 Daniel W. Sirkis1*
Daniel W. Sirkis1* Alexis P. Oddi1
Alexis P. Oddi1 Caroline Jonson1,2,3
Caroline Jonson1,2,3 Luke W. Bonham1,4
Luke W. Bonham1,4 Phuong T. Hoang5
Phuong T. Hoang5 Jennifer S. Yokoyama1,4,6*
Jennifer S. Yokoyama1,4,6*- 1Memory and Aging Center, Department of Neurology, Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, CA, United States
- 2Center for Alzheimer’s and Related Dementias, National Institutes of Health, Bethesda, MD, United States
- 3DataTecnica LLC, Washington, DC, United States
- 4Department of Radiology and Biomedical Imaging, University of California, San Francisco, San Francisco, CA, United States
- 5Movement Disorders and Neuromodulation Center, Department of Neurology, Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, CA, United States
- 6Global Brain Health Institute, University of California, San Francisco, San Francisco, CA, United States
Recent advances in transcriptomics research have uncovered heightened interferon (IFN) responses in neurodegenerative diseases including Alzheimer’s disease, primary tauopathy, Parkinson’s disease, TDP-43 proteinopathy, and related mouse models. Augmented IFN signaling is now relatively well established for microglia in these contexts, but emerging work has highlighted a novel role for IFN-responsive T cells in the brain and peripheral blood in some types of neurodegeneration. These findings complement a body of literature implicating dysregulated IFN signaling in neuropsychiatric disorders including major depression and post-traumatic stress disorder. In this review, we will characterize and integrate advances in our understanding of IFN responses in neurodegenerative and neuropsychiatric disease, discuss how sex and ancestry modulate the IFN response, and examine potential mechanistic explanations for the upregulation of antiviral-like IFN signaling pathways in these seemingly non-viral neurological and psychiatric disorders.
Introduction
The last five years have witnessed impressive growth in the number of publications dissecting the role of interferon (IFN) signaling in neurodegenerative disease. Although the earliest explorations of IFN production in Alzheimer’s disease (AD) occurred more than 40 years ago (1), in this review we will focus primarily on research conducted in the past decade, using the pioneering work of Roy and colleagues (2) as a starting point. We will summarize recent advances in our understanding of the role of dysregulated IFN signaling in AD, primary tauopathy, TAR DNA-binding protein 43 (TDP-43) proteinopathy including cases associated with C9orf72 hexanucleotide repeat expansion (HRE), and Parkinson’s disease (PD). We will also explore evidence for augmented IFN responses in several neuropsychiatric disorders and discuss whether IFN signaling is likely to play a role in cognitive dysfunction in either neurodegenerative or neuropsychiatric disease. Finally, we will highlight important yet understudied biological modulators of the IFN response, including sex and ancestry. We will conclude by highlighting the most important open questions facing the field, such as which IFN-responsive cell types (e.g., microglia, T cells) are most likely to contribute to pathogenesis and whether targeting IFN signaling represents a promising avenue for modifying pathobiology in neurodegenerative or neuropsychiatric disease (see accompanying Figure 1, Table 1 for a summary of topics discussed and callouts to references of special importance).
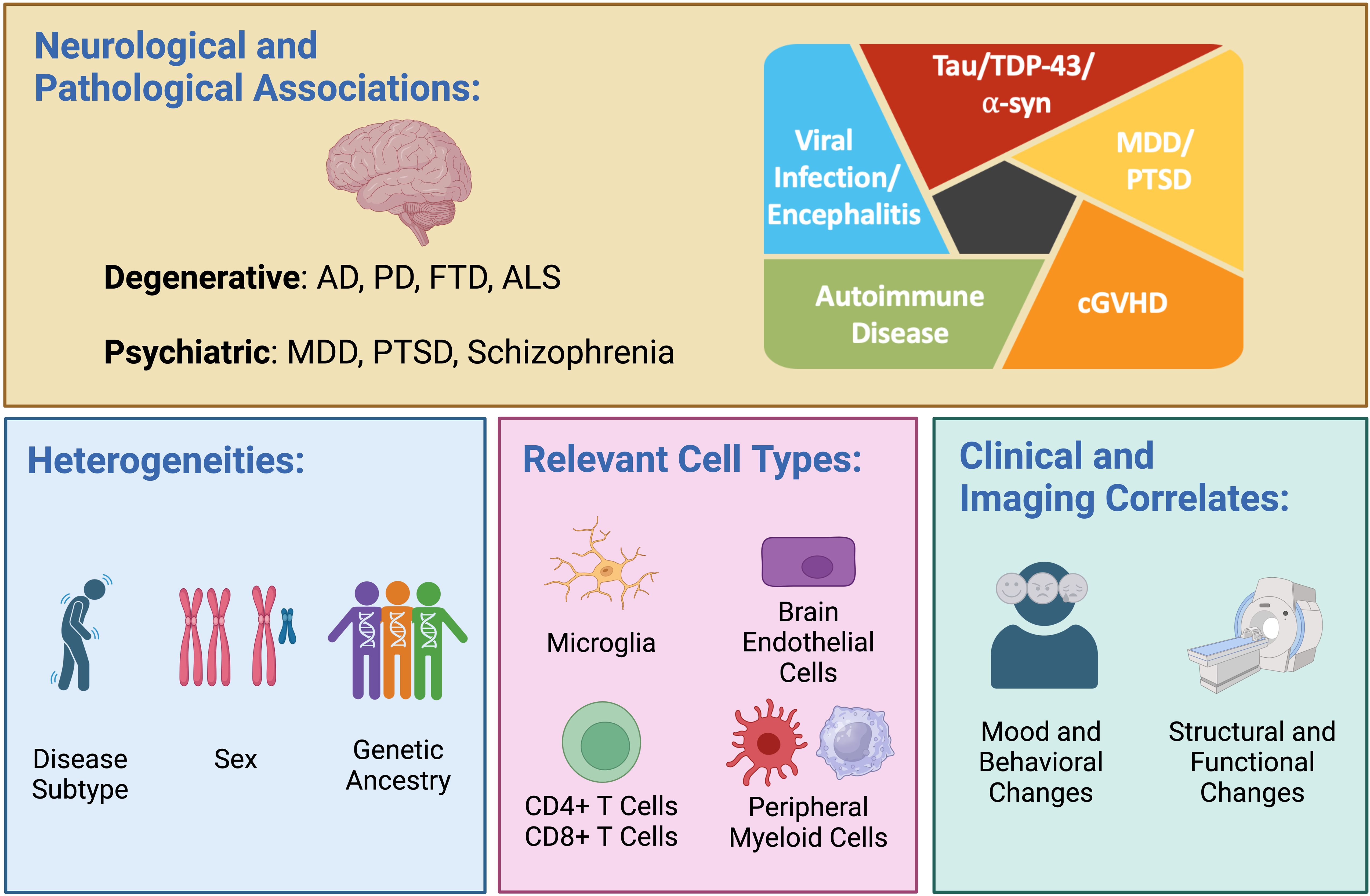
Figure 1. Dysregulated interferon (IFN) responses are observed in a variety of neurodegenerative diseases—including Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS)—and across multiple proteinopathies, including those driven by tau, TDP-43 and α-synuclein (α-syn). Altered IFN signaling is also observed in psychiatric diseases, including major depressive disorder (MDD), post-traumatic stress disorder (PTSD), and schizophrenia (upper panel). A heightened IFN response manifesting as expansion of highly IFN-responsive T cells has been observed in early-onset AD, viral encephalitis, autoimmune disease, and chronic graft versus host disease (cGVHD; upper panel, right). Important biological variables that may modulate the IFN response include disease subtype, sex, and genetic ancestry (lower panel, left). Relevant cell types mediating dysregulated IFN signaling in disease include brain-resident microglia, brain endothelial cells, CD4 and CD8 T cells, and peripheral myeloid cells (lower panel, middle). The use of recombinant type I IFN is associated with cognitive and behavioral changes as well as structural and functional neuroimaging changes (lower panel, right). Created in BioRender. Yokoyama, J. (2024) BioRender.com/n80o218.
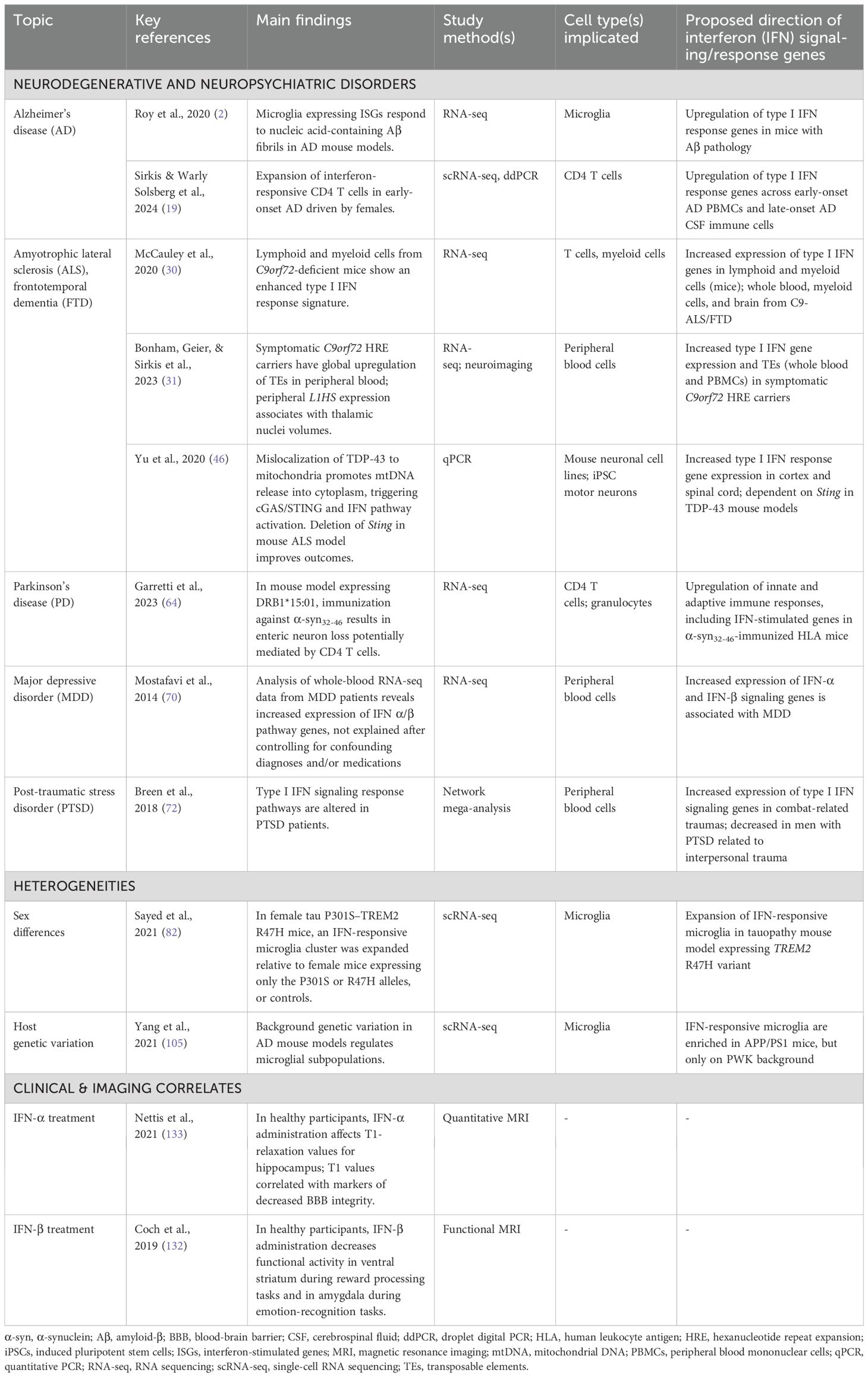
Table 1. Key studies implicating altered interferon responses in neurodegenerative diseases and neuropsychiatric disorders.
