 Mustafa A. Salih
Mustafa A. Salih Albandary AlBakheet
Albandary AlBakheet Rawan Almass3
Rawan Almass3 Namik Kaya
Namik Kaya- 1Consultant Pediatric Neurologist, Health Sector, King Abdulaziz City for Science and Technology, Riyadh, Saudi Arabia
- 2Translational Genomic Department, Center for Genomic Medicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 3Medical Genetics Department, Center for Genomic Medicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 4Consultant Pediatric Neurologist, Faculty of Medicine, University of Khartoum, Khartoum, Sudan
- 5Research and Innovation, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
Background: Pathogenic variants in ALDH7A1 are associated with pyridoxine-dependent epilepsy (PDE), a rare autosomal recessive disorder characterized by epileptic seizures, unresponsiveness to standard antiseizure medications (ASM), and a response only to pyridoxine. Here, we report two patients (from a consanguineous family) with neonatal seizures and developmental delay.
Case presentation: Patient 1 (a 13-year-old girl) was born normally at term. Her pregnancy was complicated by antiphospholipid syndrome, and persistent vomiting was managed with several medications, including pyridoxine (40 mg daily). Seizures occurred 6 h after birth and did not respond to antiseizure medications. However, they ceased 2 days later when pyridoxine (40 mg daily) was administered. She continued her medications and had delayed early milestones. Phenobarbitone was discontinued at 18 months, and pyridoxine was increased to 100 mg daily at 8 years of age. She was able to join a regular school and performed well. Patient 2, a 12-year-old boy, was delivered normally at term. Seizures started 10 h after birth, and he immediately received 40 mg of pyridoxine. Seizures have been controlled since then, and he experienced delayed milestones. Pyridoxine was increased to 100 mg daily at 7 years of age. He is currently in fifth grade and has dyslexia. Whole exome sequencing (WES) revealed that both patients 1 and 2 harbor a novel homozygous missense variant in ALDH7A1 (NM_001202404: exon 12: c.1168G>C; (p.Gly390Arg)).
Conclusion: The present study reports a novel ALDH7A1 variant causing PDE and highlights the associated developmental delay and intellectual disability, despite early seizure control treatment.
1 Introduction
Variants in the ALDH7A1 gene are associated with pyridoxine-dependent epilepsy (PDE), a rare autosomal recessive disorder characterized by etiology-specific developmental and epileptic seizures specific to the condition. These seizures typically occur in the first hours of life, are unresponsive to standard antiseizure medications (ASM), and respond only to pyridoxine administration (1). The incidence of PDE ranges between 1:65,000 and 250,000 live births (2, 3). Timely diagnosis is imperative to prevent uncontrolled seizures, which may jeopardize neurocognitive development (4). Atypical presentations are known to occur, and seizures may manifest after the neonatal period (5). In some patients, seizures may initially respond to standard ASM (6). Global developmental delay and/or intellectual disability have been recorded in 75% of pyridoxine-dependent epilepsy patients, even with early treatment initiation and seizure control (7–9).
The ALDH7A1 gene, also known as aldehyde dehydrogenase 7 family member A1, encodes an enzyme within the aldehyde dehydrogenase (ALDH) family. ALDH enzymes are essential for metabolizing aldehydes, toxic byproducts produced during various cellular processes, such as alcohol metabolism and lipid peroxidation. ALDH7A1 specifically catalyzes the conversion of these aldehydes into less harmful carboxylic acids. Specifically, it acts on a variety of aldehydes, including acetaldehyde and 4-hydroxybutyric acid. ALDH7A1 is involved in several metabolic pathways, notably the breakdown of lysine, an essential amino acid, in the mitochondrial matrix. In detoxification, ALDH7A1 helps prevent the accumulation of toxic aldehydes, protecting cells from potential damage. A deficiency in ALDH7A1 can also lead to hyperammonemia due to lysine accumulation. The enzyme is expressed in various tissues, including the liver, brain, and kidney. One or more of the accumulated biomarkers are thought to contribute to the developmental delay and/or intellectual disability observed in patients who are treated late, and lysine reduction therapies (LRT) have been implemented to reduce these consequences (10).
