 Joanne Ryan
Joanne Ryan Peter Fransquet
Peter Fransquet Paul Lacaze
Paul Lacaze- Department of Epidemiology and Preventive Medicine, Monash University, Melbourne, VIC, Australia
Dementia can result from a number of distinct diseases with differing etiology and pathophysiology. Even within the same disease, there is considerable phenotypic heterogeneity with varying symptoms and disease trajectories. Dementia diagnosis is thus very complex, time-consuming, and expensive and can only be made definitively post-mortem with histopathological confirmation. These inherent difficulties combined with the overlap of some symptoms and even neuropathological features, present a challenging problem for research in the field. This has likely hampered progress in epidemiological studies of risk factors and preventative interventions, as well as genetic and biomarker research. Resource limitations in large epidemiologically studies mean that limited diagnostic criteria are often used, which can result in phenotypically heterogeneous disease states being grouped together, potentially resulting in misclassification bias. When biomarkers are identified for etiologically heterogeneous diseases, they will have low specificity for any utility in clinical practice, even if their sensitivity is high. We highlight several challenges in in the field which must be addressed for the success of future genetic and biomarker studies, and may be key to the development of the most effective treatments. As a step toward achieving this goal, defining the dementia as a biological construct based on the presence of specific pathological features, rather than clinical symptoms, will enable more precise predictive models. It has the potential to lead to the discovery of novel genetic variants, as well as the identification of individuals at heightened risk of the disease, even prior to the appearance of clinical symptoms.
Introduction
Dementia is a major public health problem, with enormous social and economic costs, and substantial burden for the individual, their caregiver and families (1). By 2050, it is estimated that over 130 million people will be living with dementia (2). This sharp increase from the 2015 estimates of 48 million reflects not only the aging population worldwide, but the current lack of effective treatments or cures. The results of drug trials to slow or halt the progression of dementia have so far been unsuccessful (3). This emphasizes the need for more research into the etiology of the diseases which cause dementia, with better characterization of genetic and environmental risk factors (4). There is also an increasing push to identify valid disease biomarkers, which would aid in diagnosis, and could be used to predict individuals at future risk (5).
Challenges With Diagnosis
Dementia is an overarching term used to describe a group of symptoms that results in severe long-term decline in cognitive function that is significant enough to affect daily function (6). Dementia can result from a number of complex disorders which damage the brain. The most common includes Alzheimer's disease (AD), vascular dementia, frontotemporal dementia, dementia with Lewy bodies, and Parkinson's disease. Typical symptoms of dementia can include a decline in memory, language deficits, and impaired visuospatial skills, as well as a loss of executive function and attention. Associated mood and behavioral disturbances, including delusions, are also frequent (6). However the exact symptoms a person experiences depends on the disease that is causing dementia, as they are distinct diseases with differing etiology and pathophysiology. Symptoms also depend on the parts of the brain that are damaged and the complexity of these conditions is such that even within common underlying conditions, presentation of symptoms differs between individuals (7). For example, there are now classifications of both typical and atypical AD (8). Further, these diseases exist on a continuum of severity and with varying disease trajectories (9). When mild, dementia can be dismissed as “normal” age-related cognitive decline, and some individuals are able to mask symptoms in the early stages (7). The extent to which dementia progresses is also highly variable. Further, given the common behavioral and mood disturbances, dementia can also be misdiagnosed as symptoms of a psychiatric disorder (10). This presents an important challenge for the field (7).
In the absence of clear biomarkers, dementia diagnosis is very challenging. Neuropsychological evaluation with profiles of cognitive strengths and weaknesses are used by both clinicians and researchers to define the likely form of dementia. This information is used in combination with reports of clinical symptoms, the results of blood tests and neuroimaging, and is in accordance with diagnostic criteria which are continually evolving (11). As such, diagnosis is often a very expensive, long and time-consuming process which does not always result in a clear outcome. The heterogeneity in symptoms within different diseases, combined with the overlapping features (both symptoms and neuropathology) across many of the diseases (Table 1) further complicates the issue. However the importance of early and accurate differential diagnosis of the underlying dementia condition is crucial. It has implications for prognosis, longer term health planning, and heritability, as well as symptom management, which could potentially be made worse by the use of incorrect treatment (20, 21). Given the continual advances in disease-modifying treatments, it also will have implications for future therapeutics (22).
