
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Public Health , 21 August 2019
Sec. Infectious Diseases – Surveillance, Prevention and Treatment
Volume 7 - 2019 | https://doi.org/10.3389/fpubh.2019.00235
This article is part of the Research Topic Pathogen Genomics: Empowering Infectious Disease Surveillance and Outbreak Investigations View all 14 articles
 Helena M. B. Seth-Smith1,2,3
Helena M. B. Seth-Smith1,2,3 Adrian Egli1,2*
Adrian Egli1,2*Corynebacterium diphtheriae (C. diphtheriae) is a relatively rare pathogen in most Western countries. While toxin producing strains can cause pharyngeal diphtheria with potentially fatal outcomes, the more common presentation is wound infections. The diphtheria toxin is encoded on a prophage and can also be carried by Corynebacterium ulcerans and Corynebacterium pseudotuberculosis. Currently, across Europe, infections are mainly diagnosed in travelers and refugees from regions where diphtheria is more endemic, patients from urban areas with poor hygiene, and intravenous drug users. About half of the cases are non-toxin producing isolates. Rapid identification of the bacterial pathogen and toxin production is a critical element of patient and outbreak management. Beside the immediate clinical management of the patient, public health agencies should be informed of toxigenic C. diphtheriae diagnoses as soon as possible. The collection of case-related epidemiological data from the patient is often challenging due to language barriers and social circumstances. However, information on patient contacts, vaccine status and travel/refugee route, where appropriate, is critical, and should be documented. In addition, isolates should be characterized using high resolution typing, in order to identify transmissions and outbreaks. In recent years, whole genome sequencing (WGS) has become the gold standard of high-resolution typing methods, allowing detailed investigations of pathogen transmissions. De-centralized sequencing strategies with redundancy in sequencing capacities, followed by data exchange may be a valuable future option, especially since WGS becomes more available and portable. In this context, the sharing of sequence data, using public available platforms, is essential. A close interaction between microbiology laboratories, treating physicians, refugee centers, social workers, and public health officials is a key element in successful management of suspected outbreaks. Analyzing bacterial isolates at reference centers may further help to provide more specialized microbiological techniques and to standardize information, but this is also more time consuming during an outbreak. Centralized communication strategies between public health agencies and laboratories helps considerably in establishing and coordinating effective surveillance and infection control. We review the current literature on high-resolution typing of C. diphtheriae and share our own experience with the coordination of a Swiss-German outbreak.
In recent years, rare but hypervirulent pathogens have been increasingly reported in specific geographic regions (1, 2), often associated with refugees and asylum seekers (3), but also in other high-risk populations including hospitalized patients (4, 5), the elderly, and newborns (6, 7). Reports of infections in refugees over the past decade have included Borrelia recurrentis (8), methicillin resistant Staphylococcus aureus (S. aureus) (MRSA) (9, 10), and toxigenic Corynebacterium diphtheriae (11). In 2016, the European Center for Disease Control (ECDC) warned about increased rates of cutaneous C. diphtheriae infections in Europe due to the refugee crisis (12). This pathogen came back into the focus of attention as it is (i) associated with severe infections in humans, including respiratory diphtheria (13–15); (ii) highly transmittable, indicated by the basis reproduction number with mean 7.2 (16); and (iii) known to cause larger outbreaks (17–19). For nearly two decades, in most high-income countries, cases have been reported rarely, occasionally in travel returners (20–24), drug users and homeless people (25–29). In the last few years, in contrast, cutaneous, and respiratory infections have predominantly been reported in refugees (16, 30–38).
Providing state-of-the-art diagnostics for rare and unexpected pathogens can be a challenge for the clinician (39) and the routine microbiology laboratory (40–42). Often specific diagnostic tests are only available in reference laboratories, thus further delaying efficient therapy, surveillance reporting, and outbreak management. Once the pathogen is cultured and identified, molecular typing technologies, such as whole genome sequencing (WGS), allow a detailed comparison on the genomic level with high resolution (43–45). In the case of C. diphtheriae, high-resolution typing is helpful to (i) provide the epidemiological broader context (35) and (ii) include or exclude transmission events between patients (30, 31).
WGS specifically, gives the highest resolution typing, and can help to identify potential sources and transmission routes as part of modern surveillance technologies. Recent comparisons using WGS data analyzed by core genome MLST (cgMLST) or single nucleotide polymorphisms (SNP)-based methods have shown significant improvements over older technologies (46, 47). The advantages of using WGS for high-resolution typing has been seen in several pathogens, being particularly helpful in settings with (i) highly similar isolates over a long time period e.g., Legionella pneumophila within a city (48) or C. difficile (49, 50), (ii) a low endemic epidemiological background, but multiple clusters of patients from high endemic region with potential transmission events e.g., C. diphtheriae (31) or M. tuberculosis (51), and (iii) high endemic burden, where transmission events cannot easily be separated based on classical epidemiological information alone.
Alongside the availability of rapid diagnostic tests and high-resolution typing, surveillance programs are an important cornerstone of public health, as the associated framework allows data collection, communication, and coordination of public health interventions. Of note, to date no global or European surveillance network exists which integrates both classical and molecular epidemiological data into a single real-time updated platform. Future surveillance programs may not only incorporate baseline features of an isolate such as sequence type and presence or absence of the tox gene, but also more detailed genomic analysis and a virulence factor profile. The aim of this would be to better assess the potential of a strain to cause outbreaks with more severe clinical phenotypes. In this review article, we will focus on C. diphtheriae as a re-emerging but rare pathogen, and will discuss the various aspects of classical and molecular epidemiology utilizing new sequencing technologies for surveillance.
Corynebacterium diphtheriae was first isolated in 1884 by Loeffler (52). The classical presentation is pharyngeal diphtheria, a toxin-mediated infectious disease of the upper respiratory tract. The hallmark feature is an inflamed pseudo-membrane on the pharynx, potentially causing asphyxia (13). Beside respiratory infections, C. diphtheriae may cause skin infections and other invasive diseases such as endocarditis, osteomyelitis, and septic arthritis (53–58). At the moment, non-toxigenic cutaneous diphtheria is the most prevalent clinical presentation (24, 39, 57, 59, 60). Wound infections often occur with other skin pathogens, such as Streptococcus pyogenes or S. aureus (28, 31). Cutaneous diphtheria may be a source of toxigenic pathogens and may be transferred to other body sites then potentially causing respiratory diphtheria. Therefore, even wound infections with non-toxigenic strains might ideally be considered to be reported to surveillance programs in order to identify carriers, clusters of potential transmissions, and high-risk groups.
The species C. diphtheriae is divided into four biochemical biovars—belfanti, gravis, intermedius, and mitis (15, 61). Although the biochemical distinctions are not reliable, for historical reasons reference laboratories still use them. Recently, two distinct subspecies have been proposed based on genomic features: C. diphtheriae subsp. diphtheriae and C. diphtheriae subsp. lausannense. Of interest, members of the newly described subspecies lausannense show a larger genome size and are enriched in genes related to transport and metabolism of lipids and inorganic ion (62). On the other hand, the new subspecies lacks all genes involved in the synthesis of pili, molybdenum cofactor, and nitrate reductase. Closely related to C. diphtheriae are two zoonotic pathogens, C. ulcerans and C. pseudotuberculosis (63), both of which can acquire the toxin gene via a phage (64). Increasing numbers of toxigenic C. ulcerans infections have been reported (65, 66) e.g., in the UK (67), but these pathogens remain rare in the clinic. Host jumps from domesticated and wild animals to humans have been postulated (63, 68, 69). If either C. ulcerans or C. pseudotuberculosis is diagnosed, the isolate should be tested for the presence of the toxin and reported in surveillance programs.
The ß-corynephage encodes the diphtheria toxin, and can be transmitted between isolates. The ß-corynephage may pose a survival benefit for the bacterium by increasing the effectiveness of transmission by helping to cause local tissue damage (14, 70). The DtxR regulator is present elsewhere in the genome, and controls the transcription of the toxin gene (tox). This regulator is a key determinant for iron homeostasis (71). Iron is crucial for a number of cellular functions and the expression of a toxin in situations with low iron concentrations might help pathogens to compete with the host for iron or release iron via lysis of host cells. Of particular importance are pili encoded by spa operons (spaABC, spaDEF, and spaHIG), which contribute to the interaction with the host. Gain or loss of the function of these genes correlate to the number and expression of pili on the cell surface—especially the major pilin genes spaA, spaD, and spaH. The spaA- spaD- and spaH-type pili interact with the pharyngeal, laryngeal, and lung epithelial cell types, respectively (72). Pilus expression may strongly influence the virulence of a strain (73–78), especially in combination with the presence of the tox gene.
Specific culture media such as tellurite agar improves the culture of C. diphtheriae (61, 79)—although the agar adds some selection, most diagnostic laboratories do not carry the agar as part of routine stock. The three species of interest, C. diphtheriae, C. ulcerans, and C. pseudotuberculosis, can be reliable identified with matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) (80–82). More specialized laboratories have the capacity to detect the presence of the diphtheria toxin either by PCR or measurement of toxin production by a modified Elek test (31, 83, 84). Other virulence factors such as pili are generally not determined in routine diagnostics. A survey of the diphtheria surveillance network (DIPNET) indicated that many centers were not able to isolate the target organisms, and most found difficulties differentiating them from specimens that contained Corynebacterium striatum, a commensal contaminant (85). More recently, an ECDC technical report on the diagnostic gaps has been published (86). Regular workshops and external quality assessments are important aspects in maintaining diagnostic quality for rare pathogens in the context of a surveillance program.
In 2016 the EDSN reported 47 laboratory confirmed cases of C. diphtheriae and C. ulcerans in European countries—corresponding to an overall notification rate below 0.01 cases per 100,000 people (66). In contrast, worldwide, 7097 diphtheria cases were reported in 2016, mainly in low-income countries (www.who.it). In the 1900s−1950s, infections with C. diphtheriae were among the most severe infections during childhood, especially in pre-school children where case fatality rates of 2–25% were reported (87, 88). Prior to the availability of toxoid-vaccines, nearly 70% of the cases were in children younger than 15 years of age (89). With the introduction of vaccines in the 1940s and 1950s, a significant decrease in incidence was observed (87, 88, 90–93), although no controlled clinical trial to evaluate the efficacy of the toxoid-vaccines in preventing diphtheria has ever been performed.
