 Timo Jendrik Faustmann
Timo Jendrik Faustmann Franco Corvace2†
Franco Corvace2† Fatme Seval Ismail
Fatme Seval Ismail
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Psychiatry , 18 March 2025
Sec. Psychopathology
Volume 16 - 2025 | https://doi.org/10.3389/fpsyt.2025.1522128
This article is part of the Research Topic Immunity and Inflammation in Neuropsychiatric Disorders View all 8 articles
Schizophrenia is a severe mental disorder with a strong lifetime impact on patients’ health and wellbeing. Usually, symptomatic treatment includes typical or atypical antipsychotics. Study findings show an involvement of low-grade inflammation (blood, brain parenchyma, and cerebrospinal fluid) in schizophrenia. Moreover, experimental and neuropathological evidence suggests that reactive microglia, which are the main resident immune cells of the central nervous system (CNS), have a negative impact on the differentiation and function of oligodendrocytes, glial progenitor cells, and astrocytes, which results in the disruption of neuronal networks and dysregulated synaptic transmission, contributing to the pathophysiology of schizophrenia. Here, the role of microglial cells related to neuroinflammation in schizophrenia was discussed to be essential. This review aims to summarize the evidence for the influence of antipsychotics on microglial inflammatory mechanisms in schizophrenia. Furthermore, we propose an established astrocyte–microglia co-culture model for testing regulatory mechanisms and examining the effects of antipsychotics on glia-mediated neuroinflammation. This could lead to a better understanding of how typical and atypical antipsychotics can be used to address positive and negative symptoms in schizophrenia and comorbidities like inflammatory diseases or the status of low-grade inflammation.
Schizophrenia is a severe mental disorder associated with increased mortality and significant morbidity (1). The symptoms of schizophrenia can be divided into positive symptoms (e.g., hallucinations and delusions), negative symptoms (e.g., social withdrawal and anhedonia), mood deficits (e.g., depressed/irritated mood), cognitive deficits (e.g., attention and memory deficits), and motor symptoms (1–3). The pathophysiology of schizophrenia is multifactorial. Genome-wide association studies have identified numerous genetic variants associated with an increased risk of schizophrenia (e.g., DISC1, NRG1, and DTNBP1) by influencing synaptic functions, neurotransmission, and neuronal development and plasticity (4–8). Furthermore, biochemical dysregulation has been discussed considering “the dopamine hypothesis of schizophrenia,” which postulates that overactivity of the mesolimbic dopamine system results in positive symptoms of schizophrenia, while underactivity of the mesocortical dopamine system causes the negative symptoms (9). Another hypothesis postulates the hypofunction of the N-methyl-d-aspartate (NMDA) receptor, a subset of the glutamate receptor (10–13). This hypothesis is underlined by NMDA receptor antagonists such as ketamine and phencyclidine (PCP), which can induce symptoms that resemble those of schizophrenia (14–20). Importantly, the inflammatory mechanisms and status of low-grade inflammation have been discussed including the terms “mild encephalitis” (21) and “autoimmune psychosis” (22), the role of prenatal infections (23) and infections during early childhood (24), and the role of peripheral (25) and cytokines in cerebrospinal fluid (CSF) (26, 27). Furthermore, the interrelation between impaired social interactions and inflammatory reactions in schizophrenia has been recognized (28). Moreover, the role of brain microvascular endothelial cells and blood–brain barrier (BBB) dysfunction has been found relevant in inflammation in schizophrenia (29). Structural and functional abnormalities in the brains of patients, which are often present before the onset of clinical symptoms (30, 31), and chronic stress and traumatic experiences have been further discussed (32–34) (Figure 1A).
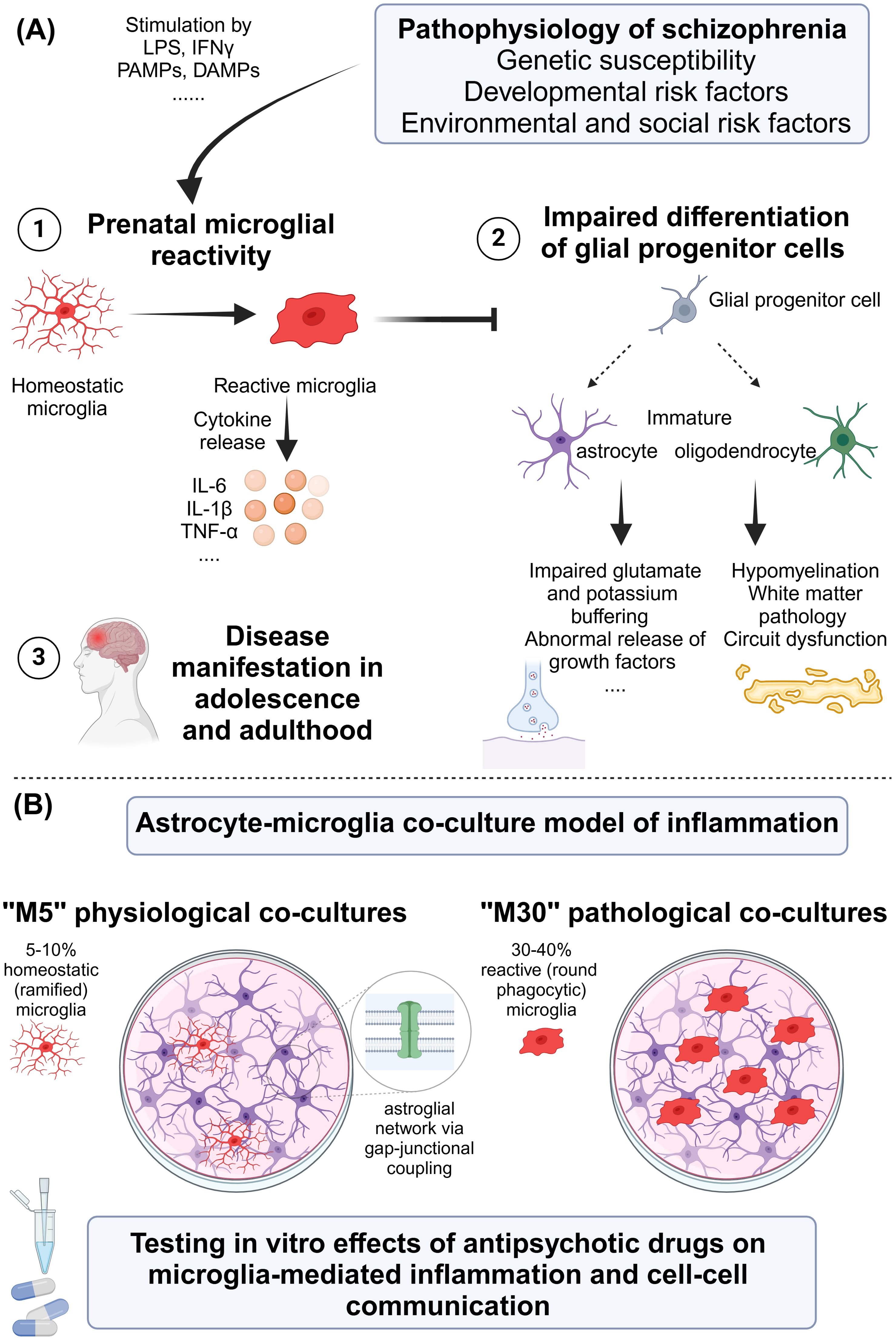
Figure 1. (A) Pathophysiology of schizophrenia with focus on microglia. (B) Proposal for an in vitro astrocyte–microglia co-culture model of inflammation for studying the effects of antipsychotic drugs. DAMP, damage-associated molecular pattern; IFN, interferon; IL, interleukin; LPS, lipopolysaccharide; PAMP, pathogen-associated molecular pattern; TNF, tumor necrosis factor. Created in BioRender. Ismail, F. (2024) https://BioRender.com/z92a369.
Glial cells (astrocytes, microglia, and oligodendrocytes) perform a variety of supportive functions for neurons (35). In schizophrenia, various dysfunctions in glial cells have been identified (36, 37).
Astrocytes, the most abundant glial cells in the central nervous system (CNS), have numerous essential roles (e.g., supporting ion and transmitter homeostasis). Findings indicate that astrocytes in schizophrenia exhibit dysregulated glutamate homeostasis. Normally, astrocytes uptake glutamate from the synaptic cleft to prevent neurotoxicity, but in schizophrenia, reduced expression of the glutamate transporters excitatory amino acid transporter 1 (EAAT1) and excitatory amino acid transporter 2 (EAAT2) has been observed, leading to elevated extracellular glutamate concentrations and neuronal hyperexcitability (38–40). Furthermore, in schizophrenia, there is evidence of a dysregulated astrocyte-dependent release of d-serine, a co-agonist for NMDA receptors (41–43). Oligodendrocytes, responsible for the myelination of neurons, also exhibit dysfunctions in schizophrenia. Reduced myelin-associated gene expression and impaired myelination have been observed in the white matter of patients with schizophrenia, which can lead to deficits in neuronal connectivity and signal transmission. These myelination-related deficits have been suggested to contribute to cognitive impairment in schizophrenia (44–48). Additionally, microglial cells have been found to influence oligodendrocytes and their progenitor cells, and as a result, myelination in schizophrenia (49, 50).
