
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 23 April 2024
Sec. Autism
Volume 15 - 2024 | https://doi.org/10.3389/fpsyt.2024.1364858
This article is part of the Research Topic Autism Spectrum Disorders: Developmental Trajectories, Neurobiological Basis, Treatment Update, Volume III View all 6 articles
 Junzi Long1,2,3
Junzi Long1,2,3 Hui Li2,4
Hui Li2,4 Ying Liu1,2
Ying Liu1,2 Xingxing Liao1,2,3
Xingxing Liao1,2,3 Zhiqing Tang1,2
Zhiqing Tang1,2 Kaiyue Han1,2Jiarou Chen5
Kaiyue Han1,2Jiarou Chen5 Hao Zhang1,2,3,4,5*
Hao Zhang1,2,3,4,5*The hippocampus is one of the brain areas affected by autism spectrum disorder (ASD). Individuals with ASD typically have impairments in hippocampus-dependent learning, memory, language ability, emotional regulation, and cognitive map creation. However, the pathological changes in the hippocampus that result in these cognitive deficits in ASD are not yet fully understood. In the present review, we will first summarize the hippocampal involvement in individuals with ASD. We will then provide an overview of hippocampal structural and functional abnormalities in genetic, environment-induced, and idiopathic animal models of ASD. Finally, we will discuss some pharmacological and non-pharmacological interventions that show positive impacts on the structure and function of the hippocampus in animal models of ASD. A further comprehension of hippocampal aberrations in ASD might elucidate their influence on the manifestation of this developmental disorder and provide clues for forthcoming diagnostic and therapeutic innovation.
Autism spectrum disorder (ASD) is a developmental disability resulting from a combination of genetic and environmental factors, with an estimated prevalence of 0.72% globally (1). The most characteristic symptoms of ASD are impaired social communication and stereotyped or repetitive behaviors (2). In addition, ASD is associated with a typical profile of difficulties across various domains of cognition, including memory, learning, language ability, emotion, and cognitive map creation (3, 4). It has long been recognized that impaired memory for faces, working, and social scenes in individuals with ASD contributes to clinical manifestations of the disorder, such as executive dysfunction (5, 6). The co-occurrence of ASD and learning disability is common, with previous estimates suggesting that as many as three-quarters of individuals with ASD have impaired learning abilities, including language learning skills (7). Language delay is an important feature of autistic children – word and speech learning difficulties are noticeable early in development and continue throughout the school-aged years (8, 9). Individuals with ASD often experience emotional dysregulation, including symptoms of anxiety, which are associated with a range of negative mental and physical health outcomes (10). Data from several navigation and search tasks suggest that people with ASD were slower at learning spatial distribution regularities, less efficient in exploring an environment, and more likely to revisit the area they have already explored (11–13).
The hippocampus has consistently been a brain structure of interest in the seek for physiopathological mechanisms and rehabilitation treatment of ASD, given its important role in memory (14), learning (15), language ability (16), emotional regulation (17), and cognitive map creation (14). For example, recently researchers found that increased recruitment of the hippocampus compensated for decreased connectivity between medial temporal lobes and the posterior medial network during relational encoding tasks, which supported preserved episodic memory in individuals with ASD (18). Previous observations in ASD adults have also suggested that altered hippocampal functioning contributes to difficulties with structural learning that are likely to underlie more complex cognitive processes, including episodic memory, learning processes, and cognitive map creation (19). The language impairments observed in some individuals with ASD are linked to abnormalities in the hippocampal regions (20). Moreover, recent evidence indicates that altered neuronal projection and chemical metabolites in the amygdala-hippocampus region modulate emotional regulation in ASD (21, 22).
Herein, we will first review structural and functional abnormalities in the hippocampus of individuals with ASD and discuss discrepancies in the results of existing studies. In the next part, we will discuss the known genetic, environment-induced, and idiopathic animal models of ASD, mostly in mice and rats, that exhibit hippocampal synaptic plasticity impairments and hippocampus-dependent behavioral deficits. Then, we will summarize the main findings from pharmacological and non-pharmacological interventions for ASD that have been shown to positively affect the structure and function of the hippocampus. Directing more attention to the involvement of hippocampal pathology in the aetiology of ASD and ASD-related behaviors will provide new insights into pathogenic mechanisms, contributing to innovative molecularly or cellularly targeted therapies.
The adult human hippocampus, a prominent part of the limbic system, has a three-dimensional tortuous structure that resembles a seahorse because of its arched shape in the axial plane. Based on the cellular composition, the hippocampus can be anatomically divided into four subregions, namely cornu ammonis one (CA1) to cornu ammonis four (CA4) (23). In addition to the hippocampus, the hippocampal formation comprises the dentate gyrus, subiculum, presubiculum, parasubiculum, and entorhinal cortex. The dentate gyrus retains the capacity for neurogenesis throughout an individual’s lifespan, which is widely believed to play a pivotal role in cognitive processes such as learning and memory (Figure 1A) (24). The fornix is a primary outflow bundle of the hippocampus and wraps around the thalamus. The hippocampus extends its axons into the fornix to establish synaptic connections with neurons in the mammillary body, which then project fibers via the mammillothalamic tract toward the anterior thalamic nucleus. Subsequently, the cingulate gyrus that contains a large number of myelinated fibers receives a connection from the anterior thalamic nucleus. The cingulate gyrus curves over the corpus callosum and stretches into the parahippocampal region and the entorhinal cortex. In this way, it forms a closed loop by returning to the hippocampal formation. This intrinsic neural pathway, known as the Papez circuit, has long been considered to be responsible for regulating emotions, memories, and learning processes (Figure 1B) (25, 26).
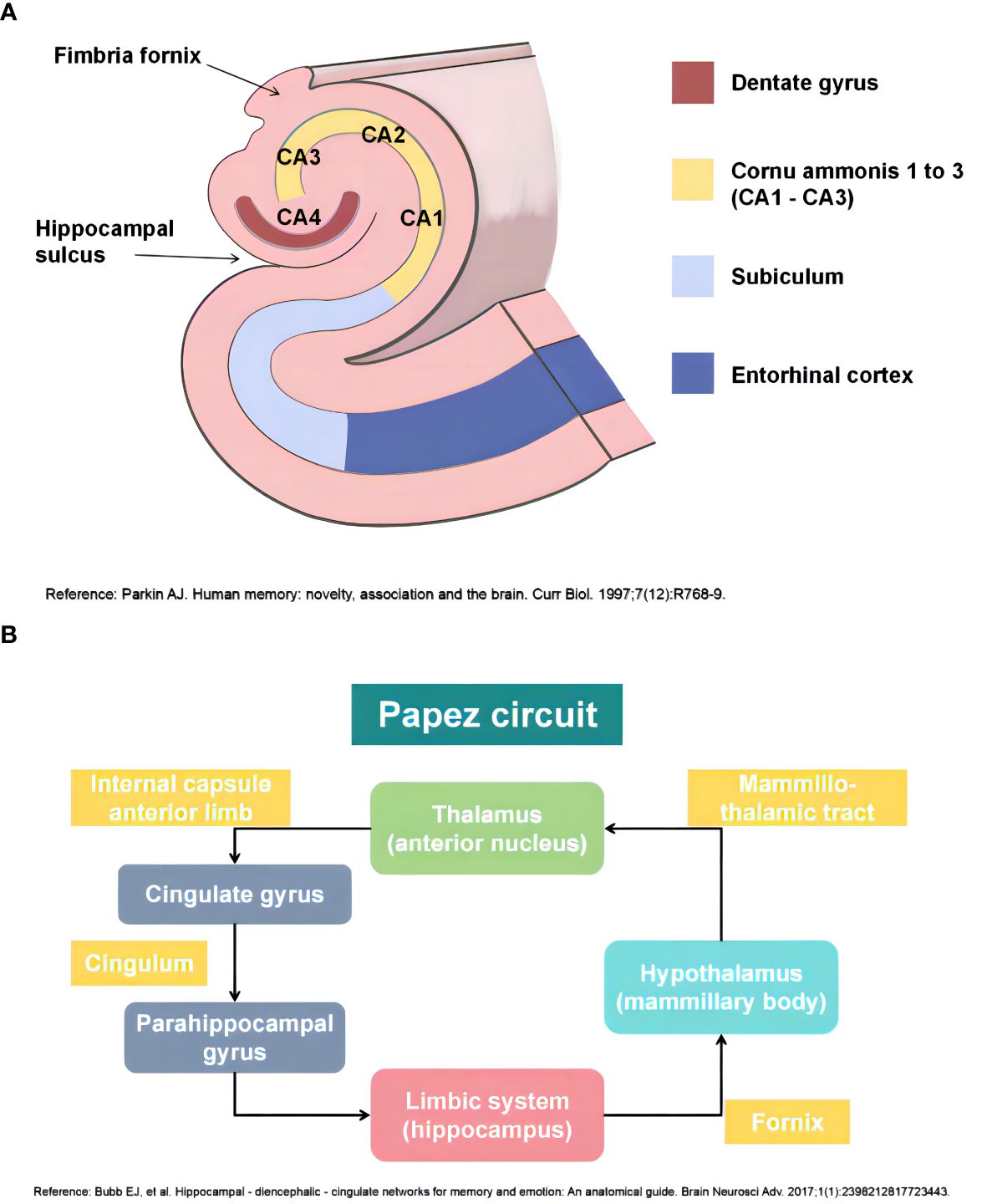
Figure 1 (A) Anatomy of the adult human hippocampus (Ref: Parkin AJ. Human memory: novelty, association and the brain. Curr Biol. 1997;7(12):R768-9); (B) Papez circuit of the human hippocampus (Ref: Bubb EJ, Kinnavane L, Aggleton JP. Hippocampal - diencephalic - cingulate networks for memory and emotion: An anatomical guide. Brain Neurosci Adv. 2017;1(1):2398212817723443). This figure is created by using Adobe Photoshop and PowerPoint.
The volume of the human hippocampus increases linearly throughout gestation and the first two years after birth, and continues to increase slowly thereafter, suggesting that the crucial developmental period of the human hippocampus is likely to be before the age of two (27, 28). Extensive research has proven that social and behavioral impairments specific to individuals with ASD also emerge between the ages of one-and-a-half and two years, which lead to diagnostic delays until at least three to four years of age (29, 30). It is therefore argued that the developmental timeline of the hippocampus is probably consistent with the behavioral developmental milestones of ASD (31). To sum up, considering the importance of the hippocampus in learning, memory, language ability, emotional regulation, and cognitive map creation, it could conceivably be hypothesized that gaining an understanding of the structural and functional characteristics of the hippocampus in ASD may offer novel perspectives on the pathogenesis and therapeutic targets for ASD.
The advancement of neuroimaging methods has significantly increased the ability to observe alterations in the morphology and brain activation patterns of individuals diagnosed with ASD over the past decades. In particular, since the late 1980s, when the first research of ASD using magnetic resonance imaging (MRI) was published, various MRI modalities, including different types of functional MRI, structural MRI, and diffusion tensor imaging, have been reported to effectively facilitate the non-invasive clinical diagnosis of ASD. These publications emphasize that the clinical symptoms of ASD are correlated with volumetric abnormalities in different areas of the brain, including the hippocampus. For instance, extensive MRI research has demonstrated abnormalities in hippocampal volume in individuals with ASD when compared to typically developing people, which is proven to be associated with aberrant autobiographical memory, impaired language skills, social communication deficit, and emotional problems in individuals with ASD (32–34).
Sussman and co-workers (35) have observed a decreased relative volume of the left hippocampus in 72 children and adolescents with ASD. Similarly, a study comparing autistic adults without intellectual disability and healthy community volunteers, found that the reduction in hippocampal volume in the autistic subjects was apparent when corrected for total brain volume (36). Another research based on the ABIDE database also found decreased gray-matter volume in the posterior hippocampus among a large sample of participants with ASD as compared to typically developing controls (32). As these studies all included high-functioning autistic individuals (Full-Scale IQ > 80), the reduction in hippocampal volume is likely to reflect features of autism itself, rather than features of the intellectual deficits that typically accompany autism (36). In other words, autistic traits have a robust correspondence with reduced brain volume in the hippocampus that is related to social processing, working memory, perceptual reasoning, and spatial learning. One possible hypothesis is that high-functioning autistic populations may experience an adaptive reduction in hippocampal volume as a result of their decreased specific functional demands on the hippocampus and altered interactions with the environment (37). Decreased hippocampal volume in ASD individuals was also related to impaired language skills, including verbal learning and memory (32, 38). It has also been suggested that left hippocampal volume is a positive predictor of early language development in females, but not in males (39). Furthermore, it is worth mentioning that in many studies that have reported reductions in hippocampal volume among ASD subjects, only reductions in relative hippocampal volume (percentage of total brain volume) have been observed rather than the absolute volume (35, 36, 40). Numerous studies have now firmly established that individuals with ASD have a significantly larger head circumference and a higher prevalence of macrocephaly than age-matched healthy subjects (41), which could explain the reduced relative hippocampal volume in the ASD group.
Surprisingly, in contrast to the aforementioned findings, an MRI study discovered larger volumes of the hippocampus bilaterally in ASD adults with IQ > 100 when compared to healthy controls (42). Likewise, Xu et al. (43) found evidence of larger absolute hippocampal volume in adults with ASD and adequate intelligence than that of healthy adults based on the ABIDE datasets. The Intense World Theory posits that ASD traits may be attributed to the activation of a molecular syndrome, which sensitizes gene expression pathways to excessively respond to environmental stimulation. Under normal circumstances, these pathways facilitate brain development through enriched environments; however, when sensitized, they can lead to accelerated brain development in response to environmental stimuli (44). This theory focuses on the neocortex and the amygdala, but it may also apply to other brain regions, such as the hippocampus. For example, the chronic stress process in ASD enhances amygdala activity, which may initially result in hypertrophy of the hippocampus as it moderates the amygdala activity through multiple reciprocal connections (34). The amygdala and hippocampus also play a major role in olfaction. Research has shown that the severity of taste and smell dysfunction is inversely related to the hippocampal volume, and females with ASD have more severe taste and olfactory impairment than males (45). These theories reflect characteristic behavioral variations in ASD and remain to be elucidated in the future. Another possible hypothesis suggests that the increased size of the hippocampus in people with ASD may be due to an enhancement in experience-dependent function (43, 46). Indeed, individuals with ASD have been reported to excel in certain cognitive domains for which the hippocampus is responsible (e.g. visuospatial abilities) compared to typically developing individuals, but these differences may be driven by task demands (31). Current scientific opinion still holds that ASD can lead to a range of hippocampus-related dysfunctions, which are linked to abnormalities in the volume of the hippocampus (32–34). The assertion that individuals with ASD are more proficient than their neurotypical peers in a specific hippocampus-dependent function is a limited occurrence and cannot be generalized to the entire autistic population (4).
In addition to these observations, a number of researches have reported no difference in the hippocampal volume between ASD individuals and healthy volunteers (47–49). The above inconsistent findings on hippocampal size can actually be attributed to the heterogeneity of ASD individuals. For example, the age range is a significant confounding factor because individuals with ASD appear to consistently have larger hippocampus than their healthy peers from childhood to adolescence (46), but in adulthood, their hippocampus begins to decrease (36), or in some cases, remain stable (47). Although several studies have reported no correlation between age and hippocampal volume in individuals with ASD (35, 42), this may be because of the broad age range of participants, which appears to neutralize the disparities. Second, sex is another important determinant of hippocampal volume in people with ASD. Possibly due to the higher prevalence of ASD in males, existing studies have included a larger sample of male participants. It has been reported that male ASD subjects had faster hippocampal volume growth and greater hippocampal asymmetry than female subjects (50, 51). Relative volumes of both the left and right hippocampus were smaller in females with ASD than in age-matched males (35, 39). Third, many studies have included autism, Asperger’s syndrome, and pervasive developmental disorder (32, 46–48), and a mixed population with varying levels of intellectual disabilities (33, 46, 48) and seizure disorders (52). There are also significant variations in medication statuses, brain MRI scanners, measurement approaches, controls for total brain volume, and anatomical definitions of the hippocampus in related studies. Furthermore, hippocampal abnormalities in ASD could potentially be genetically based, as evidenced by the larger hippocampal volume observed in parents of children with ASD compared to control subjects (53). Due to the high variability in hippocampal volume in typical individuals (54), and the current prevalence of cross-sectional studies with limited sample sizes, considerably more MRI studies will need to be done to determine hippocampal volume changes in ASD, particularly for a specific autistic group with large sample sizes and a longitudinal design.
Postmortem examinations were conducted in ten cases of ASD, comprising eight males and two females, with eight individuals presenting intellectual disability and five individuals exhibiting seizure disorder, ranging from 4 to 29 years old. The findings revealed reduced neural size and increased neuronal density within the hippocampal formation (55–57), similar to typical hippocampal developmental trends. It has previously been observed in an immunohistochemical study, that the density of parvalbumin-, calbindin-, and calretinin-immunoreactive interneurons within the subfields of the hippocampus was increased in cases of ASD compared to controls (58). Increased gray matter density was found in the hippocampal formation and peri-hippocampal cortex of children with ASD, correlating with symptoms of impaired social interaction and mnemonic function in ASD (59, 60). These observations of heightened neuron density in the hippocampal formation could be attributed to incorrect neuronal migration (59). The distorted shape of the dentate granule layer, forming irregular circles and loops, was indicative of abnormal neuronal migration and seemed to be another evidence of the phenomenon described above (61). Additionally, Golgi studies of CA4 and CA1 neurons revealed a noticeable decrease in dendritic branching complexity in two ASD children compared to their age-matched controls. Nevertheless, it remains uncertain whether increased hippocampal neuron density, fewer neuron size volumes, and dendritic branching lead to corresponding reductions in the macroscopic size of the hippocampus. If this is the case, it could account for the decrease in MRI volumes observed in the hippocampus of individuals with ASD. The dentate gyrus development distortion in ASD was manifested by granule cell migration to the molecular layer and the formation of an extra granule cell layer (61). A previous MRI evidence of ASD also suggested that significantly smaller cross-sectional areas of the area dentata than healthy subjects could be attributed to the hypoplasia of the dentate gyrus and CA4 subfield (52). However, caution should be exercised when interpreting these findings as this MRI study and prior neuroanatomical researches have included ASD individuals with seizure disorders that are known to be connected with a decrease in hippocampal volume (62). Moreover, most current autopsy studies have been conducted in males, and it is unclear whether there are sex differences in hippocampal pathological anatomy in ASD. Three-dimensional MRI measurements of hippocampal shape variation in children with ASD indicated an upward curvature in the head and tail of the hippocampus, as well as inward deformation in its medial aspect (48). In terms of morphological structure, the hippocampal regions in children with ASD may be the initial site of alteration and a significant area in brain imaging that can be utilized for diagnosing ASD in children (63). Considering that clinical autistic symptoms were associated with various structural abnormalities in hippocampal regions, additional neuropathological and imaging studies will be needed to obtain the objective criteria for early diagnosis of children with ASD based on the morphological changes of the hippocampus.
As documented through quantitative MRI and 3D pseudo-continuous arterial spin labeling, cerebral hypoperfusion has been observed in the hippocampus among children with ASD in comparison to healthy controls, which may not be related to sex differences (63–65). Proper brain oxygenation is essential not only for the early development of neurons but also for optimal functioning. Previous research has indicated a positive correlation between hippocampal blood flow and spatial memory performance (66). Therefore, measuring hippocampal blood flow may be an effective method for brain imaging diagnosis in children with ASD. The reduction in cerebral blood flow in the hippocampus also led to a decrease in hemoglobin iron and non-hemoglobin iron in the blood, which ultimately resulted in a decrease in iron content in the hippocampus (63, 65). The hippocampal growth is closely related to blood perfusion, but current findings suggest that reduced blood perfusion and iron ion levels within the hippocampal region do not have a significant impact on the hippocampal volume of ASD children (63–65). One contradictory result was that as compared to healthy controls, high-functioning ASD adults showed a significant cerebral blood flow increase in the right parahippocampal cortex (67). Differences in functional ability and sedative use among the included ASD participants may explain this inconsistency.