The current WHO recommendation states that a series of three toxoid-vaccine doses should be provided, starting at six weeks of age, with additional booster doses based on local epidemiology (94). Vaccine effectiveness is high after three or more doses, ranging from 96 to 98% (95, 96). Although not assessed in routine, an antitoxin level of 0.01 IU/mL provides the lowest level of protection, 0.1 IU/mL is considered a protective level, and levels of >1.0 IU/mL result with long term protection (61). Interestingly, two cases of fatal diphtheria in patients with antitoxin levels above 30 IU/mL have been reported, suggesting that no absolute protection exists (97). Although immunization programs of infants started in the late 1970s, the vaccine coverage rates of infants in developing countries increased only slowly from 46% in 1985 to 79% in 1992 (98). If vaccines rates in the general population are too low, herd immunity fails to protect the non-vaccinated population, resulting in outbreaks with the potential for high mortality in younger and older age groups. An assessment of the immunity against a series of pathogens in adult asylum seekers in the Netherlands showed median 82% seroprotective anti-toxin titers against diphtheria (99). Although diphtheria vaccine rates in infants range from 89 to 98% in most European countries, a recent meta-analysis showed that vaccine rates against diphtheria and tetanus toxoids, and acellular pertussis (dTap) in healthcare workers was only 45.1% in the US and 63.9% in France (100). In Luxembourg only 2.5% of individuals under the age of 20 were seronegative, while 42% of individuals over the age of 40 years were seronegative (101). Similar low seroprotection rates have been documented in China, where only 34.1% of subjects older than 40 years were seroprotected (102). The reason for low seroprotection in some population groups in countries, where the vaccine is available, may result in a decrease in circulating toxigenic C. diphtheriae isolates (89), resulting in (i) an increase in non-toxigenic cases (103), and (ii) lower natural boost effects of antibody titers against the toxin (104). Especially in the adult population, gaps in herd immunity have been described due to waning of protective antibodies either from lower natural exposure or booster-vaccination. It has been found that the diphtheria vaccination only prevents symptomatic infection, and does not inhibit carriage or transmission of the pathogen. Miller and colleagues have shown that a high percentage of C. diphtheriae carriers were fully vaccinated, suggesting that antibodies against the toxin does not inhibit nasopharyngeal colonization (93). Based on this data, we may conclude that adults and the elderly are at higher risk of C. diphtheriae infection. Regular assessment of seroprotection rates in a given population should be a part of surveillance programs.
In the 1960–1970s, any outbreaks described in high income countries were smaller (92, 105–108) in comparison to the larger outbreaks which occurred in the late 1990s and early 2000s, particularly in countries of the former Soviet Union (17–19, 109–113). A very large outbreak affected states of the former Soviet Union with more than 150,000 infected people and between 3,000 and 5,000 deaths (18). In this outbreak, a high proportion of adults were affected, potentially due to disruption of health services resulting in poor vaccine coverage (114, 115) and reduced “natural” exposure over the preceding decades, resulting in antibody titers below protective levels (116–118). In recent years, multiple outbreaks, or potential transmission clusters have been reported in: Bangladesh (119, 120), Brazil (121), Colombia (122), Germany (30, 35), India (123–125), Indonesia (126), Laos (127), Norway (128), Nigeria (129), Poland (130), Spain (38), South Africa (36, 131), Syria (132), Switzerland (31), Thailand (114), the United Kingdom (37), Venezuela (133, 134), and Yemen (135). The global list of affected countries indicates that (i) the disease is remains poorly controlled, (ii) the main burden lies in low-income countries, and (iii) local and global surveillance should be intensified in order to better control the disease.
Some of the key factors driving the spread of hypervirulent pathogens include poor vaccine rates, waning antibody titers, reduced access to healthcare, failing, or collapsing healthcare systems, poor hygiene, transfer of patients between healthcare institutions, changes in travel behaviors, increased traveling to high endemic regions, and migration from high endemic regions due to violent conflicts or for economic reasons (136–138). The development of effective preventative strategies to reduce the impact of hypervirulent bacteria should, as for multidrug resistant (MDR) pathogens, have a top global priority among public health experts, clinical microbiologists, and infectious diseases physicians. The basis for preventative strategies relies on two key elements: classical and molecular epidemiological data.
Classical epidemiological methods are used to investigate an unexpected frequency of specific pathogens clustering within a certain time and/or geographical range. Determining a case definition is an important first step. Cases have to be confirmed, background rates established, and patient data collected via, for example, structured questionnaire, and accessing detailed medical history. Thus, a hypothesis for the disease transmission can be formulated and potential sources named (139, 140). Although classical epidemiological methodologies provide tremendously important information, data collection is often challenging due to delayed or incomplete reporting of cases, lack of centralized communication strategies, especially at the beginning of an outbreak, vague medical history, language barriers, and cultural differences. Especially in the case of refugees, where classical epidemiological data are often not reliable, available or re-constructible, in many cases classical methods cannot provide the required data.
Molecular epidemiological methods are based on detailed comparison of pathogens, using some or all of the genomic information. The relatedness of pathogens can be visualized in trees, thereby helping to cluster isolates and provide information on potential molecular epidemiological links. Several genotyping approaches have been used for C. diphtheriae including ribotyping, amplified fragment length polymorphisms, PFGE, random amplified polymorphic DNA (RAPD), clustered regularly interspaced short palindromic repeat (CRISPR)-based spoligotyping and MLST (141–149). Some typing methods show better resolution than others: ribotyping outperforms PFGE and AFLP in terms of discriminatory power (143). Ribotyping was for many years considered the gold-standard before the introduction of a robust MLST approach. Many ribotypes were allocated a geographical name based on the location of the initial isolate, however some followed an arbitrary nomenclature (144). CRISPR-based spoligotyping can offer additional resolution within ribotypes, and be used successfully to further characterize outbreak-associated strains (147, 148): the epidemic strains from the former Soviet Union belonged to two ribotypes (St. Petersburg and Rossija) that could be subdivided into 45 additional spoligotypes (146, 147). Data from various outbreaks shows the relative high molecular diversity of isolates indicating that new strains are emerging regularly within this species (150).
A robust MLST scheme was developed in 2010, including the genes atpA, dnaE, dnaK, fusA, leuA, odhA, and rpoB (www.pubmlst.org/cdiphtheriae). The advantages of an MLST scheme include transferability and comparability. The sequence types were shown to be consistent with the previously determined ribotypes and offered higher resolution in most cases (141). MLST diversity has grown continuously, with 608 types currently categorized (March 2019). Of note, the MLST scheme lacks the biochemical correlation of the biovar system and STs have not been able to be associated with a more severe clinical phenotype (141, 151, 152).
Comparison of the performance of various typing techniques is important, as low resolution typing methods may overcall transmission events masking the real transmission steps and potentially delaying the identification of the source. Stucki et al. showed this for M. tuberculosis transmissions events in Switzerland, where a VNTR low-resolution typing gave evidence of a significantly higher rate of transmissions events in comparison to WGS based typing on the same set of isolates (153). Similarly, C. diphtheriae SNP-based WGS comparisons improved the typing resolution in comparison to cgMLST (35).
The first complete genome sequence of C. diphtheriae (strain NCTC13129) was analyzed in 2003, a UK clinical isolate containing a series of pathogenicity factors including iron-uptake systems, adhesins and fimbrial proteins (154). The genome of C. diphtheriae is 2.45 Mbp with a G+C content of 53.5% (154). Through WGS analysis we can determine the presence of virulence factors such as the toxin gene (and ß-corynephage) and pili, and genes encoding antimicrobial resistance determinants (62, 155, 156). During outbreak and public health investigations, WGS SNP-based typing clearly shows important benefits due to its high resolution (31). Although MLST may be more cost effective, MLST data can also be extracted from WGS data, providing the ST as well as high resolution phylogeny and additional important genetic information. WGS can identify additional toxins and adherence factors, which may allow the generation of a specific risk profile for the pathogen.
Comparative studies have shown that the species has a set of ~1,630 core genes which almost every representative of this species possesses [60% of the genome], and a relatively large, open pan-genome (155, 156). The difference in genome content across the species is largely due to the presence of genomic islands, prophages, transposons, restriction-modification systems, and CRISPR elements. Horizontal transfer substantially helps to shape the bacterial genome (62, 155). Some of the identified genomic islands carry genes for siderophore synthesis and transportation and degradation of polysaccharides, and heavy metal resistance. Interestingly, prophages are genetically more similar within specific clusters of bacterial isolates than between clusters, suggesting that prophages do not randomly mix between isolates, but rather cluster within specific clades (31, 157, 158).
While MLST analysis first suggested, that there is significant recombination within C. diphtheriae (141), this has been confirmed through analysis of whole genome sequences (159). Recombination plays an important role in bacterial evolution and has been linked to increased virulence in some pathogens (160–162). Especially in the upper respiratory tract, where C. diphtheriae can form a colonizing state, horizontal gene transfer can commonly happen (163). WGS allowed to study genetic ancestry of multiple bacterial species—including C. diphtheriae. This challenged sometimes our current understanding and groups based on biochemistry or serotypes may change. As an example, it has also been shown that biovars of C. diphtheriae do not correlate to genetic ancestry (152, 159). In recent years, several cohorts of C. diphtheriae isolates have been analyzed using WGS (30, 31, 35, 36, 62, 152, 155, 156, 164–167). Comparison of WGS data across a species generally uses one of two approaches: cgMLST, or SNP-based variant calling across the whole genome based on a reference, which provides more information and higher resolution. Dangel et al. have generated a cgMLST scheme including 1553 target loci and an extended cgMLST scheme including 2154 target loci, providing higher resolution (35).
cgMLST and SNP-based analyses of all publicly available whole genome sequences (Figures 1, 2 and Supplementary Table 1) shows vast diversity, and geographic mixing: isolates identified in Malaysia, India, Australia, and Switzerland are found throughout the trees. Relatively few cgMLST clusters are defined at the five allele cut off, yet some clades/clusters clearly show geographic association, such as those from South Africa, Belarus and Germany (35), suggestive of local outbreaks. The largest clade of highly related isolates, at the top of Figure 2, includes those from Germany, Poland, the UK and the former Soviet Union, suggesting that these may have had a common source, but spread prior to diagnosis (This clustering is not represented in the minimum spanning tree of Figure 1). However, the dates of the isolates in this clade range from 1996 to 2017, also suggesting some stability of the isolates over time. This is also evidenced as closely related isolates throughout the tree may have been isolated many decades apart.
