
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Psychiatry , 27 February 2024
Sec. Schizophrenia
Volume 14 - 2023 | https://doi.org/10.3389/fpsyt.2023.1334335
 Masanobu Kogure1
Masanobu Kogure1 Nobuhisa Kanahara2*Atsuhiro Miyazawa1,3
Nobuhisa Kanahara2*Atsuhiro Miyazawa1,3 Yuki Shiko4
Yuki Shiko4 Ikuo Otsuka5Koichi Matsuyama1,6Masayuki Takase7Makoto Kimura8,9
Ikuo Otsuka5Koichi Matsuyama1,6Masayuki Takase7Makoto Kimura8,9 Hiroshi Kimura1,10,11Kiyomitsu Ota3,12
Hiroshi Kimura1,10,11Kiyomitsu Ota3,12 Keita Idemoto1,3Masaki Tamura3,13
Keita Idemoto1,3Masaki Tamura3,13 Yasunori Oda1Taisuke Yoshida3
Yasunori Oda1Taisuke Yoshida3 Satoshi Okazaki5Fumiaki Yamasaki1Yusuke Nakata1Yoshinori Watanabe14
Satoshi Okazaki5Fumiaki Yamasaki1Yusuke Nakata1Yoshinori Watanabe14 Tomihisa Niitsu1
Tomihisa Niitsu1 Akitoyo Hishimoto5Masaomi Iyo1
Akitoyo Hishimoto5Masaomi Iyo1Background: Most genetic analyses that have attempted to identify a locus or loci that can distinguish patients with treatment-resistant schizophrenia (TRS) from those who respond to treatment (non-TRS) have failed. However, evidence from multiple studies suggests that patients with schizophrenia who respond well to antipsychotic medication have a higher dopamine (DA) state in brain synaptic clefts whereas patients with TRS do not show enhanced DA synthesis/release pathways.
Patients and methods: To examine the contribution (if any) of genetics to TRS, we conducted a genetic association analysis of DA-related genes in schizophrenia patients (TRS, n = 435; non-TRS, n = 539) and healthy controls (HC: n = 489).
Results: The distributions of the genotypes of rs3756450 and the 40-bp variable number tandem repeat on SLC6A3 differed between the TRS and non-TRS groups. Regarding rs3756450, the TRS group showed a significantly higher ratio of the A allele, whereas the non-TRS group predominantly had the G allele. The analysis of the combination of COMT and SLC6A3 yielded a significantly higher ratio of the putative low-DA type (i.e., high COMT activity + high SLC6A3 activity) in the TRS group compared to the two other groups. Patients with the low-DA type accounted for the minority of the non-TRS group and exhibited milder psychopathology.
Conclusion: The overall results suggest that (i) SLC6A3 could be involved in responsiveness to antipsychotic medication and (ii) genetic variants modulating brain DA levels may be related to the classification of TRS and non-TRS.
Approximately 30% of individuals with schizophrenia do not respond well to any standard antipsychotics, even at a sufficient dose or sufficient duration; these patients are defined as having treatment-resistant schizophrenia (TRS) (1, 2). Patients with TRS generally continue to represent severe psychiatric symptoms along with an unstable clinical course over the long term, leading to very poor outcomes. Several studies have suggested that TRS patients should be understood as those who do not respond well to antipsychotic medications, meeting the diagnostic criteria of TRS from the early treatment period (3–5). Numerous risk factors for TRS such as younger onset (6–9), poor premorbid social functioning (10–12), and autistic traits (13, 14) suggest that psychiatric abnormalities may have already begun prior to the onset of psychosis.
Magnetic resonance spectroscopy (MRS) studies have demonstrated some differences in GABAergic and glutamatergic networks between patients with TRS taking clozapine and those who are not taking clozapine (15–19). Clozapine, the only approved agent for TRS, is presumed to have effects on GABAergic and glutamatergic systems, which may be associated with its high efficacy (20, 21). These findings indicate that (i) patients with TRS may have a pathology that differs from that of non-TRS patients, and (ii) abnormalities in GABAergic and glutamatergic systems could be involved in the pathogenesis of TRS (5, 22, 23).
Alterations in the dopamine (DA) system have also been documented in patients with TRS. Several positron emission tomography (PET) studies reported that the DA synthesis capacity of TRS patients was not enhanced (i.e., it was comparable to that of healthy controls), in contrast to patients who responded to an antipsychotic with an enhanced synthesis and release of DA (24, 25). Other PET studies also suggested that patients with higher DA levels respond better to antipsychotic medication (26–28). An investigation by Amato et al. (29) using animal models indicated that an organically high DA state in the synaptic cleft is essential for the efficacy of antipsychotic drugs, and that the tyrosine hydroxylase (TH) enzyme and dopamine transporter (DAT) proteins could play important roles in the efficacy of antipsychotics.
Several genetic studies have focused on single nucleotide polymorphisms (SNPs) of candidate genes [e.g., dopamine D2 receptor gene (DRD2)] in patients with TRS (30–34), but nearly all of them failed to identify definite variant(s) relating to TRS pathology, partly due to the studies’ relatively small sample sizes. An increasing number of polygenic risk score (PRS) analyses of TRS with genome-wide association study (GWAS) data have been published, but their results have been inconsistent: some reports showed that TRS patients exhibited significantly higher PRS values compared to non-TRS patients (35–38) whereas others denied this finding (39, 40). These inconsistencies could potentially be explained by the diagnosis of TRS based on the use of clozapine as a surrogate marker and the varying rates of TRS patients in the discovery cohort used to create the PRS threshold (35, 36). Given the rich evidence of a relationship between the DA synthesis capacity and responses to antipsychotic medication, we speculated that compared to consortium genome samples, a study of TRS patients whose cases were collected at the same time as their clinical assessments (including accurate medication histories) would be more beneficial to detect variants on genes that are relevant to the response to antipsychotics.
In order to gain a deeper understanding of the mechanisms underlying TRS, we conducted the present study of patients with and without TRS to investigate molecules that modulate the amount of DA in the synaptic cleft (i.e., synthesis, degradation, and reuptake) from a genetics viewpoint. We also evaluated the status of gene variants that are known to be functionally relevant to the dopaminergic system and then compared the findings in the TRS patients with those in the non-TRS patients and a group of healthy controls.
We analyzed the cases of a total of 974 patients who were treated at one of several psychiatric hospitals in mainly Chiba prefecture, which is east of Tokyo. They were diagnosed as having schizophrenia or schizoaffective disorder by at least two experienced psychiatrists based on the Diagnostic and Statistical Manual of Disorders IV-TR or the 5th edition (DSM-5). Patients who met the criteria of substance abuse were excluded from the study. A total of 489 healthy controls also participated in the study. They were confirmed to have no history of psychiatric disorders of their own or among family members. All participants were Japanese.
At the blood sample collection, the subjects were provided a detailed explanation of the study and gave their written informed consent to participate in the study. For a patient who was judged to be compromised in understanding the study due to psychiatric symptoms, the consent was obtained from his/her family member. The study was approved by the ethics committees of Chiba University Graduate School of Medicine and the other psychiatric hospitals that participated in the study. The study was conducted in accord with the Declaration of Helsinki.
The diagnosis of TRS was made based on both the evaluation by the patient’s attending physician and the patient’s chart record. The diagnostic criteria for TRS followed the criteria of the Clozapine Patient Monitoring Service (CPMS), which defines patients with TRS as those who have not responded adequately [i.e., never reaching a Global Assessment of Functioning (GAF) score > 41 points in the 12 months prior to the blood sampling] to at least two different antipsychotic medications with a ≥ 600 mg chlorpromazine-equivalent (CP-eq.) dose and ≥ 4 weeks’ treatment with each medication. Patients who met the “intolerance to antipsychotic medication” criterion in the CPMS criteria were excluded from the study. The patients’ levels of adherence to their medications was also assessed as part of the determination of whether they met the criteria for TRS. Those who clearly did not meet the TRS criteria were judged as belonging to the non-TRS group in this study. The patients for whom sufficient information was unavailable were removed from the analysis.
As shown in Table 1, eight functional variants of genes relevant to DA synthesis, metabolism, and reuptake and DRD2 were selected for analysis, following previous genetic studies.

Table 1. Eight polymorphisms (SNP and VNTR) examined in the present study.
Genomes were extracted from each subject’s blood sample with the use of the QIAamp DNA Blood Mini Kit (250) (Qiagen, Valencia, CA, United States). The genotype of each SNP was determined by a TaqMan probe assay (Applied Biosystems, Foster City, CA, United States) using an ABI PRISM 7300 real-time polymerase chain reaction (PCR) system. This was done at 95°C for one 10-min cycle, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s.
For the amplification of the 40-bp variable number tandem repeat (VNTR) on Solute Carrier Family 6 Member 3 (dopamine transporter) gene (SLC6A3), the following primer sequences were used: upstream, 5′-TGTGGTGTAGGGAACGGCCTGAGA-3′; downstream, 5′-TGTTGGTCTGCAGGCTGCCTGCAT-3′ (60). The PCR procedure was followed by a one-cycle denaturation step at 95°C for 1 min, followed by 35 cycles of denaturation at 95°C for 15 s, annealing at 55°C for 15 s, extension at 72°C for 30 s, and finally an extension step at 72°C for 7 min. Then, 2 μL of PCR products were run on a 2% agarose gel, and the repeat allele sizes were quantified with a transilluminator: 7-repeat (330 bp), 8-repeat (370 bp), 9-repeat (410 bp), 10-repeat (450 bp), and 11-repeat (490 bp).
