 Katherine R. H. Mortimer
Katherine R. H. Mortimer Mohammed Zia Ul Haq Katshu2,3
Mohammed Zia Ul Haq Katshu2,3 Lisa Chakrabarti
Lisa Chakrabarti
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Psychiatry, 24 November 2023
Sec. Molecular Psychiatry
Volume 14 - 2023 | https://doi.org/10.3389/fpsyt.2023.1257460
This article is part of the Research TopicExploring The Association Between Metabolism and Psychiatric DisordersView all 5 articles
Psychosis is a known risk factor for developing metabolic syndrome (MetS). The risk is even greater in patients who are taking second-generation antipsychotics (SGAs). SGAs exacerbate metabolic abnormalities and lead to a 3-fold increased risk of severe weight gain, type 2 diabetes, and cardiovascular disease in patients. Mitochondrial dysfunction is a hallmark of MetS. Mitochondria process glucose and fatty acids into ATP. If these processes are impaired, it can result in dyslipidaemia, hyperglycaemia and an imbalance between nutrient input and energy output. This leads to increased adiposity, insulin resistance and atherosclerosis. It is unclear how SGAs induce MetS and how mitochondria might be involved in this process. It has been found that SGAs impair cellular glucose uptake in liver, dysregulating glucose and fatty acid metabolism which leads to an accumulation of glucose and/or lipids and an increase reactive oxygen species (ROS) which target mitochondrial proteins. This affects complexes of the electron transport chain (ETC) to reduce mitochondrial respiration. While there is a suggestion that SGAs may interact with a variety of processes that disrupt mitochondrial function, some of the results are conflicting, and a clear picture of how SGAs interact with mitochondria in different cell types has not yet emerged. Here, we outline the current evidence showing how SGAs may trigger mitochondrial dysfunction and lead to the development of MetS.
The term psychoses describes a cluster of psychiatric disorders which include acute and transient psychotic disorder, schizophrenia, schizoaffective disorder, delusional disorder, bipolar affective disorder and psychotic depression (1). Schizophrenia and psychosis have a lifetime prevalence of 1% and a high global economic burden (1, 2). The aetiology of psychosis is multifactorial, including genetics, substance misuse, and medical conditions, like infections, and autoimmune disorders (3–6). Psychosis is primarily treated with antipsychotic medications (APs) but a variety of comorbidities associated with psychosis means antidepressants or anti-anxiety medications are prescribed alongside (1). Prescription of antipsychotic medications doubled between 2000 and 2014 and the rate of first prescriptions of antipsychotics increased between 2000 and 2019 by 2% each year (7, 8). Antipsychotics are divided into two subclasses, first-generation antipsychotics, also known as typical antipsychotics, and second-generation or atypical antipsychotics.
First-generation antipsychotics (FGAs), were discovered due to their sedative effects and by chance were found to reduce the positive symptoms, hallucinations and delusions, of psychosis (9). FGAs such as chlorpromazine and haloperidol, were some of the first drugs to be developed and utilised in treating mental illnesses (10). Shortly after their use in clinical practise, extrapyramidal symptoms (EPS) such as tardive dyskinesia, parkinsonism and dystonia, were classed as side effects of FGA treatment (9). These symptoms were provoked by the dopamine receptor antagonism of FGAs, particularly their affinity for the D2 receptor (11). Following this, second-generation antipsychotics (SGAs) were developed, which reduce EPS as they have more diverse receptor affinities.