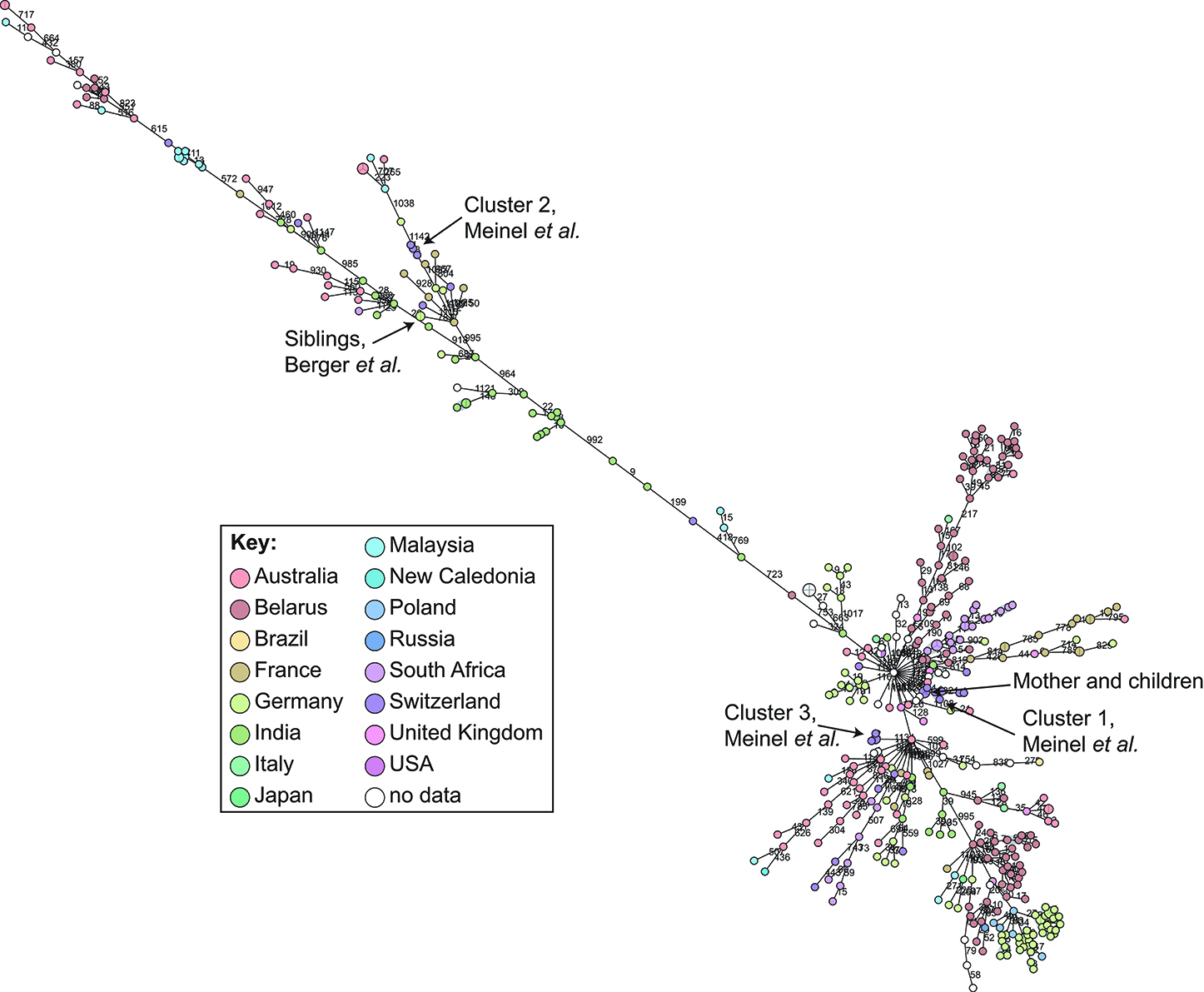
Figure 1. Minimum spanning tree showing relationship of all available Corynebacterium diphtheriae genomes by cgMLST. All C. diphtheriae assemblies available from NCBI on 27.02.2019 were retrieved, and compared to isolates from our laboratory, as well as all reads available from NCBI on 08.10.2018, and those published, which were assembled using unicycler (168), and duplicate samples removed, giving n = 419 genomes. The cgMLST scheme of (35) was used within Ridom SeqSphere+ v4.1.6, with clusters given between nodes with five or fewer differences. Nodes are colored according to country of isolation.
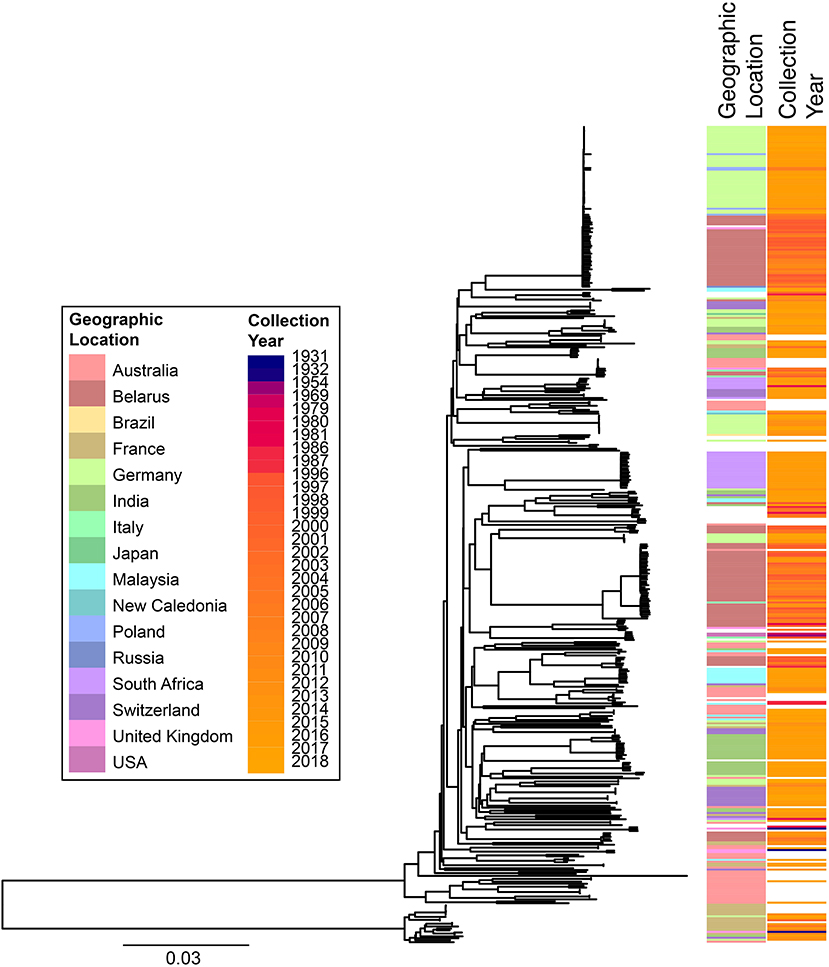
Figure 2. Phylogenetic overview of all available C. diphtheriae genomes. All C. diphtheriae reads available from NCBI on 08.10.2018 were retrieved, and those published, and compared to isolates from our laboratory, as well as all assemblies available from NCBI on 27.02.2019, which were shredded to reads using wgsim in samtools (https://github.com/lh3/wgsim), and duplicate samples removed, giving n = 419 genomes. All reads were mapped against the reference genome CP003210 (155) within CLC Genomics Workbench 10.1.1, also used to generate a single nucleotide polymorphism (SNP) phylogeny with parameters that differed from the default as: variant calling with 10x minimum coverage, 10 minimum count and 70% minimum frequency, and SNP tree creation with 10x minimum coverage, 10% minimum coverage, 0 prune distance and including multi-nucleotide variants (MNVs). Metadata was retrieved from the NCBI database and was associated with the phylogeny using phandango (169). Colors use the same key as Figure 1; years are shown on a continuous scale. The bottom clade shows the clearly separate cluster proposed as C. diphtheriae subsp. lausannense.
There is an ongoing debate about defining diversity thresholds to separate clusters of pathogens. Determining a threshold of diversity to reliably describe a transmission cluster is a question commonly asked, yet difficult to answer, particularly in recombinogenic bacteria. Dangel et al. defined a cluster in their cgMLST scheme as five or fewer allele differences, with higher resolution of subclusters analyzed through an extended cgMLST scheme (35).
In order to determine a reliable cut-off, it is beneficial to combine the genomic analysis with more classical epidemiological data, which significantly contributes to understanding the transmission risks. However, in the literature and epidemiological data associated with WGS, few such cases have been described: in one case of direct transmission between siblings, the isolates show no allele differences in the defined core genome or accessory genome (30); and one case of direct transmission from mother to twin newborns showed a single SNP between the isolates on a whole genome level (unpublished data) and zero allele differences in the cgMLST scheme (Figure 1).
During our study on isolates from refugees in Basel, we asked ourselves if the observed whole genome diversity of 50–150 SNPs within clusters could represent a recent transmission event. We considered two different mutation rates representing extremes of plausible ranges, and estimated the approximate transmission dynamic. Even using a very high mutational rate of 0.00018 substitutions/bp/year, the estimation indicated that transmission occurred more than four to 6 weeks prior to sampling. In that paper, we played with substitution rates an picked the mutation rate of Helicobacter pylori, in order to have a highly conservative estimated if the transmission occurred on European ground to trigger potential outbreak investigations. This helped to exclude a transmission event within Europe, as the affected refugees arrived 2 weeks prior in South Italy (31). Analyzing these clusters by cgMLST shows that the isolates diverge by 0–4 alleles (Figure 1), within the cluster threshold, despite possessing at least 50 SNP differences and not representing recent transmission (31). This exemplifies the increased resolution of using whole genome SNP-based methods, and the difficulty of inferring direct transmissions from cgMLST data alone. As C. diphtheriae can also undergo recombination, it is crucial to consider a recent recombination by studying the distribution of SNPs across the genome: if many SNPs cluster in one or more genomic loci, then a recombination event is likely to have occurred, bringing the putative transmission event more recent.
Although country specific surveillance systems for hypervirulent pathogens such as C. diphtheriae exist, the interoperability of data and the exchange across countries presents problems (170). In 2014, a WHO-recommended surveillance standard of diphtheria was published. This included a case definition, laboratory criteria for diagnosis, and minimum data elements which should be collected (171). Similarly, the ECDC has established a surveillance program for diphtheria. Founded in 1993 as European Laboratory Working Group in Diphtheria in 2006 it became the European Diphtheria Surveillance Network (EDSN, www.ecdc.europa.eu) (172). The network provides valuable information and aims to standardize surveillance activities and ensure availability of more comparable data between countries. It also includes laboratory components focusing on trainings and external quality assessments (EQAs), strengthening the laboratory capacity to characterize isolates and develop novel tools for molecular typing of C. diphtheriae.
While the EDSN provides an important framework for surveillance of C. diphtheriae, in the current refugee crisis, multi-national coordination of outbreak investigation is clearly a challenge. Rapid and effective mechanisms of communication are crucial. Patients may be evaluated several times on their journey, and the same pathogen may be isolated in different countries. A recent report on the tracing of an MDR M. tuberculosis cluster was very well-coordinated by a joint effort from multiple centers (51). Similarly, for C. diphtheriae, we directed an investigation with multiple refugees presenting with wound infection across different hospitals and diagnostic laboratories in Switzerland in 2015 (31). In both situations, a multi-national taskforce organized a coordinated effort to collect isolates and information, using case report forms to collect structured epidemiological information on migration routes, vaccine status, and other affected travelers.
