
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Public Health, 22 September 2020
Sec. Infectious Diseases: Epidemiology and Prevention
Volume 8 - 2020 | https://doi.org/10.3389/fpubh.2020.543898
 Rendani I. Manenzhe1†
Rendani I. Manenzhe1† Felix S. Dube1,2*†
Felix S. Dube1,2*† Meredith Wright3
Meredith Wright3 Katie Lennard4Stephanie Mounaud3Stephanie W. Lo5Heather J. Zar6
Katie Lennard4Stephanie Mounaud3Stephanie W. Lo5Heather J. Zar6 William C. Nierman3Mark P. Nicol1,7
William C. Nierman3Mark P. Nicol1,7 Clinton Moodley1,8
Clinton Moodley1,8Background: There remains a significant proportion of deaths due to pneumococcal pneumonia in infants from low- and middle-income countries despite the marginal global declines recorded in the past decade. Monitoring changes in pneumococcal carriage is key to understanding vaccination-induced shifts in the ecology of carriage, patterns of antimicrobial resistance, and impact on health. We longitudinally investigated pneumococcal carriage dynamics in PCV-13 vaccinated infants by collecting nasopharyngeal (NP) samples at 2-weekly intervals from birth through the first year of life from 137 infants. As a proof of concept, 196 NP samples were retrieved from a subset of 23 infants to explore strain-level pneumococcal colonization patterns and associated antimicrobial-resistance determinants. These were selected on the basis of changes in serotype and antibiogram over time. NP samples underwent short-term enrichment for streptococci prior to total nucleic acid extraction and whole metagenome shotgun sequencing (WMGS). Reads were assembled and aligned to pneumococcal reference genomes for the extraction of pneumococcal and non-pneumococcal bacterial reads. Pneumococcal contigs were aligned to the Antibiotic Resistance Gene-ANNOTation database of acquired AMR genes. In silico pneumococcal capsular and multilocus sequence typing were performed.
Results: Of the 196 samples sequenced, 174 had corresponding positive cultures for pneumococci, of which, 152 were assigned an in silico serotype. Metagenomic sequencing detected a single pneumococcal serotype in 85% (129/152), and co-colonization in 15% (23/152) of the samples. Twenty-two different pneumococcal serotypes were identified, with 15B/15C and 16F being the most common non-PCV13 serotypes, while 23F and 19A were the most common PCV13 serotypes. Twenty-six different sequence types (STs), including four novel STs were identified in silico. Mutations in the folA and folP genes, associated with cotrimoxazole resistance, were detected in 89% (87/98) of cotrimoxazole-non-susceptible pneumococci, as well as in the pbp1a and pbp2x genes, in penicillin non-susceptible ST705215B/15C isolates.
Conclusions: Metagenomic sequencing of NP samples is a valuable culture-independent technique for a detailed evaluation of the pneumococcal component and resistome of the NP microbiome. This method allowed for the detection of novel STs, as well as co-colonization, with a predominance of non-PCV13 serotypes in this cohort. Forty-eight resistance genes, as well as mutations associated with resistance were detected, but the correlation with phenotypic non-susceptibility was lower than expected.
Streptococcus pneumoniae (the pneumococcus) is a frequent bacterial cause of infections such as bacterial pneumonia, otitis media, meningitis, sinusitis, and bacteraemia in young children, and is a major cause of morbidity and mortality (1–4). Globally, pneumococcal pneumonia was responsible for an estimated 393,000 deaths in children <5 years of age in 2015 (5). Asymptomatic pneumococcal nasopharyngeal (NP) carriage is common among infants, a reservoir for transmission and precedes the development of infections (6). Pneumococci are classified into nearly 100 serotypes, based on the antigenic specificity of the polysaccharide capsule (7, 8). Children are often sequentially colonized by multiple different serotypes (9, 10), and may be co-colonized by different pneumococcal serotypes at the same time (11).
Immunization with the pneumococcal conjugate vaccine (PCV) has substantially reduced NP carriage and invasive pneumococcal disease caused by serotypes represented in the vaccine (12, 13). In addition, the introduction of PCV has resulted in a reduction in antimicrobial resistant pneumococci amongst circulating strains due to the inclusion of serotypes associated with high antibiotic resistance in the conjugate vaccine (14). However, non-vaccine-serotypes have emerged among both carriage and disease-causing isolates, and are increasingly associated with antimicrobial resistance (15, 16). Increasing pneumococcal resistance to different classes of antibiotics, including beta-lactams, macrolides, tetracyclines, and cotrimoxazole, has compromised the effectiveness of antibiotics to treat pneumococcal infections (17).
Pneumococci are most commonly identified from carriage and disease samples using bacterial culture followed by phenotypic and genotypic characterization of the isolates (18, 19). The Quellung method, based on antisera reactions, is currently the gold standard for pneumococcal serotyping, but requires viable isolates in pure culture. To detect colonization with multiple serotypes, typing of many individual (often identical) pneumococcal colonies from each NP sample is required (20). Microarrays are able to detect co-colonization by all known pneumococcal serotypes, and depending on the microarray panel used, and targets included, may provide additional information on virulence and resistance determinants present. This method is however limited and cannot provide information on genes not included in the array (21, 22). Sequential multiplex PCR assays are increasingly used for serotyping, and can be done directly on the NP sample, however, this is relatively laborious, costly, and limited to serotypes targeted by the PCR (23). None of these methods are able to provide a more detailed, strain-level characterization, important for pneumococci, as capsular switching often results in different serotypes within the same lineage (24).
Metagenomic approaches, where the collective genomes of all organisms recovered directly from a sample are sequenced, is a high-throughput approach to investigate the members of a microbial community in an ecological niche, and represents an alternative to culture-dependant methods for microbial characterization (25). In this study, we explored the use of shotgun metagenomic sequencing to characterize the pneumococcal component of the NP, including identification of co-colonization with multiple serotypes and antimicrobial resistance, among PCV-vaccinated children participating in a South African birth cohort.
Children were enrolled in a longitudinal birth-cohort study, the Drakenstein Child Health Study (26), in the Western Cape Province of South Africa. NP swabs were collected every second week from birth through the first year of life, from 137 infants during 2012–2013 (10). Infants received 2+1 doses of 13-valent pneumococcal conjugate vaccine (PCV13) at 6 weeks, 14 weeks, and 9 months according to the national immunization program in South Africa. Details of the study population and sampling have been previously described (26). The study was approved by the University of Cape Town, Faculty of Health Sciences Human Research Ethics Committee (Reference numbers 401/2009 and 235/2016). Written informed consent was obtained from mothers.
The collected NP swabs were immediately placed into 1 ml skim milk-tryptone-glucose-glycerol (STGG) medium, transported on ice within 2 h of collection, and stored at −80°C for batch processing within a week. A 10 μl aliquot of the NP-STGG was inoculated onto a blood agar plate (Oxoid Columbia base with 5% sheep blood) containing gentamicin (5 μg/ml) and incubated overnight at 37°C, in 5% CO2. Presumptive pneumococcal isolates were identified as previously described (27); only a single pneumococcal isolate was selected for conventional characterization from each sample. All serotypes were initially deduced by sequetyping and subsequently confirmed by Quellung, as previously described (28, 29).
