 Dora Il'yasova
Dora Il'yasova Alexander Kinev
Alexander Kinev Rose Grégoire
Rose Grégoire Craig C. Beeson
Craig C. Beeson- 1Department of Population Health Science, School of Public Health, Georgia State University, Atlanta, GA, United States
- 2Creative Scientist, Inc., Durham, NC, United States
- 3Department of Pharmaceutical and Biomedical Sciences, Medical University of South Carolina, Charleston, SC, United States
Although humans, as placental mammals, possess a highly sophisticated system protecting the embryo/fetus in uterus from the environment, prenatal development might still be negatively affected via exposures to environmental factors (1–4). Many xenobiotic compounds in mothers' circulation (e.g., toxicants, drugs) can cross the placental barrier and reach developing tissues and organs, consequently altering cellular functions within the tissues. Among different cellular functions, mitochondrial metabolism is thought to be particularly sensitive to various stressors, including those produced by environmental toxicants (5, 6). Since mitochondria play a central role in cellular energy homeostasis and apoptotic/stress responses, it is not surprising that mitochondrial dysfunction is associated with numerous chronic diseases, such as neurodegenerative diseases, obesity, type-2 diabetes, along with a variety of cancers—the conditions which have been linked to environmental exposures (7–11). Many of the chronic disease conditions occurring later in life were proposed to originate from in utero exposures (1, 12). The proposed connections between in utero exposures to a wide variety of environmental toxicants and mitochondrial dysfunction have been documented by multiple animal studies. For example, maternal exposures to small-sized particulate matter and cigarette smoke were associated with a reduction in the mitochondrial (mt)DNA copy number per cell and a lower expression of oxidative phosphorylation proteins in the offspring (13). Exposure to cadmium, a toxic component of cigarette smoke, has been shown to inhibit mitochondrial respiration while simultaneously increasing reactive oxygen species (ROS) production (14). Prenatal exposure of rats to perfluorooctane sulfonate (PFOS) induced apoptosis in offspring heart tissue through the mitochondria-mediated apoptotic pathway (15). In utero exposure to arsenic impaired mitochondrial respiration with subsequent cardiac myopathies (16) and neurotoxicity (11). It was further shown that obesogens—chemicals that increase an offspring's weight—may also act as mitochondrial toxicants (10). Established obesogens, organotin compounds inhibit mitochondrial ATP production (17). Early-life exposure to the endocrine disruptor bisphenol A (BPA) has been linked with the development of obesity (18). At the same time, exposure to BPA has been associated with hypermethylation of the master mitochondrial regulatory gene PGC-1α and reduced mitochondrial respiration and ATP production (19). All these examples summarized in Table 1 corroborate the hypothesis that prenatal exposures to xenobiotics might influence post-natal development, with mitochondrial metabolism playing an important role in such a connection.
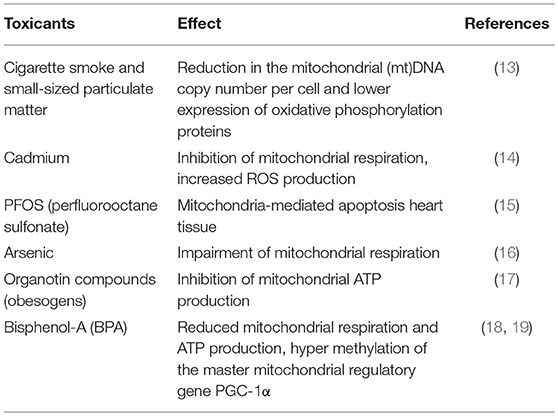
Table 1. Mitochondrial function impairment after prenatal exposure to environmental toxicants in animal studies.
The idea of a prenatal origin of childhood obesity attracts attention, because lifestyle interventions implemented amongst this age group have not been effective in lowering adiposity among corpulent children (20). If prenatal environment plays an important role in inducing individual predispositions to excessive adiposity, then typical postnatal interventions may have little-to-no effect on obesity in both adults and children. Given the rapid increase in childhood obesity prevalence, evaluation of how prenatal xenobiotic exposures affect mitochondrial metabolism in embryo-fetal development have become an important focus of biomedical research. Some studies estimate that in utero exposure to BPA alone may contribute to 12,404 cases of childhood obesity, with a U.S. societal cost of $1.49 billion (21). Similarly, prenatal exposure to BPA in the European Union has a 20–60% probability of initiating 42,400 cases of childhood obesity (22). However, the confirmatory evidence from prospective human epidemiological studies that link prenatal exposure to altered mitochondrial function is currently lacking. In this opinion manuscript, we suggest that the assessment of mitochondrial function at birth in cells isolated from the umbilical cord blood might help to identify prenatal exposures through their alteration of mitochondrial bioenergetics in newborns, which can be subsequently linked to postnatal health outcomes. Next, we will discuss both the strengths and limitations of such an approach.