Microglia are the resident immune cells of the CNS, maintaining neuronal homeostasis, defending against pathogens, and repairing tissue damage. They are highly dynamic cells that adapt their morphology and function to changes in the neuronal environment (51, 52).
Postmortem studies have demonstrated an increase in different inflammatory markers related to microglia in schizophrenia, e.g., an increase in microglial density in cortical gray matter using ionized calcium-binding adaptor molecule-1 (Iba1) (53). Furthermore, an increase in the density of cells [using staining for major histocompatibility complex class II (MHC-II)] morphologically resembling microglia and a change in interleukin-1β (IL-1β), interleukin-6 (IL-6), and interleukin-8 (IL-8) have been found (54). Additionally, the role of an increase in the human leukocyte antigen–DR isotype-positive (HLA-DR+) microglia in the frontal cortex and hippocampus of patients with schizophrenia (55, 56) and calprotectin co-expressed with the microglial marker cluster of differentiation 68 (CD68) has been discussed (57). Other studies have found a change in microglial phenotype rather than a change in the density of cells according to a meta-analysis (58). Twin studies using human induced pluripotent stem cell (iPSC)-derived microglia found an increased expression of inflammatory genes in microglia like MHC-II but no signs of hyperactivation of microglia (59). Furthermore, translocator protein (TSPO) positron emission tomography (PET) and the second-generation radioligand [(11)C]PBR28 in vivo confirmed microglial reactivity in patients with schizophrenia and subclinical symptoms (60). However, importantly, postmortem studies did not conclude microglial activation for all the cases, and further in vivo studies did not find microglial activation using PET and TSPO in patients with schizophrenia. It can be concluded that postmortem and in vivo findings are discussed to be related to brain regions and stage of the disorder and if a antipsychotic treatment was used or not at the same time (36, 37, 61–66). The described state of chronic neuroinflammation could contribute to the pathophysiology of schizophrenia, as inflammatory mediators can impair synaptic transmission and plasticity, resulting in symptoms of schizophrenia (37, 67–69). Furthermore, physiological microglia can perform synaptic pruning, which is crucial for maintaining healthy neuronal circuits. However, the “synaptic hypothesis of schizophrenia” describes a lower synaptic density, which was demonstrated in patients with schizophrenia. Here, genetic and environmental stressors could lead to a microglia-mediated and complement-dependent (among others complement protein C4) elimination of synaptic structures (70–74). Interestingly, microgliosis in patients with schizophrenia, those with depression, and matched controls who committed suicide was found in the dorsolateral prefrontal cortex (DLPC), anterior cingulate cortex (ACC), and mediodorsal thalamus (MD), pointing toward possible further connections between inflammation and psychopathology (75, 76). A further finding that could contribute to the pathophysiology of psychiatric diseases and schizophrenia is the interesting role of microglial priming after early-life infections (first hit) and consequently long-term changes, e.g., on memory after a “second hit” later in life (77, 78). These findings consider the impaired synaptic plasticity in schizophrenia (71). In addition to in vivo, postmortem, and animal studies, future directions point toward iPSC models in schizophrenia investigating microglia and neuron interactions (for review, see (65)).
In summary, dysfunctional microglia were found to contribute to the pathophysiology of schizophrenia by promoting inflammatory reactions, abnormal phagocytosis, and reactions to early life stressors. These findings offer potential targets for therapeutic interventions aimed at modulating microglial function (37, 54, 67) (Figure 1A). Interestingly, a pharmacological approach (antipsychotics) may mitigate these effects by suppressing microglial activation, offering a dual therapeutic mechanism beyond dopamine modulation (79). Cognitive deficits in schizophrenia are closely tied to microglial overactivation, which exacerbates neurotoxicity through pro-inflammatory cytokines and free radicals. Targeting microglial activation presents a promising avenue for ameliorating cognitive symptoms (80). Also, stress-induced alterations in microglial function have been shown to impact fear memory and extinction deficits, mechanisms that may overlap with schizophrenia pathology. Research has highlighted that aberrant microglial cytokine production affects fear generalization and inhibitory processes in memory, as observed in post-traumatic stress disorder (PTSD) models, and has underscored the necessity for translational studies bridging the gap between animal models and human conditions (81).
Antipsychotics are the main therapeutic strategy in schizophrenia and other disorders with psychotic symptoms and can be divided into first-generation (typical) and second-generation (atypical) antipsychotics (82–84). The most valuable difference is the reduced ability to cause extrapyramidal side effects and tardive dyskinesia when using atypical antipsychotics compared to typical antipsychotics. Typical antipsychotics primarily provide modulations of dopaminergic transmission compared to a more serotonergic transmission in cases of atypical antipsychotics. The most prominent antipsychotics are haloperidol as a typical antipsychotic and clozapine as an atypical antipsychotic, which is also useful in cases of treatment-resistant schizophrenia. Different effects on positive and negative symptoms in schizophrenia when using antipsychotics have been discussed. Clozapine has a strong effect on positive symptoms like hallucinations and delusions with an improvement in social functioning. Haloperidol was found to have an effect on positive [scale for the assessment of positive symptoms (SAPS)] and negative symptoms [scale for the assessment of negative symptoms (SANS)], and negative symptoms were found to be independent of positive ones (85–88).
Increased activity of microglia and pro-inflammatory cytokines was found to be important in the pathophysiology of schizophrenia. These inflammatory findings were potentially influenced by antipsychotics (89, 90). A cytokine imbalance in serum was found to be an important biomarker in treatment-resistant schizophrenia, and concentrations vary between acute and chronic stages (25, 91). In a meta-analysis, antipsychotic treatment was found to reduce cytokine levels in patients with schizophrenia in vivo and here, especially IL-6 levels (92). In contrast, using PET studies for TSPO as a marker for microglial activity revealed a significant increase in the marker in schizophrenia patients with antipsychotic treatment. Furthermore, the marker correlated with negative symptoms using the Positive and Negative Syndrome Scale (PANSS) (66, 93). Other data found a decrease in TSPO in antipsychotic-treated patients (94). Moreover, an increase in TSPO binding in rats upon clozapine treatment was found (95). Reviewing clinical data, no clear conclusion could be drawn about antipsychotic drugs influencing microglial cells (96). Nevertheless, microglial proliferation and morphological changes resembling microglial activation were found in vivo in rats during an 8-week treatment with haloperidol and olanzapine (97).
Haloperidol is a typical and strong dopamine D2 receptor blocker (98). It reduces pro-inflammatory action in C57/BL6 murine microglial cells (BV-2 microglia) (99) and increases brain-derived neurotrophic factor (BDNF), transforming growth factor-β (TGF-β), and neurotrophin-3 (NT-3) gene expression in microglial cells from newborn Wistar rats (100). Interestingly, haloperidol did not change microglial density in an in vivo rat model (101) and did not prevent microglial activation in a PCP model of psychosis in rats (102). Furthermore, a high-fat diet increased microglial expression in rats, and this was not found in a combination of diet and haloperidol (103). Expression of OX-42 protein and IL-6 expression was decreased, and extracellular signal-regulated kinase (ERK) and signal transducer and activator of transcription 3 (STAT3) was suppressed by haloperidol in lipopolysaccharide (LPS)-activated microglia (104). Microglial proton currents in BV-2 microglial cells were inhibited by haloperidol and could contribute to the anti-inflammatory effects of antipsychotics on microglia.
Furthermore, the same was found for chlorpromazine, another typical antipsychotic (105). Additionally, chlorpromazine reduced secretion of interleukin-2 (IL-2) and IL-1β in mixed glial cultures (106) and acts as a microglia Kv1.3 (voltage-gated potassium channel) channel inhibitor (107).
Flupentixol decreased IL-2 and IL-1β release by microglial cells, and trifluperidol reduced IL-1β and IL-2 release by mixed glial cultures (108). Both antipsychotics also reduced the nitric oxide (NO) and tumor necrosis factor-alpha (TNF-α) release from LPS-influenced microglial cultures (109).
Spiperone attenuates TNF-α production, expression of IL-1β and TNF-α, and nuclear translocation of the p65 subunit of nuclear factor kappa B (NF-κB) in BV-2 microglia and reduces microglia-mediated cell death in microglia/neuron co-cultures (110).