Previous magnetic resonance spectroscopy studies have suggested altered chemical metabolism in hippocampal regions in ASD. Most studies of the hippocampus have reported a trend toward reduced N-acetylaspartate concentrations and N-acetylaspartate/creatine ratios in children with ASD (21, 68, 69). N-acetyl-aspartate synthesis occurs exclusively in the mitochondria of neuronal cells and is widely accepted as a marker for axonal and neuronal integrity and viability (70), so a diminished level of N-acetyl-aspartate may signify an irregularity in hippocampal regional neuronal development and mitochondrial function. This hypothesis was corroborated by a previous study that demonstrated a positive correlation between N-acetyl-aspartate levels in the right hippocampal region and performance IQ in the ASD group (69). However, among adult subjects, Page et al. (71) and O’Brien et al. (72) discovered no variation in NAA levels between the ASD group and the matched comparison group. The impact of age on alterations to metabolites in distinct areas of the brain has been evidenced; for instance, NAA levels are diminished in a neonate and subsequently surge during cerebral development (70). When comparing only boys with and without ASD, there were also no differences in NAA, choline, and creatine concentrations in the hippocampal region (73). People with ASD had a significantly higher concentration of glutamate+glutamine, aka Glx in the amygdala-hippocampal complex than comparison subjects (71), which is consistent with prior data in the auditory cortex of people with ASD (74). As glutamate is the most abundant excitatory neurotransmitter, the pathogenesis of ASD is likely to be associated with increased hippocampal excitability. Previous researches have identified an association between choline/creatine ratio elevation within the hippocampus and the severity of autistic symptoms, including language impairment (69, 75). Similarly, high concentrations of creatine and phosphocreatine in the hippocampal formation of ASD individuals had a significant positive correlation with their aggressive behaviors (70, 76). In sum, it can be hypothesized that disturbances of metabolic substances in the hippocampus are associated with abnormalities in hippocampus-dependent functions.
In the study of the autistic human hippocampus, functional MRI has emerged as a powerful tool for mapping disrupted functional connectivity, which denotes the measurement of the temporal synchronization of activity between the hippocampus and other brain regions while a person is at rest or involved in a given cognitive activity. For example, in ASD children and adolescents, the resting-state functional connectivity strength between the left posterior hippocampus and the posterior cingulate cortex has a significant negative correlation with successful memory performance (77). Aberrant circuitry between the hippocampus and posterior cingulate cortex was found to be a common feature in ASD associated with reduced general and facial memory (78). A preliminary neuropsychological and neuroanatomical investigation has indicated that hippocampal and parahippocampal abnormalities are associated with memory deficits in low-functioning ASD individuals, which in turn undermines their learning and language abilities (20). Earlier data from experimental tasks suggested that adults with ASD had particular difficulties with structural learning due to changes in the function of the hippocampus, which played a critical role in the aetiology of ASD (19). During learning, strong functional connectivity was observed between the hippocampus and caudate in both ASD and typically developing adolescents, but this connectivity was positively associated with task performance in ASD and negatively associated with performance in typically developing adolescents (79). Infants with low familial risk for developing ASD exhibited stronger resting-state functional connectivity between the right posterior superior temporal gyrus and the right hippocampus, as well as parahippocampal gyrus, in comparison to high-risk infants. The atypical and immature functional connectivity in high-risk infants could cascade into their later language deficits (80). During the emotional resonance condition, boys with ASD displayed reduced responses in the bilateral hippocampus, indicating potential difficulties with emotional information integration (81). Neural abnormalities within the hippocampus area may potentially place young individuals with ASD at risk for anxiety or other emotional and behavioral dysfunctions (82). Finally, in spatial memory tasks involving the dorsolateral prefrontal-hippocampal circuit, people with ASD showed worse performance than typical controls (83). When performing the virtual reality shopping task, participants with ASD also displayed lower activation in the parahippocampal gyrus, which is implicated in scene recognition and spatial navigation (84). These studies investigate a range of functional domains linked to the hippocampus, indicating that altered functional connectivity between the hippocampal region and other brain networks may account for diverse autism symptoms.
As discussed above, changes in hippocampal volume, morphology, blood flow, metabolites, and functional connectivity may underlie altered hippocampal functions, including the formation and consolidation of memory, learning, language abilities, emotional regulation, and cognitive map creation (Figure 2).
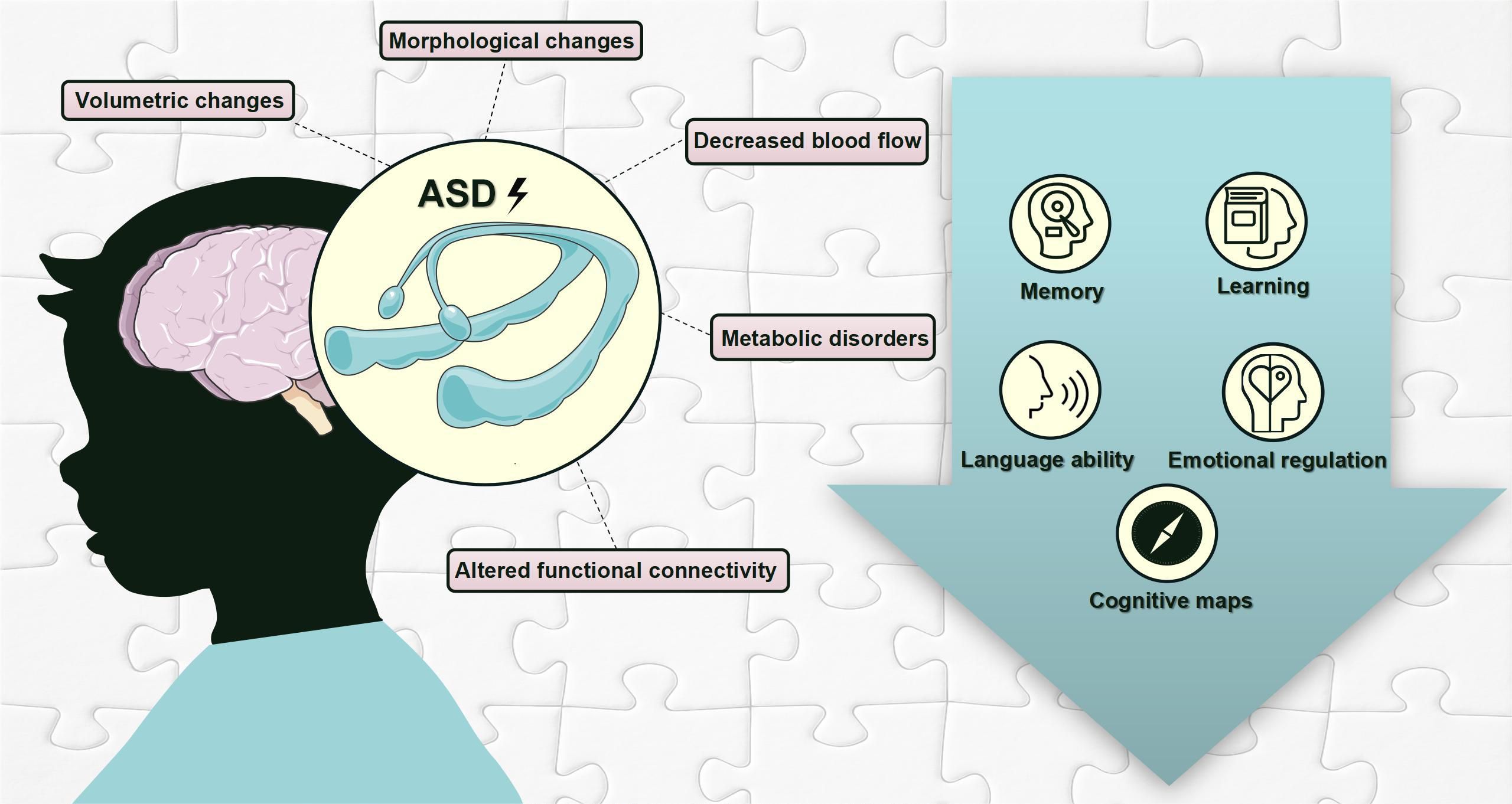
Figure 2 Hippocampal pathological changes and hippocampus-dependent behavioral deficits in individuals with ASD. This figure is created by using PowerPoint.
Over recent decades, numerous animal models have been constructed to examine the mechanisms underlying the pathophysiology of ASD, with growing evidence substantiating abnormalities in the structure and function of the hippocampus in ASD. These animal models can be generally categorized into genetic, environment-induced, and idiopathic models.
It is widely accepted that ASD is a heritable disorder with origins in copy number variants, unusual mutations of a single gene, and cumulative effects of particular gene variants (85). To date, hundreds of genetically modified models (especially monogenic models) have intentionally replicated identified human autistic syndromes, both syndromic and non-syndromic ASD. The genetic cause has been clearly defined in cases of syndromic ASD, which often presents alongside ASD-related behavioral phenotypes, and on the contrary, non-syndromic ASD lacks a distinct phenotype (86).
Transcriptional silencing of the Fragile X messenger ribonucleoprotein 1 (Fmr1) gene encoding the Fragile X Messenger Ribonucleoprotein causes fragile X syndrome, the most common monogenic cause of ASD characterized by intellectual and language disabilities (87). Compared to wild-type mice, the dendritic spines of hippocampal neurons in Fmr1 knockout (KO) mice are longer and thinner, and the density of stubby and mushroom-shaped (mature) spines is lower (88–91). Lazarov and colleagues reported a notable reduction in the number of neural progenitor/stem cells and the subsequent survival of these cells in the subgranular layer of the dentate gyrus in Fmr1-KO mice (92). Moreover, hippocampal neurons in Fmr1-KO mice had fewer functional synaptic connections, which develop at a slower rate and produce smaller excitatory synaptic currents relative to wild-type controls (93). In agreement, Klemmer et al. (94) discovered that 2-week-old Fmr1-KO mice exhibited an early-stage presynaptic phenotype marked by diminutive synaptic structures, decreased number of vesicles per cluster surface, and an overall reduction in the number of synaptic vesicles. Fmr1-KO mice also showed a prominent reduction in synaptic density and thickness of postsynaptic density, and an increase in synaptic cleft width (91). On the other hand, while baseline neurotransmitter release and short-term synaptic plasticity are unaffected at CA1 synapse in both younger and older Fmr1-KO mice (95), synaptic release probability is excessively elevated in CA1-CA3 areas during repetitive activity, resulting in abnormal short-term plasticity (96). Regarding alterations in long-term plasticity, Fmr1-KO mice have specific impairments of glutamatergic signaling in the hippocampus. Abnormally enhanced metabotropic glutamate receptor-dependent long-term depression (mGluR-LTD) in the CA1 region and defective long-term potentiation (LTP) of N-methyl-D-aspartate receptors (NMDA) in the dentate gyrus have been considered as established phenotypes of Fmr1-KO mice (95, 97).
These abnormalities observed in the hippocampus may underlie, at least in part, behavioral and cognitive changes of fragile X syndrome animal models. Extensive research has shown that fragile X syndrome murine models have significant deficits in hippocampus-dependent forms of spatial learning and memory as tested using the Morris water maze (91, 98–102). During the novel object recognition test, it was observed that Fmr1 mutant mice spent significantly more time sniffing the old object and less time exploring the novel object than wild types, suggesting visual recognition memory deficits in these mice (100, 101). Furthermore, Fmr1-KO mice had deficiencies in hippocampus-dependent fear memory, characterized by low levels of freezing behavior response to fear conditioning (91, 102). It has previously been noted that Fmr1-KO mice had circadian defects involved in hippocampus-dependent memory, which may lead to sleep disturbance (103). The abnormal hippocampal neural activity of Fmr1-KO mice during sleep likely leads to adverse consequences for memory processes (104). As mentioned before, neural abnormalities within the hippocampus also cause emotional problems, including anxiety-like behavior. Current experiment results from the open field test and elevated plus maze indicate that Fmr1-KO mice exhibit high anxiety levels (91, 105), and mice of the fragile X premutation (CGG repeat sequences between 55 and 200) demonstrate an indication of “social anxiety” (106).
Rett syndrome, one of the rare ASDs affecting mainly females, is caused by loss-of-function mutations of the X chromosome-linked gene encoding the Methyl CpG binding protein 2 (MECP2) (107, 108). Rett syndrome animal models reproduce typical neurological features of this disorder, including impairments in hippocampus-dependent memory and learning. For example, Mecp2 mutant mice present deficits in spatial memory and spatial learning in the Morris water maze task (107, 108), the Barnes maze test (109), and the object location test (110). In addition, compared to wild-type mice, Mecp2 mutant mice showed declines in contextual fear memory when subjected to the fear conditioning task. This was evidenced by their notable decreases in freezing duration, primarily observed at long but not short time scales (107, 108, 111, 112). It is worth noting that the hyperactive hippocampal network (i.e. an imbalance of synaptic excitation/inhibition in hippocampal neurons) is responsible for learning and memory impairments in Rett syndrome (111, 113, 114). Pervasive spontaneous glutamate release in the hippocampus has been considered a defining characteristic of Mecp2 KO mice, which contributes to the hyperexcitability of neurons (115). In the hippocampus of symptomatic Mecp2 mutant mice, Schaffer-collateral synapses exhibited enhanced neurotransmitter release (108), and potentiated glutamatergic synapses (e.g. high surface levels of GluA1) occluded the LTP (116). Moreover, the frequency and amplitude of spontaneous excitatory postsynaptic currents of hippocampal neurons from Mecp2 KO mice were found to be significantly decreased, revealing a loss of excitatory synaptic response in the inhibitory neurons of the hippocampus (111, 113, 117, 118). The diminished basal inhibitory rhythmic activity in the hippocampus of Mecp2-null mice can in turn give rise to a hyper-excitable state of the hippocampal network (119). In morphology, CA1 pyramidal neurons and dentate gyrus granule neurons exhibited delayed dendritic maturation and low dendritic spine density in Mecp2 mutant mice compared to wild types (114, 120), which could potentially arise from insufficient BDNF expression in hippocampal neurons (109). Taken together, these findings imply that mutation in Mecp2 causes various forms of hippocampal synaptic plasticity impairment, which in turn affects learning and memory functions.
With a high prevalence of comorbid ASD, Angelman syndrome is caused by the deletion of the maternally inherited ubiquitin protein ligase E3A (Ube3a) gene in the 15q11-q13 chromosome region, associated with impaired hippocampus-dependent learning, memory, and emotion (121–124). Several animal studies have collectively reported that learning and memory deficits in Angelman syndrome are due to a marked decrease in hippocampal LTP (121–124). Researchers have demonstrated that hippocampal pyramidal cells of Angelman syndrome mice have elongated axon initial segments (125), reduced activity-dependent calcium dynamics (122), and hyperpolarized resting membrane potentials (125), which can be attributed to increased expression of α1-NaKA in the hippocampus. These alternations in intrinsic properties of hippocampal neurons could have driven the hippocampal pathology, namely LTP impairment, and memory and learning deficits revealed by contextual fear conditioning and Morris water maze (121, 122). In addition, increased inhibitory phosphorylation of αCaMKII (123) and elevated Arc expression (124) in hippocampal slices from Angelman syndrome mice also underlie the hippocampal LTP deficits. As another type of synaptic plasticity involved in learning and memory, mGluR-LTD of excitatory synaptic transmission was enhanced in hippocampal slices of Ube3a-deficient mice, possibly due to increased synaptic small conductance calcium-activated potassium channel protein 2 levels in the hippocampus (126). As far as emotional problems are concerned, maternal Ube3a-deficient mice are under chronic stress and exhibit anxiety-like behaviors. Within the hippocampus, these mice demonstrate susceptibility to glucocorticoid exposure (127), disrupted glucocorticoid receptor signaling (127–129), and reduced number of parvalbumin-positive inhibitory interneurons (128, 129), resulting in chronic stress, hippocampal hyperactivity and ultimately increased anxiety.
Tuberous sclerosis complex is a rare form of ASD that is often accompanied by epilepsy and cognitive deficits, caused by mutations in either of the Tuberous sclerosis complex 1 or 2 (Tsc1/2) gene. These genes act as inhibitors of the mTOR signaling, and their mutations lead to hyperactivity of the pathway (130). Like other syndromic ASDs, tuberous sclerosis complex mice also have impairments in hippocampus-dependent spatial learning (131–133), contextual fear memory (131–133), and spatial working memory (131), associated with hippocampal synaptic excitation/inhibition imbalance induced by up-regulated mTORC1 signaling (131, 134). This hippocampal hyperactivity likely results from a reduced synaptic inhibition of pyramidal cells, while the excitatory transmission is unaffected (131, 134, 135). Previous studies have identified a potential link between hippocampal hyperexcitability and epilepsy phenotypes in mice deleting Tsc1 or Tsc2 (134, 136), but Koene et al. found the hippocampal excitation/inhibition imbalance only present in the epileptic state, which suggests that these changes in the hippocampus are unlikely to drive epileptogenesis (135). Additionally, dysfunctional glutamate homeostasis (137), impaired astrocytic gap junction coupling (138), and altered potassium clearance (138), as well as microgliosis (139) and astrogliosis (140) in the hippocampus of tuberous sclerosis complex mice, are correlated with seizure onset. Prior studies have noted that mGluR-LTD was not enhanced but rather reduced in tuberous sclerosis complex mutant mice (141–143), indicating divergent synaptic plasticity phenotypes from fragile X syndrome and Angelman syndrome. A possible explanation for this is that the deletion of Tsc1/Tsc2 genes heightened the expression of mGluR5 and exaggerated ERK signaling, which developed a novel mTOR-independent LTP in the CA1 hippocampus (141, 142). Several studies have also reported the enlargement of hippocampal neurons and dendritic spines in mutant mice (143, 144), whilst significantly increased basal cerebral blood flow and oxygen consumption in the hippocampus of rats with tuberous sclerosis complex could partially explain this observation (145, 146).
As major synaptic scaffolding proteins, SH3 and multiple ankyrin repeat domains protein 1/2/3 (SHANK1/2/3) are highly concentrated in the postsynaptic density of hippocampal excitatory synapses (147–149). Shank family genes (Shank1/2/3), especially the Shank 3, are well-known ASD-related genes. Deletions or mutations of the Shank3 gene can lead to Phelan-McDermid syndrome, characterized by autistic behavior, intellectual disability, and speech delay (150). It has been reported that the disruption of major Shank3 isoforms in mouse/rat models decreases levels of other post-synaptic density scaffolding components and glutamatergic receptors in the hippocampus, including the HOMER1 (147–149), PSD95 (148), mGluR5 (148), GluA1 (149), and GKAP (149). At the level of hippocampal synaptic morphology, these animals had smaller postsynaptic density structures (151), lower spine density (149, 151, 152), and longer dendritic spines (149, 151) as compared to wild types. Unlike these profound synaptic changes, however, there have been no distinct alterations in the frequency and amplitude of miniature excitatory (149, 153–155) and inhibitory (149, 154, 155) postsynaptic currents, as well as field excitatory postsynaptic potentials (147, 156) in CA1 pyramidal neurons from mice and rats lacking different Shank3 isoforms. These data indicate that basal synaptic transmission, neurotransmitter release probability, and short-term plasticity at hippocampal synapses may be preserved in Phelan-McDermid syndrome. Indeed, Shank3 is critical for long-term hippocampal synaptic plasticity. Shank3-deficient mice and rats have reduced LTP (147, 149, 155–157) but unaltered LTD (155–157), and a hippocampal excitation/inhibition imbalance (153, 154). These deficits result in impaired social recognition memory (147, 156, 158), object location memory (147, 149, 155), and spatial learning and memory (149, 152, 153, 155, 156), particularly affecting long-term memory processes. In contrast to the above observations, Peça et al. (159) have reported that the frequency and amplitude of miniature excitatory postsynaptic currents and Morris water maze performance in Shank3B mutant mice are comparable to those of controls. Similarly, Cope et al. (160) have found impaired social but not object location memory in Shank3B KO mice. Contradictory findings across these studies may be attributed to mutations of various Shank3 isoforms. This hypothesis is backed by recent transcriptomics investigating gene dosage-differential changes in the hippocampus of Shank3 mutant mice (161).
