKV), a panantiviral which shows efficacy against these other viruses would be ideal. Apart from displaying anti-CHIKV activity, sofosbuvir, suramin, favipiravir, ribavirin, 6-azauridine, and ECGC also display antiviral activity against DENV or ZIKV or both in vitro (50, 136, 213–215).
Indirect antivirals targeting host factors yielded some promising results in vitro, but in vivo tests are still outstanding. This approach bears the risk of disrupting the physiological balance of the host factors which might lead to serious adverse effects.
Although research has brought forth a number of promising compounds, most of them still have to be validated in vivo and in clinical trials. The past epidemics caused by CHIKV demonstrated the impact a neglected or (re)emerging disease may have on a naïve population. Agents that have the potential to disable a population for a longer period with possible long-term sequelae, pose a vast threat to health and the economy. With no licensed vaccine and no specific antiviral treatment against CHIKF, research in the area of antiviral therapy is of utmost importance and the effort to find a specific treatment should be continued.
Author Contributions
Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by Bundeswehr STAN 59-2016-01 Biological evaluation of antiviral compounds and DZIF TI 07.003 MD Programme-Hucke.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Virology department of the Bundeswehr Institute of Microbiology for critical reading of the manuscript.
References
1. Silva LA, Dermody TS. Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J Clin Invest. (2017) 127:737–49. doi: 10.1172/JCI84417
2. Robinson MC. An epidemic of virus disease in Southern province, tanganyika territory, in 1952-53. I. clinical features. Trans Roy Soc Trop Med Hygiene. (1955) 49:28–32. doi: 10.1016/0035-9203(55)90080-8
3. Control) EECfDPa. Autochthonous Transmission of Chikungunya Virus in EU/EEA, 2007–2020. (2020). Available online at: https://www.ecdc.europa.eu/en/all-topics-z/chikungunya-virus-disease/surveillance-and-disease-data/autochthonous-transmission (accessed August 17, 2020).
4. Control EuCfDPa. Chikungunya Worldwide Overview: Geographical Distribution of Chikungunya Cases Reported Worldwide. (2020). Available online at: https://www.ecdc.europa.eu/en/chikungunya-monthly (accessed July 17 2020).
5. Renault P, Balleydier E, D'Ortenzio E, Baville M, Filleul L. Epidemiology of Chikungunya infection on Reunion Island, Mayotte, and neighboring countries. Med Mal Infect. (2012) 42:93–101. doi: 10.1016/j.medmal.2011.12.002
6. Ruiz Silva M, van der Ende-Metselaar H, Mulder HL, Smit JM, Rodenhuis-Zybert IA. Mechanism and role of MCP-1 upregulation upon chikungunya virus infection in human peripheral blood mononuclear cells. Sci Rep. (2016) 6:32288. doi: 10.1038/srep32288
7. Matusali G, Colavita F, Bordi L, Lalle E, Ippolito G, Capobianchi MR, et al. Tropism of the Chikungunya virus. Viruses. (2019) 11:175. doi: 10.3390/v11020175
8. CDC. Chikungunya Virus: CDC Centres for Disease Control and Prevention. (2020). Available online at: https://www.cdc.gov/chikungunya/geo/index.html (accessed September 19, 2019).
9. Pietila MK, Hellstrom K, Ahola T. Alphavirus polymerase and RNA replication. Virus Res. (2017) 234:44–57. doi: 10.1016/j.virusres.2017.01.007
10. Hucke FIL, Bestehorn-Willmann M, Bugert JJ. Prophylactic strategies to control Chikungunya virus infection. Virus Genes. (2020).
11. Bourjot M, Delang L, Nguyen VH, Neyts J, Gueritte F, Leyssen P, et al. Prostratin and 12-O-tetradecanoylphorbol 13-acetate are potent and selective inhibitors of Chikungunya virus replication. J Nat Prod. (2012) 75:2183–7. doi: 10.1021/np300637t
12. Bassetto M, De Burghgraeve T, Delang L, Massarotti A, Coluccia A, Zonta N, et al. Computer-aided identification, design and synthesis of a novel series of compounds with selective antiviral activity against chikungunya virus. Antiviral Res. (2013) 98:12–8. doi: 10.1016/j.antiviral.2013.01.002
13. Nothias-Scaglia LF, Pannecouque C, Renucci F, Delang L, Neyts J, Roussi F, et al. Antiviral activity of diterpene esters on chikungunya virus and HIV replication. J Nat Prod. (2015) 78:1277–83. doi: 10.1021/acs.jnatprod.5b00073
14. Gigante A, Gomez-SanJuan A, Delang L, Li C, Bueno O, Gamo AM, et al. Antiviral activity of [1,2,3]triazolo[4,5-d]pyrimidin-7(6H)-ones against chikungunya virus targeting the viral capping nsP1. Antiviral Res. (2017) 144:216–22. doi: 10.1016/j.antiviral.2017.06.003
15. Olivon F, Allard PM, Koval A, Righi D, Genta-Jouve G, Neyts J, et al. Bioactive natural products prioritization using massive multi-informational molecular networks. ACS Chem Biol. (2017) 12:2644–51. doi: 10.1021/acschembio.7b00413
16. Scholte FE, Tas A, Martina BE, Cordioli P, Narayanan K, Makino S, et al. Characterization of synthetic Chikungunya viruses based on the consensus sequence of recent E1-226V isolates. PLoS ONE. (2013) 8:e71047. doi: 10.1371/journal.pone.0071047
17. Allard PM, Martin MT, Dau ME, Leyssen P, Gueritte F, Litaudon M. Trigocherrin A, the first natural chlorinated daphnane diterpene orthoester from Trigonostemon cherrieri. Organ Lett. (2012) 14:342–5. doi: 10.1021/ol2030907
18. Ho YJ, Wang YM, Lu JW, Wu TY, Lin LI, Kuo SC, et al. Suramin inhibits chikungunya virus entry and transmission. PLoS ONE. (2015) 10:e0133511. doi: 10.1371/journal.pone.0133511
19. Hwu JR, Gupta NK, Tsay SC, Huang WC, Albulescu IC, Kovacikova K, et al. Bis(benzofuran-thiazolidinone)s and bis(benzofuran-thiazinanone)s as inhibiting agents for chikungunya virus. Antiviral Res. (2017) 146:96–101. doi: 10.1016/j.antiviral.2017.08.008
20. Feibelman KM, Fuller BP, Li L, LaBarbera DV, Geiss BJ. Identification of small molecule inhibitors of the Chikungunya virus nsP1 RNA capping enzyme. Antiviral Res. (2018) 154:124–31. doi: 10.1016/j.antiviral.2018.03.013