Interferon responses in tauopathy: microglia and beyond
A role for microglial type I IFN signaling (occurring via the IFN-α/β receptor) in AD, primary tauopathy, and associated mouse models is now relatively well established (2–5). Activation of this pathway may be regulated in part by signaling through the microglial receptor Trem2, as loss of Trem2 in a combined mutant P301L tau–PS2APP mouse model increases the likelihood of microglial polarization toward an IFN-responsive state (6). On the other hand, work involving another model of tauopathy (P301S) suggests that mutant tau-mediated expansion of IFN-responsive microglia may not depend on Trem2 signaling in all contexts (7). Additionally, induced pluripotent stem cell (iPSC) models of trisomy 21, which is a potent risk factor for early-onset AD due to triplicated APP (8, 9), have provided independent confirmation of a role for a microglial type I IFN response to pathologic forms of tau (10). Given that the IFN-responsive microglial state coincides with synapse loss and neurodegeneration (2, 6), and that pharmacologic and genetic inhibition of type I IFN signaling ameliorates synaptic and memory deficits in the 5XFAD model (2, 11), this subpopulation of microglia most likely contributes to pathogenesis. Interestingly, AD-related IFN responses in brain-resident cells may not be limited to microglia—brain endothelial cells in APOE4 carriers with AD display a heightened IFN-response signature (12), and complementary findings have been observed in mouse models of amyloidosis (13). Beyond studies of mouse models and post-mortem AD brain tissue, recent integrative analyses of human genetic data and microglial transcriptomic data have suggested that variation in OAS1, an established IFN response gene, may modify risk for AD (14, 15).
Interferon-responsive T cells in neurodegeneration and beyond
A unique population of highly IFN-responsive human CD4 T cells has recently been described (16, 17). A closely related population of antiviral CD4 T cells is markedly expanded in the cerebrospinal fluid (CSF) in viral encephalitis (18), and, strikingly, we have recently observed significant expansion of a very similar population of highly IFN-responsive CD4 T cells in the blood of patients with sporadic, early-onset AD (19). Excitingly, diverse physiological and pathological roles for this subpopulation continue to be suggested, as recent large-scale single-cell transcriptomic analyses have shown that IFN-responsive CD4 T cells are expanded in several autoimmune diseases including systemic lupus erythematosus (SLE) and primary Sjögren syndrome (20). Intriguingly, unpublished work also suggests that IFN-responsive CD4 T cells are expanded in patients who received hematopoietic stem cell transplants that went on to develop chronic graft versus host disease (21), suggesting these cells may have pathogenic properties. Given their relatively recent description, this population has been variably referred to as IFN-responsive T cells (16, 19), IFN signaling-associated gene (ISAG)hi T cells (17, 19), antiviral CD4 T cells (18), IFN-driven inflammatory CD4 naive T cells (21), and Tnaive MX1 (20). This population appears to be at least partly naive (i.e., antigen inexperienced) by gene expression profiling (20, 21), raising the intriguing possibility that human IFN-responsive CD4 T cells may promote pathogenic responses in an innate-like manner, as has been shown to occur for subsets of naive CD8 T cells (22). Although it currently remains unclear if mice possess an equivalent population of highly IFN-driven CD4 T cells, several papers have found evidence for expansion of activated, IFN-responsive CD8 T cells in the brain in several AD models, including the tau P301S-APOE4 (“TE4”) knock-in model (5) and the 5XFAD model (23). Future work in this area is required to characterize the functional role and unique properties of these IFN-responsive T cells in neurodegenerative disease.
Interferon responses in C9orf72 expansion carriers
The most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) is a hexanucleotide repeat expansion (HRE) intronic to C9orf72 (C9-ALS/FTD). Autosomal dominant inheritance of over 30 repeats is considered pathogenic and is associated with accumulation of repeat-containing RNA, dipeptide repeat (DPR) proteins, C9orf72 haploinsufficiency, and the development of TDP-43 neuropathology (24). Although not considered pathogenic, individuals possessing an intermediate number of hexanucleotide repeats (9–30) are thought to have an increased prevalence of systemic autoimmune conditions, including SLE and rheumatoid arthritis (RA) (25). Indeed, autoimmune diseases are more broadly enriched in patients with FTD and ALS, particularly in those with underlying TDP-43 neuropathology (26, 27). Dysregulated type I IFN signaling, in turn, has long been implicated in autoimmune conditions, including SLE, Sjögren syndrome, and a set of Mendelian disorders named “type I interferonopathies” (28). As will be described below, these findings collectively suggest that type I IFN signaling may also be dysregulated in FTD and ALS.
C9orf72 and interferon signaling
Despite the observed relationship between immune dysregulation and C9orf72 HREs (29), the association between C9orf72 and IFN signaling has only recently been investigated. RNA-seq analyses of T cells from C9orf72 knockout mice have revealed upregulation of the type I IFN response pathway, and overlapping IFN-associated pathways were also found to be increased in monocyte-derived macrophages (MDMs) derived from C9-ALS patients (30). Additionally, our group has confirmed the presence of heightened type I IFN-related gene expression in peripheral blood cells in two independent cohorts of symptomatic C9-ALS/FTD patients, compared to non-carrier controls (31). Furthermore, a 2021 study found evidence that brains donated by C9-ALS/FTD patients have increased neuronal levels of cytoplasmic double-stranded RNA (cdsRNA), and in vitro experiments showed that transfection of repeat-containing dsRNA led to induction of IFN-stimulated genes (ISGs). Finally, the presence of cdsRNA in mouse neurons was associated with type I IFN signaling and cell death in vivo (32).
C9orf72 and STING pathway activation
Recent studies have investigated the molecular mechanisms connecting C9orf72 and IFN signaling via the stimulator of IFN genes (STING) pathway. Although a detailed description of the STING pathway is beyond the scope of this article, excellent reviews are available (33). When exposed to a STING inhibitor, both C9-ALS patient peripheral blood mononuclear cells (PBMCs) and MDMs exhibit a suppression of elevated ISG expression (30). Partial C9orf72 deficiency due to C9orf72 HRE may thus lead to an overactive type I IFN response via upstream activation of the STING pathway. Consistent with this possibility, an independent study recently reported elevated STING protein levels in a C9orf72-deficient macrophage cell line and in C9orf72-deficient spleen (34).
C9orf72, DNA damage, and transposable elements
C9orf72 haploinsufficiency is thought to synergize with DPR proteins in the induction of DNA damage and/or inhibition of efficient repair mechanisms (35–37). Notably, damaged nuclear DNA is an activator of the non-canonical STING pathway (38), and DNA damage is more broadly a hallmark of several neurodegenerative diseases, including C9-ALS/FTD (39). A recent study observed increased DNA damage and neuronal STING pathway activation in human post-mortem tissue and several models of C9orf72 HRE-associated toxicity (40). Taken together, studies have identified that C9-ALS/FTD patients may possess an altered immunophenotype due to both C9orf72 haploinsufficiency and HRE-associated toxicity that is associated with heightened DNA damage and subsequent activation of the STING pathway.
Beyond frank DNA damage, IFN signaling can also be triggered by the de-repression of transposable elements (TEs), including long interspersed elements (LINEs) that cause the production of cytosolic LINE-1 cDNA (41). In symptomatic C9orf72 HRE carriers, we observed a significant increase in the peripheral expression of human LINE-1 element L1HS (31). Combined with the finding that several IFN-associated signaling genes were highly upregulated in the same cohort, the dysregulation of L1HS in these patients suggests that TE de-repression is another potential mechanism for the enhanced innate immune response observed in C9orf72 HRE carriers.
Interferon responses in TDP-43 proteinopathy
Enhanced type I IFN signaling has also been observed in models of TDP-43 proteinopathy independent of C9orf72-related pathobiology. Physiologically, TDP-43 is involved in several facets of RNA regulation (42, 43). In disease states, TDP-43 can undergo mis-localization and post-translational modifications that promote its misfolding and abnormal aggregation in the cytoplasm (44). Depletion of TDP-43 has been implicated in regulating the accumulation of double-stranded RNA (dsRNA), promoting heightened IFN signaling (45). Additionally, recent in vitro experiments have shown that mislocalization of TDP-43 to mitochondria is associated with increased levels of mitochondrial reactive oxygen species (mtROS), a hallmark of mitochondrial stress (46, 47). Excessive production of mtROS in conjunction with increased mitochondrial permeability is thought to promote the errant release of mitochondrial DNA (mtDNA) into the cytoplasm (47). Cytoplasmic mtDNA, in turn, is known to precipitate the cGAS-STING cascade, initiating the activation of type I interferon signaling (46–48).