Individual responsible experts, such as representatives of the EDSN or reference laboratories, should be assigned in each country in order to keep track of potential movements of refugees. In the C. diphtheriae situation, refugees were rapidly lost to follow-up, for example due to relocation in other refugee centers. Communication to refugee center responsible personnel and physicians should be established. The molecular epidemiology of diphtheria would certainly benefit from implementation of WGS. Such analysis offers improvements over the current model of global tracing of large clonal clusters toward fine-tuned strain discrimination. At the same time, a multicenter evaluation of recently developed inexpensive and discriminatory VNTR and CRISPR methods is warranted to see if and how they could complement regional surveillance (150). Beside the molecular definition of an outbreak, a centralized database allows running the standardized bioinformatic algorithms and thereby may provide a benefit for investigations. Isolates could be registered with particular coded identifiers to avoid re-sequencing the same isolate (173).
To date, no database can integrate classical epidemiological data in the form of coded patient identification, vaccine status, potential exposures, spatiotemporal information of cases, socioeconomic and immunological data on a population level, with high-resolution molecular epidemiological data from sequenced strains. We are developing such a platform, initially for MDR pathogens (173), which could easily be expanded to hypervirulent species including C. diphtheriae. This Swiss Pathogen Surveillance Platform (www.spsp.ch) aims to integrate all relevant data in the near future, thereby providing various stakeholders with important information in real-time. Such a platform may provide a public health data sharing hub not only for Switzerland, but for European countries and beyond.
In many countries, reporting of C. diphtheriae cases to public health authorities is mandatory. Information is collected and reported back to the diagnostic laboratories and infectious diseases specialist in order to heighten awareness. Various email alerting system for surveillance exists, one of the most well-known being PROMED (https://www.promedmail.org/), a subscription service which has been in place since the early 2000s (174). Those warning systems collect information from media reports, official reports, online summaries, local observers, subscribers, and others. However, those services rely on reporting toward the service and also inaccurate interpretation and privacy issues may be an issue. Nevertheless, there is still room for faster, more targeted and international ways of communication to be established. The connection of various data sources will require the usage of standardized and specific epidemiological ontologies being used across various databases such as SNOMED CT (www.snomed.org) or IRIDA (www.irida.ca). The ethical and legal implications of such big-data driven surveillance programs need to be clarified in the near future. Clearly individual patient data should be protected, but those rights should be balanced in situations where outbreaks with hypervirulent pathogens may put the general population at risk—in the case of C. diphtheriae the risk for the general healthy population in Western countries seems rather low and therefore surveillance efforts should rather focus on at-risk populations. Social media may be used to generate epidemiological data but could also be used as a tool to inform the general public and health care specialists. We could imagine internet-based warning systems being combined with a more detailed platform allowing clinicians to assess classical and molecular epidemiological aspects.
In the near future, we can foresee interconnected databases containing epidemiological data on individual cases, incidence rates of particular infections, spatiotemporal clusters, WGS data, travel and migration information, social and print media reports, and vaccine rates in populations. These may then be used for machine learning based epidemiological surveillance, such as that recently published on prediction of dengue outbreaks (175).
Machine learning based algorithms may also be used to predict the case severity of a particular infection based on NGS and other clinical data, as similar performed by Njage et al. in the case of shigatoxigenic E. coli (176). Bacterial genome wide association studies (GWAS) using machine learning in C. diphtheriae may help to identify critical biomarkers, linking bacterial genomic features such as virulence or resistance with specific host outcomes. Such work often requires hundreds to thousands of bacterial genomes to compensate for host variability effects (177) as shown for M. tuberculosis, Campylobacter spp. and Bordetella spp. (178–180).
The advances in machine learning algorithms may allow the development of revolutionary surveillance programs, potentially providing valuable information to public health policy makers about potential epidemiological trends and risks for the general public. Although such databases are likely to first be established for more common epidemic scenarios such as annual influenza, MDR pathogens, and foodborne pathogens, particular risks may also be calculated for rare pathogens such as measles, ebola, or hypervirulent bacteria such as C. diphtheriae. As we live in an increasingly globalized world with rapid spread of pathogens, new concepts for epidemiological surveillance are needed, to enable rapid and effective interventions.
Corynebacterium diphtheriae is reemerging in clinics in high income countries, partly as a result of refugee movement, and requiring greater awareness of the issue. WGS offers the opportunity to describe potential transmission events and infection sources with the highest resolution. Data provided from molecular typing methods should, where possible, be analyzed in the context of classical epidemiological information, for which information has to be rapidly shared with local public health authorities. In addition, surveillance for C. diphtheriae and other re-emerging hypervirulent pathogens would benefit from rapid data collection and sharing platforms sharing information on classical and molecular epidemiology.
HS-S performed data analysis and wrote the manuscript. AE wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2019.00235/full#supplementary-material
1. Liu C, Guo J. Hypervirulent Klebsiella pneumoniae (hypermucoviscous and aerobactin positive) infection over 6 years in the elderly in China: antimicrobial resistance patterns, molecular epidemiology and risk factor. Ann Clin Microbiol Antimicrob. (2019) 18:4. doi: 10.1186/s12941-018-0302-9
2. Acevedo R, Bai X, Borrow R, Caugant DA, Carlos J, Ceyhan M, et al. The global meningococcal initiative meeting on prevention of meningococcal disease worldwide: epidemiology, surveillance, hypervirulent strains, antibiotic resistance and high-risk populations. Exp Rev Vaccines. (2019) 18:15–30. doi: 10.1080/14760584.2019.1557520
3. Isenring E, Fehr J, Gultekin N, Schlagenhauf P. Infectious disease profiles of Syrian and Eritrean migrants presenting in Europe: a systematic review. Travel Med Infect Dis. (2018) 25:65–76. doi: 10.1016/j.tmaid.2018.04.014
4. Couturier J, Davies K, Gateau C, Barbut F. Ribotypes and new virulent strains across Europe. Adv Exp Med Biol. (2018) 1050:45–58. doi: 10.1007/978-3-319-72799-8_4
5. Yakob L, Riley TV, Paterson DL, Marquess J, Magalhaes RJ, Furuya-Kanamori L, et al. Mechanisms of hypervirulent Clostridium difficile ribotype 027 displacement of endemic strains: an epidemiological model. Sci Rep. (2015) 5:12666. doi: 10.1038/srep12666
6. Joubrel C, Tazi A, Six A, Dmytruk N, Touak G, Bidet P, et al. Group B streptococcus neonatal invasive infections, France 2007–2012. Clin Microbiol Infect. (2015) 21:910–6. doi: 10.1016/j.cmi.2015.05.039
7. Giufre M, Accogli M, Ricchizzi E, Barbanti F, Farina C, Fazii P, et al. Multidrug-resistant infections in long-term care facilities: extended-spectrum β-lactamase-producing Enterobacteriaceae and hypervirulent antibiotic resistant Clostridium difficile. Diagn Microbiol Infect Dis. (2018) 91:275–81. doi: 10.1016/j.diagmicrobio.2018.02.018
8. Goldenberger D, Claas GJ, Bloch-Infanger C, Breidthardt T, Suter B, Martinez M, et al. Louse-borne relapsing fever (Borrelia recurrentis) in an Eritrean refugee arriving in Switzerland, August 2015. Euro surveillance. (2015) 20:2–5. doi: 10.2807/1560-7917.ES2015.20.32.21204
9. Aro T, Kantele A. High rates of meticillin-resistant Staphylococcus aureus among asylum seekers and refugees admitted to Helsinki University Hospital, 2010 to 2017. Euro surveillance. (2018) 23:1–14. doi: 10.2807/1560-7917.ES.2018.23.45.1700797
10. Piso RJ, Kach R, Pop R, Zillig D, Schibli U, Bassetti S, et al. Correction: a Cross-sectional study of colonization rates with methicillin-resistant Staphylococcus aureus (MRSA) and Extended-Spectrum Beta-Lactamase (ESBL) and carbapenemase-producing enterobacteriaceae in four swiss refugee centres. PLoS ONE. (2017) 12:e0174911. doi: 10.1371/journal.pone.0174911
11. Bloch-Infanger C, Battig V, Kremo J, Widmer AF, Egli A, Bingisser R, et al. Increasing prevalence of infectious diseases in asylum seekers at a tertiary care hospital in Switzerland. PLoS ONE. (2017) 12:e0179537. doi: 10.1371/journal.pone.0179537
13. Hadfield TL, McEvoy P, Polotsky Y, Tzinserling VA, Yakovlev AA. The pathology of diphtheria. J Infect Dis. (2000) 181(Suppl 1):S116–20. doi: 10.1086/315551
15. Funke G, von Graevenitz A, Clarridge JE III, Bernard KA. Clinical microbiology of coryneform bacteria. Clin Microbiol Rev. (1997) 10:125–59. doi: 10.1128/CMR.10.1.125
16. Matsuyama R, Akhmetzhanov AR, Endo A, Lee H, Yamaguchi T, Tsuzuki S, et al. Uncertainty and sensitivity analysis of the basic reproduction number of diphtheria: a case study of a Rohingya refugee camp in Bangladesh, November-December 2017. PeerJ. (2018) 6:e4583. doi: 10.7717/peerj.4583
17. Rakhmanova AG, Lumio J, Groundstroem K, Valova E, Nosikova E, Tanasijchuk T, et al. Diphtheria outbreak in St. Petersburg: clinical characteristics of 1860 adult patients. Scand J Infect Dis. (1996) 28:37–40. doi: 10.3109/00365549609027147
18. Markina SS, Maksimova NM, Vitek CR, Bogatyreva EY, Monisov AA. Diphtheria in the russian federation in the 1990s. J Infect Dis. (2000) 181(Suppl 1):S27–34. doi: 10.1086/315535
19. Popovic T, Kombarova SY, Reeves MW, Nakao H, Mazurova IK, Wharton M, et al. Molecular epidemiology of diphtheria in Russia, 1985–1994. J Infect Dis. (1996) 174:1064–72. doi: 10.1093/infdis/174.5.1064
20. FitzGerald RP, Rosser AJ, Perera DN. Non-toxigenic penicillin-resistant cutaneous C. diphtheriae infection: a case report and review of the literature. J Infect Public Health. (2015) 8:98–100. doi: 10.1016/j.jiph.2014.05.006
21. Lindhusen-Lindhe E, Dotevall L, Berglund M. Imported laryngeal and cutaneous diphtheria in tourists returning from western Africa to Sweden, March 2012. Euro surveillance. (2012) 17:20189.
