 Samantha E. Yohn
Samantha E. Yohn Phillip D. Harvey
Phillip D. Harvey Stephen K. Brannan
Stephen K. Brannan William P. Horan1,3
William P. Horan1,3- 1Bristol Myers Squibb, Princeton, NJ, United States
- 2Division of Psychology, University of Miami, Miami, FL, United States
- 3Department of Psychiatry & Biobehavioral Sciences, University of California, Los Angeles, Los Angeles, CA, United States
Cognitive impairment is a core symptom of schizophrenia and a major determinant of poor long-term functional outcomes. Despite considerable efforts, we do not yet have any approved pharmacological treatments for cognitive impairment associated with schizophrenia (CIAS). A combination of advances in pre-clinical research and recent clinical trial findings have led to a resurgence of interest in the cognition-enhancing potential of novel muscarinic acetylcholine receptor (mAChR) agonists in schizophrenia. This article provides an overview of the scientific rationale for targeting M1 and M4 mAChRs. We describe the evolution of neuroscience research on these receptors since early drug discovery efforts focused on the mAChR agonist xanomeline. This work has revealed that M1 and M4 mAChRs are highly expressed in brain regions that are implicated in cognition. The functional significance of M1 and M4 mAChRs has been extensively characterized in animal models via use of selective receptor subtype compounds through neuronal and non-neuronal mechanisms. Recent clinical trials of a dual M1/M4 mAChR agonist show promising, replicable evidence of potential pro-cognitive effects in schizophrenia, with several other mAChR agonists in clinical development.
1 Introduction
Schizophrenia is a complex, heterogenous psychiatric disorder characterized by an array of debilitating symptoms and one of the top 10 leading causes of disability worldwide (1). Symptoms of schizophrenia span three main domains: positive, negative and cognitive symptoms, which vary in relative severity between affected individuals (1). Positive symptoms include delusions and hallucinations as well as disorganized speech/behavior. These symptoms generally respond well to antipsychotic medications though many patients continue to experience residual symptoms and adverse side effects (e.g., weight gain, metabolic disturbances) (1, 2). Negative symptoms include social withdrawal, lack of motivation, anhedonia, and flattened affect. These symptoms typically do not respond to antipsychotic medications and contribute to chronic functional disability for many patients (3).
This review focuses on the cognitive symptoms of schizophrenia. Since the earliest description of schizophrenia by Kraeplin as “dementia praecox,” cognitive deficits have been considered a core component of this debilitating neuropsychiatric disorder (4, 5). Schizophrenia is characterized by broad impairment across multiple cognitive domains, such as learning and memory, reasoning and problem solving, speed of processing, and attention (6). The magnitude of impairment is substantial, with people with schizophrenia, on average, falling 1.5 to 2 standard deviations (SD) below healthy normative standards (7, 8). Cognitive impairment associated with schizophrenia (CIAS) is distinct and separate from positive and negative symptoms, is present prior to the initial onset of positive symptoms, and highly stable across both symptom state changes and the longitudinal course of illness (9). Importantly, like negative symptoms, CIAS is a major contributor to poor long−term functional outcomes, impeding the ability of people with schizophrenia to live independently, attain competitive employment, and develop supportive social networks (10, 11).
No efficacious pharmacological treatments for CIAS yet exist. Approved first-line treatments for schizophrenia, including the first- and second-generation antipsychotics that rely on D2 dopamine (DA) receptor blockade, do not meaningfully impact cognitive deficits (12–14). The National Institute of Mental Health developed the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) initiative in the early 2000s, which stimulated major efforts to develop novel adjunctive agents, co-administered with an antipsychotic, for CIAS. Based on their role in healthy cognition and in the pathophysiology of cognitive impairments in schizophrenia, these efforts have focused on regulating four neurotransmitter systems.
• Cortical DA signaling plays a central role in normal attention, working memory, and executive functions as well as inhibiting unrelated noise to fine-tune adaptive neural signaling. In schizophrenia, DA dysregulation is strongly linked to positive symptoms via presynaptic hyperdopaminergia in striatal regions as well as cognitive impairment via cortical hypodopaminergia (15).
• Balanced and coordinated activity between excitatory excitatory glutamate (Glu) pyramidal cells and inhibitory Gamma-aminobutyric acid (GABA) interneurons is essential for normal learning, working memory, and neuroplasticity. In schizophrenia, cortical disinhibition associated with an altered excitatory and inhibitory balance between these neurotransmitters is thought to produce discoordination in neural networks that results in cognitive deficits (16).
• The two families of Acetylcholine (ACh) receptors, nicotinic (nAChRs) and muscarinic (mAChRs), are both associated with a range of cognitive functions, including learning, sensory gating, episodic memory, working memory, spatial memory, and attention (17). In schizophrenia, most treatment development has focused on the nAChR system. Impairment in several cognitive domains has been linked to nAChRs, particularly the α7 subunit (18).
Unfortunately, dozens of trials aimed at enhancing or restoring DA, Glu and GABA, or ACh via nAChRs for CIAS through adjunctive treatments have been plagued by replication failures and have not led to any regulatory approvals (13). There remains a very significant unmet need for more efficacious treatments for cognitive impairment based on new mechanisms and modes of action.
Despite the discouraging history, there has been a recent surge of interest in and optimism about CIAS drug development efforts focused on mAChRs. This renewed excitement comes from trials indicating that M1 and M4 mAChR-targeted drugs, which do not have direct antagonist effects on D2 DA receptors, can effectively treat not only the positive symptoms of schizophrenia but possibly cognitive impairments, as well. This article describes the evolution of clinical and pre-clinical in vivo and ex vivo research that supports this promising, though not entirely new, treatment approach for CIAS. First, the story traces its origins to early clinical drug discovery efforts in the 1990s that were searching for ACh-targeted treatments to impact the cognitive impairment associated with Alzheimer’s disease (AD). Second, these clinical findings motivated decades of basic neuroscience research on M1 and M4 mAChRs, which has extensively characterized their distribution and function (e.g., neuronal and non-neuronal) in brain regions implicated in cognition via in vitro assays and animal models. Finally, current clinical research in schizophrenia in the past 5 years has demonstrated the impact of M1 and M4 mAChR activators as an entirely novel monotherapy approach for multiple symptom domains of schizophrenia, potentially including cognitive impairment.
2 Early clinical drug discovery efforts
In the 1990s, early clinical efforts to evaluate the therapeutic potential of ACh-modulating drugs focused on discovering new treatments for cognitive impairment in AD. Evidence that AD is associated with a degeneration of cholinergic neurons motivated efforts to pharmacologically enhance cholinergic signaling. Initial efforts to broadly improve cholinergic transmission and enhance cognitive function in AD focused on acetylcholinesterase (AChE) inhibitors (e.g., tacrine, physostigmine, and donepezil; for detailed review, see (19). Although AChE inhibitors are still used today for symptom treatment, their efficacy is modest at best (20). During the same period, parallel pre-clinical research (e.g., cell-based assays and animal models) led to a much deeper and refined understanding of the cholinergic system.
2.1 Muscarinic cholinergic system
The cholinergic system is one of the most important modulatory neurotransmitter systems in the brain as it controls a wide range of activities (21). Of relevance to the pathophysiology of schizophrenia, cholinergic innervation can be split into two primary networks: the hindbrain complex that projects to the midbrain, which plays an important modulatory role in neural circuits implicated in psychosis [see (22) for review], and the forebrain complex, which projects to the cortical regions involved in cognitive function (e.g., frontal cortex and hippocampus).
The neural circuit effects of ACh are mediated by two receptor types: nAChRs, which are ligand-gated ion channels, and mAChRs, a five-receptor family of G-protein coupled receptors (GPCRs) (23). Both classes of receptors are expressed in the central (CNS) and peripheral nervous system (PNS). Within the PNS, activation of mAChRs produces end-organ responses that mimic parasympathetic nervous system stimulation (e.g., salivation, urination, and increases in gastric secretion and motility) (24). Within the CNS, mAChRs play important roles in modulating neuronal activity and neurotransmitter release in many brain regions (25). For this review, we focus on M1 and M4 mAChRs as these receptor subtypes are highly expressed in brain regions implicated in cognitive function (Figure 1).
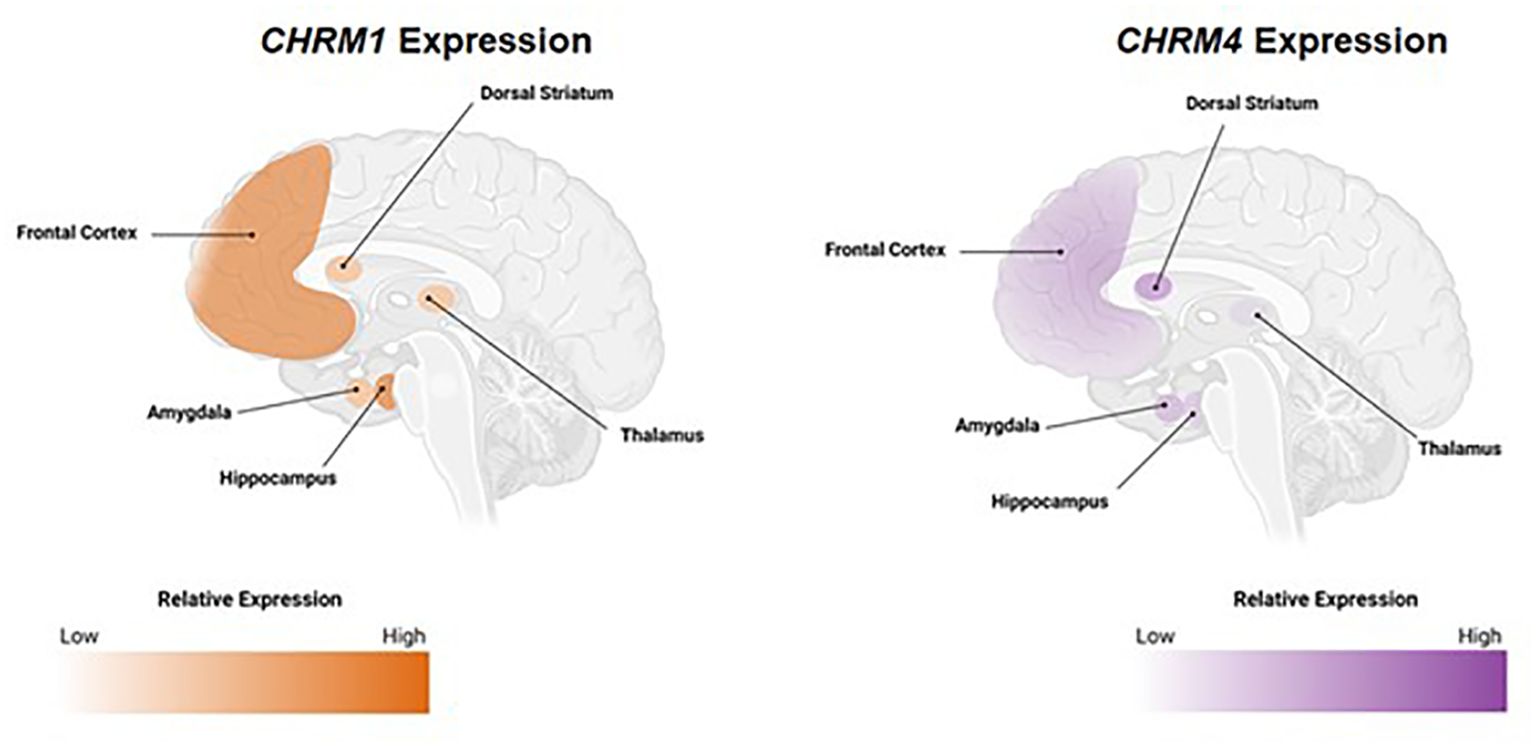
Figure 1. Expression pattern of M1 and M4 mAChRs in brain regions implicated in cognition. Relative CHRM1 and CHRM4 expression from healthy human tissue. All mAChRs can be found in the CNS, but the most prominent subtypes in brain regions implicated in cognition are M1 and M4 mAChRs. Data retrieved from GTEx Portal on September 6, 2022. Areas depicted represent classical cognition circuits. CHRM1/4, cholinergic receptor muscarinic 1/4 genes; CNS; central nervous system; mAChR, muscarinic acetylcholine receptor.