The demographic characteristics of the healthy controls (HC), schizophrenia (SCH) group, and patients with and without TRS were compared using Student’s t-test or an ANOVA for continuous variables, and the χ2 test for categorical variables. We used the same statistical methods to compare the allelic and genotype distributions of the eight studied SNPs between the schizophrenia subgroups and the HC group. As post hoc tests, Bonferroni correction was used for the ANOVA and a residual analysis was used for the χ2 test. To explore the potential impact of each SNP on the classification of TRS or non-TRS, we conducted a multivariate logistic analysis in which we used the group (TRS, non-TRS, or HC) as the response variable and the genotype of each SNP as the explanatory variables with patient’s age and sex as covariates.
The statistical significance level was set at p < 0.05, with the exception of the comparisons of SNP distribution. For the analysis of SNP distributions, a Bonferroni correction for multiple comparisons was applied, and the statistical significance level was thus set at p = 0.00625 (=0.05/8). All statistical analyses were performed using SPSS ver. 22.0 software (IBM, NY).
The SCH group had a significantly higher proportion of females (49.8%) compared to the HC group (44.1%) (Table 2). The patients’ ages at the time of blood sampling were significantly higher in the SCH group compared to the HC group. There was also a significant difference in age among the three groups: the TRS group was the oldest, followed by the non-TRS group, with the youngest group being the HC group.
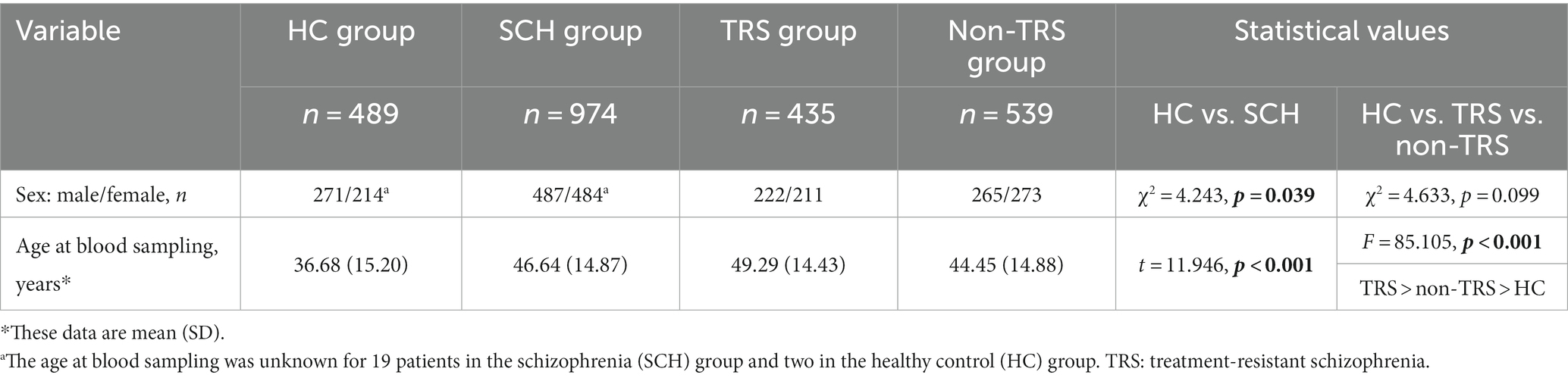
Table 2. Demographic characteristics of the healthy controls, schizophrenia group, and the patients with and without treatment-resistant schizophrenia.
The univariate analysis detected no polymorphisms that showed deviant results from the Hardy–Weinberg equilibrium in the HC or SCH groups.
None of the seven polymorphisms other than the 40-bp VNTR showed significant differences in genotype or allelic distribution between the HC and SCH groups (Supplementary Table S1). Regarding the 40-bp VNTR, there was a significantly higher ratio of subjects with the 10X genotype in the SCH group compared to the HC group.
When additional analyses separating male and female subjects were conducted, there were no significant differences between the HC and SCH groups for any of the eight variants (data not shown).
There was a significant difference in rs3756450 in both the genotype and allelic distributions; the residual analysis showed a higher ratio of A alleles in the TRS group and a higher ratio of G alleles in the non-TRS group (Table 3). These results are the same as those obtained in the analyses that were conducted separately for the male and female subjects (the residual analysis showed a higher ratio of G allele in the non-TRS group, with p values of 0.00338 in the males-only analysis and 0.0418 in the females-only analysis), although there was no significant difference in the genotype-based analyses.
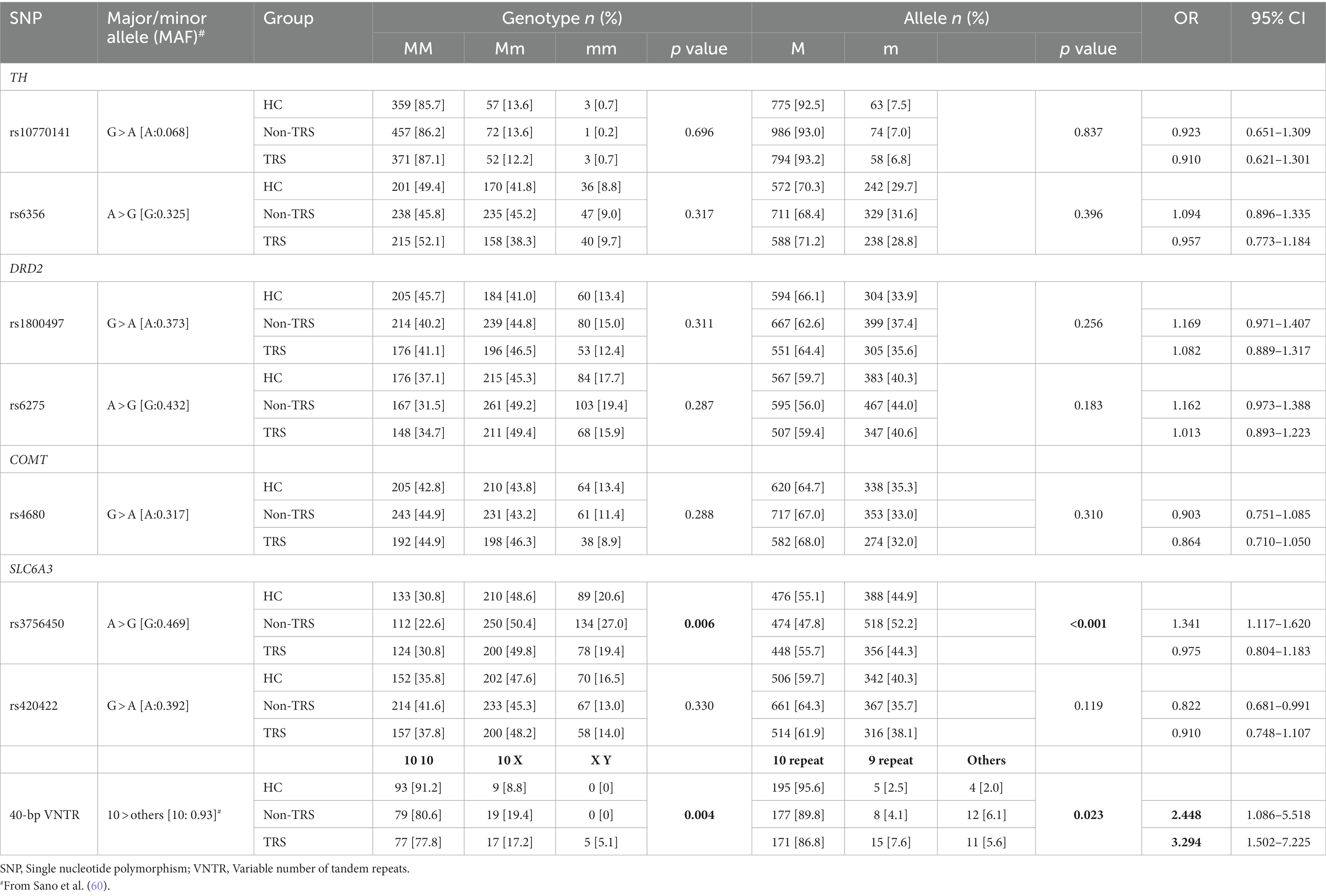
Table 3. Comparisons of the allelic and genotype distributions of the eight studied SNPs between the schizophrenia subgroups and the healthy control group.
The results for the 40-bp VNTR also showed a significant difference, with a higher ratio of XY in the TRS group and a higher ratio of homozygotes with 10 10 in the HC group (Table 3). When 10-repeat, 9-repeat, or other repeat allele sizes were examined separately, we observed that the distribution differed significantly among the three groups (Supplementary Table S2). This was also true for the analysis of the females-only group, while there was no significant difference in the analysis for the male subjects. There were no significant differences in the genotype or allelic distribution for other polymorphisms in the entire series of subjects or in the separate analyses of males and females.
By applying the seven SNPs or the seven SNPs plus the 40-bp VNTR to multivariate logistic models, we searched for significant variants affecting the classification of the TRS and non-TRS patients.
The first model including the seven SNPs revealed that the GG genotype of rs1800497 was significantly less related to TRS, with an odds ratio (OR) of 0.640 (p = 0.045) using the HC group as the reference (Table 4). When the TRS group was compared with the non-TRS (reference) group, the GG genotype of rs3756450 showed protection against TRS (OR = 0.685, p = 0.044), whereas the AA genotype was related to a risk of TRS (OR 1.430, p = 0.043).
