 Zhen-Tong Wei1†
Zhen-Tong Wei1† Gui-Lian Yang
Gui-Lian Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Public Health , 08 January 2021
Sec. Infectious Diseases: Epidemiology and Prevention
Volume 8 - 2020 | https://doi.org/10.3389/fpubh.2020.587298
Persistent infection with the carcinogenic human papillomavirus (HPV) is a prerequisite for the progression of cervical lesions and cancer. A growing body of research has focused on the functional role of the vaginal microbiota in the persistence of HPV infection. Understanding the microbial composition and structure in women with high-risk (hr)-HPV infection may help reveal associations between the vaginal microbiota and HPV infection, and identify potential biomarkers. Our study investigated the vaginal microbial community in women with and without hr-HPV infection, by using 16s rRNA gene sequencing. We found that microbial perturbations occurred in the early phase of hr-HPV infection. Lactobacillus and Sporolactobacillus were decreased, while bacteria related to bacterial vaginosis (BV), such as Gardnerella, Prevotella, Dialister, Slackia, Actinomyces, Porphyromonas, Peptoniphilus, Anaerococcus, Peptostreptococcus, Streptococcus, Ureaplasma, Megasphaera, and Mycoplasma were increased. Our results could offer insights into the correlations between hr-HPV and the vaginal microbiota in the early infection period, and provide indications that the predominance of some BV-associated bacteria during hr-HPV infection may increase the risk for cervical neoplasia.
Globally, cervical carcinoma is the fourth most common type of cancer among women, after breast cancer, colorectal cancer, and lung cancer (1). Persistent infection with the oncogenic human papillomavirus (HPV) is the main factor responsible for the progression of cervical lesions and cervical cancer (2, 3). HPV is a non-enveloped DNA virus that infects human epithelial tissues. In approximately 10% of women infected with HPV, the virus is not completely cleared from the body, leading to persistent high-risk HPV (hr-HPV) infection (4). Infection with an hr-HPV genotype is considered one of the most important causes of cervical cancer. For instance, HPV16 and HPV18, two hr-HPVs, are responsible for approximately 55 and 15% of all cases of cervical cancer, respectively. HPV16 infection is mainly related to the occurrence of cervical squamous cell carcinoma (5, 6). In addition, douching practices, sexually transmitted infections, vulvovaginitis, vulvitis, and bacterial vaginosis (BV) perturb the vaginal microenvironment and likely serve as cofactors for persistent hr-HPV infection (7, 8). Other factors correlated with HPV persistence include age, smoking, immunodeficiency, contraceptive use, and infection with Chlamydia trachomatis (9, 10).
Highly diverse microbial communities at specific sites in the human body play vital roles in physiology, immunity, nutrition, and development, and are generally considered a sign of health (11, 12). Of these microbial communities, the gut microbiota is the best characterized, and has been implicated in certain diseases as well as in carcinogenesis (13). The Human Microbiome Project investigated whether the vaginal microbiome was correlated with human health and disease (14). Lactobacillus spp. are commensal bacteria in the vaginal microbiome that can help defend against sexually transmitted infections and other pathogens by maintaining a hostile pH, producing species-specific metabolites and bacteriocins, adhering to mucous membranes, and disrupting biofilms (15, 16). Most previous studies on vaginal microbiota focused on analyzing the compositions of the vaginal microbiota in healthy women and women with BV (17, 18). Recently, however, it was found that the vaginal microbiota was correlated with HIV infection (19–21). The precise associations between the vaginal microbiota and HPV infection, and the key bacterial population changes relevant to the development of cervical carcinoma remain unclear.
The majority of studies on vaginal microbiota have relied on cultivation-dependent methods to assess microbial community composition (22). Recently, however, sequencing technologies have been used to investigate the human microbiome at various body sites, and these methods enable a more direct and rapid analysis of microbial taxa (23). In the present study, we used 16S rRNA gene sequencing to comparatively analyze the vaginal microbiota in 30 HPV-negative women and 30 HPV-positive women with normal cytology in the northeast of China, in order to reveal potential biomarkers in the early phase of HPV infection.
We recruited a total of 60 women who underwent routine gynecological examination in the Department of Oncologic Gynecology, the First Hospital of Jilin University, Changchun, China, between September and December 2018. The exclusion criteria included current pregnancy, menopause, sexual intercourse or vaginal lavage within 3 days, use of antibiotics or vaginal antimicrobials within 2 weeks, HIV infection, and autoimmune disorders. All subjects were interviewed to collect the following information: age, ethnicity, smoking habits, parity, and contraceptive use. In addition, all subjects underwent cervical ThinPrep cytology tests, regardless of HPV infection status, and were found to have normal cytology without cervical intraepithelial neoplasia (CIN). All participants provided written informed consent for the use and publication of their data prior to their enrollment into the study. The study protocol was approved by the ethics committee of the First Hospital of Jilin University, and the study was conducted in accordance with the principles of the Declaration of Helsinki.
Three swab specimens were collected from each participant during a routine pelvic examination. The first specimen was collected from the cervix with a Cytobrush and used for cervical ThinPrep cytology testing (Hologic, Marlborough, MA, USA). The second specimen was also collected from the cervix and used for hr-HPV DNA detection via the Hybrid Capture II (HC2) test (Qiagen, Gaithersburg, MD, USA), according to the manufacturer's guidelines. The HC2 test is a chemiluminescence-based assay that involves nucleic acid hybridization and signal amplification. This test can qualitatively detect 13 types of hr-HPV DNA, namely, 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 68. The third specimen was collected from the mid-vagina, and was immediately frozen to−80°C and subsequently used for bacterial DNA extraction and sequencing.