Within the cell membrane, GPCRs bind extracellular substances (e.g., the endogenous ligand, agonists, or antagonists) and transmit signals from these substances to an intracellular molecule called a G-protein (26). Based on signal transduction properties, mAChRs can be grouped into two families. Canonically, M1 mAChRs couple to the Gq/11 family (e.g., excitatory G-proteins), leading to intracellular calcium mobilization and cellular excitability, whereas M4 mAChRs preferentially signal through Gi/o (e.g., inhibitory G-proteins), leading to inhibition of adenylate cyclase and cellular inhibition.
The pharmacological characterization of therapeutic agents that act on mAChRs has not been a straightforward task due to the high level of conservation at the orthosteric site (site that binds the endogenous ligand) across the mAChR subtypes (25). Therefore, there are very few orthosteric agonists and antagonists that exhibit high selectivity for one subtype over the others. Activation of the orthosteric site can lead to unwanted activation of mAChR subtypes, and as discussed below, this was the shortcoming of many early drug discovery programs. mAChRs also contain an allosteric site that is topographically distinct and less conserved compared with the orthosteric site (27). The common allosteric binding site is located between the second and third transmembrane loops; however, computer modeling studies of allosteric ligands have revealed that allosteric binding to some mAChRs is more complicated. For instance, simulation of molecular dynamics have revealed cryptic allosteric binding pockets in the vicinity of the common allosteric binding site (28). Targeting the allosteric site has afforded the development of selective molecules that are believed to modulate efficacy via actions on critical subtypes while avoiding other subtypes that are believed to contribute to the side effect profile.
2.2 Clinical trials with mAChR agonists
Direct acting functional mAChR agonists in the cortex were of interest for many of the early drug discovery programs in AD. This was largely rooted in the etiology and progression of AD, which demonstrated that postsynaptic M1 mAChRs were less susceptible to degeneration, making this an attractive target for symptomatic treatment of AD (29). Several compounds were investigated in Phase 2 or Phase 3 clinical trials; however, development of these compounds for the treatment of AD was discontinued due to lack of efficacy, poor side-effect profiles due to stimulation of peripheral mAChRs, and unsuitable pharmacokinetic profiles (30).
Of all the compounds that were investigated for the treatment of cognitive impairment in AD, xanomeline, a dual M1/M4 mAChR orthosteric agonist initially developed by Eli Lilly & Company advanced the furthest. In one large-scale trial of safety and efficacy in people with AD, xanomeline was associated with enhanced cognition relative to placebo (31). The maximal effect on cognition was evident by 8 weeks of treatment and remained stably improved until the end of the 24-week trial. Notably, the magnitude of the xanomeline-associated cognitive benefit was substantially larger in participants with moderate than in those with mild AD (32). Interestingly, a completely unexpected finding was that xanomeline also improved psychotic-like symptoms. However, the discontinuation rate associated with xanomeline was 58.6% versus 33.3% in those receiving placebo, due primarily to gastrointestinal (GI) side effects attributable to peripheral mAChR agonism.
Based on the unexpected finding that xanomeline improved psychotic-like symptoms, a small proof-of-concept study subsequently evaluated acute schizophrenia among inpatient participants assigned to xanomeline or placebo. Those assigned to xanomeline demonstrated significant improvements in cognitive symptoms (e.g., listing learning, story recall, and delayed memory) as well as positive and negative symptoms (33). However, the peripherally mediated cholinergic side effect profile was again quite poor.
In summary, early clinical drug discovery efforts indicated that stimulation of M1 and M4 mAChRs in the CNS could produce treatment benefits for cognition and other neuropsychiatric symptoms. However, the therapeutic index for this compound was insufficient, as mAChR stimulation in the CNS was accompanied by peripherally mediated cholinergic side effects. As a result, drug development programs for xanomeline and related compounds were shelved and would remain dormant for many years.
3 Development of selective pharmacological agents
To overcome intrafamily promiscuity of mAChR orthosteric drugs, efforts have been made to target the allosteric sites of these receptors (25, 34). The classical mAChR allosteric pocket is located just above the orthosteric binding site and is partially formed by extracellular loops, which show greater sequence variation among the receptor subtypes (Figure 2, panel 1) (35). Depending on the type of allosteric ligand, binding can result in several changes, such as modifying the affinity of the orthosteric ligand (e.g., strengthening or weakening the binding affinity of ACh; Figure 2, panel 3a) (36), changing the intracellular signaling strength (Figure 2, panel 3b), or acting as a dualsteric (bitopic) ligand that simultaneously targets the orthosteric and allosteric sites (Figure 2, panel 5) (37).
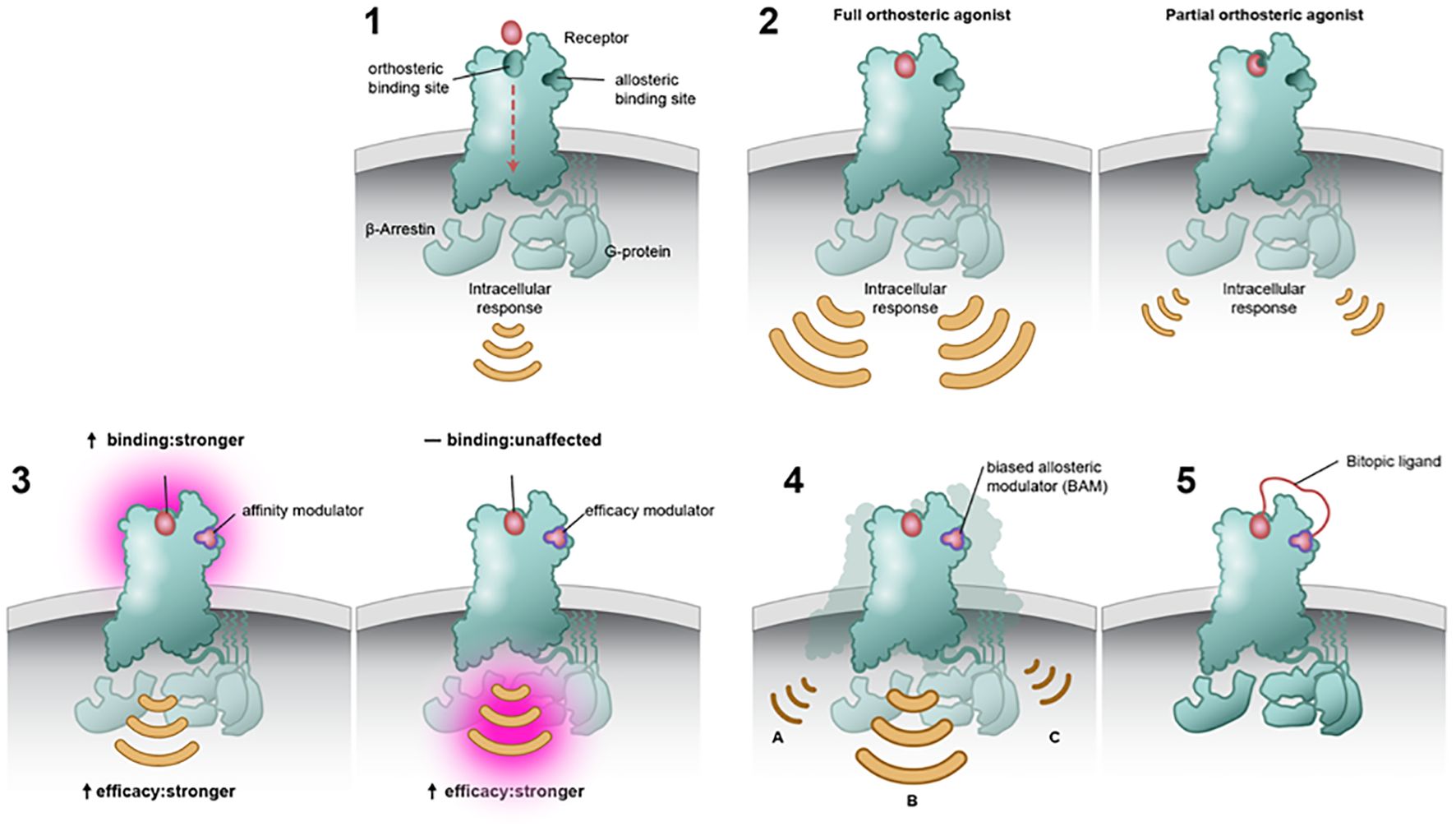
Figure 2. Modes of binding at the orthosteric and/or allosteric site. Muscarinic acetylcholine receptors (mAChRs) contain an orthosteric site, which is predominantly made up of highly conserved residues within the top third of the transmembrane domains. Orthosteric drugs bind at the active site, competing with the natural substrate or ligand. mAChRs also contain a temporal distinct allosteric binding site that is less conserved (panel 1). Full orthosteric agonists bind to and activate the mAChR with the maximum response that an agonist can elicit at the receptor (panel 2a). Partial orthosteric agonists are drugs that bind to and activate a given receptor but have only partial efficacy at the receptor relative to a full agonist. They may also be considered ligands that display both agonistic and antagonistic effects (panel 2b). Allosteric modulators can alter the affinity and efficacy of other substances acting on a receptor. Positive allosteric modulators may increase the response of the receptor by increasing the probability that an agonist will bind to a receptor (i.e., affinity; panel 3a), increasing its ability to activate the receptor (i.e., efficacy; panel 3b), or both. Biased ligands Ligands engage less well-conserved regulatory motifs outside the orthosteric pocket and exert pathway-specific effects on receptor signaling (panel 4). A bitopic ligand makes molecule makes concomitant interaction with both an orthosteric site and an allosteric site upon a single receptor (panel 5).
Allosteric modulators have two main classes: positive (PAMs) that increase response to orthosteric agonists and negative (NAMs) that inhibit responses to orthosteric agonists (27, 35). An allosteric modulator would only induce an action when the endogenous neurotransmitter is released (in this case ACh), and its action would be restricted in space and time to those synapses where signaling is currently happening.
Over the past three decades, a structurally diverse array of mAChR selective ligands have been identified and characterized for both M1 and M4 mAChRs (34, 38). PAMs may differ in their ability to confer receptor signaling, known as signal bias (Figure 2, panel 4) (39, 40). That is, activation of mAChRs may cause an effect in all second messenger signaling pathways, whereas a biased molecule would cause an effect in one second messenger pathway over the others. By imparting biased modulation, these allosteric modulators could activate therapeutically relevant signaling pathways while not acting on those pathways thought to be responsible for on-target side effects.
In summary, the unique properties of allosteric ligands necessitate a more comprehensive and nuanced approached to their pharmacological evaluation. Researchers must employ sophisticated techniques to fully understand the therapeutic potential and limitations of allosteric modulators. As discussed below, the development of M1 and M4 mAChR allosteric modulators has helped to advance our understanding of the roles of these receptors in neural nodes important for cognitive functions.
3.1 Modeling cognitive deficits in pre-clinical species
Animal models have played a vital role in investigating the physiological processes and mechanisms associated with M1 and M4 mAChRs in the neural bases of cognitive function. Most animal models currently being used are chemical- or drug-induced models, which facilitate studies of behavioral effects and neuronal effects. A particularly important animal model focuses on N-methyl-d-aspartate (NMDA) receptor dysfunction via administration of NMDA receptor antagonists or genetic mutations (41, 42). NMDA receptor hypofunction is a convergence point for the development and diverse symptoms of schizophrenia, especially cognitive deficits (43, 44). Hypofunction of NMDA receptors causes an imbalance of GABA and Glu neurotransmission in the brain (45), leading to deficits in local neural networks (e.g., hippocampus and prefrontal cortex [PFC]) as well as long-range disconnections between regions of the brain. Reduced excitatory input to NMDA receptors located on GABA inhibitory interneurons in cortical brain regions leads to reduced inhibition of excitatory pyramidal neurons and can cause an excitatory:inhibitory imbalance and perturbed network function that could explain CIAS (16). Use of NMDA receptor antagonists as tool compounds in pre-clinical species has high predictive validity (i.e., high translational potential across people with schizophrenia, healthy volunteers, and pre-clinical studies). For instance, studies have shown altered plasticity in people with schizophrenia (46) and that NMDA receptor antagonists can induce and exacerbate cognitive deficits in clinical populations (41).