Corroborating the in vitro work described above, increased levels of cytosolic mtDNA have also been detected in cells derived from the spinal cord and cortex in the TARDBP p.A315T mouse model of TDP-43 proteinopathy. Strikingly, when the mutant TDP-43 mice were crossed with a Sting-deficient model, disease progression was slowed by nearly 60% and median lifespan was extended by 40% (46). Finally, direct STING inhibition increased the viability of TDP-43-mutant, iPSC-derived motor neurons; in vivo, Sting inhibition in the TDP-43 A315T mouse model rescued neuron loss and improved performance on a motor coordination test (46). In summary, given the observed connection between aberrant, mitochondria-localized TDP-43, mtDNA release, and STING activation, further research into STING inhibition as a potential treatment for patients affected by TDP-43 neuropathology is warranted.
Interferon responses in Parkinson’s disease
Dysregulation of IFN signaling pathways is also implicated in the development and progression of Parkinson’s disease (PD). The underlying pathology of PD involves the accumulation of aggregated α-synuclein (α-syn) protein in vulnerable dopaminergic neurons in the substantia nigra (SN), leading to their degeneration. Recent studies suggest that α-syn dysregulation leads to activation of microglia and peripheral T cells—especially CD4 T cells—generating an inflammatory cascade, including a heightened IFN response, that may contribute to neurodegeneration in PD (49–51).
Viral infections are a well-known cause of parkinsonism, with the most famous example the postencephalitic parkinsonism associated with “encephalitis lethargica,” which was, in turn, potentially associated with the 1918 influenza pandemic (52). Certain viruses (e.g., influenza, HIV, West Nile virus) have been linked to increased risk of idiopathic PD, with in vitro and in vivo studies showing increased expression, decreased degradation, and altered post-translational modification and aggregation of α-syn, including in dopaminergic and dopaminergic-like neurons (53–56). Furthermore, α-syn has been shown to be required for effective immune response against viral pathogens. For example, both α-syn-deficient mice and human stem cell-derived dopaminergic neurons infected with neurotropic RNA viruses have higher viral loads, with increased mortality in α-syn-null mice (56, 57). Notably, using α-syn knockout human dopaminergic neurons, Monogue and colleagues showed that α-syn is required for the IFN response and effective control of viral infections, potentially through direct α-syn interaction with phosphorylated STAT2 to activate ISGs (56).
Both type I and II IFN responses have been linked to PD, although the results are mixed. Reduced type I IFN signaling mitigates neuronal death in vitro after treatment with rotenone, and dopamine neuron death in mice exposed to MPTP, two neurotoxins linked to PD risk (58, 59). On the other hand, mice lacking IFN-β function are reported to have increased α-syn aggregation and dopaminergic degeneration, as well as motor and cognitive deficits akin to human PD symptoms (60). Meanwhile, PD patients have elevated IFN-γ levels in the SN and striatum, as well as peripherally in plasma and CD4 T cells; IFN-γ is also secreted by subsets of PD patient CD4 T cells recognizing α-syn antigenic peptides (50, 61, 62). Overexpression of IFN-γ causes dopaminergic neuron death and nigrostriatal tract degeneration, while midbrain neuron–microglia co-cultures treated with IFN-γ neutralizing antibody show reduced microglia-mediated neuron loss when exposed to rotenone (61, 63). Altogether, these and other studies suggest that dysregulation of either type I or II IFN signaling may be a risk factor for PD.
Finally, a recent study indicates that an autoimmune response to α-syn in the gut, potentially mediated via a heightened IFN response, may directly contribute to PD symptoms (64). Circulating CD4 T cells from PD patients recognize specific α-syn epitopes, with strong affinity to α-syn32-46 peptide, which is highly restricted to certain human leukocyte antigen (HLA) alleles, including the PD-associated DRB1*15:01 allele. Using a mouse strain expressing DRB1*15:01, Garretti et al. showed that immunization against α-syn32-46 resulted in intestinal inflammation, enteric dopaminergic neuron degeneration, as well as constipation and weight loss. These changes could be partially reversed by depletion of CD4, but not CD8, T cells. Subsequent differential gene expression analysis showed upregulation of innate and adaptive immune responses, including IFN-stimulated genes (Stat1/2, Oas2, Irf7, Isg15). These results suggest that a dysregulated IFN response, acting in concert with CD4 T cells recognizing specific α-syn epitopes, might underlie both PD pathogenesis as well as clinical presentation.
Interferon responses in neuropsychiatric disorders
Recent research has begun to elucidate a significant role for IFN responses in various neuropsychiatric disorders, including major depressive disorder (MDD) and post-traumatic stress disorder (PTSD). In the context of MDD, a recent single-nucleus RNA-seq (snRNA-seq) study using samples from dorsolateral prefrontal cortex has provided compelling evidence of altered IFN-γ signaling via identification of a distinct pattern of gene expression changes associated with the IFN-γ response (65). Given the higher prevalence of MDD in women and emerging evidence suggesting distinct, sex-specific molecular mechanisms in depression, this study conducted many of its analyses in a sex-stratified manner. Specifically, the authors found significant negative enrichment in IFN-γ and NF-κB signaling pathways in microglia from female cases. This supports existing literature pointing to reduced microglial activation and enhanced synaptic connectivity in females, with the reverse observed in males (66). Furthermore, alterations in microglial gene expression were more pronounced in females than males, aligning with studies demonstrating that many microglial immune functions are influenced by gonadal hormones or exhibit sex-specific characteristics (67–69). Given the inherent limitations related to the use of post-mortem tissue for snRNA-seq studies, additional research has focused on identifying peripheral blood biomarkers of MDD. For example, a large-scale, whole-blood RNA-seq study of more than 900 individuals, including cases with recurrent MDD and controls of European ancestry, revealed a significant association of MDD with increased expression of genes involved in IFN-α/β signaling, suggesting this pathway may lead to or exacerbate depressive symptoms (70).
PTSD has also been linked to aberrant immune responses, including those mediated by IFNs. A 2015 meta-analysis (71) found that elevated levels of IFN-γ, interleukin 1β, and interleukin 6 are all robust features of PTSD. Additionally, a comprehensive 2017 mega-analysis of transcriptome-wide gene expression studies involving over 500 PTSD patients and controls aimed to clarify molecular heterogeneities related to sex and trauma types (72). This study identified significant alterations in type I IFN response pathways. Notably, IFIT3 emerged as the sole gene consistently differentially expressed across all examined trauma groups (including interpersonal-related traumas in both females and males, and combat-related traumas in males) albeit with inconsistent direction of expression change. In cases of combat-related trauma, another IFN-responsive gene, IFI44L, was consistently upregulated across three different studies. Furthermore, consensus gene co-expression network analyses highlighted a type I IFN signaling cascade module as enriched in all trauma groups. This underscores the importance of IFN-related molecular pathways in PTSD despite variability related to sex and trauma type in expression patterns among individual IFN-responsive genes.
Sex differences in interferon signaling
Several studies have recorded heterogeneities in immune responses between biological males and females, including in the context of neurodegeneration and aging (73–75). Briefly, females more commonly experience enhanced immune cell activation after viral exposure and vaccination (73, 75). Many viruses exhibit sex-based differences in disease outcomes, including SARS-CoV-2 and hepatitis B, with males typically experiencing increased disease severity (76). Despite the apparent female advantage against viral exposure, females in turn experience the highest burden of several autoimmune and immune-mediated neurodegenerative diseases, including SLE (77), primary Sjögren syndrome (78), multiple sclerosis (79), and AD (80). Given that dysregulated type I IFN signaling plays a major role in autoimmune diseases (as described above), it is not surprising that sex differences also associate with differential type I IFN responses. In particular, both the differential expression of genes located on sex chromosomes as well as the production of sex hormones have been shown to alter type I IFN responses (81), but the precise immunological mechanisms underlying sex differences in the prevalence and severity of diseases associated with IFN signaling remain unclear.
Both mouse and human studies have revealed that sex may modulate IFN responses in the context of AD. In a combined tauopathy–TREM2 R47H mouse model, only female mice demonstrated enhanced spatial memory deficits (82), and these deficits occurred amid expansion of an IFN-responsive microglia cluster. In a recent preprint, a similar mouse model was created by combining TREM2 R47H and APOE4 knock-in alleles in mice carrying a tau P301S transgene (83). Remarkably, female mice of this model displayed robust evidence of neurodegeneration, again in conjunction with expansion of an IFN-responsive microglial subpopulation. In a striking convergence with other recent work on tau- and TDP-43-mediated disease (46, 84), the heightened IFN response in these mice appeared to be driven by upstream cGAS–STING pathway activation. Taken together, these recent findings suggest that the effect of variation in TREM2 and APOE, when combined with sex-based IFN- and other immune-mediated signaling differences, may convey unique risk for neurodegeneration in females.
In humans, using single-cell RNA-seq analysis, we discovered that female patients with sporadic early-onset AD were driving a unique peripheral immune signature (19). Specifically, female patients exhibited significantly elevated levels of a population of highly IFN-responsive T cells. We confirmed this sex difference via droplet digital PCR of isolated CD4 T cells, finding that females with early-onset AD had heightened expression of the classical IFN response gene, MX1. In parallel, a meta-analysis of over 1.8 million CD4 T cells found that females have increased levels of a subpopulation of naïve T cells identified by expression of the same marker gene, MX1 (20). The exact mechanism driving sex differences in the IFN response in early-onset AD, however, remains an area of active inquiry. Interestingly, a 2022 preprint found that early-onset AD is associated with a higher prevalence of several autoimmune diseases relative to both late-onset AD and the general population (85). Whether the relative enrichment of autoimmunity in both females and early-onset AD is related to the female-driven expansion of IFN-responsive CD4 T cells in early-onset AD remains to be determined. Further investigation into the influence of sex chromosomes, hormones, and environmental factors on IFN responses in neurodegenerative and neuropsychiatric diseases is clearly warranted.