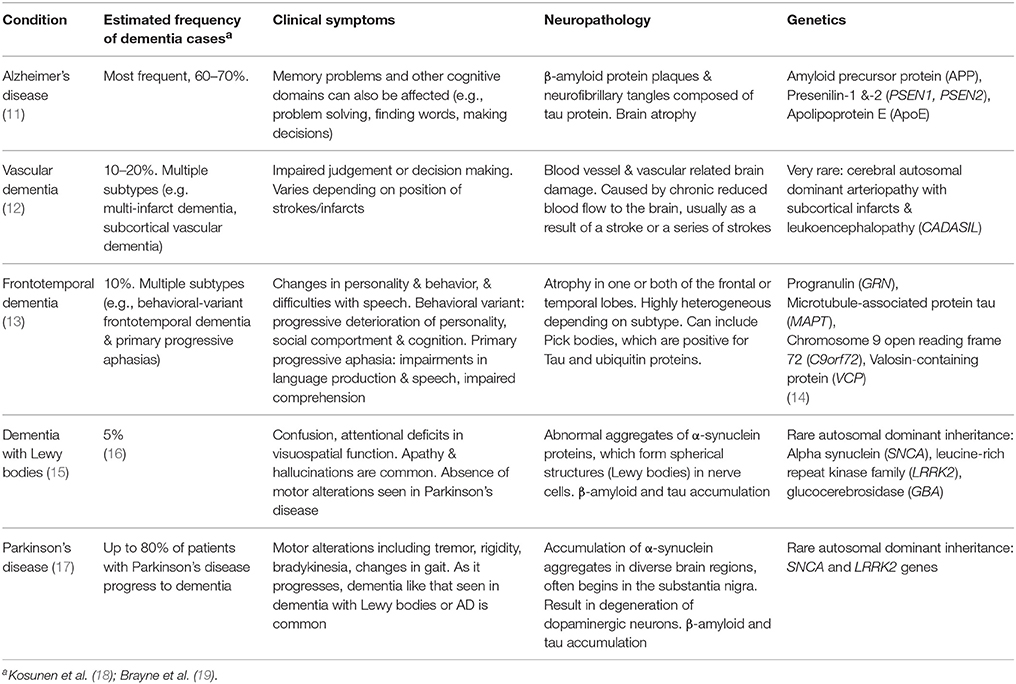
Table 1. Common neurodegenerative disorders characterized by dementia symptoms in older individuals, and characteristic features.
Alzheimer's Disease
The most common form of dementia is AD, an insidious and incapacitating neurodegenerative disorder which accounts for ~60% of all dementia cases (23). The defining pathological features of AD are the presence of two proteins in the brain, amyloid, and tau. Accumulated amyloid beta (β) peptides clump together forming extracellular neurotic plaques, while hyper phosphorylated TAU proteins form intracellular neurofibrillary tangles (24). A definitive diagnosis of AD thus requires histopathologic confirmation via post-mortem. In living individuals, AD is diagnosed as probable or possible according to set criteria (often DSM) by a panel of expert clinicians who review a range of documentation (25). The guidelines established by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association work group (NINCDS-ADRDA), updated in 2011, are the most frequently used for dementia diagnosis (11). An expert panel review the results of extensive neuropsychological testing, detailed medical history, blood tests and imaging, such as magnetic resonance imaging (MRI), positron emission tomography (PET), and/or computerized tomography (CT) and reach a consensus.
Overlapping Features and Misdiagnosis
However studies have shown that a significant proportion of individuals diagnosed with probable/possible AD by experts, do not display the hallmark neuropathological criteria for AD on post-mortem examination (26). In many other cases, more than one form of dementia is identified (“mixed dementia”), and this becomes increasingly more common in later life (27). Vascular dementia is caused by stroke and/or small vessel disease and includes a number of different sub-types (12). It occurs frequently with AD and the presence of both could exacerbate the development of dementia compared with either condition alone (28). Coexistent Parkinson's disease changes also occur relatively frequently in individuals with AD (29). Dementia with Lewy bodies sometimes co-occurs with AD or vascular dementia or can be misdiagnosed as these conditions depending on the presence of symptoms of cognitive impairment or Parkinsonism (30). In fact, Dementia with Lewy bodies and Parkinson's disease are now considered as a continuum of the same disease (Lewy body dementias), with Dementia with Lewy bodies being an early manifestation in patients with Parkinson's (15).