Susceptibility to penicillin (assessed using oxacillin disc), erythromycin, and cotrimoxazole were assessed using the disc diffusion method, and interpreted using Clinical and Laboratory Standards Institute (CLSI) guidelines (30). Penicillin minimum inhibitory concentrations (MICs) were determined using the E-test method (bioMérieux, Marcy I'Etoile, France), according to the manufacturer's instructions. Intermediate and resistant pneumococcal isolates were collectively regarded as non-susceptible.
In order to explore the utility of metagenomic sequencing to detect colonization with multiple serotypes and identify antimicrobial-resistance determinants in this exploratory study, a total of 196 NP-STGG samples were purposively selected from a subset of 23 of the 137 infants described above. These samples were selected based on changes in pneumococcal serotype and antibiogram over time identified using phenotypic methods. For the purpose of comparison and longitudinally assessing colonization dynamics with other potential pathogens, 22 out of 196 samples from a subset of 23 infants which were culture negative for S. pneumoniae, were also included. The NP-STGG samples were enriched as previously described, with minor modifications (31). Briefly, 200 μl of an NP-STGG sample was transferred to 6 ml Todd-Hewitt Broth (without antibiotics), containing 0.5% yeast extract and 17% fetal bovine serum. The liquid culture was incubated at 37°C with 5% CO2, without shaking for 6 h. The culture was then centrifuged at 9,000 rpm for 10 min at 4°C. Total nucleic acid extraction was performed on the collected pellet using the QIAsymphony SP automated platform (Qiagen, Hilden, Germany) with the QIAsymphony Virus/Bacteria Mini Kit (Cat. No. 931036) following the manufacturer's instructions. Nucleic acid concentrations and purity were determined using the NanoDrop® ND-100 (Thermo Fishers Scientific, Waltham, USA). The purified nucleic acid was stored at −20°C and sequenced within a month.
Total nucleic acid was subjected to shotgun sequencing on the MiSeq platform using the MiSeq Reagent Kit v3 (600-cycle) (Illumina, San Diego, USA) at the J. Craig Venter Institute, Rockville, USA. Metagenomic DNA sequencing protocols and the pipeline used to assemble the reads and evaluate the assembly have been previously described (31). Reads were assembled using metaSPAdes (32), and aligned to the created serotype nucleotide database using BLASTn, with an identity over 98% considered a match (31). In addition, assembly-based in silico multi-locus sequence typing (MLST) was performed using LOCUST as previously described (33, 34).
Serotypes identified by Quellung/sequetyping (29), were compared to in silico serotypes identified from metagenomic sequences. Culture-based and metagenomic-assigned serotypes were considered concordant if the serotype detected by Quellung/sequetyping was detected by shotgun sequencing (either as a single serotype or amongst co-colonizing serotypes detected).
A phylogenetic tree of pneumococcal genomes was constructed to assess strain relatedness. Assembly-based (metaSPAdes assembler) variant calling (32), was performed using The Northern Arizona Single Nucleotide Polymorphism Pipeline (NASP) (35). All completed pneumococcal genomes available on the NCBI database were included in the NASP run and the S. pneumoniae R6 genome (Accession number AE007317) was used as reference genome. The aligned base calls for each of the core variant positions identified by NASP were used for the construction of the maximum likelihood tree (36).
Pneumococcal contigs (contigs that mapped to reference pneumococcal genome R6) were compared to the Antibiotic Resistance Gene-Annotation (ARG-ANNOT) database by BLAST alignment (37). Results were filtered using ≥90% sequence identity over 80% of the sequence length of the reference antibiotic-resistance gene as cut-offs. The pbp, folA [encoding dihydropteroate synthase (DHPS)], and folP [encoding dihydrofolate reductase (DHFR)] gene mutations associated with resistance to beta-lactams, trimethoprim, and sulfonamides, respectively, were investigated by manual local alignment, as the ARG-ANNOT database does not automatically detect these mutations. The ARG-ANNOT database only included the pbp1a (JN645776) and pbp1b (AF101781) genes of wild-type pneumococcal strains. Therefore, local alignments of the pbp1a (JN645776.1), pbp2x (JN645706.1), pbp2b (DQ056780.1), folA_R6 (Gene ID 4442919), and folP_R6 (Gene ID 4443057) were performed using BLASTn (80% identity and 60% gene length coverage).
For each individual, and for each of the genes, sequences from the longitudinal samples were aligned with Multiple Alignment using Fast Fourier Transform (MAFFT) (38). Aligned sequences were viewed and translated to amino acids in AliViewer (version 1.18.1) for active site mutation analysis. The transpeptidase domains of the pbp1a, 2x, and 2b, from the wild type S. pneumoniae R6 strain were used as references. Phylogenetic trees were constructed with Molecular Evolutionary Genetic Analysis software (MEGA version 7.0.26) using a neighbor-joining method and bootstrapping 1000 replicates. pbp1a (JN645776.1), pbp2x (JN645706.1), and pbp2b (DQ056780.1) were used as reference gene sequences in the construction of the phylogenetic trees.
A subset of 196 NP samples from 23 infants was selected for shotgun sequencing to investigate the pneumococcal population structure. The number of NP samples selected for sequencing ranged from 4 to 21 samples per infant (average of 8.5 samples); selected samples and age at sampling (average 14.9 weeks) for each of the infants is shown in Figure 1. Thirteen of the infants were males (57%). The mean birth weight was 3.0 kg (range, 2.4–3.8 kg) with only one preterm infant. Eight infants were HIV exposed (born to HIV-infected mothers), but were HIV-uninfected.