Here, we investigate a novel pathogenic homozygous variant in ALDH7A1, primarily causing neonatal seizures and developmental delay in two patients from a consanguineous Sudanese family living in Saudi Arabia. The older patient (13-year-old girl) exhibited delayed early milestones but was able to attend regular school, performing well in the seventh and eighth grades. The younger patient had similar features and had dyslexia in both Arabic (his native language) and English.
2 Methods
2.1 Sample collection
Two patients from a consanguineous Sudanese family residing in Saudi Arabia were recruited with approval from the Institution’s Research Advisory Council (Ethics Statement) at King Faisal Specialist Hospital and Research Center, Riyadh, KSA (KFSHRC, IRB-approved protocols, RAC#2180004). The participants signed written informed consent forms.
2.2 Molecular genetic detection
Peripheral blood samples (5 ml) were collected in EDTA tubes from both affected and unaffected individuals within the participating family. DNA was isolated using the Gentra® Puregene DNA Purification Kit (Gentra Systems Inc., Minneapolis, MN, USA). PCR was performed with primers designed by Primer3, followed by direct sequencing on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), in accordance with the manufacturer’s instructions. WES was conducted on the patient’s DNA, and variants were filtered and analyzed for in silico pathogenicity as previously described (11, 12). DNA samples were genotyped using Affymetrix Axiom chips, following the manufacturer’s protocols (Affymetrix Inc., Santa Clara, CA, USA). The analysis was performed as previously described (11). Autozygome coordinates were considered during the WES variant filtering process.
3 Results
3.1 Clinical features
3.1.1 Patient 1 (IV:6)
Patient 1 is a 13-year-old girl, born as the third child to healthy, first-degree consanguineous Sudanese parents living in Saudi Arabia (Figure 1). She was delivered at 38 weeks via normal spontaneous vaginal delivery after a smooth course of labor. Her Apgar score was 8 at 5 min. She received naloxone due to pethidine administered to her mother and was admitted to the Neonatal Intensive Care Unit (NICU) for pethidine depression. The pregnancy was complicated by antiphospholipid syndrome, for which the mother received 81 mg of aspirin tablets daily and 40 mg of clexane subcutaneously daily until term. The mother experienced persistent vomiting, which subsided only after delivery despite attempts with metoclopramide (Primperan), promethazine (Phenergan), pyridoxine (40 mg daily), and ondansetron during pregnancy. At 34 weeks, the mother had pre-term contractions and a suspected urinary tract infection, which was treated with cephradine, ritodrine (Yutopar), and a course of dexamethasone injections. The mother had three previous miscarriages. Six hours after birth, patient 1 started to have seizures that did not respond to phenobarbitone and midazolam, and she was ultimately ventilated due to recurrent seizures. Two days later, pyridoxine (40 mg daily) was administered via nasogastric tube, after which no further seizures were recorded. She was extubated on day 5 and continued on phenobarbitone, midazolam, pyridoxine, and empirical antibiotics.