We extracted genomic DNA from a total of 60 cervical samples by using E.Z.N.A.® DNA extraction kits (Omega Bio-Tek, Norcross, GA, USA), according to the manufacturer's instructions. Next, we verified the quality of the extracted DNA by performing 1% agarose gel electrophoresis, and measured its concentration using NanoDrop spectrophotometry. The extracted DNA was then frozen to −20°C and stored until further analysis. We then performed high-throughput sequencing of the V3–V4 hypervariable regions of the bacterial 16S rRNA gene on the Illumina MiSeq PE300 sequencing platform (Illumina Inc., San Diego, CA, USA). Amplification of the V3–V4 regions was performed with the following pair of universal primers, which contained a set of 8-nucleotide barcode sequences unique to each sample: forward, 338F (5′-ACTCCTACGGGAGGCAGCAG-3′); and reverse, 806R (5′-GACTACHVGGGTWTCTAAT-3′). The polymerase chain reaction (PCR) protocol was as follows: 5 min at 95°C, 25 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C, and final extension for 10 min at 72°C. PCR was performed in a 25-μL mixture containing 2.5 μL of 10 × Pyrobest buffer, 0.4 U Pyrobest DNA Polymerase (TaKaRa, Shiga, Japan), 2 μL of 2.5 mM dNTPs, 15 ng template DNA, and 1 μL of each primer (10 μM). All PCR assays were performed in triplicate. The resulting amplicons were subjected to 2% agarose gel electrophoresis, purified (AxyPrep DNA Gel Extraction Kit, Axygen Biosciences, Union City, CA, USA), and then quantified (QuantiFluor™ -ST, Promega, Madison, WI, USA), according to the manufacturers' instructions. Finally, the purified amplicons were pooled in an equimolar ratio and paired-end sequenced (2 × 300) on the Illumina MiSeq platform.
We used QIIME (Quantitative Insights Into Microbial Ecology, version 1.2.1; http://qiime.org/) to extract high-quality sequences from the raw sequences obtained using MiSeq sequencing. Sequence selection was based on the length and quality of the sequences, tags, and primers. The set of unique sequences thus identified was categorized into operational taxonomic units (OTUs; threshold, 97% identity) by using UCLUST (version 1.2.22; https://drive5.com/usearch/manual/uclust_algo.html). Using Usearch (version 8.0.1623; https://www.drive5.com/usearch/), we identified and removed chimeric sequences. Taxonomic identification of the remaining 16S rRNA gene sequences was performed using the Silva119 16S rRNA database and UCLUST software (confidence threshold, 90%).
We evaluated the Chao1 and observed species indices, which estimate microbial richness, and the Shannon index, which measures species biodiversity (24, 25). These three indices were used to calculate the alpha diversity of the samples. Principal component analysis of OTUs was performed using the R software (version 3.6.0, Revolutions; https://www.r-project.org/), and co-occurrence network analysis was performed using the R packages “igraph” and “psych.” To identify significant differences in microbial composition, we performed linear discriminant analysis effect size (LEfSe; http://huttenhower.sph.harvard.edu/galaxy/) with the thresholds P < 0.05 and effect size > 2. Clusters of Orthologous Groups (COG) categories were assigned by performing BLAST searches to identify the closest matching sequence in the Search Tool for the Retrieval of Interacting Genes database. The raw reads were deposited into Sequence Read Archive (NCBI accession number: SRP212297).
Experimental comparisons were performed using analysis of variance and the Tukey honestly significant difference test. The results are presented as mean and standard error of the mean. P < 0.05 was considered to indicate statistically significant differences.
Of the 60 women, 30 tested negative for hr-HPV infection, while the remaining 30 tested positive for hr-HPV infection. The average ages of HPV-positive and HPV-negative women were 36.70 ± 6.30 and 34.13 ± 6.43 years, respectively. All cytological tests were normal, and no CIN lesions were detected in any of the women from either group. The HPV-positive and HPV-negative groups did not differ in terms of age, parity, ethnicity, cigarette smoking, or use of hormonal contraceptives. The demographic characteristics of each group are presented in Table 1.
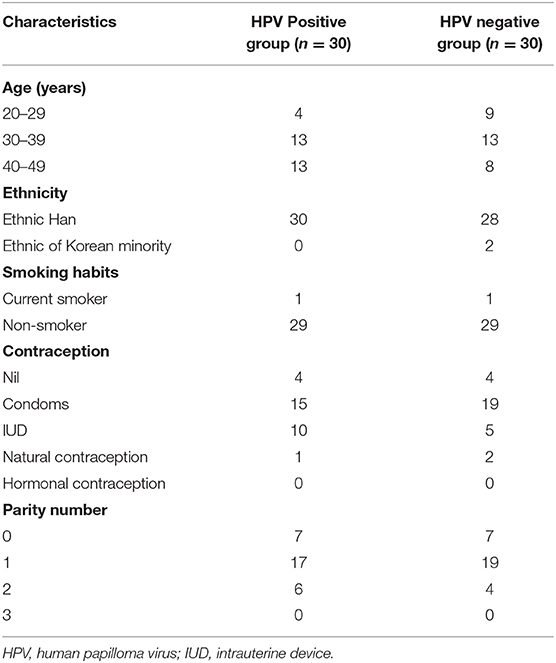
Table 1. Demographic characteristics of the HPV-positive and HPV-negative groups.
Millions of raw reads were obtained after 16S rRNA gene sequencing. After MiSeq sequencing analysis, 2,874,102 sequences were identified from the 60 samples (average sequence length, 427.8 bp; Good coverage index, >99%). In total, 138 OTUs were identified in the two study groups. The OTU rank abundance and rarefaction curves of all the samples indicated that the data sampling was sufficient, and the sequencing depth was adequate; the 16S rRNA gene sequences covered almost all microbial communities (Figures 1A,B).