In addition to behavioral studies, pre-clinical models can also incorporate electrophysiology and/or microdialysis measures, which provide researchers with a way to “peek under the hood” at what is occurring in the brain. Electrophysiology is the measurement of electrical activity of cells. Neurons in the brain form biochemical synapses with one another that facilitate the transmission of electrical signals between neurons (47). In the brain, synapses can modulate their response to various stimuli through synaptic plasticity; synaptic plasticity is driven by Glu and GABA interactions. Functionally, long-term potentiation (LTP) and long-term depression (LTD) are the two forms of synaptic plasticity (48). LTP is the strengthening of synapses following repeated synaptic activity, whereas LTD is the weakening of synaptic strengths. LTP and LTD are a neural correlate of learning and memory; that is, LTP and LTD are important for suppressing “outdated” memories to allow synapses to be “updated” with current information. Microdialysis is the measurement of neurotransmitter release in brain regions of interest; increases in certain neurotransmitters has been correlated with cognitive function. Changes in ACh and DA content (e.g., release) in hippocampal and frontal brain regions have been associated with cognitive function (49, 50). Although it remains unclear what changes induce the onset of cognitive dysfunction, it is reasonable to hypothesize that alterations to Glu and GABA balance and neurotransmitter disturbances (e.g., ACh and/or DA) could contribute to a disruption in network functioning in schizophrenia.
3.2 Behavioral effects of M1 and M4 mAChR activators: pre-clinical evidence
Pharmacological blockade or genetic deletion of the M1 mAChR produces significant learning and memory disturbances in pre-clinical animal models (51–53). M1 mAChR activation has been shown to consistently enhance memory consolidation and retrieval in various tasks (e.g., object recognition, spatial learning, and fear conditioning), executive function, attention (54), and cognitive flexibility (55). Additionally, M1 mAChR activation can attenuate cognitive deficits induced by NMDA antagonists (56) and genetic mutations in the NMDA receptor (57) (for detailed review on the development of M1 mAChR selective ligands see Nguyen et al., 2024 (58)).
In contrast to impairments in memory, learning and attentional accuracy (59) seen in global M1 mAChR knockout mice, M4 mAChR knockout mice (e.g., mice that have M1 or M4 mAChR turned off or ‘knocked out’) have robust deficits in the acquisition of both contextual and cue-dependent fear conditioning (60) but not spatial memory (60, 61), suggesting that the role of M4 mAChRs may be important for certain types of memory. M4 mAChR PAMs have demonstrated pro-cognitive benefits in rodents (60, 62–64) and nonhuman primates (65). Imaging studies have shown that M4 mAChR PAM administration can normalize amphetamine-induced changes in hippocampal activity (66). Chronic administration of M4 mAChR PAMs can enhance the rate of acquisition (63), an effect absent in mice where the M4 mAChR is removed; these findings suggest that M4 mAChR PAMs can enhance cognition. Interestingly, the efficacious dose range of M4 mAChR PAMs in pre-clinical in vivo models for antipsychotic-like activity and cognitive function have nonoverlapping minimal effective dose concentrations (e.g., lower doses are needed to improve cognitive deficits induced by an NMDA antagonist [5.6 mg/kg] versus higher doses to reverse NMDA-induced hyperlocomotor activity [10 mg/kg]) (60). However, additional studies are needed to explore the higher end of the locomotor dose response curve on cognitive function. Overall, although further research is needed to fully understand the role of M4 mAChRs in cognition, current evidence suggests their involvement in modulating cognitive processes and possibly influencing emotional memory processes.
In rodents, M1 and M4 mAChR activators have been found to improve memory in animals that performed poorly at baseline (62), and individual differences in extinction learning, but not acquisition, have been correlated with M1 mAChR signaling (67). The findings by Galloway and colleagues (62) align with prior findings which indicate that the benefit of mAChR activation on memory performance is dependent on baseline performance level (68). Future studies are needed to determine whether differences in baseline memory performance is due to individual differences in the level of endogenous ACh or mAChR receptor signaling. It is important to note that these findings (62) and others (69, 70) also support the possibility M1 and M4 mAChR beyond an optimal range has no beneficial effect and may even be detrimental to average or above average performance, suggesting that the relationship between mAChR stimulation and cognitive function is non-linear and has an inverted U-shaped curve. However, future research is required to explore these findings, particularly within the context of schizophrenia. In summary, dual activation of M1 and M4 mAChRs can also attenuate deficits in cognition in pharmacological and genetic models (71–73). Taken together, these findings suggest that M1 plus M4 mAChR activation may have pro-cognitive effects across several cognitive domains.
The impact of M1 and M4 mAChRs on cognitive function in pre-clinical in vivo models is complex and can vary depending on the specific cognitive task, brain region, and experimental conditions. Although both M1 and M4 mAChRs are implicated in pre-clinical cognitive models, they seem to have distinct roles; their specific contributions in neuronal nodes important for cognitive functioning continue to be actively investigated.
3.3 Neuronal actions of M1 and M4 mAChRs in the hippocampus
M1 and M4 mAChRs play important roles in the hippocampus, a region of the brain that is critical to cognitive processes. Cholinergic tone within the hippocampus shapes neural circuit function and subsequent behavior (74). Within the hippocampus, M1 mAChRs are abundantly expressed across all regions and cell types, and, to a lesser extent, M4 mAChRs are expressed in a more regionally specific manner (75). The main function of ACh in the hippocampus is to modulate levels of Glu and GABA (e.g., excitatory and inhibitory transmission) to drive synaptic plasticity as well as neuronal oscillations.
In the hippocampus, ACh facilitates learning and memory through cholinergic induction of neural oscillations (76). mAChRs modulate the excitability and synaptic connectivity of pyramidal neurons located in CA1 and CA3 subregions. That is, transient (short-term) activation of mAChRs within the CA1 region causes an inhibition (engagement of GABAergic interneurons) followed by an excitation (engagement of Glu-containing pyramidal neurons) (77). In contrast to the biphasic nature of mAChR activation in CA1, mAChR activation in the CA3 subregion evokes an excitatory response (e.g., activation of Glu-containing pyramidal neurons) (78).
Studies using mice that have mAChR subtypes deleted (e.g., global mAChR knockout mice) suggest that M1 is the major mAChR subtype responsible for direct cholinergic modulation of pyramidal neurons within hippocampal circuits (79–82). M1 mAChR activation is analogous to that of a switch in that the net effect of turning on the M1 mAChR is to facilitate excitatory transmission. For instance, electrophysiology studies have shown reduced or lack of cholinergic modulation of both CA1 and CA3 pyramidal neurons in M1 mAChR knockout mice (79, 81). An increase in Glu excitatory neurotransmission leads to LTP, an effect that is mediated via activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (83). This is of particular interest as M1 mAChR activation has been reported to facilitate hippocampal memory due to co-communication with AMPA receptors (84, 85). This finding highlights the central role that M1 mAChRs play in shaping excitatory synapses involved in learning and memory (Figure 3).
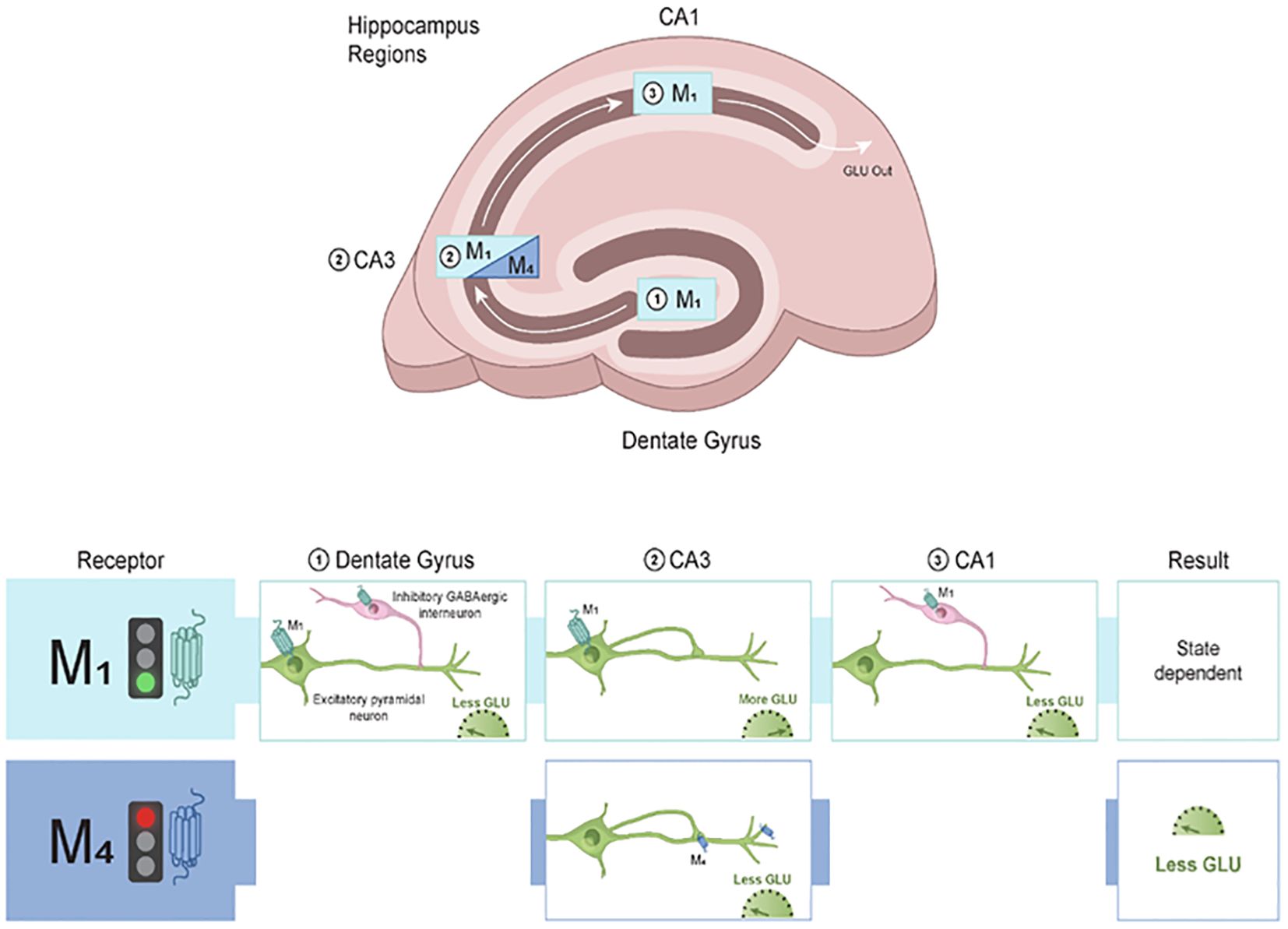
Figure 3. Modulatory role of M1 and M4 mAChRs in the hippocampus. The hippocampus is a small, curved formation located deep in the temporal lobe of the brain. Neurobiological and functional evidence strongly suggests that the hippocampus is a homologous structure across species. M1 mAChRs are distributed across the hippocampal subregions where they can modulate excitatory (Glu) and inhibitory (GABA) neurotransmission. Within the dentate gyrus (DG), activation of M1 mAChRs on inhibitory interneurons leads to a reduction of Glu output to CA3. In CA3, activation of M1 mAChRs facilitates Glu release, leading to more excitatory neurotransmission into CA1. Within CA1, M1 mAChRs are found on inhibitory interneurons and increase GABA input onto excitatory pyramidal neurons. In contrast to the broad distribution pattern of M1 mAChRs in the hippocampus, M4 mAChRs are primarily located in the CA3 region and modulate excitatory neurotransmission. M4 mAChRs synapse back onto themselves and are found on axon terminals, where they gate glutamate release onto CA1. The net output of M4 mAChR activation is to decrease excitatory neurotransmission and subsequently lead to decreased Glu output to downstream structures. Taken together, M1 and M4 mAChRs can fine-tune excitatory and inhibitory balance within the hippocampus that is critical for cognitive function. GABA, gamma-aminobutyric acid; Glu, glutamate; mAChR, muscarinic acetylcholine receptor.
GABAergic hippocampal interneurons also modulate firing frequency and neuronal excitability via M1 mAChRs (86). In CA1 pyramidal neurons, cholinergic activation of extracellular signal-regulated kinase (ERK) and mitogen-activated protein kinase (MAPK) pathways occur through stimulation of M1 mAChRs (87). This finding is important as ERK and MAPK activation is required for the formation and maintenance of LTP (88), a process involving the persistent strengthening of synapses (89). These findings suggest that M1 mAChR activation is important for cell-mediated responses.
M4 mAChRs also play a role in modulating hippocampal microcircuitry (90). M4 mAChR knockout mice treated with the nonselective cholinergic receptor agonist carbachol display a reduced suppression of excitatory postsynaptic potentials between the CA3 and CA1 regions, an effect gated by Glu neurotransmission (79). Thus, ACh can modulate pyramidal neuron excitability directly (via M1 mAChR activation; Figure 3) as well as through alterations of synaptic transmission between CA3 and CA1 pyramidal neurons (via M4 mAChR activation). In line with this, mAChR-induced hippocampal gamma oscillations in CA3 neurons are absent and carbachol-induced depression of transmission at excitatory synapses are blunted in M1 and M4 mAChR knockout mice, respectively (91, 92).