Clozapine was the first SGA to be developed by Wander Laboratories in 1959; it was discovered due to its similar structure to tricyclic antidepressants (12). When clozapine was found to have antipsychotic properties but no motor side effects, it became apparent that EPS were not necessary for the efficacy of antipsychotics (12). Clozapine has a more diverse receptor profile than FGAs, with an affinity for multiple serotonergic receptors, alpha-1 adrenergic receptors, D3 and D4 receptors and a lower affinity for D2 receptors (13, 14). The era of SGAs as superior alternatives to FGAs started in the 1970s, however it was cut short as within 6 months of clozapine administration to patients during clinical trials, cases of fatal agranulocytosis were reported (15). There were 17 cases in ~3,000 patients, which led to eight fatalities (16). While the risk is low, 0.8% of patients develop clozapine-induced agranulocytosis and low neutrophil levels shortly after starting clozapine treatment (17). In the mid to late 1980s, clozapine was tested against the commonly prescribed FGA chlorpromazine for tolerability and efficacy in patients (18). Clozapine showed increased efficacy in patients who were unresponsive to a previous antipsychotic, with particular focus on improving negative psychotic symptoms such as apathy and asocial behaviour, as well as positive symptoms (19, 20). Consequently, treatment with clozapine was reintroduced but now requires patients to undergo weekly blood testing and in recent years it has been reserved for patients who are deemed ‘treatment-resistant’. Treatment-resistant patients are defined by those who have not responded to two or more previously prescribed antipsychotics (1).
Following the incidences of clozapine-induced agranulocytosis, analogues of clozapine were explored to find a drug with similar efficacy in treating psychosis, a reduced propensity for EPS but one that did not affect neutrophil levels in the blood. Olanzapine was developed by Eli Lilly and Co., with a similar receptor binding profile to clozapine but a slightly different chemical structure and presented a promising development in the field of antipsychotic medication (21). Olanzapine has shown to be efficacious in treating first-episode psychosis with a lower discontinuation rate than haloperidol and distinctly does not induce agranulocytosis like clozapine (21). Clinical trials in the late 1990s showed olanzapine treatment produced a greater reduction in positive and negative symptoms of schizophrenia and an improved response in a variety of psychiatric symptom scales in patients, compared to treatment with haloperidol (22).
Further SGAs have been developed since olanzapine was discovered including quetiapine (AstraZeneca) and risperidone (Janssen Pharmaceuticals Inc.), which differ slightly in their receptor binding affinities (23). Aripiprazole is an SGA that was developed by Otsuka Pharmaceutical Co., Ltd. and approved for use in the early 2000s (24). It is unique from the other SGAs as it is a partial agonist of D2 and 5-HT1A receptors and an antagonist of 5-HT2A receptors (14). Aripiprazole has been labelled a ‘dopamine stabiliser’ as its partial agonistic properties allow for treatment of hypodopaminergia and hyperdopaminergia of different brain regions that contribute to negative and positive symptoms (25). Despite aripiprazole’s alternative mode of action, it has been found to have a similar efficacy to D2 receptor antagonists and a lower efficacy when compared to olanzapine, although it is better tolerated by patients (26).
While SGAs do not have adverse motor symptoms, they are not without other side effects which reduce a patient’s tolerability of the drug. SGAs can have a sedative effect on patients, they can cause endocrine issues such as sexual dysfunction as well as prominent metabolic side effects (27). The incidence of metabolic syndrome (MetS) is significantly higher in patients taking SGAs (32–68%) than in SGA-naïve patients (3.3–26%) (28). MetS describes a co-occurrence of morbidities such as hypertension, obesity, dyslipidaemia and peripheral insulin resistance which are predictors of type 2 diabetes and cardiovascular disease (CVD) (29, 30). Psychosis alone is a known risk factor for developing MetS (31). Treatment with SGAs exacerbate metabolic symptoms which, along with lifestyle changes and genetic predisposition, increase the risk of CVD (31, 32). The main cause of mortality in schizophrenia patients is CVD, suggesting treatment with SGAs contribute to a patients decline in cardiovascular health and increased risk of CVD-related mortality (33).
Olanzapine and clozapine are the most efficacious antipsychotics, proven to be the only antipsychotics to reduce the risk of suicidality in patients (34, 35), but they are also associated with the most severe risk of MetS. From a meta-analysis, clozapine was shown to be associated with the highest amount of weight gain in patients, and olanzapine with the greatest degree of BMI increase in patients (36). Aripiprazole is associated with the lowest risk of weight gain and metabolic abnormalities (36, 37). The mechanisms behind the increased propensity for SGAs to trigger MetS are largely unknown. Due to their diverse receptor binding profiles, there is the potential for SGAs to bind to receptors in the periphery or alternative off-target binding sites which leads to metabolic disturbances. This off-target binding may include receptors or proteins in the liver, pancreas, kidneys or adipose tissue.