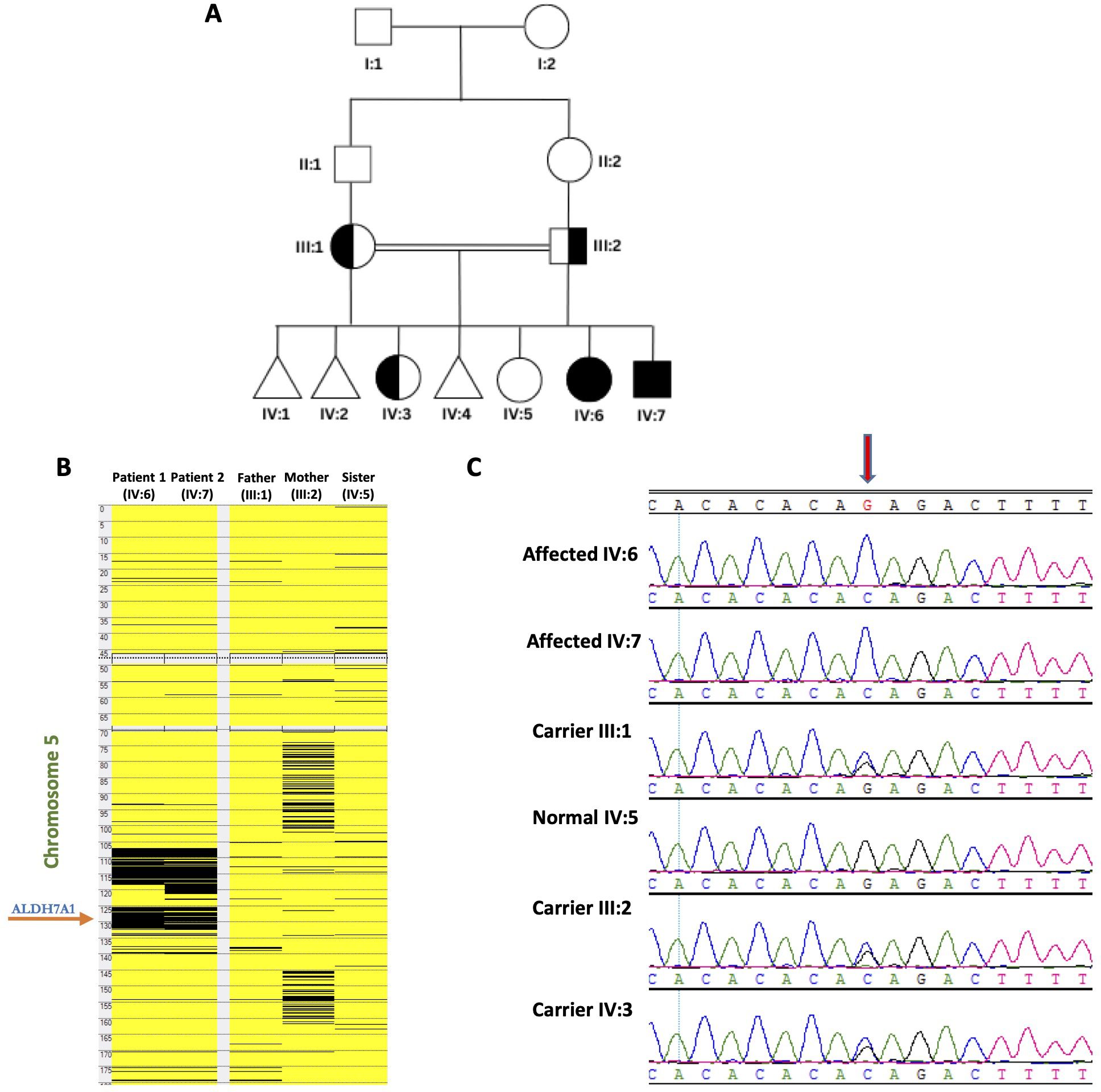
Figure 1. Genetic analyses of the families: (A) Pedigree of the studied family showing two affected members. (B) Homozygosity mapping indicating the location of the ALDH7A1 gene within the major ROH in the family. (C) The chromatograms from Sanger Sequencing revealing segregation of the mutation in the family members (The red arrow highlights the position of the mutation).
Complete blood count, biochemistry, arterial blood gas, blood culture and sensitivity, lumbar culture results, coagulation screen, and tandem mass spectrometry were normal. Electroencephalography (EEG) showed epileptiform discharges while staying in the NICU. One month after discharge, no epileptiform discharges were seen, but diffuse slowing was observed. Since then, and until now, repeated EEG examinations have been normal. Cranial magnetic resonance imaging (MRI), done for suspected cavernous sinus thrombosis on day 5, revealed normal findings. Pyridoxine metabolite tests (alpha-aminoadipic semialdehyde) were not done, as they were not accessible. She had delayed early milestones: started sitting at 14 months, crawling at 20 months, walking at 26 months, and spoke one word by 24 months. Phenobarbitone was stopped at the age of 18 months, and the dose of pyridoxine was increased to 100 mg daily at the age of 8 years following confirmatory genetic results. This was adjusted to 300 mg daily at the age of 13 years. She managed to join a regular school, performing well in seventh and eighth grades.