The NLGN family of synaptic cell adhesion molecules is fundamental in regulating excitatory and inhibitory synapses. There are four isoforms (Nlgn1-4) expressed in rodents or humans, all of which are linked with ASD symptoms. Nlgn1 is predominantly localized to glutamatergic synapses, and Nlgn1 overexpression in mice enhances the number and maturity of excitatory synapses and spines in the CA1 region (162). As for Nlgn1-KO mice, it is highly probable that their hippocampal NMDAR-LTP deficits (163, 164) result from diminished glutamate receptor functions, including reductions in NMDAR-mediated excitatory transmission (163, 164), expression levels of synaptic AMPA and NMDA (163), and the NMDAR/AMPAR ratio (164, 165) in perforant path-granule cell synapses and CA1 pyramidal neurons. In contrast, the Nlgn2 is principally localized to inhibitory GABAergic synapses with a key role in enhancing inhibitory but not excitatory synaptic function (165). Nlgn2-deficient mice displayed reduced postsynaptic gephyrin and GABAAR cluster numbers in the dentate gyrus, decreased inhibitory GABAergic synaptic transmission, and increased granule cell excitability (166). This observation matches those observed in recent studies that mice overexpressing Nlgn2 have a reduced hippocampal excitation/inhibition ratio, thereby inhibiting their aggressive behaviors and impairing spatial memory performances (167, 168). Nlgn3 is the only NLGN isoform that is found in both excitatory and inhibitory synapses. In Nlgn3 KO mice, the number of excitatory synapses in the CA1 stratum oriens (169) and neuronal excitability in the CA2 area (170) were increased. Deleting Nlgn3 also reduced hippocampal gamma oscillations and sharp wave ripples, which could lead to abnormal fear memory retention and extinction (170, 171). Nlgn3-R451C mutant mice exhibited large increases in both excitatory (172, 173) and inhibitory (174) synaptic transmission in the hippocampal CA1 region, and particularly the increased NMDA/AMPA ratio may have enhanced NMDAR-dependent LTP (172, 173). This also accords with several observations, which showed that these mice have better spatial learning and memory performance than wild-type controls (173, 174). However, it has also been shown that the Nlgn3-R451C mutation can cause loss-of-function effects in neonatal mice, characterized by premature hyperpolarizing effect of GABA at immature hippocampal MF-CA3 synapses and fail to express spike time-dependent LTP (175). The specific subcellular localization of Nlgn4 in synapses is not fully understood, but Nlgn4 is certainly expressed in the mouse hippocampus (176). A prior study has shown that the loss of Nlgn4 caused postsynaptic changes at inhibitory synapses and aberrant inhibitory neurotransmission, heavily disrupting γ-oscillations in the CA3 region of the mouse hippocampus (176). Unexpectedly, Muellerleile and colleagues discovered increased network inhibition within the dentate gyrus of adult Nlgn4 KO mice but unaltered in neonatal Nlgn4 KO mice (177). Guneykaya et al. (178) found that hippocampal γ-oscillations were disrupted and hippocampal microglia density was reduced only in male Nlgn4 KO mice. Hence, contradictory results in the hippocampal inhibitory state may partly be explained by sex-dependent and age-related impacts of Nlgn4 loss.
Like Tsc1/2, the Pten gene functions as a mTOR pathway negative regulator, and alteration of this pathway is involved in ASD pathogenesis. Activation of mTOR via Pten deletion from hippocampal dentate granule cells re-initiates additive growth, which leads to hypertrophied neurons (179–182), enlarged mossy fiber axons (179, 180, 183), elongated dendrites (179, 180, 184) and increased dendritic spine density (179–181, 184). These hippocampal morphological changes partly underlie the macrocephaly and epilepsy that are notable features of inherited Pten mutations. The observed increase in spontaneous excitatory synaptic current frequency (particularly in females) (181, 184) and field excitatory postsynaptic potential slope (185, 186) suggests increased excitatory synapses on cells and enhanced basal synaptic transmission in the hippocampus of Pten KO mice. The increased epileptogenic activity of Pten KO mice is largely due to hippocampal hyperexcitability (182, 184). More importantly, these mouse models display impaired hippocampal LTP and LTD (185, 187), as well as spatial memory (187) during postnatal development, which has been shown to precede the appearance of their morphological abnormalities (185).
Mutations in the X-linked Cdkl5 gene cause severe neurodevelopmental disorders marked by early-life autistic behaviors and intractable epilepsy (188). The Cdkl5 is highly expressed in the hippocampus, and its deficiency in mice reduces dendritic length, branches, and maturation of hippocampal pyramidal and granule neurons (189–192). Meanwhile, Cdkl5 KO mice exhibit an elevated incidence of newborn cell apoptosis within the hippocampal dentate gyrus leading to diminished granule neuron counts (189, 190), coupled with accelerated senescence and death of hippocampal neurons during the aging process (191). Strikingly, the increase in apoptosis is paralleled by the rapid proliferation of neuronal precursor cells in the dentate gyrus, which modulates the equilibrium between precursor proliferation and survival (190). Additionally, it is noted that hippocampal neurons of Cdkl5 KO mice demonstrate heightened susceptibility to neurotoxicity, excitotoxicity, and oxidative stress (192, 193), implying that the absence of Cdkl5 augments neuronal vulnerability. These neuroanatomical alterations are associated with hippocampus-dependent learning and memory impairment observed in multiple tasks (189–191). The robust seizures in Cdkl5 KO mice have been demonstrated to be correlated with microglial activation (192), BDNF-TrkB signaling enhancement (194), and postsynaptic overaccumulation of GluN2B-containing NMDAR (195) in the hippocampus.
It is clear now ASD etiology involves both genetic and environmental factors or their possible combinations. Exposure of animals to given chemicals, toxins, viruses, and other agents during gestation can induce models of ASD in their offspring.
Valproic acid (VPA) is a commonly used anti-epileptic or mood-stabilizing drug but is classified as a human teratogen. In animal studies, typical ASD models in newborn mice or rats have been simulated by exposing their mothers to VPA during pregnancy (196). Similar to the ASD traits in humans, male animals might be more susceptible to VPA-induced ASD than females. For instance, male VPA mice exhibited higher locomotor activity and lower social ability index than females, potentially attributed to increased hippocampal cell atrophy and heightened expression of the 5-HT2A receptor protein in the hippocampus (197). The VPA exposure in mice or rats can induce several autism-like behaviors related to hippocampal functions, including impairments in spatial learning and memory (198–206), visual recognition memory (198, 203, 207–209), working memory (208, 210), and emotional regulation (199, 200, 204–206, 211–213). It is widely documented that exposure to VPA notably increases levels of the pro-inflammatory markers (IL-1β, TNF-α, IL-6, IFN-γ, IL-17, TGF-β) and reduces levels of the anti-inflammatory marker (IL-10) in the hippocampus (199, 208, 211, 213–216). The hippocampal neuroinflammatory state is primarily observed in young ages, perhaps resulting from microglia and astrocyte activation that started in the early postnatal developmental stages. In adult mature VPA rats/mice, however, changes in the expression of neuroglial markers in the hippocampus seem to be mild, with an amelioration of the neuroinflammatory phenotype (212, 217, 218). After exposure to prenatal VPA, biochemical markers associated with neuronal oxidative/nitrosative stress such as MDA, TBARS, and NO were found to be significantly increased contrary to markers such as GSH, SOD, and CAT in the hippocampal regions (198, 202, 212–215, 219). At the same time, increased oxidative stress is accompanied by aberrant mitochondrial electron transport chain enzyme activity, reduced ATP levels, and ultrastructurally destructed mitochondria in the hippocampus (198, 212). Exposure of mice or rats to VPA activates mTOR and Notch signaling, which amplifies autophagic deficiency in the hippocampus, characterized by decreased expression levels of Beclin1 and LC3-II and a small number of autophagosomes (203, 210). Previous studies evaluating hippocampal excitatory/inhibitory imbalance found that VPA exposure enhanced excitatory glutamatergic and impaired inhibitory GABAergic synaptic transmission, with a decrease in the GABA/glutamate ratio (201, 206). Compared to controls, VPA-induced rats showed a significantly larger number of hippocampal apoptotic neurons, accompanied by increased levels of the apoptotic markers “Bax, caspase-3 and p53” and decreased levels of the antiapoptotic marker “BCL2” (201, 204, 219–221). Moreover, reductions in the hippocampal levels of BDNF, synapsin-IIa, DCX, and pCREB are strongly implicated in ASD as these proteins play significant roles in neuronal formation, synaptic transmission, neuroplasticity, and neurogenesis (201, 213–215, 221). Taken together, these pathophysiological processes significantly weaken hippocampal neuron viability in VPA-exposed mice and rats. Furthermore, VPA exposure significantly altered the expression of multiple ASD candidate genes in the hippocampus: Shank3 (212), Shank2 (209), Nlgn3 (212), and Pten (222). This observation further supports that high-risk ASD genes can also be altered by environmental factors – gene-environment interactions have a pivotal role in the pathophysiology of ASD.
Maternal immune activation (MIA) during pregnancy increases the risk of the unborn fetus developing ASD later in life (223). The most commonly used methods for emulating MIA models of ASD involve the intraperitoneal administration of lipopolysaccharide (LPS) and polyinosinic: polycytidylic acid (poly (I: C)) during gestation. The administration of LPS can induce sex-dependent alterations in hippocampal volume, neuronal morphology, and gliovascular maturation. Compared with female LPS-induced mice, the male LPS group showed a larger size of hippocampus (224), higher hippocampal neuronal spine density (224, 225), and lower vascular coverage of astrocytic end-feet (226) associated with their reduced interest in social novelty (224, 225). Microglial activation and astrogliosis could be functionally important in altering hippocampus-dependent learning and memory performance observed in LPS-induced rat models (227, 228). Surprisingly, at the early postnatal stage, exposure to LPS had no negative effects on hippocampal cellular or tissue morphology but instead stimulated nerve growth by promoting cell proliferation (228), increasing the number of spines (225), and raising the density of mossy fiber synapses (229) in the hippocampal area. It seems possible that these results are due to the M2-biased microglia polarization at the acute inflammatory phase, which releases excessive anti-inflammatory cytokines and growth factors (228). In contrast, MIA induced by poly (I: C) did not alter the density of Iba1+ microglia (or GFAP+ astrocytes), nor did it modify their activation phenotypes in the hippocampal formation of the offspring (230, 231). However, prenatal exposure to poly (I: C) increased hippocampal IL-6 and IL-1β levels, resulting in the promotion of hippocampal kindling epileptogenesis (230, 232). Additionally, the offspring of poly (I: C)-exposed mice displayed a substantial reduction in the relative density of hippocampal pre- and postsynaptic proteins (synaptophysin, bassoon, PSD95, and SynGap) and changed the firing traits of hippocampal place cells in adult offspring (230, 233). The hippocampal synaptic deficits could alter the electrophysiological properties of hippocampal cells, hence affecting the firing activity of hippocampal neurons. These changes may underlie the spatial memory impairments found in MIA mice following poly (I: C) injection (233, 234).
Gestational exposure to air pollution can increase the incidence of ASD in offspring (223). Hippocampal transcriptome data revealed that gestational nanosized particulate matter (PM) exposure induced multiple differentially expressed genes in young adult offspring. The stratification by sex revealed a twofold increase in the number of differentially expressed genes in males compared to females, and there was male-specific enrichment of differentially expressed genes involved in serotonin receptor signaling, cAMP-mediated signaling, and endocytosis (235). Other studies have also found that PM2.5-induced mice have aggravated hippocampal neuroinflammation, with elevation in NF-κB, TNF-α, and IL-1β levels (236) and microglial activation (237). Meanwhile, these mice exhibited impaired spatial learning and memory, associated with disrupted hippocampal synaptic ultrastructure, decreased hippocampal neurogenesis, and increased hippocampal neuronal apoptosis (236). These findings can explain the reduction in hippocampal size and structural integrity after exposure to airborne PM (237, 238).
As genetic and environment-induced models cannot accurately replicate all the pathological features of ASD, strains of mice and rats have been developed using idiopathic models, which display robust and well-replicated behavioral characteristics of ASD such as social deficits and repetitive behaviors. The BTBR T+Itpr3tf/J (BTBR) strain is one of the most valid models of idiopathic ASD, and inbred strain C57BL/6J is often used as a control for BTBR (239).
Recently, the BTBR inbred mouse strain has gained popularity as a rodent model of ASD. In addition to the core symptoms of ASD, BTBR mice also display learning and memory impairments in various settings (240–242). Several histological observations and MRI assessments support separated hippocampal commissure and increased hippocampal volume in BTBR mice relative to controls, and these anatomical changes may underlie their behavioral phenotypes (243–246). Moreover, the hippocampus has lower 5-HT, acetylcholine, dopamine, and histamine content in the BTBR animals than in the C57BL/6J strain (246–248). In particular, immunofluorescent labeling of 5-HT transporter axons revealed a reduction in the density of innervation to the hippocampus in BTBR mice (246). Premature changes in hippocampal neuronal excitability, involving elevated ERK signaling (248) and increased GABAergic neurotransmission (249) during neonatal development in the BTBR mice, may also contribute to the high susceptibility to epilepsy and aggressive behaviors observed in these mice (248, 250). As for adult and aged BTBR animals, significant reductions in mRNA or protein levels of BDNF, as well as neurogenesis in the hippocampus have previously been reported (241, 245, 250).
To sum up, hippocampal excitatory/inhibitory imbalance is one of the most important pathological mechanisms in ASD. These animal models mainly show impairments in dendrite morphology, neurogenesis, neuronal viability, LTP, and LTD in the hippocampus, leading to dysfunction in learning, memory, emotional regulation, and spatial ability. Figures 3, 4 were used to depict some common hippocampal structural and functional impairments in rodent models of ASD, but it is important to note that each model has its own characteristics of hippocampal dysfunctions and cannot be generalized. In a word, the above animal studies suggest that the hippocampus is strongly implicated in the pathophysiology of ASD and should be considered as an important target for future therapeutic approaches.
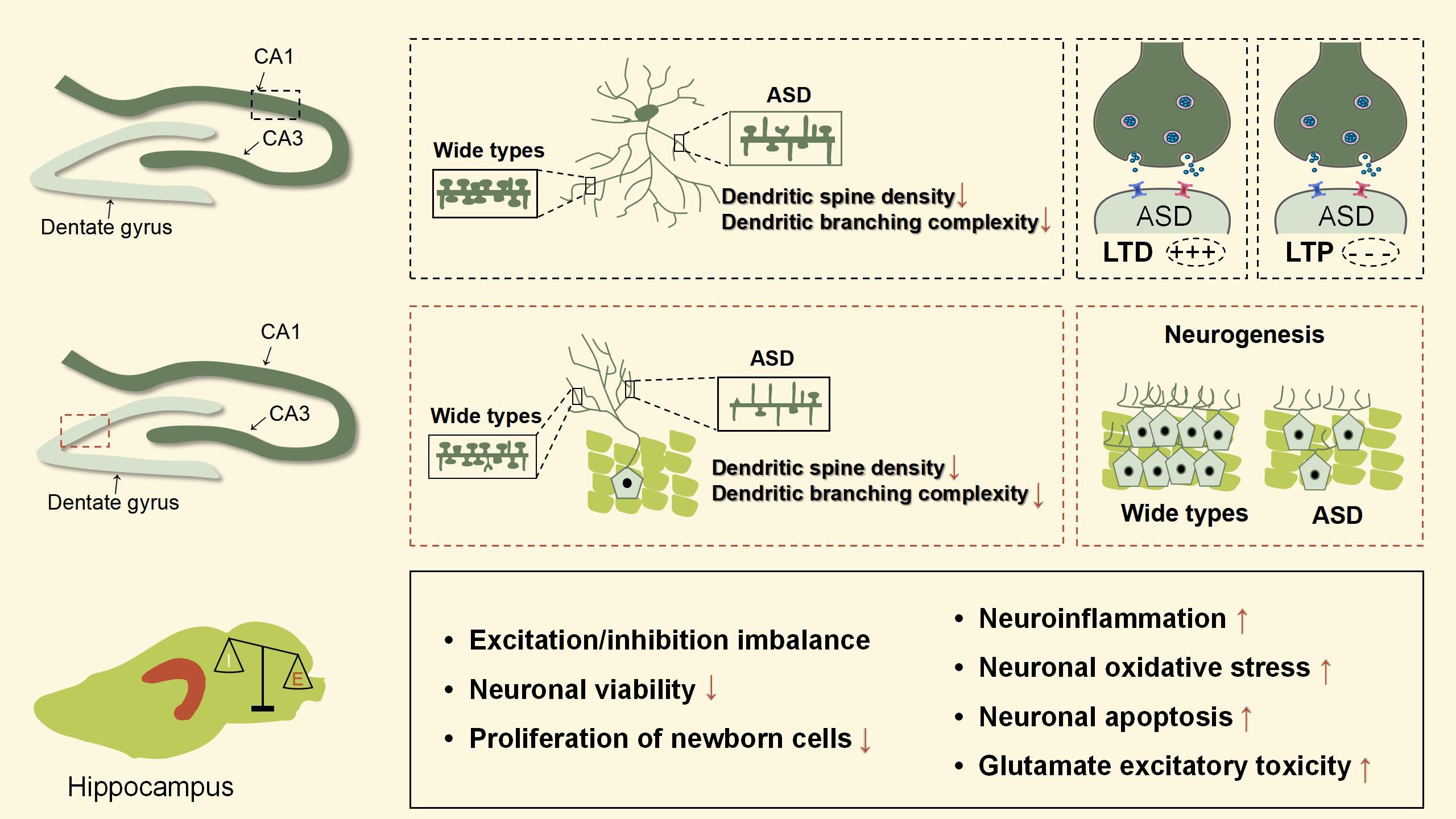
Figure 3 Hippocampal pathophysiological processes of ASD animal models. This figure is created by using PowerPoint.
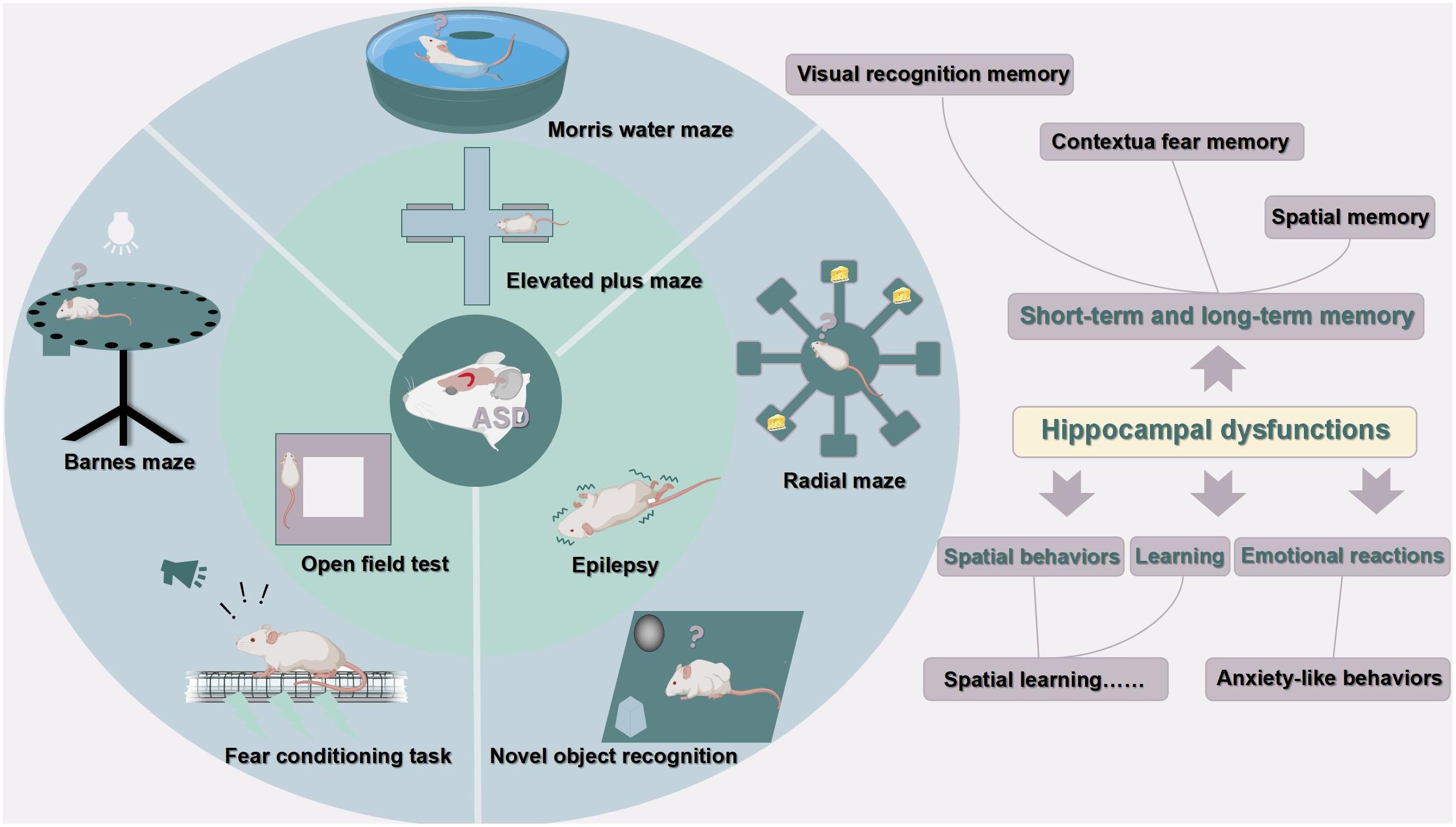
Figure 4 Hippocampal dysfunctions in ASD animal models. This figure is created by using PowerPoint.