Ancestry-associated differences in interferon signaling
In addition to sex, evidence also suggests that host genetic factors, including genetic ancestry (GA), may influence IFN signaling. GA describes the paths through which an individual inherited their DNA; in a sample, shared GA is often measured by calculating the genetic similarity between individuals to each other or to a known reference population (86). A 2021 single-cell RNA-seq study found that increasing genetic similarity to a European ancestry reference panel was associated with heightened type I IFN response after infecting PBMCs with influenza virus in vitro (87). Relatedly, a recent study that analyzed the post-mortem brains of self-identified Black Americans (most of whom have admixed GA) found that increasing European GA was associated with enrichment of various immune-related differentially expressed gene sets in the dentate gyrus, dorsolateral prefrontal cortex, and hippocampus. In contrast, in the caudate nucleus, increasing African GA was associated with enrichment of largely distinct immune-related gene ontology terms such as “response to virus” that are typically enriched for IFN-responsive genes. This suggests that the influence of GA on immune functions in the brain is complex and region-specific (88), and supports the notion that GA may influence IFN responsiveness in vivo.
Shared GA is frequently proxied by using self-reported or assumed identities that are tied to socially defined labels, such as race, ethnicity, nationality, or continental geography (89). Between these socially defined groups, multiple neurodegenerative diseases influenced by IFN signaling have disparate rates of prevalence, disease severity, and/or mortality, including AD, ALS, and multiple sclerosis (80, 90, 91). While observed health disparities may be attributed to environmental and cultural factors, including access to health care, genetic risk factors associated with shared ancestry have also been identified (92–94).
The most striking example of an IFN-associated disease with marked health disparities is SLE, which consistently displays increased prevalence and severity in non-White populations, with Black and American Indian/Alaskan Native women being the most affected groups in the United States (77). Even after controlling for socioeconomic status, studies have shown that non-White race persists as a risk factor for SLE prevalence, severity, and outcome, suggesting that there may be underlying genetic factors driving these disparities (95, 96). Furthermore, ISGs associated with SLE have higher expression levels in individuals of self-described East Asian and African American descent, in comparison to Europeans (97–99). While the exact contributions of genetic ancestry to IFN signaling in SLE remain to be elucidated, differential DNA hypomethylation of ISGs between European American and African American women with SLE has been observed. Specifically, hypomethylation of IFI44L was observed only in African American women (100). However, these patterns of DNA hypomethylation may also reflect risk for developing lupus nephritis (101), a severe disease outcome of SLE which European ancestry is thought to be protective against (102).
A major limitation of many studies of SLE has been the usage of race, ethnicity, or geography as a proxy for shared GA. Using socially-defined labels as proxies for shared GA relies on inaccurate assumptions about the genetic homogeneity of these groups (86). In order to more accurately quantify the contributions of population genetics on IFN signaling in SLE and beyond, it will be important to move towards continuous measures of genetic similarity, such as genetic distance (103). Furthermore, as noted in our recent systematic review, non-European ancestry populations have been significantly underrepresented in neurodegenerative disease research, particularly in genetic studies. The intentional inclusion of genetically diverse cohorts will be essential to our understanding of the host and population genetic factors contributing to IFN signaling, not only in autoimmune disease but also in the context of neurodegenerative and psychiatric disease (104).
Similar to human populations, wild-derived mice with distinct genetic backgrounds can be leveraged to explore the impact of genetic background on immune function. Indeed, a single-cell RNA-seq study of four genetically distinct, wild-derived strains of mice found that a cluster of microglia expressing IFN-responsive genes was significantly enriched in APP/PS1 mice, but only on the background of the PWK strain; this effect was not observed in commonly used C57BL/6J mice or other wild-derived strains (105). Taken together, multiple lines of evidence suggest that the expression of IFN- and other immune-related genes is potently influenced by genetic variation.
Interferon responses in aging
Beyond biological sex and ancestry, the process of aging is thought to exert a major effect on the IFN response. This phenomenon, explored recently in several excellent review articles (106, 107), has been studied primarily in model systems (108) but is also likely to play an important contributory role in human diseases of aging such as AD. While some studies have found elevated type I IFN responses associated with aging (e.g., at the choroid plexus (108)), others have found decreased IFN signaling potential (e.g., in peripheral myeloid cells (109)). Given that inappropriate IFN pathway hyperactivation may underlie diseases of premature aging (110), it is reasonable to hypothesize that aging-associated neurodegenerative diseases are also driven, in part, by augmented IFN signaling. How these findings can be reconciled with the known, aging-associated reduction in peripheral innate immune IFN signaling (109, 111) remains an important open question worthy of careful future investigation.
Clinical and neuroimaging features of interferon response
While there has been marked progress in our understanding of how dysregulated IFN signaling contributes to neurologic disease in model systems, there is relatively limited information on its contribution to behavioral symptoms and risk for neurodegenerative disease in human populations. In this section, we provide a focused summary of the clinical and neuroimaging features of IFN response as they relate to neuropsychiatric and neurodegenerative disease.
Clinical features of interferon treatment and response
Our clinical understanding of the behavioral symptoms of IFN-pathway dysregulation stems from two key clinical populations: patients receiving exogenous IFN therapeutically and patients with intrinsic interferonopathies. Interestingly, some of the earliest clinical evidence that IFN dysregulation caused neuropsychiatric symptoms was incidentally discovered during IFN treatment for hepatitis C, hepatitis B, and melanoma (112–114).
Several key features of IFN-related neurotoxicity are shared – depression and fatigue are the most common behavioral symptoms and occur in up to 70% of patients while rarer symptoms such as mania, seizures, and psychosis occur in less than 5% (112, 115). These symptoms have been most thoroughly characterized in the setting of IFN treatment (112), but overlap with behavioral symptoms in multiple diseases including primary immune-mediated diseases such as HIV-associated neurocognitive disorder (HAND) (116, 117) and subsets of SLE (118), neuropsychiatric diseases such as major depression (119) and schizophrenia (120), and neurodegenerative diseases like AD (80). In the case of HAND, type I IFN responses are thought to be important for maintaining virological control, both systemically and in the brain (121, 122), but may also contribute to cognitive dysfunction. Multiple mechanisms have been hypothesized to explain the connections between IFN pathway dysregulation and psychiatric symptoms including increased degradation of serotonin, decreased synthesis of dopamine, and altered glutamatergic signaling (123), all of which are key regulators of mood and behavior. Interestingly, an additional modulator of clinical symptoms has been hypothesized in neurodegenerative diseases – IFN indirectly promotes NMDA receptor activation, a possible driver of neurotoxicity and worsening symptoms (123).
Treatment of the behavioral effects of interferonopathies remains in its infancy with many promising treatment paradigms though few validated treatments. For example, tyrosine kinase inhibitors have successfully treated Aicardi–Goutières syndrome (AGS) (124–126), an inherited type I interferonopathy which presents with immune dysfunction, motor deficits, and developmental delay (127). Whether the findings from inherited syndromes can be extrapolated to IFN-related neuropsychiatric and neurodegenerative diseases is an exciting area for future research.
Neuroimaging features of interferon response
Neuroimaging is an important though underutilized tool in the study of interferonopathies, highlighting promising links between the above-described clinical symptoms and the underlying IFN-mediated pathobiology described elsewhere in this review.
Exogenous IFN administration has been shown to alter brain structure and function in both patient populations and healthy controls. Patients undergoing IFN-α treatment demonstrate decreased functional activity in basal ganglia during reward processing tasks (128), increased amygdala activity during emotion processing tasks (129), and impaired whole-brain connectivity during resting state MRI (130). Furthermore, early imaging evidence indicates that dopamine and glutamate concentrations are altered in basal ganglia and that these changes correlate with functional connectivity and depressive symptoms (128, 131). Remarkably, IFN-β administration in healthy controls also decreases functional activity in the ventral striatum during reward processing tasks and amygdala during emotion-recognition tasks (132). Interestingly, one quantitative MRI study in healthy controls demonstrated that IFN-α administration alters the intrinsic T1-relaxation values (a measure of tissue composition impacted by factors such as protein levels, water content, etc) for hippocampus and that these changes correlated with markers of decreased blood brain barrier integrity (133). Despite the methodological differences, these findings suggest that the effects of administered type I IFN are likely independent of disease status, point to neuroanatomic pathways by which IFN alters behavior, and further highlight the importance of dopamine in the brain’s response to IFN.
Compared to imaging studies of exogenous IFN administration, there are relatively few studies of intrinsic interferonopathies. While uncommon, inherited interferonopathies such as monogenic SLE and AGS most commonly present with intracerebral calcifications and T2 white matter hyperintensities (134, 135). Not surprisingly, imaging findings are variable depending on each syndrome’s underlying genetic mutation (134). For example, AGS patients with SAMHD1 mutations may present with intracranial vasculopathy while patients with ADAR1 mutations present with striatal necrosis (134). There is limited literature directly testing whether IFN levels alter brain structure or function in neurodegenerative disease, but indirect evidence suggests that IFN signaling plays an important role in brain structure in C9-FTD (31). Future studies will be required to determine whether IFN levels are associated directly with neuroanatomic correlates of neurodegenerative disease progression.
Overlapping interferon pathway dysregulation in neurodegenerative and psychiatric disease: the way forward
In summary, we have reviewed key evidence from both basic and translational scientific approaches to show that aberrantly upregulated IFN and downstream IFN signaling not only predispose patients to neurodegenerative diseases but may also modulate risk for psychiatric diseases. Indeed, the links between IFN and both neurodegenerative and neuropsychiatric diseases go far beyond overlapping clinical features and behavioral symptoms. At the molecular level, broad-ranging data from mice, human brain tissue, and even neuroimaging studies suggests that IFN-mediated neurotransmitter dysregulation, particularly dopamine and glutamate dysfunction, is a shared risk factor for psychiatric symptoms in both neurodegenerative and neuropsychiatric diseases (56, 60, 128, 131). Future studies should aim to directly address the question of whether IFN pathway dysregulation in the context of neurodegeneration drives psychiatric symptoms in addition to cognitive decline.