Adding further to these complexities is the overlap in neuroanatomical features of these disorders (Table 1). The hallmark features of AD are the accumulation of amyloid-β and tau protein, yet neither is sufficient to cause dementia nor unique to this disease (31). Tau may be present from early adulthood and could only become problematic once amyloid accumulates (32). Even then, around 30% of people may have amyloid accumulation without any obvious clinical symptoms (33, 34). Dementia with Lewy bodies also shares the neuropathology characteristics of amyloid-β and tau (REF), and the latter is also found in other neurodegenerative conditions such as chronic traumatic encephalopathy (30). Parkinson's disease and frontotemporal dementia both involve tau alterations, but these are a loss of function rather than phosphorylation (35). Likewise, hallmark characteristics of Lewy body dementias, such as α-synuclein inclusions, are also found in many cases of AD (36).
Increasingly evidence from studies investigating neuropathology and molecular genetics has demonstrated that clinical symptoms (phenotype) are not always tightly linked with etiology, as they can be influenced by a variety of other factors including prior experience, cognitive reserve, and epigenetics (37). Studies of several autosomal dominant dementias indicate that the presenting clinical phenotype may vary widely, even for those individuals with the same causative mutation. For example, mutations in the PSEN1 gene are considered almost deterministic for earlier onset AD, yet there is considerable heterogeneity in the clinical expression of neurological features (38). This can include behavioral and psychiatric symptoms which can sometimes reflect frontotemporal dementia or dementia with Lewy bodies (38). Another example is a very rare autosomal dominant neurodegenerative disorder, frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17) which has different phenotypes, even within families carrying the exact same mutation (39). The most established genetic risk factor for late-onset AD is the APOE ε4 allele, and this is also over-represented in sporadic Lewy body dementias compared with controls (40).
Other people have argued that the different dementia conditions are highly related conditions with a continuous range of abnormalities (41), although genetically and epigenetically they are distinct. Indeed, dementia with Lewy bodies has been shown to be similar genetically to AD, while AD and Parkinson's disease were only very weakly correlated (42). Similar aberrant changes in DNA methylation patterns have also been found in individuals with different forms of dementia (41). However the vast majority of genetic and epigenetic patterns are unique to each disease (37). Further, given the potential inaccuracies in diagnosing dementia, overlapping patterns may also reflect, at least in part, inaccuracies in how the conditions have been defined (discussed further below).
The inherent difficulties in diagnosing dementia, as well as the overlapping symptoms and even neuropathological features, presents a complex and challenging problem for research in the field. This is likely to have hampered progress in genetic and biomarker studies to date, as well as epidemiological studies of risk factors and preventative interventions.
Problems With Inaccurate Phenotyping
Genetic and biomarker studies rely on accurate phenotypes and diagnosis (43). Most genetic risk variants identified from such studies are either rare with moderate effect sizes or common with very small effect sizes (44). Large samples are thus needed to have sufficient power to detect true associations, especially at genome-wide significance levels (45). Mixing together diseases with different etiology, pathophysiology and potentially different genetic architecture, is obviously problematic for the investigation of novel genetic variants, diluting out any signals (43). As an example, new genetic loci identified as being associated with clinically-defined AD, were not found to be associated with AD neuropathology at postmortem (46). Similar problems are likely to be plaguing new biomarker discovery. When biomarkers are identified, if they are in fact reflective of etiological heterogeneous disease states, they will have low specificity for any utility in clinical practice, even if their sensitivity is high (47). These issues are exacerbated by the challenges in selecting unaffected controls who are without dementia. AD for example has a very long pre-symptomatic phase (48), meaning that individuals without dementia in the “control” group, may be free of clinical symptoms, but could already have the disease. Together these issues may help explain the lack of substantial progress in this field to date.
Unique Challenges for Large Cohorts
Epidemiological cohort studies of dementia, often with the aim of identifying risk and protective factors for the disease (49, 50), are confronted with many of these challenges. Risk factors identified as being associated with cognitive decline and AD diagnosed solely on the basis of clinical symptoms, may in fact not be associated with AD pathology (46). Diagnosing dementia is expensive and time-consuming, which is compounded when undertaken on a larger scale. As a result, studies often only collect relatively sparse phenotypic data, without imaging, blood measures or other biological markers (51).