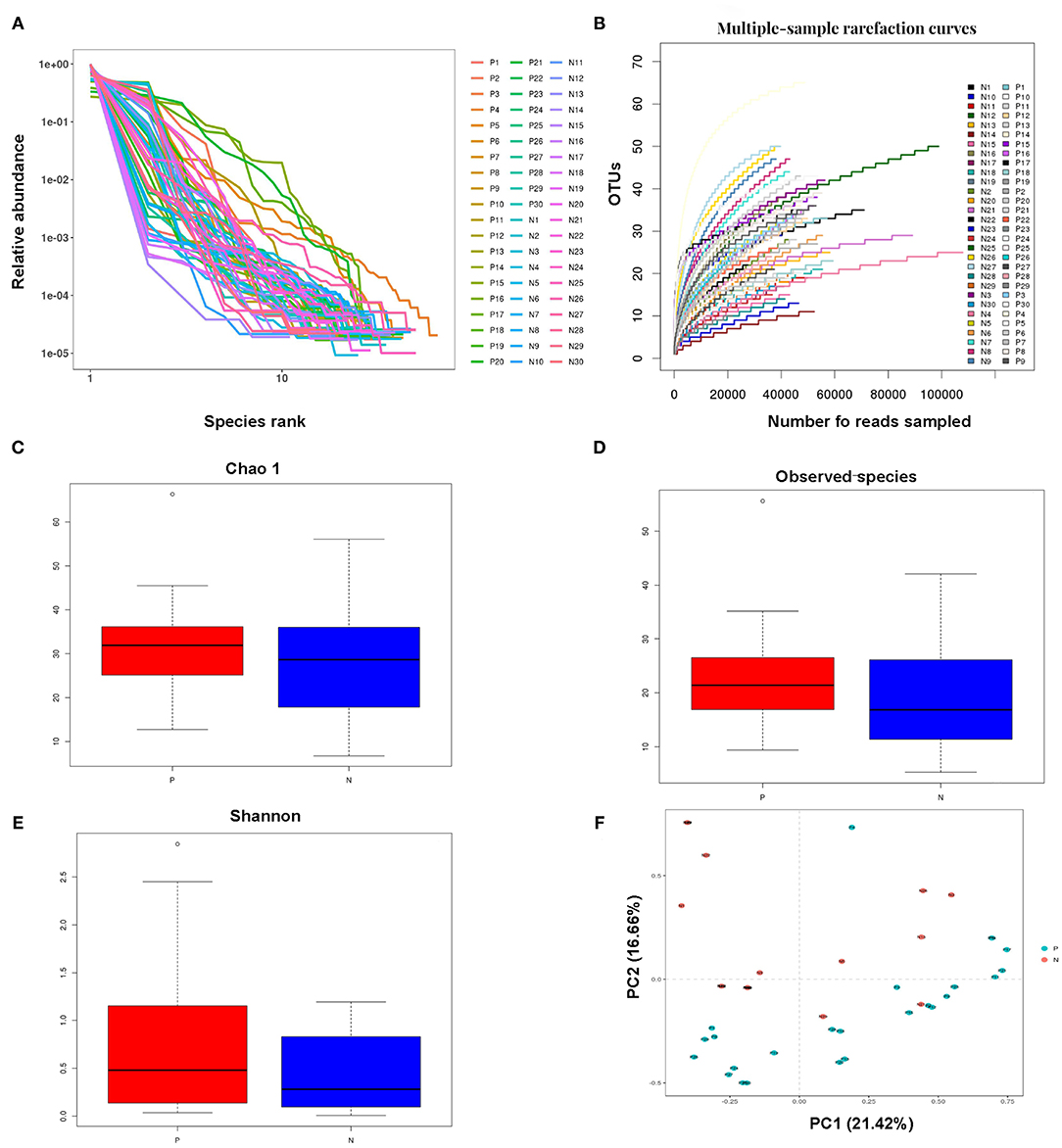
Figure 1. Analysis of the vaginal microbial compositions in the HPV-positive and HPV-negative groups. (A,B) Species accumulation curves (A) and rarefaction curves (B) for OTUs obtained from 60 cervical samples. (C–E) Analysis of alpha-diversity. The Chao1 (C) and observed species (D) indices were used as richness estimators, while the Shannon index (E) was used as a diversity estimator. (F) Principal coordinates analysis of the structure of the vaginal microbiota. HPV, human papilloma virus; OTU, operational taxonomic unit.
The Chao1, observed species, and Shannon indices were all higher, but not significantly so, in the HPV-positive group than in the HPV-negative group. The alpha diversity of the vaginal microbiota did not significantly differ between the HPV-negative and HPV-positive groups. These results indicated that microbial shifts occurred in the vagina in women with HPV infection (Figures 1C–E).
Principal component analysis indicated that the vaginal microbial composition slightly differed between the HPV-negative and HPV-positive groups. Principal components 1 and 2 explained 21.42 and 16.66% of the variance between the two study groups, respectively. The impact of HPV infection on the vaginal microbial composition was generally consistent with the results of the alpha diversity analysis (Figure 1F).
Firmicutes, Actinobacteria, Bacteroidetes, and Tenericutes were the dominant phyla in the vaginal microbiota in both the study groups, with relative abundances of 96.71, 2.82, 0.18, and 0.06%, respectively, in the HPV-negative group, and 88.99, 6.87, 2.73, and 1.12%, respectively, in the HPV-positive group. At the genus level, Lactobacillus (96.57%), Atopobium (2.1%), Gardnerella (0.72%), Prevotella (0.04%), and Mycoplasma (0.0001%) dominated the microbial community in the HPV-negative group, while in the HPV-positive group, Lactobacillus (86.34%), Gardnerella (6.42%), Prevotella (2.44%), Megasphaera (1.21%), Mycoplasma (0.80%), and Atopobium (0.37%) were the dominant microflora (Figures 2A,B).
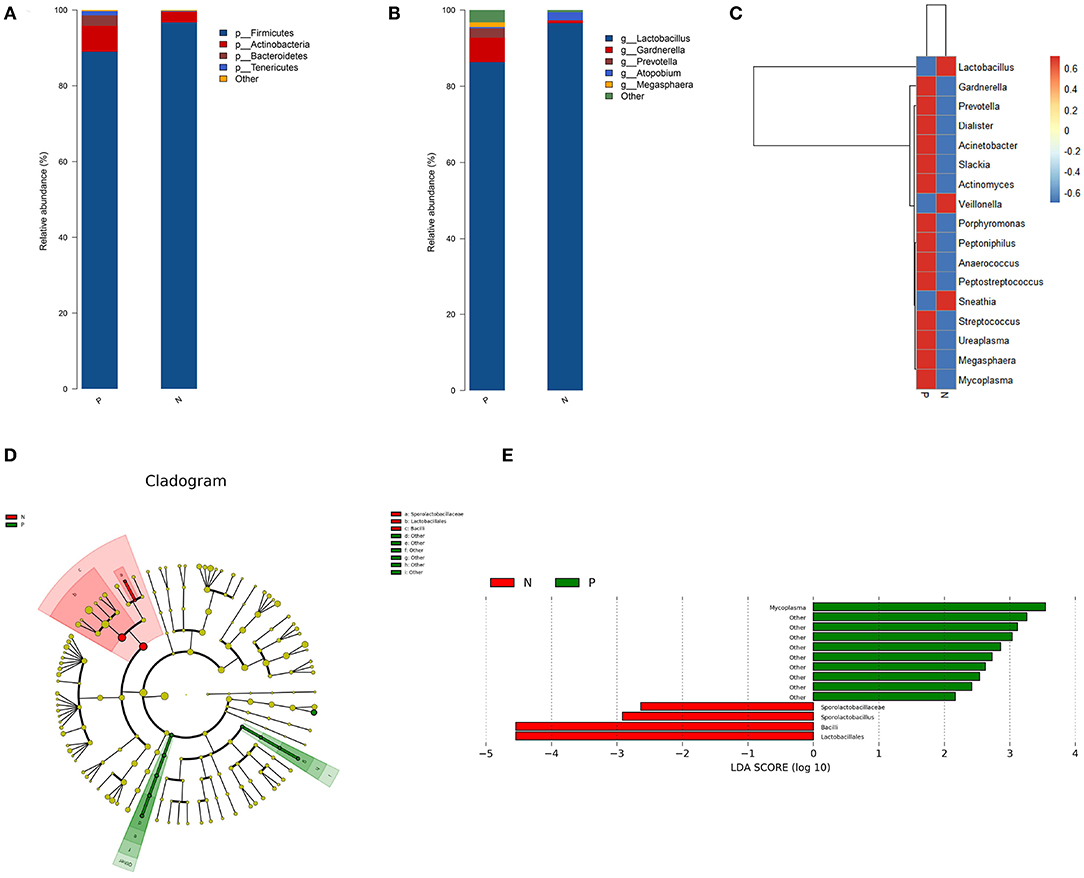
Figure 2. Bacterial taxa analysis of the HPV-positive and HPV-negative groups. (A) Relative abundances of the vaginal microbiota at the phylum level. (B) Relative abundances of the vaginal microbiota at the genus level. (C) Heatmap showing the relative abundance of each bacterial genus. (D) Cladogram of the LEfSe analysis of the vaginal microbiota in the two study groups. The microbial compositions were compared at different evolutionary levels. (E) LDA scores obtained from the LEfSe analysis of the gut microbiota in the different groups. An LDA effect size of >2 was used as a threshold for the LEfSe analysis. HPV, human papilloma virus; LDA, linear discriminant analysis; LEfSe, LDA effect size analysis.