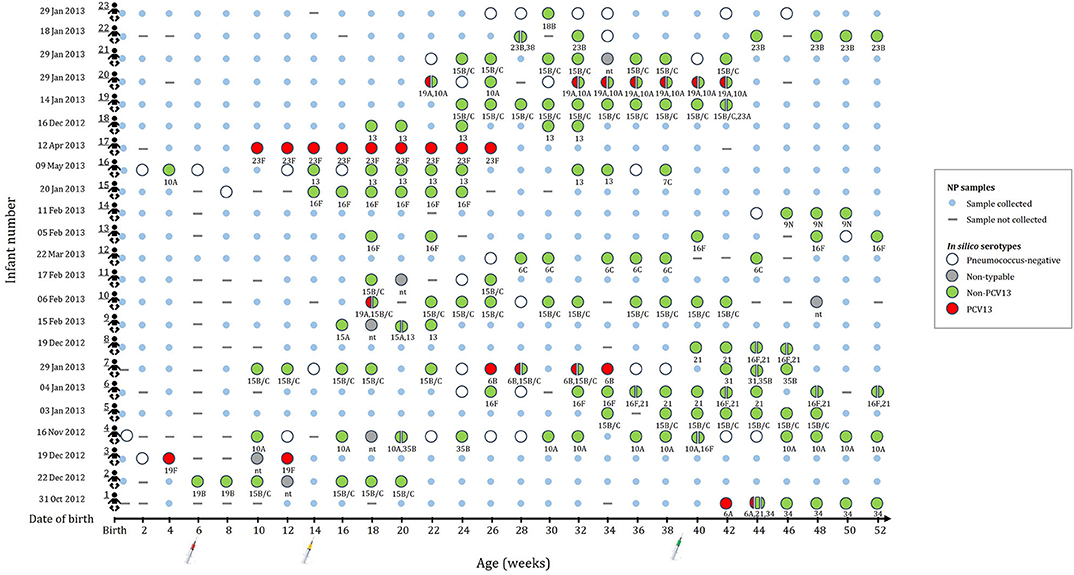
Figure 1. This figure indicates the number of samples selected for metagenomic sequencing and serotype assignments for the 196 nasopharyngeal (NP) samples selected from 23 infants (shown in rows 1 to 23). None of the infants were colonized until 4 weeks of age. Small blue circles represent the NP samples that were collected but not included for shotgun metagenomic sequencing. Large circles (colors represent serotype group) represent the collected NP samples selected for shotgun metagenomic sequencing. Split circles represent samples with co-colonization with multiple serotypes. in silico serotypes are displayed for samples selected for shotgun metagenomic sequencing. (-) NP sample not collected. (nt) Non-typeable.
The average number of reads per sample was 13 million (ranging from 80 thousand to 93 million reads per sample) despite low input DNA concentrations (data not shown). The average number of microbial reads per sample that mapped to the R6 pneumococcal reference genome was 4.5 million (range, 141 reads−32 million reads per sample) with an average coverage of 308.53X (range, 0.01X−2239.71X).
Of the 196 samples sequenced, 174 had a corresponding positive pneumococcal culture. Shotgun sequencing detected pneumococcal reads in all 174 samples, however, 15 samples had no reads covering the required genomic regions (cps, housekeeping genes, and core variant positions identified by the NASP) of the pneumococcal genomes and were therefore excluded from further analyses. In silico serotypes using shotgun sequencing were assigned in 96% (152/159) of the remaining samples and serotypes were assigned in 93% (148/159) of samples by both Quellung/sequetyping and shotgun sequencing. The concordance between Quellung/sequetyping and shotgun in silico typing was 86% (127/148) (Supplementary Table 1). Co-colonization with two (n = 22) or three (n = 1) different serotypes was detected in 15% (23/152) of the samples from ten infants using shotgun sequencing. The bioinformatic analyses of these 23 samples produced highly reliable and reproducible alignment results with high read mapping counts, negating the possibility of mosaic loci from other Streptococcus species. Since Quellung/sequetyping typing was only performed on a single colony per sample, co-colonization was not detected using this method.
Non-PCV13 serotypes were more commonly detected than PCV13 serotypes (Figure 1). Twenty-two different pneumococcal serotypes were identified in silico, with 15B/15C (n = 49), 16F (n = 21), 10A (n = 21), 13 (n = 14), and 21 (n = 12) being the most common non-PCV13 serotypes, and 23F (n = 9), and 19A (n = 8) being the most common PCV13 serotypes. Serotypes and STs identified are shown in Supplementary Table 2. MLST was assigned in 89% (142/159) of the samples representing 26 different MLST profiles (Figure 2). Eleven STs (non-PCV13-related: ST868715B/15C, ST564713, ST73456C, ST408816F, ST1085421, ST1067316F, ST1060515A, and ST88387C; and PCV13-related: ST205923F, ST206219A, and ST1082319F) have previously been described only in South Africa, while five STs associated with non-PCV13 serotypes (ST206810A, ST345016F, ST335831, ST19919B, and ST39338) had not been previously described in Africa. In addition, four novel STs (ST1379515B/15C, ST1379734, ST1379823B, and ST1379921) were identified (Figure 2 and Supplementary Table 2).
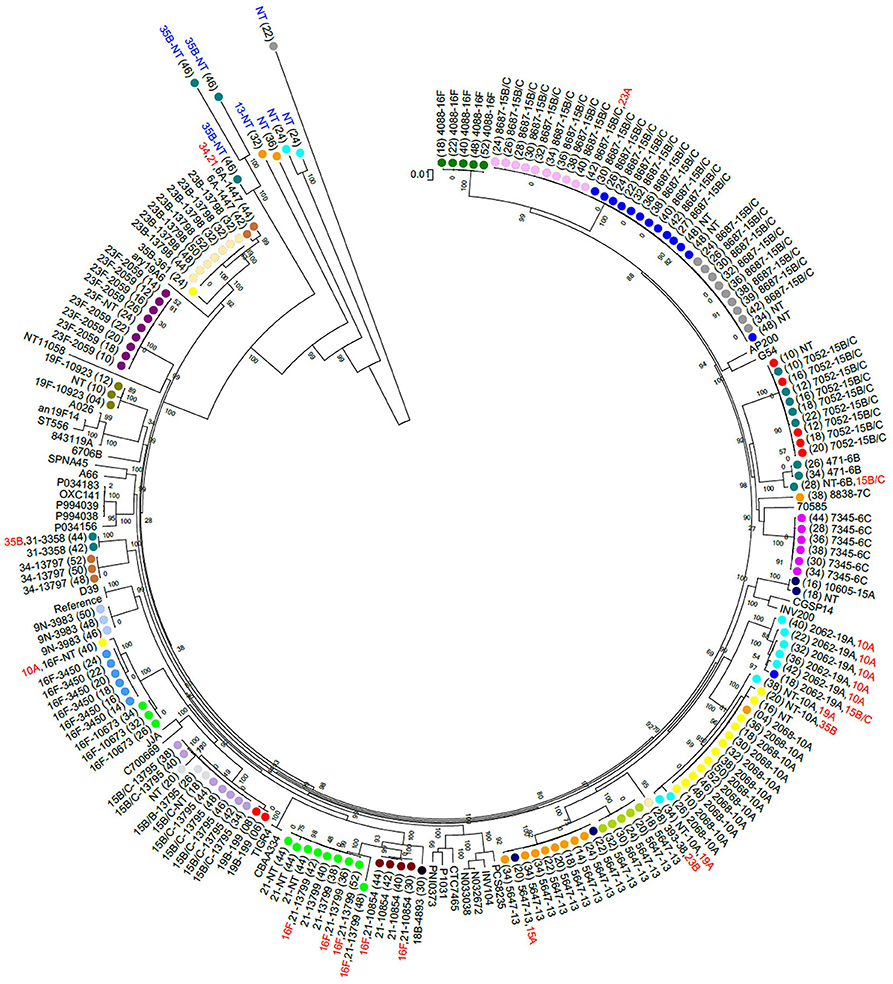
Figure 2. A maximum likelihood whole genome phylogenetic tree of pneumococcal isolates recovered from 23 infants. Bootstrap values are shown on each branch and the scale bar represents the number of SNPs. Circles with the same color represent longitudinal samples from the same infant. Numbers in brackets indicate the age in weeks at each time-point. The sequence type (ST) detected in each sample is shown as a number, followed by the associated serotype. Serotypes indicated in red were among co-colonizing strains and had genomes with lower coverage than the other co-detected strain. Five out of 15 samples that had low reads mapping to pneumococcal genomes are indicated in blue text. NT, Non-typeable. Completed pneumococcal genomes available on the NCBI database were included and the R6 genome was used as a reference. The samples clustered according to ST but not serotype. Persistent colonization with the same genotype was common.