22. May ML, McDougall RJ, Robson JM. Corynebacterium diphtheriae and the returned tropical traveler. J Travel Med. (2014) 21:39–44. doi: 10.1111/jtm.12074
23. Jakovljev A, Steinbakk M, Mengshoel AT, Sagvik E, Brugger-Synnes P, Sakshaug T, et al. Imported toxigenic cutaneous diphtheria in a young male returning from Mozambique to Norway, March 2014. Euro surveillance. (2014) 19:1–4. doi: 10.2807/1560-7917.ES2014.19.24.20835
24. Nelson TG, Mitchell CD, Sega-Hall GM, Porter RJ. Cutaneous ulcers in a returning traveller: a rare case of imported diphtheria in the UK. Clin Exp Dermatol. (2016) 41:57–9. doi: 10.1111/ced.12763
25. Gruner E, Opravil M, Altwegg M, von Graevenitz A. Nontoxigenic Corynebacterium diphtheriae isolated from intravenous drug users. Clin Infect Dis. (1994) 18:94–6. doi: 10.1093/clinids/18.1.94
26. Monsuez JJ, Mathieu D, Arnoult F, Passeron J. Cutaneous diphtheria in a homeless man. Lancet. (1995) 346:649–50. doi: 10.1016/S0140-6736(95)91490-0
28. Lowe CF, Bernard KA, Romney MG. Cutaneous diphtheria in the urban poor population of Vancouver, British Columbia, Canada: a 10-year review. J Clin Microbiol. (2011) 49:2664–6. doi: 10.1128/JCM.00362-11
29. Gubler J, Huber-Schneider C, Gruner E, Altwegg M. An outbreak of nontoxigenic Corynebacterium diphtheriae infection: single bacterial clone causing invasive infection among Swiss drug users. Clin Infect Dis. (1998) 27:1295–8. doi: 10.1086/514997
30. Berger A, Dangel A, Schober T, Schmidbauer B, Konrad R, Marosevic D, et al. Whole genome sequencing suggests transmission of Corynebacterium diphtheriae-caused cutaneous diphtheria in two siblings, Germany, 2018. Euro surveillance. (2019) 24:1–4. doi: 10.2807/1560-7917.ES.2019.24.2.1800683
31. Meinel DM, Kuehl R, Zbinden R, Boskova V, Garzoni C, Fadini D, et al. Outbreak investigation for toxigenic Corynebacterium diphtheriae wound infections in refugees from Northeast Africa and Syria in Switzerland and Germany by whole genome sequencing. Clin Microbiol Infect. (2016) 22:1003 e1–8. doi: 10.1016/j.cmi.2016.08.010
32. Reynolds GE, Saunders H, Matson A, O'Kane F, Roberts SA, Singh SK, et al. Public health action following an outbreak of toxigenic cutaneous diphtheria in an Auckland refugee resettlement centre. Commun Dis Intell Q Rep. (2016) 40:E475–81.
33. Sane J, Sorvari T, Widerstrom M, Kauma H, Kaukoniemi U, Tarkka E, et al. Respiratory diphtheria in an asylum seeker from Afghanistan arriving to Finland via Sweden, December 2015. Euro surveillance. (2016) 21:14–17. doi: 10.2807/1560-7917.ES.2016.21.2.30105
34. Scheifer C, Rolland-Debord C, Badell E, Reibel F, Aubry A, Perignon A, et al. Re-emergence of Corynebacterium diphtheriae. Med Mal Infect. (2018) doi: 10.1016/j.medmal.2018.12.001. [Epub ahead of print].
35. Dangel A, Berger A, Konrad R, Bischoff H, Sing A. Geographically diverse clusters of nontoxigenic Corynebacterium diphtheriae infection, Germany, 2016–2017. Emerging Infect Dis. (2018) 24:1239–45. doi: 10.3201/eid2407.172026
36. du Plessis M, Wolter N, Allam M, de Gouveia L, Moosa F, Ntshoe G, et al. Molecular characterization of Corynebacterium diphtheriae outbreak isolates, South Africa, March-June 2015. Emerging Infect Dis. (2017) 23:1308–15. doi: 10.3201/eid2308.162039
37. Edwards D, Kent D, Lester C, Brown CS, Murphy ME, Brown NM, et al. Transmission of toxigenic Corynebacterium diphtheriae by a fully immunised resident returning from a visit to West Africa, United Kingdom, 2017. Euro surveillance. (2018) 23:1700681. doi: 10.2807/1560-7917.ES.2018.23.39.1700681
38. Jane M, Vidal MJ, Camps N, Campins M, Martinez A, Balcells J, et al. A case of respiratory toxigenic diphtheria: contact tracing results and considerations following a 30-year disease-free interval, Catalonia, Spain, 2015. Euro surveillance. (2018) 23:1–6. doi: 10.2807/1560-7917.ES.2018.23.13.17-00183
39. Cassir N, Bagneres D, Fournier PE, Berbis P, Brouqui P, Rossi PM. Cutaneous diphtheria: easy to be overlooked. Int J Infect Dis. (2015) 33:104–5. doi: 10.1016/j.ijid.2015.01.008
40. Bloss S, Klemann C, Rother AK, Mehmecke S, Schumacher U, Mucke U, et al. Diagnostic needs for rare diseases and shared prediagnostic phenomena: results of a urvey. PLoS ONE. (2017) 12:e0172532. doi: 10.1371/journal.pone.0172532
41. Caliendo AM, Gilbert DN, Ginocchio CC, Hanson KE, May L, Quinn TC, et al. Better tests, better care: improved diagnostics for infectious diseases. Clin Infect Dis. (2013) 57(Suppl 3):S139–70. doi: 10.1093/cid/cit578
42. van Duin D. Diagnostic challenges and opportunities in older adults with infectious diseases. Clin Infect Dis. (2012) 54:973–8. doi: 10.1093/cid/cir927
43. Kozinska A, Seweryn P, Sitkiewicz I. A crash course in sequencing for a microbiologist. J Appl Genet. (2019) 60:103–11. doi: 10.1007/s13353-019-00482-2
44. McNerney R, Zignol M, Clark TG. Use of whole genome sequencing in surveillance of drug resistant tuberculosis. Exp Rev Anti Infect Ther. (2018) 16:433–42. doi: 10.1080/14787210.2018.1472577
45. Mirande C, Bizine I, Giannetti A, Picot N, van Belkum A. Epidemiological aspects of healthcare-associated infections and microbial genomics. Eur J Clin Microbiol Infect Dis. (2018) 37:823–31. doi: 10.1007/s10096-017-3170-x
46. Revez J, Espinosa L, Albiger B, Leitmeyer KC, Struelens MJ, Points ENMF, et al. Survey on the use of whole-genome sequencing for infectious diseases surveillance: rapid expansion of european national capacities, 2015–2016. Front Public Health. (2017) 5:347. doi: 10.3389/fpubh.2017.00347
47. Schurch AC, van Schaik W. Challenges and opportunities for whole-genome sequencing-based surveillance of antibiotic resistance. Ann N Y Acad Sci. (2017) 1388:108–20. doi: 10.1111/nyas.13310
48. Wuthrich D, Gautsch S, Spieler-Denz R, Dubuis O, Gaia V, Moran-Gilad J, et al. Air-conditioner cooling towers as complex reservoirs and continuous source of Legionella pneumophila infection evidenced by a genomic analysis study in 2017, Switzerland. Euro Surveill. (2019) 24:1800192. doi: 10.2807/1560-7917.ES.2019.24.4.1800192
49. Endres BT, Dotson KM, Poblete K, McPherson J, Lancaster C, Basseres E, et al. Environmental transmission of Clostridioides difficile ribotype 027 at a long-term care facility; an outbreak investigation guided by whole genome sequencing. Infect Control Hosp Epidemiol. (2018) 39:1322–9. doi: 10.1017/ice.2018.230
50. Halstead FD, Ravi A, Thomson N, Nuur M, Hughes K, Brailey M, et al. Whole genome sequencing of toxigenic Clostridium difficile in asymptomatic carriers: insights into possible role in transmission. J Hosp Infect. (2018) 2:125–134. doi: 10.1016/j.jhin.2018.10.012
51. Walker TM, Merker M, Knoblauch AM, Helbling P, Schoch OD, van der Werf MJ, et al. A cluster of multidrug-resistant Mycobacterium tuberculosis among patients arriving in Europe from the Horn of Africa: a molecular epidemiological study. Lancet Infect Dis. (2018) 18:431–40. doi: 10.1016/S1473-3099(18)30004-5
52. Loeffler F. Untersuchungen über die Bedeuteung der Mikroorganismen für die Entstehung der Diphtherie beim Menschen, bei der Taube und beim Kalbe. Mitt KJ in Gesundh. (1884) 2:421–99.
53. Barakett V, Morel G, Lesage D, Petit JC. Septic arthritis due to a nontoxigenic strain of Corynebacterium diphtheriae subspecies mitis. Clin Infect Dis. (1993) 17:520–1. doi: 10.1093/clinids/17.3.520
54. Belko J, Wessel DL, Malley R. Endocarditis caused by Corynebacterium diphtheriae: case report and review of the literature. Pediatr Infect Dis J. (2000) 19:159–63. doi: 10.1097/00006454-200002000-00015
55. Farfour E, Badell E, Zasada A, Hotzel H, Tomaso H, Guillot S, et al. Characterization and comparison of invasive Corynebacterium diphtheriae isolates from France and Poland. J Clin Microbiol. (2012) 50:173–5. doi: 10.1128/JCM.05811-11
56. Poilane I, Fawaz F, Nathanson M, Cruaud P, Martin T, Collignon A, et al. Corynebacterium diphtheriae osteomyelitis in an immunocompetent child: a case report. Eur J Pediatr. (1995) 154:381–3. doi: 10.1007/BF02072108
57. Romney MG, Roscoe DL, Bernard K, Lai S, Efstratiou A, Clarke AM. Emergence of an invasive clone of nontoxigenic Corynebacterium diphtheriae in the urban poor population of Vancouver, Canada. J Clin Microbiol. (2006) 44:1625–9. doi: 10.1128/JCM.44.5.1625-1629.2006
58. Tiley SM, Kociuba KR, Heron LG, Munro R. Infective endocarditis due to nontoxigenic Corynebacterium diphtheriae: report of seven cases and review. Clin Infect Dis. (1993) 16:271–5. doi: 10.1093/clind/16.2.271
59. Gordon CL, Fagan P, Hennessy J, Baird R. Characterization of Corynebacterium diphtheriae isolates from infected skin lesions in the Northern Territory of Australia. J Clin Microbiol. (2011) 49:3960–2. doi: 10.1128/JCM.05038-11
60. Huhulescu S, Hirk S, Zeinzinger V, Hasenberger P, Skvara H, Mullegger R, et al. Letter to the editor: cutaneous diphtheria in a migrant from an endemic country in east Africa, Austria May 2014. Euro surveillance. (2014) 19:1–2. doi: 10.2807/1560-7917.ES2014.19.26.20845
61. Efstratiou A, Maple PAC. Manual for the Laboratory Diagnosis of Diphtheria. WHO: The Expanded Programme on Immunization in the European Region of WHO (1994).