At the cellular level, converging evidence from mouse and human studies suggests that microglia play a key role in IFN pathway dysregulation and that microglial dysfunction associates broadly with risk for neurodegenerative diseases like AD (4, 82, 83) and PD (61) alongside sex-specific effects in psychiatric diseases like MDD (66). Beyond the large body of literature connecting aberrant microglial IFN signaling in neurodegenerative disease, additional evidence points to important roles for endothelial cells (12), the choroid plexus (136), and IFN-responsive CD4 and CD8 T cells (5, 19, 23) in neurodegenerative disease. IFN-responsive CD4 T cells remain poorly understood and are particularly intriguing given their expansion in sporadic early-onset AD (19), viral encephalitis (18), and autoimmune disease (20). That autoimmune disease is found at higher-than-expected prevalence in early-onset AD (85) suggests a mechanistic explanation for dysregulated peripheral IFN signaling in early-onset AD and the potential for shared immune-mediated etiology, although additional study is needed to confirm this possibility.
The primary source(s) of type I IFN for modulating T cell and microglial activity in the context of neurodegenerative and psychiatric disease represents an important yet unresolved question. Plasmacytoid dendritic cells (pDCs) are a major source of type I IFN in the context of viral infection (137), but their role in producing IFN during neurodegeneration remains unclear. Intriguing recent work suggests that a pathogenic variant in TLR9 may represent a novel, autosomal-dominant cause of early-onset AD (138). Interestingly, TLR9, while robustly expressed in pDCs, is apparently not expressed in human microglia (138). Strikingly, TLR9 is a major regulator of type I IFN production by pDCs (139). Taken together, these findings suggest that dysregulated production of type I IFN by pDCs may promote risk for early-onset AD. Further research into the potential role of TLR9 signaling in AD, including the identification of additional families with early-onset dementia harboring pathogenic TLR9 mutations, is now needed.
Beyond these connections, many psychiatric symptoms in primary autoimmune diseases such as SLE and Sjögren syndrome are also increasingly attributed to microglial dysfunction (140, 141). At the network level, early evidence has shown broad, IFN-associated dysregulation of genetic networks (3), linked inflammatory cytokines (120), and even functional neuroanatomic networks (130). Most relevant network analyses to date, however, have focused relatively narrowly on a single data modality or disease state. It stands to reason that combined analyses of multiple data modalities (e.g., genetic and functional brain networks) and disease states (e.g., neurodegenerative, primary autoimmune, and psychiatric) will reveal scientifically and clinically relevant information about the shared biology underlying these complex diseases. With large and publicly available multimodal datasets available for these diseases, integrative analyses represent a promising, even vital, area for future research.
There is now well-established evidence for type I IFN pathway dysregulation—generally manifesting as pathologic upregulation of IFN-response genes—in a range of neurodegenerative and neuropsychiatric diseases. In addition, clear evidence shows that recombinant IFN-α/β treatment can promote cognitive and behavioral dysfunction. Given the reasonable hypothesis that aberrant IFN signaling may exacerbate pathology and contribute to cognitive dysfunction and/or decline in these diseases, is it similarly reasonable to postulate that inhibiting excessive IFN signaling would represent an attractive therapeutic strategy? A monoclonal antibody that inhibits signaling through the IFN-α/β receptor has been approved by the United States Food and Drug Administration (FDA) for use in SLE and could represent a promising candidate for early clinical testing in neurodegenerative disease. Caution is warranted, however, given the reported increase in herpes zoster (142, 143) and influenza infections (144, 145) in those being treated with this antibody. Because viral infections may also increase risk for neurodegenerative disease (146), this therapeutic strategy could perversely promote incipient neurodegeneration and cognitive decline. Indeed, a precise homeostatic balance in IFN signaling may be required to enable effective antiviral responses throughout life while, at the same time, not unnecessarily promoting risk for neurodegeneration.
Author contributions
DS: Writing – review & editing, Writing – original draft, Supervision, Investigation, Conceptualization. AO: Writing – review & editing, Writing – original draft, Investigation. CJ: Writing – original draft, Visualization, Investigation. LB: Writing – review & editing, Writing – original draft, Investigation. PH: Writing – original draft, Investigation. JY: Writing – review & editing, Supervision, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. JY receives funding from NIH-NIA R01AG062588, R01AG057234, P30AG062422, P01AG019724, and U19AG079774; NIH-NINDS U54NS123985; NIH-NIDA 75N95022C00031; the Rainwater Charitable Foundation; the Bluefield Project to Cure Frontotemporal Dementia; the Alzheimer’s Association; the Global Brain Health Institute; the French Foundation; and the Mary Oakley Foundation. CJ’s participation in this project was part of a competitive contract awarded to DataTecnica by the National Institutes of Health (NIH) to support open science research. CJ was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (NIA); project number ZO1 AG000534, as well as the National Institute of Neurological Disorders and Stroke (NINDS). LB receives funding from NIH-NIBIB T32 EB001631 and RSNA R&E Foundation RR24–217. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the RSNA R&E Foundation.
Acknowledgments
The figure was created using BioRender.com.
Conflict of interest
Author CJ was employed by DataTecnica LLC; participation in this project was part of a competitive contract awarded to DataTecnica by the National Institutes of Health (NIH) to support open science research. Author JY serves on the scientific advisory board for the Epstein Family Alzheimer’s Research Collaboration.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Miller AE, Neighbour PA, Katzman R, Aronson M, Lipkowitz R. Immunological studies in senile dementia of the Alzheimer type: evidence for enhanced suppressor cell activity. Ann Neurol. (1981) 10:506–10. doi: 10.1002/ana.410100603
2. Roy ER, Wang B, Wan Y-W, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. (2020) 130:1912–30. doi: 10.1172/JCI133737
3. Rexach JE, Polioudakis D, Yin A, Swarup V, Chang TS, Nguyen T, et al. Tau pathology drives dementia risk-associated gene networks toward chronic inflammatory states and immunosuppression. Cell Rep. (2020) 33:108398. doi: 10.1016/j.celrep.2020.108398
4. Prater KE, Green KJ, Mamde S, Sun W, Cochoit A, Smith CL, et al. Human microglia show unique transcriptional changes in Alzheimer’s disease. Nat Aging. (2023) 3:894–907. doi: 10.1038/s43587-023-00424-y
5. Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. (2023) 615:668–77. doi: 10.1038/s41586-023-05788-0
6. Lee S-H, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron. (2021) 109:1283–1301.e6. doi: 10.1016/j.neuron.2021.02.010
7. Gratuze M, Schlachetzki JCM, D’Oliveira Albanus R, Jain N, Novotny B, Brase L, et al. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron. (2023) 111:202–219.e7. doi: 10.1016/j.neuron.2022.10.022
8. Lott IT, Head E. Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol. (2019) 15:135–47. doi: 10.1038/s41582-018-0132-6
9. Fortea J, Zaman SH, Hartley S, Rafii MS, Head E, Carmona-Iragui M. Alzheimer’s disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol. (2021) 20:930–42. doi: 10.1016/S1474-4422(21)00245-3
10. Jin M, Xu R, Wang L, Alam MM, Ma Z, Zhu S, et al. Type-I-interferon signaling drives microglial dysfunction and senescence in human iPSC models of Down syndrome and Alzheimer’s disease. Cell Stem Cell. (2022) 29:1135–1153.e8. doi: 10.1016/j.stem.2022.06.007
11. Roy ER, Chiu G, Li S, Propson NE, Kanchi R, Wang B, et al. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity. (2022) 55:879–894.e6. doi: 10.1016/j.immuni.2022.03.018
12. Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. (2022) 603:885–92. doi: 10.1038/s41586-021-04369-3
13. Jana A, Wang X, Leasure JW, Magana L, Wang L, Kim Y-M, et al. Increased Type I interferon signaling and brain endothelial barrier dysfunction in an experimental model of Alzheimer’s disease. Sci Rep. (2022) 12:16488. doi: 10.1038/s41598-022-20889-y
14. Salih DA, Bayram S, Guelfi S, Reynolds RH, Shoai M, Ryten M, et al. Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun. (2019) 1:fcz022. doi: 10.1093/braincomms/fcz022
15. Magusali N, Graham AC, Piers TM, Panichnantakul P, Yaman U, Shoai M, et al. A genetic link between risk for Alzheimer’s disease and severe COVID-19 outcomes via the OAS1 gene. Brain. (2021) 144:3727–41. doi: 10.1093/brain/awab337
16. Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat Commun. (2019) 10:4706. doi: 10.1038/s41467-019-12464-3