Phenotypic antibiotic susceptibility profiles were determined for the 159 pneumococcal isolates obtained from samples included for this analysis (39). Overall, 18% (29/159), 17% (28/159), and 61% (98/159) of the isolates were non-susceptible to penicillin, erythromycin, and cotrimoxazole, respectively.
Using in silico analyses, a total of 48 acquired antimicrobial-resistance (AMR) genes were identified from pneumococcal contigs in 20 samples collected from 10 infants (Figure 3). The AMR genes detected included macrolide-lincosamide-streptogramin B resistance (MLSB) (msrD, mef A and ermB), and tetracycline resistance (tetM) genes (Figure 3). The average sequence length coverage across the reference genes was 99% (range, 81–100%) while the average sequence depth was 160X (range, 1–411X).
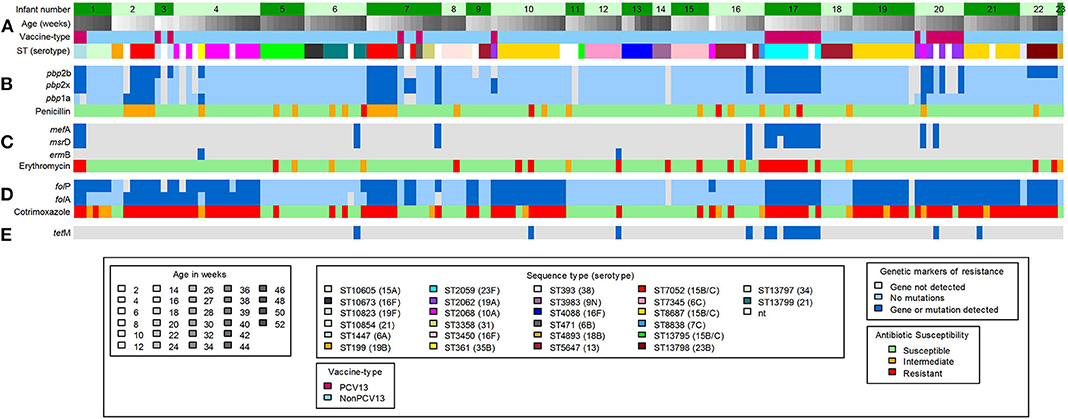
Figure 3. Shotgun sequence-derived molecular sequence type and resistome (from direct sequencing of NP swabs), and phenotypic antimicrobial susceptibility results (from pneumococcal isolates) in 159 out of 196 NP samples obtained from 23 infants (assigned 1 to 23, top row). (A) in silico sequence types (STs) and associated serotypes detected in longitudinal NP samples from each infant. (B) pbp gene mutations and associated penicillin phenotypic susceptibility profiles; light blue color indicates wild-type pbp genes; the dark blue color indicates detected mutations within or close to the conserved motifs for the pbp genes. (C) Macrolide-resistance genes and associated erythromycin phenotypic susceptibility profiles; the dark blue color indicates detected resistance genes. (D) folA and folP gene mutations and associated cotrimoxazole phenotypic susceptibility profiles; light blue color indicates wild-type folA and folP genes; dark blue color indicates detected mutations, in folA and folP, that reduce the affinity of trimethoprim or sulfamethoxazole. (E) Detected tetracycline resistance gene; dark blue color indicates detected resistance gene. Gray color in (B–E) indicates that no genes were detected.
Phenotypic non-susceptibility to erythromycin was detected in 18% (28/159) isolates (Figure 3). Among the 28 samples from which these isolates were obtained, genes predicted to confer macrolide non-susceptibility were detected in 39% (11/28) samples [msrD and mef A (n = 9), mef A (n = 1), and ermB (n = 1)] from three infants. In addition, macrolide resistance genes were detected in a further six samples, from five infants, from which erythromycin-non-susceptible pneumococci were not cultured (Figure 3); two of these samples had co-colonization with multiple serotypes.
Pneumococcal pbp genes were analyzed for mutations associated with beta-lactam resistance. No indels were identified in any of the extracted pbp gene sequences, but multiple amino acid changes were identified at various positions within the transpeptidase domains. Concordance between penicillin non-susceptibility and the presence of resistance conferring mutations was low (52%, 15/29). Interestingly, resistance conferring mutations were also identified among 14 penicillin susceptible strains (Figure 3). Pneumococcal strains with penicillin MICs between 0.064 and 8.0 μg/ml commonly carried mutations within the transpeptidase domains of the pbp1a and pbp2b genes, but not in pbp2x (Supplementary Figure 1), compared to the wild-type pneumococcal strain R6. pbp point mutations known to contribute to beta-lactam resistance are shown in Supplementary Table 3. The S351A and P432T point mutations which are within or close to the conserved motifs of the pbp1a gene were identified in penicillin-non-susceptible ST705215B/15C (n = 9), ST36135B (n = 1), and ST206810A (n = 1) isolates from four infants (Figure 3 and Supplementary Table 3). Amino acid substitution H394L, in the pbp2x gene, was only identified among PCV13 serotypes 23F (n = 9), 19A (n = 4), and 6A (n = 2), from four infants, irrespective of penicillin susceptibility (Supplementary Table 4). These serotypes (23F, 19A, and 6A) had identical amino acid sequences of the transpeptidase domain of the pbp2x gene despite having different STs (Supplementary Figure 3). Pneumococcal strains identified in this study had a higher divergence in the transpeptidase domain sequences of the pbp1a and pbp2x genes than in the pbp2b gene (Supplementary Figures 2–4).
Ninety-eight out of 159 (61%) isolates were phenotypically non-susceptible to cotrimoxazole (Figure 3). The folA (I100L) and folP (6-bp insertion in the region encoding amino acid 58 to 67) gene mutations, known to confer resistance to cotrimoxazole (40, 41), were detected in 89% (87/98) of the samples from which the non-susceptible isolates were obtained (Figure 3 and Supplementary Table 4). Combinations of resistance mutations including I100L plus R59P60 (n = 45), and I100L plus S62Y63 (n = 24) were commonly detected (Supplementary Table 4).
This study explored the use of shotgun metagenomic sequencing as an alternative approach to culture-based testing to investigate pneumococcal NP colonization and associated antimicrobial-resistance determinants, in a South African birth cohort. We were able to derive pneumococcal serotypes and sequence types, and to identify co-colonization and antimicrobial-resistance determinants directly from shotgun sequence data. Since NP samples from apparently healthy individuals generally have low numbers of bacterial cells, we used short-term broth enrichment, which has previously been shown to successfully enrich streptococci (31). Since antibiotics may select for resistant strains, the current study used broth enrichment without antibiotics to encourage growth of all S. pneumoniae present without selective pressure (42).