Mitochondrial dysfunction is a hallmark of metabolic dysregulation and the development of MetS (38). Fundamentally, the disparity between energy production and utilisation can lead to metabolic disorders (39). Mitochondria are responsible for energy production via ATP synthesis, the main energy source for cells. Disruption of this energy production, potentially by inhibition of mitochondrial proteins responsible for ATP synthesis, is likely to either lead to metabolic disorders or may be a consequence of such. Oxidative stress in mitochondria may be at least partly responsible for the pathogenesis of insulin resistance, weight gain and CVDs (40). Other disruptions to regular mitochondrial functioning include scarcity of substrates required to fuel glycolysis or oxidative phosphorylation (OXPHOS) which are processes by which ATP is generated. This may be due to a reduced capacity for uptake of nutrients into cells or inhibition of enzymes involved in the synthesis of substrates (41) (summarised in Figure 1). Therefore, one theory is that SGAs impair mitochondrial function which leads to the development of metabolic disorders by disrupted energy metabolism (42).
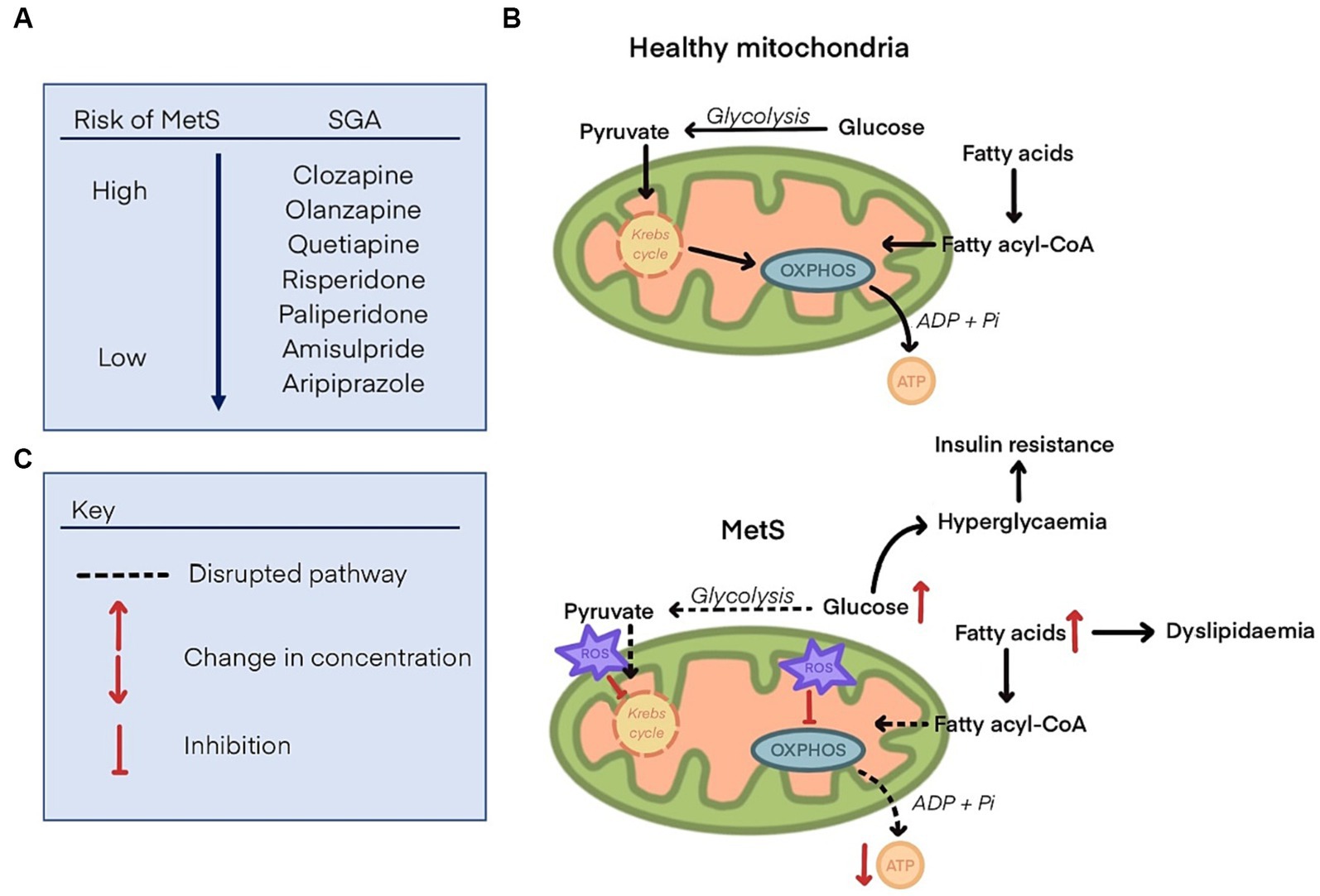
Figure 1. (A) List of second-generation antipsychotics and their propensity to cause metabolic syndrome, ranging from the highest risk (clozapine), to the lowest (aripiprazole). (B) Glucose and fatty acid metabolism to generate substrates that fuel oxidative phosphorylation (OXPHOS) and produce ATP in health mitochondria. (C) Disrupted glucose and fatty acid metabolism in mitochondria with metabolic syndrome. Reactive oxygen species (ROS) are increased and inhibit components of the Krebs cycle and OXPHOS, leading to a decline in ATP synthesis. Glucose and fatty acids concentrations are increased, leading to dyslipidaemia, hyperglycaemia, and insulin resistance.
Glucose metabolism is shown to be impaired in patients taking SGAs independent of adiposity and age. In one study, the highest blood glucose and insulin resistant levels were found in patients taking olanzapine and clozapine, while risperidone and FGA-treated patients did not show increased insulin resistance but did have elevated glucose levels (43). As patients were matched by adiposity and age, this decline in cardiovascular health and changes in glucose metabolism can be primarily attributed to treatment with olanzapine and clozapine. Further evidence for this was shown when mice were administered olanzapine and they showed an increase in body weight and adiposity that was not concurrent with the rate of food consumption (44).