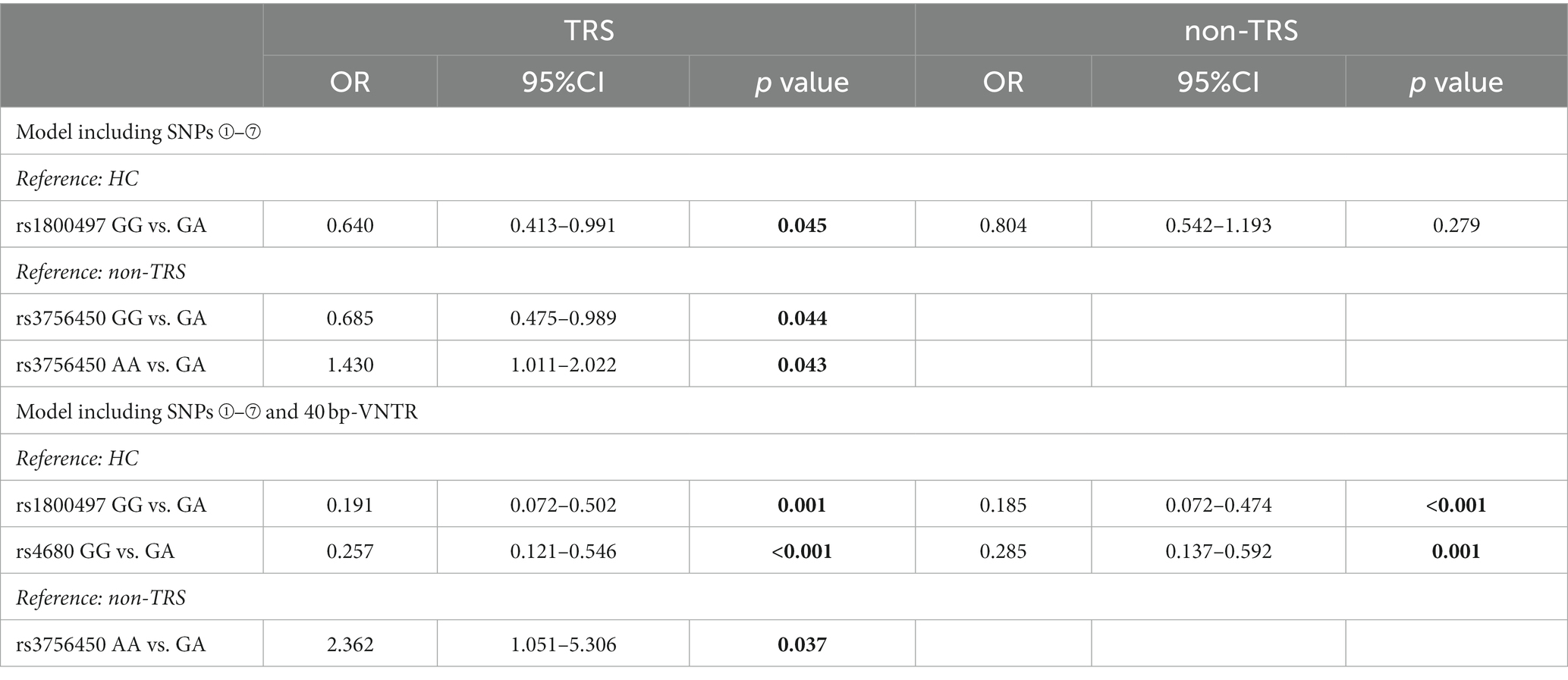
Table 4. Multivariate logistic regression analysis discriminating the TRS and non-TRS groups.
The second model including the seven SNPs plus the 40-bp VNTR indicated that the GG genotype of rs1800497 and the GG(ValVal)genotype of rs4680 carried lower risks for TRS and non-TRS relative to the HC (reference) group, suggesting that there can be protective genotypes against schizophrenia per se (Table 4). When the non-TRS group was the reference group, the AA genotype of rs3756450 was significantly related to TRS, with an OR of 2.362 (p = 0.037).
We next examined the effect of the combination of rs4680 on Catechol-O-methyltransferase gene (COMT) and rs3756450 (SLC6A3), which we speculated may alter the activity of the two proteins and thus modulate the amount of DA in the synaptic cleft; our hypothesis was that patients with the combination of variants exhibiting a low-DA state would show a poorer response to antipsychotic medications. In this analysis, we defined patients with the combination of the GG (ValVal) of rs4680 and the AA of rs3756450 as those with a “low-DA combination” (in place of the actual measurement of DA levels) and the patients with other combinations as “others.”
The results revealed that the distribution of this genotype combination differed between the TRS and non-TRS groups: the TRS group more frequently had a low-DA combination and less frequently had other combinations (χ2 = 5.304, p = 0.021). These results were the same for the females-only analysis, whereas the male-only analysis showed no significant difference. Additional analyses of the TRS vs. non-TRS vs. HC groups supported these results, revealing that the low-DA combination was significantly less common in the non-TRS group (χ2 = 6.000, p = 0.0498) compared to the TRS group, whereas the distribution in the TRS and HC groups did not differ significantly (Table 5).
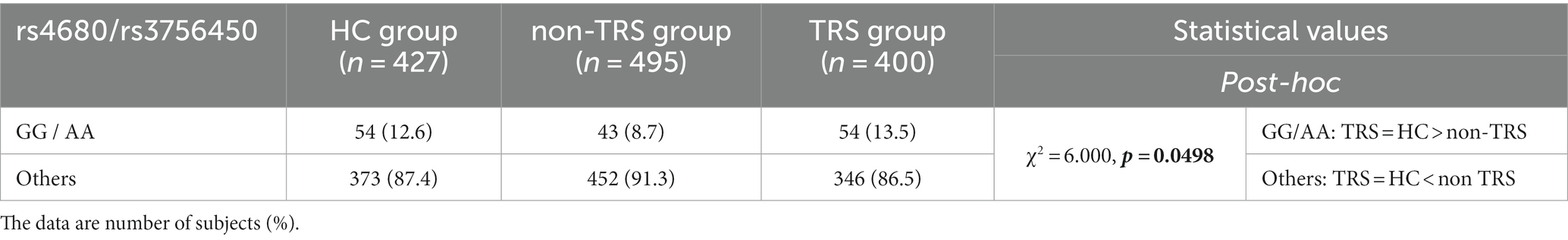
Table 5. Genotypic analysis of the COMT gene rs4680 combined with the SLC6A3 gene rs3756450.
We also examined the potential effects of the combination of rs4680 and rs3756450, rs4680 alone, rs3756450 alone, and the 40-bp VNTR on clinical indicators. We observed that in the TRS group, there were no significant effects of the combination of the two SNPs, rs4680, rs3756450, or 40-bp VNTR on any clinical measurements.
In the non-TRS group, the analyses of the two-SNP combination and rs3756450 showed significant differences in the subjects’ Clinical Global Impression-Severity (CGI-S) scale score, employment, and antipsychotic dosage: the patients with the low-DA combination among the combined two-SNP or G allele carriers of rs3756450 exhibited more severe psychopathology in terms of the CGI-S and employment and were being treated with higher doses of antipsychotics compared to the patients with other combinations and compared to the patients without the G allele (Table 6; Supplementary Table S3).
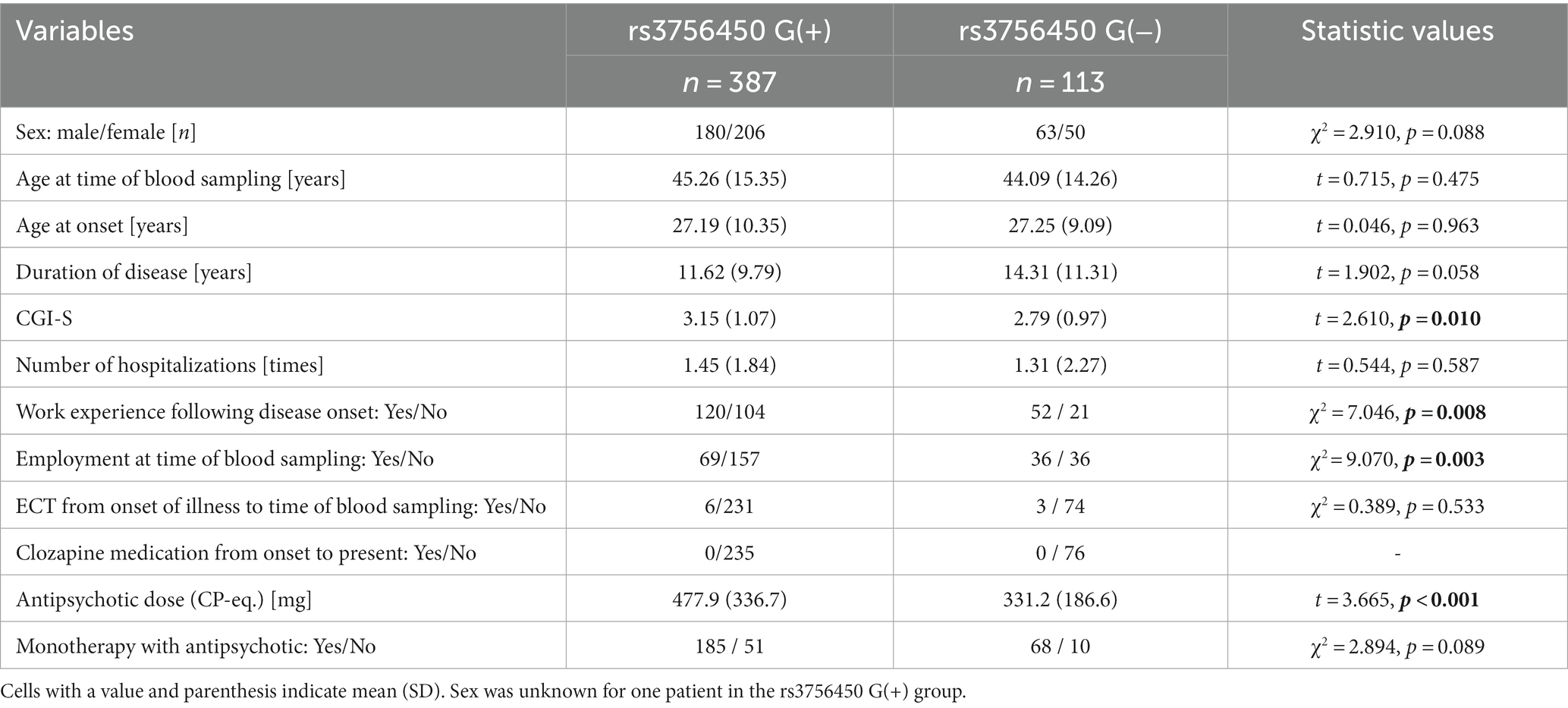
Table 6. Comparison of clinically related measures for rs3756450 G(+) carriers and G(−) carriers in the non-TRS groups.