In recent years there have been widely commended efforts to increase uniformity around the diagnostic criteria for dementia and the underlying construct. The vast majority of publications in good quality journals now define probable AD using clinical criteria by the National Institute on Aging-Alzheimer's Association (NIA-AA) (11). However this criteria predominantly lists recommendations rather than requirements, with the acknowledgment that not all clinicians will have access to the results of the full range of tests, which are time consuming and expensive to obtain. This criteria also includes evidence of neurodegeneration, and thus recommends where possible, that MRI is used to assess cerebral atrophy, but there are no strong criteria regarding other neuropathological changes. With published studies, there is rarely detailed information concerning the information that was obtained to support a dementia diagnosis, and thus difficult for the reader to assess the strength of evidence for these diagnoses. Many studies instead broadly define dementia, and determine risk factors for this heterogeneous condition, which has obvious limitations (as discussed above).
A large number of other studies use less reliable measures of dementia, such as self-reports, linkage data (52), or community diagnoses, with no additional clinical evidence sought to confirm and establish dementia diagnosis (51, 53). This has obvious problems and would increase both the false positives and false negatives. ICD coding is also still frequently used, but has well-documented limitations (54). In other cases, exact diagnostic criteria is not stated (41). Together such studies are likely to be plagued by misclassification bias which would make it more difficult to identify true associations.
Future Directions
Currently the methods for identifying and delineating different dementia sub-types are imperfect and not scalable. For research to advance in this area there is a need for better definitions, with clearly established guidelines for the minimal information which must be collected data, and diagnostic markers are required to improve classification of the underlying form of dementia and at a level which is standardized and scalable for large studies.
Deep phenotyping is considered to be the key to advancing genetic studies (55), and this is not just unique to dementia, although it may be one of the most challenging areas. Descriptions of disease phenotypes often do not capture the full diversity of clinical and even pathophysiological manifestations. Advancing research in this area may require sub-categorization of the disease into more homogenous groups or disease states, which would permit increased precision (18, 46). Indeed, very recently there have been calls from the NIA-AA working group to establish a new research framework where AD is defined as a pathophysiological construct, rather than a clinical syndrome (56). While AD is often described by its clinical symptoms, it was identified and initially defined by its neuropathological features, namely the build-up in the brain of β-amyloid (Aβ) protein plaques and neurofibrillary tangles composed of aggregates of hyperphosphorylated TAU protein (11). The presence of these protein enables a definitive diagnosis of AD to be made post-mortem and there are now validated in vivo biomarkers for these. Using PET combined with MRI (to assess brain atrophy), the accumulation of amyloid-β and phosphorylated tau can be ascertained (57, 58). Defining AD as a biological construct based on the presence of these imaging biomarkers, will enable the generation of more precise predictive models for this specific neuropathological processes. This will shift away from the focus on clinical symptoms of the disease which are phenotypically heterogeneous, as discussed above, and thus problematic for biomarker and epidemiological studies.
Conclusion
The results of drug trials to slow or halt the progression of dementia have so far been unsuccessful (3), raising at least two important issues. Current treatments and interventions are unlikely to be effective in individuals with overt disease symptoms. However they could be effective if targeted very early in the disease process, before the appearance of clinical signs. Hence the need for clear biomarkers which would permit timely diagnosis and accurate characterization of the underlying condition resulting in dementia. Secondly, disease prevention is recognized as increasingly important, given the current lack of therapeutics. This is particularly pertinent for individuals identified at high-risk of the disease. This stresses the need for accurate risk prediction models, and thus the identification of the full range of genetic risk variants, as well as environmental factors through large epidemiological studies. This will also facilitate the categorization of subgroups within the population most suited for studies of new pharmacological and non-pharmacologic interventions. Adding to this is the increasing focus on precision medicine more generally.
Accurately determining the condition resulting in dementia is critical for research, including epidemiological, genetic, and biomarker studies (46). It is also of particular importance for treatment and prevention trials. Currently there are many challenges with diagnosing dementia, and as such it is a long, complicated and costly task, and misdiagnosis remains an issue. The emergence of new disease biomarkers will have a considerable impact on clinical diagnostic procedures. However, advances in biomarker research have been limited the inability to define a “clear” homogenous dementia phenotype with current biomarkers having considerable overlap with a number of dementia conditions. This creates a circularity problem which is difficult to resolve. However these challenges must be addressed if the likelihood of success for future genetic and biomarker studies is to increase. As an initial step, the focus on neuropathological markers of dementia and defining dementia as a biological construct will enable more accurate characterization of risk factors specific for this disease, shifting the definition from syndromal to biological (56, 59). It has the potential to lead to the discovery of novel genetic variants, the identification of readily accessible peripheral biomarkers reflective of these neuropathological processes, as well as the identification of individuals at heightened risk of the disease, even prior to the appearance of clinical symptoms (60). This will also become an increasingly important issue as new drug treatments are developed (61).