The correct balance of inhibitory and excitatory neurotransmission in the hippocampus is a key feature of learning and memory processes. A disruption in this balance can result in cognitive impairments (93, 94). M1 and M4 mAChRs can exert modulatory effects on synaptic plasticity processes within the hippocampus to ensure appropriate filtering of information and changes in downstream areas (Table 1) (95). This perspective seems consistent with clinical data that suggest an increased signal-to-noise ratio within the hippocampus can improve memory encoding accuracy (96) via preserving the excitatory and inhibitory balance and gating activity in hippocampal subregions.

Table 1. Summary of activation of M1 and M4 mAChRs on neuronal circuits.
3.4 Neuronal actions of M1 and M4 mAChRs in the frontal cortex
The functional microcircuitry of the PFC is shaped by cholinergic input from midbrain regions (97). Within the PFC, cholinergic tone is a crucial component of cognitive function, and cholinergic input acts as a gatekeeper to modulate synaptic plasticity and tone various other neurotransmitter systems. Similar to its actions within the hippocampus, ACh activity within the PFC is primarily modulated via M1 and M4 mAChRs. As discussed below, the location of these receptors within the PFC allows them to exert a variety of effects on cognitive function; thus, understanding the role of M1 and M4 mAChRs in shaping cognitive functions is crucial for developing potential therapeutic strategies for cognitive impairment.
Transient ACh release onto pyramidal neurons causes a depolarization of layer 5 pyramidal neurons via M1 mAChR activation, causing them to fire Glu to downstream structures, whereas constant presence of ACh does the opposite (98). In this sense, M1 mAChR activation gates neuronal activity through two approaches: sustained tonic stimulation, which leads to reduced excitability in output structures, versus transient phasic stimulation, which leads to increased excitability in output structures (Figure 4). These two modes represent a form of communication to sharpen the signal-to-noise ratio within the local microcircuits of the PFC. This is particularly important because in states characterized by NMDA hypofunction, like schizophrenia, the signal-to-noise ratio is disrupted (99). M1 mAChR activation can restore burst activity and accentuate the signal transmission efficiency of PFC pyramidal neurons in NMDA hypofunction states (100).
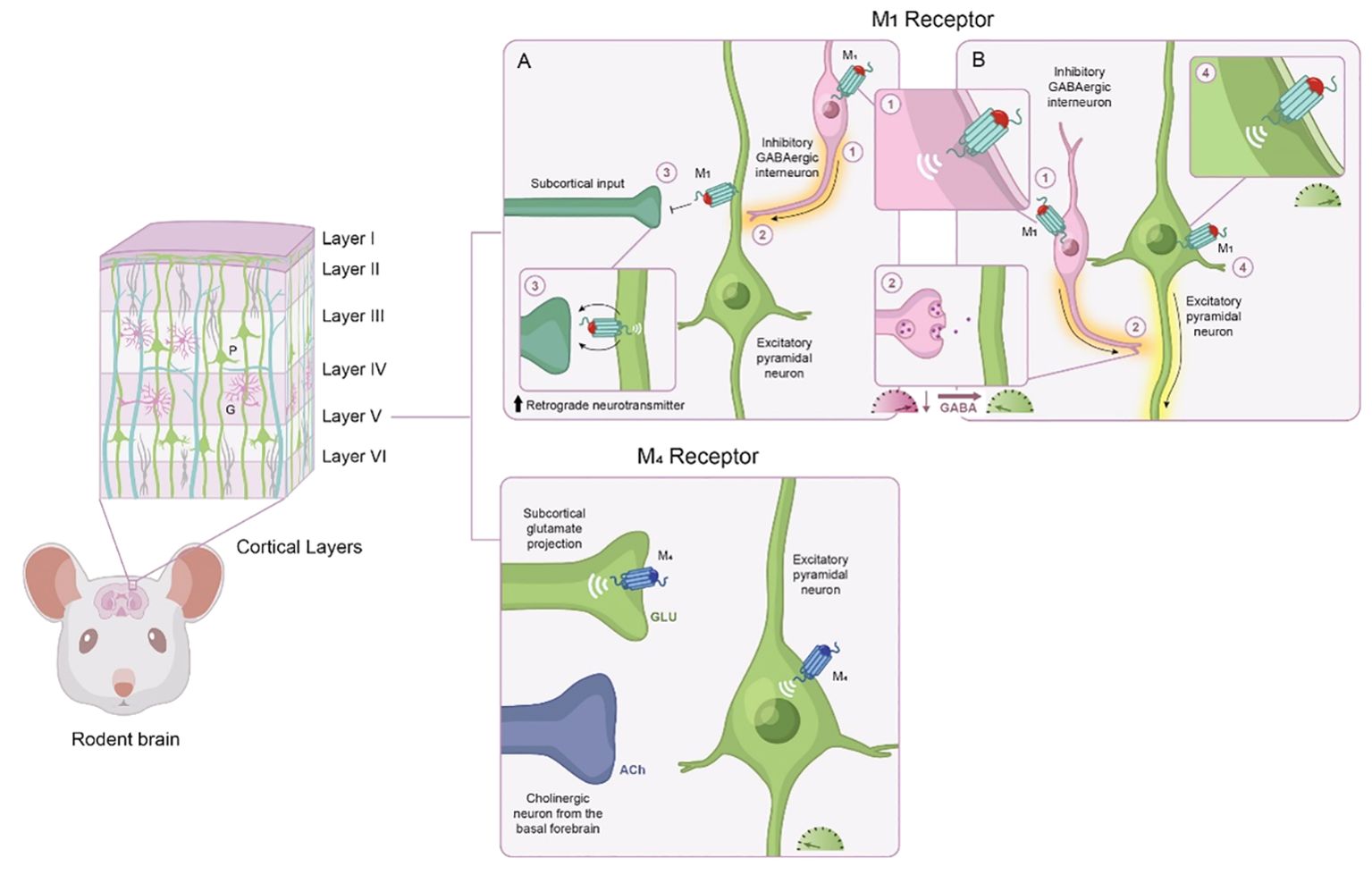
Figure 4. Modulatory role of M1 and M4 mAChR activation in the frontal cortex. The frontal cortex has some degree of laminar organization, with different layers composed of neurons with distinct connectivity patterns and molecular profiles. Based on cytoarchitectural different, the rodent frontal cortex is classified into four distinct neuroanatomical subregions along a dorsal to ventral axis. The frontal cortex neural networks consist predominately of excitatory pyramidal neurons (green) and inhibitory GABAergic interneurons (purple). Based on their physiological and molecular properties, interneurons can be divided into several subpopulations. M1 mAChRs are positioned to modulate a wide variety of neuronal activity; the actions of M1 mAChR activation depend on which cell type M1 mAChRs are expressed, the location of M1 mAChRs, and the receiving neuron. (A) M1 mAChR activation induces a form of long-term depression of glutamatergic inputs from subcortical areas, such as the ventral hippocampus, via activation of retrograde neurotransmitters. (B) M1 mAChR activation also increases the excitability of pyramidal neurons (i.e., more glutamate release to downstream structures) and GABAergic interneurons (i.e., increased inhibition onto glutamatergic neurons, meaning less glutamate output to downstream structures). Activation of M1 mAChRs via GABAergic interneurons also can increase gamma oscillation synchrony in the cortex. Within layer V, M4 mAChRs are located on subcortical glutamate projections into the prefrontal cortex where they can modulate glutamate release within layer V. M4 mAChRs are also found on Layer V principal output neurons of the prefrontal cortex, where they contribute to hyperpolarization. GABA, gamma-aminobutyric acid; Glu, glutamate; mAChR, muscarinic acetylcholine receptor.
Pre-clinical in vivo and ex vivo evidence suggests that phasic and tonic ACh release can occur concurrently during performance of cognitive tasks (101). Further, since M1 mAChRs are coupled to diverse signaling pathways, both excitatory and inhibitory responses may co-occur in the same neuron (102). The weight and direction of the response is an important feature underlying learning and memory, as alterations in behavior (attentional switching, reaction time, etc.) are caused by changes in neuronal firing. It is also important to note that the activity of M1 mAChRs may be layer specific. For instance, differences in cholinergic responsiveness of pyramidal neurons have been reported between layers 2/3 and layer 5, an effect most likely attributed to calcium-activated potassium channels (103).
Although the M4 mAChR has not been traditionally associated with actions in the PFC, emerging data has shed light on the modulatory role of M4 mAChRs within cognition microcircuits. M4 mAChR activation has been reported to decrease excitatory Glu transmission at corticostriatal synapses, resulting in LTD (104), which gates downstream activity and therefore shapes contextual representations (Figure 4). The output gating dynamic for working memory depends on interactions between the cortex and the striatum (105). The excitatory drive from corticostriatal glutamatergic afferents activates striatal neurons, which, in turn, alters the activity of neurons throughout the entire basal ganglia. Neuroimaging studies demonstrate that schizophrenia is associated with aberrant connectivity of the corticostriatal network (106). Although future studies are needed to investigate the relationship between abnormal connectivity and cognitive function, these findings suggest that M4 mAChR activation shapes corticostriatal network activity and modifying M4 mAChR activity could have beneficial effects on cognitive processes dependent on this network.
In addition to shaping plasticity, M1 and M4 mAChR activation in the PFC has been shown to facilitate ACh neurotransmission (66, 72, 107, 108). In fact, this idea was the driving factor for early drug development programs targeting the cholinergic system in AD; that is, an increase in ACh in the PFC was expected to improve cognitive function (30). It is hypothesized that ACh enhances the encoding of memory by facilitating feedforward, top-down output.
The PFC has reciprocal connectivity with several neuromodulatory systems, including the midbrain DA system (109). In the rodent brain, ventral tegmental area (VTA) DA neurons send sparse projections to frontal regions (110). It is important to note that there is considerable variation of DA integration of the PFC across mammalian species. Pre-clinical literature supports the idea that mesocortical DA is functionally distinct from mesolimbic DA (for detailed review, see (111). Cholinergic neurotransmission can orchestrate DA dynamics within the PFC. For instance, dual M1/M4 mAChR activation has been shown to increase DA release in the PFC in rodent models (72, 108). This increase in PFC DA release may be one of the ways that dual M1/M4 mAChR activation facilitates elements of cognitive functioning.
Postsynaptically, DA exerts its actions in the PFC via D1 DA receptor activation (112, 113). Hypofunction of the D1 DA receptor pathway may underlie cognitive dysfunction (114, 115). Within the PFC, signal transduction crosstalk between M1 mAChRs and D1 DA receptors within pyramidal cell dendrites has been reported (116). This interaction may be relevant for optimizing the level of D1 DA receptor stimulation that is required for working memory performance (117, 118). Previously, D1 DA and mAChR interplay has been characterized in the striatum, where activation of M4 mAChRs inhibits D1 DA receptor second messengers (119, 120). These findings suggest that a dual M1/M4 mAChR agonist, like xanomeline, may be beneficial in restoring aberrant D1 DA receptor signaling in the striatum (associated with antipsychotic-like activity) via M4 mAChR activation and the frontal cortex via M1 mAChR activation (associated with cognitive improvement).
Taken together these findings suggest that M1 and M4 mAChR activation can shape neuronal activity of the PFC in multiple ways. The interplay between M1 and M4 mAChRs can modulate the balance of excitatory and inhibitory signaling in neural circuits, ultimately shaping neuronal activity and cognitive processes. It is important to note that the roles of M1 and M4 mAChRs in the PFC and hippocampus can be complex and context dependent (Table 2).