Clozapine is an atypical antipsychotic with a strong influence on serotonergic transmission [blockade of 5-HT(2A) receptors] and reduced blockade of dopamine D2 receptors in the ventral and dorsal striatum (111). In a mouse model of experimental autoimmune encephalomyelitis (EAE), clozapine regulated the iron-impaired microglial function and reduced the release of IL-6 and neuronal phagocytosis (112). Furthermore, it reduced the inflammatory NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) pathway in a polyriboinosinic–polyribocytidylic acid (poly(I:C))-stimulated primary microglial cell culture model (113). Clozapine reduces the inhibition of calcium/calmodulin/Akt-mediated NF-κB activation in microglia (114), and clozapine-induced neuronal protection was microglial mediated in an LPS-induced model of inflammatory neurodegeneration using neuron–glia cultures (115). Interestingly, clozapine can reduce proton currents in BV-2 microglial cells, which could be considered as an anti-inflammatory effect (116).
Aripiprazole is a partial agonist at the dopamine D2 and serotonin 5-HT(1A) receptor and an antagonist at the serotonin 5-HT(2A) receptor (117). Aripiprazole inhibited inflammatory mechanisms in a poly(I:C)-induced microglial activation model in mice (118). An interferon-gamma (IFN-γ)-induced microglial activation was found to be attenuated by aripiprazole via intracellular calcium regulation in vitro (119). In BV-2 microglial cells, aripiprazole reduced the pro-inflammatory action and expression of anti-inflammatory markers (99). Interestingly, aripiprazole and minocycline inhibited damage of oligodendrocytes via the inhibition of IFN-γ-activated microglia (50).
Quetiapine inhibited NO generation and TNF-α release from activated microglia (120). In a transgenic mouse model of Alzheimer’s disease, it decreased β-amyloid-(1-42) (Aβ(1-42))-induced activation of primary microglia by attenuating pro-inflammatory cytokines and in primary microglia stimulated by Aβ(1-42) via activation of the NF-κB pathway (121). Furthermore, quetiapine inhibits microglial activation via neutralization of abnormal intercellular calcium homeostasis in a cuprizone mouse model (122).
Risperidone reduced the pro-inflammatory activation of BV-2 microglial cells (99) and deactivated IFN-γ-induced microglia (123). Moreover, risperidone reduced the expression of OX-42 protein and decreased the IL-6 and TNF-α production via STAT3 in LPS-activated microglia (104).
Olanzapine reduced NO release in an LPS-activated mouse microglia cell line N9 (124). A high-fat diet increased microglial expression in rats, and this was not found in a combination of diet and olanzapine (103).
In summary, mitogen-activated protein kinase (MAPK) was found to be important in the activation of BV2 microglia by LPS and ERK in IFN-γ activated BV2 microglia (125, 126). As previously described, antipsychotics seem to modulate intracellular signals like MAPK, calcium homeostasis, NF-κB, and protein kinase C (PKC), which further inhibit nuclear activation and cytokine production and release by microglia (89). These discussions could contribute to the described decrease in pro-inflammatory cytokines when using antipsychotics. Ca2+ is a main point of interest, as it is an activator of PKC and found to be dysregulated in schizophrenia (127). It could also be influenced by aripiprazole, for example (89).
Taken together, psychopathologies in psychiatric diseases have been known to be associated with the sensitization of glial and particularly microglial cells, which could be influenced by psychotropic drugs like antipsychotics, contributing to the microglia hypothesis of schizophrenia. Here, the involvement of microglia and oligodendrocytes in negative and cognitive symptoms in schizophrenia was discussed (37, 46, 48, 66, 128, 129). As described above, the known antipsychotics (typical and atypical) can contribute to a possible anti-inflammatory effect concerning microglial cells (Table 1). Furthermore, in this vein, specific effects on positive and negative symptoms of schizophrenia when using antipsychotic drugs were discussed. In contrast, microglial reactivity could not be concluded as ubiquitous in schizophrenia and psychiatric diseases concerning postmortem and in vivo studies in humans. Postmortem microglial markers were found to be increased or unchanged, and the reason for this is not clear; similar findings were found to be related to PET studies. The contradictory findings in postmortem studies and PET investigations may be attributed to differences in the sensitivity of the methods capturing different stages of microglial activity (36, 62, 130). Furthermore, the role of antipsychotics in the treatment of psychiatric diseases and their role in glial reactivity were discussed. The interrelations among neurons, astrocytes as part of the tripartite synapse, and microglia underline the complex glial mechanisms influenced by antipsychotics (131). Nevertheless, using antipsychotics could be one way of regulating a microglial immune response.
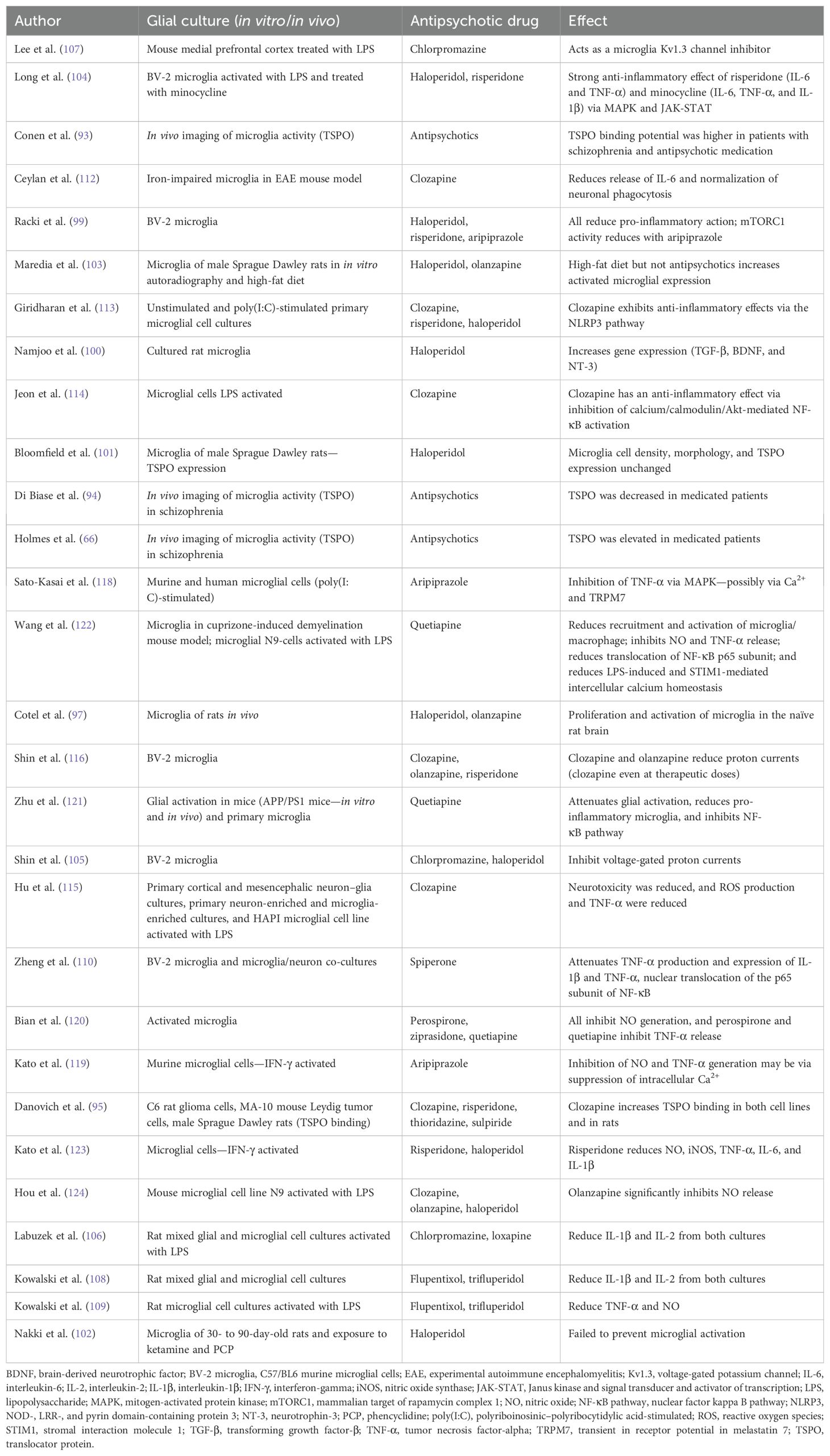
Table 1. Literature on antipsychotic drugs influencing microglia.