21. Gigante A, Canela MD, Delang L, Priego EM, Camarasa MJ, Querat G, et al. Identification of [1,2,3]triazolo[4,5-d]pyrimidin-7(6H)-ones as novel inhibitors of Chikungunya virus replication. J Med Chem. (2014) 57:4000–8. doi: 10.1021/jm401844c
22. Das PK, Puusepp L, Varghese FS, Utt A, Ahola T, Kananovich DG, et al. Design and validation of novel chikungunya virus protease inhibitors. Antimicrob Agents Chemother. (2016) 60:7382–95. doi: 10.1128/AAC.01421-16
23. Lucas-Hourani M, Lupan A, Despres P, Thoret S, Pamlard O, Dubois J, et al. A phenotypic assay to identify Chikungunya virus inhibitors targeting the nonstructural protein nsP2. J Biomol Screen. (2013) 18:172–9. doi: 10.1177/1087057112460091
24. Ferreira AC, Reis PA, de Freitas CS, Sacramento CQ, Villas Boas Hoelz L, Bastos MM, et al. Beyond members of the flaviviridae family, sofosbuvir also inhibits chikungunya virus replication. Antimicrob Agents Chemother. (2019) 63:e01389–18. doi: 10.1101/360305
25. Franco EJ, Rodriquez JL, Pomeroy JJ, Hanrahan KC, Brown AN. The effectiveness of antiviral agents with broad-spectrum activity against chikungunya virus varies between host cell lines. Antiviral Chem Chemother. (2018) 26:2040206618807580. doi: 10.1177/2040206618807580
26. Rothan HA, Bahrani H, Mohamed Z, Teoh TC, Shankar EM, Rahman NA, et al. A combination of doxycycline and ribavirin alleviated chikungunya infection. PLoS ONE. (2015) 10:e0126360. doi: 10.1371/journal.pone.0126360
27. Briolant S, Garin D, Scaramozzino N, Jouan A, Crance JM. In vitro inhibition of Chikungunya and Semliki Forest viruses replication by antiviral compounds: synergistic effect of interferon-alpha and ribavirin combination. Antiviral Res. (2004) 61:111–7. doi: 10.1016/j.antiviral.2003.09.005
28. Gallegos KM, Drusano GL, DZ DA, Brown AN. Chikungunya virus: in vitro response to combination therapy with ribavirin and interferon Alfa 2a. J Infect Dis. (2016) 214:1192–7. doi: 10.1093/infdis/jiw358
29. Kaur P, Thiruchelvan M, Lee RC, Chen H, Chen KC, Ng ML, et al. Inhibition of chikungunya virus replication by harringtonine, a novel antiviral that suppresses viral protein expression. Antimicrob Agents Chemother. (2013) 57:155–67. doi: 10.1128/AAC.01467-12
30. Ehteshami M, Tao S, Zandi K, Hsiao HM, Jiang Y, Hammond E, et al. Characterization of β-d-N(4)-Hydroxycytidine as a novel inhibitor of chikungunya virus. Antimicrob Agents Chemother. (2017) 61:16. doi: 10.1128/AAC.02395-16
31. Delang L, Segura Guerrero N, Tas A, Querat G, Pastorino B, Froeyen M, et al. Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J Antimicrob Chemother. (2014) 69:2770–84. doi: 10.1093/jac/dku209
32. Cruz DJ, Bonotto RM, Gomes RG, da Silva CT, Taniguchi JB, No JH, et al. Identification of novel compounds inhibiting chikungunya virus-induced cell death by high throughput screening of a kinase inhibitor library. PLoS Negl Trop Dis. (2013) 7:e2471. doi: 10.1371/journal.pntd.0002471
33. Abdelnabi R, Amrun SN, Ng LF, Leyssen P, Neyts J, Delang L. Protein kinases C as potential host targets for the inhibition of chikungunya virus replication. Antiviral Res. (2017) 139:79–87. doi: 10.1016/j.antiviral.2016.12.020
34. Corlay N, Delang L, Girard-Valenciennes E, Neyts J, Clerc P, Smadja J, et al. Tigliane diterpenes from Croton mauritianus as inhibitors of chikungunya virus replication. Fitoterapia. (2014) 97:87–91. doi: 10.1016/j.fitote.2014.05.015
35. Ho YJ, Liu FC, Yeh CT, Yang CM, Lin CC, Lin TY, et al. Micafungin is a novel anti-viral agent of chikungunya virus through multiple mechanisms. Antiviral Res. (2018) 159:134–42. doi: 10.1016/j.antiviral.2018.10.005
36. Varghese FS, Kaukinen P, Glasker S, Bespalov M, Hanski L, Wennerberg K, et al. Discovery of berberine, abamectin and ivermectin as antivirals against chikungunya and other alphaviruses. Antiviral Res. (2016) 126:117–24. doi: 10.1016/j.antiviral.2015.12.012
37. Hwu JR, Huang WC, Lin SY, Tan KT, Hu YC, Shieh FK, et al. Chikungunya virus inhibition by synthetic coumarin-guanosine conjugates. Eur J Med Chem. (2019) 166:136–43. doi: 10.1016/j.ejmech.2019.01.037
38. Abdelnabi R, Neyts J, Delang L. Chikungunya virus infections: time to act, time to treat. Curr Opin Virol. (2017) 24:25–30. doi: 10.1016/j.coviro.2017.03.016
39. Subudhi BB, Chattopadhyay S, Mishra P, Kumar A. Current strategies for inhibition of chikungunya infection. Viruses. (2018) 10:235. doi: 10.3390/v10050235
40. da Silva-Junior EF, Leoncini GO, Rodrigues EES, Aquino TM, Araujo-Junior JX. The medicinal chemistry of Chikungunya virus. Bioorg Med Chem. (2017) 25:4219–44. doi: 10.1016/j.bmc.2017.06.049
41. Bugert JJ, Hucke F, Zanetta P, Bassetto M, Brancale A. Antivirals in medical biodefense. Virus Genes. (2020) 56:150–67. doi: 10.1007/s11262-020-01737-5
42. Rainsford KD, Parke AL, Clifford-Rashotte M, Kean WF. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology. (2015) 23:231–69. doi: 10.1007/s10787-015-0239-y
43. Khan M, Santhosh SR, Tiwari M, Lakshmana Rao PV, Parida M. Assessment of in vitro prophylactic and therapeutic efficacy of chloroquine against Chikungunya virus in vero cells. Med Virol. (2010) 82:817–24. doi: 10.1002/jmv.21663
44. Bernard E, Solignat M, Gay B, Chazal N, Higgs S, Devaux C, et al. Endocytosis of chikungunya virus into mammalian cells: role of clathrin and early endosomal compartments. PLoS ONE. (2010) 5:e11479. doi: 10.1371/journal.pone.0011479
45. Allard PM, Leyssen P, Martin MT, Bourjot M, Dumontet V, Eydoux C, et al. Antiviral chlorinated daphnane diterpenoid orthoesters from the bark and wood of Trigonostemon cherrieri. Phytochemistry. (2012) 84:160–8. doi: 10.1016/j.phytochem.2012.07.023