3.1.2 Patient 2 (IV:7)
Patient 2, the younger brother of patient 1, is currently aged 12 years old (Figure 1). He was delivered at term via normal spontaneous vaginal delivery. Seizures started 10 h after birth, and he immediately received 40 mg of pyridoxine. Since then, the seizures have been controlled. Electroencephalography performed 2 days later revealed no abnormalities, and magnetic resonance imaging (done at the age of 1 month) was normal. Pyridoxine metabolite tests (alpha-aminoadipic semialdehyde) were not performed, as they were not accessible. He had delayed milestones, walked at 18 months, and was able to make sentences by 3 years of age. The dose of pyridoxine was increased to 100 mg daily at the age of 7 years following confirmatory genetic results. This was adjusted to 300 mg daily at the age of 12 years. He is currently in fifth grade and has dyslexia in both Arabic (his native language) and English.
3.2 Molecular genetic results
WES analysis identified a novel homozygous variant in ALDH7A1 (NM_001202404: exon 12: c.1168G>C; p.Gly390Arg) as the only strong candidate underlying the disorder. The gene is in one of the major runs of homozygosity (ROH) block detected on chromosome 5q31 (Figure 1). Confirmatory Sanger sequencing demonstrated complete segregation of this variant within the family (Figure 1). Based on this, an elder sister (IV:3) was found to be a heterozygous carrier and is clinically normal. The c.1168G>C variant causes a missense change involving the alteration of a conserved nucleotide. The variant was absent in control chromosomes in the GnomAD project, as analyzed by MutationTaster and Genebe. According to the American College of Medical Genetics (ACMG), the c.1168G>C variant is classified as likely pathogenic based on the following criteria: scores: 11, CADD: pathogenic (31), PolyPhen: damaging (D), PM1: moderate, PM2: moderate, and PP3: strong. No clinical diagnostic laboratories have submitted clinical-significance assessments for this variant to ClinVar.
4 Discussion
The present study highlights a rare, treatable genetic disorder associated with intellectual disability, namely PDE, where timely diagnosis is imperative. It describes the clinical phenotype of a novel familial ALDH7A1 mutation causing neonatal seizures and developmental delay in two patients from a consanguineous family. Both patients manifested the classic presentation of ALDH7A1-related pyridoxine-dependent epilepsy, with seizures starting shortly after birth (13). Patient 1 did not respond to antiseizure medications and was ultimately placed on a ventilator due to recurrent seizures. She responded to pyridoxine (40 mg daily), administered 2 days later via nasogastric tube, after which no further seizures were recorded. Seizures started 10 h after birth in the younger brother (patient 2), who immediately received 40 mg of pyridoxine. No further seizures occurred since then. Following confirmatory genetic results, pyridoxine was increased to 100 mg daily in both patients. However, this is lower than the current recommended dose for children, adolescents, and adults (range 5–30 mg/kg/day), along with dietary modifications aimed at reducing lysine intake (4, 13). Pyridoxine was recently adjusted for both patients to comply with the recommended dose. An EEG performed on patient 1 during their stay in NICU showed epileptiform discharges. No epileptiform discharges were seen 1 month after discharge, but diffuse slowing was noted, and repeated EEG examinations have been normal. EEG recordings are known to be variable and nonspecific in pyridoxine-dependent epilepsy caused by ALDH7A1 (4, 14). Reported abnormalities include high-voltage delta waves, burst-suppression patterns, and hypsarrhythmia (6, 15, 16). On the other hand, a cranial MRI performed on day 5 for patient 1 revealed normal findings. Reported MRI abnormalities in ALDH7A1-associated pyridoxine-dependent epilepsy include hypoplasia of corpus callosum, cortical dysplasia, mega cisterna magna, cerebellar hypoplasia, ventriculomegaly, and hydrocephalus (13, 17–19).