The astrocyte–microglia co-culture model of inflammation was developed in 2003 by Faustmann et al. to investigate the physiological as well as pathological inflammatory conditions in the brain in relation to the percentage and activation state of microglia (132) (Figure 1B). The astrocytes and microglia were obtained from the brains of postnatal Wistar rats (postnatal days 0–2) and were prepared according to an established protocol (132, 133).
The physiological astrocyte–microglia co-culture model (so-called M5) contains 5%–10% microglia with the predominantly homeostatic ramified phenotype (formerly known as resting ramified). The pathological, inflammatory astrocyte–microglia co-culture model (so-called M30) contains 30%–40% microglia with predominantly reactive phenotype (formerly known as activated, round phagocytic) (132, 134). Treatment of M5 co-cultures with the pro-inflammatory cytokines TNF-α, IL-1β, IL-6, and IFN-γ led to microglial reactivity, whereas treatment of M30 co-cultures with the main anti-inflammatory cytokine transforming growth factor-β1 (TGF-β1) caused a reduction of microglial reactivity. In addition, treatment of M5 co-cultures with the anti-inflammatory cytokine interferon-beta (IFN-β) prevented the pro-inflammatory effects of TNF-α, IL-1β, and IFN-γ (134). Further, the study findings of our co-culture model revealed a functional relationship between microglial activation states and astroglial coupling (132).
Interestingly, previous findings point to the fact that functional abnormalities of tripartite synapses, contributing to dynamic signaling between pre- and post-synaptic neurons as well as astrocytes, can be involved in the pathophysiology of schizophrenia and associated cognitive impairments (135). The gliotransmitter release through vesicles, hemichannels, and reverse transport of astrocytes plays an important role in tripartite synaptic transmission. Astrocytes are connected with each other and other cell types via connexin (Cx)-based gap junctions, mainly consisting of Cx43 and Cx30 (136). Connexins can also form hemichannels that enable the connection between intra- and extracellular spaces. Study findings show that atypical antipsychotics such as clozapine, brexpiprazole, and quetiapine increased astroglial Cx43 containing hemichannel activities, resulting in enhanced tripartite synaptic glutamatergic transmission (135). In vivo chronic treatment of mice with clozapine caused increased Cx43 expression in the prefrontal cortex; however, haloperidol led to a decrease in Cx43 (137).
The inflammatory astrocyte–microglia co-culture model offers the possibility to examine the inflammatory states including microglial morphology and Cx-based gap junctional coupling, which are also involved in the pathophysiology of schizophrenia. Consequently, the co-culture model can have a long-term impact on the treatment of schizophrenia, such as in the development of new drugs. For example, connexin-based channels could serve as a target for new drugs in schizophrenia, modulating the gap junctional coupling, and indirectly, the microglial reactivity. Study findings suggest that astrocytic deficits in the dorsolateral prefrontal cortex can disrupt the neuron–glia interactions, resulting in a dysfunctional effect on prefronto-striatal circuits in schizophrenia (138). Repairing astrocytic dysfunction could offer new therapeutic options for schizophrenia. Our astrocyte–microglia co-culture model has more advantages for studying cellular interactions compared to only astrocyte or microglia monocultures (133). Our co-culture model mimics natural inflammation because of the preparation method, allowing concomitant proliferation of astrocytes and microglia (132). In contrast to this, other astrocyte–microglia co-cultures consist of a mixture of two primary cultures (astrocytes and microglia) cultivated together in different ratios. Of course, there is a limitation of our model to tricultures, including neurons in addition to astrocytes and microglia (139). Nevertheless, our model is robust and suitable for schizophrenia research in the first step before further steps are taken in animal models.
In further studies, the in vitro model was already used for the investigation of pharmaceutical effects on glia-mediated neuroinflammation and cellular interactions (133). Different groups of neuropsychiatric drugs including anti-seizure medication and mood-stabilizing drugs/antidepressants were already investigated in the co-culture model (139–147). For example, venlafaxine as well as doxepin and amitriptyline reduced the microglial reactivity, leading to the attenuation of microglia-mediated neuroinflammation (141, 146). Next, the co-culture model can be suitable for testing the possible pro-/anti-inflammatory effects of antipsychotic drugs.
Psychotic disorders encompass a broad spectrum of psychiatric conditions including schizophrenia as one of the leading psychotic disorders with a strong lifetime impact on patients’ health and wellbeing. In recent years, studies have been conducted to better understand the cellular neurobiology of schizophrenia, particularly with regard to cellular-mediated neuroinflammation (28, 79, 148). The low-level inflammation concept of schizophrenia was linked to risk genes promoting inflammation, prenatal maternal and early childhood infections with microglial reactivity, and an increase in cytokines, environmental stress factors, and alterations of the immune system (69). In addition, autoimmune factors such as anti-NMDA receptor antibodies contributing to psychosis were discussed (22). Experimental and neuropathological evidence suggests that reactive microglia have a negative impact on the differentiation and function of oligodendrocytes, glial progenitor cells, and astrocytes, which results in the disruption of neuronal networks and dysregulated synaptic transmission, contributing to the pathophysiology of schizophrenia (148) (Figure 1A). Following this, research focusing on therapeutic approaches modulating microglia-mediated neuroinflammation has potential. However, it is currently even more exciting to consider whether antipsychotics used in everyday clinical practice have anti-inflammatory properties in relation to microglia-mediated neuroinflammation.
Research focusing on inflammatory disease mechanisms in many neuropsychiatric disorders has recently expanded. Also, various immunotherapeutic approaches have been developed, e.g., immunotherapy for Alzheimer’s disease with monoclonal antibodies removing abnormal β-amyloid (Aβ) from the brain and preventing disease progression (149–153). Preliminary data have been about the effects of monoclonal antibodies acting by directly neutralizing cytokines or by binding cytokine receptors in schizophrenia (154). Subsequently, aiming at the involvement of microglia in schizophrenia, the use of in vitro models such as our astrocyte–microglia co-culture model of inflammation can help to better understand the underlying pathomechanism by testing the effect of antipsychotic and anti-inflammatory drugs (Figure 1B). This could lead to a better understanding of how typical and atypical antipsychotics can be used to address positive and negative symptoms in schizophrenia and comorbidities like inflammatory diseases or the status of low-grade inflammation.