46. Chopra A, Saluja M, Venugopalan A. Effectiveness of chloroquine and inflammatory cytokine response in patients with early persistent musculoskeletal pain and arthritis following chikungunya virus infection. Arthritis Rheumatol. (2014) 66:319–26. doi: 10.1002/art.38221
47. Roques P, Thiberville SD, Dupuis-Maguiraga L, Lum FM, Labadie K, Martinon F, et al. Paradoxical effect of chloroquine treatment in enhancing Chikungunya virus infection. Viruses. (2018) 10:268. doi: 10.3390/v10050268
48. Weber C, Sliva K, von Rhein C, Kummerer BM, Schnierle BS. The green tea catechin, epigallocatechin gallate inhibits chikungunya virus infection. Antiviral Res. (2015) 113:1–3. doi: 10.1016/j.antiviral.2014.11.001
49. Lu JW, Hsieh PS, Lin CC, Hu MK, Huang SM, Wang YM, et al. Synergistic effects of combination treatment using EGCG and suramin against the chikungunya virus. Biochem Biophys Res Commun. (2017) 491:595–602. doi: 10.1016/j.bbrc.2017.07.157
50. Basavannacharya C, Vasudevan SG. Suramin inhibits helicase activity of NS3 protein of dengue virus in a fluorescence-based high throughput assay format. Biochem Biophys Res Commun. (2014) 453:539–44. doi: 10.1016/j.bbrc.2014.09.113
51. Madsen C, Hooper I, Lundberg L, Shafagati N, Johnson A, Senina S, et al. Small molecule inhibitors of Ago2 decrease Venezuelan equine encephalitis virus replication. Antiviral Res. (2014) 112:26–37. doi: 10.1016/j.antiviral.2014.10.002
52. Albulescu IC, van Hoolwerff M, Wolters LA, Bottaro E, Nastruzzi C, Yang SC, et al. Suramin inhibits chikungunya virus replication through multiple mechanisms. Antiviral Res. (2015) 121:39–46. doi: 10.1016/j.antiviral.2015.06.013
53. Henß L, Beck S, Weidner T, Biedenkopf N, Sliva K, Weber C, et al. Suramin is a potent inhibitor of Chikungunya and Ebola virus cell entry. Virol J. (2016) 13:149. doi: 10.1186/s12985-016-0607-2
54. Chijioke CP, Umeh RE, Mbah AU, Nwonu P, Fleckenstein LL, Okonkwo PO. Clinical pharmacokinetics of suramin in patients with onchocerciasis. Eur J Clin Pharmacol. (1998) 54:249–51. doi: 10.1007/s002280050454
55. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature. (1998) 391:806–11. doi: 10.1038/35888
56. Bitko V, Barik S. Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild type negative-strand RNA viruses. BMC Microbiol. (2001) 1:34. doi: 10.1186/1471-2180-1-34
57. Dana H, Chalbatani GM, Mahmoodzadeh H, Karimloo R, Rezaiean O, Moradzadeh A, et al. Molecular mechanisms and biological functions of siRNA. Int J Biomed Sci. (2017) 13:48–57.
58. Presloid JB, Novella IS. RNA viruses and RNAi: quasispecies implications for viral escape. Viruses. (2015) 7:3226–40. doi: 10.3390/v7062768
59. Lam JK, Chow MY, Zhang Y, Leung SW. siRNA versus miRNA as therapeutics for gene silencing. Mol Ther Nucl Acids. (2015) 4:e252. doi: 10.1038/mtna.2015.23
60. Dash PK, Tiwari M, Santhosh SR, Parida M, Lakshmana Rao PV. RNA interference mediated inhibition of Chikungunya virus replication in mammalian cells. Biochem Biophys Res Commun. (2008) 376:718–22. doi: 10.1016/j.bbrc.2008.09.040
61. Parashar D, Paingankar MS, Kumar S, Gokhale MD, Sudeep AB, Shinde SB, et al. Administration of E2 and NS1 siRNAs inhibit chikungunya virus replication in vitro and protects mice infected with the virus. PLoS Negl Trop Dis. (2013) 7:e2405. doi: 10.1371/journal.pntd.0002405
62. Lam S, Chen KC, Ng MM, Chu JJ. Expression of plasmid-based shRNA against the E1 and nsP1 genes effectively silenced Chikungunya virus replication. PLoS ONE. (2012) 7:e46396. doi: 10.1371/journal.pone.0046396
63. Bhomia M, Sharma A, Gayen M, Gupta P, Maheshwari RK. Artificial microRNAs can effectively inhibit replication of Venezuelan equine encephalitis virus. Antiviral Res. (2013) 100:429–34. doi: 10.1016/j.antiviral.2013.08.010
64. Saha A, Bhagyawant SS, Parida M, Dash PK. Vector-delivered artificial miRNA effectively inhibited replication of Chikungunya virus. Antiviral Res. (2016) 134:42–9. doi: 10.1016/j.antiviral.2016.08.019
65. Das I, Basantray I, Mamidi P, Nayak TK, Pratheek BM, Chattopadhyay S, et al. Heat shock protein 90 positively regulates Chikungunya virus replication by stabilizing viral non-structural protein nsP2 during infection. PLoS ONE. (2014) 9:e100531. doi: 10.1371/journal.pone.0100531
66. Rathore AP, Haystead T, Das PK, Merits A, Ng ML, Vasudevan SG. Chikungunya virus nsP3 & nsP4 interacts with HSP-90 to promote virus replication: HSP-90 inhibitors reduce CHIKV infection and inflammation in vivo. Antiviral Res. (2014) 103:7–16. doi: 10.1016/j.antiviral.2013.12.010
67. Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. (2007) 448:39–43. doi: 10.1038/nature05901
68. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. (2002) 296:550–3. doi: 10.1126/science.1068999
69. Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Disc. (2010) 9:57–67. doi: 10.1038/nrd3010
70. Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. (2005) 23:457–62. doi: 10.1038/nbt1081
71. Lampio A, Kilpelainen I, Pesonen S, Karhi K, Auvinen P, Somerharju P, et al. Membrane binding mechanism of an RNA virus-capping enzyme. J Biol Chem. (2000) 275:37853–9. doi: 10.1074/jbc.M004865200
72. Ahola T, Kujala P, Tuittila M, Blom T, Laakkonen P, Hinkkanen A, et al. Effects of palmitoylation of replicase protein nsP1 on alphavirus infection. J Virol. (2000) 74:6725–33. doi: 10.1128/JVI.74.15.6725-6733.2000
73. Karo-Astover L, Sarova O, Merits A, Zusinaite E. The infection of mammalian and insect cells with SFV bearing nsP1 palmitoylation mutations. Virus Res. (2010) 153:277–87. doi: 10.1016/j.virusres.2010.08.019
74. Zhang N, Zhao H, Zhang L. Fatty acid synthase promotes the palmitoylation of chikungunya virus nsP1. J Virol. (2019) 93:e01747–18. doi: 10.1128/JVI.01747-18
75. Ahola T, Kaariainen L. Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc Natl Acad Sci USA. (1995) 92:507–11. doi: 10.1073/pnas.92.2.507