62. Tagini F, Pillonel T, Croxatto A, Bertelli C, Koutsokera A, Lovis A, et al. Distinct genomic features characterize two clades of Corynebacterium diphtheriae: proposal of Corynebacterium diphtheriae Subsp. diphtheriae Subsp. nov. and Corynebacterium diphtheriae Subsp. lausannense Subsp. nov. Front Microbiol. (2018) 9:1743. doi: 10.3389/fmicb.2018.01743
63. Meinel DM, Konrad R, Berger A, Konig C, Schmidt-Wieland T, Hogardt M, et al. Zoonotic transmission of toxigenic Corynebacterium ulcerans strain, Germany, 2012. Emerg Infect Dis. (2015) 21:356–8. doi: 10.3201/eid2102.141160
64. Bardsdale WL, Pappenheimer AM Jr. Phage-host relationships in nontoxigenic and toxigenic diphtheria bacilli. J Bacteriol. (1954) 67:220–32.
65. Sangal V, Nieminen L, Weinhardt B, Raeside J, Tucker NP, Florea CD, et al. Diphtheria-like disease caused by toxigenic Corynebacterium ulcerans strain. Emerg Infect Dis. (2014) 20:1257–8. doi: 10.3201/eid2007.140216
67. Wagner KS, White JM, Crowcroft NS, De Martin S, Mann G, Efstratiou A. Diphtheria in the United Kingdom, 1986–2008: the increasing role of Corynebacterium ulcerans. Epidemiol Infect. (2010) 138:1519–30. doi: 10.1017/S0950268810001895
68. Berger A, Boschert V, Konrad R, Schmidt-Wieland T, Hormansdorfer S, Eddicks M, et al. Two cases of cutaneous diphtheria associated with occupational pig contact in Germany. Zoonoses Public Health. (2013) 60:539–42. doi: 10.1111/zph.12031
69. Sing A, Konrad R, Meinel DM, Mauder N, Schwabe I, Sting R. Corynebacterium diphtheriae in a free-roaming red fox: case report and historical review on diphtheria in animals. Infection. (2016) 44:441–5. doi: 10.1007/s15010-015-0846-y
70. Pappenheimer AM Jr, Gill DM. Diphtheria. Science. (1973) 182:353–8. doi: 10.1126/science.182.4110.353
71. Brune I, Werner H, Huser AT, Kalinowski J, Puhler A, Tauch A. The DtxR protein acting as dual transcriptional regulator directs a global regulatory network involved in iron metabolism of Corynebacterium glutamicum. BMC Genom. (2006) 7:21. doi: 10.1186/1471-2164-7-21
72. Mandlik A, Swierczynski A, Das A, Ton-That H. Corynebacterium diphtheriae employs specific minor pilins to target human pharyngeal epithelial cells. Mol Microbiol. (2007) 64:111–24. doi: 10.1111/j.1365-2958.2007.05630.x
73. Bertuccini L, Baldassarri L, von Hunolstein C. Internalization of non-toxigenic Corynebacterium diphtheriae by cultured human respiratory epithelial cells. Microb Pathog. (2004) 37:111–8. doi: 10.1016/j.micpath.2004.06.002
74. Colombo AV, Hirata R Jr, de Souza CM, Monteiro-Leal LH, Previato JO, Formiga LC, et al. Corynebacterium diphtheriae surface proteins as adhesins to human erythrocytes. FEMS Microbiol Lett. (2001) 197:235–9. doi: 10.1016/S0378-1097(01)00113-6
75. Hirata R, Napoleao F, Monteiro-Leal LH, Andrade AF, Nagao PE, Formiga LC, et al. Intracellular viability of toxigenic Corynebacterium diphtheriae strains in HEp-2 cells. FEMS Microbiol Lett. (2002) 215:115–9. doi: 10.1016/S0378-1097(02)00930-8
76. Peixoto RS, Antunes CA, Louredo LS, Viana VG, Santos CSD, Fuentes Ribeiro da Silva J, et al. Functional characterization of the collagen-binding protein DIP2093 and its influence on host-pathogen interaction and arthritogenic potential of Corynebacterium diphtheriae. Microbiology. (2017) 163:692–701. doi: 10.1099/mic.0.000467
77. Peixoto RS, Pereira GA, Sanches Dos Santos L, Rocha-de-Souza CM, Gomes DLR, Silva Dos Santos C, et al. Invasion of endothelial cells and arthritogenic potential of endocarditis-associated Corynebacterium diphtheriae. Microbiology. (2014) 160(Pt 3):537–46. doi: 10.1099/mic.0.069948-0
78. Mandlik A, Swierczynski A, Das A, Ton-That H. Pili in Gram-positive bacteria: assembly, involvement in colonization and biofilm development. Trends Microbiol. (2008) 16:33–40. doi: 10.1016/j.tim.2007.10.010
79. Hoyle L, Leeds MB. A tellurite blood-agar medium for the rapid diagnosis of diphtheria. Lancet. (1941) 237:175–6. doi: 10.1016/S0140-6736(00)77533-7
80. Alibi S, Ferjani A, Gaillot O, Marzouk M, Courcol R, Boukadida J. Identification of clinically relevant Corynebacterium strains by Api Coryne, MALDI-TOF-mass spectrometry and molecular approaches. Pathol Biol. (2015) 63:153–7. doi: 10.1016/j.patbio.2015.07.007
81. Barberis C, Almuzara M, Join-Lambert O, Ramirez MS, Famiglietti A, Vay C. Comparison of the Bruker MALDI-TOF mass spectrometry system and conventional phenotypic methods for identification of Gram-positive rods. PLoS ONE. (2014) 9:e106303. doi: 10.1371/journal.pone.0106303
82. Konrad R, Berger A, Huber I, Boschert V, Hormansdorfer S, Busch U, et al. Matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry as a tool for rapid diagnosis of potentially toxigenic Corynebacterium species in the laboratory management of diphtheria-associated bacteria. Euro surveillance. (2010) 15:1–5. doi: 10.2807/ese.15.43.19699-en
83. Engler KH, Glushkevich T, Mazurova IK, George RC, Efstratiou A. A modified Elek test for detection of toxigenic corynebacteria in the diagnostic laboratory. J Clin Microbiol. (1997) 35:495–8.
84. Schuhegger R, Lindermayer M, Kugler R, Heesemann J, Busch U, Sing A. Detection of toxigenic Corynebacterium diphtheriae and Corynebacterium ulcerans strains by a novel real-time PCR. J Clin Microbiol. (2008) 46:2822–3. doi: 10.1128/JCM.01010-08
85. Neal SE, Efstratiou A, International Diphtheria Reference Laboratories. International external quality assurance for laboratory diagnosis of diphtheria. J Clin Microbiol. (2009) 47:4037–42. doi: 10.1128/JCM.00473-09
86. Czumbel I, Efstratiou A, Head V, Trindall A, West D, M. R. European Centre for Disease Prevention and Control. Gap analysis on securing diphtheria diagnostic capacity and diphtheria antitoxin availability in the EU/EEA: European Center for Disease Prevention and Control (2017). p. 33.
87. Wheeler SM, Morton AR. Epidemiological observations in the halifax epidemic. Am J Public Health Nations Health. (1942) 32:947–56. doi: 10.2105/AJPH.32.9.947
88. Stuart G. Diphtheria Incidence in European Countries. Br Med J. (1945) 2:613–5. doi: 10.1136/bmj.2.4426.613
89. Galazka AM, Robertson SE. Diphtheria: changing patterns in the developing world and the industrialized world. Eur J Epidemiol. (1995) 11:107–17. doi: 10.1007/BF01719955
90. Pezzotti P, Bellino S, Prestinaci F, Iacchini S, Lucaroni F, Camoni L, et al. The impact of immunization programs on 10 vaccine preventable diseases in Italy: 1900–2015. Vaccine. (2018) 36:1435–43. doi: 10.1016/j.vaccine.2018.01.065
91. van Wijhe M, Tulen AD, Korthals Altes H, McDonald SA, de Melker HE, Postma MJ, et al. Quantifying the impact of mass vaccination programmes on notified cases in the Netherlands. Epidemiol Infect. (2018) 146:716–22. doi: 10.1017/S0950268818000481
92. Marcuse EK, Grand MG. Epidemiology of diphtheria in San Antonio, Tex., 1970. JAMA. (1973) 224:305–10. doi: 10.1001/jama.224.3.305
93. Miller LW, Older JJ, Drake J, Zimmerman S. Diphtheria immunization. Effect upon carriers and the control of outbreaks. Am J Dis Child. (1972) 123:197–9. doi: 10.1001/archpedi.1972.02110090067004
95. Bisgard KM, Rhodes P, Hardy IR, Litkina IL, Filatov NN, Monisov AA, et al. Diphtheria toxoid vaccine effectiveness: a case-control study in Russia. J Infect Dis. (2000) 181(Suppl 1):S184–7. doi: 10.1086/315562
96. Chen RT, Hardy IR, Rhodes PH, Tyshchenko DK, Moiseeva AV, Marievsky VF. Ukraine, 1992: first assessment of diphtheria vaccine effectiveness during the recent resurgence of diphtheria in the Former Soviet Union. J Infect Dis. (2000) 181(Suppl 1):S178–83. doi: 10.1086/315561