Antipsychotics have also been shown to disrupt fatty acid metabolism (45, 46). Fatty acid oxidation yields more ATP than glucose but consumes more oxygen (47). A series of studies in mice and rats found that olanzapine lowered plasma free fatty acid (FFA) concentrations and lowered respiratory exchange ratio (RER) prior to fasting, suggesting that olanzapine treatment triggers a shift towards fatty acid oxidation to produce fuel substrates for respiration (45, 46). This switch to fatty acid oxidation as a fuel source rather than glucose, leads to accumulation of glucose and glucose intolerance. C57BL/6 mice were treated a single dose of olanzapine, clozapine, risperidone and aripiprazole (doses between 1 and 10 mg/kg) and the general rate of oxygen consumption and RER was measured using indirect calorimetry (45). Olanzapine, clozapine and risperidone reduced the rate of oxygen consumption and RER dose-dependently, and in olanzapine-treated mice this was associated with an increase in plasma glucose concentration, while aripiprazole showed a much more reduced effect on RER lowering. Similar results were found in Sprague–Dawley rats who were given 10 mg/kg of olanzapine over 24 h (46).
The effects of SGAs on AMP-activated protein kinase (AMPK) signalling has been investigated as an explanation for increases in plasma glucose levels and weight gain (48–52). AMPK senses energy in response to the AMP/ATP ratio and an increase in AMP suggests that ATP is being used up for energy processes in cells at a faster rate than it is being synthesised. AMPK regulates cellular metabolism and in its active form, phosphorylated AMPK, it stimulates glucose uptake and fatty acid oxidation in the liver but AMPK signalling in the hypothalamus reduces glucose uptake and lipid metabolism (53, 54). The differential downstream effects of AMPK, according to the effected tissue, may provide an insight into the off-target metabolic effects of SGAs. SGAs have been shown to have a varying effect on AMPK expression (summarised in Table 1), depending on the tissue studied. Overall olanzapine, clozapine and quetiapine increase AMPK signalling in brain regions such as the hypothalamus and frontal cortex (49–52). As for the effect of SGAs on AMPK signalling in peripheral tissues such as the liver and adipocytes, the results are conflicting (44, 48, 55, 56).