17. Wang X, Shen X, Chen S, Liu H, Hong N, Zhong H, et al. Reinvestigation of classic T cell subsets and identification of novel cell subpopulations by single-cell RNA sequencing. J Immunol. (2022) 208:396–406. doi: 10.4049/jimmunol.2100581
18. Heming M, Li X, Räuber S, Mausberg AK, Börsch A-L, Hartlehnert M, et al. Neurological manifestations of COVID-19 feature T cell exhaustion and dedifferentiated monocytes in cerebrospinal fluid. Immunity. (2021) 54:164–175.e6. doi: 10.1016/j.immuni.2020.12.011
19. Sirkis DW, Warly Solsberg C, Johnson TP, Bonham LW, Oddi AP, Geier EG, et al. Expansion of highly interferon-responsive T cells in early-onset Alzheimer’s disease. Alzheimers Dement. (2024) 20:5062–70. doi: 10.1002/alz.13892
20. Yasumizu Y, Takeuchi D, Morimoto R, Takeshima Y, Okuno T, Kinoshita M, et al. Single-cell transcriptome landscape of circulating CD4 T cell populations in autoimmune diseases. Cell Genom. (2024) 4:100473. doi: 10.1016/j.xgen.2023.100473
21. Albanese A, Chen E, Zavistaski J, Betz K, Suessmuth Y, Cagnin L, et al. Single-cell RNA-seq reveals an interferon-driven inflammatory CD4 naïve T cell subpopulation at day 100 in hematopoietic stem cell transplant patients that ultimately develop chronic GvHD. Blood. (2022) 140:270–1. doi: 10.1182/blood-2022-170214
22. Watson NB, Patel RK, Kean C, Veazey J, Oyesola OO, Laniewski N, et al. The gene regulatory basis of bystander activation in CD8 T cells. Sci Immunol. (2024) 9:eadf8776. doi: 10.1126/sciimmunol.adf8776
23. Fernando N, Gopalakrishnan J, Behensky A, Reich L, Liu C, Bass V, et al. Single-cell multiomic analysis reveals the involvement of Type I interferon-responsive CD8+ T cells in amyloid beta-associated memory loss. bioRxiv. (2023). doi: 10.1101/2023.03.18.533293
24. Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. (2018) 14:544–58. doi: 10.1038/s41582-018-0047-2
25. Fredi M, Cavazzana I, Biasiotto G, Filosto M, Padovani A, Monti E, et al. C9orf72 intermediate alleles in patients with amyotrophic lateral sclerosis, systemic lupus erythematosus, and rheumatoid arthritis. Neuromol Med. (2019) 21:150–9. doi: 10.1007/s12017-019-08528-8
26. Miller ZA, Rankin KP, Graff-Radford NR, Takada LT, Sturm VE, Cleveland CM, et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J Neurol Neurosurg Psychiatry. (2013) 84:956–62. doi: 10.1136/jnnp-2012-304644
27. Miller ZA, Sturm VE, Camsari GB, Karydas A, Yokoyama JS, Grinberg LT, et al. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts: Completing the picture. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e301. doi: 10.1212/NXI.0000000000000301
28. Crow MK, Olferiev M, Kirou KA. Type I interferons in autoimmune disease. Annu Rev Pathol. (2019) 14:369–93. doi: 10.1146/annurev-pathol-020117-043952
29. Masrori P, Beckers J, Gossye H, Van Damme P. The role of inflammation in neurodegeneration: novel insights into the role of the immune system in C9orf72 HRE-mediated ALS/FTD. Mol Neurodegener. (2022) 17:22. doi: 10.1186/s13024-022-00525-z
30. McCauley ME, O’Rourke JG, Yáñez A, Markman JL, Ho R, Wang X, et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. (2020) 585:96–101. doi: 10.1038/s41586-020-2625-x
31. Bonham LW, Geier EG, Sirkis DW, Leong JK, Ramos EM, Wang Q, et al. Radiogenomics of C9orf72 expansion carriers reveals global transposable element derepression and enables prediction of thalamic atrophy and clinical impairment. J Neurosci. (2023) 43:333–45. doi: 10.1523/JNEUROSCI.1448-22.2022
32. Rodriguez S, Sahin A, Schrank BR, Al-Lawati H, Costantino I, Benz E, et al. Genome-encoded cytoplasmic double-stranded RNAs, found in ALS-FTD brain, propagate neuronal loss. Sci Transl Med. (2021) 13. doi: 10.1126/scitranslmed.aaz4699
33. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
34. Pang W, Hu F. C9ORF72 suppresses JAK-STAT mediated inflammation. iScience. (2023) 26:106579. doi: 10.1016/j.isci.2023.106579
35. Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, et al. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. (2016) 92:383–91. doi: 10.1016/j.neuron.2016.09.015
36. Farg MA, Konopka A, Soo KY, Ito D, Atkin JD. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet. (2017) 26:2882–96. doi: 10.1093/hmg/ddx170
37. He L, Liang J, Chen C, Chen J, Shen Y, Sun S, et al. C9orf72 functions in the nucleus to regulate DNA damage repair. Cell Death Differ. (2023) 30:716–30. doi: 10.1038/s41418-022-01074-0
38. Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. (2015) 42:332–43. doi: 10.1016/j.immuni.2015.01.012
39. Konopka A, Atkin JD. DNA damage, defective DNA repair, and neurodegeneration in amyotrophic lateral sclerosis. Front Aging Neurosci. (2022) 14:786420. doi: 10.3389/fnagi.2022.786420
40. Marques C, Held A, Dorfman K, Sung J, Song C, Kavuturu AS, et al. Neuronal STING activation in amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. (2024) 147:56. doi: 10.1007/s00401-024-02688-z
41. De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. (2019) 566:73–8. doi: 10.1038/s41586-018-0784-9
42. Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. (2010) 19:R46–64. doi: 10.1093/hmg/ddq137
43. Mehta PR, Brown A-L, Ward ME, Fratta P. The era of cryptic exons: implications for ALS-FTD. Mol Neurodegener. (2023) 18:16. doi: 10.1186/s13024-023-00608-5
44. Keating SS, San Gil R, Swanson MEV, Scotter EL, Walker AK. TDP-43 pathology: From noxious assembly to therapeutic removal. Prog Neurobiol. (2022) 211:102229. doi: 10.1016/j.pneurobio.2022.102229
45. Dunker W, Ye X, Zhao Y, Liu L, Richardson A, Karijolich J. TDP-43 prevents endogenous RNAs from triggering a lethal RIG-I-dependent interferon response. Cell Rep. (2021) 35:108976. doi: 10.1016/j.celrep.2021.108976
46. Yu C-H, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. (2020) 183:636–649.e18. doi: 10.1016/j.cell.2020.09.020
47. Kim J, Kim H-S, Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. (2023) 55:510–9. doi: 10.1038/s12276-023-00965-7
48. Paul BD, Snyder SH, Bohr VA. Signaling by cGAS-STING in neurodegeneration, neuroinflammation, and aging. Trends Neurosci. (2021) 44:83–96. doi: 10.1016/j.tins.2020.10.008
49. Liscovitch N, French L. Differential co-expression between α-synuclein and IFN-γ Signaling genes across development and in Parkinson’s disease. PloS One. (2014) 9:e115029. doi: 10.1371/journal.pone.0115029
50. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. (2017) 546:656–61. doi: 10.1038/nature22815
51. Lindestam Arlehamn CS, Dhanwani R, Pham J, Kuan R, Frazier A, Rezende Dutra J, et al. [amp]]alpha;-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun. (2020) 11:1875. doi: 10.1038/s41467-020-15626-w
52. Hoffman LA, Vilensky JA. Encephalitis lethargica: 100 years after the epidemic. Brain. (2017) 140:2246–51. doi: 10.1093/brain/awx177
53. Jang H, Boltz D, McClaren J, Pani AK, Smeyne M, Korff A, et al. Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci. (2012) 32:1545–59. doi: 10.1523/JNEUROSCI.5123-11.2012
54. Marreiros R, Müller-Schiffmann A, Trossbach SV, Prikulis I, Hänsch S, Weidtkamp-Peters S, et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc Natl Acad Sci U.S.A. (2020) 117:6741–51. doi: 10.1073/pnas.1906466117
55. Santerre M, Arjona SP, Allen CN, Callen S, Buch S, Sawaya BE. HIV-1 Vpr protein impairs lysosome clearance causing SNCA/alpha-synuclein accumulation in neurons. Autophagy. (2021) 17:1768–82. doi: 10.1080/15548627.2021.1915641
56. Monogue B, Chen Y, Sparks H, Behbehani R, Chai A, Rajic AJ, et al. Alpha-synuclein supports type 1 interferon signalling in neurons and brain tissue. Brain. (2022) 145:3622–36. doi: 10.1093/brain/awac192
57. Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE, et al. Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol. (2015) 90:2767–82. doi: 10.1128/JVI.02949-15
58. Main BS, Zhang M, Brody KM, Ayton S, Frugier T, Steer D, et al. Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson’s disease. Glia. (2016) 64:1590–604. doi: 10.1002/glia.23028
59. Main BS, Zhang M, Brody KM, Kirby FJ, Crack PJ, Taylor JM. Type-I interferons mediate the neuroinflammatory response and neurotoxicity induced by rotenone. J Neurochem. (2017) 141:75–85. doi: 10.1111/jnc.13940
60. Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjørn M, Kuss M, et al. Lack of neuronal IFN-β-IFNAR causes lewy body- and parkinson’s disease-like dementia. Cell. (2015) 163:324–39. doi: 10.1016/j.cell.2015.08.069
61. Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, et al. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J Neurosci. (2007) 27:3328–37. doi: 10.1523/JNEUROSCI.5321-06.2007
62. Kustrimovic N, Comi C, Magistrelli L, Rasini E, Legnaro M, Bombelli R, et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naïve and drug-treated patients. J Neuroinflamm. (2018) 15:205. doi: 10.1186/s12974-018-1248-8
63. Chakrabarty P, Ceballos-Diaz C, Lin W-L, Beccard A, Jansen-West K, McFarland NR, et al. Interferon-γ induces progressive nigrostriatal degeneration and basal ganglia calcification. Nat Neurosci. (2011) 14:694–6. doi: 10.1038/nn.2829
64. Garretti F, Monahan C, Sloan N, Bergen J, Shahriar S, Kim SW, et al. Interaction of an α-synuclein epitope with HLA-DRB115:01 triggers enteric features in mice reminiscent of prodromal Parkinson’s disease. Neuron. (2023) 111:3397–3413.e5. doi: 10.1016/j.neuron.2023.07.015