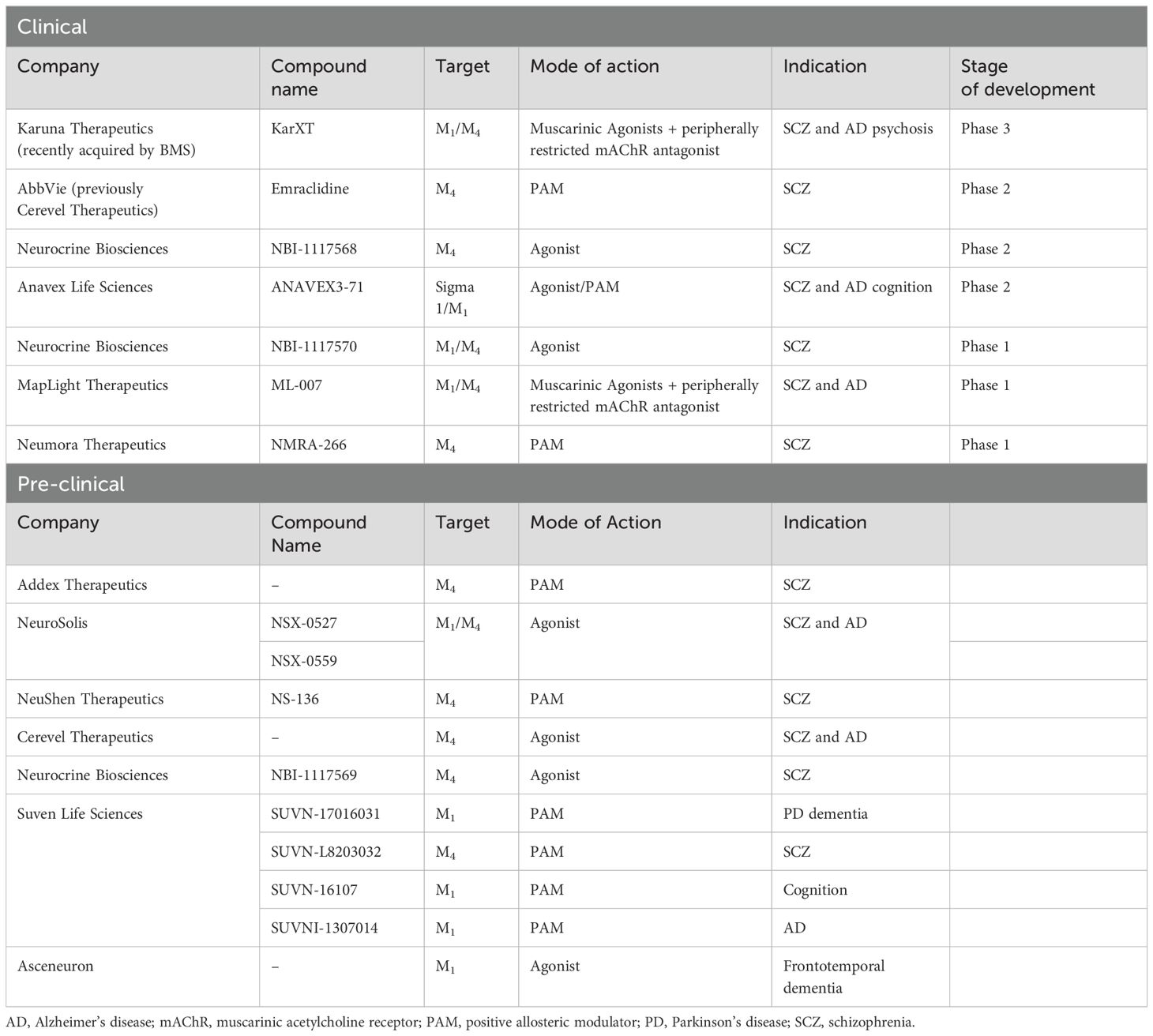
Table 2. Active clinical and pre-clinical mAChR programs for schizophrenia.
3.5 Non-neuronal actions of M1 and M4 mAChRs in memory
There is considerable support for the role of cytokine release from microglia in the modulation of memory. Administration of inflammatory cytokines causes deficits in spatial memory in pre-clinical behavioral models (121); an altered immune profile has been reported in people with mild cognitive impairment (122), and associations between cytokine levels and cognition in people with chronic and first-episode schizophrenia has been reported (123, 124). In pre-clinical models, the dual M1/M4 mAChR agonist xanomeline has been shown to suppress excessive pro-inflammatory cytokine responses (125), and recent data suggests that M4 mAChR activation alone can reduce pro-inflammatory cytokines (126); however, additional work is needed. Additional trials in clinical populations are needed to confirm non-neuronal actions of mAChR activators.
4 Clinical neuroscience support for mAChR drug development in schizophrenia
In the past 5 years, there has been a resurgence of interest in the mACh system for the treatment of CIAS. This renewed interest has been catalyzed by a much deeper neuroscience-based understanding of the mAChRs in cognitive function based on evidence from in vitro and in vivo pre-clinical studies. Additionally, several lines of clinical research evidence have implicated cholinergic functioning in the pathophysiology of cognitive impairment in schizophrenia.
- Postmortem studies demonstrate reductions of M1 mAChRs in the dorsolateral PFC and M4 mAChRs in the hippocampus, with relative sparing of M2 and M3 mAChRs, in schizophrenia (127).
● In medication free subjects with psychosis, the reduction in M1 mAChRs in the dorsolateral PFC and hippocampus was shown to be related to overall performance in verbal learning and delay in recognition of verbal memory (128).
- Molecular neuroimaging in medication-free individuals with early psychosis provide in vivo evidence of reduced M1/M4 mAChRs. A decrease in M1/M4 mAChRs provides preliminary in vivo support for a disbalance in M1/M4 mAChR expression in schizophrenia that might directly impact clinical outcomes (129).
- Exposure to medications with higher anticholinergic (e.g., mAChR antagonist pharmacology) burden is associated with impaired cognition in people with schizophrenia (130), although this is a wide-ranging effect across conditions. Patients exposed to high anticholinergic burden have lower brain activity in the frontoparietal network, a flexible hub for cognitive control, and lower performance during working memory tasks as compared with patients with low anticholinergic medication exposure (131).
● The cognition impairing effects observed with mAChR antagonist pharmacology are due to activation of CNS mAChRs (132). For instance, a prior study found that patients with schizophrenia on maintenance treatment plus centrally active anticholinergic antiparkinsonian drugs (e.g, benztropine) had significant impairment on free recall compared to the placebo group.
- There is some evidence that the mechanistic uniqueness of the antipsychotic clozapine may be due to its active metabolite N-desmethylclozapine (NDMC), a partial M1 mAChR agonist. NMDC increases cortical ACh and DA release (133). In clinical populations, lower clozapine:NDMC ratios are associated with improvements in working memory and executive function, whereas higher ratios are associated with cognitive deficits (134).
As discussed below, there are data currently available from one mAChR agent, KarXT, in relation to cognitive function in individuals with schizophrenia, and several other mAChR agents are currently in clinical development.
4.1 KarXT clinical development program
Despite xanomeline’s promising efficacy profile described above (see Section 2.2), the development of xanomeline was discontinued because of significant levels of cholinergic adverse events (AEs), namely nausea, vomiting, diarrhea, excessive sweating, and salivary hypersecretion (31, 33), due to stimulation of peripheral M1, M2, and M3 mAChRs. KarXT is a combined formulation of two drugs, xanomeline and trospium chloride, that was designed to mitigate the peripheral mAChR side effects observed with xanomeline. Trospium is a quaternary ammonium compound with a permanent cationic charge that limits its ability to meaningfully cross the blood-brain barrier (135). Thus, trospium competes with xanomeline for binding at peripheral, but not central, mAChRs, thereby reducing the negative mAChR side effects of xanomeline without impacting the potential therapeutic effects of xanomeline in the brain (136).
KarXT was developed as a monotherapy for the treatment of schizophrenia in adults. Across three pivotal, 5-week, Phase 2 and Phase 3 trials (NCT03697252, NCT04659161, and NCT04738123) with acutely symptomatic inpatient participants, KarXT demonstrated a significant improvement compared with placebo on the Positive and Negative Syndrome Scale (PANSS) total score primary efficacy endpoint, and results for secondary endpoints (PANSS positive and negative subscale scores) were typically significant and reproducible (137, 138). KarXT was generally well tolerated and not associated with many of the AEs typically associated with current antipsychotics. These trials supported the submission of a New Drug Application in September 2023 for KarXT, which has the potential to be the first of a new class of medicines based on activating mAChRs, as opposed to the D2 DA receptor blocking activity associated with current antipsychotic medications.
Based on the strong mechanistic link between M1 and M4 mAChR stimulation and cognition, all three pivotal KarXT trials evaluated cognition as an exploratory outcome. In the Phase 2 trial, participants completed an abbreviated computerized battery at baseline and end of treatment. Sample-wide, cognitive improvement was numerically but not statistically greater with KarXT than with placebo. However, a post hoc analysis of participants with clinically significant cognitive impairment at baseline, defined as performing at least 1 SD below healthy normative standards, indicated that those treated with KarXT showed a robust, significant cognitive improvement compared with placebo (Cohen’s d = 0.50). Further, cognitive improvements were minimally associated with PANSS total symptom changes (139). Prespecified analyses of the exploratory cognitive endpoint in the combined sample from the Phase 3 trials (completed at baseline, week 3, and end of treatment) replicated these findings. There was, again, no significant treatment effect across the entire sample (N = 307); however, in the cognitively impaired subgroup (n = 137), participants taking KarXT showed significantly greater improvement in cognition compared with placebo (Cohen’s d = 0.54) (140). The improvement in cognition was fully independent of changes in PANSS total, positive subscale, and negative subscale scores.
Collectively, the KarXT clinical trials reflect the first time a monotherapy for the treatment of schizophrenia has shown a replicable cognitive benefit across Phase 2 and Phase 3 trials. Although the KarXT effect on cognition did not appear to be secondary to symptom changes (i.e., it was not “pseudo-specific”), the MATRICS CIAS trial guidelines, which focus on testing adjunctive or cotreatment agents in stabilized people with schizophrenia, recommend that assessment of pro-cognitive effects for broad-spectrum agents should also utilize people who are stable. Thus, although these initial findings are encouraging, replication in a longer, well-controlled trial with clinically stable people is needed to fully characterize the potential benefit of KarXT for CIAS.
4.2 Other mAChR compounds in development for schizophrenia
At the time of this review several orthosteric and allosteric mAChR compounds have been identified as having potential antipsychotic activity and cognition-enhancing properties in clinical populations and pre-clinical drug development pipelines (Table 2).
4.2.1 Emraclidine
Emraclidine (CVL-231) is an M4 mAChR PAM (e.g., it selectively activates M4 mAChRs) currently under development by AbbVie (previously Cerevel Therapeutics). In a Phase 1b trial (NCT04136873), emraclidine demonstrated a clinically meaningful and statistically significant improvement in PANSS total score at week 6 in participants with schizophrenia compared with placebo (141). At present, three Phase 3 clinical trials are ongoing to confirm the efficacy, safety, and tolerability of emraclidine.
4.2.2 ML-007, ANAVEX3-71, and Neurocrine
ML-007, currently under development by MapLight Therapeutics, is a dual M1/M4 mAChR agonist paired with a peripherally restricted mAChR antagonist. ML-007 has completed three Phase 1 trials (one trial with an extended-release formulation [ML-007C-MA]) in healthy volunteers. Phase 2 trials with the extended-release formulation are anticipated to begin later this year.
There are currently two other clinical development programs harnessing the potential of mAChR activation for schizophrenia, namely CIAS. Earlier this year, Anavex Life Sciences announced it is recruiting for a Phase 2 trial with ANAVEX3-71, a dual sigma 1 agonist/M1 mAChR PAM, in participants with schizophrenia (NCT06245213). This trial aims to assess the benefit of ANAVEX3-71 on positive, negative, and cognitive symptoms of schizophrenia. Originally developed for AD, ANAVEX3-71 has demonstrated efficacy in animal models of cognitive impairment (e.g., transgenic disease models (142) and deficit states (143)). Additionally, although no data are currently available, Neurocrine Biosciences has announced an interest in M1 and M4 mAChR activators for treating the cognitive symptoms of schizophrenia.
There has been a resurgence of interest in the mAChR system for various neurological and neuropsychiatry disorders, including CIAS. Notably, a handful of other mAChR agents, with various pharmacology flavors, are being investigated in AD (for detailed review, see Johnson et al., 2022 (34) and Felder et al., 2018 (144)). Continued research in this area is needed to deepen our knowledge and lead to the development of innovative therapies and, as such, many novel mAChR agents are in early development (Table 2).
5 Future directions and conclusions
Schizophrenia is characterized by an array of symptoms that vary in their response to treatment. Even when positive symptoms are effectively managed, negative and cognitive symptoms frequently persist. It is widely recognized that available antipsychotic medications inadequately treat these functionally disabling symptoms (145, 146). Thus, effective new treatments for negative and cognitive symptom domains that target different neural pathways are urgently needed.