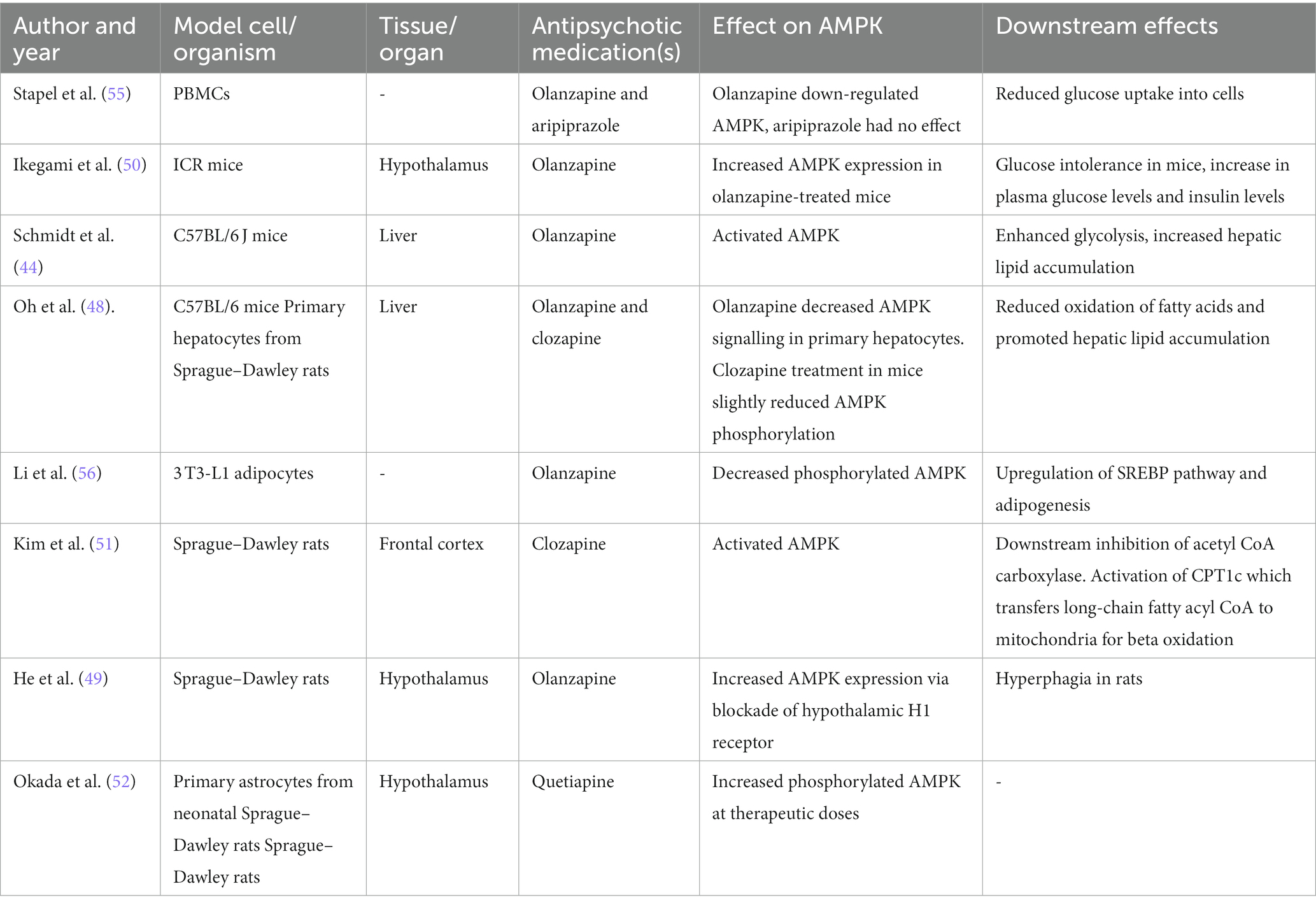
Table 1. Summary of the effects of SGAs on AMPK expression.