To date, amounts of pharmacological and non-pharmacological interventions that positively impact hippocampus-dependent cognitive functions have been identified through animal experimentation as potential treatments for ASD (Figure 5). Pharmacological interventions mainly include hormones, vitamins and minerals, atypical antipsychotic drugs, phosphodiesterase inhibitors, selective serotonin reuptake inhibitors, mTOR inhibitors, mGluR antagonists, NMDAR antagonists, histamine H3 receptor (H3R) antagonists, and insulin-like growth factor. Non-pharmacological interventions mainly include aerobic exercise and environmental enrichment. Table 1 presents elaborate cellular and molecular modifications in the hippocampus of animal models of ASD following each intervention.
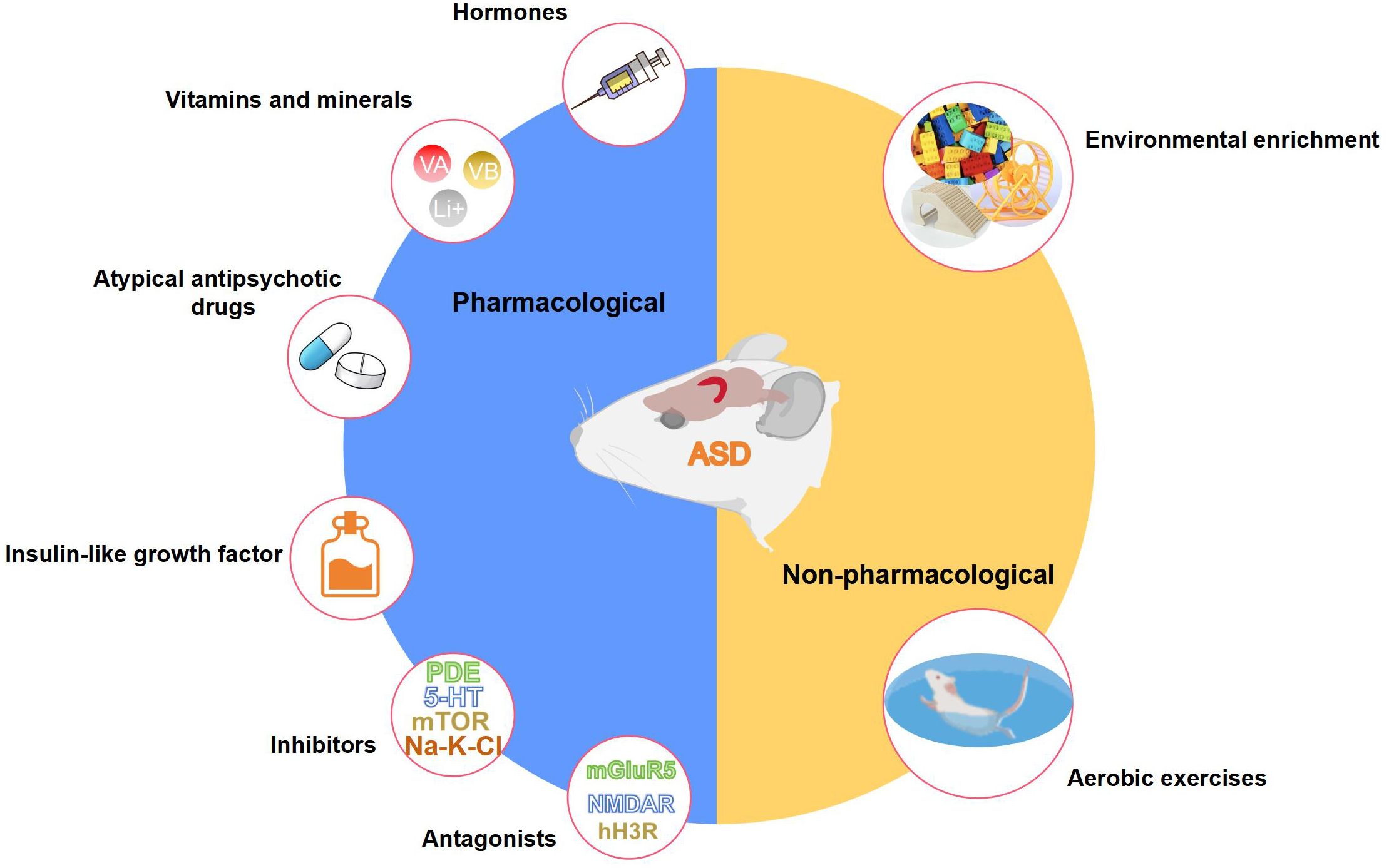
Figure 5 Pharmacological and non-pharmacological interventions that positively affect the structure and function of the hippocampus in ASD animal models. This figure is created by using PowerPoint.
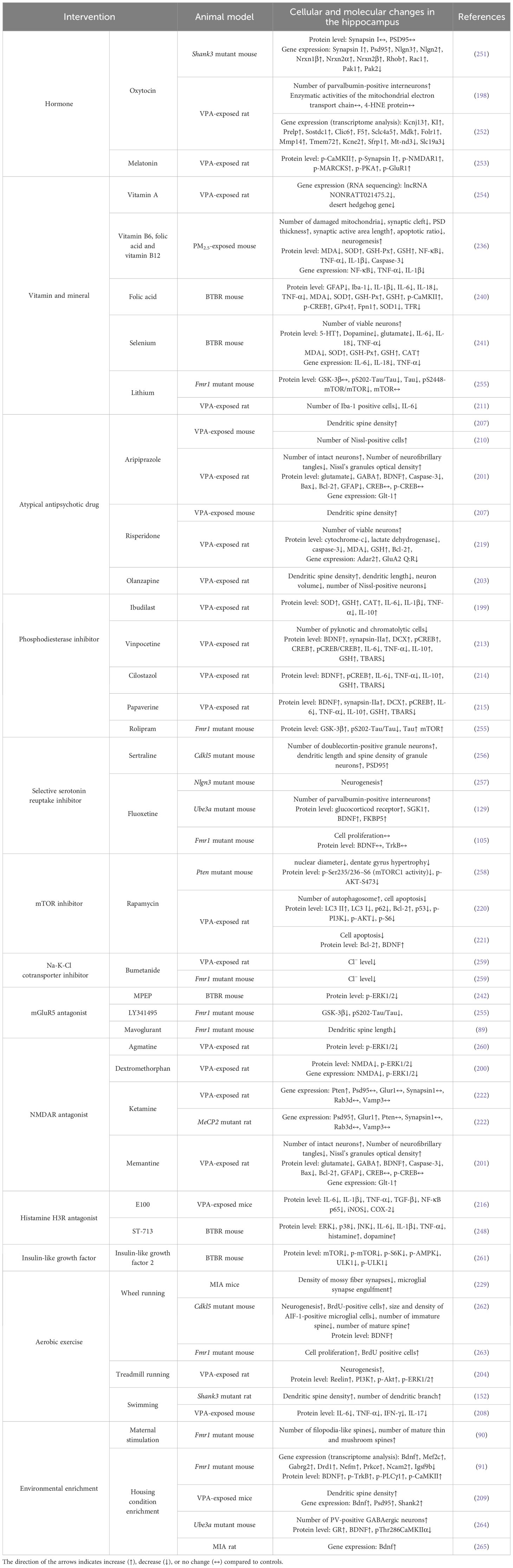
Table 1 Main effects of each intervention on cellular and molecular changes in the hippocampus of ASD animal models.
Oxytocin has been proposed as a possible therapeutic agent for ASD due to its potent regulation of mammalian social behavior. The injection of oxytocin into the left lateral ventricle specifically improved the long-term social recognition memory and LTP at hippocampal synapses in Shank3-deficient rats (147). In vivo, subcutaneous oxytocin injections induced upregulation of hippocampal postsynaptic proteins PSD95 and Nlgn3 of Shank3 deficient mice (251). In rats exposed to VPA prenatally, chronic intranasal oxytocin administration rescued ASD-like behaviors including learning and memory impairments (198), and enhanced the expression of multiple genes in the hippocampus linked to synaptic function, learning, memory, and neurodevelopment (252). Erythropoietin, a glycoprotein hormone, has recently been reported to inhibit the astrogliosis in the hippocampal CA1 subfield in both LPS (227) and VPA (266) induced rat models of ASD, contributing to the enhanced learning and memory task performance (227). A preliminary study in VPA-treated rats has shown that melatonin treatment restored hippocampal CaMKII/PKA/PKC phosphorylation and LTP reduction, which might correlate with amelioration of hippocampus-dependent memory and learning skills (253).
Recent RNA sequencing research has found that vitamin A supplementation significantly alleviated VPA-induced anxiety behaviors, possibly by regulating lncRNA-mRNA co-expression networks (particularly lncRNA NONRATT021475.2 and Desert hedgehog gene) in the hippocampus of ASD rats (254). Gestational B-vitamin supplementation (vitamin B6, folic acid, and vitamin B12) has the potential to mitigate PM2.5-induced spatial learning and memory defects in mice offspring by ameliorating hippocampal inflammation, oxidative stress, mitochondrial damage, neuronal apoptosis, and synaptic dysfunction (236). Likewise, using solely folic acid rescued hippocampal neuron death and spatial learning and memory impairments in BTBR mice as it suppressed oxidative stress, inflammation, and ferroptosis in the hippocampus (240). Regarding mineral supplementation, selenium has a protective effect on the hippocampus of BTBR mice, with a comparable mechanism to that of folic acid (241). A six-week zinc water supplementation led to a decrease in anxiety-like behavior and seizure susceptibility in BTBR mice. This effect may be related to the restoration of neural progenitor cell proliferation and excitation/inhibition balance in the hippocampus (250). Lithium treatment can rescue olfactory-based learning and memory defects in drosophila fragile X model, and increase the cAMP signaling by inhibiting GSK-3β activity in the hippocampus of fragile X mice (255). In addition, lithium exerts an anti-inflammatory role probably by reducing microglial activation and inflammatory cytokine release and increasing levels of H3K9 acetylation in the hippocampus of VPA-exposed rats, with beneficial implications for improving social memory and anxiety levels (211).
The third-generation, atypical antipsychotic drugs aripiprazole, and risperidone are the only medications approved by the American FDA for ASD treatment. Data from several studies suggest that chronic treatments with aripiprazole attenuated VPA-induced visual recognition memory (207) and spatial learning and memory (201) impairments in mouse/rat offspring with ASD. More importantly, maternal treatment with aripiprazole prevents working memory deficits and hippocampal cell death in juvenile mice exposed to VPA during the prenatal period (210). Chronic administrations of risperidone improved VPA-induced memory impairment and reductions in hippocampal dendritic spine density, but there was no improvement with acute administrations (207). In addition, risperidone impeded hippocampal glutamate excitotoxicity in the VPA rat model of ASD, ultimately promoting neuronal survival (219). Similarly, the antipsychotic olanzapine can alleviate VPA-induced impairments in recognition and spatial memory by mitigating neuroplastic alterations in the hippocampus, including neuronal hypotrophy, reduced spine density, and elongated dendritic length (203).
The phosphodiesterase class of enzymes is responsible for the degradation of cAMP, which affects various neurobiological processes from neuroinflammation to learning and memory formation (267). For instance, ibudilast, a phosphodiesterase-4 inhibitor, was found to elevate levels of oxidative stress markers (SOD, GSH, CAT) and lower levels of pro-inflammatory markers (IL-1β, TNF-α, IL-6) in the hippocampus of VPA exposed rats. Meanwhile, these ASD rats administered with two doses of ibudilast showed significantly reduced deficits in learning/memory and anxious behaviors (199). In the same way, Luhach and colleagues have demonstrated that vinpocetine, cilostazol, and papaverine, all of which are phosphodiesterase inhibitors, positively influenced neurogenesis, neuronal survival, synaptic transmission, neuronal transcription, neuronal inflammation, and neuronal oxidative stress in the hippocampus of VPA-exposed rat models (213–215). Both rolipram (phosphodiesterase-4 inhibitor) and BAY-60-7550 (phosphodiesterase-2 inhibitor) treatment abrogated the exaggerated hippocampal mGluR-LTD observed in fragile X mice, which can be attributed to significantly increased cAMP levels (88, 255, 268). In like manner, rolipram treatment rescued the LTP of hippocampal CA1 neurons to a significant level in Rett syndrome mice (269). Lastly, rolipram rescued olfactory-based long-term memory defects of drosophila fragile X models (255, 268).
Sertraline and fluoxetine function as primary selective serotonin reuptake inhibitors, impeding 5-hydroxytryptamine uptake into presynaptic vesicles from the synaptic cleft in the central nervous system. Chronic treatment with sertraline improved autistic-like features in Cdkl5 KO mice, including hippocampus-dependent spatial learning and memory deficiency. This positive behavioral effect was associated with restored neuronal survival, dendritic development, and synaptic connectivity in the dentate gyrus and CA1 pyramidal neurons (256). Recent evidence suggests that fluoxetine can ameliorate social behavior in Nlgn3-KO mice, at least in part, by promoting adult hippocampal neurogenesis (257). More typically, long-term fluoxetine treatment normalizes hippocampal parvalbumin-positive interneurons number and glucocorticoid signaling of Angelman syndrome model mice, which is important for the restoration of anxiety-like behaviors (129). On the contrary, however, Fmr1 KO mice after fluoxetine treatment have reduced anxiety but enhanced explorative activity during the open field test, with abnormal changes of BDNF/TrkB signaling in the hippocampus (105). Activation with selective serotonin reuptake inhibitors has been observed in people with fragile X syndrome, which can manifest as mood changes and disinhibited behavior (270). What calls for special attention is the subsequent risk of ASD with exposure to selective serotonin reuptake inhibitors during pregnancy. Recent findings underscored that maternal fluoxetine exposure impaired hippocampal LTP, spatial discrimination, and spatial learning in adult offspring (271), accompanied by decreased hippocampal neurogenesis and hippocampal IL-10, IFN-γ and IL-13 levels (272).
Dysregulation of mTOR signaling is strongly associated with ASD, and the inhibition of mTOR can prevent the binding of mTOR with other protein components and reduce mTOR phosphorylation (273). For example, rapamycin, as a prominent mTOR inhibitor, has been shown to effectively block the hippocampal mTORC1 signaling of Pten mutant mice (258). Besides, rapamycin plays a crucial role in promoting autophagy (220) and decreasing apoptosis (220, 221) in the hippocampus of VPA-induced neonatal rats of ASD, thereby improving learning and memory ability (221). As discussed earlier, tuberous sclerosis complex models had increased hippocampal neuron volume, blood flow, and oxygen consumption. Correspondingly, the administration of rapamycin has been reported to lower cerebral blood flow and oxygen consumption in hippocampal regions of the Tsc2 mutant rat, potentially by downregulating Akt signals (146). This also accords with previous observations, which showed that pharmacological inhibition of mTORC suppressed granule cell hypertrophy in Pten mutant mice (258) and inhibited the PI3K/AKT/mTOR signaling pathway in VPA-induced rats (220).
Bumetanide, a loop diuretic that inhibits the Na-K-Cl cotransporter, has been reported to improve core symptoms of ASD in children over recent years (274). In VPA-treated rats, even a brief maternal bumetanide treatment can prevent hippocampal overgrowth in their offspring (275). In addition, maternal pretreatment with bumetanide effectively restored elevated hippocampal intracellular chloride levels, increased hippocampal excitatory GABA, and enhanced hippocampal gamma oscillations in offspring of VPA-induced rats and Fmr1 mutant mice (259). Recently, a new selective Na-K-Cl cotransporter inhibitor called ARN23746 has been reported to improve sociability in VPA-induced mice, similar to bumetanide (276). The oral administration of torasemide, a diuretic that also acts as a Na-K-Cl cotransporter inhibitor, has the potential to enhance neuronal viability and reduce astrogliosis in the hippocampus of ASD rat models (277).
The dysregulated signaling mediated via mGluR5 contributes to the pathophysiology of ASD since it acts as a crucial regulator of excitatory and inhibitory signaling in the hippocampus (278). After treatment with the mGluR5 antagonist MPEP, epileptiform activity and ERK signaling in the hippocampal slices of Tsc2 mutant mice can be suppressed. The blocked mGluR-LTD in the CA3 hippocampus via antagonism of mGluR5 also improved reversal learning performance between these mice (142). Similarly, for the BTBR mouse model, MPEP treatment facilitated hippocampus-dependent object location memory and decreased synaptic p-ERK1/2 levels (242). Like lithium and rolipram, fragile X syndrome flies treated with mGluR5 antagonist LY341495 demonstrated an enhanced long-term memory paradigm compared to controls (255). Long-term antagonism of mGluR5 also rescued immature spine phenotype (89) and decreased GSK-3β activity (255) in the hippocampus of Fmr1-KO mice.
NMDAR-mediated excitation and inhibition imbalance is one of the primary theories explaining the neurotoxicity in ASD. Data from several studies on rats exposed to VPA indicate that the NMDAR antagonist, agmatine (260) and dextromethorphan (200), normalize the overly active ERK1/2 phosphorylation in the hippocampus, which serves as an indicative marker of hippocampal hyperexcitability state. Besides, spatial memory and learning deficits induced by Fmr1-KO and VPA were mitigated following agmatine (100) and dextromethorphan (200) administration, respectively. For both rats exposed to VPA and those with a Mecp2-KO, the administration of ketamine produces a positive effect on their autistic-like behaviors by improving synaptic molecule levels in the dentate gyrus of the hippocampus (222). Memantine, a non-competitive antagonist of NMDAR, was reported to alleviate anxiety and improve learning and memory deficits in VPA-exposed rats, which could be mediated via the restoration of hippocampal GABA/glutamate balance and inhibition of hippocampal neurofibrillary tangles formation and neuronal apoptosis (201).
The histamine H3R, as a presynaptic autoreceptor can regulate the production and release of histamine as well as numerous brain neurotransmitters like dopamine and acetylcholine (279). Thus, selective H3R antagonists can improve the cognitive impairment in ASD. It is evident from the observation that the administration of histamine H3R antagonists ST-2223 (247) and ST-713 (248) significantly elevated the levels of histamine, dopamine, and acetylcholine in the hippocampal tissue of BTBR mice with anxiolytic-like effects. Administration of ciproxifan could improve the VPA-induced LTP decline in the CA1 area and hippocampus-dependent learning and memory capacity (202). Moreover, the histamine H3R antagonist E100 mitigates VPA-induced hippocampal inflammation by reducing the levels of IL-6, IL-1β, TNF-α, and TGF-β and by suppressing the expression of NF-κB, iNOS, and COX-2 (216).
The insulin-like growth factor (IGF) system comprising two activating ligands (IGF-1 and IGF-2) greatly impacts the development of the central nervous system. It has been discovered that levels of IGF-1 are reduced in the hippocampus of Rett syndrome mouse model (280). The active peptide derivative of IGF-1 can cross the blood-brain barrier and rescue Rett syndrome symptoms in MeCP2 mutant mice (281). In addition, daily intraperitoneal injections of IGF-1 for 2 weeks reversed deficits in hippocampal LTP in Shank3-deficient mice (282). Previous data revealed that Nlgn3 KO mice treated with IGF-2 fully recovered the social novelty discrimination. This effect was not accompanied by any alteration in spontaneous glutamatergic synaptic transmission within the CA2 region, but rather by enhanced CA2 neuronal excitability (283). Similarly, the administration of IGF-2 ameliorated social interaction deficits in BTBR mice and enhanced their social novelty memory via hippocampal IGF-2 receptor (261). In the Angelman syndrome mouse model, the impaired contextual and recognition memories as well as working memory deficits were restored following subcutaneous injection of IGF-2 (284).