There was complete concordance between detection of pneumococcal sequences and positive culture for pneumococci. However, of the 174 culture positive samples sequenced, 15 samples produced poor sequence read mapping to the reference pneumococcal genomes, despite good overall read counts. These 15 samples were excluded from further analyses as they were likely other Streptococcus species, which would have been enriched in culture, but map poorly to the reference sequences used, despite having high read counts. There was good correlation (86%) between conventional typing methods and shotgun sequencing in assigning pneumococcal serotypes. Discordant serotype results were predominantly from samples where only one serotype was identified by shotgun sequencing, and were therefore not due to detectable co-colonization. Both Quellung and sequetyping are not infallible in assigning pneumococcal serotypes (23). Discordant serotypes between conventional and in silico typing were likely due to the increased resolution on shotgun sequencing over conventional methods, which may be less specific (43). Shotgun metagenomic sequencing produced robust serotyping results, based on sequence coverage and depth, but this technique is, at present, relatively expensive, time consuming, and computationally intensive for routine typing (44). Whole-genome sequencing of cultured pneumococcal isolates produces reliable serotyping results and can generate additional genetic information, but is typically performed on single isolates, and may thus fail to detect co-colonization, or exclude samples with non-viable bacteria (45). On the other hand, microarray techniques which may or may not require a culture-enrichment step have the potential to detect multiple serotypes within a sample, however, this technique can only detect serotypes and other genetic elements included in the array, and can be technically challenging to distinguish closely related serotypes (46).
Eleven percent (17/159) of the samples produced good alignments of most alleles, but could not be assigned a multilocus sequence type due to a lack of resolution at all the loci necessary for typing. This was observed in four out of 23 samples where co-colonization with multiple serotypes was detected, and in samples with low estimated sequencing coverage (31). Bioinformatic analyses indicated this was due to low read depth or coverage of certain alleles, primarily at the terminal nucleotide positions which are needed for assigning a locus identity. In samples where sequence types were assigned, we observed a strong association between certain pneumococcal serotypes and multilocus sequence types. However, serotypes 15B/15C (ST7052, ST8687, and ST13795) and 16F (ST4088 and ST3450/ST10673) were associated with multiple STs. Serotypes 15B/15C and 16F were the predominant serotypes detected in our cohort, and highly prevalent serotypes tend to be more diverse (47). No other ST was associated with more than one serotype, in line with previous observations (48).
Shotgun sequencing detected co-colonization with multiple serotypes in 15% of the samples; these would have been difficult or laborious to detect using the Quellung method, since multiple colonies would have to be tested individually, or pooled colonies tested with multiple reagents (45). This rate of co-colonization was lower than that reported in Malawi among children aged 0–13 years (40%, 46/116) (49), but comparable to that reported among children <2 years of age in Tehran (17%, 225/1302) (50). In the current study, serotype 19A isolates (from two infants) were only detected using shotgun sequencing (and not culture), in samples with multiple serotypes. Detection of circulating serotype 19A is important in epidemiological studies, since 19A is included in the PCV13 administered to infants. Culture-dependent techniques are biased to detect the most abundant serotype in a sample and are likely to miss co-colonization with less abundant serotypes (51). Carriage of multiple pneumococcal serotypes is also important since it provides an opportunity for horizontal gene transfer, which is one of the most common mechanisms driving pneumococcal evolution (52).
Eleven STs identified in this study, including ST205923F, ST206219A, and ST1082319F, matched other STs which have only been described among isolates from South Africa, and may therefore be endemic. These strains are associated with serotypes included in the PCV13, which is currently administered in the South African schedule, and which all infants in this study received. Five STs identified here are reported for the first time in an African country. Additionally, four novel STs were identified, namely: ST1379515B/15C, ST1379734, ST1379823B, and ST1379921.
The AMR genes identified by pneumococcal resistome analysis were msrD, mef A, ermB, and tetM. Macrolide-resistance genes (msrD, mef A, or ermB), predicted to confer resistance, were detected in 11/28 samples with erythromycin-non-susceptible pneumococci. Acquired AMR genes were not detected in the remaining samples, and the resistance observed could be due to other mechanisms of macrolide resistance not investigated here (53). The frequency of msrD or mef A was higher than that of ermB, and this is in contrast to what has been reported in other studies (54, 55). TetM gene was detected in eight samples but this could not be associated with phenotypic resistance as isolates in this study were not tested for susceptibility to tetracycline, since it is not routinely used to treat pneumococcal infections.
A high proportion of cotrimoxazole-non-susceptibility (61%, 98/159) was observed in pneumococcal isolates present in samples included for shotgun sequencing. Cotrimoxazole, which is a combination of trimethoprim and sulfamethoxazole, inhibits folic acid biosynthesis, and non-susceptibility to this drug is conferred by the acquisition of mutations in folA and folP (40). The majority of cotrimoxazole-non-susceptible isolates (81%) in the current study possessed the I100L amino acid substitution in DHFR, and this mutation has been shown to be sufficient to confer high-level cotrimoxazole resistance (40, 41). The most common insertions detected in DHPS led to the duplication of R58P59 and S62Y63 in 56 and 27% of cotrimoxazole-non-susceptible isolates, respectively. These insertions have been shown to confer low-level cotrimoxazole resistance (40, 41). Most instances (71/86) of high-level cotrimoxazole resistance observed were due to both folA I100L substitution and folP insertion, and this has been previously described (56). Lack of associations in other isolates might be due to other mechanisms of resistance or loss of expression of the detected mutations (Supplementary Table 4) (56).
Pbp genes code for the penicillin-binding proteins (PBPs) which are essential for cell envelope bio-synthesis and are the target for beta-lactam antibiotics (57). Gene mutations occurring within or close to the pbp-conserved motifs within the transpeptidase domain are known to confer resistance to beta-lactams (58). Amino acid alterations in pbp1a, pbp2b, and pbp2x have been shown to be the most reliable markers for beta-lactam-resistance in pneumococci (59). A higher level of variation in the transpeptidase domain sequences of pbp1a and pbp2x, than in pbp2b was observed (Supplementary Figures 2–4). The pbp1a and pbp2x genes flank the capsule (cps) locus, which is prone to frequent recombination events (60), and recombination events involving the cps locus and one or both pbp1a and/or pbp2x genes have been observed (61). This could account for the higher level of variation observed in the pbp1a and pbp2x genes among the strains in this study.