Intellectual disability, primarily affecting expressive language, is common in this disease, with a higher risk correlated with the length of the delay in diagnosis and initiation of appropriate pyridoxine therapy (7, 13, 20). The neurodevelopmental disability, affecting approximately 75%, ranges from mild to severe intellectual disability and includes autistic features (4, 7, 21, 22). The longer the time between the initial administration of antiseizure medication, before suspecting PDE, and the subsequent administration of pyridoxine, the worse the neurological outcome appears to be at follow-up (23). To avoid missing cases, a trial of pyridoxine as a first-line treatment in all patients with seizures of unknown etiology has been suggested (24). The availability of newborn screening and rapid molecular genetic testing are expected to improve the neurological outcome in ALDH7A1-associated pyridoxine-dependent epilepsy (4). Deficiency of alpha-aminoadipic semialdehyde in PDE results in impaired lysine degradation due to deficiency of alpha-aminoadipic semialdehyde dehydrogenase. This leads to the accumulation of toxic metabolites from impaired lysine metabolism (22). To reduce the accumulation of these neurotoxic metabolites, a lysine-restricted diet and pharmacologic doses of arginine, which acts as a competitive inhibitor of lysine transport, are currently employed in addition to pyridoxine for the management of PDE (25). A significant increase in developmental testing scores was reported with pyridoxine and LRT treatment provided during the first 6 months of life (25). It is noteworthy that patient 1 received 40 mg of pyridoxine daily prenatally, which was administered to her mother due to persistent vomiting. This might have influenced the neurological outcome (16), as, despite early delayed development, she was able to join a regular school with good performance. Her younger brother, who also received 40 mg of pyridoxine immediately after his first seizure, also experienced delayed early development. He was also able to join a regular school but has dyslexia. The influence of genotype might explain the relatively favorable outcome in these siblings (26).
We identified a novel pathogenic homozygous variant in ALDH7A1 (c.1168G>C) in two patients, which has not been previously reported. This variant affects a highly conserved amino acid position, potentially impacting protein functionality. Bioinformatic prediction tools suggest the variant’s pathogenicity. The parents of these siblings also carry the same mutation. Collectively, this evidence supports the interpretation that this variant is disease-causing. To date, a total of 143 variants have been reported for ALDH7A1, with most being missense/nonsense (n = 82), along with 26 splicing, 14 small deletions, seven small insertions, two small indels, 21 gross deletions, and one gross insertion. Fang et al. (26) explored the genotype–phenotype associations concerning variant site, type, and zygosity. The findings showed that patients with homozygous L455P mutations were more likely to experience clonic seizures, while those with homozygous E427Q mutations had a higher likelihood of developing ventriculomegaly, cysts, and abnormal white matter signals (26). Furthermore, patients with two in-frame deletion variants were more prone to myoclonic seizures, whereas those with homozygous variants were more likely to manifest a severe phenotypic abnormality including early-neonatal seizures, status epilepticus, and epileptic encephalopathy (26).
Identifying mutations in ALDH7A1 through genetic testing confirms the diagnosis of PDE, allowing for the timely initiation of pyridoxine therapy, which is critical for preventing ongoing seizures and associated neurological damage. Moreover, genetic testing helps distinguish PDE from other forms of epilepsy, guides treatment decisions, and provides important information for family planning and genetic counseling. Understanding the role of ALDH7A1 in PDE has been essential for diagnosing and managing this condition, emphasizing the importance of genetic testing in patients with unexplained epilepsy (23).
5 Conclusions
In conclusion, we reported a novel homozygous variant in the ALDH7A1 mutation causing neonatal seizures and developmental delay in two patients from a consanguineous family. We also highlighted the associated developmental delay and intellectual disability despite early treatment and seizure control.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Office of Research Affairs, Research Advisory Council (RAC#2180004) at KFSHRC. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
MS: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. AbA: Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. RA: Formal analysis, Investigation, Writing – review & editing. AH: Investigation, Writing – review & editing. AA: Supervision, Writing – review & editing. NK: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the King Salman Center for Disability Research, Grant No: RAC# 2180004 (NK).