The beneficial effects on memory functions of aerobic exercise, as a non-pharmacological intervention, have been well-documented in individuals with ASD (285). Compared to sedentary controls, both Cdkl5 KO mice (262) and VPA-induced rats (204) after one month of wheel/treadmill running showed increased hippocampal neurogenesis, improved memory performance, and reduced anxious and impulsive behaviors. One-month voluntary wheel running not only can decrease hippocampal microglia overactivation in Cdkl5 KO mice (262), but also stimulate microglia-mediated engulfment of surplus synapses in the granule cell axons observed in MIA mice (229). On the other hand, however, high-intensity exercise may produce negative effects on ASD symptoms. For instance, two-month treadmill training with relatively high speed led to impaired social memory in ASD rats with mercury exposure (286). Voluntary running for seven days increased cell proliferation in the hippocampal dentate gyrus of Fmr1 KO mice, but this effect was not observed when running for 28 days (263). Swimming is another beneficial aerobic exercise that has been shown to increase hippocampal gray matter volume and dendritic spine density in Shank3-KO rats (152) and to prevent hippocampal neuroinflammation in VPA-exposed mice (208). It also contributes to improvements in spatial, working, social, and visual recognition memory (152, 208).
Environmental enrichment is a novel living condition with increased social exploration opportunities and sensory, cognitive, and motor stimulations, which positively affect the hippocampus-dependent learning, memory, and anxiety behavior of children with ASD (287). Living with the mother and an additional non-lactating female (enhanced maternal stimulation) can rescue the spatial and contextual fear memory deficits displayed in adulthood by Fmr1-KO mice (90). In addition to this “social environmental enrichment”, enrichment in housing conditions (running wheel, toys, tunnels, ladders, etc.) also ameliorates impaired anxiety-like behavior (91, 209, 264), visual recognition memory (209, 264), spatial learning and memory (91, 265), and fear memory (91), potentially by acting on the hippocampal BDNF/TrkB-PLCγ1-CaMKII pathway (91, 209, 264, 265), as observed in Fmr1-KO mice (91), VPA-exposed mice (209), Ube3a-KO mice (264), and MIA rats (265).
In summary, hippocampal involvement in the pathophysiology of ASD is now an acquired knowledge. While the majority of current structural and functional neuroimaging studies concentrate on the social brain networks in ASD, hippocampal formation should not be overlooked as it plays a crucial role in higher non-social cognitive functions that are also significantly impaired in most ASD individuals. Numerous lines of clinical evidence on hippocampal volume, morphology, blood flow, metabolism, and functional connectivity seem to converge toward the hypothesis of a hippocampal neurofunctional deficit in ASD, which concerns learning, memory, language ability, emotional regulation, and cognitive map creation. Furthermore, researches based on different ASD animal models are rapidly enhancing our understanding of the neural mechanisms underlying hippocampus-dependent behavioral deficits. In general, typical hallmarks of hippocampal deviations in ASD involve impairments in neurogenesis, dendritic morphology, neuronal viability, neuronal excitation/inhibition balance, LTP, and LTD. Results from the recent therapeutic approaches for ASD are encouraging, since some behavioral alterations such as learning and memory deficits, could be reversed even when treatment was performed on adult mice/rats, potentially by influencing the structure and function of the hippocampus. By targeting therapy at the site of hippocampal pathology, more effective pharmacological and non-pharmacological approaches may be developed in the future.
Despite these promising results, several questions remain unanswered at present. Firstly, as we noted above it is necessary to further confirm whether hippocampal abnormalities, particularly changes in volume, are present in all individuals with ASD or only in a specific subgroup. To date, comparable anatomical and morphological alterations have been detected in the hippocampus of individuals with ASD and ASD animal models, but human postmortem analysis is still limited by the relatively small numbers of individual brains. Besides, the developmental profile of hippocampal pathology in ASD remains unknown. It cannot be discounted that alternation in hippocampal structure may be secondary due to the disease process, the emotional stress, or the various treatments of ASD. Integrating the neuropathology of the hippocampus in ASD individuals with the aetiology and pathophysiology of ASD is a major challenge for the coming years. Other issues to be addressed involve ascertaining the precision of diagnosing ASD by detecting hippocampal lesions and determining the significance of hippocampal pathology compared to other affected regions. Finally, it is beyond dispute that individuals with ASD exhibit whole-brain functional connectivity deficits, and it is not possible to simply attribute the symptoms of ASD to specific regions of the brain. More specifically, as discussed above atypical connectivity at the local network level between the hippocampus and other brain regions could potentially account for impaired non-social behaviors in ASD. The utilization of multimodal neuroimaging data, such as CT, MRI, fNIRS, EEG, MEG, etc., is progressively increasing in both scientific research and clinical settings nowadays. In future investigations, it is hoped that this multilevel approach will provide insight into the neural circuits behind the hippocampus-dependent functional deficits in those with ASD, which can in turn elucidate the developmental mechanisms underlying hippocampal pathology in ASD.
JL: Writing – original draft, Conceptualization. HL: Writing – original draft. YL: Software, Visualization, Writing – review & editing. XL: Software, Visualization, Writing – review & editing. ZT: Software, Visualization, Writing – review & editing. KH: Software, Visualization, Writing – review & editing. JC: Writing – review & editing, Software, Visualization. HZ: Conceptualization, Funding acquisition, Supervision, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Key Research and Development Program of China (2018YFC2001703) and Changping laboratory (2021B-01-01-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Talantseva OI, Romanova RS, Shurdova EM, Dolgorukova TA, Sologub PS, Titova OS, et al. The global prevalence of autism spectrum disorder: A three-level meta-analysis. Front Psychiatry. (2023) 14:1071181. doi: 10.3389/fpsyt.2023.1071181
2. Lord C, Brugha TS, Charman T, Cusack J, Dumas G, Frazier T, et al. Autism spectrum disorder. Nat Rev Dis Primers. (2020) 6:5. doi: 10.1038/s41572-019-0138-4
3. Dawson G, Webb S, Schellenberg GD, Dager S, Friedman S, Aylward E, et al. Defining the broader phenotype of autism: genetic, brain, and behavioral perspectives. Dev Psychopathol. (2002) 14:581–611. doi: 10.1017/S0954579402003103
4. Smith AD. Spatial navigation in autism spectrum disorders: a critical review. Front Psychol. (2015) 6:31. doi: 10.3389/fpsyg.2015.00031
5. Williams DL, Goldstein G, Minshew NJ. Impaired memory for faces and social scenes in autism: clinical implications of memory dysfunction. Arch Clin Neuropsychol. (2005) 20:1–15. doi: 10.1016/j.acn.2002.08.001
6. Wang Y, Zhang YB, Liu LL, Cui JF, Wang J, Shum DH, et al. A meta-analysis of working memory impairments in autism spectrum disorders. Neuropsychol Rev. (2017) 27:46–61. doi: 10.1007/s11065-016-9336-y
7. O'Brien G, Pearson J. Autism and learning disability. Autism. (2004) 8:125–40. doi: 10.1177/1362361304042718
8. Luyster R, Lord C. Word learning in children with autism spectrum disorders. Dev Psychol. (2009) 45:1774–86. doi: 10.1037/a0016223
9. Norrelgen F, Fernell E, Eriksson M, Hedvall Å, Persson C, Sjölin M, et al. Children with autism spectrum disorders who do not develop phrase speech in the preschool years. Autism. (2015) 19:934–43. doi: 10.1177/1362361314556782
10. Kirsch AC, Huebner ARS, Mehta SQ, Howie FR, Weaver AL, Myers SM, et al. Association of comorbid mood and anxiety disorders with autism spectrum disorder. JAMA Pediatr. (2020) 174:63–70. doi: 10.1001/jamapediatrics.2019.4368
11. Lind SE, Williams DM, Raber J, Peel A, Bowler DM. Spatial navigation impairments among intellectually high-functioning adults with autism spectrum disorder: exploring relations with theory of mind, episodic memory, and episodic future thinking. J Abnorm Psychol. (2013) 122:1189–99. doi: 10.1037/a0034819
12. Lind SE, Bowler DM, Raber J. Spatial navigation, episodic memory, episodic future thinking, and theory of mind in children with autism spectrum disorder: evidence for impairments in mental simulation? Front Psychol. (2014) 5:1411. doi: 10.3389/fpsyg.2014.01411
13. Pellicano E, Smith AD, Cristino F, Hood BM, Briscoe J, Gilchrist ID. Children with autism are neither systematic nor optimal foragers. Proc Natl Acad Sci USA. (2011) 108:421–6. doi: 10.1073/pnas.1014076108
14. Lisman J, Buzsáki G, Eichenbaum H, Nadel L, Ranganath C, Redish AD. Viewpoints: how the hippocampus contributes to memory, navigation and cognition. Nat Neurosci. (2017) 20:1434–47. doi: 10.1038/nn.4661
15. Izquierdo I. The hippocampus and learning. Prog Neurobiol. (1975) 5:37–75. doi: 10.1016/0301-0082(75)90007-6
16. Covington NV, Duff MC. Expanding the language network: direct contributions from the hippocampus. Trends Cognit Sci. (2016) 20:869–70. doi: 10.1016/j.tics.2016.10.006
17. Qasim SE, Mohan UR, Stein JM, Jacobs J. Neuronal activity in the human amygdala and hippocampus enhances emotional memory encoding. Nat Hum Behav. (2023) 7:754–64. doi: 10.1038/s41562-022-01502-8
18. Hogeveen J, Krug MK, Geddert RM, Ragland JD, Solomon M. Compensatory hippocampal recruitment supports preserved episodic memory in autism spectrum disorder. Biol Psychiatry Cognit Neurosci Neuroimaging. (2020) 5:97–109. doi: 10.1016/j.bpsc.2019.08.009
19. Ring M, Derwent CLT, Gaigg SB, Bowler DM. Structural learning difficulties implicate altered hippocampal functioning in adults with autism spectrum disorder. J Abnorm Psychol. (2017) 126:793–804. doi: 10.1037/abn0000277
20. Boucher J, Cowell P, Howard M, Broks P, Farrant A, Roberts N, et al. A combined clinical, neuropsychological, and neuroanatomical study of adults with high functioning autism. Cognit Neuropsychiatry. (2005) 10:165–213. doi: 10.1080/13546800444000038
21. Endo T, Shioiri T, Kitamura H, Kimura T, Endo S, Masuzawa N, et al. Altered chemical metabolites in the amygdala-hippocampus region contribute to autistic symptoms of autism spectrum disorders. Biol Psychiatry. (2007) 62:1030–7. doi: 10.1016/j.biopsych.2007.05.015
22. Allsop SA, Vander Weele CM, Wichmann R, Tye KM. Optogenetic insights on the relationship between anxiety-related behaviors and social deficits. Front Behav Neurosci. (2014) 8:241. doi: 10.3389/fnbeh.2014.00241
23. Destrieux C, Bourry D, Velut S. Surgical anatomy of the hippocampus. Neurochirurgie. (2013) 59:149–58. doi: 10.1016/j.neuchi.2013.08.003
24. El-Falougy H, Benuska J. History, anatomical nomenclature, comparative anatomy and functions of the hippocampal formation. Bratisl Lek Listy. (2006) 107:103–6.
25. Rajmohan V, Mohandas E. The limbic system. Indian J Psychiatry. (2007) 49:132–9. doi: 10.4103/0019-5545.33264
26. Weininger J, Roman E, Tierney P, Barry D, Gallagher H, Murphy P, et al. Papez's forgotten tract: 80 years of unreconciled findings concerning the thalamocingulate tract. Front Neuroanat. (2019) 13:14. doi: 10.3389/fnana.2019.00014
27. Gindes L, Weissmann-Brenner A, Weisz B, Zajicek M, Geffen KT, Achiron R. Identification of the fetal hippocampus and fornix and role of 3-dimensional sonography. J Ultrasound Med. (2011) 30:1613–8. doi: 10.7863/jum.2011.30.12.1613
28. Utsunomiya H, Takano K, Okazaki M, Mitsudome A. Development of the temporal lobe in infants and children: analysis by MR-based volumetry. AJNR Am J Neuroradiol. (1999) 20:717–23.
29. Hazlett HC, Gu H, Munsell BC, Kim SH, Styner M, Wolff JJ, et al. Early brain development in infants at high risk for autism spectrum disorder. Nature. (2017) 542:348–51. doi: 10.1038/nature21369
30. Mandell DS, Novak MM, Zubritsky CD. Factors associated with age of diagnosis among children with autism spectrum disorders. Pediatrics. (2005) 116:1480–6. doi: 10.1542/peds.2005-0185
31. Banker SM, Gu X, Schiller D, Foss-Feig JH. Hippocampal contributions to social and cognitive deficits in autism spectrum disorder. Trends Neurosci. (2021) 44:793–807. doi: 10.1016/j.tins.2021.08.005
32. Eilam-Stock T, Wu T, Spagna A, Egan LJ, Fan J. Neuroanatomical alterations in high-functioning adults with autism spectrum disorder. Front Neurosci. (2016) 10:237. doi: 10.3389/fnins.2016.00237
33. Arutiunian V, Davydova E, Pereverzeva D, Sorokin A, Tyushkevich S, Mamokhina U, et al. Reduced grey matter volume of amygdala and hippocampus is associated with the severity of autistic symptoms and language abilities in school-aged children with Autism Spectrum Disorder: an exploratory study. Brain Struct Funct. (2023) 228:1573–9. doi: 10.1007/s00429-023-02660-9
34. Groen W, Teluij M, Buitelaar J, Tendolkar I. Amygdala and hippocampus enlargement during adolescence in autism. J Am Acad Child Adolesc Psychiatry. (2010) 49:552–60. doi: 10.1016/j.jaac.2009.12.023
35. Sussman D, Leung RC, Vogan VM, Lee W, Trelle S, Lin S, et al. The autism puzzle: Diffuse but not pervasive neuroanatomical abnormalities in children with ASD. NeuroImage Clin. (2015) 8:170–9. doi: 10.1016/j.nicl.2015.04.008
36. Aylward EH, Minshew NJ, Goldstein G, Honeycutt NA, Augustine AM, Yates KO, et al. MRI volumes of amygdala and hippocampus in non-mentally retarded autistic adolescents and adults. Neurology. (1999) 53:2145–50. doi: 10.1212/WNL.53.9.2145
37. Nees F, Banaschewski T, Bokde ALW, Desrivières S, Grigis A, Garavan H, et al. Global and regional structural differences and prediction of autistic traits during adolescence. Brain Sci. (2022) 12:1187. doi: 10.3390/brainsci12091187
38. Pagni BA, Walsh MJM, Ofori E, Chen K, Sullivan G, Alvar J, et al. Effects of age on the hippocampus and verbal memory in adults with autism spectrum disorder: Longitudinal versus cross-sectional findings. Autism Res. (2022) 15:1810–23. doi: 10.1002/aur.2797
39. Lee JK, Nordahl CW, Amaral DG, Lee A, Solomon M, Ghetti S. Assessing hippocampal development and language in early childhood: Evidence from a new application of the Automatic Segmentation Adapter Tool. Hum Brain Mapp. (2015) 36:4483–96. doi: 10.1002/hbm.22931
40. Nicolson R, DeVito TJ, Vidal CN, Sui Y, Hayashi KM, Drost DJ, et al. Detection and mapping of hippocampal abnormalities in autism. Psychiatry Res. (2006) 148:11–21. doi: 10.1016/j.pscychresns.2006.02.005
41. Sacco R, Gabriele S, Persico AM. Head circumference and brain size in autism spectrum disorder: A systematic review and meta-analysis. Psychiatry Res. (2015) 234:239–51. doi: 10.1016/j.pscychresns.2015.08.016
42. Maier S, Tebartz van Elst L, Beier D, Ebert D, Fangmeier T, Radtke M, et al. Increased hippocampal volumes in adults with high functioning autism spectrum disorder and an IQ>100: A manual morphometric study. Psychiatry Res. (2015) 234:152–5. doi: 10.1016/j.pscychresns.2015.08.002
43. Xu Q, Zuo C, Liao S, Long Y, Wang Y. Abnormal development pattern of the amygdala and hippocampus from childhood to adulthood with autism. J Clin Neurosci. (2020) 78:327–32. doi: 10.1016/j.jocn.2020.03.049
44. Markram K, Markram H. The intense world theory - a unifying theory of the neurobiology of autism. Front Hum Neurosci. (2010) 4:224. doi: 10.3389/fnhum.2010.00224
45. Habata K, Cheong Y, Kamiya T, Shiotsu D, Omori IM, Okazawa H, et al. Relationship between sensory characteristics and cortical thickness/volume in autism spectrum disorders. Transl Psychiatry. (2021) 11:616. doi: 10.1038/s41398-021-01743-7
46. Schumann CM, Hamstra J, Goodlin-Jones BL, Lotspeich LJ, Kwon H, Buonocore MH, et al. The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J Neurosci. (2004) 24:6392–401. doi: 10.1523/JNEUROSCI.1297-04.2004
47. Murphy CM, Deeley Q, Daly EM, Ecker C, O'Brien FM, Hallahan B, et al. Anatomy and aging of the amygdala and hippocampus in autism spectrum disorder: an in vivo magnetic resonance imaging study of Asperger syndrome. Autism Res. (2012) 5:3–12. doi: 10.1002/aur.227
48. Dager SR, Wang L, Friedman SD, Shaw DW, Constantino JN, Artru AA, et al. Shape mapping of the hippocampus in young children with autism spectrum disorder. AJNR Am J Neuroradiol. (2007) 28:672–7.