In total, 29 out of 159 (18%) pneumococcal isolates were phenotypically non-susceptible to penicillin. Concordance between penicillin non-susceptibility and the presence of resistance conferring mutations was observed in 52% of the isolates. No known resistance conferring mutations were identified in the remaining penicillin non-susceptible strains, and the resistance observed may be due to the combination of other pbp and/or non-pbp mutations which may confer resistance in pneumococci (62). The presence of resistance conferring mutations among the 14 penicillin susceptible strains may indicate the poor predictability of these mutations for phenotypic resistance. Only P432T (in pbp1a) and T338P (in pbp2x) mutations were associated with phenotypic penicillin non-susceptibility in this study (Supplementary Table 3). The P432T and T338P mutations were detected in all nine penicillin-non-susceptible, ST705215B/15C isolates, from two infants. The P432T mutation, which is close to the 428SRN430 conserved motif (59), and the T338P mutation, occurring within the active 337STMK340 motif (63), decrease beta-lactam-binding affinity of PBP1a and PBP2x, respectively. The contribution of other mutations to penicillin non-susceptibility among our strains was unclear (Supplementary Table 3).
This study was limited by a small sample size and the use of metagenomic sequencing in samples obtained from a larger population is warranted. NP samples were enriched using broth culture without antibiotics which may have favored the growth of other bacteria over pneumococci, leading to low sequence reads counts and the inability to assemble the pneumococcal genomes in these samples.
This study indicated the utility of direct metagenomic sequencing of NP samples to privide in-depth understanding of pneumococcal carriage and antimicrobial resistance determinants. There was complete concordance between culture and shotgun sequencing, with a high concordance between in silico and conventional serotyping, indicating a predominance of non-PCV13 serotypes in this cohort. This approach was also able to identify co-colonization. Serotypes endemic to South Africa, several not reported locally before, and four novel serotypes were also identified. There was however a poor correlation between phenotypic antimicrobial non-susceptibility and the detection of certain resistance determinants. This technique will contribute to understanding increasing vaccine failure rates, by providing stain-level data for vaccine design strategies which are effective in controlling carriage, preventing invasive disease, and limiting the potential spread of resistance.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories: https://www.ebi.ac.uk/ena/browser/view/PRJEB37312.
The studies involving human participants were reviewed and approved by Human Research Ethics Committee of the Faculty of Health Sciences, University of Cape Town and the Western Cape Provincial Child Health Research Committee. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
CM, FD, MN, HZ, and WN designed the study. RM, FD, CM, and SM performed data acquisition. RM, FD, CM, SL, MW, KL, and SM analyzed the data. RM, FD, and CM produced the first draft. All authors revised the work, approved the final version, and took accountability for all aspects of the work.
This work was funded by grants from the Bill and Melinda Gates Foundation Global Health Grant (OPP1017641), H3Africa funded through the Office of Strategic Coordination/Office of the NIH Director, National Institute of Environmental Health Sciences and National Human Genome Institute of Health of the National Institutes of Health under Award Number U54HG009824 and 1U01HG006961, National Research Foundation of South Africa Thuthuka Grant (UID107410), and by a Pfizer Investigator Initiated Award (WI208622). RM was supported by the National Research Foundation of South Africa (UID95257 and UID111570). FD (UID99696) was supported by the National Research Foundation of South Africa, FIDSSA-GlaxoSmithKline Research Fellowships and received funding from the University of Cape Town Faculty Research Committee (FRC) Post-graduate Publication Incentive; HZ was supported by the SAMRC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Anne von Gottberg, Linda de Gouveia and the staff of the Center for Respiratory Diseases and Meningitis (CRDM), National Institute for Communicable Diseases (NICD) of the National Health Laboratory Service for training, sharing of standard operating procedures, supplying control isolates and performing Quellung serotyping. We thank the clinical research staff involved in the Drakenstein Child Lung Health Study for collection of samples and the children and parents for participating in the study. We further wish to thank Charmaine Barthus, Widaad Zemanay, Whitney Barnett, and the Drakenstein Child Lung Health Study team for their help and technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2020.543898/full#supplementary-material
1. Liu L, Johnson HL, Cousens S, Perin J, Scott S, Lawn JE, et al. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet. (2012) 379:2151–61. doi: 10.1016/S0140-6736(12)60560-1
2. Lawn JE, Cousens S, Zupan J. 4 million neonatal deaths: when? where? why? Lancet. (2005) 365:891–900. doi: 10.1016/S0140-6736(05)71048-5
3. Debbia EA, Schito GC, Zoratti A, Gualco L, Tonoli E, Marchese A. Epidemiology of major respiratory pathogens. J Chemother. (2001) 13(Suppl. 2):205–10. doi: 10.1179/joc.2001.13.Supplement-2.205
4. Rodríguez L, Cervantes E, Ortiz R. Malnutrition and gastrointestinal and respiratory infections in children: a public health problem. Int J Environ Res Public Health. (2011) 8:1174–205. doi: 10.3390/ijerph8041174
5. GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the global burden of disease study 2015. Lancet. (2016) 388:1459–544. doi: 10.1016/S0140-6736(16)31012-1
6. Simell B, Auranen K, Käyhty H, Goldblatt D, Dagan R, O'Brien KL, et al. The fundamental link between pneumococcal carriage and disease. Expert Rev Vaccines. (2012) 11:841–55. doi: 10.1586/erv.12.53
7. Mostowy RJ, Croucher NJ, De Maio N, Chewapreecha C, Salter SJ, Turner P, et al. Pneumococcal capsule synthesis locus cps as evolutionary hotspot with potential to generate novel serotypes by recombination. Mol Biol Evol. (2017) 34:2537–54. doi: 10.1093/molbev/msx173