Regarding both rs4680 and the 40-bp VNTR, there were no significant differences between polymorphisms or clinical measurements among the SCH, TRS, and non-TRS groups.
Our findings demonstrated that the distributions of the genotypes of rs3756450 and the 40-bp VNTR of SLC6A3 differed between the TRS and non-TRS groups in the following three ways: (i) regarding rs3756450, the A allele was significantly less abundant in the non-TRS patients and significantly more abundant in the TRS patients (Tables 3, 4). Concerning the 40-bp VNTR, the TRS group had significantly fewer carriers of 10-repeat homozygotes compared to the non-TRS and HC groups (Table 3; Supplementary Table S2). (ii) The analysis of the combination of COMT and SLC6A3 revealed the TRS group’s significantly higher ratio of patients with the low-DA type (i.e., high COMT activity + high DAT activity) compared to both the HC and non-TRS groups (Table 5). (iii) In the non-TRS group, patients with the low-DA type were a minority and exhibited milder psychopathology compared to those with other genotype combinations (Supplementary Table S3; Table 6).
Several neuroimaging studies have indicated that the capacity to synthesize DA is not enhanced in patients with TRS and that a lower level of DA in the synaptic cleft may contribute to their nonresponsiveness to antipsychotics (24, 25). Our present results are the first to provide evidence that SLC6A3 may be involved in the responsiveness to antipsychotic medication or the classification of TRS/non-TRS and that the specific genotype leading to the low-DA state could be related to patients with TRS.
In vitro studies have revealed that several variants spanning from the 5′-UTR to exon 1 on SLC6A3 affect the expression of DAT in a combined manner (61–63). These variants are in a relatively high-linkage disequilibrium (LD) relationship (64), and their alteration allele uniformly leads to lower expression relative to their respective reference allele (61, 64, 65). The same difference in expression was true for rs3756450: the alteration allele (A) showed lower expression (55, 65), except in a single study (62). The binding site with transcription factors is defined uniquely to the specific allele in the 5′-UTR on SLC6A3, which might be a reason for the differential expression by alleles (65).
Regarding the association with schizophrenia, two studies have demonstrated that rs3756450 was not the top-hit polymorphism of SLC6A3; however, it showed a highly significant signal in an association analysis between patients with schizophrenia and healthy subjects (55, 64). This finding was supported by a meta-analysis of the association between rs3756450 and schizophrenia (66). In the present study, this SNP was not significantly related to the patients with schizophrenia as a whole (Supplementary Table S1). However, when we divided the patients into TRS and non-TRS groups, a significant difference was observed: the non-TRS group had a significantly higher ratio of the G allele of rs3756450 (Table 3). In contrast, the TRS group did not differ from the HC group in the genotype or allelic distributions of rs3756450, but the TRS group had a significantly higher ratio of AA genotype carriers than the non-TRS group. In fact, the AA genotype was identified as the only risk factor in the multivariate logistic analysis including all other polymorphisms as independent variables (Table 4). These results suggest that in addition to the potential involvement of this SNP is might not only be involved in the vulnerability to schizophrenia (i.e., significant relationship with the non-TRS group), it may also be involved in the development of TRS following the onset of the disease.
Our present results showed a significantly higher ratio of the 9-repeat of the 40-bp VNTR in the TRS group compared to the non-TRS and HC groups (Supplementary Table S2). The 40-bp VNTR polymorphism in SLC6A3 can affect its transcription (57, 67). Extensive in vivo and in vitro studies have obtained highly controversial findings regarding the effect of the 40-bp VNTR polymorphism in SLC6A3 on DAT expression: higher expression in 10-repeat relative to 9-repeat (68–72); higher expression in 9-repeat compared to 10-repeat (73, 74); and no difference between them (75–78). This uncertain situation remains as of this writing, and there is some speculation that these variants are unlikely to be involved in schizophrenia (58, 59).
As our results concerning the 40-bp VNTR came from a small sample size, no conclusion can be drawn. However, the significant difference in the distribution of the 40-bp VNTR between schizophrenia subtypes provides an important insight into its relation to the responsiveness to antipsychotic medication among patients with schizophrenia, although two studies have denied the association of the 40-bp VNTR with responses to antipsychotics (79, 80). The regulation of the expression of DAT has not yet been clarified, and several regions such as the 5′-UTR (including rs3756450), the 3′-UTR (including the 40-bp VNTR), and other variants are thought to work together to determine expression levels (81). A complete understanding of the regulation of DAT expression is strongly desired.
In our additional analysis of the combined effects with the Val/Met polymorphism (rs4680) of COMT and rs3756450 of SLC6A3, which are suspected to more strongly affect the amounts of DA in the synaptic cleft, we observed that the TRS group’s frequency of this combination did not differ significantly from that of the HC group, but the ratio of “low-DA types” (the combination of GG of rs4680 and AA of rs3756450) was significantly lower in the non-TRS group (Table 5). This difference was even more evident in the two-group (TRS and non-TRS groups) comparison: the TRS group had a higher ratio of low-DA types than the non-TRS group.
Catechol-O-methyltransferase gene has been shown to be related to multiple symptom domains as well as cognitive impairment in schizophrenia patients (82, 83). Most of the relevant studies have consistently shown that patients with the Val allele (leading to low DA) have severe psychopathology or poor cognitive function (84–87), although a meta-analysis did not support a relationship with working memory (88). In many areas of the human brain such as the striatum, thalamus, and hippocampus, both COMT and DAT provide DA clearance (89, 90), and it has been indicated that DAT could be involved more closely than COMT in the synaptic clefts (91–93). In the prefrontal cortex where DAT is less abundant, DA is broken down by COMT and other transporters such as norepinephrine transporter, and DAT plays a role in the volume transmission of DA away from the synaptic clefts (94, 95), suggesting that the effect of a specific genotype on brain function might differ depending on the brain region.
Several fMRI studies used the N-back task performed by healthy subjects, and with the classification of their genotypes the authors reported that the interacting effect of COMT (rs4680) and SLC6A3 (40-bp VNTR) on brain activation was linear in the dorsolateral prefrontal cortex (96–98), whereas the effect was nonlinear (i.e., an inverted-U-shape curve) in the hippocampus (97). Notably, the lowest-DA genetic combination (defined as the Val allele of COMT and the 10-repeat of SLC6A3) did not exhibit the lowest DA signal or the highest blood oxygen level-dependent (BOLD) signal, indicating the veracity of the inverted-U-shape. It has been speculated that DA in the prefrontal cortex is modulated in a multi-layered way at both the synaptic level (i.e., by COMT) and the network level in the cortical-striatal-thalamus-cortex pathway (i.e., by both COMT and DAT) (99), leading to differential relationships between molecule functions and brain activation.
Our results after the selection of the 5′-UTR on SLC6A3 instead of the 40-bp VNTR in the 3′-UTR, demonstrated that the 5′-UTR (rs3475860) alone and the combination of 5′-UTR on SLC6A3 and COMT could be significantly related to TRS (Table 5). Our findings might support a linear interacting effect of COMT and SLC6A3 on the responsiveness to antipsychotic(s) in the striatum, in contrast to the nonlinear interaction (inverted U-shape curve) in the prefrontal cortex under cognitive tasks shown in fMRI studies.
Although several PET studies have reported that DA synthesis is not enhanced in individuals with TRS (24, 25), our present analyses showed that the two SNPs on the TH gene were not significantly associated with assignment to the TRS group, or the non-TRS group, or to schizophrenia patients as a whole. There are several potential reasons for this discrepancy between our result and those of the PET studies. Since no SNP with a significant impact on the expression of TH has been found to date, it is possible that a polymorphism other than the two SNPs we selected (rs10770141 and rs6356) may have a greater impact on the enzyme’s function (100, 101). Significant associations of rs10840491, rs10840489, rs11042978, and rs11564717 on TH gene in individuals with schizophrenia were reported in association studies of schizophrenia (102, 103), but other reports denied these associations. The synthesis of DA by TH may be influenced by the methylation of TH or another upstream region of this molecule (104). In addition, the degree of DA synthesis in patients with TRS observed in the PET studies (i.e., a lack of enhanced DA synthesis) is similar to that in healthy subjects, and in this sense, this finding is not inconsistent with our present results.
Our analyses considering the subjects’ clinically related measures revealed that among the non-TRS patients, the patients with a low-DA type showed only slightly but nevertheless significantly lower values than the patients with other types in terms of psychopathology and antipsychotic doses, in both the single analysis of rs3756450 and the analysis combining rs4680 and rs3756450 (Supplementary Table S3; Table 6). These results in the non-TRS group were in contrast to the TRS-group findings showing that the low-DA type(s) could be risk genotypes for TRS with severe psychopathology (Table 5). However, the results concerning the non-TRS group were not contradictory since patients in the non-TRS group respond to antipsychotic medication by definition: the schizophrenia of the patients with the low-DA types in particular would be controlled with lower doses of antipsychotics, and these patients would thus experience better social lives, including employment.