Clozapine and olanzapine have been shown to increase production of reactive oxygen species (ROS) and oxidise key proteins involved in energy metabolism (57–61). ROS are produced by complex I and III during oxidative phosphorylation when leaked electrons react with oxygen to form superoxide ions (62). ROS damage protein structure and function by oxidation. Complex I of the electron transport chain has been shown to be particularly sensitive to ROS attack (63). An increase in the production of ROS leads to damage of mitochondrial elements, a positive feedback of ROS production and consequently, impaired metabolism. As antipsychotics interact with dopamine receptors, their interference with normal dopamine metabolism has been shown to favour the formation of hydrogen peroxidase through monoamine oxidase (58). Using 6-iodoacetamid fluorescein (IAF) labelling which is not incorporated into cysteine residues following protein oxidation, clozapine-treated neuroblastoma cell line SKNSH cells showed increased protein oxidation (57). The proteins were identified using HPLC-electrospray ionisation tandem mass spectrometry and were shown to be a variety of mitochondrial enzymes including malate dehydrogenase, pyruvate kinase and 3-oxoacid CoA. Malate dehydrogenase and pyruvate kinase were also found to be oxidised by olanzapine using cDNA microarray technology on olanzapine-treated rat frontal cortices (59). These enzymes catalyse the oxidation of malate and production of pyruvate which are substrates in the Krebs cycle that facilitate the reduction of NAD+ to NADH to feed electrons into complex I.
Clozapine treatment induces morphological changes in mitochondria in neuroblastoma cells, insulin-responsive and obesity-associated cell types when cultured with TMRM and visualised with confocal microscopy (60). The morphological changes, including mitochondrial swelling and depolarisation, observed with clozapine treatment were similar to those reported in obese rodent models and patients with MetS. It was suggested that the mitochondrial swelling may be in response to an increase in ROS production as a protective mechanism and that the swelling led to depolarisation which was consistent with a depletion of ATP production.
Mitochondrial membrane potential (MMP) loss suggests components of the ETC are dysfunctional, potentially due to the accumulation and damage by ROS (64). In isolated rat hepatocytes, olanzapine induced overproduction of ROS as indicated by DCFH-DA dye, which hydrolyses to DCFH in the liver and fluorescent DCF when interacting with ROS. Fluorescent intensity was then measured using a spectrofluorometer revealing significantly increased fluorescence in olanzapine-treated hepatocytes (61). The cationic fluorescent dye rhodamine 123 was used to measure mitochondrial membrane potential (MMP). This dye accumulates in intact mitochondria and an increase in fluorescence indicates the loss of MMP. MMP loss was found in olanzapine-treated hepatocytes using this method. MMP is a result of the chemiosmotic proton gradient supported by complexes of the ETC and is important for ATP synthesis through ATP synthase.
The effects of antipsychotics on complexes of the ETC has shown differing results. One study found a range of antipsychotics inhibited respiration through specific complexes (65). By measuring oxygen consumption of isolated pig brain mitochondria exposed to antipsychotics, they identified a range of antipsychotics that inhibited complex I-linked respiration. Haloperidol, risperidone, quetiapine and aripiprazole fully inhibited complex I-linked respiration over a range of doses and clozapine was reported to almost fully inhibit complex-I linked respiration. A smaller range of doses also inhibited the activity of complex II, III and IV. However, amongst these antipsychotics, olanzapine, one of the most potent APs associated with metabolic side effects, did not significantly inhibit mitochondrial respiration in this study. There is numerous evidence to support that Haloperidol and other FGAs inhibit complex I of the ETC and this has been implicated in the development of EPS (66). However, whether metabolic abnormalities triggered by SGA treatment is as a result of ETC complex inhibition has not been fully explored.
Lymphoblastoid cell lines (LCLs) isolated from schizophrenia patients taking clozapine, olanzapine, aripiprazole, quetiapine and healthy controls have also yielded metabolic information (42). LCLs treated with clozapine, olanzapine and quetiapine significantly reduced complex I and II activity and decreased mitochondrial respiration, with olanzapine having the most severe effect (42). There was reduced (though not statistically significant) activity of complex I and II in untreated cells from schizophrenia patients compared to controls. This study produced very different results and suggests that metabolic dysfunction in schizophrenia patients may be associated with olanzapine reducing efficiency of mitochondrial respiration. It also suggests that schizophrenia patients may be predisposed to developing MetS and subsequent treatment with SGAs triggers further metabolic decline through inhibition of mitochondrial complexes. Indeed, a common genetic risk factor for schizophrenia, a deletion at chromosome 3q29, has been associated with mitochondrial dysfunction (67). There was a reduced ability to transition from glycolysis to OXPHOS during neuronal maturation in human cortical organoids, downregulation of complex II and IV of the ETC in mice brains with the 3q29 deletion and an overall increased ‘vulnerability to metabolic challenges’ in these models.