Author Contributions
JR and PL conceived the idea. JR wrote the first draft of the manuscript. All authors contributed to revising the final manuscript.
Funding
JR is funded by a NHMRC Dementia Research Leadership Fellowship (APP1135727), PF is funded by RTP stipend Ph.D. scholarship, awarded by Monash University and the Australian Government.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. The Lancet. Dementia burden coming into focus. Lancet (2017) 390:2606. doi: 10.1016/S0140-6736(17)33304-4
2. Prince MJ, Wimo A, Guerchet M, Ali C, Wu YT, Prina M. World Alzheimer Report 2015, The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost Trends. Alzheimer's Disease International (ADI), London. (2015). Available online at: https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf
3. Lancet ET. Alzheimer's disease: expedition into the unknown. Lancet (2016) 388:2713. doi: 10.1016/S0140-6736(16)32457-6
4. Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer's disease prevalence. Lancet Neurol. (2011) 10:819–28. doi: 10.1016/S1474-4422(11)70072-2
5. Fransquet PD, Lacaze P, Saffery R, McNeil J, Woods R, Ryan J. Blood DNA methylation as a potential biomarker of dementia: a systematic review. Alzheimers Dement. (2018) 14:81–103. doi: 10.1016/j.jalz.2017.10.002
6. WHO. Dementia Fact Sheet. Available online at: http://www.who.int/mediacentre/factsheets/fs362/en/ (Accessed October 10, 2017).
7. Karantzoulis S, Galvin JE. Distinguishing Alzheimer's disease from other major forms of dementia. Expert Rev Neurother. (2011) 11:1579–91. doi: 10.1586/ern.11.155
8. Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. (2014) 13:614–29. doi: 10.1016/S1474-4422(14)70090-0
9. Jack CR, Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
10. Johnson J, Sims R, Gottlieb G. Differential diagnosis of dementia, delirium and depression. Implications for drug therapy. Drugs Aging (1994) 5:431–45. doi: 10.2165/00002512-199405060-00005
11. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:263–9. doi: 10.1016/j.jalz.2011.03.005
12. O'Brien JT, Thomas A. Vascular dementia. Lancet (2015) 386:1698–706. doi: 10.1016/S0140-6736(15)00463-8
13. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet (2015) 386:1672–82. doi: 10.1016/S0140-6736(15)00461-4
14. Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. (2015) 14:253–62. doi: 10.1016/S1474-4422(14)70324-2
15. Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet (2015) 386:1683–97. doi: 10.1016/S0140-6736(15)00462-6
16. Kane JPM, Surendranathan A, Bentley A, Barker AH, Taylor JP, Thomas AJ, et al. Clinical prevalence of Lewy body dementia. Alzheimers Res Ther. (2018) 10:19. doi: 10.1186/s13195-018-0350-6
17. Klingelhoefer L, Reichmann H. Pathogenesis of Parkinson disease–the gut-brain axis and environmental factors. Nat Rev Neurol. (2015) 11:625–36. doi: 10.1038/nrneurol.2015.197
18. Kosunen O, Soininen H, Paljarvi L, Heinonen O, Talasniemi S, Riekkinen PJ, Sr. Diagnostic accuracy of Alzheimer's disease: a neuropathological study. Acta Neuropathol. (1996) 91:185–93. doi: 10.1007/s004010050412
19. Brayne C, Richardson K, Matthews FE, Fleming J, Hunter S, Xuereb JH, et al. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J Alzheimers Dis. (2009) 18:645–58. doi: 10.3233/JAD-2009-1182
20. Zweig YR, Galvin JE. Lewy body dementia: the impact on patients and caregivers. Alzheimers Res Ther. (2014) 6:21. doi: 10.1186/alzrt251
21. Ballard C, Margallo-Lana M, Juszczak E, Douglas S, Swann A, Thomas A, et al. Quetiapine and rivastigmine and cognitive decline in Alzheimer's disease: randomised double blind placebo controlled trial. BMJ (2005) 330:874. doi: 10.1136/bmj.38369.459988.8F
22. Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. (2010) 6:108–19. doi: 10.1038/nrneurol.2009.219
23. Fratiglioni L, Launer LJ, Andersen K, Breteler MM, Copeland JR, Dartigues JF, et al. Incidence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology (2000) 54(11 Suppl 5): S10–5.