This study has several strengths over other similar association studies of TRS patients. The sample size was relatively large. In addition, the majority of the prior similar studies reported that the diagnosis of TRS was based on a threshold antipsychotic dosage (e.g., >400 or > 600 mg), whereas we conducted a thorough review of the patients’ medical records to determine the diagnosis of TRS. The results of that review also reflect the patients’ clinical courses to the greatest extent possible, and the validity of the TRS diagnosis is high, which is another study strength.
However, there are several study limitations to address. The first is that although the sample size is relatively large, it was still small enough to have insufficient statistical power. A second limitation is that the patients’ responses to antipsychotic medication were not evaluated with standard assessment tools such as the Positive and Negative Syndrome Scale (PANSS) or the Brief Psychiatric Rating Scale (BPRS). Patients with TRS are heterogeneous in their response to antipsychotic(s) and other non-genetic factors such as adherence to medication and the duration of untreated psychosis (4, 5). Several research groups recently proposed that TRS could be classified into an early subtype and a late subtype (and others), and the late subtype of TRS is speculated to be related to the development of dopamine supersensitivity psychosis (DSP) (5). Patients with DSP respond well to initial antipsychotics for their first episode of psychosis by definition, and thus the dopaminergic function of this subtype could be similar to that of non-TRS patients, but not to that of an early subtype of TRS. However, we could not collect data relevant to DSP for all of the present patients, and we did not performed such an analysis of DSP in the present study. To more precisely examine the relationships between genetic-based phenotypes and responsiveness to medication even among only patients with TRS, more objective and thorough assessments are necessary.
In addition, our analysis strategy might be slightly arbitrary; we did not measure DA in the patients’ brains, and relationships between each variant and their presumed DA level are only hypothetical, particularly in the analysis of the COMT and DAT combination, which did not reflect the actual DA state in brains. Lastly, the targeted genes and polymorphisms were greatly restricted. Other genes (e.g., MAO-B and VMAT2) or functional/non-coding variants could have influenced our findings. Resequencing analyses for the relevant genes are required to obtain more comprehensive results.
In conclusion, the results of our analyses revealed significant difference in allelic and genotypic distributions of rs3756450 between schizophrenia patients with and without a treatment-resistant phenotype. These results partially support those of several PET studies showing non-enhancement of the DA synthesis pathway in patients with TRS. Our analysis of the COMT rs4680 and SLC6A3 rs3756450 combination suggests that a presumptive low-DA state is more frequently present in the TRS patients compared to non-TRS patients. These findings suggest that DA modulation in the synaptic cleft by DAT could affect patients’ responses to antipsychotics medication, ultimately leading to the development of TRS.
The datasets presented in this study can be found in online repositories. ClinVar (the accession number 509431) can be found here: https://www.ncbi.nlm.nih.gov/clinvar/submitters/509431.
The studies involving humans were approved by the Biomedical Research Ethics Committee of the Graduate School of Medicine of Chiba University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants or thier legal guardians.
MKo: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. NK: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AM: Investigation, Resources, Writing – review & editing. YS: Formal Analysis, Methodology, Software, Writing – review & editing. IO: Investigation, Resources, Writing – review & editing. KM: Investigation, Resources, Writing – review & editing. MTak: Investigation, Resources, Writing – review & editing. MKi: Investigation, Resources, Writing – review & editing. HK: Investigation, Resources, Writing – review & editing. KO: Investigation, Resources, Writing – review & editing. KI: Investigation, Resources, Writing – review & editing. MTam: Investigation, Resources, Writing – review & editing. YO: Investigation, Resources, Writing – review & editing. TY: Investigation, Resources, Writing – review & editing. SO: Investigation, Resources, Writing – review & editing. FY: Investigation, Resources, Writing – review & editing. YN: Investigation, Resources, Writing – review & editing. YW: Investigation, Resources, Writing – review & editing. TN: Investigation, Resources, Writing – review & editing. AH: Conceptualization, Investigation, Resources, Writing – review & editing. MI: Conceptualization, Investigation, Resources, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a Grant-in-Aid for Scientific Research (21K07476, NK) from the Japan Society for the Promotion of Science (JSPS), and also supported by a Grand (MKo) from the Osaka Medical Foundation for Intractable Diseases grant no. (28-2-10).
The authors thank Mayumi Hiruma (Himorogi Psychiatric Institute, Tokyo, Japan), Kaoru Ikeda, Chisako Fujishiro, Reiko Suzuki, Akiko Yamamoto, and Komako Ito (Department of Psychiatry, Chiba University Hospital) for collecting samples and relevant information.
NK reports honoraria from Otsuka Pharmaceutical Co., Ltd., Sumitomo Pharma Co., Ltd., Janssen Pharmaceutical K.K., Meiji Seika Pharma Co., Ltd., Mitsubishi Tanabe Pharma Co., Ltd., Yoshitomi Pharmaceutical Industries, Ltd., and Eisai Co. Ltd. HK reports honoraria from Otsuka Pharmaceutical Co., Ltd., Meiji Seika Pharma Co., Ltd., MSD K.K., and Teijin Ltd. YN reports honoraria from Otsuka Pharmaceutical Co., Ltd., Meiji Seika Pharma Co., Ltd., Sumitomo Pharma Co., Ltd., and Takeda Pharmaceutical Co., Ltd. MI received consultant fees from Eli Lilly Japan K.K., Sumitomo Pharma Co., Ltd., Pfizer Japan Inc., Abbott Japan Co., Ltd., and Janssen Pharmaceutical K.K., and reports honoraria from Janssen Pharmaceutical K.K., Eli Lilly Japan K.K., Otsuka Pharmaceutical Co., Ltd., Meiji Seika Pharma Co., Ltd., Astellas Pharma Inc., Sumitomo Pharma Co., Ltd., Ono Pharmaceutical Co., Ltd., GlaxoSmithKline K.K., Takeda Pharmaceutical Co., Ltd., Mochida Pharmaceutical Co., Ltd., Kyowa Kirin Co., Ltd., MSD K.K., Eisai Co. Ltd., Daiichi-Sankyo Co. Ltd., Novartis Pharma K.K., Teijin Ltd., Shionogi & Co., Ltd., Hisamitsu Pharmaceutical Co., Inc., and Asahi Kasei Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2023.1334335/full#supplementary-material
1. Kane, J, Honigfeld, G, Singer, J, and Meltzer, H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. (1988) 45:789–96. doi: 10.1001/archpsyc.1988.01800330013001
2. Elkis, H. Treatment-Resistant Schizophrenia. Psychiatrr Clin North Am. (2007) 30:511–33. doi: 10.1016/j.psc.2007.04.001
3. Lally, J, Ajnakina, O, di Forti, M, Trotta, A, Demjaha, A, Kolliakou, A, et al. Two distinct patterns of treatment resistance: clinical predictors of treatment resistance in first-episode schizophrenia spectrum psychoses. Psychol Med. (2016) 46:3231–40. doi: 10.1017/S0033291716002014
4. Demjaha, A, Lappin, JM, Stahl, D, Patel, MX, MacCabe, JH, Howes, OD, et al. Antipsychotic treatment resistance in first-episode psychosis: prevalence, subtypes and predictors. Psychol Med. (2017) 47:1981–9. doi: 10.1017/S0033291717000435
5. Kane, JM, Agid, O, Baldwin, ML, Howes, O, Lindenmayer, JP, Marder, S, et al. Clinical guidance on the identification and management of treatment-resistant schizophrenia. J Clin Psychiatry. (2019) 80:18com12123. doi: 10.4088/JCP.18com12123
6. Meltzer, HY, Rabinowitz, J, Lee, MA, Cola, PA, Ranjan, R, Findling, RL, et al. Age at onset and gender of schizophrenic patients in relation to neuroleptic resistance. Am J Psychiatry. (1997) 154:475–82. doi: 10.1176/ajp.154.4.475
7. Hollis, C. Adult outcomes of child- and adolescent-onset schizophrenia: diagnostic stability and predictive validity. Am J Psychiatry. (2000) 157:1652–9. doi: 10.1176/appi.ajp.157.10.1652