A heat map was constructed based on the abundances of the detected species and genera. The heat map revealed that in the HPV-positive group, the abundances of the following species and genera were increased: Gardnerella, Slackia, and Actinomyces belonging to Actinobacteria; Prevotella and Porphyromonas belonging to Bacteroidetes; Dialister, Peptoniphilus, Anaerococcus, Peptostreptococcus, Streptococcus, and Megasphaera belonging to Firmicutes; Ureaplasma and Mycoplasma belonging to Tenericutes; and Acinetobacter (Proteobacteria). In contrast, the abundances of Lactobacillus and Veillonella (Firmicutes) and Sneathia (Fusobacteria) were decreased in the HPV-positive group (Figure 2C).
The LEfSe analysis revealed that the abundance of Mycoplasma was significantly increased while those of Lactobacillales, Sporolactobacillaceae, and Sporolactobacillus were significantly decreased in the HPV-positive group as compared to the HPV-negative group (Figures 2D,E).
To explore the bacterial interactions of key genera in the vaginal ecosystem, we performed a bacterial community network analysis. In both the HPV-positive and HPV-negative groups, Lactobacillus was negatively correlated with Dialister, Gardnerella, Ureaplasma, Prevotella, and Peptoniphilus, while positive correlations were identified among the genera Anaerococcus, Dialister, Gardnerella, Ureaplasma, Prevotella, Peptoniphilus, Porphyromonas, and Peptostreptococcus (Figure 3).
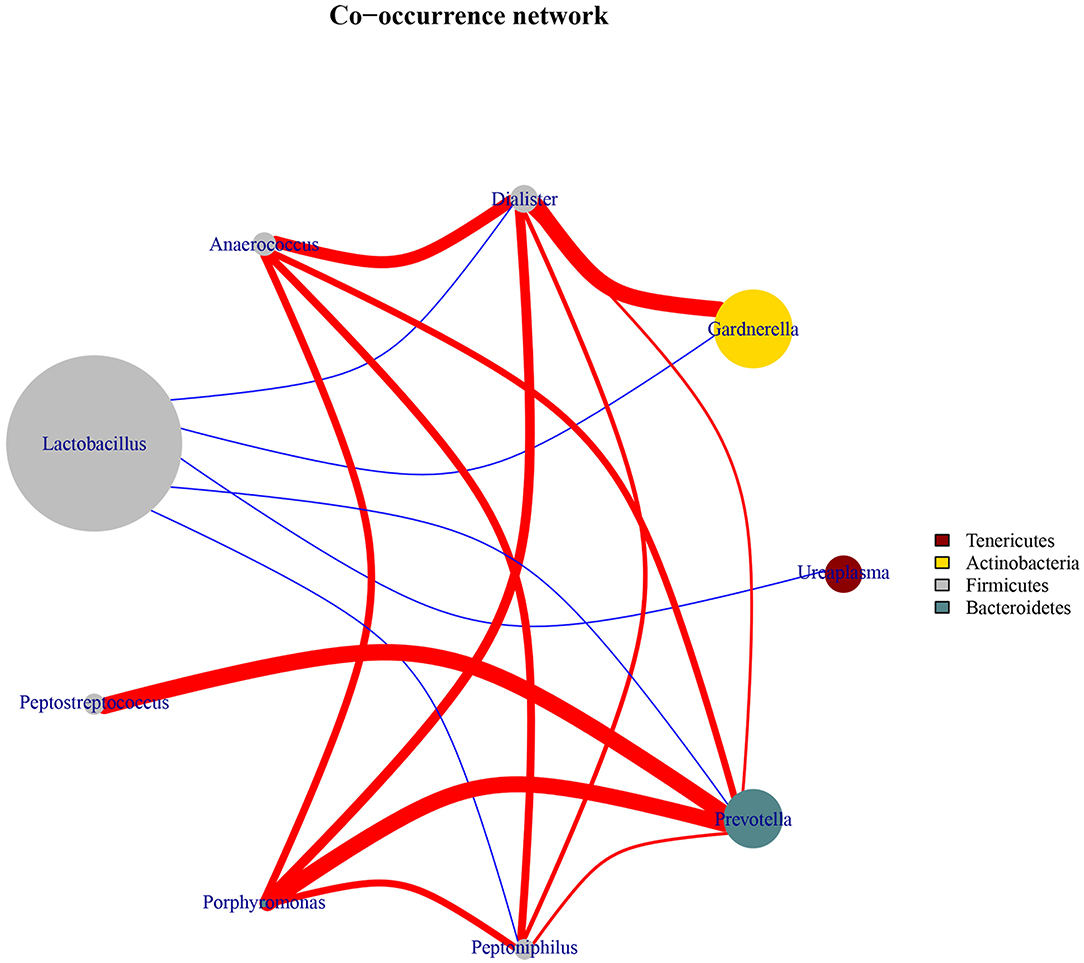
Figure 3. Co-occurrence network diagram of the core vaginal microbiota. The edges between the nodes represent correlations between the nodes they connect, with edge color indicating positive (red) and negative (blue) correlations, and edge shade indicating the magnitude of the correlation.