Proposed Strengths
One of the important considerations in the human assessment of mitochondrial health is accessibility to a sufficient amount of biological material. This issue is especially pertinent to the development of biomarkers in the pediatric population and is even more challenging for epidemiological studies in human embryos, fetuses, or neonates (23). Umbilical cord blood can be obtained in an entirely non-invasive manner, and it represents a rich source of fetal cells—especially the highly proliferative stem and progenitor cells. Our previous work indicates that a population-wide collection of donor-specific endothelial colony-forming cells (or ECFC's) is possible to obtain because ECFCs can be isolated from cord blood with a high success rate (>90%) (24). ECFCs can be expanded in vitro, thereby providing sufficient amounts of biological material for multiple cell-based assays, including those for mitochondrial function (25). Moreover, a collection of donor-specific ECFCs can be viewed as a panel of tissues representing human neonatal population to enable assessment of individual variability in responses to mitochondrial toxicants.
In the past, mitochondrial health was assessed indirectly from changes in mtDNA and enzymatic activity related to different mitochondrial functions. Such measurements in peripheral blood mononuclear cells provided the first evidence that mitochondrial health can be associated with many chronic health conditions, including but not limited to diabetes, cardiovascular disease, and common neurodegenerative disorders (26–31). Donor-specific cells isolated from cord blood provide an opportunity to use the current high-resolution respirometry technology (32) to directly assess mitochondrial bioenergetic profiles within newborns. The sequential additions of different metabolic modulators, known commonly as a “stress test,” determines several parameters in one experiment: the glycolytic activity, the ATP-linked respiration, proton leak, basal and maximal respirations, and finally the non-mitochondrial respiration. Each of these parameters presents interpretable measurements that are the biomarkers of mitochondrial health (6). For example, when cells are placed in identical experimental conditions, lower ATP-linked respiration could indicate damage to substrate uptake or oxidative phosphorylation. A greater proton leak could indicate increased uncoupling protein (UCP) activity that has been related to the “less economical” bioenergetics with a higher intensity of fat oxidation and a lower predisposition to obesity and diabetes (32). Alternatively, a high proton leak may indicate a response to oxidative stress (32) or even damage to the inner mitochondrial membrane. Furthermore, the difference between the basal and maximal respiration presents the reserve bioenergetic capacity, a sensitive parameter correlated to cellular stress (6). Recently, a combination of these parameters were fused into a single Bioenergetic Health Index (BHI), which has been proposed to represent the donor's composite mitochondrial profile. Such an index could provide a dynamic indicator of bioenergetic health measured in platelets and leukocytes. Ultimately, the BHI has the potential to be a new biomarker in the assessment of patient health via presentation of both the prognostic and diagnostic values (6). However, even peripheral blood measurements that are considered minimally invasive can be invasive for newborns. Fortunately, using umbilical cord blood cells presents a non-invasive alternative. Thus, using cord blood cells as a platform to assess a newborn's mitochondrial health provides the direct measurements of mitochondrial bioenergetics in this otherwise difficult-to-study population.
Unanswered Questions and Limitations
When assessing mitochondrial health, the major question is as follows: which human cells should be used to assess mitochondrial function in an individual? It has been argued that the mitochondrial respiration profile of circulating leukocytes and platelets can serve to assess mitochondrial function in other tissues, or more specifically, in that of a skeletal muscle (33, 34). Similarly, it is not certain whether the bioenergetic profile of umbilical cord blood cells correlates with cells comprising other fetal/newborn tissues. In humans, these correlations are practically impossible to assess due to ethical barriers; however, such research can be conducted in animal models.