8. Reichert, A, Kreiker, S, Mehler-Wex, C, and Warnke, A. The psychopathological and psychosocial outcome of early-onset schizophrenia: preliminary data of a 13-year follow-up. Child Adolesc Psychiatry Ment Health. (2008) 2:6. doi: 10.1186/1753-2000-2-6
9. Legge, SE, Dennison, CA, Pardiñas, AF, Rees, E, Lynham, AJ, Hopkins, L, et al. Clinical indicators of treatment-resistant psychosis. Br J Psychiatry. (2020) 216:259–66. doi: 10.1192/bjp.2019.120
10. WIERSMA, D, WANDERLING, J, DRAGOMIRECKA, E, GANEV, K, HARRISON, G, AN der HEIDEN, W, et al. Social disability in schizophrenia: its development and prediction over 15 years in incidence cohorts in six European centres. Psychol Med. (2000) 30:1155–67. doi: 10.1017/S0033291799002627
11. Schennach, R, Obermeier, M, Meyer, S, Jäger, M, Schmauss, M, Laux, G, et al. Predictors of relapse in the year after hospital discharge among patients with schizophrenia. Psychiatr Serv. (2012) 63:87–90. doi: 10.1176/appi.ps.201100084
12. Legge, SE, Cardno, AG, Allardyce, J, Dennison, C, Hubbard, L, Pardiñas, AF, et al. Associations between schizophrenia polygenic liability, symptom dimensions, and cognitive ability in schizophrenia. JAMA Psychiatry. (2021) 78:1143–51. doi: 10.1001/jamapsychiatry.2021.1961
13. Asarnow, JR, Tompson, MC, and McGrath, EP. Annotation: childhood-onset schizophrenia: clinical and treatment issues. Psychol Psychiatry. (2004) 45:180–94. doi: 10.1111/j.1469-7610.2004.00213.x
14. Nakata, Y, Kanahara, N, Kimura, A, Niitsu, T, Komatsu, H, Oda, Y, et al. Autistic traits and cognitive profiles of treatment-resistant schizophrenia. Schizophr Res Cogn. (2020) 22:100186. doi: 10.1016/j.scog.2020.100186
15. Bustillo, JR, Rowland, LM, Mullins, P, Jung, R, Chen, H, Qualls, C, et al. 1H-MRS at 4 tesla in minimally treated early schizophrenia. Mol Psychiatry. (2010) 15:629–36. doi: 10.1038/mp.2009.121
16. Marsman, A, van den Heuvel, MP, Klomp, DW, Kahn, RS, Luijten, PR, and Hulshoff Pol, HE. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophr Bull. (2013) 39:120–9. doi: 10.1093/schbul/sbr069
17. Goldstein, ME, Anderson, VM, Pillai, A, Kyd, RR, and Russell, BR. Glutamatergic 17.Neurometabolites in clozapine-responsive and -resistant schizophrenia. J Neuro-Oncol. (2015) 18:pyu117. doi: 10.1093/ijnp/pyu117
18. Merritt, K, Egerton, A, Kempton, MJ, Taylor, MJ, and McGuire, PK. Nature of glutamate alterations in schizophrenia: a Meta-analysis of proton magnetic resonance spectroscopy studies. JAMA Psychiatry. (2016) 73:665–74. doi: 10.1001/jamapsychiatry.2016.0442
19. Iwata, Y, Nakajima, S, Plitman, E, Caravaggio, F, Kim, J, Shah, P, et al. Glutamatergic neurometabolite levels in patients with ultra-treatment-resistant schizophrenia: a cross-sectional 3T proton magnetic resonance spectroscopy study. Biol Psychiatry. (2019) 85:596–605. doi: 10.1016/j.biopsych.2018.09.009
20. O’Connor, WT, and O’Shea, SD. Clozapine and GABA transmission in schizophrenia disease models: establishing principles to guide treatments. Pharmacol Ther. (2015) 150:47–80. doi: 10.1016/j.pharmthera.2015.01.005
21. de Bartolomeis, A, Vellucci, L, Barone, A, Manchia, M, de Luca, V, Iasevoli, F, et al. Clozapine’s multiple cellular mechanisms: what do we know after more than fifty years? A systematic review and critical assessment of translational mechanisms relevant for innovative strategies in treatment-resistant schizophrenia. Pharmacol Ther. (2022) 236:108236. doi: 10.1016/j.pharmthera.2022.108236
22. Mouchlianitis, E, Bloomfield, MA, Law, V, Beck, K, Selvaraj, S, Rasquinha, N, et al. Treatment-resistant schizophrenia patients show elevated anterior cingulate cortex glutamate compared to treatment-responsive. Schizophr Bull. (2016) 42:744–52. doi: 10.1093/schbul/sbv151
23. Potkin, SG, Kane, JM, Correll, CU, Lindenmayer, JP, Agid, O, Marder, SR, et al. The neurobiology of treatment-resistant schizophrenia: paths to antipsychotic resistance and a roadmap for future research. NPJ Schizophr. (2020) 6:1. doi: 10.1038/s41537-019-0090-z
24. Demjaha, A, Egerton, A, Murray, RM, Kapur, S, Howes, OD, Stone, JM, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry. (2014) 75:e11–3. doi: 10.1016/j.biopsych.2013.06.011
25. Kim, E, Howes, OD, Veronese, M, Beck, K, Seo, S, Park, JW, et al. Presynaptic dopamine capacity in patients with treatment-resistant schizophrenia taking clozapine: an [18F]DOPA PET study. Neuropsychopharmacology. (2017) 42:941–50. doi: 10.1038/npp.2016.258
26. Abi-Dargham, A, Rodenhiser, J, Printz, D, Zea-Ponce, Y, Gil, R, Kegeles, LS, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A. (2000) 97:8104–9. doi: 10.1073/pnas.97.14.8104
27. Jauhar, S, Veronese, M, Nour, MM, Rogdaki, M, Hathway, P, Turkheimer, FE, et al. Determinants of treatment response in first-episode psychosis: an 18F-DOPA PET study. Mol Psychiatry. (2019) 24:1502–12. doi: 10.1038/s41380-018-0042-4
28. Sigvard, AK, Nielsen, MØ, Gjedde, A, Bojesen, KB, Fuglø, D, Tangmose, K, et al. Dopaminergic activity in antipsychotic-Naïve patients assessed with positron emission tomography before and after partial dopamine D2 receptor agonist treatment: association with psychotic symptoms and treatment response. Biol Psychiatry. (2022) 91:236–45. doi: 10.1016/j.biopsych.2021.08.023
29. Amato, D, Canneva, F, Cumming, P, Maschauer, S, Groos, D, Dahlmanns, JK, et al. A dopaminergic mechanism of antipsychotic drug efficacy, failure, and failure reversal: the role of the dopamine transporter. Mol Psychiatry. (2020) 25:2101–18. doi: 10.1038/s41380-018-0114-5
30. Inada, T, Nakamura, A, and Iijima, Y. Relationship between catechol-O-methyltransferase polymorphism and treatment-resistant schizophrenia. Am J Med Genet B Neuropsychiatr Genet. (2003) 120B:35–9. doi: 10.1002/ajmg.b.20023
31. Kohlrausch, FB, Gama, CS, Lobato, MI, Belmonte-de-Abreu, P, Callegari-Jacques, SM, Gesteira, A, et al. Naturalistic pharmacogenetic study of treatment resistance to typical neuroleptics in European-Brazilian schizophrenics. Pharmacogenet Genomics. (2008) 18:599–609. doi: 10.1097/FPC.0b013e328301a763
32. Ota, VK, Spíndola, LN, Gadelha, A, Santos Filho,, Santoro, ML, Christofolini, DM, et al. DRD1 rs4532 polymorphism: a potential pharmacogenomic marker for treatment response to antipsychotic drugs. Schizophr Res (2012) 142: 206–208. doi: 10.1016/j.schres.2012.08.003
33. Bilic, P, Jukic, V, Vilibic, M, Savic, A, and Bozina, N. Treatment-resistant schizophrenia and DAT and SERT polymorphisms. Gene. (2014) 543:125–32. doi: 10.1016/j.gene.2014.03.050
34. Kogure, M, Kanahara, N, Miyazawa, A, Oishi, K, Nakata, Y, Oda, Y, et al. Interacting roles of COMT and GAD1 genes in patients with treatment-resistant schizophrenia: a genetic association study of schizophrenia patients and healthy controls. J Mol Neurosci. (2021) 71:2575–82. doi: 10.1007/s12031-021-01866-y
35. Frank, J, Lang, M, Witt, SH, Strohmaier, J, Rujescu, D, Cichon, S, et al. Identification of increased genetic risk scores for schizophrenia in treatment-resistant patients. Mol Psychiatry. (2015) 20:150–1. doi: 10.1038/mp.2014.56
36. Gasse, C, Wimberley, T, Wang, Y, Mors, O, Børglum, A, Als, TD, et al. Schizophrenia polygenic risk scores, urbanicity and treatment-resistant schizophrenia. Schizophr Res. (2019) 212:79–85. doi: 10.1016/j.schres.2019.08.008
37. Werner, MCF, Wirgenes, KV, Haram, M, Bettella, F, Lunding, SH, Rødevand, L, et al. Indicated association between polygenic risk score and treatment-resistance in a naturalistic sample of patients with schizophrenia spectrum disorders. Schizophr Res. (2020) 218:55–62. doi: 10.1016/j.schres.2020.03.006
38. Pardiñas, AF, Smart, SE, Willcocks, IR, Holmans, PA, Dennison, CA, Lynham, AJ, et al. Interaction testing and polygenic risk scoring to estimate the Association of Common Genetic Variants with Treatment Resistance in schizophrenia. JAMA Psychiatry. (2022) 79:260–9. doi: 10.1001/jamapsychiatry.2021.3799
39. Martin, AK, and Mowry, B. Increased rare duplication burden genomewide in patients with treatment-resistant schizophrenia. Psychol Med. (2016) 46:469–76. doi: 10.1017/S0033291715001701