76. Ghosh A, Lima CD. Enzymology of RNA cap synthesis. Wiley Interdiscip Rev RNA. (2010) 1:152–72. doi: 10.1002/wrna.19
77. Lampio A, Ahola T, Darzynkiewicz E, Stepinski J, Jankowska-Anyszka M, Kaariainen L. Guanosine nucleotide analogs as inhibitors of alphavirus mRNA capping enzyme. Antiviral Res. (1999) 42:35–46. doi: 10.1016/S0166-3542(99)00011-X
78. Bullard-Feibelman KM, Fuller BP, Geiss BJ. A sensitive and robust high-throughput screening assay for inhibitors of the chikungunya virus nsp1 capping enzyme. PLoS ONE. (2016) 11:e0158923. doi: 10.1371/journal.pone.0158923
79. Delang L, Li C, Tas A, Querat G, Albulescu IC, De Burghgraeve T, et al. The viral capping enzyme nsP1: a novel target for the inhibition of chikungunya virus infection. Sci Rep. (2016) 6:31819. doi: 10.1038/srep31819
80. Jones PH, Maric M, Madison MN, Maury W, Roller RJ, Okeoma CM. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology. (2013) 438:37–49. doi: 10.1016/j.virol.2013.01.010
81. Wan JJ, Ooi YS, Kielian M. Mechanism of tetherin inhibition of alphavirus release. J Virol. (2019) 93(7). doi: 10.1128/JVI.02165-18
82. Fros JJ, van der Maten E, Vlak JM, Pijlman GP. The C-terminal domain of chikungunya virus nsP2 independently governs viral RNA replication, cytopathicity, and inhibition of interferon signaling. J Virol. (2013) 87:10394–400. doi: 10.1128/JVI.00884-13
83. Garmashova N, Gorchakov R, Volkova E, Paessler S, Frolova E, Frolov I. The old world and new world alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J Virol. (2007) 81:2472–84. doi: 10.1128/JVI.02073-06
84. Mathur K, Anand A, Dubey SK, Sanan-Mishra N, Bhatnagar RK, Sunil S. Analysis of chikungunya virus proteins reveals that non-structural proteins nsP2 and nsP3 exhibit RNA interference (RNAi) suppressor activity. Sci Rep. (2016) 6:38065. doi: 10.1038/srep38065
85. de Leuw P, Stephan C. Protease inhibitors for the treatment of hepatitis C virus infection. GMS Infect Dis. (2017) 5:34. doi: 10.3205/id000034
86. Jadav SS, Sinha BN, Hilgenfeld R, Pastorino B, de Lamballerie X, Jayaprakash V. Thiazolidone derivatives as inhibitors of chikungunya virus. Eur J Med Chem. (2015) 89:172–8. doi: 10.1016/j.ejmech.2014.10.042
87. Das PK, Merits A, Lulla A. Functional cross-talk between distant domains of chikungunya virus non-structural protein 2 is decisive for its RNA-modulating activity. J Biol Chem. (2014) 289:5635–53. doi: 10.1074/jbc.M113.503433
88. Matayoshi ED, Wang GT, Krafft GA, Erickson J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science. (1990) 247:954–8. doi: 10.1126/science.2106161
89. Rausalu K, Utt A, Quirin T, Varghese FS, Zusinaite E, Das PK, et al. Chikungunya virus infectivity, RNA replication and non-structural polyprotein processing depend on the nsP2 protease's active site cysteine residue. Sci Rep. (2016) 6:37124. doi: 10.1038/srep37124
90. Rupp JC, Sokoloski KJ, Gebhart NN, Hardy RW. Alphavirus RNA synthesis and non-structural protein functions. J Gen Virol. (2015) 96:2483–500. doi: 10.1099/jgv.0.000249
91. Chen MW, Tan YB, Zheng J, Zhao Y, Lim BT, Cornvik T, et al. Chikungunya virus nsP4 RNA-dependent RNA polymerase core domain displays detergent-sensitive primer extension and terminal adenylyltransferase activities. Antiviral Res. (2017) 143:38–47. doi: 10.1016/j.antiviral.2017.04.001
92. Zou G, Chen YL, Dong H, Lim CC, Yap LJ, Yau YH, et al. Functional analysis of two cavities in flavivirus NS5 polymerase. J Biol Chem. (2011) 286:14362–72. doi: 10.1074/jbc.M110.214189
93. Eyer L, Nencka R, de Clercq E, Seley-Radtke K, Ruzek D. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antiviral Chem Chemother. (2018) 26:2040206618761299. doi: 10.1177/2040206618761299
94. Slusarczyk M, Serpi M, Pertusati F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antiviral Chem Chemother. (2018) 26:2040206618775243. doi: 10.1177/2040206618775243
95. Feng JY. Addressing the selectivity and toxicity of antiviral nucleosides. Antiviral Chem Chemother. (2018) 26:2040206618758524. doi: 10.1177/2040206618758524
96. Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Disc. (2013) 12:447–64. doi: 10.1038/nrd4010
97. Varga A, Lionne C, Roy B. Intracellular metabolism of nucleoside/nucleotide analogues: a bottleneck to reach active drugs on HIV reverse transcriptase. Curr Drug Metab. (2016) 17:237–52. doi: 10.2174/1389200217666151210141903
98. De Clercq E. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates: the magic of the phosphonate bond. Biochem Pharmacol. (2011) 82:99–109. doi: 10.1016/j.bcp.2011.03.027
99. Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, et al. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem. (2001) 276:40847–57. doi: 10.1074/jbc.M106743200
100. McGuigan C, Sutton PW, Cahard D, Turner K, O'Leary G, Wang Y, et al. Synthesis, anti-human immunodeficiency virus activity and esterase lability of some novel carboxylic ester-modified phosphoramidate derivatives of stavudine (d4T). Antiviral Chem Chemother. (1998) 9:473–9. doi: 10.1177/095632029800900603
101. McGuigan C, Tsang HW, Sutton PW, De Clercq E, Balzarini J. Synthesis and anti-HIV activity of some novel chain-extended phosphoramidate derivatives of d4T (stavudine): esterase hydrolysis as a rapid predictive test for antiviral potency. Antiviral Chem Chemother. (1998) 9:109–15. doi: 10.1177/095632029800900202
102. Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowski JT, Robins RK. Broad-spectrum antiviral activity of virazole: 1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science. (1972) 177:705–6. doi: 10.1126/science.177.4050.705
103. Cooper AC, Banasiak NC, Allen PJ. Management and prevention strategies for respiratory syncytial virus (RSV) bronchiolitis in infants and young children: a review of evidence-based practice interventions. Pediatr Nursing. (2003) 29:452–6.