Another question is whether a mitochondrial stress test profile has the ability to measure an individual characteristic that is stable over time. For epidemiological assessment, low within-person variability of a biomarker is especially important in evaluating its association with a disease of long latency (35). If within-person variability is large compared to the overall between-person variability, such a biomarker cannot represent an individual characteristic, differentiating one study participant from another. With respect to mitochondrial health, within-person variability can be assessed by comparing cells derived from the same donor vs. variability between cells derived from different donors using a mitochondrial stress test profiles, individual stress parameters and/or the composite BHI measurements. With respect to peripheral blood cells, within- and between-person variability can be assessed from blood specimens obtained from the same donors procured at different time points. One important caveat is that such an evaluation must be performed carefully, especially considering the circadian differences in circulating nutrients and/or physical activity that may influence the bioenergetic profiles of cells isolated from peripheral blood. Since peripheral blood cells cannot produce viable primary in vitro cell lines, the omission of the confounding effect of recent exposures would be problematic. In contrast, using primary cultures of cord blood progenitor cells could help to bypass the confounding influence of recent exposures, as these cells produce viable donor-specific cell lines and can be cultured in identical conditions. However, there is only one chance to obtain a cord blood specimen—during a delivery. How, then, will within-person variability of mitochondrial health can be evaluated in cord blood-derived cells? To bypass such a limitation, we propose to develop multiple cell lines from a single donor/newborn for the assessment of within-person variability.
Finally, feasibility considerations for large-scale epidemiological studies bring up a question of utilization of the existing cord blood biobanks. The current cell isolation methods do not consistently provide viable, proliferating cells from either frozen whole blood or partially purified blood samples. Therefore, fresh umbilical cord blood samples should be used to isolate cells, which then can be stored for a long period of time (several years in our experience) for cell-based assays. Thus, the need to use fresh cord blood specimens considerably limits the use of the existing children cohorts, for which the frozen cord-blood specimens are available.
Conclusions
Mitochondrial health can be assessed at birth in cell-based assays using cells that are isolated from umbilical cord blood. Animal studies present enough evidence connecting prenatal exposure to environmental toxicants and mitochondrial health in different tissues of the offspring, demonstrating that such a connection might play an important role in human health. Animal studies can answer questions about correlations between mitochondrial health biomarkers obtained from cord-blood cells and other tissues in the offspring, ultimately proving or disproving that the assessment of mitochondrial health in one tissue can be extrapolated to the entire individual organism. However, only human studies can characterize within- and between-person variability of mitochondrial health biomarkers. With respect to the hypothesis of mitochondrial involvement in early origins of disease, using cord blood cells presents a unique opportunity for studying the connections between prenatal exposures, mitochondrial health, and long-term health outcomes.
Further Readings
For further readings on the topic, we suggest the following references: (36–53).
Author Contributions
DI and AK conceived the idea of this opinion and wrote the initial text. CB provided his expertise in mitochondrial science. RG helped to edit the initial text, researched the published literature for correct references, and wrote the part of the manuscript about animal models.