49. Zuo C, Wang D, Tao F, Wang Y. Changes in the development of subcortical structures in autism spectrum disorder. Neuroreport. (2019) 30:1062–7. doi: 10.1097/WNR.0000000000001300
50. Reinhardt VP, Iosif AM, Libero L, Heath B, Rogers SJ, Ferrer E, et al. Understanding hippocampal development in young children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. (2020) 59:1069–79. doi: 10.1016/j.jaac.2019.08.008
51. Richards R, Greimel E, Kliemann D, Koerte IK, Schulte-Körne G, Reuter M, et al. Increased hippocampal shape asymmetry and volumetric ventricular asymmetry in autism spectrum disorder. NeuroImage Clin. (2020) 26:102207. doi: 10.1016/j.nicl.2020.102207
52. Saitoh O, Karns CM, Courchesne E. Development of the hippocampal formation from 2 to 42 years: MRI evidence of smaller area dentata in autism. Brain. (2001) 124:1317–24. doi: 10.1093/brain/124.7.1317
53. Rojas DC, Smith JA, Benkers TL, Camou SL, Reite ML, Rogers SJ. Hippocampus and amygdala volumes in parents of children with autistic disorder. Am J Psychiatry. (2004) 161:2038–44. doi: 10.1176/appi.ajp.161.11.2038
54. Ferreira D, Hansson O, Barroso J, Molina Y, MaChado A, Hernández-Cabrera JA, et al. The interactive effect of demographic and clinical factors on hippocampal volume: A multicohort study on 1958 cognitively normal individuals. Hippocampus. (2017) 27:653–67. doi: 10.1002/hipo.22721
55. Kemper TL, Bauman ML. The contribution of neuropathologic studies to the understanding of autism. Neurol Clin. (1993) 11:175–87. doi: 10.1016/S0733-8619(18)30176-2
56. Raymond GV, Bauman ML, Kemper TL. Hippocampus in autism: a Golgi analysis. Acta Neuropathol. (1996) 91:117–9. doi: 10.1007/s004010050401
57. Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, et al. A clinicopathological study of autism. Brain. (1998) 121:889–905. doi: 10.1093/brain/121.5.889
58. Lawrence YA, Kemper TL, Bauman ML, Blatt GJ. Parvalbumin-, calbindin-, and calretinin-immunoreactive hippocampal interneuron density in autism. Acta Neurol Scand. (2010) 121:99–108. doi: 10.1111/j.1600-0404.2009.01234.x
59. Salmond CH, Ashburner J, Connelly A, Friston KJ, Gadian DG, Vargha-Khadem F. The role of the medial temporal lobe in autistic spectrum disorders. Eur J Neurosci. (2005) 22:764–72. doi: 10.1111/j.1460-9568.2005.04217.x
60. Sun F, Chen Y, Gao Q, Zhao Z. Abnormal gray matter structure in children and adolescents with high-functioning autism spectrum disorder. Psychiatry Res Neuroimaging. (2022) 327:111564. doi: 10.1016/j.pscychresns.2022.111564
61. Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. (2010) 119:755–70. doi: 10.1007/s00401-010-0655-4
62. Allebone J, Kanaan R, Maller J, O'Brien T, Mullen SA, Cook M, et al. Bilateral volume reduction in posterior hippocampus in psychosis of epilepsy. J Neurol Neurosurg Psychiatry. (2019) 90:688–94. doi: 10.1136/jnnp-2018-319396
63. Tang S, Nie L, Liu X, Chen Z, Zhou Y, Pan Z, et al. Application of quantitative magnetic resonance imaging in the diagnosis of autism in children. Front Med. (2022) 9:818404. doi: 10.3389/fmed.2022.818404
64. Tang S, Liu X, Ran Q, Nie L, Wu L, Pan Z, et al. Application of three-dimensional pseudocontinuous arterial spin labeling perfusion imaging in the brains of children with autism. Front Neurol. (2022) 13:851430. doi: 10.3389/fneur.2022.851430
65. Tang S, Liu X, Nie L, Chen Z, Ran Q, He L. Diagnosis of children with attention-deficit/hyperactivity disorder. (ADHD) comorbid autistic traits. (ATs) by applying quantitative magnetic resonance imaging techniques. Front Psychiatry. (2022) 13:1038471. doi: 10.3389/fpsyt.2022.1038471
66. Heo S, Prakash RS, Voss MW, Erickson KI, Ouyang C, Sutton BP, et al. Resting hippocampal blood flow, spatial memory and aging. Brain Res. (2010) 1315:119–27. doi: 10.1016/j.brainres.2009.12.020
67. Pagani M, Manouilenko I, Stone-Elander S, Odh R, Salmaso D, Hatherly R, et al. Brief Report: alterations in cerebral blood flow as assessed by PET/CT in adults with autism spectrum disorder with normal IQ. J Autism Dev Disord. (2012) 42:313–18. doi: 10.1007/s10803-011-1240-y
68. Otsuka H, Harada M, Mori K, Hisaoka S, Nishitani H. Brain metabolites in the hippocampus-amygdala region and cerebellum in autism: an 1H-MR spectroscopy study. Neuroradiology. (1999) 41:517–19. doi: 10.1007/s002340050795
69. Gabis L, Huang W, Azizian A, DeVincent C, Tudorica A, Kesner-Baruch Y, et al. 1H-magnetic resonance spectroscopy markers of cognitive and language ability in clinical subtypes of autism spectrum disorders. J Child Neurol. (2008) 23:766–74. doi: 10.1177/0883073808315423
70. Liserre R, Pinelli L, Gasparotti R. MR spectroscopy in pediatric neuroradiology. Transl Pediatr. (2021) 10:1169–200. doi: 10.21037/tp-20-445
71. Page LA, Daly E, Schmitz N, Simmons A, Toal F, Deeley Q, et al. In vivo 1H-magnetic resonance spectroscopy study of amygdala-hippocampal and parietal regions in autism. Am J Psychiatry. (2006) 163:2189–192. doi: 10.1176/ajp.2006.163.12.2189
72. O'Brien FM, Page L, O'Gorman RL, Bolton P, Sharma A, Baird G, et al. Maturation of limbic regions in Asperger syndrome: a preliminary study using proton magnetic resonance spectroscopy and structural magnetic resonance imaging. Psychiatry Res. (2010) 184:77–85. doi: 10.1016/j.pscychresns.2010.08.007
73. Zeegers M, van der Grond J, van Daalen E, Buitelaar J, van Engeland H. Proton magnetic resonance spectroscopy in developmentally delayed young boys with or without autism. J Neural Transm. (2007) 114:289–95. doi: 10.1007/s00702-006-0501-y
74. Brown MS, Singel D, Hepburn S, Rojas DC. Increased glutamate concentration in the auditory cortex of persons with autism and first-degree relatives: a. (1)H-MRS study. Autism Res. (2013) b6:1–10. doi: 10.1002/aur.1260
75. Sokol DK, Dunn DW, Edwards-Brown M, Feinberg J. Hydrogen proton magnetic resonance spectroscopy in autism: preliminary evidence of elevated choline/creatine ratio. J Child Neurol. (2002) 17:245–9. doi: 10.1177/088307380201700401
76. Suzuki K, Nishimura K, Sugihara G, Nakamura K, Tsuchiya KJ, Matsumoto K, et al. Metabolite alterations in the hippocampus of high-functioning adult subjects with autism. Int J Neuropsychopharmacol. (2010) 13:529–34. doi: 10.1017/S1461145709990952
77. Hashimoto T, Yokota S, Matsuzaki Y, Kawashima R. Intrinsic hippocampal functional connectivity underlying rigid memory in children and adolescents with autism spectrum disorder: A case-control study. Autism. (2021) 25:1901–12. doi: 10.1177/13623613211004058
78. Liu J, Chen L, Chang H, Rudoler J, Al-Zughoul AB, Kang JB, et al. Replicable patterns of memory impairments in children with autism and their links to hyperconnected brain circuits. Biol Psychiatry Cognit Neurosci Neuroimaging. (2023) 8:1113–23. doi: 10.1016/j.bpsc.2023.05.002
79. Solomon M, Ragland JD, Niendam TA, Lesh TA, Beck JS, Matter JC, et al. Atypical learning in autism spectrum disorders: A functional magnetic resonance imaging study of transitive inference. J Am Acad Child Adolesc Psychiatry. (2015) 54:947–55. doi: 10.1016/j.jaac.2015.08.010
80. Liu J, Okada NJ, Cummings KK, Jung J, Patterson G, Bookheimer SY, et al. Emerging atypicalities in functional connectivity of language-related networks in young infants at high familial risk for ASD. Dev Cognit Neurosci. (2020) 45:100814. doi: 10.1016/j.dcn.2020.100814
81. Klapwijk ET, Aghajani M, Colins OF, Marijnissen GM, Popma A, van Lang ND, et al. Different brain responses during empathy in autism spectrum disorders versus conduct disorder and callous-unemotional traits. J Child Psychol Psychiatry. (2016) 57:737–47. doi: 10.1111/jcpp.12498
82. Green SA, Rudie JD, Colich NL, Wood JJ, Shirinyan D, Hernandez L, et al. Overreactive brain responses to sensory stimuli in youth with autism spectrum disorders. J Am Acad Child Adolesc Psychiatry. (2013) 52:1158–72. doi: 10.1016/j.jaac.2013.08.004
83. Loveland KA, Bachevalier J, Pearson DA, Lane DM. Fronto-limbic functioning in children and adolescents with and without autism. Neuropsychologia. (2008) 46:49–62. doi: 10.1016/j.neuropsychologia.2007.08.017
84. Mouga S, Duarte IC, Café C, Sousa D, Duque F, Oliveira G, et al. Parahippocampal deactivation and hyperactivation of central executive, saliency and social cognition networks in autism spectrum disorder. J Neurodev Disord. (2022) 14:9. doi: 10.1186/s11689-022-09417-1
85. Beaudet AL. Autism: highly heritable but not inherited. Nat Med. (2007) 13:534–6. doi: 10.1038/nm0507-534
86. Li Z, Zhu YX, Gu LJ, Cheng Y. Understanding autism spectrum disorders with animal models: applications, insights, and perspectives. Zool Res. (2021) 42:800–24. doi: 10.24272/j.issn.2095-8137.2021.251
87. Fyke W, Velinov M. FMR1 and autism, an intriguing connection revisited. Genes. (2021) 12:1218. doi: 10.3390/genes12081218
88. Maurin T, Melancia F, Jarjat M, Castro L, Costa L, Delhaye S, et al. Involvement of phosphodiesterase 2A activity in the pathophysiology of fragile X syndrome. Cereb Cortex. (2019) 29:3241–52. doi: 10.1093/cercor/bhy192
89. Pop AS, Levenga J, de Esch CE, Buijsen RA, Nieuwenhuizen IM, Li T, et al. Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology. (2014) 231:1227–35. doi: 10.1007/s00213-012-2947-y
90. Oddi D, Subashi E, Middei S, Bellocchio L, Lemaire-Mayo V, Guzmán M, et al. Early social enrichment rescues adult behavioral and brain abnormalities in a mouse model of fragile X syndrome. Neuropsychopharmacology. (2015) 40:1113–22. doi: 10.1038/npp.2014.291
91. Chen YS, Zhang SM, Yue CX, Xiang P, Li JQ, Wei Z, et al. Early environmental enrichment for autism spectrum disorder Fmr1 mice models has positive behavioral and molecular effects. Exp Neurol. (2022) 352:114033. doi: 10.1016/j.expneurol.2022.114033
92. Lazarov O, Demars MP, Zhao Kda T, Ali HM, Grauzas V, Kney A, et al. Impaired survival of neural progenitor cells in dentate gyrus of adult mice lacking fMRP. Hippocampus. (2012) 22:1220–24. doi: 10.1002/hipo.20989
93. Braun K, Segal M. FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb Cortex. (2000) 10:1045–52. doi: 10.1093/cercor/10.10.1045
94. Klemmer P, Meredith RM, Holmgren CD, Klychnikov OI, Stahl-Zeng J, Loos M, et al. Proteomics, ultrastructure, and physiology of hippocampal synapses in a fragile X syndrome mouse model reveal presynaptic phenotype. J Biol Chem. (2011) 286:25495–504. doi: 10.1074/jbc.M110.210260
95. Zhang J, Hou L, Klann E, Nelson DL. Altered hippocampal synaptic plasticity in the Fmr1 gene family knockout mouse models. J Neurophysiol. (2009) 101:2572–80. doi: 10.1152/jn.90558.2008
96. Wang XS, Peng CZ, Cai WJ, Xia J, Jin D, Dai Y, et al. Activity-dependent regulation of release probability at excitatory hippocampal synapses: a crucial role of fragile X mental retardation protein in neurotransmission. Eur J Neurosci. (2014) 39:1602–12. doi: 10.1111/ejn.12546
97. Yun SH, Trommer BL. Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J Neurosci Res. (2011) 89:176–82. doi: 10.1002/jnr.22546
98. Tian Y, Yang C, Shang S, Cai Y, Deng X, Zhang J, et al. Loss of FMRP impaired hippocampal long-term plasticity and spatial learning in rats. Front Mol Neurosci. (2017) 10:269. doi: 10.3389/fnmol.2017.00269
99. Arbab T, Pennartz CMA, Battaglia FP. Impaired hippocampal representation of place in the Fmr1-knockout mouse model of fragile X syndrome. Sci Rep. (2018) 8:8889. doi: 10.1038/s41598-018-26853-z
100. Jeon SJ, Kwon H, Bae HJ, Gonzales EL, Kim J, Chung HJ, et al. Agmatine relieves behavioral impairments in Fragile X mice model. Neuropharmacology. (2022) 219:109234. doi: 10.1016/j.neuropharm.2022.109234
101. Prieto M, Folci A, Poupon G, Schiavi S, Buzzelli V, Pronot M, et al. Missense mutation of Fmr1 results in impaired AMPAR-mediated plasticity and socio-cognitive deficits in mice. Nat Commun. (2021) 12:1557. doi: 10.1038/s41467-021-21820-1
102. Reinhard SM, Rais M, Afroz S, Hanania Y, Pendi K, Espinoza K, et al. Reduced perineuronal net expression in Fmr1 KO mice auditory cortex and amygdala is linked to impaired fear-associated memory. Neurobiol Learn Mem. (2019) 164:107042. doi: 10.1016/j.nlm.2019.107042
103. Sawicka K, Hale CR, Park CY, Fak JJ, Gresack JE, Van Driesche SJ, et al. FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. Elife. (2019) 8:e46919. doi: 10.7554/eLife.46919
104. Boone CE, Davoudi H, Harrold JB, Foster DJ. Abnormal sleep architecture and hippocampal circuit dysfunction in a mouse model of fragile X syndrome. Neuroscience. (2018) 384:275–89. doi: 10.1016/j.neuroscience.2018.05.012
105. Uutela M, Lindholm J, Rantamäki T, Umemori J, Hunter K, Võikar V, et al. Distinctive behavioral and cellular responses to fluoxetine in the mouse model for Fragile X syndrome. Front Cell Neurosci. (2014) 8:150. doi: 10.3389/fncel.2014.00150
106. Qin M, Entezam A, Usdin K, Huang T, Liu ZH, Hoffman GE, et al. A mouse model of the fragile X premutation: effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol Dis. (2011) 42:85–98. doi: 10.1016/j.nbd.2011.01.008
107. Hao S, Tang B, Wu Z, Ure K, Sun Y, Tao H, et al. Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature. (2015) 526:430–34. doi: 10.1038/nature15694
108. Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, et al. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. (2006) 26:319–27. doi: 10.1523/JNEUROSCI.2623-05.2006
109. Bertoldi ML, Zalosnik MI, Fabio MC, Aja S, Roth GA, Ronnett GV, et al. MeCP2 deficiency disrupts kainate-induced presynaptic plasticity in the mossy fiber projections in the hippocampus. Front Cell Neurosci. (2019) 13:286. doi: 10.3389/fncel.2019.00286
110. Li W, Bellot-Saez A, Phillips ML, Yang T, Longo FM, Pozzo-Miller L. A small-molecule TrkB ligand restores hippocampal synaptic plasticity and object location memory in Rett syndrome mice. Dis Model Mech. (2017) 10:837–45. doi: 10.1242/dmm.029959
111. He L, Caudill MS, Jing J, Wang W, Sun Y, Tang J, et al. A weakened recurrent circuit in the hippocampus of Rett syndrome mice disrupts long-term memory representations. Neuron. (2022) 110:1689–1699.e6. doi: 10.1016/j.neuron.2022.02.014
112. Kee SE, Mou X, Zoghbi HY, Ji D. Impaired spatial memory codes in a mouse model of Rett syndrome. Elife. (2018) 7:e31451. doi: 10.7554/eLife.31451
113. Calfa G, Li W, Rutherford JM, Pozzo-Miller L. Excitation/inhibition imbalance and impaired synaptic inhibition in hippocampal area CA3 of Mecp2 knockout mice. Hippocampus. (2015) 25:159–68. doi: 10.1002/hipo.22360
114. Sun Y, Gao Y, Tidei JJ, Shen M, Hoang JT, Wagner DF, et al. Loss of MeCP2 in immature neurons leads to impaired network integration. Hum Mol Genet. (2019) 28:245–57. doi: 10.1093/hmg/ddy338
115. Balakrishnan S, Mironov SL. Regenerative glutamate release in the hippocampus of Rett syndrome model mice. PloS One. (2018) 13:e0202802. doi: 10.1371/journal.pone.0202802
116. Li W, Xu X, Pozzo-Miller L. Excitatory synapses are stronger in the hippocampus of Rett syndrome mice due to altered synaptic trafficking of AMPA-type glutamate receptors. Proc Natl Acad Sci USA. (2016) 113:E1575–84. doi: 10.1073/pnas.1517244113
117. Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. (2006) 16:710–6. doi: 10.1016/j.cub.2006.02.062
118. Lu H, Ash RT, He L, Kee SE, Wang W, Yu D, et al. Loss and gain of meCP2 cause similar hippocampal circuit dysfunction that is rescued by deep brain stimulation in a rett syndrome mouse model. Neuron. (2016) 91:739–47. doi: 10.1016/j.neuron.2016.07.018
119. Zhang L, He J, Jugloff DG, Eubanks JH. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus. (2008) 18:294–309. doi: 10.1002/hipo.20389
120. Chapleau CA, Boggio EM, Calfa G, Percy AK, Giustetto M, Pozzo-Miller L. Hippocampal CA1 pyramidal neurons of Mecp2 mutant mice show a dendritic spine phenotype only in the presymptomatic stage. Neural Plast. (2012) 2012:976164. doi: 10.1155/2012/976164
121. Kaphzan H, Buffington SA, Ramaraj AB, Lingrel JB, Rasband MN, Santini E, et al. Genetic reduction of the α1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep. (2013) 4:405–12. doi: 10.1016/j.celrep.2013.07.005
122. Rayi PR, Koyavski L, Chakraborty D, Bagrov A, Kaphzan H. α1-Na/K-ATPase inhibition rescues aberrant dendritic calcium dynamics and memory deficits in the hippocampus of an Angelman syndrome mouse model. Prog Neurobiol. (2019) 182:101676. doi: 10.1016/j.pneurobio.2019.101676
123. van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, et al. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. (2007) 10:280–2. doi: 10.1038/nn1845
124. Cao C, Rioult-Pedotti MS, Migani P, Yu CJ, Tiwari R, Parang K, et al. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PloS Biol. (2013) 11:e1001478. doi: 10.1371/journal.pbio.1001478
125. Kaphzan H, Buffington SA, Jung JI, Rasband MN, Klann E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J Neurosci. (2011) 31:17637–48. doi: 10.1523/JNEUROSCI.4162-11.2011
126. Sun J, Zhu G, Liu Y, Standley S, Ji A, Tunuguntla R, et al. UBE3A regulates synaptic plasticity and learning and memory by controlling SK2 channel endocytosis. Cell Rep. (2015) 12:449–61. doi: 10.1016/j.celrep.2015.06.023
127. Viho EMG, Punt AM, Distel B, Houtman R, Kroon J, Elgersma Y, et al. The hippocampal response to acute corticosterone elevation is altered in a mouse model for angelman syndrome. Int J Mol Sci. (2022) 24:303. doi: 10.3390/ijms24010303
128. Godavarthi SK, Dey P, Maheshwari M, Jana NR. Defective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndrome. Hum Mol Genet. (2012) 21:1824–34. doi: 10.1093/hmg/ddr614
129. Godavarthi SK, Sharma A, Jana NR. Reversal of reduced parvalbumin neurons in hippocampus and amygdala of Angelman syndrome model mice by chronic treatment of fluoxetine. J Neurochem. (2014) 130:444–54. doi: 10.1111/jnc.12726
130. Curatolo P, Scheper M, Emberti Gialloreti L, Specchio N, Aronica E. Is tuberous sclerosis complex-associated autism a preventable and treatable disorder? World J Pediatr. (2024) 20:40–53. doi: 10.1007/s12519-023-00762-2
131. Haji N, Riebe I, Aguilar-Valles A, Artinian J, Laplante I, Lacaille JC. Tsc1 haploinsufficiency in Nkx2.1 cells upregulates hippocampal interneuron mTORC1 activity, impairs pyramidal cell synaptic inhibition, and alters contextual fear discrimination and spatial working memory in mice. Mol Autism. (2020) 11:29. doi: 10.1186/s13229-020-00340-7
132. Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. (2008) 14:843–8. doi: 10.1038/nm1788
133. Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann Neurol. (2007) 62:648–55. doi: 10.1002/ana.21317
134. Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron. (2013) 78:510–22. doi: 10.1016/j.neuron.2013.03.017
135. Koene LM, Niggl E, Wallaard I, Proietti-Onori M, Rotaru DC, Elgersma Y. Identifying the temporal electrophysiological and molecular changes that contribute to TSC-associated epileptogenesis. JCI Insight. (2021) 6:e150120. doi: 10.1172/jci.insight.150120
136. Lasarge CL, Danzer SC. Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front Mol Neurosci. (2014) 7:18. doi: 10.3389/fnmol.2014.00018
137. Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, et al. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol. (2003) 54:251–6. doi: 10.1002/ana.10648
138. Xu L, Zeng LH, Wong M. Impaired astrocytic gap junction coupling and potassium buffering in a mouse model of tuberous sclerosis complex. Neurobiol Dis. (2009) 34:291–9. doi: 10.1016/j.nbd.2009.01.010
139. Zhang B, Zou J, Han L, Rensing N, Wong M. Microglial activation during epileptogenesis in a mouse model of tuberous sclerosis complex. Epilepsia. (2016) 57:1317–25. doi: 10.1111/epi.13429
140. Deng Y, Yang Q, Yang Y, Li Y, Peng H, Wu S, et al. Conditional knockout of Tsc1 in RORγt-expressing cells induces brain damage and early death in mice. J Neuroinflamm. (2021) 18:107. doi: 10.1186/s12974-021-02153-8
141. Chévere-Torres I, Kaphzan H, Bhattacharya A, Kang A, Maki JM, Gambello MJ, et al. Metabotropic glutamate receptor-dependent long-term depression is impaired due to elevated ERK signaling in the ΔRG mouse model of tuberous sclerosis complex. Neurobiol Dis. (2012) 45:1101–10. doi: 10.1016/j.nbd.2011.12.028
142. Potter WB, Basu T, O'Riordan KJ, Kirchner A, Rutecki P, Burger C, et al. Reduced juvenile long-term depression in tuberous sclerosis complex is mitigated in adults by compensatory recruitment of mGluR5 and Erk signaling. PloS Biol. (2013) 11:e1001627. doi: 10.1371/journal.pbio.1001627
143. Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J Neurosci. (2011) 31:8862–9. doi: 10.1523/JNEUROSCI.1617-11.2011
144. Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. (2005) 8:1727–34. doi: 10.1038/nn1566
145. Weiss HR, Liu X, Chi OZ. Cerebral O(2) consumption in young Eker rats, effects of GABA blockade: implications for autism. Int J Dev Neurosci. (2008) 26:517–21. doi: 10.1016/j.ijdevneu.2008.01.002
146. Chi OZ, Wu CC, Liu X, Rah KH, Jacinto E, Weiss HR. Restoration of normal cerebral oxygen consumption with rapamycin treatment in a rat model of autism-tuberous sclerosis. Neuromol Med. (2015) 17:305–13. doi: 10.1007/s12017-015-8359-5
147. Harony-Nicolas H, Kay M, du Hoffmann J, Klein ME, Bozdagi-Gunal O, Riad M, et al. Oxytocin improves behavioral and electrophysiological deficits in a novel Shank3-deficient rat. Elife. (2017) 6:e18904. doi: 10.7554/eLife.18904
148. Atanasova E, Arévalo AP, Graf I, Zhang R, Bockmann J, Lutz AK, et al. Immune activation during pregnancy exacerbates ASD-related alterations in Shank3-deficient mice. Mol Autism. (2023) 14:1. doi: 10.1186/s13229-022-00532-3
149. Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet. (2011) 20:3093–108. doi: 10.1093/hmg/ddr212
150. Oberman LM, Boccuto L, Cascio L, Sarasua S, Kaufmann WE. Autism spectrum disorder in Phelan-McDermid syndrome: initial characterization and genotype-phenotype correlations. Orphanet J Rare Dis. (2015) 10:105. doi: 10.1186/s13023-015-0323-9
151. Urrutia-Ruiz C, Rombach D, Cursano S, Gerlach-Arbeiter S, Schoen M, Bockmann J, et al. Deletion of the autism-associated protein SHANK3 abolishes structural synaptic plasticity after brain trauma. Int J Mol Sci. (2022) 23:6081. doi: 10.3390/ijms23116081
152. Xu D, Meng Y, An S, Meng W, Li H, Zhang W, et al. Swimming exercise is a promising early intervention for autism-like behavior in Shank3 deletion rats. CNS Neurosci Ther. (2023) 29:78–90. doi: 10.1111/cns.13920
153. Lee J, Chung C, Ha S, Lee D, Kim DY, Kim H, et al. Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front Cell Neurosci. (2015) 9:94. doi: 10.3389/fncel.2015.00094
154. Yoo YE, Yoo T, Lee S, Lee J, Kim D, Han HM, et al. Shank3 mice carrying the human Q321R mutation display enhanced self-grooming, abnormal electroencephalogram patterns, and suppressed neuronal excitability and seizure susceptibility. Front Mol Neurosci. (2019) 12:155. doi: 10.3389/fnmol.2019.00155
155. Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Liu S, Powell CM. Altered striatal synaptic function and abnormal behaviour in shank3 exon4-9 deletion mouse model of autism. Autism Res. (2016) 9:350–75. doi: 10.1002/aur.1529
156. Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Escamilla CO, Weaver TP, et al. Novel Shank3 mutant exhibits behaviors with face validity for autism and altered striatal and hippocampal function. Autism Res. (2017) 10:42–65. doi: 10.1002/aur.1664
157. Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. (2010) 1:15. doi: 10.1186/2040-2392-1-15
158. Tao K, Chung M, Watarai A, Huang Z, Wang MY, Okuyama T. Disrupted social memory ensembles in the ventral hippocampus underlie social amnesia in autism-associated Shank3 mutant mice. Mol Psychiatry. (2022) 27:2095–105. doi: 10.1038/s41380-021-01430-5
159. Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. (2011) 472:437–42. doi: 10.1038/nature09965
160. Cope EC, Wang SH, Waters RC, Gore IR, Vasquez B, Laham BJ, et al. Activation of the CA2-ventral CA1 pathway reverses social discrimination dysfunction in Shank3B knockout mice. Nat Commun. (2023) 14:1750. doi: 10.1038/s41467-023-37248-8
161. Yoo T, Yoo YE, Kang H, Kim E. Age, brain region, and gene dosage-differential transcriptomic changes in Shank3-mutant mice. Front Mol Neurosci. (2022) 15:1017512. doi: 10.3389/fnmol.2022.1017512
162. Dahlhaus R, Hines RM, Eadie BD, Kannangara TS, Hines DJ, Brown CE, et al. Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus. (2010) 20:305–22. doi: 10.1002/hipo.20630
163. Jedlicka P, Vnencak M, Krueger DD, Jungenitz T, Brose N, Schwarzacher SW. Neuroligin-1 regulates excitatory synaptic transmission, LTP and EPSP-spike coupling in the dentate gyrus. vivo. Brain Struct Funct. (2015) 220:47–58. doi: 10.1007/s00429-013-0636-1
164. Jiang M, Polepalli J, Chen LY, Zhang B, Südhof TC, Malenka RC. Conditional ablation of neuroligin-1 in CA1 pyramidal neurons blocks LTP by a cell-autonomous NMDA receptor-independent mechanism. Mol Psychiatry. (2017) 22:375–83. doi: 10.1038/mp.2016.80
165. Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, et al. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. (2007) 54:919–31. doi: 10.1016/j.neuron.2007.05.029
166. Jedlicka P, Hoon M, Papadopoulos T, Vlachos A, Winkels R, Poulopoulos A, et al. Increased dentate gyrus excitability in neuroligin-2-deficient mice. vivo. Cereb Cortex. (2011) 21:357–67. doi: 10.1093/cercor/bhq100
167. Kohl C, Riccio O, Grosse J, Zanoletti O, Fournier C, Schmidt MV, et al. Hippocampal neuroligin-2 overexpression leads to reduced aggression and inhibited novelty reactivity in rats. PloS One. (2013) 8:e56871. doi: 10.1371/journal.pone.0056871
168. Van Zandt M, Weiss E, Almyasheva A, Lipior S, Maisel S, Naegele JR. Adeno-associated viral overexpression of neuroligin 2 in the mouse hippocampus enhances GABAergic synapses and impairs hippocampus-dependent behaviors. Behav Brain Res. (2019) 362:7–20. doi: 10.1016/j.bbr.2018.12.052
169. Koganezawa N, Hanamura K, Schwark M, Krueger-Burg D, Kawabe H. Super-resolved 3D-STED microscopy identifies a layer-specific increase in excitatory synapses in the hippocampal CA1 region of Neuroligin-3 KO mice. Biochem Biophys Res Commun. (2021) 582:144–9. doi: 10.1016/j.bbrc.2021.10.003
170. Modi B, Pimpinella D, Pazienti A, Zacchi P, Cherubini E, Griguoli M. Possible implication of the CA2 hippocampal circuit in social cognition deficits observed in the neuroligin 3 knock-out mouse, a non-syndromic animal model of autism. Front Psychiatry. (2019) 10:513. doi: 10.3389/fpsyt.2019.00513
171. Polepalli JS, Wu H, Goswami D, Halpern CH, Südhof TC, Malenka RC. Modulation of excitation on parvalbumin interneurons by neuroligin-3 regulates the hippocampal network. Nat Neurosci. (2017) 20:219–29. doi: 10.1038/nn.4471
172. Etherton M, Földy C, Sharma M, Tabuchi K, Liu X, Shamloo M, et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci USA. (2011) 108:13764–9. doi: 10.1073/pnas.1111093108
173. Tabuchi K, Chang W, Asgar NF, Pramanik G. Synapse maturation and autism: learning from neuroligin model mice. Nihon Shinkei Seishin Yakurigaku Zasshi. (2014) 34:1–4.
174. Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. (2007) 318:71–6. doi: 10.1126/science.1146221
175. Sgritta M, Vignoli B, Pimpinella D, Griguoli M, Santi S, Bialowas A, et al. Impaired synaptic plasticity in an animal model of autism exhibiting early hippocampal GABAergic-BDNF/TrkB signaling alterations. iScience. (2022) 26:105728. doi: 10.1016/j.isci.2022.105728
176. Hammer M, Krueger-Burg D, Tuffy LP, Cooper BH, Taschenberger H, Goswami SP, et al. Perturbed hippocampal synaptic inhibition and γ-oscillations in a neuroligin-4 knockout mouse model of autism. Cell Rep. (2015) 13:516–23. doi: 10.1016/j.celrep.2015.09.011
177. Muellerleile J, Vnencak M, Sethi MVA, Jungenitz T, Schwarzacher SW, Jedlicka P. Increased network inhibition in the dentate gyrus of adult neuroligin-4 knock-out mice. eNeuro. (2023) 10:ENEURO.0471–22.2023. doi: 10.1523/ENEURO.0471-22.2023
178. Guneykaya D, Ugursu B, Logiacco F, Popp O, Feiks MA, Meyer N, et al. Sex-specific microglia state in the Neuroligin-4 knock-out mouse model of autism spectrum disorder. Brain Behav Immun. (2023) 111:61–75. doi: 10.1016/j.bbi.2023.03.023
179. Yonan JM, Steward O. Vector-mediated PTEN deletion in the adult dentate gyrus initiates new growth of granule cell bodies and dendrites and expansion of mossy fiber terminal fields that continues for months. Neurobiol Dis. (2023) 184:106190. doi: 10.1016/j.nbd.2023.106190
180. Amiri A, Cho W, Zhou J, Birnbaum SG, Sinton CM, McKay RM, et al. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J Neurosci. (2012) 32:5880–90. doi: 10.1523/JNEUROSCI.5462-11.2012
181. Luikart BW, Schnell E, Washburn EK, Bensen AL, Tovar KR, Westbrook GL. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci. (2011) 31:4345–54. doi: 10.1523/JNEUROSCI.0061-11.2011
182. Matsushita Y, Sakai Y, Shimmura M, Shigeto H, Nishio M, Akamine S, et al. Hyperactive mTOR signals in the proopiomelanocortin-expressing hippocampal neurons cause age-dependent epilepsy and premature death in mice. Sci Rep. (2016) 6:22991. doi: 10.1038/srep22991
183. LaSarge CL, Santos VR, Danzer SC. PTEN deletion from adult-generated dentate granule cells disrupts granule cell mossy fiber axon structure. Neurobiol Dis. (2015) 75:142–50. doi: 10.1016/j.nbd.2014.12.029
184. Santos VR, Pun RYK, Arafa SR, LaSarge CL, Rowley S, Khademi S, et al. PTEN deletion increases hippocampal granule cell excitability in male and female mice. Neurobiol Dis. (2017) 108:339–51. doi: 10.1016/j.nbd.2017.08.014
185. Takeuchi K, Gertner MJ, Zhou J, Parada LF, Bennett MV, Zukin RS. Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc Natl Acad Sci USA. (2013) 110:4738–43. doi: 10.1073/pnas.1222803110
186. LaSarge CL, Pun RY, Muntifering MB, Danzer SC. Disrupted hippocampal network physiology following PTEN deletion from newborn dentate granule cells. Neurobiol Dis. (2016) 96:105–14. doi: 10.1016/j.nbd.2016.09.004
187. Sperow M, Berry RB, Bayazitov IT, Zhu G, Baker SJ, Zakharenko SS. Phosphatase and tensin homologue. (PTEN) regulates synaptic plasticity independently of its effect on neuronal morphology and migration. J Physiol. (2012) 590:777–92. doi: 10.1113/jphysiol.2011.220236
188. Kind PC, Bird A. CDKL5 deficiency disorder: a pathophysiology of neural maintenance. J Clin Invest. (2021) 131:e153606. doi: 10.1172/JCI153606
189. Fuchs C, Rimondini R, Viggiano R, Trazzi S, De Franceschi M, Bartesaghi R, et al. Inhibition of GSK3β rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol Dis. (2015) 82:298–310. doi: 10.1016/j.nbd.2015.06.018
190. Fuchs C, Trazzi S, Torricella R, Viggiano R, De Franceschi M, Amendola E, et al. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3β signaling. Neurobiol Dis. (2014) 70:53–68. doi: 10.1016/j.nbd.2014.06.006
191. Gennaccaro L, Fuchs C, Loi M, Pizzo R, Alvente S, Berteotti C, et al. Age-related cognitive and motor decline in a mouse model of CDKL5 deficiency disorder is associated with increased neuronal senescence and death. Aging Dis. (2021) 12:764–85. doi: 10.14336/AD.2020.0827
192. Galvani G, Mottolese N, Gennaccaro L, Loi M, Medici G, Tassinari M, et al. Inhibition of microglia overactivation restores neuronal survival in a mouse model of CDKL5 deficiency disorder. J Neuroinflamm. (2021) 18:155. doi: 10.1186/s12974-021-02204-0
193. Fuchs C, Medici G, Trazzi S, Gennaccaro L, Galvani G, Berteotti C, et al. CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling. Brain Pathol. (2019) 29:658–74. doi: 10.1111/bpa.12716
194. Zhu ZA, Li YY, Xu J, Xue H, Feng X, Zhu YC, et al. CDKL5 deficiency in adult glutamatergic neurons alters synaptic activity and causes spontaneous seizures via TrkB signaling. Cell Rep. (2023) 42:113202. doi: 10.1016/j.celrep.2023.113202
195. Okuda K, Kobayashi S, Fukaya M, Watanabe A, Murakami T, Hagiwara M, et al. CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol Dis. (2017) 106:158–70. doi: 10.1016/j.nbd.2017.07.002
196. Nicolini C, Fahnestock M. The valproic acid-induced rodent model of autism. Exp Neurol. (2018) 299:217–27. doi: 10.1016/j.expneurol.2017.04.017
197. Gouda B, Sinha SN, Chalamaiah M, Vakdevi V, Shashikala P, Veeresh B, et al. Sex differences in animal models of sodium-valproate-induced autism in postnatal BALB/c mice: whole-brain histoarchitecture and 5-HT2A receptor biomarker evidence. Biology. (2022) 11:79. doi: 10.3390/biology11010079
198. Matsuo K, Yabuki Y, Fukunaga K. 5-aminolevulinic acid inhibits oxidative stress and ameliorates autistic-like behaviors in prenatal valproic acid-exposed rats. Neuropharmacology. (2020) 168:107975. doi: 10.1016/j.neuropharm.2020.107975
199. Sandhu A, Rawat K, Gautam V, Sharma A, Kumar A, Saha L. Phosphodiesterase inhibitor, ibudilast alleviates core behavioral and biochemical deficits in the prenatal valproic acid exposure model of autism spectrum disorder. Brain Res. (2023) 1815:148443. doi: 10.1016/j.brainres.2023.148443
200. Singla R, Mishra A, Joshi R, Kumar R, Sarma P, Sharma AR, et al. Inhibition of the ERK1/2 phosphorylation by dextromethorphan protects against core autistic symptoms in VPA induced autistic rats: in silico and in vivo drug repurposition study. ACS Chem Neurosci. (2021) 12:1749–67. doi: 10.1021/acschemneuro.0c00672
201. Zohny SM, Habib MZ, Mohamad MI, Elayat WM, Elhossiny RM, El-Salam MFA, et al. Memantine/aripiprazole combination alleviates cognitive dysfunction in valproic acid rat model of autism: hippocampal CREB/BDNF signaling and glutamate homeostasis. Neurotherapeutics. (2023) 20:464–83. doi: 10.1007/s13311-023-01360-w
202. Taheri F, Esmaeilpour K, Sepehri G, Sheibani V, Ur Rehman N, Maneshian M. Histamine H3 receptor antagonist, ciproxifan, alleviates cognition and synaptic plasticity alterations in a valproic acid-induced animal model of autism. Psychopharmacology. (2022) 239:2673–93. doi: 10.1007/s00213-022-06155-z
203. Lima-Castañeda LÁ, Bringas ME, Aguilar-Hernandez L, Garcés-Ramírez L, Morales-Medina JC, Flores G. The antipsychotic olanzapine reduces memory deficits and neuronal abnormalities in a male rat model of Autism. J Chem Neuroanat. (2023) 132:102317. doi: 10.1016/j.jchemneu.2023.102317
204. Seo TB, Cho HS, Shin MS, Kim CJ, Ji ES, Baek SS. Treadmill exercise improves behavioral outcomes and spatial learning memory through up-regulation of reelin signaling pathway in autistic rats. J Exerc Rehabil. (2013) 9:220–9. doi: 10.12965/jer.130003
205. Zhang Y, Xiang Z, Jia Y, He X, Wang L, Cui W. The Notch signaling pathway inhibitor Dapt alleviates autism-like behavior, autophagy and dendritic spine density abnormalities in a valproic acid-induced animal model of autism. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 94:109644. doi: 10.1016/j.pnpbp.2019.109644
206. Hou Q, Wang Y, Li Y, Chen D, Yang F, Wang S. A developmental study of abnormal behaviors and altered GABAergic signaling in the VPA-treated rat model of autism. Front Behav Neurosci. (2018) 12:182. doi: 10.3389/fnbeh.2018.00182
207. Hara Y, Ago Y, Taruta A, Hasebe S, Kawase H, Tanabe W, et al. Risperidone and aripiprazole alleviate prenatal valproic acid-induced abnormalities in behaviors and dendritic spine density in mice. Psychopharmacology. (2017) 234:3217–28. doi: 10.1007/s00213-017-4703-9
208. Barzegari A, Amouzad Mahdirejei H, Hanani M, Esmaeili MH, Salari AA. Adolescent swimming exercise following maternal valproic acid treatment improves cognition and reduces stress-related symptoms in offspring mice: Role of sex and brain cytokines. Physiol Behav. (2023) 269:114264. doi: 10.1016/j.physbeh.2023.114264
209. Yamaguchi H, Hara Y, Ago Y, Takano E, Hasebe S, Nakazawa T, et al. Environmental enrichment attenuates behavioral abnormalities in valproic acid-exposed autism model mice. Behav Brain Res. (2017) 333:67–73. doi: 10.1016/j.bbr.2017.06.035
210. de Oliveira Ferreira E, Pessoa Gomes JM, Neves KRT, Lima FAV, de Barros Viana GS, de Andrade GM. Maternal treatment with aripiprazole prevents the development of a valproic acid-induced autism-like phenotype in juvenile male mice. Behav Pharmacol. (2023) 34:154–68. doi: 10.1097/FBP.0000000000000718
211. Wang J, Xu C, Liu C, Zhou Q, Chao G, Jin Y. Effects of different doses of lithium on the central nervous system in the rat valproic acid model of autism. Chem Biol Interact. (2023) 370:110314. doi: 10.1016/j.cbi.2022.110314