24. Chui HC, Tierney M, Zarow C, Lewis A, Sobel E, Perlmutter LS. Neuropathologic diagnosis of Alzheimer disease: interrater reliability in the assessment of senile plaques and neurofibrillary tangles. Alzheimer Dis Assoc Disord. (1993) 7:48–54. doi: 10.1097/00002093-199307010-00006
25. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), 4th Edn. Washington, DC: American Psychiatric Association (1994).
26. Nelson PT, Head E, Schmitt FA, Davis PR, Neltner JH, Jicha GA, et al. Alzheimer's disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. (2011) 121:571–87. doi: 10.1007/s00401-011-0826-y
27. Neuropathology Group. Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet (2001) 357:169–75. doi: 10.1016/S0140-6736(00)03589-3
28. Petrovitch H, White LR, Ross GW, Steinhorn SC, Li CY, Masaki KH, et al. Accuracy of clinical criteria for AD in the Honolulu-Asia Aging Study, a population-based study. Neurology (2001) 57:226–34. doi: 10.1212/WNL.57.2.226
29. Gearing M, Mirra SS, Hedreen JC, Sumi SM, Hansen LA, Heyman A. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer's disease. Neurology (1995) 45(3 Pt 1):461–6. doi: 10.1212/WNL.45.3.461
30. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology (2005) 65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1
31. Dong A, Toledo JB, Honnorat N, Doshi J, Varol E, Sotiras A, et al. Heterogeneity of neuroanatomical patterns in prodromal Alzheimer's disease: links to cognition, progression and biomarkers. Brain (2017). 140:735–47. doi: 10.1093/brain/aww319
32. Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. (2013) 12:207–16. doi: 10.1016/S1474-4422(12)70291-0
33. Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BN, et al. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA (2015) 313:1939–49. doi: 10.1001/jama.2015.4669
34. Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging (2010) 31:1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007
35. Galpern WR, Lang AE. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol. (2006) 59:449–58. doi: 10.1002/ana.20819
36. Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases: the prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science (2015) 349:1255555. doi: 10.1126/science.1255555
37. Delgado-Morales R, Esteller M. Opening up the DNA methylome of dementia. Mol Psychiatry (2017) 22:485–96. doi: 10.1038/mp.2016.242
38. Larner AJ, Doran M. Genotype-phenotype relationships of presenilin-1 mutations in Alzheimer's disease: an update. J Alzheimers Dis. (2009) 17:259–65. doi: 10.3233/JAD-2009-1042
39. Wszolek ZK, Tsuboi Y, Ghetti B, Pickering-Brown S, Baba Y, Cheshire WP. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet J Rare Dis. (2006) 1:30. doi: 10.1186/1750-1172-1-30
40. Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. (2013) 70:223–8. doi: 10.1001/jamaneurol.2013.600
41. Sanchez-Mut JV, Heyn H, Vidal E, Moran S, Sayols S, Delgado-Morales R, et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry (2016) 6:e718. doi: 10.1038/tp.2015.214
42. Guerreiro R, Escott-Price V, Darwent L, Parkkinen L, Ansorge O, Hernandez DG, et al. Genome-wide analysis of genetic correlation in dementia with Lewy bodies, Parkinson's and Alzheimer's diseases. Neurobiol Aging (2016) 38:214.e7–214.e10. doi: 10.1016/j.neurobiolaging.2015.10.028
43. Manchia M, Cullis J, Turecki G, Rouleau GA, Uher R, Alda M. The impact of phenotypic and genetic heterogeneity on results of genome wide association studies of complex diseases. PLoS ONE (2013) 8:e76295. doi: 10.1371/journal.pone.0076295
44. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature (2009) 461:747–53. doi: 10.1038/nature08494
45. Spencer CC, Su Z, Donnelly P, Marchini J. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. (2009) 5:e1000477. doi: 10.1371/journal.pgen.1000477
46. Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias. PLoS Genet. (2014) 10:e1004606. doi: 10.1371/journal.pgen.1004606
47. Takacs A, Koncz R, Mohan A, Sachdev P. Forgetfulness, stress or mild dementia? Cognitive assessment of older patients. Med Today (2017) 18:14–22.