TJF: Conceptualization, Project administration, Data curation, Formal analysis, Supervision, Writing – original draft, Writing – review & editing. FC: Data curation, Writing – original draft, Writing – review & editing. PMF: Writing – review & editing. FSI: Conceptualization, Data curation, Writing – original draft, Visualization, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This open access publication was funded by the Open-Access-Fonds of the Heinrich Heine University, Düsseldorf, Germany.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr Res. (2009) 110:1–23. doi: 10.1016/j.schres.2009.03.005
2. Arciniegas DB. Psychosis. Continuum (Minneap Minn). (2015) 21:715–36. doi: 10.1212/01.CON.0000466662.89908.e7
3. Lieberman JA, First MB. Psychotic disorders. N Engl J Med. (2018) 379:270–80. doi: 10.1056/NEJMra1801490
4. Stefansson H, Sarginson J, Kong A, Yates P, Steinthorsdottir V, Gudfinnsson E, et al. Association of neuregulin 1 with schizophrenia confirmed in a Scottish population. Am J Hum Genet. (2003) 72:83–7. doi: 10.1086/345442
5. Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. (2014) 511:421–7. doi: 10.1038/nature13595
6. Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. (2013) 45:1150–9. doi: 10.1038/ng.2742
7. Wang H, Xu J, Lazarovici P, Zheng W. Dysbindin-1 involvement in the etiology of schizophrenia. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18102044
8. Dahoun T, Trossbach SV, Brandon NJ, Korth C, Howes OD. The impact of Disrupted-in-Schizophrenia 1 (DISC1) on the dopaminergic system: a systematic review. Transl Psychiatry. (2017) 7:e1015. doi: 10.1038/tp.2016.282
9. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bull. (2009) 35:549–62. doi: 10.1093/schbul/sbp006
10. Nakazawa K, Sapkota K. The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol Ther. (2020) 205:107426. doi: 10.1016/j.pharmthera.2019.107426
11. Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. (1999) 33:523–33. doi: 10.1016/s0022-3956(99)00029-1
12. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. (2012) 37:4–15. doi: 10.1038/npp.2011.181
13. Uno Y, Coyle JT. Glutamate hypothesis in schizophrenia. Psychiatry Clin Neurosci. (2019) 73:204–15. doi: 10.1111/pcn.12823
14. Faustmann T, Theiss S, Görtz P, Lange-Asschenfeldt C. Differential protective effects of typical versus atypical antipsychotic drugs in a phencyclidine microelectrode array model of schizophrenia. Authorea (Preprint). (2024). doi: 10.22541/au.172646104.48099324/v1
15. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. (1994) 51:199–214. doi: 10.1001/archpsyc.1994.03950030035004
16. Morgan CJ, Curran HV. Acute and chronic effects of ketamine upon human memory: a review. Psychopharmacol (Berl). (2006) 188:408–24. doi: 10.1007/s00213-006-0572-3
17. Li D, Pan Q, Xiao Y, Hu K. Advances in the study of phencyclidine-induced schizophrenia-like animal models and the underlying neural mechanisms. Schizophr (Heidelb). (2024) 10:65. doi: 10.1038/s41537-024-00485-x
18. Anticevic A, Gancsos M, Murray JD, Repovs G, Driesen NR, Ennis DJ, et al. NMDA receptor function in large-scale anticorrelated neural systems with implications for cognition and schizophrenia. Proc Natl Acad Sci U.S.A. (2012) 109:16720–5. doi: 10.1073/pnas.1208494109
19. Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. (2012) 62:1574–83. doi: 10.1016/j.neuropharm.2011.01.022
20. Adell A. Brain NMDA receptors in schizophrenia and depression. Biomolecules. (2020) 10. doi: 10.3390/biom10060947
21. Bechter K. Schizophrenia–a mild encephalitis]? Fortschr Neurol Psychiatr. (2013) 81:250–9. doi: 10.1055/s-0033-1335253
22. Pollak TA, Lennox BR, Muller S, Benros ME, Pruss H, Tebartz van Elst L, et al. Autoimmune psychosis: an international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin. Lancet Psychiatry. (2020) 7:93–108. doi: 10.1016/S2215-0366(19)30290-1
23. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. (2010) 167:261–80. doi: 10.1176/appi.ajp.2009.09030361
24. Dalman C, Allebeck P, Gunnell D, Harrison G, Kristensson K, Lewis G, et al. Infections in the CNS during childhood and the risk of subsequent psychotic illness: a cohort study of more than one million Swedish subjects. Am J Psychiatry. (2008) 165:59–65. doi: 10.1176/appi.ajp.2007.07050740
25. Halstead S, Siskind D, Amft M, Wagner E, Yakimov V, Shih-Jung Liu Z, et al. Alteration patterns of peripheral concentrations of cytokines and associated inflammatory proteins in acute and chronic stages of schizophrenia: a systematic review and network meta-analysis. Lancet Psychiatry. (2023) 10:260–71. doi: 10.1016/S2215-0366(23)00025-1
26. Garver DL, Tamas RL, Holcomb JA. Elevated interleukin-6 in the cerebrospinal fluid of a previously delineated schizophrenia subtype. Neuropsychopharmacology. (2003) 28:1515–20. doi: 10.1038/sj.npp.1300217
27. Orlovska-Waast S, Kohler-Forsberg O, Brix SW, Nordentoft M, Kondziella D, Krogh J, et al. Cerebrospinal fluid markers of inflammation and infections in schizophrenia and affective disorders: a systematic review and meta-analysis. Mol Psychiatry. (2019) 24:869–87. doi: 10.1038/s41380-018-0220-4
28. Faustmann TJ, Kamp D, Rauber S, Dukart J, Melzer N, Schilbach L. Social interaction, psychotic disorders and inflammation: A triangle of interest. Prog Neuropsychopharmacol Biol Psychiatry. (2023) 122:110697. doi: 10.1016/j.pnpbp.2022.110697
29. Pong S, Karmacharya R, Sofman M, Bishop JR, Lizano P. The role of brain microvascular endothelial cell and blood-brain barrier dysfunction in schizophrenia. Complex Psychiatry. (2020) 6:30–46. doi: 10.1159/000511552
31. Fatemi SH, Folsom TD. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull. (2009) 35:528–48. doi: 10.1093/schbul/sbn187
32. Popovic D, Schmitt A, Kaurani L, Senner F, Papiol S, Malchow B, et al. Childhood trauma in schizophrenia: current findings and research perspectives. Front Neurosci. (2019) 13:274. doi: 10.3389/fnins.2019.00274
33. van Winkel R, Stefanis NC, Myin-Germeys I. Psychosocial stress and psychosis. A review of the neurobiological mechanisms and the evidence for gene-stress interaction. Schizophr Bull. (2008) 34:1095–105. doi: 10.1093/schbul/sbn101
34. Misiak B, Moustafa AA, Kiejna A, Frydecka D. Childhood traumatic events and types of auditory verbal hallucinations in first-episode schizophrenia patients. Compr Psychiatry. (2016) 66:17–22. doi: 10.1016/j.comppsych.2015.12.003
35. Jakel S, Dimou L. Glial cells and their function in the adult brain: A journey through the history of their ablation. Front Cell Neurosci. (2017) 11:24. doi: 10.3389/fncel.2017.00024
36. Trepanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP. Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry. (2016) 21:1009–26. doi: 10.1038/mp.2016.90
37. Monji A, Kato T, Kanba S. Cytokines and schizophrenia: Microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci. (2009) 63:257–65. doi: 10.1111/j.1440-1819.2009.01945.x
38. Parkin GM, Udawela M, Gibbons A, Dean B. Glutamate transporters, EAAT1 and EAAT2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J Psychiatry. (2018) 8:51–63. doi: 10.5498/wjp.v8.i2.51
39. Shan D, Lucas EK, Drummond JB, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal expression of glutamate transporters in temporal lobe areas in elderly patients with schizophrenia. Schizophr Res. (2013) 144:1–8. doi: 10.1016/j.schres.2012.12.019
40. Bauer D, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal glycosylation of EAAT1 and EAAT2 in prefrontal cortex of elderly patients with schizophrenia. Schizophr Res. (2010) 117:92–8. doi: 10.1016/j.schres.2009.07.025
41. Koh W, Park M, Chun YE, Lee J, Shim HS, Park MG, et al. Astrocytes render memory flexible by releasing D-serine and regulating NMDA receptor tone in the hippocampus. Biol Psychiatry. (2022) 91:740–52. doi: 10.1016/j.biopsych.2021.10.012
42. Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. (2003) 60:572–6. doi: 10.1001/archpsyc.60.6.572
43. Mei YY, Wu DC, Zhou N. Astrocytic regulation of glutamate transmission in schizophrenia. Front Psychiatry. (2018) 9:544. doi: 10.3389/fpsyt.2018.00544
44. Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, et al. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. (2003) 362:798–805. doi: 10.1016/S0140-6736(03)14289-4
45. Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR, et al. White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry. (2003) 60:443–56. doi: 10.1001/archpsyc.60.5.443
46. Hoistad M, Segal D, Takahashi N, Sakurai T, Buxbaum JD, Hof PR. Linking white and grey matter in schizophrenia: oligodendrocyte and neuron pathology in the prefrontal cortex. Front Neuroanat. (2009) 3:9.2009. doi: 10.3389/neuro.05.009.2009
47. Cassoli JS, Guest PC, Malchow B, Schmitt A, Falkai P, Martins-de-Souza D. Disturbed macro-connectivity in schizophrenia linked to oligodendrocyte dysfunction: from structural findings to molecules. NPJ Schizophr. (2015) 1:15034. doi: 10.1038/npjschz.2015.34
48. Takahashi N, Sakurai T, Davis KL, Buxbaum JD. Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog Neurobiol. (2011) 93:13–24. doi: 10.1016/j.pneurobio.2010.09.004
49. Chew LJ, Fusar-Poli P, Schmitz T. Oligodendroglial alterations and the role of microglia in white matter injury: relevance to schizophrenia. Dev Neurosci. (2013) 35:102–29. doi: 10.1159/000346157
50. Seki Y, Kato TA, Monji A, Mizoguchi Y, Horikawa H, Sato-Kasai M, et al. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-gamma-stimulated microglia in co-culture model. Schizophr Res. (2013) 151:20–8. doi: 10.1016/j.schres.2013.09.011
51. Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. (2013) 77:10–8. doi: 10.1016/j.neuron.2012.12.023
52. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
53. Gober R, Ardalan M, Shiadeh SMJ, Duque L, Garamszegi SP, Ascona M, et al. Microglia activation in postmortem brains with schizophrenia demonstrates distinct morphological changes between brain regions. Brain Pathol. (2022) 32:e13003. doi: 10.1111/bpa.13003
54. Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. (2013) 18:206–14. doi: 10.1038/mp.2012.110
55. Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett. (1999) 271:126–8. doi: 10.1016/s0304-3940(99)00545-5
56. Busse S, Busse M, Schiltz K, Bielau H, Gos T, Brisch R, et al. Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual versus paranoid schizophrenia: further evidence for disease course-related immune alterations? Brain Behav Immun. (2012) 26:1273–9. doi: 10.1016/j.bbi.2012.08.005
57. Foster R, Kandanearatchi A, Beasley C, Williams B, Khan N, Fagerhol MK, et al. Calprotectin in microglia from frontal cortex is up-regulated in schizophrenia: evidence for an inflammatory process? Eur J Neurosci. (2006) 24:3561–6. doi: 10.1111/j.1460-9568.2006.05219.x
58. Snijders G, van Zuiden W, Sneeboer MAM, Berdenis van Berlekom A, van der Geest AT, Schnieder T, et al. A loss of mature microglial markers without immune activation in schizophrenia. Glia. (2021) 69:1251–67. doi: 10.1002/glia.23962
59. Koskuvi M, Porsti E, Hewitt T, Rasanen N, Wu YC, Trontti K, et al. Genetic contribution to microglial activation in schizophrenia. Mol Psychiatry. (2024) 29:2622–33. doi: 10.1038/s41380-024-02529-1
60. Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [(11)C]PBR28 PET brain imaging study. Am J Psychiatry. (2016) 173:44–52. doi: 10.1176/appi.ajp.2015.14101358
61. van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. (2008) 64:820–2. doi: 10.1016/j.biopsych.2008.04.025
62. Sneeboer MAM, van der Doef T, Litjens M, Psy NBB, Melief J, Hol EM, et al. Microglial activation in schizophrenia: Is translocator 18kDa protein (TSPO) the right marker? Schizophr Res. (2020) 215:167–72. doi: 10.1016/j.schres.2019.10.045
63. Hafizi S, Tseng HH, Rao N, Selvanathan T, Kenk M, Bazinet RP, et al. Imaging microglial activation in untreated first-episode psychosis: A PET study with [(18)F]FEPPA. Am J Psychiatry. (2017) 174:118–24. doi: 10.1176/appi.ajp.2016.16020171
64. Ottoy J, De Picker L, Kang MS. Microglial positron emission tomography imaging in vivo: positron emission tomography radioligands: utility in research and clinical practice. Adv Neurobiol. (2024) 37:579–89. doi: 10.1007/978-3-031-55529-9_32
65. Hartmann SM, Heider J, Wust R, Fallgatter AJ, Volkmer H. Microglia-neuron interactions in schizophrenia. Front Cell Neurosci. (2024) 18:1345349. doi: 10.3389/fncel.2024.1345349
66. Holmes SE, Hinz R, Drake RJ, Gregory CJ, Conen S, Matthews JC, et al. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: a [(11)C](R)-PK11195 positron emission tomography study. Mol Psychiatry. (2016) 21:1672–9. doi: 10.1038/mp.2016.180
67. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry. (2008) 63:801–8. doi: 10.1016/j.biopsych.2007.09.024
68. Khandaker GM, Dantzer R, Jones PB. Immunopsychiatry: important facts. Psychol Med. (2017) 47:2229–37. doi: 10.1017/S0033291717000745
69. Muller N. Inflammation in schizophrenia: pathogenetic aspects and therapeutic considerations. Schizophr Bull. (2018) 44:973–82. doi: 10.1093/schbul/sby024
70. Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci. (2019) 22:374–85. doi: 10.1038/s41593-018-0334-7
71. Howes OD, Onwordi EC. The synaptic hypothesis of schizophrenia version III: a master mechanism. Mol Psychiatry. (2023) 28:1843–56. doi: 10.1038/s41380-023-02043-w
72. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. (2016) 530:177–83. doi: 10.1038/nature16549
73. Comer AL, Jinadasa T, Sriram B, Phadke RA, Kretsge LN, Nguyen TPH, et al. Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PloS Biol. (2020) 18:e3000604. doi: 10.1371/journal.pbio.3000604
74. Druart M, Nosten-Bertrand M, Poll S, Crux S, Nebeling F, Delhaye C, et al. Elevated expression of complement C4 in the mouse prefrontal cortex causes schizophrenia-associated phenotypes. Mol Psychiatry. (2021) 26:3489–501. doi: 10.1038/s41380-021-01081-6
75. Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res. (2008) 42:151–7. doi: 10.1016/j.jpsychires.2006.10.013
76. Steiner J, Gos T, Bogerts B, Bielau H, Drexhage HA, Bernstein HG. Possible impact of microglial cells and the monocyte-macrophage system on suicidal behavior. CNS Neurol Disord Drug Targets. (2013) 12:971–9. doi: 10.2174/18715273113129990099
77. Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci. (2009) 3:14.2009. doi: 10.3389/neuro.08.014.2009
78. Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and memory: modulation by early-life infection. J Neurosci. (2011) 31:15511–21. doi: 10.1523/JNEUROSCI.3688-11.2011
79. Monji A, Kato TA, Mizoguchi Y, Horikawa H, Seki Y, Kasai M, et al. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry. (2013) 42:115–21. doi: 10.1016/j.pnpbp.2011.12.002
80. Zhuo C, Tian H, Song X, Jiang D, Chen G, Cai Z, et al. Microglia and cognitive impairment in schizophrenia: translating scientific progress into novel therapeutic interventions. Schizophr (Heidelb). (2023) 9:42. doi: 10.1038/s41537-023-00370-z
81. Enomoto S, Kato TA. Stress mediated microglial hyper-activation and psychiatric diseases. Brain Nerve. (2021) 73:795–802. doi: 10.11477/mf.1416201837
82. Bruijnzeel D, Suryadevara U, Tandon R. Antipsychotic treatment of schizophrenia: an update. Asian J Psychiatr. (2014) 11:3–7. doi: 10.1016/j.ajp.2014.08.002
83. Efthimiou O, Taipale H, Radua J, Schneider-Thoma J, Pinzon-Espinosa J, Ortuno M, et al. Efficacy and effectiveness of antipsychotics in schizophrenia: network meta-analyses combining evidence from randomised controlled trials and real-world data. Lancet Psychiatry. (2024) 11:102–11. doi: 10.1016/S2215-0366(23)00366-8
84. Faustmann TJ, Paschali M, Kojda G, Schilbach L, Kamp D. Antipsychotic treatment of alcohol withdrawal syndrome with focus on delirium tremens: a systematic review. Fortschr Neurol Psychiatr. (2023). doi: 10.1055/a-2029-6387
85. Meltzer HY, Matsubara S, Lee JC. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J Pharmacol Exp Ther. (1989) 251:238–46. doi: 10.1016/S0022-3565(25)20710-8
86. Meltzer HY. Update on typical and atypical antipsychotic drugs. Annu Rev Med. (2013) 64:393–406. doi: 10.1146/annurev-med-050911-161504
87. Buchanan RW, Breier A, Kirkpatrick B, Ball P, Carpenter WT Jr. Positive and negative symptom response to clozapine in schizophrenic patients with and without the deficit syndrome. Am J Psychiatry. (1998) 155:751–60. doi: 10.1176/ajp.155.6.751