We further analyzed the inferred metagenomics to identify functional differences in the vaginal microbiota between HPV-positive and HPV-negative women. In the HPV-positive group, LEfSe analysis showed that the abundances of the categories “RNA processing and modification” and “cytoskeleton” were significantly increased in the vaginal microbiota. In contrast, the categories of “Chromatin structure and dynamics” and “Carbohydrate transport and metabolism” were significantly enriched in the vaginal microbiota of the HPV-negative group (Figure 4).
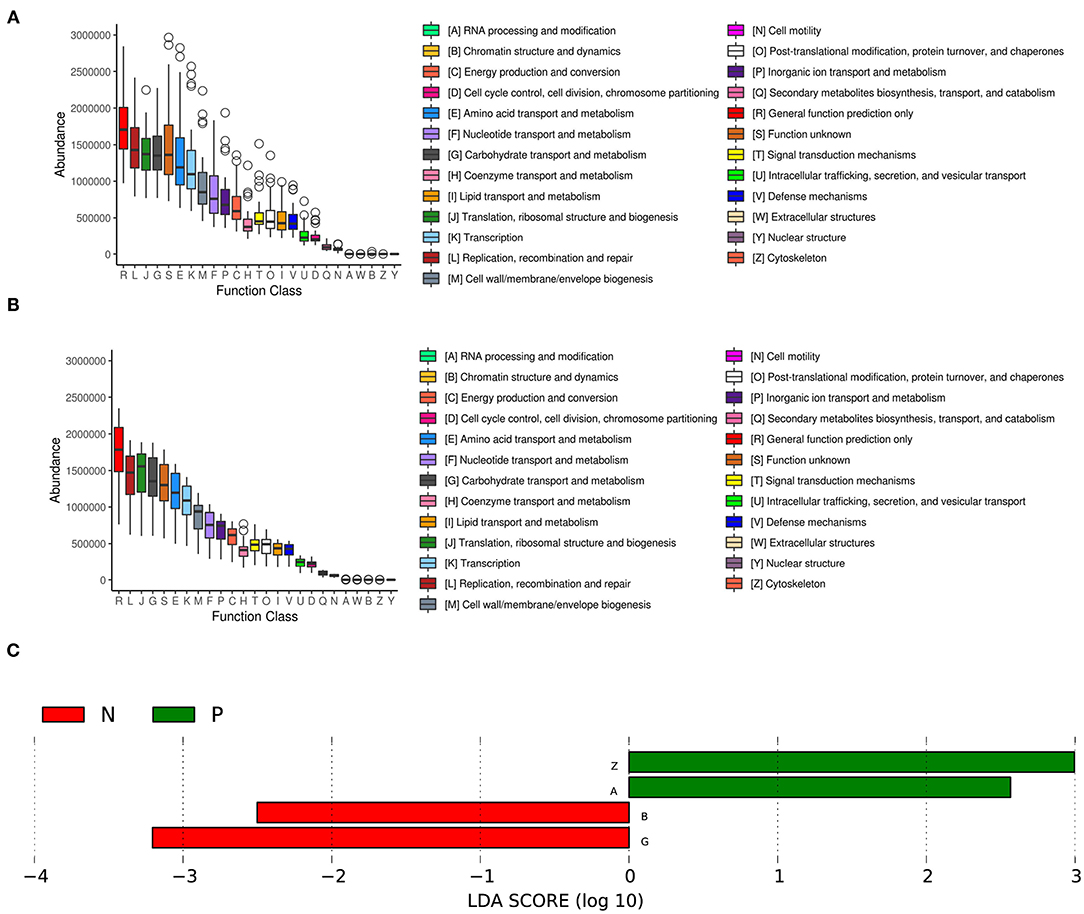
Figure 4. COG functional classification in the HPV-positive group (A) and HPV-negative group (B). The boxes represent the interquartile range (IQR) between the first and third quartiles. The lines and spots inside the boxes represent the median and mean, respectively. The whiskers denote the lowest and highest values within 1.5 × IQR from the first and third quartiles, respectively. The ring above the box indicates the outliers. (C) LEfSe analysis of functional differences in the vaginal microbiota in the two study groups. An LDA effect size of >2 was used as a threshold for the LEfSe analysis. HPV, human papilloma virus.
The present study showed that perturbations in the vaginal microbiota occurred in the early stage of hr-HPV infection in the absence of CIN. High-throughput sequencing of the 16s rRNA gene revealed that Lactobacillus, Sporolactobacillus, Atopobium, Veillonella, and Sneathia were decreased in the vaginal microbiota of HPV-positive women, as compared with HPV-negative women. In contrast, bacteria related to BV, such as Gardnerella, Prevotella, Dialister, Slackia, Actinomyces, Porphyromonas, Peptoniphilus, Anaerococcus, Peptostreptococcus, Streptococcus, Ureaplasma, Megasphaera, and Mycoplasma, were more abundant in HPV-positive women than in the HPV-negative women. These results suggest that the early phase of HPV infection is relevant to the homeostasis of the vaginal microbiota, and the predominance of some pathogenic bacteria during early HPV infection may contribute to the subsequent development of CIN lesions.