Funding
This research was supported by the National Institute of Environmental Health under award number 1R43ES027716 and by NC SBIR/STTR Matching Funds Program.
Conflict of Interest Statement
AK is a founder and executive director of Creative Scientist, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. (2008) 359:61–73. doi: 10.1056/NEJMra0708473
2. Woodruff TJ, Burke TA, Zeise L. The need for better public health decisions on chemicals released into our environment. Health Affairss. (2011) 30:957–67. doi: 10.1377/hlthaff.2011.0194
3. Landrigan PJ, Kimmel CA, Correa A, Eskenazi B. Children's health and the environment: public health issues and challenges for risk assessment. Environ Health Perspect. (2004) 112:257–65. doi: 10.1289/ehp.6115
4. Gitterman BA, Bearer CF. A developmental approach to pediatric environmental health. Pediatr Clin North Am. (2001) 48:1071–83. doi: 10.1016/S0031-3955(05)70361-8
5. Shaughnessy DT, McAllister KA, Worth L, Haugen AC, Meyer JN, Domann FE. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. (2014) 122:1271–8. doi: 10.1289/ehp.1408418
6. Chacko BK, Kramer PA, Ravi S, Benavides GA, Mitchell T, Dranka BP, et al. The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clinical Science. (2014) 127(Pt 6):367–73. doi: 10.1042/CS20140101
7. Eze IC, Imboden M, Kumar A, Adam M, von Eckardstein A, Stolz D, et al. A common functional variant on the pro-inflammatory Interleukin-6 gene may modify the association between long-term PM10 exposure and diabetes. Environ Health. (2016) 15:39. doi: 10.1186/s12940-016-0120-5
8. Balti EV, Echouffo-Tcheugui JB, Yako YY, Kengne AP. Air pollution and risk of type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Res Clin Pract. (2014) 106:161–72. doi: 10.1016/j.diabres.2014.08.010
9. Heindel JJ, vom Saal FS, Blumberg B, Bovolin P, Calamandrei G, Ceresini G, et al. Parma consensus statement on metabolic disruptors. Environ Health. (2015) 14:54. doi: 10.1186/s12940-015-0042-7
10. Thayer KA, Heindel JJ, Bucher JR, Gallo MA. Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect. (2012) 120:779–89. doi: 10.1289/ehp.1104597
11. Prakash C, Sony M, Kumar V. Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: a review. J Appl Toxicol. (2016) 36:179–88. doi: 10.1002/jat.3256
12. Barker DJ. The origins of the developmental origins theory. J Inter Med. (2007) 261:412–7. doi: 10.1111/j.1365-2796.2007.01809.x
13. Stephenson EJ, Ragauskas A, Jaligama S, Redd JR, Parvathareddy J, Peloquin MJ, et al. Exposure to environmentally persistent free radicals during gestation lowers energy expenditure and impairs skeletal muscle mitochondrial function in adult mice. Am J Physiol Endocrinol Metab. (2016) 310:E1003–15. doi: 10.1152/ajpendo.00521.2015
14. Gobe G, Crane D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol Lett. (2010) 198:49–55. doi: 10.1016/j.toxlet.2010.04.013
15. Zeng HC, He QZ, Li YY, Wu CQ, Wu YM, Xu SQ. Prenatal exposure to PFOS caused mitochondria-mediated apoptosis in heart of weaned rat. Environ Toxicol. (2015) 30:1082–90. doi: 10.1002/tox.21981
16. Ambrosio F, Brown E, Stolz D, Ferrari R, Goodpaster B, Deasy B, et al. Arsenic induces sustained impairment of skeletal muscle and muscle progenitor cell ultrastructure and bioenergetics. Free Radical Biol Med. (2014) 74:64–73. doi: 10.1016/j.freeradbiomed.2014.06.012
17. Aldridge WN. The biochemistry of organotin compounds: trialkyltins and oxidative phosphorylation. Biochem J. (1958) 69:367–76. doi: 10.1042/bj0690367
18. Hoepner L, Whyatt R, Widen E, Hassoun A, Oberfield S, Mueller N, et al. Bisphenol A and Adiposity in an Inner-City Birth Cohort. Environ Health Perspect. (2016) 124:1644–50. doi: 10.1289/EHP205
19. Jiang Y, Xia W, Yang J, Zhu Y, Chang H, Liu J, et al. BPA-induced DNA hypermethylation of the master mitochondrial gene PGC-1alpha contributes to cardiomyopathy in male rats. Toxicology. (2015) 329:21–31. doi: 10.1016/j.tox.2015.01.001