97. Ipsen J. Circulating antitoxin at the onset of diphtheria in 425 patients. J Immunol. (1946) 54:325–47.
98. Galazka AM, Robertson SE, Oblapenko GP. Resurgence of diphtheria. Eur J Epidemiol. (1995) 11:95–105. doi: 10.1007/BF01719954
99. Freidl GS, Tostmann A, Curvers M, Ruijs WLM, Smits G, Schepp R, et al. Immunity against measles, mumps, rubella, varicella, diphtheria, tetanus, polio, hepatitis A and hepatitis B among adult asylum seekers in the Netherlands, 2016. Vaccine. (2018) 36:1664–72. doi: 10.1016/j.vaccine.2018.01.079
100. Randi BA, Sejas ONE, Miyaji KT, Infante V, Lara AN, Ibrahim KY, et al. A systematic review of adult tetanus-diphtheria-acellular (Tdap) coverage among healthcare workers. Vaccine. (2019) 37:1030–7. doi: 10.1016/j.vaccine.2018.12.046
101. Mossong J, Putz L, Shkedy Z, Schneider F. Seroepidemiology of diphtheria and pertussis in Luxembourg in 2000. Epidemiol Infect. (2006) 134:573–8. doi: 10.1017/S0950268805005662
102. Li X, Chen M, Zhang T, Li J, Zeng Y, Lu L. Seroepidemiology of diphtheria and pertussis in Beijing, China: a cross-sectional study. Hum Vaccin Immunother. (2015) 11:2434–9. doi: 10.1080/21645515.2015.1062954
103. Zasada AA, Baczewska-Rej M, Wardak S. An increase in non-toxigenic Corynebacterium diphtheriae infections in Poland–molecular epidemiology and antimicrobial susceptibility of strains isolated from past outbreaks and those currently circulating in Poland. Int J Infect Dis. (2010) 14:e907–12. doi: 10.1016/j.ijid.2010.05.013
104. Galazka AM, Robertson SE. Immunization against diphtheria with special emphasis on immunization of adults. Vaccine. (1996) 14:845–57. doi: 10.1016/0264-410X(96)00021-7
105. Anderson GS, Penfold JB. An outbreak of diphtheria in a hospital for the mentally subnormal. J Clin Pathol. (1973) 26:606–15. doi: 10.1136/jcp.26.8.606
106. Belsey MA, Sinclair M, Roder MR, LeBlanc DR. Corynebacterium diphtheriae skin infections in Alabama and Louisiana. A factor in the epidemiology of diphtheria. N Engl J Med. (1969) 280:135–41. doi: 10.1056/NEJM196901162800304
107. Sinclair MC, Overton R, Donald WJ. Alabama diphtheria outbreak, 1967. HSMHA Health Rep. (1971) 86:1107–11. doi: 10.2307/4594395
108. Zalma VM, Older JJ, Brooks GF. The Austin, Texas, diphtheria outbreak. Clinical and epidemiological aspects. JAMA. (1970) 211:2125–9. doi: 10.1001/jama.211.13.2125
109. Filonov VP, Zakharenko DF, Vitek CR, Romanovsky AA, Zhukovski VG. Epidemic diphtheria in Belarus, 1992–1997. J Infect Dis. (2000) 181(Suppl 1):S41–6. doi: 10.1086/315537
110. Griskevica A, Ching P, Russo G, Kreysler J. Diphtheria in Latvia, 1986–1996. J Infect Dis. (2000) 181(Suppl 1):S60–4. doi: 10.1086/315540
111. Jogiste A, Ching P, Trei T, Kreysler J. Diphtheria in Estonia, 1991–1996. J Infect Dis. (2000) 181(Suppl 1):S65–8. doi: 10.1086/315541
112. Nekrassova LS, Chudnaya LM, Marievski VF, Oksiuk VG, Gladkaya E, Bortnitska II, et al. Epidemic diphtheria in Ukraine, 1991–1997. J Infect Dis. (2000) 181(Suppl 1):S35–40. doi: 10.1086/315536
113. Usonis V, Bakasenas V, Morkunas B, Valentelis R, Ching P, Kreysler J. Diphtheria in Lithuania, 1986–1996. J Infect Dis. (2000) 181(Suppl 1):S55–9. doi: 10.1086/315539
114. Wanlapakorn N, Yoocharoen P, Tharmaphornpilas P, Theamboonlers A, Poovorawan Y. Diphtheria outbreak in Thailand, (2012) seroprevalence of diphtheria antibodies among Thai adults and its implications for immunization programs. South Asian J Trop Med Public Health. (2014) 45:1132–41.
115. Pool V, Tomovici A, Johnson DR, Greenberg DP, Decker MD. Humoral immunity 10years after booster immunization with an adolescent and adult formulation combined tetanus, diphtheria, and 5-component acellular pertussis vaccine in the USA. Vaccine. (2018) 36:2282–7. doi: 10.1016/j.vaccine.2018.03.029
116. Golaz A, Hardy IR, Glushkevich TG, Areytchiuk EK, Deforest A, Strebel P, et al. Evaluation of a single dose of diphtheria-tetanus toxoids among adults in Odessa, Ukraine, 1995: immunogenicity and adverse reactions. J Infect Dis. (2000) 181(Suppl 1):S203–7. doi: 10.1086/315558
117. Ronne T, Valentelis R, Tarum S, Griskevica A, Wachmann CH, Aggerbeck H, et al. Immune response to diphtheria booster vaccine in the Baltic states. J Infect Dis. (2000) 181(Suppl 1):S213–9. doi: 10.1086/315560
118. Sutter RW, Hardy IR, Kozlova IA, Tchoudnaia LM, Gluskevich TG, Marievsky V, et al. Immunogenicity of tetanus-diphtheria toxoids (Td) among Ukrainian adults: implications for diphtheria control in the Newly Independent States of the Former Soviet Union. J Infect Dis. (2000) 181(Suppl 1):S197–202. doi: 10.1086/315557
119. Finger F, Funk S, White K, Siddiqui MR, Edmunds WJ, Kucharski AJ. Real-time analysis of the diphtheria outbreak in forcibly displaced Myanmar nationals in Bangladesh. BMC Med. (2019) 17:58. doi: 10.1186/s12916-019-1288-7
120. Rahman MR, Islam K. Massive diphtheria outbreak among Rohingya refugees: lessons learnt. J Travel Med. (2019) 26:tay141. doi: 10.1093/jtm/tay122
121. Santos LS, Sant'anna LO, Ramos JN, Ladeira EM, Stavracakis-Peixoto R, Borges LL, et al. Diphtheria outbreak in Maranhao, Brazil: microbiological, clinical and epidemiological aspects. Epidemiol Infect. (2015) 143:791–8. doi: 10.1017/S0950268814001241
122. Landazabal Garcia N, Burgos Rodriguez MM, Pastor D. Diphtheria outbreak in Cali, Colombia, August-October 2000. Epidemiol Bull. (2001) 22:13–5.
123. Saikia L, Nath R, Saikia NJ, Choudhury G, Sarkar M. A diphtheria outbreak in Assam, India. Southeast Asian J Trop Med Public Health. (2010) 41:647–52.
124. Das PP, Patgiri SJ, Saikia L, Paul D. Recent outbreaks of diphtheria in Dibrugarh District, Assam, India. J Clin Diagn Res. (2016) 10:DR01–3. doi: 10.7860/JCDR/2016/20212.8144
125. Parande MV, Roy S, Mantur BG, Parande AM, Shinde RS. Resurgence of diphtheria in rural areas of North Karnataka, India. Indian J Med Microbiol. (2017) 35:247–51. doi: 10.4103/ijmm.IJMM_17_48
126. Hughes GJ, Mikhail AF, Husada D, Irawan E, Kafatos G, Bracebridge S, et al. Seroprevalence and determinants of immunity to diphtheria for children living in two districts of contrasting incidence during an outbreak in east Java, Indonesia. Pediatr Infect Dis J. (2015) 34:1152–6. doi: 10.1097/INF.0000000000000846
127. Nanthavong N, Black AP, Nouanthong P, Souvannaso C, Vilivong K, Muller CP, et al. Diphtheria in Lao PDR: insufficient coverage or ineffective vaccine? PLoS ONE. (2015) 10:e0121749. doi: 10.1371/journal.pone.0121749
128. Rasmussen I, Wallace S, Mengshoel AT, Hoiby EA, Brandtzaeg P. Diphtheria outbreak in Norway: lessons learned. Scand J Infect Dis. (2011) 43:986–9. doi: 10.3109/00365548.2011.600326
129. Besa NC, Coldiron ME, Bakri A, Raji A, Nsuami MJ, Rousseau C, et al. Diphtheria outbreak with high mortality in northeastern Nigeria. Epidemiol Infect. (2014) 142:797–802. doi: 10.1017/S0950268813001696
130. Czajka U, Wiatrzyk A, Mosiej E, Forminska K, Zasada AA. Changes in MLST profiles and biotypes of Corynebacterium diphtheriae isolates from the diphtheria outbreak period to the period of invasive infections caused by nontoxigenic strains in Poland (1950–2016). BMC Infect Dis. (2018) 18:121. doi: 10.1186/s12879-018-3020-1
131. Mahomed S, Archary M, Mutevedzi P, Mahabeer Y, Govender P, Ntshoe G, et al. An isolated outbreak of diphtheria in South Africa, 2015. Epidemiol Infect. (2017) 145:2100–8. doi: 10.1017/S0950268817000851
132. Raad I, Chaftari AM, Dib RW, Graviss EA, Hachem R. Emerging outbreaks associated with conflict and failing healthcare systems in the Middle East. Infect Control Hosp Epidemiol. (2018) 39:1230–6. doi: 10.1017/ice.2018.177
133. Lodeiro-Colatosti A, Reischl U, Holzmann T, Hernandez-Pereira CE, Risquez A, Paniz-Mondolfi AE. Diphtheria outbreak in amerindian communities, Wonken, Venezuela, 2016–2017. Emerg Infect Dis. (2018) 24:1340–4. doi: 10.3201/eid2407.171712
134. Paniz-Mondolfi AE, Tami A, Grillet ME, Marquez M, Hernandez-Villena J, Escalona-Rodriguez MA, et al. Resurgence of vaccine-preventable diseases in venezuela as a regional public health threat in the americas. Emerg Infect Dis. (2019) 25:625–32. doi: 10.3201/eid2504.181305
135. Dureab F, Muller O, Jahn A. Resurgence of diphtheria in Yemen due to population movement. J Travel Med. (2018) 25:1–2. doi: 10.1093/jtm/tay094
136. Doi Y, Iovleva A, Bonomo RA. The ecology of extended-spectrum β-lactamases (ESBLs) in the developed world. J Travel Med. (2017) 24(Suppl 1):S44–51. doi: 10.1093/jtm/taw102
137. Navon-Venezia S, Kondratyeva K, Carattoli A. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev. (2017) 41:252–75. doi: 10.1093/femsre/fux013
138. Vilches TN, Bonesso MF, Guerra HM, Fortaleza C, Park AW, Ferreira CP. The role of intra and inter-hospital patient transfer in the dissemination of heathcare-associated multidrug-resistant pathogens. Epidemics. (2018) 3:362–72. doi: 10.1016/j.epidem.2018.11.001
139. Goodman RA, Buehler JW, Koplan JP. The epidemiologic field investigation: science and judgment in public health practice. Am J Epidemiol. (1990) 132:9–16. doi: 10.1093/oxfordjournals.aje.a115647
140. Reingold AL. Outbreak investigations–a perspective. Emerg Infect Dis. (1998) 4:21–7. doi: 10.3201/eid0401.980104
141. Bolt F, Cassiday P, Tondella ML, Dezoysa A, Efstratiou A, Sing A, et al. Multilocus sequence typing identifies evidence for recombination and two distinct lineages of Corynebacterium diphtheriae. J Clin Microbiol. (2010) 48:4177–85. doi: 10.1128/JCM.00274-10
142. Damian M, Grimont F, Narvskaya O, Straut M, Surdeanu M, Cojocaru R, et al. Study of Corynebacterium diphtheriae strains isolated in Romania, northwestern Russia and the Republic of Moldova. Res Microbiol. (2002) 153:99–106. doi: 10.1016/S0923-2508(01)01294-3
143. De Zoysa A, Hawkey P, Charlett A, Efstratiou A. Comparison of four molecular typing methods for characterization of Corynebacterium diphtheriae and determination of transcontinental spread of C. diphtheriae based on BstEII rRNA gene profiles. J Clin Microbiol. (2008) 46:3626–35. doi: 10.1128/JCM.00300-08
144. Grimont PA, Grimont F, Efstratiou A, De Zoysa A, Mazurova I, Ruckly C, et al. International nomenclature for Corynebacterium diphtheriae ribotypes. Res Microbiol. (2004) 155:162–6. doi: 10.1016/j.resmic.2003.12.005
145. Kolodkina V, Titov L, Sharapa T, Grimont F, Grimont PA, Efstratiou A. Molecular epidemiology of C. diphtheriae strains during different phases of the diphtheria epidemic in Belarus. BMC Infect Dis. (2006) 6:129. doi: 10.1186/1471-2334-6-129
146. Mokrousov I, Limeschenko E, Vyazovaya A, Narvskaya O. Corynebacterium diphtheriae spoligotyping based on combined use of two CRISPR loci. Biotechnol J. (2007) 2:901–6. doi: 10.1002/biot.200700035
147. Mokrousov I, Narvskaya O, Limeschenko E, Vyazovaya A. Efficient discrimination within a Corynebacterium diphtheriae epidemic clonal group by a novel macroarray-based method. J Clin Microbiol. (2005) 43:1662–8. doi: 10.1128/JCM.43.4.1662-1668.2005
148. Mokrousov I, Vyazovaya A, Kolodkina V, Limeschenko E, Titov L, Narvskaya O. Novel macroarray-based method of Corynebacterium diphtheriae genotyping: evaluation in a field study in Belarus. Eur J Clin Microbiol Infect Dis. (2009) 28:701–3. doi: 10.1007/s10096-008-0674-4
149. Titov L, Kolodkina V, Dronina A, Grimont F, Grimont PA, Lejay-Collin M, et al. Genotypic and phenotypic characteristics of Corynebacterium diphtheriae strains isolated from patients in belarus during an epidemic period. J Clin Microbiol. (2003) 41:1285–8. doi: 10.1128/JCM.41.3.1285-1288.2003
150. Mokrousov I. Resolution threshold of current molecular epidemiology of diphtheria. Emerg Infect Dis. (2014) 20:1937–8. doi: 10.3201/eid2011.140094
151. Fourel G, Phalipon A, Kaczorek M. Evidence for direct regulation of diphtheria toxin gene transcription by an Fe2+-dependent DNA-binding repressor, DtoxR, in Corynebacterium diphtheriae. Infect Immun. (1989) 57:3221–5.