65. Maitra M, Mitsuhashi H, Rahimian R, Chawla A, Yang J, Fiori LM, et al. Cell type specific transcriptomic differences in depression show similar patterns between males and females but implicate distinct cell types and genes. Nat Commun. (2023) 14:2912. doi: 10.1038/s41467-023-38530-5
66. Seney ML, Glausier J, Sibille E. Large-scale transcriptomics studies provide insight into sex differences in depression. Biol Psychiatry. (2022) 91:14–24. doi: 10.1016/j.biopsych.2020.12.025
67. Hanamsagar R, Bilbo SD. Sex differences in neurodevelopmental and neurodegenerative disorders: Focus on microglial function and neuroinflammation during development. J Steroid Biochem Mol Biol. (2016) 160:127–33. doi: 10.1016/j.jsbmb.2015.09.039
68. Kodama L, Gan L. Do microglial sex differences contribute to sex differences in neurodegenerative diseases? Trends Mol Med. (2019) 25:741–9. doi: 10.1016/j.molmed.2019.05.00125:741–9
69. Doust YV, King AE, Ziebell JM. Implications for microglial sex differences in tau-related neurodegenerative diseases. Neurobiol Aging. (2021) 105:340–8. doi: 10.1016/j.neurobiolaging.2021.03.010
70. Mostafavi S, Battle A, Zhu X, Potash JB, Weissman MM, Shi J, et al. Type I interferon signaling genes in recurrent major depression: increased expression detected by whole-blood RNA sequencing. Mol Psychiatry. (2014) 19:1267–74. doi: 10.1038/mp.2013.161
71. Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. (2015) 2:1002–12. doi: 10.1016/S2215-0366(15)00309-0
72. Breen MS, Tylee DS, Maihofer AX, Neylan TC, Mehta D, Binder EB, et al. PTSD blood transcriptome mega-analysis: shared inflammatory pathways across biological sex and modes of trauma. Neuropsychopharmacology. (2018) 43:469–81. doi: 10.1038/npp.2017.220
73. Wilkinson NM, Chen H-C, Lechner MG, Su MA. Sex differences in immunity. Annu Rev Immunol. (2022) 40:75–94. doi: 10.1146/annurev-immunol-101320-125133
74. Calabrò A, Accardi G, Aiello A, Caruso C, Candore G. Sex and gender affect immune aging. Front Aging. (2023) 4:1272118. doi: 10.3389/fragi.2023.1272118
75. Forsyth KS, Jiwrajka N, Lovell CD, Toothacre NE, Anguera MC. The conneXion between sex and immune responses. Nat Rev Immunol. (2024) 24:487–502. doi: 10.1038/s41577-024-00996-9
76. Jacobsen H, Klein SL. Sex differences in immunity to viral infections. Front Immunol. (2021) 12:720952. doi: 10.3389/fimmu.2021.720952
77. Izmirly PM, Parton H, Wang L, McCune WJ, Lim SS, Drenkard C, et al. Prevalence of systemic lupus erythematosus in the United States: estimates from a meta-analysis of the centers for disease control and prevention national lupus registries. Arthritis Rheumatol. (2021) 73:991–6. doi: 10.1002/art.41632
78. Punnanitinont A, Kramer JM. Sex-specific differences in primary Sjögren’s disease. Front Dent Med. (2023) 4:1168645. doi: 10.3389/fdmed.2023.1168645
79. Briggs FB, Hill E. Estimating the prevalence of multiple sclerosis using 56.6 million electronic health records from the United States. Mult Scler. (2020) 26:1948–52. doi: 10.1177/1352458519864681
80. 2024 Alzheimer’s disease facts and figures. Alzheimers Dement. (2024) 20:3708–821. doi: 10.1002/alz.13809
81. Pujantell M, Altfeld M. Consequences of sex differences in Type I IFN responses for the regulation of antiviral immunity. Front Immunol. (2022) 13:986840. doi: 10.3389/fimmu.2022.986840
82. Sayed FA, Kodama L, Fan L, Carling GK, Udeochu JC, Le D, et al. AD-linked R47H- mutation induces disease-enhancing microglial states via AKT hyperactivation. Sci Transl Med. (2021) 13:eabe3947. doi: 10.1126/scitranslmed.abe3947
83. Carling GK, Fan L, Foxe NR, Norman K, Ye P, Wong MY, et al. Alzheimer’s disease-linked risk alleles elevate microglial cGAS-associated senescence and neurodegeneration in a tauopathy model. bioRxiv. (2024). doi: 10.1101/2024.01.24.577107
84. Udeochu JC, Amin S, Huang Y, Fan L, Torres ERS, Carling GK, et al. Tau activation of microglial cGAS-IFN reduces MEF2C-mediated cognitive resilience. Nat Neurosci. (2023) 26:737–50. doi: 10.1038/s41593-023-01315-6
85. Miller ZA, Ossenkoppele R, Graff-Radford NR, Allen IE, Shwe W, Rosenberg L, et al. Left-handedness, learning disability, autoimmune disease, and seizure history influence age at onset and phenotypical targeting of Alzheimer’s disease. bioRxiv. (2022). doi: 10.1101/2022.12.17.22283307
86. National Academies of Sciences, Engineering, and Medicine, Division of Behavioral and Social Sciences and Education, Health and Medicine Division, Committee on Population, Board on Health Sciences Policy, Committee on the Use of Race, Ethnicity, and Ancestry as Population Descriptors in Genomics Research. Using Population Descriptors in Genetics and Genomics Research: A New Framework for an Evolving Field. Washington (DC: National Academies Press (US (2023).
87. Randolph HE, Fiege JK, Thielen BK, Mickelson CK, Shiratori M, Barroso-Batista J, et al. Genetic ancestry effects on the response to viral infection are pervasive but cell type specific. Science. (2021) 374:1127–33. doi: 10.1126/science.abg0928
88. Benjamin KJM, Chen Q, Eagles NJ, Huuki-Myers LA, Collado-Torres L, Stolz JM, et al. Analysis of gene expression in the postmortem brain of neurotypical Black Americans reveals contributions of genetic ancestry. Nat Neurosci. (2024) 27:1064–74. doi: 10.1038/s41593-024-01636-0
89. Dauda B, Molina SJ, Allen DS, Fuentes A, Ghosh N, Mauro M, et al. Ancestry: How researchers use it and what they mean by it. Front Genet. (2023) 14:1044555. doi: 10.3389/fgene.2023.1044555
90. Roberts AL, Johnson NJ, Chen JT, Cudkowicz ME, Weisskopf MG. Race/ethnicity, socioeconomic status, and ALS mortality in the United States. Neurology. (2016) 87:2300–8. doi: 10.1212/WNL.0000000000003298
91. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult Scler. (2020) 26:1816–21. doi: 10.1177/1352458520970841
92. Costa B, Manzoni C, Bernal-Quiros M, Kia DA, Aguilar M, Alvarez I, et al. C9orf72, age at onset, and ancestry help discriminate behavioral from language variants in FTLD cohorts. Neurology. (2020) 95:e3288–302. doi: 10.1212/WNL.0000000000010914
93. Beydon M, Seror R, Le Guern V, Chretien P, Mariette X, Nocturne G. Impact of patient ancestry on heterogeneity of Sjögren’s disease. RMD Open. (2023) 9. doi: 10.1136/rmdopen-2022-002955
94. Barrie W, Yang Y, Irving-Pease EK, Attfield KE, Scorrano G, Jensen LT, et al. Elevated genetic risk for multiple sclerosis emerged in steppe pastoralist populations. Nature. (2024) 625:321–8. doi: 10.1038/s41586-023-06618-z
95. Lewis MJ, Jawad AS. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology. (2017) 56:i67–77. doi: 10.1093/rheumatology/kew399
96. Barber MRW, Drenkard C, Falasinnu T, Hoi A, Mak A, Kow NY, et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol. (2021) 17:515–32. doi: 10.1038/s41584-021-00668-1
97. Catalina MD, Bachali P, Yeo AE, Geraci NS, Petri MA, Grammer AC, et al. Patient ancestry significantly contributes to molecular heterogeneity of systemic lupus erythematosus. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.140380
98. Siddiqi KZ, Wilhelm TR, Ulff-Møller CJ, Jacobsen S. Cluster of highly expressed interferon-stimulated genes associate more with African ancestry than disease activity in patients with systemic lupus erythematosus. A systematic review of cross-sectional studies. Transl Res. (2021) 238:63–75. doi: 10.1016/j.trsl.2021.07.006
99. Rector I, Owen KA, Bachali P, Hubbard E, Yazdany J, Dall’era M, et al. Differential regulation of the interferon response in systemic lupus erythematosus distinguishes patients of Asian ancestry. RMD Open. (2023) 9. doi: 10.1136/rmdopen-2023-003475
100. Joseph S, George NI, Green-Knox B, Treadwell EL, Word B, Yim S, et al. Epigenome-wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon-related genes based on ethnicity and SLEDAI. J Autoimmun. (2019) 96:147–57. doi: 10.1016/j.jaut.2018.09.007
101. Allen PC, Roberts K, Rubio JE, Tiwari HK, Absher DM, Cooper SJ, et al. Genome-wide DNA methylation analysis implicates enrichment of interferon pathway in African American patients with Systemic Lupus Erythematosus and European Americans with lupus nephritis. J Autoimmun. (2023) 139:103089. doi: 10.1016/j.jaut.2023.103089
102. Lanata CM, Nititham J, Taylor KE, Chung SA, Torgerson DG, Seldin MF, et al. Genetic contributions to lupus nephritis in a multi-ethnic cohort of systemic lupus erythematous patients. PloS One. (2018) 13:e0199003. doi: 10.1371/journal.pone.0199003
103. Ding Y, Hou K, Xu Z, Pimplaskar A, Petter E, Boulier K, et al. Polygenic scoring accuracy varies across the genetic ancestry continuum. Nature. (2023) 618:774–81. doi: 10.1038/s41586-023-06079-4
104. Jonson C, Levine KS, Lake J, Hertslet L, Jones L, Patel D, et al. Assessing the lack of diversity in genetics research across neurodegenerative diseases: A systematic review of the GWAS Catalog and literature. Alzheimers Dement. (2024) 20:5740–56. doi: 10.1002/alz.13873
105. Yang HS, Onos KD, Choi K, Keezer KJ, Skelly DA, Carter GW, et al. Natural genetic variation determines microglia heterogeneity in wild-derived mouse models of Alzheimer’s disease. Cell Rep. (2021) 34:108739. doi: 10.1016/j.celrep.2021.108739
106. Cao W. IFN-aging: Coupling aging with interferon response. Front Aging. (2022) 3:870489. doi: 10.3389/fragi.2022.870489