In the present study, Firmicutes, Actinobacteria, Bacteroidetes, and Tenericutes were the major phyla in the vaginal microbiota, which is in agreement with a previous study (26). Lactobacillus, an important member of the phylum Firmicutes, was the most abundant bacterial species in the vagina; this species helps maintain a low environmental pH through its metabolic activities, which inhibits the growth of pathogens and disrupts biofilms (27, 28). The obvious decline in the abundance of Lactobacillus in the HPV-positive group as compared to the HPV-negative group indicated that hr-HPV infection was associated with a deteriorated vaginal environment. The conspicuous decline in Lactobacillus was also consistent with the changes observed in the microbiota composition at the phylum level. Atopobium vaginae, a newly identified species belonging to the Coriobacteriaceae family, was associated with ~80% of all cases of BV in one study (29). A. vaginae activates the proinflammatory transcription factor NF-κB in the cervicovaginal epithelial cells, triggering abundant inflammation and innate immune responses (30–32). In a study of South African women, Atopobium was found to be among the most abundant taxa in cervicovaginal microbial communities associated with high levels of inflammatory mediators (33). Sneathia sanguinegens and Sneathia amnii, the only two species of Sneathia, secrete fibrinolysins and collagenase, which disrupt the mucosal barrier and promote vaginal epithelial cell detachment (34, 35). In addition, S. amnii and S. sanguinegens act as inflammatory modulators and upregulate interleukin (IL)-8, IL-α, IL-β, and tumor necrosis factor (TNF)-α; both species were among the 6 most prevalent proinflammatory taxa in the vaginal microbiota in one South African study (33). Gardnerella vaginalis is a facultative anaerobe belonging to the Bifidobacteriaceae family. G. vaginalis adheres to the vaginal epithelium, providing a scaffold for biofilm formation and facilitating the growth of bacteria associated with BV (36, 37). G. vaginalis was also among the six most prevalent proinflammatory taxa identified in the abovementioned South African study (33). Peptoniphilus and Anaerococcus, two newly identified members of the Peptoniphilaceae family, are significantly associated with BV (38). Similar to Peptoniphilus and Anaerococcus, Peptostreptococcus (Peptostreptococcaceae) is a common Gram-positive anaerobe that has not been reported to adhere to vaginal epithelial cells, but does usually adhere to epithelial cells at other body sites (39). Mycoplasma and Ureaplasma belong to the Mycoplasmataceae family. Mycoplasma genitalium has been associated with symptomatic urethritis and cervicitis, while Mycoplasma hominis has been associated with BV (40). Ureaplasma urealyticum induces cervicovaginal epithelial cells to produce proinflammatory cytokines and significantly increases the risk of hr-HPV infection (41, 42). The variations in genera such as Gardnerella, Slackia, and Actinomyces (Actinobacteria), Prevotella and Porphyromonas (Bacteroidetes), and Ureaplasma and Mycoplasma (Tenericutes) were consistent with the variations observed in the microbiota composition at the phylum level.
LEfSe analysis indicated that Mycoplasma (Mycoplasmataceae), Lactobacillales, Sporolactobacillaceae, and Sporolactobacillus (Sporolactobacillaceae) were candidate biomarkers for HPV infection. The rate of Mycoplasma infection is higher among women with high-grade squamous intraepithelial lesions (HSILs) than among women with low-grade squamous intraepithelial lesions (LSILs); however, in patients with cervical cancer, no Mycoplasma (M. hominis or M. genitalium) infections were detected (43). Thus, it can be inferred that Mycoplasma plays a major role in the early phase of HPV infection, rather than affecting the development of cervical carcinoma. A significant increase in Mycoplasma abundance may facilitate virus penetration, survival, and persistence (42, 43). Sporolactobacillus resembles Lactobacillus in appearance and metabolism, except that it produces endospores and is aerobic (44). Spore-forming Sporolactobacillus has been extensively studied and has even been commercialized as a probiotic. It has also been modified as a surface expression vector to deliver immunogenic HPV compositions to prepare an HPV vaccine (45). Significant reduction in Sporolactobacillus abundance may indicate a worse vaginal environment that promotes the growth of pathogenic bacteria (46).
The complex interactions between the vaginal microbiota, HPV, CIN, and cancer have recently been investigated. Mitra et al. (47) reported that the progression of CIN lesions was correlated with increased diversity of the vaginal microbiota and decreased abundance of Lactobacillus spp.; the authors also identified S. sanguinegens, Anaerococcus tetradius, and Peptostreptococcus anaerobius as biomarkers of HSILs. In the present study, the genera Anaerococcus and Peptostreptococcus showed the same increasing trends in the early phase of HPV infection; however, the relative abundance of Sneathia decreased in the HPV-positive group. Oh et al. (48) reported that the increased abundance of G. vaginalis, A. vaginae, and L. iners in the cervical microbiota along with the reduced abundance of L. crispatus was linked to a risk of CIN, which indicates that bacterial dysbiosis in the presence of hr-HPV infection might be a risk factor for cervical neoplasia. G. vaginalis showed similar results in our research, while A. vaginae showed a contradictory result: in the early phase of HPV infection, the abundances of the proinflammatory bacteria Sneathia and Atopobium decreased. It is difficult to elucidate the dynamics and mechanisms of these two genera in disease. As the COG category “RNA processing and modification” was significantly enriched in the HPV-positive group, we speculated that in the initial period of hr-HPV infection, the virus invaded epithelial cells and induced a proinflammatory environment, which facilitated the integration of viral DNA into the host cells (49, 50) and caused excessive burden for the host. This led to a robust host immune response, which probably resulted in the observed decrease in the abundances of the proinflammatory genera Sneathia and Atopobium.
Our study focused on analyzing the differences in the vaginal microbiota between HPV-negative women and HPV-positive women with normal cytology. We found that hr-HPV infection was associated with vaginal bacterial dysbiosis. Certain bacteria associated with BV showed increased abundance, while Lactobacillus spp. showed decreased in the early phase of HPV infection, leading to alterations in the vaginal microbiome. We did not investigate the interplay between the vaginal microbiota, hr-HPV infection, CIN lesions, and cervical cancer. In the future, we plan to identify causal connections among HPV infection, CIN status (1+, 2+, 3+), cervical cancer, and the vaginal microbiota.
The present study demonstrated that vaginal microbial perturbations occurred in the early phase of hr-HPV infection when no CIN lesions had developed. The abundance of probiotics such as Lactobacillus and Sporolactobacillus decreased, while the abundance of BV-related pathogenic bacterial genera increased. By studying the early effects of hr-HPV infection on the vaginal microbiota, we provide candidate microbial markers to monitor persistent HPV infection and the development of cervical neoplasms.
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: (NCBI Sequence Read Archive database and SRA accession number-SRP212297, BioProject accession number-PRJNA551647).
The studies involving human participants were reviewed and approved by the ethics committee of the First Hospital of Jilin University. The patients/participants provided their written informed consent to participate in this study.