152. Sangal V, Burkovski A, Hunt AC, Edwards B, Blom J, Hoskisson PA. A lack of genetic basis for biovar differentiation in clinically important Corynebacterium diphtheriae from whole genome sequencing. Infect Genet Evol. (2013) 21:54–7. doi: 10.1016/j.meegid.2013.10.019
153. Stucki D, Ballif M, Egger M, Furrer H, Altpeter E, Battegay M, et al. Standard genotyping overestimates transmission of Mycobacterium tuberculosis among immigrants in a low-incidence country. J Clin Microbiol. (2016) 54:1862–70. doi: 10.1128/JCM.00126-16
154. Cerdeno-Tarraga AM, Efstratiou A, Dover LG, Holden MT, Pallen M, Bentley SD, et al. The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res. (2003) 31:6516–23. doi: 10.1093/nar/gkg874
155. Trost E, Blom J, Soares Sde C, Huang IH, Al-Dilaimi A, Schroder J, et al. Pangenomic study of Corynebacterium diphtheriae that provides insights into the genomic diversity of pathogenic isolates from cases of classical diphtheria, endocarditis, and pneumonia. J Bacteriol. (2012) 194:3199–215. doi: 10.1128/JB.00183-12
156. Sangal V, Blom J, Sutcliffe IC, von Hunolstein C, Burkovski A, Hoskisson PA. Adherence and invasive properties of Corynebacterium diphtheriae strains correlates with the predicted membrane-associated and secreted proteome. BMC Genom. (2015) 16:765. doi: 10.1186/s12864-015-1980-8
157. Arnold JW, Koudelka GB. The Trojan Horse of the microbiological arms race: phage-encoded toxins as a defence against eukaryotic predators. Environ Microbiol. (2014) 16:454–66. doi: 10.1111/1462-2920.12232
158. Mokrousov I. Corynebacterium diphtheriae: genome diversity, population structure and genotyping perspectives. Infect Genet Evol. (2009) 9:1–15. doi: 10.1016/j.meegid.2008.09.011
159. Grosse-Kock S, Kolodkina V, Schwalbe EC, Blom J, Burkovski A, Hoskisson PA, et al. Genomic analysis of endemic clones of toxigenic and non-toxigenic Corynebacterium diphtheriae in Belarus during and after the major epidemic in 1990s. BMC Genom. (2017) 18:873. doi: 10.1186/s12864-017-4276-3
160. Chen J, Ram G, Penades JR, Brown S, Novick RP. Pathogenicity island-directed transfer of unlinked chromosomal virulence genes. Mol Cell. (2015) 57:138–49. doi: 10.1016/j.molcel.2014.11.011
161. Joseph B, Schwarz RF, Linke B, Blom J, Becker A, Claus H, et al. Virulence evolution of the human pathogen Neisseria meningitidis by recombination in the core and accessory genome. PLoS ONE. (2011) 6:e18441. doi: 10.1371/journal.pone.0018441
162. Sabharwal V, Stevenson A, Figueira M, Orthopoulos G, Trzcinski K, Pelton SI. Capsular switching as a strategy to increase pneumococcal virulence in experimental otitis media model. Microbes Infect. (2014) 16:292–9. doi: 10.1016/j.micinf.2013.12.002
163. Marks LR, Reddinger RM, Hakansson AP. High levels of genetic recombination during nasopharyngeal carriage and biofilm formation in Streptococcus pneumoniae. MBio. (2012) 3:e00200–12. doi: 10.1128/mBio.00200-12
164. Sangal V, Tucker NP, Burkovski A, Hoskisson PA. Draft genome sequence of Corynebacterium diphtheriae biovar intermedius NCTC 5011. J Bacteriol. (2012) 194:4738. doi: 10.1128/JB.00939-12
165. Sangal V, Tucker NP, Burkovski A, Hoskisson PA. The draft genome sequence of Corynebacterium diphtheriae bv. mitis NCTC 3529 reveals significant diversity between the primary disease-causing biovars. J Bacteriol. (2012) 194:3269. doi: 10.1128/JB.00503-12
166. Hong KW, Asmah Hani AW, Nurul Aina Murni CA, Pusparani RR, Chong CK, Verasahib K, et al. Comparative genomic and phylogenetic analysis of a toxigenic clinical isolate of Corynebacterium diphtheriae strain B-D-16–78 from Malaysia. Infect Genet Evol. (2017) 54:263–70. doi: 10.1016/j.meegid.2017.07.015
167. Timms VJ, Nguyen T, Crighton T, Yuen M, Sintchenko V. Genome-wide comparison of Corynebacterium diphtheriae isolates from Australia identifies differences in the Pan-genomes between respiratory and cutaneous strains. BMC Genom. (2018) 19:869. doi: 10.1186/s12864-018-5147-2
168. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computat Biol. (2017) 13:e1005595. doi: 10.1371/journal.pcbi.1005595
169. Hadfield J, Croucher NJ, Goater RJ, Abudahab K, Aanensen DM, Harris SR. Phandango: an interactive viewer for bacterial population genomics. Bioinformatics. (2017) doi: 10.1093/bioinformatics/btx610. [Epub ahead of print].
170. Lawpoolsri S, Kaewkungwal J, Khamsiriwatchara A, Sovann L, Sreng B, Phommasack B, et al. Data quality and timeliness of outbreak reporting system among countries in Greater Mekong subregion: challenges for international data sharing. PLoS Negl Trop Dis. (2018) 12:e0006425. doi: 10.1371/journal.pntd.0006425
172. Neal S, Efstratiou A. DIPNET - establishment of a dedicated surveillance network for diphtheria in Europe. Euro surveillance. (2007) 12:E9–10. doi: 10.2807/esm.12.12.00754-en
173. Egli A, Blanc DS, Greub G, Keller PM, Lazarevic V, Lebrand A, et al. Improving the quality and workflow of bacterial genome sequencing and analysis: paving the way for a Switzerland-wide molecular epidemiological surveillance platform. Swiss Med Wkly. (2018) 148:w14693. doi: 10.4414/smw.2018.14693
174. Choi J, Cho Y, Shim E, Woo H. Web-based infectious disease surveillance systems and public health perspectives: a systematic review. BMC Public Health. (2016) 16:1238. doi: 10.1186/s12889-016-3893-0
175. Jain R, Sontisirikit S, Iamsirithaworn S, Prendinger H. Prediction of dengue outbreaks based on disease surveillance, meteorological and socio-economic data. BMC Infect Dis. (2019) 19:272. doi: 10.1186/s12879-019-3874-x
176. Njage PMK, Leekitcharoenphon P, Hald T. Improving hazard characterization in microbial risk assessment using next generation sequencing data and machine learning: predicting clinical outcomes in shigatoxigenic Escherichia coli. Int J Food Microbiol. (2019) 292:72–82. doi: 10.1016/j.ijfoodmicro.2018.11.016
177. Power RA, Parkhill J, de Oliveira T. Microbial genome-wide association studies: lessons from human GWAS. Nat Rev Genet. (2017) 18:41–50. doi: 10.1038/nrg.2016.132
178. Coll F, Phelan J, Hill-Cawthorne GA, Nair MB, Mallard K, Ali S, et al. Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat Genet. (2018) 50:307–16.
179. Park J, Zhang Y, Buboltz AM, Zhang X, Schuster SC, Ahuja U, et al. Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens. BMC Genom. (2012) 13:545. doi: 10.1186/1471-2164-13-545
Keywords: Corynebacterium diphtheriae, diphtheria, surveillance, public health, whole genome sequencing, molecular epidemiology, toxin
Citation: Seth-Smith HMB and Egli A (2019) Whole Genome Sequencing for Surveillance of Diphtheria in Low Incidence Settings. Front. Public Health 7:235. doi: 10.3389/fpubh.2019.00235
Received: 14 May 2019; Accepted: 06 August 2019;
Published: 21 August 2019.
Edited by:
Marc Jean Struelens, European Centre for Disease Prevention and Control (ECDC), SwedenReviewed by:
Andreas Sing, Bavarian State Office for Health and Food Safety, GermanyCopyright © 2019 Seth-Smith and Egli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrian Egli, YWRyaWFuLmVnbGlAdXNiLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.