Acknowledgments
We thank the patients and their families for participating in this study. The authors extend their appreciation to the King Salman Center for Disability Research for funding this work (RAC#2180004).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. Ilae classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ilae task force on nosology and definitions. Epilepsia. (2022) 63:1349–97. doi: 10.1111/epi.17239
2. Been JV, Bok LA, Andriessen P, Renier WO. Epidemiology of pyridoxine dependent seizures in the Netherlands. Arch Dis Child. (2005) 90:1293–6. doi: 10.1136/adc.2005.075069
3. Coughlin CR 2nd, Swanson MA, Spector E, Meeks NJL, Kronquist KE, Aslamy M, et al. The genotypic spectrum of aldh7a1 mutations resulting in pyridoxine dependent epilepsy: A common epileptic encephalopathy. J Inherit Metab Dis. (2019) 42:353–61. doi: 10.1002/jimd.12045
4. Coughlin CR 2nd, Tseng LA, Abdenur JE, Ashmore C, Boemer F, Bok LA, et al. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to alpha-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. (2021) 44:178–92. doi: 10.1002/jimd.12332
5. Mastrangelo M, Gasparri V, Bernardi K, Foglietta S, Ramantani G, Pisani F. Epilepsy phenotypes of vitamin B6-dependent diseases: an updated systematic review. Children (Basel). (2023) 10:553. doi: 10.3390/children10030553
6. Salih MM, Kabiraj M, Gascon G, Al Jarallah A, Al-Zamil F. Typical and atypical presentations of pyridoxine-dependent seizures. Saudi Med J. (1995) 16:347–51.
7. Basura GJ, Hagland SP, Wiltse AM, Gospe SM Jr. Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 north american cases submitted to a patient registry. Eur J Pediatr. (2009) 168:697–704. doi: 10.1007/s00431-008-0823-x
8. Bok LA, Halbertsma FJ, Houterman S, Wevers RA, Vreeswijk C, Jakobs C, et al. Long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol. (2012) 54:849–54. doi: 10.1111/j.1469-8749.2012.04347.x
9. Alfadhel M, Sirrs S, Waters PJ, Szeitz A, Struys E, Coulter-Mackie M, et al. Variability of phenotype in two sisters with pyridoxine dependent epilepsy. Can J Neurol Sci. (2012) 39:516–9. doi: 10.1017/s0317167100014050
10. Dixon M, Millington C, Bernstein L, Coughlin CR 2nd, Drumm M, Gaughan S, et al. Dietary management for pyridoxine-dependent epilepsy due to alpha-aminoadipic semialdehyde dehydrogenase deficiency, a follow-on from the international consortium guidelines. JIMD Rep. (2024) 65:188–203. doi: 10.1002/jmd2.12418
11. AlBakheet A, AlQudairy H, Alkhalifah J, Almoaily S, Kaya N, Rahbeeni Z. Detailed genetic and clinical analysis of a novel de novo variant in hprt1: case report of a female patient from Saudi Arabia with lesch-nyhan syndrome. Front Genet. (2022) 13:1044936. doi: 10.3389/fgene.2022.1044936
12. Aldhalaan H, AlBakheet A, AlRuways S, AlMutairi N, AlNakiyah M, AlGhofaili R, et al. A novel gemin4 variant in a consanguineous family leads to neurodevelopmental impairment with severe microcephaly, spastic quadriplegia, epilepsy, and cataracts. Genes (Basel). (2021) 13:92. doi: 10.3390/genes13010092
13. Gospe SM Jr. Pyridoxine-Dependent Epilepsy - Aldh7a1. Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al, editors. Seattle (WA: Genereviews((R (1993).