Nearly three decades have passed since the cholinergic hypothesis first motivated early drug discovery efforts to become an approach toward the improvement of cognitive systems in AD. Since that time, our understanding of the regional expression and functional roles of M1 and M4 mAChRs in cognitive circuits has grown dramatically. Similar to what was observed in AD, cognitive functioning in people with schizophrenia was improved by treatment with the dual M1/M4 mAChR preferring agonist xanomeline (33). As a result of these findings, several novel mAChR therapeutic strategies have emerged, including combining xanomeline with the peripherally restricted pan-mAChR antagonist trospium (KarXT) to reduce peripheral cholinergic side effects as well as developing more subtype selective orthosteric and allosteric agents targeting either M1 or M4 mAChRs. Recent pivotal placebo-controlled clinical trials demonstrate that KarXT is an effective, well-tolerated monotherapy for positive symptoms, and possibly for cognitive impairment as well. However, future trials will be required to confirm the potential efficacy of KarXT in treating cognitive symptoms. Several other compounds that target M1 and/or M4 mAChRs more selectively are in earlier stages of clinical development. This emerging new class of mAChR therapies may provide long-awaited breakthroughs in the treatment of CIAS.
Author contributions
SY: Visualization, Writing – review & editing, Writing – original draft, Conceptualization. PH: Writing – review & editing. SB: Writing – review & editing. WH: Writing – original draft, Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Karuna Therapeutics, a Bristol Myers Squibb Company, supplier of KarXT. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
Editorial support in the preparation of this article was provided by Shannon Davis of Apollo Medical Communications, part of Helios Global Group, and funded by Karuna Therapeutics, a Bristol Myers Squibb Company. Graphical support was provided by Mao Miyamoto and Michael Konomos of Mao Miyamoto Medical Media, Inc.
Conflict of interest
SY and WH are employees of Karuna Therapeutics, a Bristol Myers Squibb Company, supplier of KarXT. SB is a consultant of Karuna Therapeutics, a Bristol Myers Squibb Company, supplier of KarXT. PH is a consultant for Alkermes, Boehringer Ingelheim, Karuna Therapeutics, a Bristol Myers Squibb Company, Merck, Minerva Neuroscience, and Sunovion/DSP and has received royalties from WCG.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. McCutcheon RA, Reis Marques T, Howes OD. Schizophrenia-an overview. JAMA Psychiatry. (2020) 77:201–10. doi: 10.1001/jamapsychiatry.2019.3360
2. Kahn RS, Sommer IE, Murray RM, Meyer-Lindenberg A, Weinberger DR, Cannon TD, et al. Schizophrenia. Nat Rev Dis Primers. (2015) 1:15067. doi: 10.1038/nrdp.2015.67
3. Correll CU, Schooler NR. Negative symptoms in schizophrenia: a review and clinical guide for recognition, assessment, and treatment. Neuropsychiatr Dis Treat. (2020) 16:519–34. doi: 10.2147/NDT.S225643
4. Kahn RS, Keefe RS. Schizophrenia is a cognitive illness: time for a change in focus. JAMA Psychiatry. (2013) 70:1107–12. doi: 10.1001/jamapsychiatry.2013.155
5. Reichenberg A, Harvey PD. Neuropsychological impairments in schizophrenia: Integration of performance-based and brain imaging findings. Psychol Bull. (2007) 133:833–58. doi: 10.1037/0033-2909.133.5.833
6. Marder SR. The NIMH-MATRICS project for developing cognition-enhancing agents for schizophrenia. Dialogues Clin Neurosci. (2006) 8:109–13. doi: 10.31887/DCNS.2006.8.1/smarder
7. Fatouros-Bergman H, Cervenka S, Flyckt L, Edman G, Farde L. Meta-analysis of cognitive performance in drug-naïve patients with schizophrenia. Schizophr Res. (2014) 158:156–62. doi: 10.1016/j.schres.2014.06.034
8. Weinberg D, Lenroot R, Jacomb I, Allen K, Bruggemann J, Wells R, et al. Cognitive subtypes of schizophrenia characterized by differential brain volumetric reductions and cognitive decline. JAMA Psychiatry. (2016) 73:1251–9. doi: 10.1001/jamapsychiatry.2016.2925
9. Reichenberg A. The assessment of neuropsychological functioning in schizophrenia. Dialogues Clin Neurosci. (2010) 12:383–92. doi: 10.31887/DCNS.2010.12.3/areichenberg
10. Green MF, Horan WP, Lee J. Nonsocial and social cognition in schizophrenia: current evidence and future directions. World Psychiatry. (2019) 18:146–61. doi: 10.1002/wps.20624
11. McCutcheon RA, Keefe RSE, McGuire PK. Cognitive impairment in schizophrenia: aetiology, pathophysiology, and treatment. Mol Psychiatry. (2023) 28:1902–18. doi: 10.1038/s41380-023-01949-9
12. Hill SK, Bishop JR, Palumbo D, Sweeney JA. Effect of second-generation antipsychotics on cognition: current issues and future challenges. Expert Rev Neurother. (2010) 10:43–57. doi: 10.1586/ern.09.143
13. Horan WP, Catalano LT, Green MF. An update on treatment of cognitive impairment associated with schizophrenia. Curr Top Behav Neurosci. (2023) 63:407–36. doi: 10.1007/7854_2022_382
14. Sinkeviciute I, Begemann M, Prikken M, Oranje B, Johnsen E, Lei WU, et al. Efficacy of different types of cognitive enhancers for patients with schizophrenia: a meta-analysis. NPJ Schizophr. (2018) 4:22. doi: 10.1038/s41537-018-0064-6
15. Conn KA, Burne THJ, Kesby JP. Subcortical dopamine and cognition in schizophrenia: looking beyond psychosis in preclinical models. Front Neurosci. (2020) 14:542. doi: 10.3389/fnins.2020.00542
16. Reddy-Thootkur M, Kraguljac NV, Lahti AC. The role of glutamate and GABA in cognitive dysfunction in schizophrenia and mood disorders - A systematic review of magnetic resonance spectroscopy studies. Schizophr Res. (2022) 249:74–84. doi: 10.1016/j.schres.2020.02.001
17. Newman EL, Gupta K, Climer JR, Monaghan CK, Hasselmo ME. Cholinergic modulation of cognitive processing: insights drawn from computational models. Front Behav Neurosci. (2012) 6:24. doi: 10.3389/fnbeh.2012.00024
18. Terry AV Jr., Callahan PM. [amp]]alpha;7 nicotinic acetylcholine receptors as therapeutic targets in schizophrenia: Update on animal and clinical studies and strategies for the future. Neuropharmacology. (2020) 170:108053. doi: 10.1016/j.neuropharm.2020.108053
19. Moreta MP, Burgos-Alonso N, Torrecilla M, Marco-Contelles J, Bruzos-Cidón C. Efficacy of acetylcholinesterase inhibitors on cognitive function in Alzheimer’s disease. Rev Rev Biomedicines. (2021) 9(11):1689. doi: 10.3390/biomedicines9111689
20. Barten DM, Albright CF. Therapeutic strategies for Alzheimer’s disease. Mol Neurobiol. (2008) 37:171–86. doi: 10.1007/s12035-008-8031-2
21. Bertrand D, Wallace TL. A review of the cholinergic system and therapeutic approaches to treat brain disorders. Curr Top Behav Neurosci. (2020) 45:1–28.
22. Paul SM, Yohn SE, Brannan SK, Neugebauer NM, Breier A. Muscarinic receptor activators as novel treatments for schizophrenia. Biol Psychiatry. (2024) S0006-3223(24)01173-9. doi: 10.1016/j.biopsych.2024.03.014
23. Sam C, Bordoni B. Physiology, acetylcholine. In: StatPearls. Treasure Island, Florida: StatPearls Publishing (2023).
24. Kudlak M, Tadi P. Physiology, muscarinic receptor. In: StatPearls. Treasure Island, Florida: StatPearls Publishing (2023).
25. Yohn SE, Weiden PJ, Felder CC, Stahl SM. Muscarinic acetylcholine receptors for psychotic disorders: bench-side to clinic. Trends Pharmacol Sci. (2022) 43:1098–112. doi: 10.1016/j.tips.2022.09.006
26. Felder CC. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J. (1995) 9:619–25. doi: 10.1096/fasebj.9.8.7768353
27. Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discovery. (2009) 8:41–54. doi: 10.1038/nrd2760
28. Jakubik J, El-Fakahany EE. Current advances in allosteric modulation of muscarinic receptors. Biomolecules. (2020) 10(2):325. doi: 10.3390/biom10020325
29. Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother. (2008) 8:1703–18. doi: 10.1586/14737175.8.11.1703
30. McArthur RA, Gray J, Schreiber R. Cognitive effects of muscarinic M1 functional agonists in non-human primates and clinical trials. Curr Opin Investig Drugs. (2010) 11:740–60.
31. Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. (1997) 54:465–73. doi: 10.1001/archneur.1997.00550160091022
32. Veroff AE, Bodick NC, Offen WW, Sramek JJ, Cutler NR. Efficacy of xanomeline in Alzheimer disease: cognitive improvement measured using the Computerized Neuropsychological Test Battery (CNTB). Alzheimer Dis Assoc Disord. (1998) 12:304–12. doi: 10.1097/00002093-199812000-00010
33. Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. (2008) 165:1033–9. doi: 10.1176/appi.ajp.2008.06091591
34. Johnson CR, Kangas BD, Jutkiewicz EM, Bergman J, Coop A. Drug design targeting the muscarinic receptors and the implications in central nervous system disorders. Biomedicines. (2022) 10(2):398. doi: 10.3390/biomedicines10020398
35. Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. (2013) 504:101–6. doi: 10.1038/nature12735
36. Nussinov R, Tsai CJ. The different ways through which specificity works in orthosteric and allosteric drugs. Curr Pharm Des. (2012) 18:1311–6. doi: 10.2174/138161212799436377
37. Kamal M, Jockers R. Bitopic ligands: all-in-one orthosteric and allosteric. F1000 Biol Rep. (2009) 1:77. doi: 10.3410/B1-77
38. Teal LB, Gould RW, Felts AS, Jones CK. Selective allosteric modulation of muscarinic acetylcholine receptors for the treatment of schizophrenia and substance use disorders. Adv Pharmacol. (2019) 86:153–96. doi: 10.1016/bs.apha.2019.05.001
39. Slosky LM, Caron MG, Barak LS. Biased allosteric modulators: new frontiers in GPCR drug discovery. Trends Pharmacol Sci. (2021) 42:283–99. doi: 10.1016/j.tips.2020.12.005
40. Kaoullas MG, Thal DM, Christopoulos A, Valant C. Ligand bias at the muscarinic acetylcholine receptor family: opportunities and challenges. Neuropharmacology. (2024), 110092. doi: 10.1016/j.neuropharm.2024.110092
41. Gilmour G, Dix S, Fellini L, Gastambide F, Plath N, Steckler T, et al. NMDA receptors, cognition and schizophrenia–testing the validity of the NMDA receptor hypofunction hypothesis. Neuropharmacology. (2012) 62:1401–12. doi: 10.1016/j.neuropharm.2011.03.015
42. Kruse AO, Bustillo JR. Glutamatergic dysfunction in schizophrenia. Transl Psychiatry. (2022) 12:500. doi: 10.1038/s41398-022-02253-w
43. Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. (2006) 26:365–84. doi: 10.1007/s10571-006-9062-8
44. Nakazawa K, Jeevakumar V, Nakao K. Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr. (2017) 3:7. doi: 10.1038/s41537-016-0003-3
45. Kehrer C, Maziashvili N, Dugladze T, Gloveli T. Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front Mol Neurosci. (2008) 1:6. doi: 10.3389/neuro.02.006.2008
46. Bhandari A, Voineskos D, Daskalakis ZJ, Rajji TK, Blumberger DM. A review of impaired neuroplasticity in schizophrenia investigated with non-invasive brain stimulation. Front Psychiatry. (2016) 7:45. doi: 10.3389/fpsyt.2016.00045
47. Lovinger DM. Communication networks in the brain: neurons, receptors, neurotransmitters, and alcohol. Alcohol Res Health. (2008) 31:196–214.
48. Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. (2008) 33:18–41. doi: 10.1038/sj.npp.1301559
49. Huang Q, Liao C, Ge F, Ao J, Liu T. Acetylcholine bidirectionally regulates learning and memory. J Neurorestoratology. (2022) 10:100002. doi: 10.1016/j.jnrt.2022.100002
50. Westbrook A, Braver TS. Dopamine does double duty in motivating cognitive effort. Neuron. (2016) 89:695–710. doi: 10.1016/j.neuron.2015.12.029
51. Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, et al. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. (2003) 6:51–8. doi: 10.1038/nn992
52. Drinkenburg WH, Sondag HN, Coenders CJ, Andrews JS, Vossen JM. Effects of selective antagonism or depletion of the cholinergic system on visual discrimination performance in rats. Behav Pharmacol. (1995) 6:695–702. doi: 10.1097/00008877-199511000-00005
53. Hunter AJ, Roberts FF. The effect of pirenzepine on spatial learning in the Morris Water Maze. Pharmacol Biochem Behav. (1988) 30:519–23. doi: 10.1016/0091-3057(88)90490-X
54. Bubser M, Byun N, Wood MR, Jones CK. Muscarinic receptor pharmacology and circuitry for the modulation of cognition. Handb Exp Pharmacol. (2012) (208):121–66. doi: 10.1007/978-3-642-23274-9_7
55. Hassani SA, Neumann A, Russell J, Jones CK, Womelsdorf T. M(1)-selective muscarinic allosteric modulation enhances cognitive flexibility and effective salience in nonhuman primates. Proc Natl Acad Sci U.S.A. (2023) 120:e2216792120. doi: 10.1073/pnas.2216792120
56. Yohn SE, Conn PJ. Positive allosteric modulation of M(1) and M(4) muscarinic receptors as potential therapeutic treatments for schizophrenia. Neuropharmacology. (2018) 136:438–48. doi: 10.1016/j.neuropharm.2017.09.012
57. Kurimoto E, Yamada R, Hirakawa T, Kimura H. Therapeutic potential of TAK-071, a muscarinic M(1) receptor positive allosteric modulator with low cooperativity, for the treatment of cognitive deficits and negative symptoms associated with schizophrenia. Neurosci Lett. (2021) 764:136240. doi: 10.1016/j.neulet.2021.136240
58. Nguyen HTM, van der Westhuizen ET, Langmead CJ, Tobin AB, Sexton PM, Christopoulos A, et al. Opportunities and challenges for the development of M(1) muscarinic receptor positive allosteric modulators in the treatment for neurocognitive deficits. Br J Pharmacol. (2024) 181:2114–42. doi: 10.1111/bph.v181.14
59. Bartko SJ, Romberg C, White B, Wess J, Bussey TJ, Saksida LM. Intact attentional processing but abnormal responding in M1 muscarinic receptor-deficient mice using an automated touchscreen method. Neuropharmacology. (2011) 61:1366–78. doi: 10.1016/j.neuropharm.2011.08.023
60. Bubser M, Bridges TM, Dencker D, Gould RW, Grannan M, Noetzel MJ, et al. Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem Neurosci. (2014) 5:920–42. doi: 10.1021/cn500128b
61. Koshimizu H, Leiter LM, Miyakawa T. M4 muscarinic receptor knockout mice display abnormal social behavior and decreased prepulse inhibition. Mol Brain. (2012) 5:10. doi: 10.1186/1756-6606-5-10
62. Galloway CR, Lebois EP, Shagarabi SL, Hernandez NA, Manns JR. Effects of selective activation of M1 and M4 muscarinic receptors on object recognition memory performance in rats. Pharmacology. (2014) 93:57–64. doi: 10.1159/000357682
63. Gould RW, Grannan MD, Gunter BW, Ball J, Bubser M, Bridges TM, et al. Cognitive enhancement and antipsychotic-like activity following repeated dosing with the selective M(4) PAM VU0467154. Neuropharmacology. (2018) 128:492–502. doi: 10.1016/j.neuropharm.2017.07.013
64. Wu L, Chen D, Jia H, Zhao L, Liu N, Shen H. NS-136, a selective muscarinic receptor positive allosteric modulator (PAM) for the treatment of schizophrenia, Neuroscience 2023. Washington, DC (2023).
65. Lange HS, Vardigan JD, Cannon CE, Puri V, Henze DA, Uslaner JM. Effects of a novel M4 muscarinic positive allosteric modulator on behavior and cognitive deficits relevant to Alzheimer’s disease and schizophrenia in rhesus monkey. Neuropharmacology. (2021) 197:108754. doi: 10.1016/j.neuropharm.2021.108754
66. Byun NE, Grannan M, Bubser M, Barry RL, Thompson A, Rosanelli J, et al. Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100. Neuropsychopharmacology. (2014) 39:1578–93. doi: 10.1038/npp.2014.2
67. Jones GC. Individual differences in markers of cholinergic signaling correlating to fear and extinction learning. (Master’s thesis). University of South Carolina. (2017).
68. Malkova L, Kozikowski AP, Gale K. The effects of huperzine A and IDRA 21 on visual recognition memory in young macaques. Neuropharmacology. (2011) 60:1262–8. doi: 10.1016/j.neuropharm.2010.12.018
69. Vijayraghavan S, Major AJ, Everling S. Muscarinic M1 receptor overstimulation disrupts working memory activity for rules in primate prefrontal cortex. Neuron. (2018) 98:1256–1268.e4. doi: 10.1016/j.neuron.2018.05.027
70. Galvin VC, Yang ST, Paspalas CD, Yang Y, Jin LE, Datta D, et al. Muscarinic M1 receptors modulate working memory performance and activity via KCNQ potassium channels in the primate prefrontal cortex. Neuron. (2020) 106:649–661.e4. doi: 10.1016/j.neuron.2020.02.030
71. Baker S, Chin CL, Basso AM, Fox GB, Marek GJ, Day M. Xanomeline modulation of the blood oxygenation level-dependent signal in awake rats: development of pharmacological magnetic resonance imaging as a translatable pharmacodynamic biomarker for central activity and dose selection. J Pharmacol Exp Ther. (2012) 341:263–73. doi: 10.1124/jpet.111.188797
72. Shannon HE, Rasmussen K, Bymaster FP, Hart JC, Peters SC, Swedberg MD, et al. Xanomeline, an M(1)/M(4) preferring muscarinic cholinergic receptor agonist, produces antipsychotic-like activity in rats and mice. Schizophr Res. (2000) 42:249–59. doi: 10.1016/S0920-9964(99)00138-3
73. Si W, Zhang X, Niu Y, Yu H, Lei X, Chen H, et al. A novel derivative of xanomeline improves fear cognition in aged mice. Neurosci Lett. (2010) 473:115–9. doi: 10.1016/j.neulet.2010.02.031
74. Haam J, Yakel JL. Cholinergic modulation of the hippocampal region and memory function. J Neurochem. (2017) 142 Suppl 2:111–21. doi: 10.1111/jnc.14052
75. Levey AI. Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci U.S.A. (1996) 93:13541–6. doi: 10.1073/pnas.93.24.13541
76. Hasselmo ME. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. (2006) 16:710–5. doi: 10.1016/j.conb.2006.09.002
77. Gulledge AT, Kawaguchi Y. Phasic cholinergic signaling in the hippocampus: functional homology with the neocortex? Hippocampus. (2007) 17:327–32. doi: 10.1002/hipo.20279
78. Dannenberg H, Young K, Hasselmo M. Modulation of hippocampal circuits by muscarinic and nicotinic receptors. Front Neural Circuits. (2017) 11:102. doi: 10.3389/fncir.2017.00102
79. Dasari S, Gulledge AT. M1 and M4 receptors modulate hippocampal pyramidal neurons. J Neurophysiol. (2011) 105:779–92. doi: 10.1152/jn.00686.2010
80. Dennis SH, Pasqui F, Colvin EM, Sanger H, Mogg AJ, Felder CC, et al. Activation of muscarinic M1 acetylcholine receptors induces long-term potentiation in the hippocampus. Cereb Cortex. (2016) 26:414–26. doi: 10.1093/cercor/bhv227
81. Kremin T, Gerber D, Giocomo LM, Huang SY, Tonegawa S, Hasselmo ME. Muscarinic suppression in stratum radiatum of CA1 shows dependence on presynaptic M1 receptors and is not dependent on effects at GABA(B) receptors. Neurobiol Learn Mem. (2006) 85:153–63. doi: 10.1016/j.nlm.2005.09.005
82. Leaderbrand K, Chen HJ, Corcoran KA, Guedea AL, Jovasevic V, Wess J, et al. Muscarinic acetylcholine receptors act in synergy to facilitate learning and memory. Learn Mem. (2016) 23:631–8. doi: 10.1101/lm.043133.116
83. Pereyra M, Medina JH. AMPA receptors: a key piece in the puzzle of memory retrieval. Front Hum Neurosci. (2021) 15:729051. doi: 10.3389/fnhum.2021.729051
84. Zhao L-X, Ge Y-H, Li J-B, Xiong C-H, Law P-Y, Xu J-R, et al. M1 muscarinic receptors regulate the phosphorylation of AMPA receptor subunit GluA1 via a signaling pathway linking cAMP-PKA and PI3K-Akt. FASEB J. (2019) 33:6622–31. doi: 10.1096/fj.201802351R
85. Zhao LX, Ge YH, Xiong CH, Tang L, Yan YH, Law PY, et al. M1 muscarinic receptor facilitates cognitive function by interplay with AMPA receptor GluA1 subunit. FASEB J. (2018) 32:4247–57. doi: 10.1096/fj.201800029R
86. Yi F, Ball J, Stoll KE, Satpute VC, Mitchell SM, Pauli JL, et al. Direct excitation of parvalbumin-positive interneurons by M1 muscarinic acetylcholine receptors: roles in cellular excitability, inhibitory transmission and cognition. J Physiol. (2014) 592:3463–94. doi: 10.1113/tjp.2014.592.issue-16
87. Berkeley JL, Gomeza J, Wess J, Hamilton SE, Nathanson NM, Levey AI. M1 muscarinic acetylcholine receptors activate extracellular signal-regulated kinase in CA1 pyramidal neurons in mouse hippocampal slices. Mol Cell Neurosci. (2001) 18:512–24. doi: 10.1006/mcne.2001.1042
88. Peng S, Zhang Y, Zhang J, Wang H, Ren B. ERK in learning and memory: a review of recent research. Int J Mol Sci. (2010) 11:222–32. doi: 10.3390/ijms11010222
89. Albert-Gascó H, Ros-Bernal F, Castillo-Gómez E, Olucha-Bordonau FE. MAP/ERK signaling in developing cognitive and emotional function and its effect on pathological and neurodegenerative processes. Int J Mol Sci. (2020) 21(12):4471. doi: 10.3390/ijms21124471
90. Thorn CA, Moon J, Bourbonais CA, Harms J, Edgerton JR, Stark E, et al. Striatal, hippocampal, and cortical networks are differentially responsive to the M4- and M1-muscarinic acetylcholine receptor mediated effects of xanomeline. ACS Chem Neurosci. (2019) 10:1753–64. doi: 10.1021/acschemneuro.8b00625
91. Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, et al. Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron. (2002) 33:615–24. doi: 10.1016/S0896-6273(02)00587-1
92. Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, et al. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. (2008) 4:42–50. doi: 10.1038/nchembio.2007.55
93. Spurny B, Seiger R, Moser P, Vanicek T, Reed MB, Heckova E, et al. Hippocampal GABA levels correlate with retrieval performance in an associative learning paradigm. Neuroimage. (2020) 204:116244. doi: 10.1016/j.neuroimage.2019.116244
94. Tamminga CA, Southcott S, Sacco C, Wagner AD, Ghose S. Glutamate dysfunction in hippocampus: relevance of dentate gyrus and CA3 signaling. Schizophr Bull. (2012) 38:927–35. doi: 10.1093/schbul/sbs062
95. Thomsen M, Sørensen G, Dencker D. Physiological roles of CNS muscarinic receptors gained from knockout mice. Neuropharmacology. (2018) 136:411–20. doi: 10.1016/j.neuropharm.2017.09.011
96. Hazon O, Minces VH, Tomàs DP, Ganguli S, Schnitzer MJ, Jercog PE. Noise correlations in neural ensemble activity limit the accuracy of hippocampal spatial representations. Nat Commun. (2022) 13:4276. doi: 10.1038/s41467-022-31254-y
97. Bloem B, Poorthuis RB, Mansvelder HD. Cholinergic modulation of the medial prefrontal cortex: the role of nicotinic receptors in attention and regulation of neuronal activity. Front Neural Circuits. (2014) 8:17. doi: 10.3389/fncir.2014.00017
98. Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ. Heterogeneity of phasic cholinergic signaling in neocortical neurons. J Neurophysiol. (2007) 97:2215–29. doi: 10.1152/jn.00493.2006
99. Saunders JA, Gandal MJ, Siegel SJ. NMDA antagonists recreate signal-to-noise ratio and timing perturbations present in schizophrenia. Neurobiol Dis. (2012) 46:93–100. doi: 10.1016/j.nbd.2011.12.049
100. Moran SP, Maksymetz J, Conn PJ. Targeting muscarinic acetylcholine receptors for the treatment of psychiatric and neurological disorders. Trends Pharmacol Sci. (2019) 40:1006–20. doi: 10.1016/j.tips.2019.10.007
101. Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. (2007) 56:141–54. doi: 10.1016/j.neuron.2007.08.025
102. Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH. M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci. (2009) 29:9888–902. doi: 10.1523/JNEUROSCI.1366-09.2009
103. Sailer CA, Hu H, Kaufmann WA, Trieb M, Schwarzer C, Storm JF, et al. Regional differences in distribution and functional expression of small-conductance Ca2+-activated K+ channels in rat brain. J Neurosci. (2002) 22:9698–707. doi: 10.1523/JNEUROSCI.22-22-09698.2002
104. Pancani T, Bolarinwa C, Smith Y, Lindsley CW, Conn PJ, Xiang Z. M4 mAChR-mediated modulation of glutamatergic transmission at corticostriatal synapses. ACS Chem Neurosci. (2014) 5:318–24. doi: 10.1021/cn500003z
105. Chatham CH, Frank MJ, Badre D. Corticostriatal output gating during selection from working memory. Neuron. (2014) 81:930–42. doi: 10.1016/j.neuron.2014.01.002
106. Cai M, Wang R, Liu M, Du X, Xue K, Ji Y, et al. Disrupted local functional connectivity in schizophrenia: An updated and extended meta-analysis. Schizophr (Heidelb). (2022) 8:93. doi: 10.1038/s41537-022-00311-2
107. Dwomoh L, Tejeda GS, Tobin AB. Targeting the M1 muscarinic acetylcholine receptor in Alzheimer’s disease. Neuronal Signal. (2022) 6:Ns20210004. doi: 10.1042/NS20210004
108. Perry KW, Nisenbaum LK, George CA, Shannon HE, Felder CC, Bymaster FP. The muscarinic agonist xanomeline increases monoamine release and immediate early gene expression in the rat prefrontal cortex. Biol Psychiatry. (2001) 49:716–25. doi: 10.1016/S0006-3223(00)01017-9
109. Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. (2013) 493:537–41. doi: 10.1038/nature11740
110. Haber SN, Fudge JL. The primate substantia nigra and VTA: integrative circuitry and function. Crit Rev Neurobiol. (1997) 11:323–42. doi: 10.1615/CritRevNeurobiol.v11.i4.40
111. Weele CMV, Siciliano CA, Tye KM. Dopamine tunes prefrontal outputs to orchestrate aversive processing. Brain Res. (2019) 1713:16–31. doi: 10.1016/j.brainres.2018.11.044
112. Müller U, von Cramon DY, Pollmann S. D1- versus D2-receptor modulation of visuospatial working memory in humans. J Neurosci. (1998) 18:2720–8. doi: 10.1523/JNEUROSCI.18-07-02720.1998
113. Takahashi YK, Roesch MR, Stalnaker TA, Haney RZ, Calu DJ, Taylor AR, et al. The orbitofrontal cortex and ventral tegmental area are necessary for learning from unexpected outcomes. Neuron. (2009) 62:269–80. doi: 10.1016/j.neuron.2009.03.005
114. Cai JX, Arnsten AF. Dose-dependent effects of the dopamine D1 receptor agonists A77636 or SKF81297 on spatial working memory in aged monkeys. J Pharmacol Exp Ther. (1997) 283:183–9.