104. Mangia A, Santoro R, Minerva N, Ricci GL, Carretta V, Persico M, et al. Peginterferon alfa-2b and ribavirin for 12 vs. 24 weeks in HCV genotype 2 or 3. N Engl J Med. (2005). 352:2609–17. doi: 10.1056/NEJMoa042608
105. Huggins JW, Hsiang CM, Cosgriff TM, Guang MY, Smith JI, Wu ZO, et al. Prospective, double-blind, concurrent, placebo-controlled clinical trial of intravenous ribavirin therapy of hemorrhagic fever with renal syndrome. J Infect Dis. (1991) 164:1119–27. doi: 10.1093/infdis/164.6.1119
106. Ortac Ersoy E, Tanriover MD, Ocal S, Ozisik L, Inkaya C, Topeli A. Severe measles pneumonia in adults with respiratory failure: role of ribavirin and high-dose vitamin A. Clin Respir J. (2016) 10:673–5. doi: 10.1111/crj.12269
107. Huggins JW, Robins RK, Canonico PG. Synergistic antiviral effects of ribavirin and the C-nucleoside analogs tiazofurin and selenazofurin against togaviruses, bunyaviruses, and arenaviruses. Antimicrob Agents Chemother. (1984) 26:476–80. doi: 10.1128/AAC.26.4.476
108. Leyssen P, Balzarini J, De Clercq E, Neyts J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J Virol. (2005) 79:1943–7. doi: 10.1128/JVI.79.3.1943-1947.2005
109. Tam RC, Pai B, Bard J, Lim C, Averett DR, Phan UT, et al. Ribavirin polarizes human T cell responses towards a type 1 cytokine profile. J Hepatol. (1999) 30:376–82. doi: 10.1016/S0168-8278(99)80093-2
110. Scheidel LM, Stollar V. Mutations that confer resistance to mycophenolic acid and ribavirin on Sindbis virus map to the nonstructural protein nsP1. Virology. (1991) 181:490–9. doi: 10.1016/0042-6822(91)90881-B
111. Vo NV, Young KC, Lai MM. Mutagenic and inhibitory effects of ribavirin on hepatitis C virus RNA polymerase. Biochemistry. (2003) 42:10462–71. doi: 10.1021/bi0344681
112. Rozen-Gagnon K, Stapleford KA, Mongelli V, Blanc H, Failloux AB, Saleh MC, et al. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. (2014) 10:e1003877. doi: 10.1371/journal.ppat.1003877
113. Crotty S, Cameron CE, Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci USA. (2001) 98:6895–900. doi: 10.1073/pnas.111085598
114. Thomas E, Feld JJ, Li Q, Hu Z, Fried MW, Liang TJ. Ribavirin potentiates interferon action by augmenting interferon-stimulated gene induction in hepatitis C virus cell culture models. Hepatology. (2011) 53:32–41. doi: 10.1002/hep.23985
115. Ravichandran R, Manian M. Ribavirin therapy for Chikungunya arthritis. J Infect Dev Countries. (2008) 2:140–2. doi: 10.3855/T2.2.140
116. Mishra P, Kumar A, Mamidi P, Kumar S, Basantray I, Saswat T, et al. Inhibition of chikungunya virus replication by 1-[(2-Methylbenzimidazol-1-yl) methyl]-2-oxo-indolin-3-ylidene] amino] thiourea(MBZM-N-IBT). Sci Rep. (2016) 6:20122. doi: 10.1038/srep20122
117. Sung H, Chang M, Saab S. Management of hepatitis c antiviral therapy adverse effects. Curr Hepatitis Rep. (2011) 10:33–40. doi: 10.1007/s11901-010-0078-7
118. Pfeiffer JK, Kirkegaard K. Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in the replicon RNA. J Virol. (2005) 79:2346–55. doi: 10.1128/JVI.79.4.2346-2355.2005
119. Urakova N, Kuznetsova V, Crossman DK, Sokratian A, Guthrie DB, Kolykhalov AA, et al. beta-d-N (4)-Hydroxycytidine is a potent anti-alphavirus compound that induces a high level of mutations in the viral genome. J Virol. (2018) 92:e01965–17. doi: 10.1128/JVI.01965-17
120. Tejero H, Montero F, Nuno JC. Theories of lethal mutagenesis: from error catastrophe to lethal defection. Curr Topics Microbiol Immunol. (2016) 392:161–79. doi: 10.1007/82_2015_463
121. Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res. (2013) 100:446–54. doi: 10.1016/j.antiviral.2013.09.015
122. Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, et al. Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother. (2005) 49:981–6. doi: 10.1128/AAC.49.3.981-986.2005
123. Gowen BB, Wong MH, Jung KH, Sanders AB, Mendenhall M, Bailey KW, et al. In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob Agents Chemother. (2007) 51:3168–76. doi: 10.1128/AAC.00356-07
124. Julander JG, Smee DF, Morrey JD, Furuta Y. Effect of T-705 treatment on western equine encephalitis in a mouse model. Antiviral Res. (2009) 82:169–71. doi: 10.1016/j.antiviral.2009.02.201
125. Abdelnabi R, Jochmans D, Verbeken E, Neyts J, Delang L. Antiviral treatment efficiently inhibits chikungunya virus infection in the joints of mice during the acute but not during the chronic phase of the infection. Antiviral Res. (2018) 149:113–7. doi: 10.1016/j.antiviral.2017.09.016
126. Abdelnabi R, Morais ATS, Leyssen P, Imbert I, Beaucourt S, Blanc H, et al. Understanding the mechanism of the broad-spectrum antiviral activity of favipiravir (t-705): key role of the f1 motif of the viral polymerase. J Virol. (2017) 91:e00487-17. doi: 10.1128/JVI.00487-17
127. Mentre F, Taburet AM, Guedj J, Anglaret X, Keita S, de Lamballerie X, et al. Dose regimen of favipiravir for Ebola virus disease. Lancet Infect Dis. (2015) 15:150–1. doi: 10.1016/S1473-3099(14)71047-3
128. Arias A, Thorne L, Goodfellow I. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. eLife. (2014) 3:e03679. doi: 10.7554/eLife.03679
129. Sangawa H, Komeno T, Nishikawa H, Yoshida A, Takahashi K, Nomura N, et al. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob Agents Chemother. (2013) 57:5202–8. doi: 10.1128/AAC.00649-13
130. Iglesias NG, Filomatori CV, Gamarnik AV. The F1 motif of dengue virus polymerase NS5 is involved in promoter-dependent RNA synthesis. J Virol. (2011) 85:5745–56. doi: 10.1128/JVI.02343-10
131. Peersen OB. Picornaviral polymerase structure, function, and fidelity modulation. Virus Res. (2017) 234:4–20. doi: 10.1016/j.virusres.2017.01.026
132. Keating GM, Vaidya A. Sofosbuvir: first global approval. Drugs. (2014) 74:273–82. doi: 10.1007/s40265-014-0179-7
133. Bhatia HK, Singh H, Grewal N, Natt NK. Sofosbuvir: A novel treatment option for chronic hepatitis C infection. J Pharmacol Pharmacother. (2014) 5:278–84. doi: 10.4103/0976-500X.142464
134. de Freitas CS, Higa LM, Sacramento CQ, Ferreira AC, Reis PA, Delvecchio R, et al. Yellow fever virus is susceptible to sofosbuvir both in vitro and in vivo. PLoS Negl Trop Dis. (2019) 13:e0007072. doi: 10.1371/journal.pntd.0007072
135. Ferreira AC, Zaverucha-do-Valle C, Reis PA, Barbosa-Lima G, Vieira YR, Mattos M, et al. Sofosbuvir protects Zika virus-infected mice from mortality, preventing short- and long-term sequelae. Sci Rep. (2017) 7:9409. doi: 10.1038/s41598-017-09797-8
136. Bullard-Feibelman KM, Govero J, Zhu Z, Salazar V, Veselinovic M, Diamond MS, et al. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antiviral Res. (2017) 137:134–40. doi: 10.1016/j.antiviral.2016.11.023
137. Lam AM, Espiritu C, Bansal S, Micolochick Steuer HM, Niu C, Zennou V, et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob Agents Chemother. (2012) 56:3359–68. doi: 10.1128/AAC.00054-12
138. Gosio B. Ricerche batteriologiche e chimiche sulle alterazoni del mais. Rivista d'Igiene e Sanita Publica Ann. (1896) 7:825–68.