14. Bok LA, Maurits NM, Willemsen MA, Jakobs C, Teune LK, Poll-The BT, et al. The eeg response to pyridoxine-iv neither identifies nor excludes pyridoxine-dependent epilepsy. Epilepsia. (2010) 51:2406–11. doi: 10.1111/j.1528-1167.2010.02747.x
15. Arntsen V, Jamali A, Sikiric A, Kristensen E, Tangeraas T, Kupliauskiene G, et al. Utility and limitations of eeg in the diagnosis and management of aldh7a1-related pyridoxine-dependent epilepsy. A Retrospective Observational Study. Front Neurol. (2024) 15:1355861. doi: 10.3389/fneur.2024.1355861
16. Kaminiow K, Pajak M, Pajak R, Paprocka J. Pyridoxine-dependent epilepsy and antiquitin deficiency resulting in neonatal-onset refractory seizures. Brain Sci. (2021) 12:65. doi: 10.3390/brainsci12010065
17. Mills PB, Footitt EJ, Mills KA, Tuschl K, Aylett S, Varadkar S, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (Aldh7a1 deficiency). Brain. (2010) 133:2148–59. doi: 10.1093/brain/awq143
18. Friedman SD, Ishak GE, Poliachik SL, Poliakov AV, Otto RK, Shaw DW, et al. Callosal alterations in pyridoxine-dependent epilepsy. Dev Med Child Neurol. (2014) 56:1106–10. doi: 10.1111/dmcn.12511
19. Oesch G, Maga AM, Friedman SD, Poliachik SL, Budech CB, Wright JN, et al. Geometric morphometrics reveal altered corpus callosum shape in pyridoxine-dependent epilepsy. Neurology. (2018) 91:e78–86. doi: 10.1212/WNL.0000000000005748
20. Baxter P. Pyridoxine-dependent and pyridoxine-responsive seizures. Dev Med Child Neurol. (2001) 43:416–20. doi: 10.1017/s0012162201000779
21. van Karnebeek CD, Tiebout SA, Niermeijer J, Poll-The BT, Ghani A, Coughlin CR 2nd, et al. Pyridoxine-dependent epilepsy: an expanding clinical spectrum. Pediatr Neurol. (2016) 59:6–12. doi: 10.1016/j.pediatrneurol.2015.12.013
22. Coughlin CR, Gospe SM Jr. Pyridoxine-dependent epilepsy: current perspectives and questions for future research. Ann Child Neurol Soc. (2023) 1:24–37. doi: 10.1002/cns3.20016
23. Falsaperla R, Sciacca M, Collotta AD, Tardino LG, Marino S, Marino SD, et al. Pyridoxine-dependent epilepsy (Pde): an observational study of neonatal cases on the role of pyridoxine in patients treated with standard anti-seizure medications. Seizure. (2024) 118:156–63. doi: 10.1016/j.seizure.2024.04.012
24. Scalia B, Falsaperla R. Early pyridoxine administration to avoid late diagnosis of pyridoxine-dependent epilepsy; comment on the ilae guidelines proposal on the treatment of seizures in the neonate. J Pediatr Epilepsy. (2024) 13:047. doi: 10.1055/s-0043-1778010
25. Coughlin CR, Tseng LA, Bok LA, Hartmann H, Footitt E, Striano P, et al. Association between lysine reduction therapies and cognitive outcomes in patients with pyridoxine-dependent epilepsy. Neurology. (2022) 99:e2627–e36. doi: 10.1212/WNL.0000000000201222
Keywords: ALDH7A1, novel variant, pyridoxine-dependent epilepsy, intellectual disability, neonatal seizures
Citation: Salih MA, AlBakheet A, Almass R, Hamed AAA, AlOdaib A and Kaya N (2024) Case report: Clinical and genetic characterization of a novel ALDH7A1 variant causing pyridoxine-dependent epilepsy, developmental delay, and intellectual disability in two siblings. Front. Psychiatry 15:1501238. doi: 10.3389/fpsyt.2024.1501238
Received: 24 September 2024; Accepted: 27 November 2024;
Published: 23 December 2024.
Edited by:
Curtis R. Coughlin II, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Monica Gagliardi, University of Magna Graecia, ItalyBrahim Tabarki Melaiki, University of Sousse, Tunisia
Copyright © 2024 Salih, AlBakheet, Almass, Hamed, AlOdaib and Kaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Namik Kaya, bmtheWFAa2ZzaHJjLmVkdS5zYQ==