212. Gąssowska-Dobrowolska M, Cieślik M, Czapski GA, Jęśko H, Frontczak-Baniewicz M, Gewartowska M, et al. Prenatal exposure to valproic acid affects microglia and synaptic ultrastructure in a brain-region-specific manner in young-adult male rats: relevance to autism spectrum disorders. Int J Mol Sci. (2020) 21:3576. doi: 10.3390/ijms21103576
213. Luhach K, Kulkarni GT, Singh VP, Sharma B. Vinpocetine amended prenatal valproic acid induced features of ASD possibly by altering markers of neuronal function, inflammation, and oxidative stress. Autism Res. (2021) 14:2270–86. doi: 10.1002/aur.2597
214. Luhach K, Kulkarni GT, Singh VP, Sharma B. Cilostazol attenuated prenatal valproic acid-induced behavioural and biochemical deficits in a rat model of autism spectrum disorder. J Pharm Pharmacol. (2021) 73:1460–9. doi: 10.1093/jpp/rgab115
215. Luhach K, Kulkarni GT, Singh VP, Sharma B. Attenuation of neurobehavioural abnormalities by papaverine in prenatal valproic acid rat model of ASD. Eur J Pharmacol. (2021) 890:173663. doi: 10.1016/j.ejphar.2020.173663
216. Eissa N, Azimullah S, Jayaprakash P, Jayaraj RL, Reiner D, Ojha SK, et al. The dual-active histamine H3 receptor antagonist and acetylcholine esterase inhibitor E100 ameliorates stereotyped repetitive behavior and neuroinflammmation in sodium valproate induced autism in mice. Chem Biol Interact. (2019) 312:108775. doi: 10.1016/j.cbi.2019.108775
217. Gifford JJ, Deshpande P, Mehta P, Wagner GC, Kusnecov AW. The effect of valproic acid exposure throughout development on microglia number in the prefrontal cortex, hippocampus and cerebellum. Neuroscience. (2022) 481:166–77. doi: 10.1016/j.neuroscience.2021.11.012
218. Bronzuoli MR, Facchinetti R, Ingrassia D, Sarvadio M, Schiavi S, Steardo L, et al. Neuroglia in the autistic brain: evidence from a preclinical model. Mol Autism. (2018) 9:66. doi: 10.1186/s13229-018-0254-0
219. Habib MZ, Elnahas EM, Aboul-Ela YM, Ebeid MA, Tarek M, Sadek DR, et al. Risperidone impedes glutamate excitotoxicity in a valproic acid rat model of autism: Role of ADAR2 in AMPA GluA2 RNA editing. Eur J Pharmacol. (2023) 955:175916. doi: 10.1016/j.ejphar.2023.175916
220. Zhang J, Zhang JX, Zhang QL. PI3K/AKT/mTOR-mediated autophagy in the development of autism spectrum disorder. Brain Res Bull. (2016) 125:152–8. doi: 10.1016/j.brainresbull.2016.06.007
221. Zhang J, Liu LM, Ni JF. Rapamycin modulated brain-derived neurotrophic factor and B-cell lymphoma 2 to mitigate autism spectrum disorder in rats. Neuropsychiatr Dis Treat. (2017) 13:835–42. doi: 10.2147/NDT.S125088
222. Choi M, Ko SY, Seo JY, Kim DG, Lee H, Chung H, et al. Autistic-like social deficits in hippocampal MeCP2 knockdown rat models are rescued by ketamine. BMB Rep. (2022) 55:238–43. doi: 10.5483/BMBRep.2022.55.5.038
223. Yu X, Mostafijur Rahman M, Carter SA, Lin JC, Zhuang Z, Chow T, et al. Prenatal air pollution, maternal immune activation, and autism spectrum disorder. Environ Int. (2023) 179:108148. doi: 10.1016/j.envint.2023.108148
224. Ardalan M, Chumak T, Quist A, Hermans E, Hoseinpoor Rafati A, Gravina G, et al. Reelin cells and sex-dependent synaptopathology in autism following postnatal immune activation. Br J Pharmacol. (2022) 179:4400–22. doi: 10.1111/bph.15859
225. Fernández de Cossío L, Guzmán A, van der Veldt S, Luheshi GN. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav Immun. (2017) 63:88–98. doi: 10.1016/j.bbi.2016.09.028
226. Ardalan M, Chumak T, Quist A, Jabbari Shiadeh SM, Mallard AJ, Rafati AH, et al. Sex-dependent gliovascular interface abnormality in the hippocampus following postnatal immune activation in mice. Dev Neurosci. (2022) 44:320–30. doi: 10.1159/000525478
227. Solmaz V, Erdoğan MA, Alnak A, Meral A, Erbaş O. Erythropoietin shows gender dependent positive effects on social deficits, learning/memory impairments, neuronal loss and neuroinflammation in the lipopolysaccharide induced rat model of autism. Neuropeptides. (2020) 83:102073. doi: 10.1016/j.npep.2020.102073
228. Pang Y, Dai X, Roller A, Carter K, Paul I, Bhatt AJ, et al. Early postnatal lipopolysaccharide exposure leads to enhanced neurogenesis and impaired communicative functions in rats. PloS One. (2016) 11:e0164403. doi: 10.1371/journal.pone.0164403
229. Andoh M, Shibata K, Okamoto K, Onodera J, Morishita K, Miura Y, et al. Exercise reverses behavioral and synaptic abnormalities after maternal inflammation. Cell Rep. (2019) 27:2817–2825.e5. doi: 10.1016/j.celrep.2019.05.015
230. Giovanoli S, Weber-Stadlbauer U, Schedlowski M, Meyer U, Engler H. Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav Immun. (2016) 55:25–38. doi: 10.1016/j.bbi.2015.09.015
231. Smolders S, Smolders SM, Swinnen N, Gärtner A, Rigo JM, Legendre P, et al. Maternal immune activation evoked by polyinosinic:polycytidylic acid does not evoke microglial cell activation in the embryo. Front Cell Neurosci. (2015) 9:301. doi: 10.3389/fncel.2015.00301
232. Pineda E, Shin D, You SJ, Auvin S, Sankar R, Mazarati A. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann Neurol. (2013) 74:11–9. doi: 10.1002/ana.23898
233. Wolff AR, Bilkey DK. Prenatal immune activation alters hippocampal place cell firing characteristics in adult animals. Brain Behav Immun. (2015) 48:232–43. doi: 10.1016/j.bbi.2015.03.012
234. Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. (2008) 22:469–86. doi: 10.1016/j.bbi.2007.09.012
235. Haghani A, Johnson RG, Woodward NC, Feinberg JI, Lewis K, Ladd-Acosta C, et al. Adult mouse hippocampal transcriptome changes associated with long-term behavioral and metabolic effects of gestational air pollution toxicity. Transl Psychiatry. (2020) 10:218. doi: 10.1038/s41398-020-00907-1
236. Wang T, Zhang T, Sun L, Li W, Zhang C, Yu L, et al. Gestational B-vitamin supplementation alleviates PM2.5-induced autism-like behavior and hippocampal neurodevelopmental impairment in mice offspring. Ecotoxicol Environ Saf. (2019) 185:109686. doi: 10.1016/j.ecoenv.2019.109686
237. Klocke C, Allen JL, Sobolewski M, Mayer-Pröschel M, Blum JL, Lauterstein D, et al. Neuropathological consequences of gestational exposure to concentrated ambient fine and ultrafine particles in the mouse. Toxicol Sci. (2017) 156:492–508. doi: 10.1093/toxsci/kfx010
238. Nephew BC, Nemeth A, Hudda N, Beamer G, Mann P, Petitto J, et al. Traffic-related particulate matter affects behavior, inflammation, and neural integrity in a developmental rodent model. Environ Res. (2020) 183:109242. doi: 10.1016/j.envres.2020.109242
239. Meyza KZ, Blanchard DC. The BTBR mouse model of idiopathic autism - Current view on mechanisms. Neurosci Biobehav Rev. (2017) 76:99–110. doi: 10.1016/j.neubiorev.2016.12.037
240. Zhang Q, Wu H, Zou M, Li L, Li Q, Sun C, et al. Folic acid improves abnormal behavior via mitigation of oxidative stress, inflammation, and ferroptosis in the BTBR T+ tf/J mouse model of autism. J Nutr Biochem. (2019) 71:98–109. doi: 10.1016/j.jnutbio.2019.05.002
241. Wu H, Zhao G, Liu S, Zhang Q, Wang P, Cao Y, et al. Supplementation with selenium attenuates autism-like behaviors and improves oxidative stress, inflammation and related gene expression in an autism disease model. J Nutr Biochem. (2022) 107:109034. doi: 10.1016/j.jnutbio.2022.109034
242. Seese RR, Maske AR, Lynch G, Gall CM. Long-term memory deficits are associated with elevated synaptic ERK1/2 activation and reversed by mGluR5 antagonism in an animal model of autism. Neuropsychopharmacology. (2014) 39:1664–73. doi: 10.1038/npp.2014.13
243. Martin LA, Hsu FW, Herd B, Gregg M, Sample H, Kaplan J. Executive functions in agenesis of the corpus callosum: Working memory and sustained attention in the BTBR inbred mouse strain. Brain Behav. (2021) 11:e01933. doi: 10.1002/brb3.1933
244. Reshetnikov VV, Ayriyants KA, Ryabushkina YA, Sozonov NG, Bondar NP. Sex-specific behavioral and structural alterations caused by early-life stress in C57BL/6 and BTBR mice. Behav Brain Res. (2021) 414:113489. doi: 10.1016/j.bbr.2021.113489
245. Stephenson DT, O'Neill SM, Narayan S, Tiwari A, Arnold E, Samaroo HD, et al. Histopathologic characterization of the BTBR mouse model of autistic-like behavior reveals selective changes in neurodevelopmental proteins and adult hippocampal neurogenesis. Mol Autism. (2011) 2:7. doi: 10.1186/2040-2392-2-7
246. Guo YP, Commons KG. Serotonin neuron abnormalities in the BTBR mouse model of autism. Autism Res. (2017) 10:66–77. doi: 10.1002/aur.1665
247. Eissa N, Venkatachalam K, Jayaprakash P, Yuvaraju P, Falkenstein M, Stark H, et al. Experimental studies indicate that ST-2223, the antagonist of histamine H3 and dopamine D2/D3 receptors, restores social deficits and neurotransmission dysregulation in mouse model of autism. Pharmaceuticals. (2022) 15:929. doi: 10.3390/ph15080929
248. Eissa N, Awad MA, Thomas SD, Venkatachalam K, Jayaprakash P, Zhong S, et al. Simultaneous antagonism at H3R/D2R/D3R reduces autism-like self-grooming and aggressive behaviors by mitigating MAPK activation in mice. Int J Mol Sci. (2022) 24:526. doi: 10.3390/ijms24010526
249. Cellot G, Maggi L, Di Castro MA, Catalano M, Migliore R, Migliore M, et al. Premature changes in neuronal excitability account for hippocampal network impairment and autistic-like behavior in neonatal BTBR T+tf/J mice. Sci Rep. (2016) 6:31696. doi: 10.1038/srep31696
250. Zhang L, Xu X, Ma L, Wang X, Jin M, Li L, et al. Zinc water prevents autism-like behaviors in the BTBR mice. Biol Trace Elem Res. (2023) 201:4779–92. doi: 10.1007/s12011-022-03548-1
251. Reichova A, Bacova Z, Bukatova S, Kokavcova M, Meliskova V, Frimmel K, et al. Abnormal neuronal morphology and altered synaptic proteins are restored by oxytocin in autism-related SHANK3 deficient model. Mol Cell Endocrinol. (2020) 518:110924. doi: 10.1016/j.mce.2020.110924
252. Matsuo K, Shinoda Y, Abolhassani N, Nakabeppu Y, Fukunaga K. Transcriptome analysis in hippocampus of rats prenatally exposed to valproic acid and effects of intranasal treatment of oxytocin. Front Psychiatry. (2022) 13:859198. doi: 10.3389/fpsyt.2022.859198
253. Tian Y, Yabuki Y, Moriguchi S, Fukunaga K, Mao PJ, Hong LJ, et al. Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J Pineal Res. (2014) 56:1–11. doi: 10.1111/jpi.12081
254. Liu Z, Wang J, Xu Q, Wu Z, You L, Hong Q, et al. Vitamin A supplementation ameliorates prenatal valproic acid-induced autism-like behaviors in rats. Neurotoxicology. (2022) 91:155–65. doi: 10.1016/j.neuro.2022.05.008
255. Choi CH, Schoenfeld BP, Bell AJ, Hinchey J, Rosenfelt C, Gertner MJ, et al. Multiple drug treatments that increase cAMP signaling restore long-term memory and aberrant signaling in fragile X syndrome models. Front Behav Neurosci. (2016) 10:136. doi: 10.3389/fnbeh.2016.00136
256. Fuchs C, Gennaccaro L, Ren E, Galvani G, Trazzi S, Medici G, et al. Pharmacotherapy with sertraline rescues brain development and behavior in a mouse model of CDKL5 deficiency disorder. Neuropharmacology. (2020) 167:107746. doi: 10.1016/j.neuropharm.2019.107746
257. Gioia R, Seri T, Diamanti T, Fimmanò S, Vitale M, Ahlenius H, et al. Adult hippocampal neurogenesis and social behavioural deficits in the R451C Neuroligin3 mouse model of autism are reverted by the antidepressant fluoxetine. J Neurochem. (2023) 165:318–33. doi: 10.1111/jnc.15753
258. Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. (2009) 29:1773–83. doi: 10.1523/JNEUROSCI.5685-08.2009
259. Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. (2014) 343:675–9. doi: 10.1126/science.1247190
260. Kim JW, Seung H, Kim KC, Gonzales ELT, Oh HA, Yang SM, et al. Agmatine rescues autistic behaviors in the valproic acid-induced animal model of autism. Neuropharmacology. (2017) 113:71–81. doi: 10.1016/j.neuropharm.2016.09.014
261. Steinmetz AB, Stern SA, Kohtz AS, Descalzi G, Alberini CM. Insulin-like growth factor II targets the mTOR pathway to reverse autism-like phenotypes in mice. J Neurosci. (2018) 38:1015–29. doi: 10.1523/JNEUROSCI.2010-17.2017
262. Mottolese N, Uguagliati B, Tassinari M, Cerchier CB, Loi M, Candini G, et al. Voluntary running improves behavioral and structural abnormalities in a mouse model of CDKL5 deficiency disorder. Biomolecules. (2023) 13:1396. doi: 10.3390/biom13091396
263. Pinar C, Yau SY, Sharp Z, Shamei A, Fontaine CJ, Meconi AL, et al. Effects of voluntary exercise on cell proliferation and neurogenesis in the dentate gyrus of adult FMR1 knockout mice. Brain Plast. (2018) 4:185–95. doi: 10.3233/BPL-170052
264. Jamal I, Kumar V, Vatsa N, Singh BK, Shekhar S, Sharma A, et al. Environmental enrichment improves behavioral abnormalities in a mouse model of angelman syndrome. Mol Neurobiol. (2017) 54:5319–26. doi: 10.1007/s12035-016-0080-3
265. Kentner AC, Khoury A, Lima Queiroz E, MacRae M. Environmental enrichment rescues the effects of early life inflammation on markers of synaptic transmission and plasticity. Brain Behav Immun. (2016) 57:151–60. doi: 10.1016/j.bbi.2016.03.013
266. Haratizadeh S, Ranjbar M, Basiri M, Nozari M. Astrocyte responses to postnatal erythropoietin and nano-erythropoietin treatments in a valproic acid-induced animal model of autism. J Chem Neuroanat. (2023) 130:102257. doi: 10.1016/j.jchemneu.2023.102257
267. Blokland A, Heckman P, Vanmierlo T, Schreiber R, Paes D, Prickaerts J. Phosphodiesterase type 4 inhibition in CNS diseases. Trends Pharmacol Sci. (2019) 40:971–85. doi: 10.1016/j.tips.2019.10.006
268. Choi CH, Schoenfeld BP, Weisz ED, Bell AJ, Chambers DB, Hinchey J, et al. PDE-4 inhibition rescues aberrant synaptic plasticity in Drosophila and mouse models of fragile X syndrome. J Neurosci. (2015) 35:396–408. doi: 10.1523/JNEUROSCI.1356-12.2015
269. Balakrishnan S, Niebert M, Richter DW. Rescue of cyclic AMP mediated long term potentiation impairment in the hippocampus of mecp2 knockout. (Mecp2(-/y) ) mice by rolipram. Front Cell Neurosci. (2016) 10:15. doi: 10.3389/fncel.2016.00015
270. Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, et al. Advances in the treatment of fragile X syndrome. Pediatrics. (2009) 123:378–90. doi: 10.1542/peds.2008-0317
271. Arzuaga AL, Edmison DD, Mroczek J, Larson J, Ragozzino ME. Prenatal stress and fluoxetine exposure in mice differentially affect repetitive behaviors and synaptic plasticity in adult male and female offspring. Behav Brain Res. (2023) 436:114114. doi: 10.1016/j.bbr.2022.114114
272. Qiu W, Go KA, Wen Y, Duarte-Guterman P, Eid RS, Galea LAM. Maternal fluoxetine reduces hippocampal inflammation and neurogenesis in adult offspring with sex-specific effects of periadolescent oxytocin. Brain Behav Immun. (2021) 97:394–409. doi: 10.1016/j.bbi.2021.06.012
273. Winden KD, Ebrahimi-Fakhari D, Sahin M. Abnormal mTOR activation in autism. Annu Rev Neurosci. (2018) 41:1–23. doi: 10.1146/annurev-neuro-080317-061747
274. Dai Y, Zhang L, Yu J, Zhou X, He H, Ji Y, et al. Improved symptoms following bumetanide treatment in children aged 3-6 years with autism spectrum disorder: a randomized, double-blind, placebo-controlled trial. Sci Bull. (2021) 66:1591–8. doi: 10.1016/j.scib.2021.01.008
275. Cloarec R, Riffault B, Dufour A, Rabiei H, Gouty-Colomer LA, Dumon C, et al. Pyramidal neuron growth and increased hippocampal volume during labor and birth in autism. Sci Adv. (2019) 5:eaav0394. doi: 10.1126/sciadv.aav0394
276. Savardi A, Borgogno M, Narducci R, La Sala G, Ortega JA, Summa M, et al. Discovery of a small molecule drug candidate for selective NKCC1 inhibition in brain disorders. Chem. (2020) 6:2073–96. doi: 10.1016/j.chempr.2020.06.017
277. Doğan M, Albayrak Y, Erbaş O. Torasemide improves the propionic acid-induced autism in rats: A histopathological and imaging study. Alpha Psychiatry. (2023) 24:22–31. doi: 10.5152/alphapsychiatry.2023.22975
278. Purgert CA, Izumi Y, Jong YJ, Kumar V, Zorumski CF, O'Malley KL. Intracellular mGluR5 can mediate synaptic plasticity in the hippocampus. J Neurosci. (2014) 34:4589–98. doi: 10.1523/JNEUROSCI.3451-13.2014
279. Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. (1983) 302:832–7. doi: 10.1038/302832a0
280. Schaevitz LR, Moriuchi JM, Nag N, Mellot TJ, Berger-Sweeney J. Cognitive and social functions and growth factors in a mouse model of Rett syndrome. Physiol Behav. (2010) 100:255–63. doi: 10.1016/j.physbeh.2009.12.025
281. Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, et al. Sur M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA. (2009) 106:2029–34. doi: 10.1073/pnas.0812394106
282. Bozdagi O, Tavassoli T, Buxbaum JD. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol Autism. (2013) 4:9. doi: 10.1186/2040-2392-4-9
283. Pizzarelli R, Pimpinella D, Jacobs C, Tartacca A, Kullolli U, Monyer H, et al. Insulin-like growth factor 2 (IGF-2) rescues social deficits in NLG3-/y mouse model of ASDs. Front Cell Neurosci. (2024) 17:1332179. doi: 10.3389/fncel.2023.1332179
284. Cruz E, Descalzi G, Steinmetz A, Scharfman HE, Katzman A, Alberini CM. CIM6P/IGF-2 receptor ligands reverse deficits in angelman syndrome model mice. Autism Res. (2021) 14:29–45. doi: 10.1002/aur.2418
285. Ji C, Yang J, Lin L, Chen S. Executive function improvement for children with autism spectrum disorder: A comparative study between virtual training and physical exercise methods. Children. (2022) 9:507. doi: 10.3390/children9040507
286. Alipour V, Shabani R, Rahmani-Nia F, Vaseghi S, Nasehi M, Zarrindast MR. Effects of treadmill exercise on social behavior in rats exposed to thimerosal with respect to the hippocampal level of gluN1, gluN2A, and gluN2B. J Mol Neurosci. (2022) 72:1345–57. doi: 10.1007/s12031-022-02027-5
Keywords: autism spectrum disorder (ASD), hippocampus, learning, memory, cognitive map, anxiety, animal models
Citation: Long J, Li H, Liu Y, Liao X, Tang Z, Han K, Chen J and Zhang H (2024) Insights into the structure and function of the hippocampus: implications for the pathophysiology and treatment of autism spectrum disorder. Front. Psychiatry 15:1364858. doi: 10.3389/fpsyt.2024.1364858
Received: 03 January 2024; Accepted: 10 April 2024;
Published: 23 April 2024.
Edited by:
Roberto Canitano, Siena University Hospital, ItalyReviewed by:
Marilena Griguoli, National Research Council (CNR), ItalyCopyright © 2024 Long, Li, Liu, Liao, Tang, Han, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Zhang, Y3JyY3poMjAyMEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.