115. Winterer G, Weinberger DR. Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci. (2004) 27:683–90. doi: 10.1016/j.tins.2004.08.002
116. Zhang ZW, Burke MW, Calakos N, Beaulieu JM, Vaucher E. Confocal analysis of cholinergic and dopaminergic inputs onto pyramidal cells in the prefrontal cortex of rodents. Front Neuroanat. (2010) 4:21. doi: 10.3389/fnana.2010.00021
117. Goldman-Rakic PS, Muly 3EC, Williams GV. D(1) receptors in prefrontal cells and circuits. Brain Res Brain Res Rev. (2000) 31:295–301. doi: 10.1016/S0165-0173(99)00045-4
118. Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. (2007) 10:376–84. doi: 10.1038/nn1846
119. Foster DJ, Wilson JM, Remke DH, Mahmood MS, Uddin MJ, Wess J, et al. Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of dopamine release. Neuron. (2016) 91:1244–52. doi: 10.1016/j.neuron.2016.08.017
120. Onali P, Olianas MC. Muscarinic M4 receptor inhibition of dopamine D1-like receptor signalling in rat nucleus accumbens. Eur J Pharmacol. (2002) 448:105–11. doi: 10.1016/S0014-2999(02)01910-6
121. Bourgognon JM, Cavanagh J. The role of cytokines in modulating learning and memory and brain plasticity. Brain Neurosci Adv. (2020) 4:2398212820979802. doi: 10.1177/2398212820979802
122. Magaki S, Mueller C, Dickson C, Kirsch W. Increased production of inflammatory cytokines in mild cognitive impairment. Exp Gerontol. (2007) 42:233–40. doi: 10.1016/j.exger.2006.09.015
123. Miller AH, Haroon E, Raison CL, Felger JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety. (2013) 30:297–306. doi: 10.1002/da.2013.30.issue-4
124. Momtazmanesh S, Zare-Shahabadi A, Rezaei N. Cytokine alterations in schizophrenia: an updated review. Front Psychiatry. (2019) 10:892. doi: 10.3389/fpsyt.2019.00892
125. Rosas-Ballina M, Valdés-Ferrer SI, Dancho ME, Ochani M, Katz D, Cheng KF, et al. Xanomeline suppresses excessive pro-inflammatory cytokine responses through neural signal-mediated pathways and improves survival in lethal inflammation. Brain Behav Immun. (2015) 44:19–27. doi: 10.1016/j.bbi.2014.07.010
126. Gurba K, Vogt S, Gereau R. (373) Muscarinic M4 positive allosteric modulators are antinociceptive in mouse inflammatory pain models. J Pain. (2019) 20:S67. doi: 10.1016/j.jpain.2019.02.069
127. Dean B, Bakker G, Ueda HR, Tobin AB, Brown A, Kanaan RAA. A growing understanding of the role of muscarinic receptors in the molecular pathology and treatment of schizophrenia. Front Cell Neurosci. (2023) 17:1124333. doi: 10.3389/fncel.2023.1124333
128. Bakker G, Vingerhoets C, Boucherie D, Caan M, Bloemen O, Eersels J, et al. Relationship between muscarinic M(1) receptor binding and cognition in medication-free subjects with psychosis. NeuroImage Clin. (2018) 18:713–9. doi: 10.1016/j.nicl.2018.02.030
129. Raedler TJ, Knable MB, Jones DW, Urbina RA, Gorey JG, Lee KS, et al. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am J Psychiatry. (2003) 160:118–27. doi: 10.1176/appi.ajp.160.1.118
130. Joshi YB, Thomas ML, Braff DL, Green MF, Gur RC, Gur RE, et al. Anticholinergic medication burden-associated cognitive impairment in schizophrenia. Am J Psychiatry. (2021) 178:838–47. doi: 10.1176/appi.ajp.2020.20081212
131. Selvaggi P, Fazio L, Toro VD, Mucci A, Rocca P, Martinotti G, et al. Effect of anticholinergic burden on brain activity during Working Memory and real-world functioning in patients with schizophrenia. Schizophr Res. (2023) 260:76–84. doi: 10.1016/j.schres.2023.08.015
132. McEvoy JP, Freter S. The dose-response relationship for memory impairment by anticholinergic drugs. Compr Psychiatry. (1989) 30:135–8. doi: 10.1016/0010-440X(89)90065-5
133. Li Z, Huang M, Ichikawa J, Dai J, Meltzer HY. N-desmethylclozapine, a major metabolite of clozapine, increases cortical acetylcholine and dopamine release in vivo via stimulation of M1 muscarinic receptors. Neuropsychopharmacology. (2005) 30:1986–95. doi: 10.1038/sj.npp.1300768
134. Molins C, Carceller-Sindreu M, Navarro H, Carmona C, Piñeiro M, Martínez E, et al. Plasma ratio of clozapine to N-desmethylclozapine can predict cognitive performance in treatment-resistant psychotic patients. Psychiatry Res. (2017) 258:153–7. doi: 10.1016/j.psychres.2017.10.010
135. Pak RW, Petrou SP, Staskin DR. Trospium chloride: a quaternary amine with unique pharmacologic properties. Curr Urol Rep. (2003) 4:436–40. doi: 10.1007/s11934-003-0023-1
136. Breier A, Brannan SK, Paul SM, Miller AC. Evidence of trospium’s ability to mitigate cholinergic adverse events related to xanomeline: phase 1 study results. Psychopharmacol (Berl). (2023) 240:1191–8. doi: 10.1007/s00213-023-06362-2
137. Brannan SK, Sawchak S, Miller AC, Lieberman JA, Paul SM, Breier A. Muscarinic cholinergic receptor agonist and peripheral antagonist for schizophrenia. N Engl J Med. (2021) 384:717–26. doi: 10.1056/NEJMoa2017015
138. Kaul I, Sawchak S, Correll CU, Kakar R, Breier A, Zhu H, et al. Efficacy and safety of the muscarinic receptor agonist KarXT (xanomeline-trospium) in schizophrenia (EMERGENT-2) in the USA: results from a randomised, double-blind, placebo-controlled, flexible-dose phase 3 trial. Lancet. (2024) 403:160–70. doi: 10.1016/S0140-6736(23)02190-6
139. Sauder C, Allen LA, Baker E, Miller AC, Paul SM, Brannan SK. Effectiveness of KarXT (xanomeline-trospium) for cognitive impairment in schizophrenia: post hoc analyses from a randomised, double-blind, placebo-controlled phase 2 study. Transl Psychiatry. (2022) 12:491. doi: 10.1038/s41398-022-02254-9
140. Horan W, Sauder C, Harvey PD, Ramsay IS, Paul SM, Brannan SK. The Impact of KarXT on cognitive impairment in acute schizophrenia: replication in pooled data from phase 3 trials Schizophrenia International Research Society 2024 Congress. Florence, Italy (2024).
141. Krystal JH, Kane JM, Correll CU, Walling DP, Leoni M, Duvvuri S, et al. Emraclidine, a novel positive allosteric modulator of cholinergic M4 receptors, for the treatment of schizophrenia: a two-part, randomised, double-blind, placebo-controlled, phase 1b trial. Lancet. (2022) 400:2210–20. doi: 10.1016/S0140-6736(22)01990-0
142. Hall H, Iulita MF, Gubert P, Flores Aguilar L, Ducatenzeiler A, Fisher A, et al. AF710B, an M1/sigma-1 receptor agonist with long-lasting disease-modifying properties in a transgenic rat model of Alzheimer’s disease. Alzheimers Dement. (2018) 14:811–23. doi: 10.1016/j.jalz.2017.11.009
143. Orciani C, Do Carmo S, Foret MK, Hall H, Bonomo Q, Lavagna A, et al. Early treatment with an M1 and sigma-1 receptor agonist prevents cognitive decline in a transgenic rat model displaying Alzheimer-like amyloid pathology. Neurobiol Aging. (2023) 132:220–32. doi: 10.1016/j.neurobiolaging.2023.09.010
144. Felder CC, Goldsmith PJ, Jackson K, Sanger HE, Evans DA, Mogg AJ, et al. Current status of muscarinic M1 and M4 receptors as drug targets for neurodegenerative diseases. Neuropharmacology. (2018) 136:449–58. doi: 10.1016/j.neuropharm.2018.01.028
145. Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. (2009) 373:31–41. doi: 10.1016/S0140-6736(08)61764-X
Keywords: muscarinic, cognition, acetylcholine, cognitive impairment associated with schizophrenia, schizophrenia, M1 receptor, M4 receptor
Citation: Yohn SE, Harvey PD, Brannan SK and Horan WP (2024) The potential of muscarinic M1 and M4 receptor activators for the treatment of cognitive impairment associated with schizophrenia. Front. Psychiatry 15:1421554. doi: 10.3389/fpsyt.2024.1421554
Received: 23 April 2024; Accepted: 03 September 2024;
Published: 04 October 2024.
Edited by:
Emilio Merlo Pich, Gelf Health SRL, ItalyReviewed by:
Morgan Thomsen, Region Hovedstad Psychiatry, DenmarkDavid Thal, Monash University, Australia
Copyright © 2024 Yohn, Harvey, Brannan and Horan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samantha E. Yohn, c2FtYW50aGEueW9obkBibXMuY29t