88. Palao DJ, Arauxo A, Brunet M, Marquez M, Bernardo M, Ferrer J, et al. Positive versus negative symptoms in schizophrenia: response to haloperidol. Prog Neuropsychopharmacol Biol Psychiatry. (1994) 18:155–64. doi: 10.1016/0278-5846(94)90032-9
89. Kato TA, Monji A, Mizoguchi Y, Hashioka S, Horikawa H, Seki Y, et al. Anti-Inflammatory properties of antipsychotics via microglia modulations: are antipsychotics a ‘fire extinguisher’ in the brain of schizophrenia? Mini Rev Med Chem. (2011) 11:565–74. doi: 10.2174/138955711795906941
90. Na KS, Jung HY, Kim YK. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. (2014) 48:277–86. doi: 10.1016/j.pnpbp.2012.10.022
91. Shnayder NA, Khasanova AK, Strelnik AI, Al-Zamil M, Otmakhov AP, Neznanov NG, et al. Cytokine imbalance as a biomarker of treatment-resistant schizophrenia. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms231911324
92. Romeo B, Brunet-Lecomte M, Martelli C, Benyamina A. Kinetics of cytokine levels during antipsychotic treatment in schizophrenia: A meta-analysis. Int J Neuropsychopharmacol. (2018) 21:828–36. doi: 10.1093/ijnp/pyy062
93. Conen S, Gregory CJ, Hinz R, Smallman R, Corsi-Zuelli F, Deakin B, et al. Neuroinflammation as measured by positron emission tomography in patients with recent onset and established schizophrenia: implications for immune pathogenesis. Mol Psychiatry. (2021) 26:5398–406. doi: 10.1038/s41380-020-0829-y
94. Di Biase MA, Zalesky A, O'Keefe G, Laskaris L, Baune BT, Weickert CS, et al. PET imaging of putative microglial activation in individuals at ultra-high risk for psychosis, recently diagnosed and chronically ill with schizophrenia. Transl Psychiatry. (2017) 7:e1225. doi: 10.1038/tp.2017.193
95. Danovich L, Veenman L, Leschiner S, Lahav M, Shuster V, Weizman A, et al. The influence of clozapine treatment and other antipsychotics on the 18 kDa translocator protein, formerly named the peripheral-type benzodiazepine receptor, and steroid production. Eur Neuropsychopharmacol. (2008) 18:24–33. doi: 10.1016/j.euroneuro.2007.04.005
96. Dinesh AA, Islam J, Khan J, Turkheimer F, Vernon AC. Effects of antipsychotic drugs: cross talk between the nervous and innate immune system. CNS Drugs. (2020) 34:1229–51. doi: 10.1007/s40263-020-00765-x
97. Cotel MC, Lenartowicz EM, Natesan S, Modo MM, Cooper JD, Williams SC, et al. Microglial activation in the rat brain following chronic antipsychotic treatment at clinically relevant doses. Eur Neuropsychopharmacol. (2015) 25:2098–107. doi: 10.1016/j.euroneuro.2015.08.004
98. Fitzgerald PB, Kapur S, Remington G, Roy P, Zipursky RB. Predicting haloperidol occupancy of central dopamine D2 receptors from plasma levels. Psychopharmacol (Berl). (2000) 149:1–5. doi: 10.1007/s002139900333
99. Racki V, Marcelic M, Stimac I, Petric D, Kucic N. Effects of haloperidol, risperidone, and aripiprazole on the immunometabolic properties of BV-2 microglial cells. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22094399
100. Namjoo E, Shekari M, Piruozi A, Forouzandeh H, Khalafkhany D, Vahedi A, et al. Haloperidol’s effect on the expressions of TGFB, NT-3, and BDNF genes in cultured rat microglia. Basic Clin Neurosci. (2020) 11:49–58. doi: 10.32598/bcn.11.1.1272.1
101. Bloomfield PS, Bonsall D, Wells L, Dormann D, Howes O, De Paola V. The effects of haloperidol on microglial morphology and translocator protein levels: An in vivo study in rats using an automated cell evaluation pipeline. J Psychopharmacol. (2018) 32:1264–72. doi: 10.1177/0269881118788830
102. Nakki R, Nickolenko J, Chang J, Sagar SM, Sharp FR. Haloperidol prevents ketamine- and phencyclidine-induced HSP70 protein expression but not microglial activation. Exp Neurol. (1996) 137:234–41. doi: 10.1006/exnr.1996.0022
103. Maredia M, Hamilton J, Thanos PK. A high-fat diet, but not haloperidol or olanzapine administration, increases activated microglial expression in the rat brain. Neurosci Lett. (2021) 757:135976. doi: 10.1016/j.neulet.2021.135976
104. Long Y, Wang Y, Shen Y, Huang J, Li Y, Wu R, et al. Minocycline and antipsychotics inhibit inflammatory responses in BV-2 microglia activated by LPS via regulating the MAPKs/JAK-STAT signaling pathway. BMC Psychiatry. (2023) 23:514. doi: 10.1186/s12888-023-05014-1
105. Shin H, Song JH. Antipsychotics, chlorpromazine and haloperidol inhibit voltage-gated proton currents in BV2 microglial cells. Eur J Pharmacol. (2014) 738:256–62. doi: 10.1016/j.ejphar.2014.05.049
106. Labuzek K, Kowalski J, Gabryel B, Herman ZS. Chlorpromazine and loxapine reduce interleukin-1beta and interleukin-2 release by rat mixed glial and microglial cell cultures. Eur Neuropsychopharmacol. (2005) 15:23–30. doi: 10.1016/j.euroneuro.2004.04.002
107. Lee HY, Lee Y, Chung C, Park SI, Shin HJ, Joe EH, et al. The antipsychotic chlorpromazine reduces neuroinflammation by inhibiting microglial voltage-gated potassium channels. Glia. (2025) 73:210–27. doi: 10.1002/glia.24629
108. Kowalski J, Labuzek K, Herman ZS. Flupentixol and trifluperidol reduce interleukin-1 beta and interleukin-2 release by rat mixed glial and microglial cell cultures. Pol J Pharmacol. (2004) 56:563–70.
109. Kowalski J, Labuzek K, Herman ZS. Flupentixol and trifluperidol reduce secretion of tumor necrosis factor-alpha and nitric oxide by rat microglial cells. Neurochem Int. (2003) 43:173–8. doi: 10.1016/s0197-0186(02)00163-8
110. Zheng LT, Hwang J, Ock J, Lee MG, Lee WH, Suk K. The antipsychotic spiperone attenuates inflammatory response in cultured microglia via the reduction of proinflammatory cytokine expression and nitric oxide production. J Neurochem. (2008) 107:1225–35. doi: 10.1111/j.1471-4159.2008.05675.x
111. Meltzer HY, Huang M. In vivo actions of atypical antipsychotic drug on serotonergic and dopaminergic systems. Prog Brain Res. (2008) 172:177–97. doi: 10.1016/S0079-6123(08)00909-6
112. Ceylan U, Haupeltshofer S, Kamper L, Dann J, Ambrosius B, Gold R, et al. Clozapine regulates microglia and is effective in chronic experimental autoimmune encephalomyelitis. Front Immunol. (2021) 12:656941. doi: 10.3389/fimmu.2021.656941
113. Giridharan VV, Scaini G, Colpo GD, Doifode T, Pinjari OF, Teixeira AL, et al. Clozapine prevents poly (I:C) induced inflammation by modulating NLRP3 pathway in microglial cells. Cells. (2020) 9. doi: 10.3390/cells9030577
114. Jeon S, Kim SH, Shin SY, Lee YH. Clozapine reduces Toll-like receptor 4/NF-kappaB-mediated inflammatory responses through inhibition of calcium/calmodulin-dependent Akt activation in microglia. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 81:477–87. doi: 10.1016/j.pnpbp.2017.04.012
115. Hu X, Zhou H, Zhang D, Yang S, Qian L, Wu HM, et al. Clozapine protects dopaminergic neurons from inflammation-induced damage by inhibiting microglial overactivation. J Neuroimmune Pharmacol. (2012) 7:187–201. doi: 10.1007/s11481-011-9309-0
116. Shin H, Kim J, Song JH. Clozapine and olanzapine inhibit proton currents in BV2 microglial cells. Eur J Pharmacol. (2015) 755:74–9. doi: 10.1016/j.ejphar.2015.03.003
117. McGavin JK, Goa KL. Aripiprazole. CNS Drugs. (2002) 16:779–86;discussion787-8. doi: 10.2165/00023210-200216110-00008
118. Sato-Kasai M, Kato TA, Ohgidani M, Mizoguchi Y, Sagata N, Inamine S, et al. Aripiprazole inhibits polyI:C-induced microglial activation possibly via TRPM7. Schizophr Res. (2016) 178:35–43. doi: 10.1016/j.schres.2016.08.022
119. Kato T, Mizoguchi Y, Monji A, Horikawa H, Suzuki SO, Seki Y, et al. Inhibitory effects of aripiprazole on interferon-gamma-induced microglial activation via intracellular Ca2+ regulation in vitro. J Neurochem. (2008) 106:815–25. doi: 10.1111/j.1471-4159.2008.05435.x
120. Bian Q, Kato T, Monji A, Hashioka S, Mizoguchi Y, Horikawa H, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. (2008) 32:42–8. doi: 10.1016/j.pnpbp.2007.06.031
121. Zhu S, Shi R, Li V, Wang J, Zhang R, Tempier A, et al. Quetiapine attenuates glial activation and proinflammatory cytokines in APP/PS1 transgenic mice via inhibition of nuclear factor-kappaB pathway. Int J Neuropsychopharmacol. (2014) 18. doi: 10.1093/ijnp/pyu022