8. Lo SW, Gladstone RA, van Tonder AJ, Hawkins PA, Kwambana-Adams B, Cornick JE, et al. Global distribution of invasive serotype 35D Streptococcus pneumoniae isolates following introduction of 13-valent pneumococcal conjugate vaccine. J Clin Microbiol. (2018) 56:e00228–18. doi: 10.1128/JCM.00228-18
9. Sá-Leão R, Nunes S, Brito-Avô A, Alves CR, Carriço JA, Saldanha J, et al. High rates of transmission of and colonization by Streptococcus pneumoniae and Haemophilus influenzae within a day care center revealed in a longitudinal study. J Clin Microbiol. (2008) 46:225–34. doi: 10.1128/JCM.01551-07
10. Dube FS, Ramjith J, Gardner-Lubbe S, Nduru P, Robberts FJL, Wolter N, et al. Longitudinal characterization of nasopharyngeal colonization with Streptococcus pneumoniae in a South African birth cohort post 13-valent pneumococcal conjugate vaccine implementation. Sci Rep. (2018) 8:12497. doi: 10.1038/s41598-018-30345-5
11. Kandasamy R, Gurung M, Thapa A, Ndimah S, Adhikari N, Murdoch DR, et al. Multi-serotype pneumococcal nasopharyngeal carriage prevalence in vaccine naïve Nepalese children, assessed using molecular serotyping. PLoS ONE. (2015) 10:e0114286. doi: 10.1371/journal.pone.0114286
12. Arvas A, Çokugraş H, Gür E, Gönüllü N, Taner Z, Tokman HB. Pneumococcal nasopharyngeal carriage in young healthy children after pneumococcal conjugate vaccine in Turkey. Balkan Med J. (2017) 34:362–6. doi: 10.4274/balkanmedj.2016.1256
13. Weinberger DM, Malley R, Lipsitch M. Serotype replacement in disease after pneumococcal vaccination. Lancet. (2011) 378:1962–73. doi: 10.1016/S0140-6736(10)62225-8
14. Song J-H. Advances in pneumococcal antibiotic resistance. Expert Rev Respir Med. (2013) 7:491–8. doi: 10.1586/17476348.2013.816572
15. Klugman KP. The significance of serotype replacement for pneumococcal disease and antibiotic resistance. Adv Exp Med Biol. (2009) 634:121–8. doi: 10.1007/978-0-387-79838-7_11
16. Lee S, Kim J-H, Kim S-H, Park M, Bae S. Prevalent multidrug-resistant nonvaccine serotypes in pneumococcal carriage of healthy Korean children associated with the low coverage of the seven-valent pneumococcal conjugate vaccine. Osong Public Heal Res Perspect. (2013) 4:316–22. doi: 10.1016/j.phrp.2013.10.004
17. Appelbaum PC. Resistance among Streptococcus pneumoniae: implications for drug selection. Clin Infect Dis. (2002) 34:1613–20. doi: 10.1086/340400
18. Behzad H, Gojobori T, Mineta K. Challenges and opportunities of airborne metagenomics. Genome Biol Evol. (2015) 7:1216–26. doi: 10.1093/gbe/evv064
19. Abdeldaim G, Herrmann B, Mölling P, Holmberg H, Blomberg J, Olcén P, et al. Usefulness of real-time PCR for lytA, ply, and Spn9802 on plasma samples for the diagnosis of pneumococcal pneumonia. Clin Microbiol Infect. (2010) 16:1135–41. doi: 10.1111/j.1469-0691.2009.03069.x
20. Habib M, Porter BD, Satzke C. Capsular serotyping of Streptococcus pneumoniae using the quellung reaction. J Vis Exp. (2014) 84:e51208. doi: 10.3791/51208
21. Satzke C, Dunne EM, Porter BD, Klugman KP, Mulholland EK, PneuCarriage Project Group. The pneucarriage project: a multi-center comparative study to identify the best serotyping methods for examining pneumococcal carriage in vaccine evaluation studies. PLoS Med. (2015) 12:e1001903. doi: 10.1371/journal.pmed.1001903
22. McAllister LJ, Ogunniyi AD, Stroeher UH, Leach AJ, Paton JC. Contribution of serotype and genetic background to virulence of serotype 3 and serogroup 11 pneumococcal isolates. Infect Immun. (2011) 79:4839–49. doi: 10.1128/IAI.05663-11
23. Thoendel M, Jeraldo P, Greenwood-Quaintance KE, Yao J, Chia N, Hanssen AD, et al. Impact of contaminating DNA in whole-genome amplification kits used for metagenomic shotgun sequencing for infection diagnosis. J Clin Microbiol. (2017) 55:1789–801. doi: 10.1128/JCM.02402-16
24. Wyres KL, Lambertsen LM, Croucher NJ, McGee L, von Gottberg A, Liñares J, et al. Pneumococcal capsular switching: a historical perspective. J Infect Dis. (2013) 207:439–49. doi: 10.1093/infdis/jis703
25. Neelakanta G, Sultana H. The use of metagenomic approaches to analyze changes in microbial communities. Microb Insights. (2013) 6:37–48. doi: 10.4137/MBI.S10819
26. Zar HJ, Barnett W, Myer L, Stein DJ, Nicol MP. Investigating the early-life determinants of illness in Africa: the drakenstein child health study. Thorax. (2014) 70:592–4. doi: 10.1136/thoraxjnl-2014-206242
27. Russell FM, Carapetis JR, Ketaiwai S, Kunabuli V, Taoi M, Biribo S, et al. Pneumococcal nasopharyngeal carriage and patterns of penicillin resistance in young children in Fiji. Ann Trop Paediatr. (2006) 26:187–97. doi: 10.1179/146532806X120273
28. Leung MH, Bryson K, Freystatter K, Pichon B, Edwards G, Charalambous BM, et al. Sequetyping: serotyping Streptococcus pneumoniae by a single PCR sequencing strategy. J Clin Microbiol. (2012) 50:2419–27. doi: 10.1128/JCM.06384-11
29. Dube FS, van Mens SP, Robberts L, Wolter N, Nicol P, Mafofo J, et al. Comparison of a real-time multiplex PCR and sequetyping assay for pneumococcal serotyping. PLoS ONE. (2015) 10:e0137349. doi: 10.1371/journal.pone.0137349
31. Wright MS, McCorrison J, Gomez AM, Beck E, Harkins D, Shankar J, et al. Strain level Streptococcus colonization patterns during the first year of life. Front Microbiol. (2017) 8:1661. doi: 10.3389/fmicb.2017.01661
32. Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. (2017) 27:824–34. doi: 10.1101/gr.213959.116
33. Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinf . (2010) 11:595. doi: 10.1186/1471-2105-11-595
34. Brinkac LM, Beck E, Inman J, Venepally P, Fouts DE, Sutton G. LOCUST: a custom sequence locus typer for classifying microbial isolates. Bioinformatics. (2017) 33:btx045. doi: 10.1093/bioinformatics/btx045
35. Sahl JW, Lemmer D, Travis J, Schupp J, Gillece J, Aziz M, et al. The Northern Arizona SNP pipeline (NASP): accurate, flexible, and rapid identification of SNPs in WGS datasets. Microb. Genom. (2016) 2. doi: 10.1101/037267
36. Liu K, Linder CR, Warnow T. RAxML and FastTree: comparing two methods for large-scale maximum likelihood phylogeny estimation. PLoS ONE. (2011) 6:e27731. doi: 10.1371/journal.pone.0027731
37. Gupta SK, Padmanabhan BR, Diene SM, Lopez-Rojas R, Kempf M, Landraud L, et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. (2014) 58:212–20. doi: 10.1128/AAC.01310-13
38. Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. (2002) 30:3059–66. doi: 10.1093/nar/gkf436
39. Manenzhe RI, Moodley C, Abdulgader SM, Robberts FJL, Zar HJ, Nicol MP, et al. Nasopharyngeal carriage of antimicrobial-resistant pneumococci in an intensively sampled South African birth cohort. Front Microbiol. (2019) 10:610. doi: 10.3389/fmicb.2019.00610