139. Allison AC, Eugui EM. Preferential suppression of lymphocyte proliferation by mycophenolic acid and predicted long-term effects of mycophenolate mofetil in transplantation. Transpl Proc. (1994) 26:3205–10.
140. Lowe JK, Brox L, Henderson JF. Consequences of inhibition of guanine nucleotide synthesis by mycophenolic acid and virazole. Cancer Res. (1977) 37:736–43.
141. Smee DF, Bray M, Huggins JW. Antiviral activity and mode of action studies of ribavirin and mycophenolic acid against orthopoxviruses in vitro. Antiviral Chem Chemother. (2001) 12:327–35. doi: 10.1177/095632020101200602
142. Diamond MS, Zachariah M, Harris E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA. Virology. (2002) 304:211–21. doi: 10.1006/viro.2002.1685
143. Khan M, Dhanwani R, Patro IK, Rao PV, Parida MM. Cellular IMPDH enzyme activity is a potential target for the inhibition of Chikungunya virus replication and virus induced apoptosis in cultured mammalian cells. Antiviral Res. (2011) 89:1–8. doi: 10.1016/j.antiviral.2010.10.009
144. Rashad AA, Neyts J, Leyssen P, Keller PA. A reassessment of mycophenolic acid as a lead compound for the development of inhibitors of chikungunya virus replication. Tetrahedron. (2018) 74:1294–306. doi: 10.1016/j.tet.2017.12.053
145. Siebert A, Prejs M, Cholewinski G, Dzierzbicka K. New analogues of mycophenolic acid. Mini Rev Med Chem. (2017) 17:734–45. doi: 10.2174/1389557516666161129160001
146. LaStarza MW, Lemm JA, Rice CM. Genetic analysis of the nsP3 region of Sindbis virus: evidence for roles in minus-strand and subgenomic RNA synthesis. J Virol. (1994) 68:5781–91. doi: 10.1128/JVI.68.9.5781-5791.1994
147. Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, et al. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol. (2012) 86:10873–9. doi: 10.1128/JVI.01506-12
148. Saul S, Ferguson M, Cordonin C, Fragkoudis R, Ool M, Tamberg N, et al. Differences in processing determinants of nonstructural polyprotein and in the sequence of nonstructural protein 3 affect neurovirulence of semliki forest virus. J Virol. (2015) 89:11030–45. doi: 10.1128/JVI.01186-15
149. Atkins GJ, Sheahan BJ. Molecular determinants of alphavirus neuropathogenesis in mice. J Gen Virol. (2016) 97:1283–96. doi: 10.1099/jgv.0.000467
150. Malet H, Coutard B, Jamal S, Dutartre H, Papageorgiou N, Neuvonen M, et al. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J Virol. (2009) 83:6534–45. doi: 10.1128/JVI.00189-09
151. Gorbalenya AE, Koonin EV, Lai MM. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. (1991) 288:201–5. doi: 10.1016/0014-5793(91)81034-6
152. Fehr AR, Jankevicius G, Ahel I, Perlman S. Viral macrodomains: unique mediators of viral replication and pathogenesis. Trends Microbiol. (2018) 26:598–610. doi: 10.1016/j.tim.2017.11.011
153. Eckei L, Krieg S, Butepage M, Lehmann A, Gross A, Lippok B, et al. The conserved macrodomains of the non-structural proteins of Chikungunya virus and other pathogenic positive strand RNA viruses function as mono-ADP-ribosylhydrolases. Sci Rep. (2017) 7:41746. doi: 10.1038/srep41746
154. McPherson RL, Abraham R, Sreekumar E, Ong SE, Cheng SJ, Baxter VK, et al. ADP-ribosylhydrolase activity of Chikungunya virus macrodomain is critical for virus replication and virulence. Proc Natl Acad Sci USA. (2017) 114:1666–71. doi: 10.1073/pnas.1621485114
155. Nguyen PT, Yu H, Keller PA. Discovery of in silico hits targeting the nsP3 macro domain of chikungunya virus. J Mol Model. (2014) 20:2216. doi: 10.1007/s00894-014-2216-6
156. Varjak M, Zusinaite E, Merits A. Novel functions of the alphavirus nonstructural protein nsP3 C-terminal region. J Virol. (2010) 84:2352–64. doi: 10.1128/JVI.01540-09
157. Panas MD, Schulte T, Thaa B, Sandalova T, Kedersha N, Achour A, et al. Viral and cellular proteins containing FGDF motifs bind G3BP to block stress granule formation. PLoS Pathog. (2015) 11:e1004659. doi: 10.1371/journal.ppat.1004659
158. Gotte B, Liu L, McInerney GM. The enigmatic alphavirus non-structural protein 3 (nsp3) revealing its secrets at last. Viruses. (2018) 10:105. doi: 10.3390/v10030105
159. Kim C, Kang H, Kim DE, Song JH, Choi M, Kang M, et al. Antiviral activity of micafungin against enterovirus 71. Virol J. (2016) 13:99. doi: 10.1186/s12985-016-0557-8
160. Kim DY, Reynaud JM, Rasalouskaya A, Akhrymuk I, Mobley JA, Frolov I, et al. New world and old world alphaviruses have evolved to exploit different components of stress granules, fxr and g3bp proteins, for assembly of viral replication complexes. PLoS Pathog. (2016) 12:e1005810. doi: 10.1371/journal.ppat.1005810
161. Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, et al. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell. (2012) 23:4701–12. doi: 10.1091/mbc.e12-08-0619
162. Scholte FE, Tas A, Albulescu IC, Zusinaite E, Merits A, Snijder EJ, et al. Stress granule components G3BP1 and G3BP2 play a proviral role early in Chikungunya virus replication. J Virol. (2015) 89:4457–69. doi: 10.1128/JVI.03612-14
163. Goertz GP, Lingemann M, Geertsema C, Abma-Henkens MHC, Vogels CBF, Koenraadt CJM, et al. Conserved motifs in the hypervariable domain of chikungunya virus nsP3 required for transmission by aedes aegypti mosquitoes. PLoS Negl Trop Dis. (2018) 12:e0006958. doi: 10.1371/journal.pntd.0006958
164. Saxton-Shaw KD, Ledermann JP, Borland EM, Stovall JL, Mossel EC, Singh AJ, et al. O'nyong nyong virus molecular determinants of unique vector specificity reside in non-structural protein 3. PLoS Negl Trop Dis. (2013) 7:e1931. doi: 10.1371/journal.pntd.0001931