20. Kamath C, Vickers K, Ehrlich A, McGovern L, Johnson J, Singhal V, et al. Clinical review: Behavioral interventions to prevent childhood obesity: A systematic review and metaanalyses of randomized trials. J Clin Endocrinol Metab. (2008) 93:4606–15. doi: 10.1210/jc.2006-2411
21. Trasande L. Further limiting Bisphenol A in food uses could provide health and economic benefits. Health Affairs. (2014) 33:316–23. doi: 10.1377/hlthaff.2013.0686
22. Woodruff TJ. Making it real—the environmental burden of disease. what does it take to make people pay attention to the environment and health? J Clin Endocrinol Metab. (2015) 100:1241–4. doi: 10.1210/jc.2015-1622
23. Shores RS, Allen DE. Children as biomarker orphans: progress in the field of pediatric biomarkers. J Pediatr. (2018) 193:14–20. doi: 10.1016/j.jpeds.2017.08.077
24. Il'yasova D, Kloc N, Kinev A. Cord blood cells for developmental toxicology and environmental health. Front Public Health. (2015) 3:265. doi: 10.3389/fpubh.2015.00265
25. Kinev AV, Levering V, Young K, Ali-Osman F, Truskey GA, Dewhirst MW, et al. Endothelial colony forming cells (ECFCs) as a model for studying effects of low-dose ionizing radiation: growth inhibition by a single dose. Cancer Invest. (2013) 31:359–64. doi: 10.3109/07357907.2013.789903
26. Mancuso M, Orsucci D, LoGerfo A, Calsolaro V, Siciliano G. Clinical features and pathogenesis of Alzheimer's disease: involvement of mitochondria and mitochondrial DNA. Adv Exp Med Biol. (2010) 685:34–44. doi: 10.1007/978-1-4419-6448-9_4
27. Avila C, Huang RJ, Stevens MV, Aponte AM, Tripodi D, Kim KY, et al. Platelet mitochondrial dysfunction is evident in type 2 diabetes in association with modifications of mitochondrial anti-oxidant stress proteins, Exp. Clin Endocrinol Diabetes. (2012) 120:248–51. doi: 10.1055/s-0031-1285833
28. Hartman ML, Shirihai OS, Holbrook M, Xu G, Kocherla M, Shah A, et al. Relation of mitochondrial oxygen consumption in peripheral blood mononuclear cells to vascular function in type 2 diabetes mellitus. Vasc Med. (2014) 19:67–74. doi: 10.1177/1358863X14521315
29. Widlansky MEJ, Wang SM, Shenouda TM, Hagen AR, Smith TJ, Kizhakekuttu MA, et al. Altered mitochondrial membrane potential, mass, and morphology in the mononuclear cells of humans with type 2 diabetes. Transl Res. (2010) 156:15–25. doi: 10.1016/j.trsl.2010.04.001
30. Shi CK, Guo DT, Yew Z, Yao EL, Forster H, Wang J, et al. Effects of ageing and Alzheimer's disease on mitochondrial function of human platelets. Exp Gerontol. (2008) 43:589–94. doi: 10.1016/j.exger.2008.02.004
31. Chen CM, Wu YR, Cheng ML, Liu JL, Lee YM, Lee PW, et al. (2007). Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington's disease patients. Biochem Biophys Res Commun. 359:335–40. doi: 10.1016/j.bbrc.2007.05.093
32. Hill BG, Benavides GA, Lancaster JR, Ballinger S, Dell'Italia L, Zhang J, et al. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. (2012) 393:1485–512. doi: 10.1515/hsz-2012-0198
33. Tyrrell DJ, Bharadwaj MS, Van Horn CG, Kritchevsky SB, Nicklas BJ, Molina AJA. respirometric profiling of muscle mitochondria and blood cells are associated with differences in gait speed among community-dwelling older adults. J Gerontol Series A. (2015) 70:1394–9. doi: 10.1093/gerona/glu096
34. Tyrrell DJ, Bharadwaj MS, Jorgensen MJ, Register TC, Molina AJA. Blood cell respirometry is associated with skeletal and cardiac muscle bioenergetics: Implications for a minimally invasive biomarker of mitochondrial health. Redox Biol. (2016) 10:65–77. doi: 10.1016/j.redox.2016.09.009
35. Tworoger S, Hankinson S. Use of biomarkers in epidemiologic studies: Minimizing the influence of measurement error in the study design and analysis. Cancer Causes Control. (2006) 17:889–99. doi: 10.1007/s10552-006-0035-5
36. Abdo N, Xia M, Brown CC, Kosyk O, Huang R, Sakamuru S, et al. Population based in vitro hazard and concentration-response assessment of chemicals: the 1000 genomes high-throughput screening study. Environ Health Perspect. (2015) 123:458–66. doi: 10.1289/ehp.1408775
37. Blue EK, Sheehan BM, Nuss ZV, Boyle FA, Hocutt CM, Gohn CR, et al. Epigenetic Regulation of placenta-specific 8 contributes to altered function of endothelial colony-forming cells exposed to intrauterine gestational diabetes mellitus. Diabetes. (2015) 64:2664–75. doi: 10.2337/db14-1709
38. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. (2011) 435:297–312. doi: 10.1042/BJ20110162
39. Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. (2015) 10:138–146. doi: 10.1038/nrc2791
40. Carmeliet P. Angiogenesis in health and disease. Nat Med. (2003) 9:653–60. doi: 10.1038/nm0603-653
41. Dincer UD. Fetal exposure to a diabetic intrauterine environment resulted in a failure of cord blood endothelial progenitor cell adaptation against chronic hypoxia. Stem Cells Cloning. (2015) 8:1–14. doi: 10.2147/SCCAA.S73658
42. Gui J, Potthast A, Rohrbach A, Borns K, Das AM, von Versen-Hoynck F. Gestational diabetes induces alterations of sirtuins in fetal endothelial cells. Pediatric Res. (2016) 79:788–98. doi: 10.1038/pr.2015.269
43. Il'yasova D, Kinev A, Melton CD, Davis FG. Donor-specific cell-based assays in studying sensitivity to low-dose radiation: a population-based perspective. Front Public Health. (2014) 2:244. doi: 10.3389/fpubh.2014.00244
44. Mordwinkin NM, Ouzounian JG, Yedigarova L, Montoro MN, Louie SG, Rodgers KE. Alteration of endothelial function markers in women with gestational diabetes and their fetuses. J Matern Fetal Neonatal Med. (2013) 26:507–12. doi: 10.3109/14767058.2012.736564
45. Munoz-Hernandez R, Miranda ML, Stiefel P, Lin RZ, Praena-Fernandez JM, Dominguez-Simeon MJ, et al. Decreased level of cord blood circulating endothelial colony-forming cells in preeclampsia. Hypertension. (2014) 64:165–71. doi: 10.1161/HYPERTENSIONAHA.113.03058
46. National Institutes of Health. Stem Cell Information. Stem Cell Basics. Bethesda, MD: National Institutes of Health; U.S. Department of Health and Human Services (2015). Available online at: https://stemcells.nih.gov/sites/default/files/SCprimer2009.pdf
47. Onoyama S, Qiu L, Low HP, Chang CI, Strohsnitter WC, Norwitz ER, et al. Prenatal maternal physical activity and stem cells in umbilical cord blood. Med Sci Sports Exerc. (2016) 48:82–9. doi: 10.1249/MSS.0000000000000731
48. Qiu L, Onoyama S, Low HP, Chang CI, Strohsnitter WC, Norwitz ER, et al. Effect of preeclampsia on umbilical cord blood stem cells in relation to breast cancer susceptibility in the offspring. Carcinogenesis. (2015) 36:94–8. doi: 10.1093/carcin/bgu231
49. Shaughnessy DT, Worth L Jr, Lawler CP, McAllister KA, Longley MJ, Copelandb WC. Meeting report: identification of bio-markers for early detection of mitochondrial dysfunction. Mitochondrion. (2010) 10:579–81. doi: 10.1016/j.mito.2010.02.001
50. Sipos PI, Bourque SL, Hubel CA, Baker PN, Sibley CP, Davidge ST, et al. Endothelial colony-forming cells derived from pregnancies complicated by intrauterine growth restriction are fewer and have reduced vasculogenic capacity. J Clin Endocrinol Metabol. (2013) 98:4953–60. doi: 10.1210/jc.2013-2580
51. Stangenberg S, Nguyen LT, Chen H, Al-OData I, Killingsworth MC, Gosnell ME, et al. Oxidative stress, mitochondrial perturbations and fetal programming of renal disease induced by maternal smoking. Int J Biochem Cell Biol. (2015) 64:81–90. doi: 10.1016/j.biocel.2015.03.017
52. Von Balloons C, Brunner J, Dimroth P. The ion channel of F-ATP synthase is the target of toxic organotin compounds. Proc Natl Acad Sci USA. (2004) 101:11239–44. doi: 10.1073/pnas.0402869101
Keywords: biomarker, prenatal exposure, mitochondria, epidemiology, cell-based assay
Citation: Il'yasova D, Kinev A, Grégoire R and Beeson CC (2019) A Cell-Based Approach to Study the Associations Between Mitochondrial Health, Early Life Exposures, and Consequent Health Outcomes. Front. Public Health 7:36. doi: 10.3389/fpubh.2019.00036
Received: 01 October 2018; Accepted: 12 February 2019;
Published: 13 March 2019.
Edited by:
Lalage A. Katunga, Saint Louis University, United StatesReviewed by:
Richard Eugene Frye, Phoenix Children's Hospital, United StatesCopyright © 2019 Il'yasova, Kinev, Grégoire and Beeson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dora Il'yasova, ZGlseWFzb3ZhQGdzdS5lZHU=