40. Cornick JE, Harris SR, Parry CM, Moore MJ, Jassi C, Kamng'ona A, et al. Genomic identification of a novel co-trimoxazole resistance genotype and its prevalence amongst Streptococcus pneumoniae in Malawi. J Antimicrob Chemother. (2014) 69:368–74. doi: 10.1093/jac/dkt384
41. Haasum Y, Strom K, Wehelie R, Luna V, Roberts MC, Maskell JP, et al. Amino acid repetitions in the dihydropteroate synthase of Streptococcus pneumoniae lead to sulfonamide resistance with limited effects on substrate Km. Antimicrob Agents Chemother. (2001) 45:805–9. doi: 10.1128/AAC.45.3.805-809.2001
42. Manenzhe RI, Dube FS, Wright M, Lennard K, Zar HJ, Mounaud S, et al. Longitudinal changes in the nasopharyngeal resistome of South African infants using shotgun metagenomic sequencing. PLoS ONE. (2020) 15:e0231887. doi: 10.1371/journal.pone.0231887
43. Mauffrey F, Fournier É, Demczuk W, Martin I, Mulvey M, Martineau C, et al. Comparison of sequential multiplex PCR, sequetyping and whole genome sequencing for serotyping of Streptococcus pneumoniae. PLoS ONE. (2017) 12:e0189163. doi: 10.1371/journal.pone.0189163
44. Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. (2017) 35:833–44. doi: 10.1038/nbt.3935
45. Ottesen A, Ramachandran P, Reed E, White JR, Hasan N, Subramanian P, et al. Enrichment dynamics of Listeria monocytogenes and the associated microbiome from naturally contaminated ice cream linked to a listeriosis outbreak. BMC Microbiol. (2016) 16:275. doi: 10.1186/s12866-016-0894-1
46. Murad C, Dunne EM, Sudigdoadi S, Fadlyana E, Tarigan R, Pell CL, et al. Pneumococcal carriage, density, and co-colonization dynamics: a longitudinal study in Indonesian infants. Int J Infect Dis. (2019) 86:73–81. doi: 10.1016/j.ijid.2019.06.024
47. Ndlangisa KM, du Plessis M, Wolter N, de Gouveia L, Klugman KP, von Gottberg A, et al. Population snapshot of Streptococcus pneumoniae causing invasive disease in south africa prior to introduction of pneumococcal conjugate vaccines. PLoS ONE. (2014) 9:e107666. doi: 10.1371/journal.pone.0107666
48. Nurse-Lucas M, McGee L, Hawkins PA, Swanston WH, Akpaka PE. Serotypes and genotypes of Streptococcus pneumoniae isolates from trinidad and tobago. Int J Infect Dis. (2016) 46:100–6. doi: 10.1016/j.ijid.2016.04.005
49. Kamng'ona AW, Hinds J, Bar-Zeev N, Gould KA, Chaguza C, Msefula C, et al. High multiple carriage and emergence of Streptococcus pneumoniae vaccine serotype variants in Malawian children. BMC Infect Dis. (2015) 15:234. doi: 10.1186/s12879-015-0980-2
50. Tabatabaei SR, Karimi A, Fallah F, Shiva F, Shamshiri M, Gooya MM, et al. Rate of co-colonization with serotypes of strep pneumonia isolated from nasopharyngeal swab. Int J Infect Dis. (2014) 21:418. doi: 10.1016/j.ijid.2014.03.1284
51. Brugger SD, Frey P, Aebi S, Hinds J, Mühlemann K. Multiple colonization with S. pneumoniae before and after introduction of the seven-valent conjugated pneumococcal polysaccharide vaccine. PLoS ONE. (2010) 5:e11638. doi: 10.1371/journal.pone.0011638
52. Spratt BG, Hanage WP, Feil EJ. The relative contributions of recombination and point mutation to the diversification of bacterial clones. Curr Opin Microbiol. (2001) 4:602–6. doi: 10.1016/S1369-5274(00)00257-5
53. Vester B, Douthwaite S. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob Agents Chemother. (2001) 45:1–12. doi: 10.1128/AAC.45.1.1-12.2001
54. Diawara I, Zerouali K, Katfy K, Barguigua A, Belabbes H, Timinouni M, et al. Phenotypic and genotypic characterization of Streptococcus pneumoniae resistant to macrolide in Casablanca, Morocco. Infect Genet Evol. (2016) 40:200–4. doi: 10.1016/j.meegid.2016.03.003
55. Farrell DJ, Couturier C, Hryniewicz W. Distribution and antibacterial susceptibility of macrolide resistance genotypes in Streptococcus pneumoniae: PROTEKT year 5 (2003–2004). Int J Antimicrob Agents. (2008) 31:245–9. doi: 10.1016/j.ijantimicag.2007.10.022
56. Metcalf BJ, Gertz RE, Gladstone RA, Walker H, Sherwood LK, Jackson D, et al. Strain features and distributions in pneumococci from children with invasive disease before and after 13-valent conjugate vaccine implementation in the USA. Clin Microbiol Infect. (2016) 22:60.e9–29. doi: 10.1016/j.cmi.2015.08.027
57. Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol Rev. (2006) 30:673–91. doi: 10.1111/j.1574-6976.2006.00024.x
58. Nagai K, Davies TA, Jacobs MR, Appelbaum PC. Effects of amino acid alterations in penicillin-binding proteins (PBPs) 1a, 2b, and 2x on PBP affinities of penicillin, ampicillin, amoxicillin, cefditoren, cefuroxime, cefprozil, and cefaclor in 18 clinical isolates of penicillin-susceptible, -intermediate, and -resistant pneumococci. Antimicrob Agents Chemother. (2002) 46:1273–80. doi: 10.1128/AAC.46.5.1273-1280.2002
59. Biçmen M, Gülay Z, Ramaswamy SV, Musher DM, Gür D. Analysis of mutations in the pbp genes of penicillin-non-susceptible pneumococci from Turkey. Clin Microbiol Infect. (2006) 12:150–5. doi: 10.1111/j.1469-0691.2005.01334.x
60. Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. (2001) 293:498–506. doi: 10.1126/science.1061217
61. Trzciński K, Thompson CM, Lipsitch M. Single-step capsular transformation and acquisition of penicillin resistance in Streptococcus pneumoniae. J Bacteriol. (2004) 186:3447–52. doi: 10.1128/JB.186.11.3447-3452.2004
62. Granger D, Boily-Larouche G, Turgeon P, Weiss K, Roger M. Genetic analysis of pbp2x in clinical Streptococcus pneumoniae isolates in Quebec, Canada. J Antimicrob Chemother. (2005) 55:832–9. doi: 10.1093/jac/dki118
Keywords: nasopharyngeal, Streptococcus pneumoniae, pneumococcal conjugate vaccine, serotypes, shotgun metagenomic sequencing, resistance determinants, multi locus sequence typing
Citation: Manenzhe RI, Dube FS, Wright M, Lennard K, Mounaud S, Lo SW, Zar HJ, Nierman WC, Nicol MP and Moodley C (2020) Characterization of Pneumococcal Colonization Dynamics and Antimicrobial Resistance Using Shotgun Metagenomic Sequencing in Intensively Sampled South African Infants Front. Public Health 8:543898. doi: 10.3389/fpubh.2020.543898
Received: 20 March 2020; Accepted: 17 August 2020;
Published: 22 September 2020.
Edited by:
Kai Zhou, Zhejiang University, ChinaReviewed by:
Raquel Sá-Leão, New University of Lisbon, PortugalCopyright © 2020 Manenzhe, Dube, Wright, Lennard, Mounaud, Lo, Zar, Nierman, Nicol and Moodley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felix S. Dube, c2l6d2UuZHViZUB1Y3QuYWMuemE=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.