All persons who meet authorship criteria are listed as authors, and all authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the manuscript. Furthermore, each author certifies that this material or similar material has not been and will not be submitted to or published in any other publication before its appearance in the Frontiers in public health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Arbyn M, Weiderpass E, Bruni L, de Sanjose S, Saraiya M, Ferlay J, et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Global Health. (2020) 8: e191–203. doi: 10.1016/S2214-109X(19)30482-6
2. Mitra A, MacIntyre DA, Marchesi JR, Lee YS, Bennett PR, Kyrgiou M. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: what do we know and where are we going next? Microbiome. (2016) 4:58. doi: 10.1186/s40168-016-0203-0
3. Koshiol J, Lindsay L, Pimenta JM, Poole C, Jenkins D, Smith JS. Persistent human papillomavirus infection and cervical neoplasia: a systematic review and meta-analysis. Am J Epidemiol. (2008) 168:123–37. doi: 10.1093/aje/kwn036
4. Tartaglia E, Falasca K, Vecchiet J, Sabusco GP, Picciano G, Di Marco R, et al. Prevalence of HPV infection among HIV-positive and HIV-negative women in Central/Eastern Italy: strategies of prevention. Oncol Lett. (2017) 14:7629–35. doi: 10.3892/ol.2017.7140
5. Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol. (2010) 117:S5–10. doi: 10.1016/j.ygyno.2010.01.024
6. Munagala R, Dona MG, Rai SN, Jenson AB, Bala N, Ghim SJ, et al. Significance of multiple HPV infection in cervical cancer patients and its impact on treatment response. Int J Oncol. (2009) 34:263–71. doi: 10.3892/ijo_00000148
7. Gillet E, Meys JF, Verstraelen H, Bosire C, De Sutter P, Temmerman M, et al. Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: a meta-analysis. BMC Infect Dis. (2011) 11:10. doi: 10.1186/1471-2334-11-10
8. Desanjose S, Munoz N, Bosch FX, Reimann K, Pedersen NS, Orfila J, et al. Sexually-transmitted agents and cervical neoplasia in colombia and Spain. Int J Cancer. (1994) 56:358–63. doi: 10.1002/ijc.2910560311
9. Wheeler CM. Natural history of human papillomavirus infections, cytologic and histologic abnormalities, and cancer. Obstet Gynecol Clin Noth Am. (2008) 35:519–36. doi: 10.1016/j.ogc.2008.09.006
10. Vriend HJ, Bogaards JA, van Bergen JEAM, Brink AATP, van den Broek IVF, Hoebe CJPA, et al. Incidence and persistence of carcinogenic genital human papillomavirus infections in young women with or without Chlamydia trachomatis co-infection. Cancer Med. (2015) 4:1589–98. doi: 10.1002/cam4.496
11. Flores GE, Caporaso JG, Henley JB, Rideout JR, Domogala D, Chase J, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. (2014) 15:531. doi: 10.1186/s13059-014-0531-y
12. Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. (2006) 124:837–48. doi: 10.1016/j.cell.2006.02.017
13. Feng Q, Liang SS, Jia HJ, Stadlmayr A, Tang LQ, Lan Z, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. (2015) 6:6528. doi: 10.1038/ncomms7528
14. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. (2007) 449:804–10. doi: 10.1038/nature06244
15. Boris S, Barbes C. Role played by lactobacilli in controlling the population of vaginal pathogens. Microbes Infect. (2000) 2:543–6. doi: 10.1016/S1286-4579(00)00313-0
16. Ocana VS, Holgado AAPD, Nader-Macias ME. Characterization of a bacteriocin-like substance produced by a vaginal Lactobacillus salivarius strain. Appl Environ Microb. (1999) 65:5631–5. doi: 10.1128/AEM.65.12.5631-5635.1999
17. Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. (2011) 108:4680–7. doi: 10.1073/pnas.1002611107
18. Yamamoto T, Zhou X, Williams CJ, Hochwalt A, Forney LJ. Bacterial Populations in the vaginas of healthy adolescent women. J Pediatr Adolesc Gynecol. (2009) 22:11–8. doi: 10.1016/j.jpag.2008.01.073
19. Hummelen R, Fernandes AD, Macklaim JM, Dickson RJ, Changalucha J, Gloor GB, et al. Deep sequencing of the vaginal microbiota of women with HIV. PLoS ONE. (2010) 5:e12078. doi: 10.1371/journal.pone.0012078
20. McDonald AC, Denny L, Wang CH, Tsai WY, Wright TC, Kuhn L. Distribution of high-risk human papillomavirus genotypes among HIV-negative women with and without cervical intraepithelial neoplasia in South Africa. PLoS ONE. (2012) 7:e0044332. doi: 10.1371/journal.pone.0044332
21. Dols JAM, Smit PW, Kort R, Reid G, Schuren FHJ, Tempelman H, et al. Microarray-based identification of clinically relevant vaginal bacteria in relation to bacterial vaginosis. Am J Obstet Gynecol. (2011) 204:305.e1–.e7. doi: 10.1016/j.ajog.2010.11.012
22. Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. (1998) 180:4765–74. doi: 10.1128/JB.180.18.4765-4774.1998
23. Morgan J. Bacteriology of humans: an ecological perspective. Homo. (2010) 61:285. doi: 10.1016/j.jchb.2010.06.002
24. Chao A, Bunge J. Estimating the number of species in a stochastic abundance model. Biometrics. (2002) 58:531–9. doi: 10.1111/j.0006-341X.2002.00531.x
25. Chao A, Shen TJ. Nonparametric estimation of Shannon's index of diversity when there are unseen species in sample. Environ Ecol Stat. (2003) 10:429–43. doi: 10.1023/A:1026096204727
26. Lee JE, Lee S, Lee H, Song YM, Lee K, Han MJ, et al. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS ONE. (2013) 8:e63514. doi: 10.1371/journal.pone.0063514
27. Hawes SE, Hillier SL, Benedetti J, Stevens CE, Koutsky LA, Wolner-Hanssen P, et al. Hydrogen peroxide-producing lactobacilli and acquisition of vaginal infections. J Infect Dis. (1996) 174:1058–63. doi: 10.1093/infdis/174.5.1058
28. Brotman RM, Shardell MD, Gajer P, Tracy JK, Zenilman JM, Ravel J, et al. Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. J Infect Dis. (2014) 210:1723–33. doi: 10.1093/infdis/jiu330