40. Wimberley, T, Gasse, C, Meier, SM, Agerbo, E, MacCabe, JH, and Horsdal, HT. Polygenic risk score for schizophrenia and treatment-resistant schizophrenia. Schizophr Bull. (2017) 43:1064–9. doi: 10.1093/schbul/sbx0007
41. Horiguchi, M, Ohi, K, Hashimoto, R, Hao, Q, Yasuda, Y, Yamamori, H, et al. Functional polymorphism (C-824T) of the tyrosine hydroxylase gene affects IQ in schizophrenia. Psychiatry Clin Neurosci. (2014) 68:456–62. doi: 10.1111/pcn.12157
42. Kumer, SC, and Vrana, KE. Intricate regulation of tyrosine hydroxylase activity and gene expression. J Neurochem. (1996) 67:443–62. doi: 10.1046/j.1471-4159.1996.67020443.x
43. Wang, K, Zhang, H, Ji, J, Zhang, R, Dang, W, Xie, Q, et al. Expression quantitative trait locus rs6356 is associated with susceptibility to heroin addiction by potentially influencing TH gene expression in the Hippocampus and nucleus Accumbens. J Mol Neurosci. (2022) 72:1108–15. doi: 10.1007/s12031-022-01992-1
44. Thompson, J, Thomas, N, Singleton, A, Piggot, M, Lloyd, S, Perry, EK, et al. D2 dopamine receptor gene (DRD2) Taq1 a polymorphism: reduced dopamine D2 receptor binding in the human striatum associated with the A1 allele. Pharmacogenetics. (1997) 7:479–84. doi: 10.1097/00008571-199712000-00006
45. Pohjalainen, T, Rinne, JO, Någren, K, Lehikoinen, P, Anttila, K, Syvälahti, EKG, et al. The A1 allele of the human D2 dopamine receptor gene predicts low D2 receptor availability in healthy volunteers. Mol Psychiatry. (1998) 3:256–60. doi: 10.1038/sj.mp.4000350
46. Jönsson, EG, Nöthen, MM, Grünhage, F, Farde, L, Nakashima, Y, Propping, P, et al. Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers. Mol Psychiatry. (1999) 4:290–6. doi: 10.1038/sj.mp.4000532
47. Yao, J, Pan, Y, Ding, M, Pang, H, and Wang, B. Association between DRD2 (rs1799732 and rs1801028) and ANKK1 (rs1800497) polymorphisms and schizophrenia: a meta-analysis. Am J Med Genet B Neuropsychiatr Genet. (2015) 168B:1–13. doi: 10.1002/ajmg.b.32281
48. Zhang, L, Hu, L, Li, X, Zhang, J, and Chen, B. The DRD2 rs1800497 polymorphism increase the risk of mood disorder: evidence from an update meta-analysis. J Affect Disord. (2014) 158:71–7. doi: 10.1016/j.jad.2014.01.015
49. Kukreti, R, Tripathi, S, Bhatnagar, P, Gupta, S, Chauhan, C, Kubendran, S, et al. Association of DRD2 gene variant with schizophrenia. Neurosci Lett. (2006) 392:68–71. doi: 10.1016/j.neulet.2005.08.059
50. Monakhov, M, Golimbet, V, Abramova, L, Kaleda, V, and Karpov, V. Association study of three polymorphisms in the dopamine D2 receptor gene and schizophrenia in the Russian population. Schizophr Res. (2008) 100:302–7. doi: 10.1016/j.schres.2008.01.007
51. Nkam, I, Ramoz, N, Breton, F, Mallet, J, Gorwood, P, and Dubertret, C. Impact of DRD2/ANKK1 and COMT polymorphisms on attention and cognitive functions in schizophrenia. PLoS One. (2017) 12:e0170147. doi: 10.1371/journal.pone.0170147
52. Lachman, HM, Papolos, DF, Saito, T, Yu, YM, Szumlanski, CL, and Weinshilboum, RM. Human catechol-O-methyltransferase pharmacogenetics: description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics. (1996) 6:243–50. doi: 10.1097/00008571-199606000-00007
53. Chen, J, Lipska, BK, Halim, N, Ma, QD, Matsumoto, M, Melhem, S, et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. (2004) 75:807–21. doi: 10.1086/425589
54. Reith, MEA, Kortagere, S, Wiers, CE, Sun, H, Kurian, MA, Galli, A, et al. The dopamine transporter gene SLC6A3: multidisease risks. Mol Psychiatry. (2022) 27:1031–46. doi: 10.1038/s41380-021-01341-5
55. Talkowski, ME, Kirov, G, Bamne, M, Georgieva, L, Torres, G, Mansour, H, et al. A network of dopaminergic gene variations implicated as risk factors for schizophrenia. Hum Mol Genet. (2008) 17:747–58. doi: 10.1093/hmg/ddm347
56. Talkowski, ME, McCann, KL, Chen, M, McClain, L, Bamne, M, Wood, J, et al. Fine-mapping reveals novel alternative splicing of the dopamine transporter. Am J Med Genet B Neuropsychiatr Genet. (2010) 153B:1434–47. doi: 10.1002/ajmg.b.31125
57. Kang, AM, Palmatier, MA, and Kidd, KK. Global variation of a 40-bp VNTR in the 3′-untranslated region of the dopamine transporter gene (SLC6A3). Biol Psychiatry. (1999) 46:151–60. doi: 10.1016/S0006-3223(99)00101-8
58. Costa, A, Riedel, M, Müller, U, Möller, HJ, and Ettinger, U. Relationship between SLC6A3 genotype and striatal dopamine transporter availability: a meta-analysis of human single photon emission computed tomography studies. Synapse. (2011) 65:998–1005. doi: 10.1002/syn.20927
59. Faraone, SV, Spencer, TJ, Madras, BK, Zhang-James, Y, and Biederman, J. Functional effects of dopamine transporter gene genotypes on in vivo dopamine transporter functioning: a meta-analysis. Mol Psychiatry. (2014) 19:880–9. doi: 10.1038/mp.2013.126
60. Sano, A, Kondoh, K, Kakimoto, Y, and Kondo, I. A 40-nucleotide repeat polymorphism in the human dopamine transporter gene. Hum Genet. (1993) 91:405–6. doi: 10.1007/BF00217369
61. Greenwood, TA, and Kelsoe, JR. Promoter and intronic variants affect the transcriptional regulation of the human dopamine transporter gene. Genomics. (2003) 82:511–20. doi: 10.1016/S0888-7543(03)00142-3
62. Kelada, SN, Costa-Mallen, P, Checkoway, H, Carlson, CS, Weller, TS, Swanson, PD, et al. Dopamine transporter (SLC6A3) 5′ region haplotypes significantly affect transcriptional activity in vitro but are not associated with Parkinson’s disease. Pharmacogenet Genomics. (2005) 15:659–68. doi: 10.1097/01.fpc.0000170917.04275.d6
63. Zhao, Y, Xiong, N, Liu, Y, Zhou, Y, Li, N, Qing, H, et al. Human dopamine transporter gene: differential regulation of 18-kb haplotypes. Pharmacogenomics. (2013) 14:1481–94. doi: 10.2217/pgs.13.141
64. Kennedy, JL, Xiong, N, Yu, J, Zai, CC, Pouget, JG, Li, J, et al. Increased Nigral SLC6A3 activity in schizophrenia patients: findings from the Toronto-McLean cohorts. Schizophr Bull. (2016) 42:772–81. doi: 10.1093/schbul/sbv191
65. Bamne, MN, Talkowski, ME, Chowdari, KV, and Nimgaonkar, VL. Functional analysis of upstream common polymorphisms of the dopamine transporter gene. Schizophr Bull. (2010) 36:977–82. doi: 10.1093/schbul/sbp005
66. Xu, FL, Ding, M, Wu, X, Liu, YP, Xia, X, Yao, J, et al. A Meta-analysis of the association between SLC6A3 gene polymorphisms and schizophrenia. J Mol Neurosci. (2020) 70:155–66. doi: 10.1007/s12031-019-01399-5
67. Michelhaugh, SK, Fiskerstrand, C, Lovejoy, E, Bannon, MJ, and Quinn, JP. The dopamine transporter gene (SLC6A3) variable number of tandem repeats domain enhances transcription in dopamine neurons. J Neurochem. (2001) 79:1033–8. doi: 10.1046/j.1471-4159.2001.00647.x
68. Heinz, A, Goldman, D, Jones, DW, Palmour, R, Hommer, D, Gorey, JG, et al. Genotype influences in vivo dopamine transporter availability in human striatum. Neuropsychopharmacology. (2000) 22:133–9. doi: 10.1016/S0893-133X(99)00099-8
69. Fuke, S, Suo, S, Takahashi, N, Koike, H, Sasagawa, N, and Ishiura, S. The VNTR polymorphism of the human dopamine transporter (DAT1) gene affects gene expression. Pharm J. (2001) 1:152–6. doi: 10.1038/sj.tpj.6500026
70. Mill, J, Asherson, P, Browes, C, D’Souza, U, and Craig, I. Expression of the dopamine transporter gene is regulated by the 3’ UTR VNTR: evidence from brain and lymphocytes using quantitative RT-PCR. Am J Med Genet. (2002) 114:975–9. doi: 10.1002/ajmg.b.10948
71. Miller, GM, and Madras, BK. Polymorphisms in the 3′-untranslated region of human and monkey dopamine transporter genes affect reporter gene expression. Mol Psychiatry. (2002) 7:44–55. doi: 10.1038/sj.mp.4000921
72. VanNess, SH, Owens, MJ, and Kilts, CD. The variable number of tandem repeats element in DAT1 regulates in vitro dopamine transporter density. BMC Genet. (2005) 6:55. doi: 10.1186/1471-2156-6-55
73. Jacobsen, LK, Staley, JK, Zoghbi, SS, Seibyl, JP, Kosten, TR, Innis, RB, et al. Prediction of dopamine transporter binding availability by genotype: a preliminary report. Am J Psychiatry. (2000) 157:1700–3. doi: 10.1176/appi.ajp.157.10.1700
74. van Dyck, CH, Malison, RT, Jacobsen, LK, Seibyl, JP, Staley, JK, Laruelle, M, et al. Increased dopamine transporter availability associated with the 9-repeat allele of the SLC6A3 gene. J Nucl Med. (2005) 46:745–51.