48. Ritchie K, Carriere I, Berr C, Amieva H, Dartigues JF, Ancelin ML, et al. The clinical picture of Alzheimer's disease in the decade before diagnosis: clinical and biomarker trajectories. J Clin Psychiatry (2016) 77:e305–11. doi: 10.4088/JCP.15m09989
49. Tang EY, Harrison SL, Errington L, Gordon MF, Visser PJ, Novak G, et al. Current developments in dementia risk prediction modelling: an updated systematic review. PLoS ONE (2015) 10:e0136181. doi: 10.1371/journal.pone.0136181
50. Barnes DE, Beiser AS, Lee A, Langa KM, Koyama A, Preis SR, et al. Development and validation of a brief dementia screening indicator for primary care. Alzheimers Dement. (2014) 10:656–65.e1. doi: 10.1016/j.jalz.2013.11.006
51. Sibbett RA, Russ TC, Deary IJ, Starr JM. Dementia ascertainment using existing data in UK longitudinal and cohort studies: a systematic review of methodology. BMC Psychiatry (2017) 17:239. doi: 10.1186/s12888-017-1401-4
52. Ji H, Wang Y, Liu G, Chang L, Chen Z, Zhou D, et al. Elevated OPRD1 promoter methylation in Alzheimer's disease patients. PLoS ONE (2017) 12:e0172335. doi: 10.1371/journal.pone.0172335
53. Yamazaki K, Yoshino Y, Mori T, Yoshida T, Ozaki Y, Sao T, et al. Gene expression and methylation analysis of ABCA7 in patients with Alzheimer's disease. J Alzheimers Dis. (2017) 57:171–81. doi: 10.3233/JAD-161195
54. Naik M, Nygaard HA. Diagnosing dementia – ICD-10 not so bad after all: a comparison between dementia criteria according to DSM-IV and ICD-10. Int J Geriatr Psychiatry (2008) 23:279–82. doi: 10.1002/gps.1874
55. Delude CM. Deep phenotyping: the details of disease. Nature (2015) 527:S14–5. doi: 10.1038/527S14a
56. Jack CR, Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. 2017 NIA-AA Research Framework to Investigate the Alzheimer's disease continuum. Alzheimers Dement. (2017) 13:890–1. doi: 10.1016/j.jalz.2017.07.294
57. Hostetler ED, Walji AM, Zeng Z, Miller P, Bennacef I, Salinas C, et al. Preclinical characterization of 18F-MK-6240, a promising PET tracer for in vivo quantification of human neurofibrillary tangles. J Nucl Med. (2016) 57:1599–606. doi: 10.2967/jnumed.115.171678
58. Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer's Association. Alzheimers Dement. (2013) 9:e1–16. doi: 10.1016/j.jalz.2013.01.002
59. Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. (2012) 367:795–804. doi: 10.1056/NEJMoa1202753
60. Sperling RA, Karlawish J, Johnson KA. Preclinical Alzheimer disease-the challenges ahead. Nat Rev Neurol. (2013) 9:54–8. doi: 10.1038/nrneurol.2012.241
Keywords: Alzheimer's disease, biomarkers, clinical symptoms, diagnosis, dementia, heterogeneity, pathophysiology
Citation: Ryan J, Fransquet P, Wrigglesworth J and Lacaze P (2018) Phenotypic Heterogeneity in Dementia: A Challenge for Epidemiology and Biomarker Studies. Front. Public Health 6:181. doi: 10.3389/fpubh.2018.00181
Received: 02 April 2018; Accepted: 04 June 2018;
Published: 19 June 2018.
Edited by:
Charles B. Hall, Albert Einstein College of Medicine, United StatesReviewed by:
Terri Kang Johnson, Dexcom, Inc., United StatesCharalampos Socrates Siristatidis, National and Kapodistrian University of Athens, Greece
Copyright © 2018 Ryan, Fransquet, Wrigglesworth and Lacaze. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanne Ryan, am9hbm5lLnJ5YW5AbW9uYXNoLmVkdQ==