122. Wang H, Liu S, Tian Y, Wu X, He Y, Li C, et al. Quetiapine inhibits microglial activation by neutralizing abnormal STIM1-mediated intercellular calcium homeostasis and promotes myelin repair in a cuprizone-induced mouse model of demyelination. Front Cell Neurosci. (2015) 9:492. doi: 10.3389/fncel.2015.00492
123. Kato T, Monji A, Hashioka S, Kanba S. Risperidone significantly inhibits interferon-gamma-induced microglial activation in vitro. Schizophr Res. (2007) 92:108–15. doi: 10.1016/j.schres.2007.01.019
124. Hou Y, Wu CF, Yang JY, He X, Bi XL, Yu L, et al. Effects of clozapine, olanzapine and haloperidol on nitric oxide production by lipopolysaccharide-activated N9 cells. Prog Neuropsychopharmacol Biol Psychiatry. (2006) 30:1523–8. doi: 10.1016/j.pnpbp.2006.05.006
125. Kim HS, Whang SY, Woo MS, Park JS, Kim WK, Han IO. Sodium butyrate suppresses interferon-gamma-, but not lipopolysaccharide-mediated induction of nitric oxide and tumor necrosis factor-alpha in microglia. J Neuroimmunol. (2004) 151:85–93. doi: 10.1016/j.jneuroim.2004.02.006
126. Park JS, Woo MS, Kim SY, Kim WK, Kim HS. Repression of interferon-gamma-induced inducible nitric oxide synthase (iNOS) gene expression in microglia by sodium butyrate is mediated through specific inhibition of ERK signaling pathways. J Neuroimmunol. (2005) 168:56–64. doi: 10.1016/j.jneuroim.2005.07.003
127. Lidow MS. Calcium signaling dysfunction in schizophrenia: a unifying approach. Brain Res Brain Res Rev. (2003) 43:70–84. doi: 10.1016/s0165-0173(03)00203-0
128. Rahimian R, Wakid M, O'Leary LA, Mechawar N. The emerging tale of microglia in psychiatric disorders. Neurosci Biobehav Rev. (2021) 131:1–29. doi: 10.1016/j.neubiorev.2021.09.023
129. Kato TA, Yamauchi Y, Horikawa H, Monji A, Mizoguchi Y, Seki Y, et al. Neurotransmitters, psychotropic drugs and microglia: clinical implications for psychiatry. Curr Med Chem. (2013) 20:331–44. doi: 10.2174/0929867311320030003
130. Mondelli V, Vernon AC, Turkheimer F, Dazzan P, Pariante CM. Brain microglia in psychiatric disorders. Lancet Psychiatry. (2017) 4:563–72. doi: 10.1016/S2215-0366(17)30101-3
131. Schmitz I, da Silva A, Bobermin LD, Goncalves CA, Steiner J, Quincozes-Santos A. The Janus face of antipsychotics in glial cells: Focus on glioprotection. Exp Biol Med (Maywood). (2023) 248:2120–30. doi: 10.1177/15353702231222027
132. Faustmann PM, Haase CG, Romberg S, Hinkerohe D, Szlachta D, Smikalla D, et al. Microglia activation influences dye coupling and Cx43 expression of the astrocytic network. Glia. (2003) 42:101–8. doi: 10.1002/glia.10141
133. Ismail FS, Corvace F, Faustmann PM, Faustmann TJ. Pharmacological investigations in glia culture model of inflammation. Front Cell Neurosci. (2021) 15:805755. doi: 10.3389/fncel.2021.805755
134. Hinkerohe D, Smikalla D, Haghikia A, Heupel K, Haase CG, Dermietzel R, et al. Effects of cytokines on microglial phenotypes and astroglial coupling in an inflammatory coculture model. Glia. (2005) 52:85–97. doi: 10.1002/glia.20223
135. Fukuyama K, Okada M. Effects of atypical antipsychotics, clozapine, quetiapine and brexpiprazole on astroglial transmission associated with connexin43. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22115623
136. Moinfar Z, Dambach H, Faustmann PM. Influence of drugs on gap junctions in glioma cell lines and primary astrocytes in vitro. Front Physiol. (2014) 5:186. doi: 10.3389/fphys.2014.00186
137. Fatemi SH, Folsom TD, Reutiman TJ, Pandian T, Braun NN, Haug K. Chronic psychotropic drug treatment causes differential expression of connexin 43 and GFAP in frontal cortex of rats. Schizophr Res. (2008) 104:127–34. doi: 10.1016/j.schres.2008.05.016
138. Bernstein HG, Nussbaumer M, Vasilevska V, Dobrowolny H, Nickl-Jockschat T, Guest PC, et al. Glial cell deficits are a key feature of schizophrenia: implications for neuronal circuit maintenance and histological differentiation from classical neurodegeneration. Mol Psychiatry. (2024). doi: 10.1038/s41380-024-02861-6
139. Corvace F, Faustmann TJ, Faustmann PM, Ismail FS. Anti-inflammatory properties of lacosamide in an astrocyte-microglia co-culture model of inflammation. Eur J Pharmacol. (2022) 915:174696. doi: 10.1016/j.ejphar.2021.174696
140. Haghikia A, Ladage K, Hinkerohe D, Vollmar P, Heupel K, Dermietzel R, et al. Implications of antiinflammatory properties of the anticonvulsant drug levetiracetam in astrocytes. J Neurosci Res. (2008) 86:1781–8. doi: 10.1002/jnr.21639
141. Vollmar P, Haghikia A, Dermietzel R, Faustmann PM. Venlafaxine exhibits an anti-inflammatory effect in an inflammatory co-culture model. Int J Neuropsychopharmacol. (2008) 11:111–7. doi: 10.1017/S1461145707007729
142. Stienen MN, Haghikia A, Dambach H, Thone J, Wiemann M, Gold R, et al. Anti-inflammatory effects of the anticonvulsant drug levetiracetam on electrophysiological properties of astroglia are mediated via TGFbeta1 regulation. Br J Pharmacol. (2011) 162:491–507. doi: 10.1111/j.1476-5381.2010.01038.x
143. Dambach H, Hinkerohe D, Prochnow N, Stienen MN, Moinfar Z, Haase CG, et al. Glia and epilepsy: experimental investigation of antiepileptic drugs in an astroglia/microglia co-culture model of inflammation. Epilepsia. (2014) 55:184–92. doi: 10.1111/epi.12473
144. Faustmann TJ, Corvace F, Faustmann PM, Ismail FS. Effects of lamotrigine and topiramate on glial properties in an astrocyte-microglia co-culture model of inflammation. Int J Neuropsychopharmacol. (2022) 25:185–96. doi: 10.1093/ijnp/pyab080
145. Ismail FS, Faustmann PM, Kummel ML, Forster E, Faustmann TJ, Corvace F. Brivaracetam exhibits mild pro-inflammatory features in an in vitro astrocyte-microglia co-culture model of inflammation. Front Cell Neurosci. (2022) 16:995861. doi: 10.3389/fncel.2022.995861
146. Faustmann TJ, Wawrzyniak M, Faustmann PM, Corvace F, Ismail FS. Inhibition of microglial activation by amitriptyline and doxepin in interferon-beta pre-treated astrocyte-microglia co-culture model of inflammation. Brain Sci. (2023) 13. doi: 10.3390/brainsci13030493
147. Ismail FS, Faustmann PM, Forster E, Corvace F, Faustmann TJ. Tiagabine and zonisamide differentially regulate the glial properties in an astrocyte-microglia co-culture model of inflammation. Naunyn Schmiedebergs Arch Pharmacol. (2023) 396:3253–67. doi: 10.1007/s00210-023-02538-x
148. Dietz AG, Goldman SA, Nedergaard M. Glial cells in schizophrenia: a unified hypothesis. Lancet Psychiatry. (2020) 7:272–81. doi: 10.1016/S2215-0366(19)30302-5
149. Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. (2016) 537:50–6. doi: 10.1038/nature19323
150. van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early alzheimer’s disease. N Engl J Med. (2023) 388:9–21. doi: 10.1056/NEJMoa2212948
151. Jucker M, Walker LC. Alzheimer’s disease: From immunotherapy to immunoprevention. Cell. (2023) 186:4260–70. doi: 10.1016/j.cell.2023.08.021
152. Perneczky R, Jessen F, Grimmer T, Levin J, Floel A, Peters O, et al. Anti-amyloid antibody therapies in Alzheimer’s disease. Brain. (2023) 146:842–9. doi: 10.1093/brain/awad005
153. Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, et al. Donanemab in early symptomatic alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. (2023) 330:512–27. doi: 10.1001/jama.2023.13239
Keywords: antipsychotic drugs, glia, neuroinflammation, astrocyte-microglia co-culture model, psychotic disorders, schizophrenia
Citation: Faustmann TJ, Corvace F, Faustmann PM and Ismail FS (2025) Influence of antipsychotic drugs on microglia-mediated neuroinflammation in schizophrenia: perspectives in an astrocyte–microglia co-culture model. Front. Psychiatry 16:1522128. doi: 10.3389/fpsyt.2025.1522128
Received: 04 November 2024; Accepted: 12 February 2025;
Published: 18 March 2025.
Edited by:
Cong Yu, Chongqing Medical University, ChinaReviewed by:
Bernhard Bogerts, Leonardo Büro Sachsen Anhalt, GermanyCopyright © 2025 Faustmann, Corvace, Faustmann and Ismail. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timo Jendrik Faustmann, dGltby5mYXVzdG1hbm5AdW5pLWR1ZXNzZWxkb3JmLmRl
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.