75. Martinez, D, Gelernter, J, Abi-Dargham, A, van Dyck, C, Kegeles, L, Innis, RB, et al. The variable number of tandem repeats polymorphism of the dopamine transporter gene is not associated with significant change in dopamine transporter phenotype in humans. Neuropsychopharmacology. (2001) 24:553–60. doi: 10.1016/S0893-133X(00)00216-5
76. Inoue-Murayama, M, Adachi, S, Mishima, N, Mitani, H, Takenaka, O, Terao, K, et al. Variation of variable number of tandem repeat sequences in the 3′-untranslated region of primate dopamine transporter genes that affects reporter gene expression. Neurosci Lett. (2002) 334:206–10. doi: 10.1016/S0304-3940(02)01125-4
77. Lynch, DR, Mozley, PD, Sokol, S, Maas, NMC, Balcer, LJ, and Siderowf, AD. Lack of effect of polymorphisms in dopamine metabolism related genes on imaging of TRODAT-1 in striatum of asymptomatic volunteers and patients with Parkinson’s disease. Mov Disord. (2003) 18:804–12. doi: 10.1002/mds.10430
78. Mill, J, Xu, X, Ronald, A, Curran, S, Price, T, Knight, J, et al. Quantitative trait locus analysis of candidate gene alleles associated with attention deficit hyperactivity disorder (ADHD) in five genes: DRD4, DAT1, DRD5, SNAP-25, and 5HT1B. Am J Med Genet B Neuropsychiatr Genet. (2005) 133B:68–73. doi: 10.1002/ajmg.b.30107
79. Pae, CU, Chiesa, A, Patkar, AA, and Serretti, A. No influence of SLC6A3 40 base VNTR polymorphism on the response to risperidone. Int J Psychiatry Clin Pract. (2010) 14:228–32. doi: 10.3109/13651501.2010.486902
80. Szekeres, G, Kéri, S, Juhász, A, Rimanóczy, Á, Szendi, I, Czimmer, C, et al. Role of dopamine D3 receptor (DRD3) and dopamine transporter (DAT) polymorphism in cognitive dysfunction and therapeutic response to atypical antipsychotics in patients with schizophrenia. Am J Med Genet B. (2004) 124B:1–5. doi: 10.1002/ajmg.b.20045
81. Zhao, Y, Zhou, Y, Xiong, N, and Lin, Z. Identification of an intronic cis-acting element in the human dopamine transporter gene. Mol Biol Rep. (2012) 39:5393–9. doi: 10.1007/s11033-011-1339-4
82. Apud, JA, and Weinberger, DR. Treatment of cognitive deficits associated with schizophrenia: potential role of catechol-O-methyltransferase inhibitors. CNS Drugs. (2007) 21:535–57. doi: 10.2165/00023210-200721070-00002
83. Nogueira, NGHM, Bacelar, MFB, Ferreira, BP, Parma, JO, and Lage, GM. Association between the catechol-O-methyltransferase (COMT) Val158Met polymorphism and motor behavior in healthy adults: a study review. Brain Res Bull. (2019) 144:223–32. doi: 10.1016/j.brainresbull.2018.11.002
84. Egan, MF, Goldberg, TE, Kolachana, BS, Callicott, JH, Mazzanti, CM, Straub, RE, et al. Effect of COMT Val108/158Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. (2001) 98:6917–22. doi: 10.1073/pnas.111134598
85. Pelayo-Terán, JM, Crespo-Facorro, B, Carrasco-Marín, E, Pérez-Iglesias, R, Mata, I, Arranz, MJ, et al. Catechol-O-methyltransferase Val158Met polymorphism and clinical characteristics in first episode non-affective psychosis. Am J Med Genet B Neuropsychiatr Genet. (2008) 147B:550–6. doi: 10.1002/ajmg.b.30639
86. Nelson, CLM, Amsbaugh, HM, Reilly, JL, Rosen, C, Marvin, RW, Ragozzino, ME, et al. Beneficial and adverse effects of antipsychotic medication on cognitive flexibility are related to COMT genotype in first episode psychosis. Schzophr Res. (2018) 202:212–6. doi: 10.1016/j.schres.2018.06.029
87. Goghari, VM, and Sponheim, SR. Differential association of the COMT Val158Met polymorphism with clinical phenotypes in schizophrenia and bipolar disorder. Schzophr Res. (2008) 103:186–91. doi: 10.1016/j.schres.2008.05.015
88. Barnett, JH, Jones, PB, Robbins, TW, and Müller, U. Effects of the catechol-O-methyltransferase Val158Met polymorphism on executive function: a meta-analysis of the Wisconsin card Sort test in schizophrenia and healthy controls. Mol Psychiatry. (2007) 12:502–9. doi: 10.1038/sj.mp.4001973
89. Lewis, DA, Melchitzky, DS, Sesack, SR, Whitehead, RE, Auh, S, and Sampson, A. Dopamine transporter immunoreactivity in monkey cerebral cortex: regional, laminar, and ultrastructural localization. J Comp Neurol. (2001) 432:119–36. doi: 10.1002/cne.1092
90. Huotari, M, Gogos, JA, Karayiorgou, M, Koponen, O, Forsberg, M, Raasmaja, A, et al. Brain catecholamine metabolism in catechol-O-methyltransferase (COMT)-deficient mice. Eur J Neurosci. (2002) 15:246–56. doi: 10.1046/j.0953-816x.2001.01856.x
91. Gogos, JA, Morgan, M, Luine, V, Santha, M, Ogawa, S, Pfaff, D, et al. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc Natl Acad Sci U S A. (1998) 95:9991–6. doi: 10.1073/pnas.95.17.9991
92. Sesack, SR, Hawrylak, VA, Melchitzky, DS, and Lewis, DA. Dopamine innervation of a subclass of local circuit neurons in monkey prefrontal cortex: ultrastructural analysis of tyrosine hydroxylase and parvalbumin immunoreactive structures. Cereb Cortex. (1998) 8:614–22. doi: 10.1093/cercor/8.7.614
93. Tunbridge, EM, Bannerman, DM, Sharp, T, and Harrison, PJ. Catechol-o-methyltransferase inhibition improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J Neurosci. (2004) 24:5331–5. doi: 10.1523/JNEUROSCI.1124-04.2004
94. Cragg, SJ, Baufreton, J, Xue, Y, Bolam, JP, and Bevan, MD. Synaptic release of dopamine in the subthalamic nucleus. Eur J Neurosci. (2004) 20:1788–802. doi: 10.1111/j.1460-9568.2004.03629.x
95. Wayment, HK, Schenk, JO, and Sorg, BA. Characterization of extracellular dopamine clearance in the medial prefrontal cortex: role of monoamine uptake and monoamine oxidase inhibition. J Neurosci. (2001) 21:35–44. doi: 10.1523/JNEUROSCI.21-01-00035.2001
96. Bertolino, A, Blasi, G, Latorre, V, Rubino, V, Rampino, A, Sinibaldi, L, et al. Additive effects of genetic variation in dopamine regulating genes on working memory cortical activity in human brain. J Neurosci. (2006) 26:3918–22. doi: 10.1523/JNEUROSCI.4975-05.2006
97. Bertolino, A, di Giorgio, A, Blasi, G, Sambataro, F, Caforio, G, Sinibaldi, L, et al. Epistasis between dopamine regulating genes identifies a nonlinear response of the human hippocampus during memory tasks. Biol Psychiatry. (2008) 64:226–34. doi: 10.1016/j.biopsych.2008.02.001
98. Caldú, X, Vendrell, P, Bartrés-Faz, D, Clemente, I, Bargalló, N, Jurado, MÁ, et al. Impact of the COMT Val108/158 met and DAT genotypes on prefrontal function in healthy subjects. NeuroImage. (2007) 37:1437–44. doi: 10.1016/j.neuroimage.2007.06.021
99. Bilder, RM, Volavka, J, Lachman, HM, and Grace, AA. The catechol-O-methyltransferase polymorphism: relations to the tonic-phasic dopamine hypothesis and neuropsychiatric phenotypes. Neuropsychopharmacology. (2004) 29:1943–61. doi: 10.1038/sj.npp.1300542
100. Kunugi, H, Kawada, Y, Hattori, M, Ueki, A, Otsuka, M, and Nanko, S. Association study of structural mutations of the tyrosine hydroxylase gene with schizophrenia and Parkinson’s disease. Am J Med Genet. (1998) 81:131–3. doi: 10.1002/(SICI)1096-8628(19980328)81:2<131::AID-AJMG2>3.0.CO;2-Z
101. Ota, M, Nakashima, A, Ikemoto, K, Nojima, S, Tanaka, M, Okuda, M, et al. Exon 3 of tyrosine hydroxylase gene: lack of association with Japanese schizophrenic patients. Mol Psychiatry. (2001) 6:315–9. doi: 10.1038/sj.mp.4000840
102. Andreou, D, Söderman, E, Axelsson, T, Sedvall, GC, Terenius, L, Agartz, I, et al. Polymorphisms in genes implicated in dopamine, serotonin and noradrenalin metabolism suggest association with cerebrospinal fluid monoamine metabolite concentrations in psychosis. Behav Brain Funct. (2014) 10:26. doi: 10.1186/1744-9081-10-26
103. Hu, J, Chan, LF, Souza, RP, Tampakeras, M, Kennedy, JL, Zai, C, et al. The role of tyrosine hydroxylase gene variants in suicide attempt in schizophrenia. Neurosci Lett. (2014) 559:39–43. doi: 10.1016/j.neulet.2013.11.025
Keywords: antipsychotic, dopamine, psychosis, single nucleotide polymorphism, variable number of tandem repeats
Citation: Kogure M, Kanahara N, Miyazawa A, Shiko Y, Otsuka I, Matsuyama K, Takase M, Kimura M, Kimura H, Ota K, Idemoto K, Tamura M, Oda Y, Yoshida T, Okazaki S, Yamasaki F, Nakata Y, Watanabe Y, Niitsu T, Hishimoto A and Iyo M (2024) Association of SLC6A3 variants with treatment-resistant schizophrenia: a genetic association study of dopamine-related genes in schizophrenia. Front. Psychiatry. 14:1334335. doi: 10.3389/fpsyt.2023.1334335
Edited by:
Massoud Stephane, Oregon Health and Science University, United StatesReviewed by:
Yuji Ozeki, Shiga University of Medical Science, JapanCopyright © 2024 Kogure, Kanahara, Miyazawa, Shiko, Otsuka, Matsuyama, Takase, Kimura, Kimura, Ota, Idemoto, Tamura, Oda, Yoshida, Okazaki, Yamasaki, Nakata, Watanabe, Niitsu, Hishimoto and Iyo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobuhisa Kanahara, a2FuYWhhcmFAZmFjdWx0eS5jaGliYS11Lmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.