29. Perlamutrov Y, Gomberg M, Chernova N, Bochkova O, Shcherbo S. A Comparative efficacy of nifuratel and metronidazole in therapy of bacterial vaginosis associated with Atopobium vaginae. Sex Transm Infect. (2013) 89:A113–A. doi: 10.1136/sextrans-2013-051184.0347
30. Libby EK, Pascal KE, Mordechai E, Adelson ME, Trama JP. Atopobium vaginae triggers an innate immune response in an in vitro model of bacterial vaginosis. Microbes Infect. (2008) 10:439–46. doi: 10.1016/j.micinf.2008.01.004
31. Doerflinger SY, Throop AL, Herbst-Kralovetz MM. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J Infect Dis. (2014) 209:1989–99. doi: 10.1093/infdis/jiu004
32. Fichorova RN, Yamamoto HS, Delaney ML, Onderdonk AB, Doncel GF. Novel vaginal microflora colonization model providing new insight into microbicide mechanism of action. MBio. (2011) 2:e00168-11. doi: 10.1128/mBio.00168-11
33. Anahtar MN, Byme EH, Doherty KE, Bowman BA, Yamamoto HS, Soumillon M, et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity. (2015) 42:965–76. doi: 10.1016/j.immuni.2015.04.019
34. Africa CWJ, Nel J, Stemmet M. Anaerobes and bacterial vaginosis in pregnancy: virulence factors contributing to vaginal colonisation. Int J Environ Res Public Health. (2014) 11:6979–7000. doi: 10.3390/ijerph110706979
35. Harwich MD, Serrano MG, Fettweis JM, Alves JMP, Reimers MA, Buck GA, et al. Genomic sequence analysis and characterization of Sneathia amnii sp. nov. BMC Genomics. (2012) 13:S4. doi: 10.1186/1471-2164-13-S8-S4
36. Fethers K, Twin J, Fairley CK, Fowkes FJ, Garland SM, Fehler G, et al. Bacterial vaginosis (BV) candidate bacteria: associations with BV and behavioural practices in sexually-experienced and inexperienced women. PLoS ONE. (2012) 7:e30633. doi: 10.1371/journal.pone.0030633
37. Harwich MD, Alves JM, Buck GA, Strauss JF, Patterson JL, Oki AT, et al. Drawing the line between commensal and pathogenic Gardnerella vaginalis through genome analysis and virulence studies. BMC Genomics. (2010) 11:7. doi: 10.1186/1471-2164-11-375
38. Diop K, Diop A, Michelle C, Richez M, Rathored J, Bretelle F, et al. Description of three new Peptoniphilus species cultured in the vaginal fluid of a woman diagnosed with bacterial vaginosis: Peptoniphilus pacaensis sp. nov., Peptoniphilus raoultii sp. nov., and Peptoniphilus vaginalis sp. nov. Microbiologyopen. (2019) 8:e00661. doi: 10.1002/mbo3.661
39. Marrazzo JM, Thomas KK, Fiedler TL, Ringwood K, Fredricks DN. Relationship of specific vaginal bacteria and bacterial vaginosis treatment failure in women who have sex with women. Ann Intern Med. (2008) 149:20–8. doi: 10.7326/0003-4819-149-1-200807010-00006
40. Patel MA, Nyirjesy P. Role of Mycoplasma and Ureaplasma species in female lower genital tract infections. Curr Infect Dis Rep. (2010) 12:417–22. doi: 10.1007/s11908-010-0136-x
41. Peltier MR, Berlin Y, Tee SC, Smulian JC. Does progesterone inhibit bacteria-stimulated interleukin-8 production by lower genital tract epithelial cells? J Perinat Med. (2009) 37:328–33. doi: 10.1515/JPM.2009.064
42. Ye H, Song TG, Xi Z, Li L, Hou MM, Xi MR. Association between genital mycoplasmas infection and human papillomavirus infection, abnormal cervical cytopathology, and cervical cancer: a systematic review and meta-analysis. Arch Gynecol Obstet. (2018) 297:1377–87. doi: 10.1007/s00404-018-4733-5
43. Biernat-Sudolska M, Szostek S, Rojek-Zakrzewska D, Klimek M, Kosz-Vnenchak M. Concomitant infections with human papillomavirus and various Mycoplasma and Ureaplasma species in women with abnormal cervical cytology. Adv Med Sci Poland. (2011) 56:299–303. doi: 10.2478/v10039-011-0028-9
44. Margulis L, Chapman MJ. Chapter One - kingdom prokaryotae (Bacteria, Monera, Prokarya). In: Margulis L, Chapman MJ, editors. Kingdoms and Domains, 4th edn. London: Academic Press (2009). p. 35–107. doi: 10.1016/B978-0-12-373621-5.00001-5
45. Sanders ME, Morelli L, Tompkins TA. Sporeformers as human probiotics: Bacillus, Sporolactobacillus, and Brevibacillus. Compr Rev Food Sci Food Safety. (2003) 2:101–10. doi: 10.1111/j.1541-4337.2003.tb00017.x
46. Amabebe E, Anumba DOC. The vaginal microenvironment: the physiologic role of Lactobacilli. Front Med. (2018) 5:181. doi: 10.3389/fmed.2018.00181
47. Mitra A, MacIntyre DA, Lee YS, Smith A, Marchesi JR, Lehne B, et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci Rep. (2015) 5:16865. doi: 10.1038/srep16865
48. Oh HY, Kim BS, Seo SS, Kong JS, Lee JK, Park SY, et al. The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea. Clin Microbiol Infect. (2015) 21:674 e1–9. doi: 10.1016/j.cmi.2015.02.026
49. Garcea G, Dennison AR, Steward WP, Berry DP. Role of inflammation in pancreatic carcinogenesis and the implications for future therapy. Pancreatology. (2005) 5:514–29. doi: 10.1159/000087493
Keywords: persistent infection, 16s rRNA, bacterial vaginosis, vaginal microbiota, human papillomavirus
Citation: Wei Z-T, Chen H-L, Wang C-F, Yang G-L, Han S-M and Zhang S-L (2021) Depiction of Vaginal Microbiota in Women With High-Risk Human Papillomavirus Infection. Front. Public Health 8:587298. doi: 10.3389/fpubh.2020.587298
Received: 25 July 2020; Accepted: 08 December 2020;
Published: 08 January 2021.
Edited by:
Lin Yang, Hong Kong Polytechnic University, Hong KongReviewed by:
Polly H. M. Leung, Hong Kong Polytechnic University, Hong KongCopyright © 2021 Wei, Chen, Wang, Yang, Han and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song-Ling Zhang, c2x6aGFuZ0BqbHUuZWR1LmNu; Shu-Mei Han, NzczNzQ1MjQ1QHFxLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.