107. Cao W. In sickness and in health—Type I interferon and the brain. Front Aging Neurosci. (2024) 16:1403142. doi: 10.3389/fnagi.2024.1403142
108. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. (2014) 346:89–93. doi: 10.1126/science.1252945
109. Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. (2016) 352:463–6. doi: 10.1126/science.aaf3926
110. Kreienkamp R, Graziano S, Coll-Bonfill N, Bedia-Diaz G, Cybulla E, Vindigni A, et al. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin. Cell Rep. (2018) 22:2006–15. doi: 10.1016/j.celrep.2018.01.090
111. Molony RD, Nguyen JT, Kong Y, Montgomery RR, Shaw AC, Iwasaki A. Aging impairs both primary and secondary RIG-I signaling for interferon induction in human monocytes. Sci Signal. (2017) 10. doi: 10.1126/scisignal.aan2392
112. Trask PC, Esper P, Riba M, Redman B. Psychiatric side effects of interferon therapy: prevalence, proposed mechanisms, and future directions. J Clin Oncol. (2000) 18:2316–26. doi: 10.1200/JCO.2000.18.11.2316
113. Hosoda S, Takimura H, Shibayama M, Kanamura H, Ikeda K, Kumada H. Psychiatric symptoms related to interferon therapy for chronic hepatitis C: clinical features and prognosis. Psychiatry Clin Neurosci. (2000) 54:565–72. doi: 10.1046/j.1440-1819.2000.00754.x
114. Kraus MR, Schäfer A, Faller H, Csef H, Scheurlen M. Psychiatric symptoms in patients with chronic hepatitis C receiving interferon alfa-2b therapy. J Clin Psychiatry. (2003) 64:708–14. doi: 10.4088/JCP.v64n0614
115. Viengkhou B, Hofer MJ. Breaking down the cellular responses to type I interferon neurotoxicity in the brain. Front Immunol. (2023) 14:1110593. doi: 10.3389/fimmu.2023.1110593
116. Schrier RD, Hong S, Crescini M, Ellis R, Pérez-Santiago J, Spina C, et al. Cerebrospinal fluid (CSF) CD8+ T-cells that express interferon-gamma contribute to HIV associated neurocognitive disorders (HAND). PloS One. (2015) 10:e0116526. doi: 10.1371/journal.pone.0116526
117. Anderson AM, Lennox JL, Mulligan MM, Loring DW, Zetterberg H, Blennow K, et al. Cerebrospinal fluid interferon alpha levels correlate with neurocognitive impairment in ambulatory HIV-Infected individuals. J Neurovirol. (2017) 23:106–12. doi: 10.1007/s13365-016-0466-z
118. Tanaka Y, Kusuda M, Yamaguchi Y. Interferons and systemic lupus erythematosus: Pathogenesis, clinical features, and treatments in interferon-driven disease. Mod Rheumatol. (2023) 33:857–67. doi: 10.1093/mr/roac140
119. Himmerich H, Patsalos O, Lichtblau N, Ibrahim MAA, Dalton B. Cytokine research in depression: principles, challenges, and open questions. Front Psychiatry. (2019) 10:30. doi: 10.3389/fpsyt.2019.00030
120. Halstead S, Siskind D, Amft M, Wagner E, Yakimov V, Shih-Jung Liu Z, et al. Alteration patterns of peripheral concentrations of cytokines and associated inflammatory proteins in acute and chronic stages of schizophrenia: a systematic review and network meta-analysis. Lancet Psychiatry. (2023) 10:260–71. doi: 10.1016/S2215-0366(23)00025-1
121. He H, Sharer LR, Chao W, Gu C-J, Borjabad A, Hadas E, et al. Enhanced human immunodeficiency virus type 1 expression and neuropathogenesis in knockout mice lacking type I interferon responses. J Neuropathol Exp Neurol. (2014) 73:59–71. doi: 10.1097/NEN.0000000000000026
122. Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder — pathogenesis and prospects for treatment. Nat Rev Neurol. (2016) 12:234–48. doi: 10.1038/nrneurol.2016.27
123. Lai JY, Ho JX, Kow ASF, Liang G, Tham CL, Ho Y-C, et al. Interferon therapy and its association with depressive disorders - A review. Front Immunol. (2023) 14:1048592. doi: 10.3389/fimmu.2023.1048592
124. Vanderver A, Adang L, Gavazzi F, McDonald K, Helman G, Frank DB, et al. Janus kinase inhibition in the aicardi-goutières syndrome. N Engl J Med. (2020) 383:986–9. doi: 10.1056/NEJMc2001362
125. Kataoka S, Kawashima N, Okuno Y, Muramatsu H, Miwata S, Narita K, et al. Successful treatment of a novel type I interferonopathy due to a de novo PSMB9 gene mutation with a Janus kinase inhibitor. J Allergy Clin Immunol. (2021) 148:639–44. doi: 10.1016/j.jaci.2021.03.010
126. Cattalini M, Galli J, Zunica F, Ferraro RM, Carpanelli M, Orcesi S, et al. Case report: the JAK-inhibitor ruxolitinib use in aicardi-goutieres syndrome due to mutation. Front Pediatr. (2021) 9:725868. doi: 10.3389/fped.2021.725868
127. Dell’Isola GB, Dini G, Culpepper KL, Portwood KE, Ferrara P, Di Cara G, et al. Clinical spectrum and currently available treatment of type I interferonopathy Aicardi-Goutières syndrome. World J Pediatr. (2023) 19:635–43. doi: 10.1007/s12519-022-00679-2
128. Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ, et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry. (2012) 69:1044–53. doi: 10.1001/archgenpsychiatry.2011.2094
129. Davies KA, Cooper E, Voon V, Tibble J, Cercignani M, Harrison NA. Interferon and anti-TNF therapies differentially modulate amygdala reactivity which predicts associated bidirectional changes in depressive symptoms. Mol Psychiatry. (2021) 26:5150–60. doi: 10.1038/s41380-020-0790-9
130. Dipasquale O, Cooper EA, Tibble J, Voon V, Baglio F, Baselli G, et al. Interferon-α acutely impairs whole-brain functional connectivity network architecture - A preliminary study. Brain Behav Immun. (2016) 58:31–9. doi: 10.1016/j.bbi.2015.12.011
131. Haroon E, Woolwine BJ, Chen X, Pace TW, Parekh S, Spivey JR, et al. IFN-alpha-induced cortical and subcortical glutamate changes assessed by magnetic resonance spectroscopy. Neuropsychopharmacology. (2014) 39:1777–85. doi: 10.1038/npp.2014.25
132. Coch C, Viviani R, Breitfeld J, Münzer K, Dassler-Plencker J, Holdenrieder S, et al. Interferon-beta-induced changes in neuroimaging phenotypes of appetitive motivation and reactivity to emotional salience. NeuroImage Clin. (2019) 24:102020. doi: 10.1016/j.nicl.2019.102020
133. Nettis MA, Lawrence AJ, Wood T, Mariani N, Nikkheslat N, Lombardo G, et al. Using quantitative MRI to study brain responses to immune challenge with interferon-α. Brain Behav Immun Health. (2021) 18:100376. doi: 10.1016/j.bbih.2021.100376
134. Benjamin P, Sudhakar S, D’Arco F, Löbel U, Carney O, Roux C-J, et al. Spectrum of neuroradiologic findings associated with monogenic interferonopathies. AJNR Am J Neuroradiol. (2022) 43:2–10. doi: 10.3174/ajnr.A7362
135. Kurokawa M, Kurokawa R, Baba A, Gomi T, Cho S, Yoshioka K, et al. Neuroimaging features of cytokine-related diseases. Radiographics. (2024) 44:e230069. doi: 10.1148/rg.230069
136. Mesquita SD, Ferreira AC, Gao F, Coppola G, Geschwind DH, Sousa JC, et al. The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer’s disease. Brain Behav Immun. (2015) 49:280–92. doi: 10.1016/j.bbi.2015.06.008
137. Reizis B. Plasmacytoid dendritic cells: Development, regulation, and function. Immunity. (2019) 50:37–50. doi: 10.1016/j.immuni.2018.12.027
138. Cacace R, Zhou L, Hendrickx Van de Craen E, Buist A, Hoogmartens J, Sieben A, et al. Mutated Toll-like receptor 9 increases Alzheimer’s disease risk by compromising innate immunity protection. Mol Psychiatry. (2023) 28:5380–9. doi: 10.1038/s41380-023-02166-0
139. Di Domizio J, Dorta-Estremera S, Gagea M, Ganguly D, Meller S, Li P, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci U.S.A. (2012) 109:14550–5. doi: 10.1073/pnas.1206923109
140. Moore E, Huang MW, Putterman C. Advances in the diagnosis, pathogenesis and treatment of neuropsychiatric systemic lupus erythematosus. Curr Opin Rheumatol. (2020) 32:152–8. doi: 10.1097/BOR.0000000000000682
141. Salehi M, Zamiri A, Kim J, Texeira C, Shah K, Gunturu S. Exploring the psychiatric manifestations of primary Sjögren’s syndrome: A narrative review. Int J Rheumatol. (2024) 2024:5520927. doi: 10.1155/2024/5520927
142. Furie RA, Morand EF, Bruce IN, Manzi S, Kalunian KC, Vital EM, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. (2019) 1:e208–19. doi: 10.1016/S2665-9913(19)30076-1
143. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
144. Jayne D, Rovin B, Mysler EF, Furie RA, Houssiau FA, Trasieva T, et al. Phase II randomised trial of type I interferon inhibitor anifrolumab in patients with active lupus nephritis. Ann Rheum Dis. (2022) 81:496–506. doi: 10.1136/annrheumdis-2021-221478
145. Jayne D, Rovin B, Mysler E, Furie R, Houssiau F, Trasieva T, et al. Anifrolumab in lupus nephritis: results from second-year extension of a randomised phase II trial. Lupus Sci Med. (2023) 10. doi: 10.1136/lupus-2023-000910
Keywords: interferon, neurodegeneration, Alzheimer’s disease, Parkinson’s disease, TDP-43, C9orf72, neuropsychiatric disease, autoimmune disease
Citation: Sirkis DW, Oddi AP, Jonson C, Bonham LW, Hoang PT and Yokoyama JS (2024) The role of interferon signaling in neurodegeneration and neuropsychiatric disorders. Front. Psychiatry 15:1480438. doi: 10.3389/fpsyt.2024.1480438
Received: 14 August 2024; Accepted: 09 September 2024;
Published: 03 October 2024.
Edited by:
Wang Zheng, National Institutes of Health (NIH), United StatesReviewed by:
Yiwei Wang, University of Virginia, United StatesJuan Moisés De La Serna, International University of La Rioja, Spain
Copyright © 2024 Sirkis, Oddi, Jonson, Bonham, Hoang and Yokoyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel W. Sirkis, ZGFuaWVsLnNpcmtpc0B1Y3NmLmVkdQ==; Jennifer S. Yokoyama, amVubmlmZXIueW9rb3lhbWFAdWNzZi5lZHU=