165. Lastarza MW, Grakoui A, Rice CM. Deletion and duplication mutations in the C-terminal nonconserved region of Sindbis virus nsP3: effects on phosphorylation and on virus replication in vertebrate and invertebrate cells. Virology. (1994) 202:224–32. doi: 10.1006/viro.1994.1338
166. Jadav SS, Sinha BN, Hilgenfeld R, Jayaprakash V. Computer-aided structure based drug design approaches for the discovery of new anti-CHIKV agents. Curr Comput Aid Drug Design. (2017) 13:346–61. doi: 10.2174/1573409913666170309145308
167. Alvandi F, Kwitkowski VE, Ko CW, Rothmann MD, Ricci S, Saber H, et al. U.S. Food and drug administration approval summary: omacetaxine mepesuccinate as treatment for chronic myeloid leukemia. Oncologist. (2014) 19:94–9. doi: 10.1634/theoncologist.2013-0077
168. Abcam. Abcam safety data sheet of harringtonine [Abcam safety data sheet of harringtonine]. Available online at: https://www.abcam.com/ps/Products/141/ab141941/Msds/ab141941_CLP1_EN.pdf (accessed October 22, 2019).
169. Wong KZ, Chu J. The Interplay of Viral and Host Factors in Chikungunya Virus Infection: Targets for Antiviral Strategies. Viruses. (2018) 10:294. doi: 10.3390/v10060294 (accessed August 13, 2019)
170. Seo JY, Yaneva R, Cresswell P. Viperin: a multifunctional, interferon-inducible protein that regulates virus replication. Cell Host microbe. (2011) 10:534–9. doi: 10.1016/j.chom.2011.11.004
171. Teng TS, Foo SS, Simamarta D, Lum FM, Teo TH, Lulla A, et al. Viperin restricts chikungunya virus replication and pathology. J Clin Invest. (2012) 122:4447–60. doi: 10.1172/JCI63120
172. Carissimo G, Teo T-H, Chan Y-H, Lee CY-P, Lee B, Torres-Ruesta A, et al. Viperin controls chikungunya virus-specific pathogenic T cell IFNγ Th1 stimulation in mice. Life Sci Alliance. (2019) 2:e201900298. doi: 10.26508/lsa.201900298
173. Geller R, Taguwa S, Frydman J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim Biophys Acta. (2012) 1823:698–706. doi: 10.1016/j.bbamcr.2011.11.007
174. Geller R, Vignuzzi M, Andino R, Frydman J. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. (2007) 21:195–205. doi: 10.1101/gad.1505307
175. Wang Y, Jin F, Wang R, Li F, Wu Y, Kitazato K, et al. HSP90: a promising broad-spectrum antiviral drug target. Arch Virol. (2017) 162:3269–82. doi: 10.1007/s00705-017-3511-1
176. Hoter A, El-Sabban ME, Naim HY. The HSP90 family: structure, regulation, function, and implications in health and disease. Int J Mol Sci. (2018) 19:2560. doi: 10.3390/ijms19092560
177. Jhaveri K, Modi S. Ganetespib: research and clinical development. OncoTargets Ther. (2015) 8:1849–58. doi: 10.2147/OTT.S65804
178. Lillsunde KE, Tomasic T, Kikelj D, Tammela P. Marine alkaloid oroidin analogues with antiviral potential: A novel class of synthetic compounds targeting the cellular chaperone Hsp90. Chem Biol Drug Design. (2017) 90:1147–54. doi: 10.1111/cbdd.13034
179. Li SF, Gong MJ, Zhao FR, Shao JJ, Xie YL, Zhang YG, et al. Type I interferons: distinct biological activities and current applications for viral infection. Cell Physiol Biochem. (2018) 51:2377–96. doi: 10.1159/000495897
180. Hoffmann HH, Schneider WM, Rice CM. Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol. (2015) 36:124–38. doi: 10.1016/j.it.2015.01.004
181. Lin FC, Young HA. Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev. (2014) 25:369–76. doi: 10.1016/j.cytogfr.2014.07.015
182. Zhang Y, Burke CW, Ryman KD, Klimstra WB. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J Virol. (2007) 81:11246–55. doi: 10.1128/JVI.01282-07
183. Couderc T, Chretien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, et al. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. (2008) 4:e29. doi: 10.1371/journal.ppat.0040029
184. Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, et al. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. J Virol. (2012) 86:9888–98. doi: 10.1128/JVI.00956-12
185. Brehin AC, Casademont I, Frenkiel MP, Julier C, Sakuntabhai A, Despres P. The large form of human 2',5′-Oligoadenylate Synthetase (OAS3) exerts antiviral effect against Chikungunya virus. Virology. (2009) 384:216–22. doi: 10.1016/j.virol.2008.10.021
186. Ooi YS, Dube M, Kielian M. BST2/tetherin inhibition of alphavirus exit. Viruses. (2015) 7:2147–67. doi: 10.3390/v7042147
187. Cocka LJ, Bates P. Identification of alternatively translated Tetherin isoforms with differing antiviral and signaling activities. PLoS Pathog. (2012) 8:e1002931. doi: 10.1371/journal.ppat.1002931
188. Lee SK, Cui B, Mehta RR, Kinghorn AD, Pezzuto JM. Cytostatic mechanism and antitumor potential of novel 1H-cyclopenta[b]benzofuran lignans isolated from Aglaia elliptica. Chem Biol Inter. (1998) 115:215–28. doi: 10.1016/S0009-2797(98)00073-8
189. Biedenkopf N, Lange-Grunweller K, Schulte FW, Weisser A, Muller C, Becker D, et al. The natural compound silvestrol is a potent inhibitor of Ebola virus replication. Antiviral Res. (2017) 137:76–81. doi: 10.1016/j.antiviral.2016.11.011
190. Muller C, Schulte FW, Lange-Grunweller K, Obermann W, Madhugiri R, Pleschka S, et al. Broad-spectrum antiviral activity of the eIF4A inhibitor silvestrol against corona- and picornaviruses. Antiviral Res. (2018) 150:123–9. doi: 10.1016/j.antiviral.2017.12.010
191. Henß L, Scholz T, Grunweller A, Schnierle BS. Silvestrol inhibits chikungunya virus replication. Viruses. (2018) 10:592. doi: 10.3390/v10110592
192. Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to (2014). J Nat Prod. (2016) 79:629–61. doi: 10.1021/acs.jnatprod.5b01055
193. Feher M, Schmidt JM. Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J Chem Inform Comput Sci. (2003) 43:218–27. doi: 10.1021/ci0200467
194. Mathur S, Hoskins C. Drug development: Lessons from nature. Biomed Rep. (2017) 6:612–4. doi: 10.3892/br.2017.909
195. Leyssen P, Smadja J, Rasoanaivo P, Gurib-Fakim A, Mahomoodally MF, Canard B, et al. Biodiversity as a source of potent and selective inhibitors of chikungunya virus replication. In: Gurib-Fakim A, editor. Novel Plant Bioresources: Applications in Food, Medicine and Cosmetics. 1 edition. Hoboken, NJ: John Wiley & Sons, Ltd. (2014). p. 151–61. doi: 10.1002/9781118460566.ch11
196. Vasas A, Hohmann J. Euphorbia diterpenes: isolation, structure, biological activity, and synthesis (2008-2012). Chem Rev. (2014) 114:8579–2. doi: 10.1021/cr400541j
197. Goel G, Makkar HP, Francis G, Becker K. Phorbol esters: structure, biological activity, and toxicity in animals. Int J. Toxicol. (2007) 26:279–88. doi: 10.1080/10915810701464641
198. Remy S, Litaudon M. Macrocyclic diterpenoids from euphorbiaceae as a source of potent and selective inhibitors of chikungunya virus replication. Molecules. (2019) 24:e24122336. doi: 10.3390/molecules24122336
199. Newton AC. Protein kinase C as a tumor suppressor. Semin Cancer Biol. (2018) 48:18–26. doi: 10.1016/j.semcancer.2017.04.017
200. Bourjot M, Leyssen P, Neyts J, Dumontet V, Litaudon M. Trigocherrierin A, a potent inhibitor of chikungunya virus replication. Molecules. (2014) 19:3617–27. doi: 10.3390/molecules19033617
201. Nothias-Scaglia LF, Retailleau P, Paolini J, Pannecouque C, Neyts J, Dumontet V, et al. Jatrophane diterpenes as inhibitors of chikungunya virus replication: structure-activity relationship and discovery of a potent lead. J Nat Prod. (2014) 77:1505–12. doi: 10.1021/np500271u
202. Wang QJ, Bhattacharyya D, Garfield S, Nacro K, Marquez VE, Blumberg PM. Differential localization of protein kinase C delta by phorbol esters and related compounds using a fusion protein with green fluorescent protein. J Biol Chem. (1999) 274:37233–9. doi: 10.1074/jbc.274.52.37233
203. Wang QJ, Fang TW, Fenick D, Garfield S, Bienfait B, Marquez VE, et al. The lipophilicity of phorbol esters as a critical factor in determining the pattern of translocation of protein kinase C delta fused to green fluorescent protein. J Biol Chem. (2000) 275:12136–46. doi: 10.1074/jbc.275.16.12136
204. European Medicines Agency. EPAR Summary for the Public; EMA/598388/2011: European Medicines Agency; (2011). Available online at: https://www.ema.europa.eu/en/documents/overview/mycamine-epar-summary-public_en.pdf (accessed May 29, 2019).
205. European Medicines Agency. Assessment Report for Mycamine: European Medicines Agency;. (2008). Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000734/WC500031079.pdf (accessed June 2, 2019).
206. Douglas CM. Fungal beta(1,3)-D-glucan synthesis. Med Mycol. (2001) 39(Suppl 1):55–66. doi: 10.1080/mmy.39.1.55.66
207. Dong S, Kang S, Dimopoulos G. Identification of anti-flaviviral drugs with mosquitocidal and anti-Zika virus activity in Aedes aegypti. PLoS Negl Trop Dis. (2019) 13:e0007681. doi: 10.1371/journal.pntd.0007681
208. Mastrangelo E, Pezzullo M, De Burghgraeve T, Kaptein S, Pastorino B, Dallmeier K, et al. Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: new prospects for an old drug. J Antimicrob Chemother. (2012) 67:1884–94. doi: 10.1093/jac/dks147
209. Ortiz LM, Lombardi P, Tillhon M, Scovassi AI. Berberine, an epiphany against cancer. Molecules. (2014) 19:12349–67. doi: 10.3390/molecules190812349
210. Varghese FS, Thaa B, Amrun SN, Simarmata D, Rausalu K, Nyman TA, et al. The antiviral alkaloid berberine reduces chikungunya virus-induced mitogen-activated protein kinase signaling. J. Virol. (2016) 90:9743–57. doi: 10.1128/JVI.01382-16
211. Srikrishna D, Godugu C, Dubey PK. A review on pharmacological properties of coumarins. Mini Rev Med Chem. (2018) 18:113–41. doi: 10.2174/1389557516666160801094919
212. Hwu JR, Kapoor M, Tsay SC, Lin CC, Hwang KC, Horng JC, et al. Benzouracil-coumarin-arene conjugates as inhibiting agents for chikungunya virus. Antiviral Res. (2015) 118:103–9. doi: 10.1016/j.antiviral.2015.03.013
213. Qiu L, Patterson SE, Bonnac LF, Geraghty RJ. Nucleobases and corresponding nucleosides display potent antiviral activities against dengue virus possibly through viral lethal mutagenesis. PLoS Negl Trop Dis. (2018) 12:e0006421. doi: 10.1371/journal.pntd.0006421
214. Crance JM, Scaramozzino N, Jouan A, Garin D. Interferon, ribavirin, 6-azauridine and glycyrrhizin: antiviral compounds active against pathogenic flaviviruses. Antiviral Res. (2003) 58:73–9. doi: 10.1016/S0166-3542(02)00185-7
Keywords: antiviral design, CHIKV therapy, direct antiviral action, host-targeting antiviral, comparison of in vitro efficacies, favipiravir, ribavirin
Citation: Hucke FIL and Bugert JJ (2020) Current and Promising Antivirals Against Chikungunya Virus. Front. Public Health 8:618624. doi: 10.3389/fpubh.2020.618624
Received: 17 October 2020; Accepted: 19 November 2020;
Published: 15 December 2020.
Edited by:
Roger Hewson, Public Health England, United KingdomReviewed by:
Pouya Hassandarvish, University of Malaya, MalaysiaAdam Taylor, Griffith University, Australia
Copyright © 2020 Hucke and Bugert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friederike I. L. Hucke, ZnJfaHVja2VAZnJlZW5ldC5kZQ==
orcid.org/0000-0001-5396-723X