Here, we have reviewed whether the prominent metabolic side effects seen with SGA treatment, primarily with olanzapine and clozapine, are related to mitochondrial dysfunction that results in impaired metabolism. Considering the efficacy of clozapine and olanzapine in treating psychosis relative to drug alternatives and non-pharmacological interventions, determining the cause of the metabolic impairments is of significant interest. Establishing the best method for treatment through optimal polypharmacy or personalised medicine, will have a considerably beneficial outcome on psychosis patients’ quality of life in the short term. In the long term, the development of new antipsychotics with a reduced risk of metabolic side effects should be the aim.
KM: Conceptualization, Writing – review & editing, Writing – original draft. MK: Conceptualization, Supervision, Writing – review & editing. LC: Conceptualization, Writing – review & editing, Funding acquisition, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. KM is funded by the Biotechnology and Biological Sciences Research Council (grant number BB/J014508/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. National Institute for Health and Care Excellence (2019). 2019 exceptional surveillance of psychosis and schizophrenia in adults: Prevention and management (NICE guideline CG178). National Institute for Health and Care Excellence.
2. Chaiyakunapruk, N, Chong, HY, Teoh, SL, Wu, DBC, Kotirum, S, and Chiou, CF. Global economic burden of schizophrenia: a systematic review. Neuropsychiatr Dis Treat. (2016) 12:357–73. doi: 10.2147/NDT.S96649
3. Tan, EJ, Neill, E, and Rossell, SL. Psychotic disorders amid the COVID-19 pandemic: a consideration of risks and impacts. Curr Opin Psychiatry. (2022) 35:140–5. doi: 10.1097/YCO.0000000000000784
4. on behalf of the Maternal Morbidity Working Group VanderKruik, R, Barreix, M, Chou, D, Allen, T, Say, L, et al. The global prevalence of postpartum psychosis: a systematic review. BMC Psychiatry. (2017) 17:272–9. doi: 10.1186/s12888-017-1427-7
5. Arseneault, L, Cannon, M, Witton, J, and Murray, RM. Causal association between cannabis and psychosis: examination of the evidence. Br J Psychiatry. (2004) 184:110–7. doi: 10.1192/bjp.184.2.110
6. Byrne, P. Managing the acute psychotic episode. BMJ. (2007) 334:686–92. doi: 10.1136/bmj.39148.668160.80
7. Shoham, N, Cooper, C, Lewis, G, Bebbington, P, and McManus, S. Temporal trends in psychotic symptoms: repeated cross-sectional surveys of the population in England 2000–14. Schizophr Res. (2021) 228:97–102. doi: 10.1016/j.schres.2020.11.057
8. Radojčić, MR, Pierce, M, Hope, H, Senior, M, Taxiarchi, VP, Trefan, L, et al. Trends in antipsychotic prescribing to children and adolescents in England: cohort study using 2000–19 primary care data. The lancet. Psychiatry. (2023) 10:119–28. doi: 10.1016/S2215-0366(22)00404-7
9. Cunningham Owens, D, and Johnstone, EC. The development of antipsychotic drugs. Brain Neurosci Adv. (2018) 2:2398212818817498. doi: 10.1177/2398212818817498
10. Weston-Green, K. Antipsychotic drug development: from historical evidence to fresh perspectives. Front Psychol. (2022) 13:903156. doi: 10.3389/fpsyt.2022.903156
11. Sykes, DA, Moore, H, Stott, L, Holliday, N, Javitch, JA, Lane, JR, et al. Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat Commun. (2017) 8:763. doi: 10.1038/s41467-017-00716-z
12. Braslow, JT, and Marder, SR. History of psychopharmacology. Annu Rev Clin Psychol. (2019) 15:25–50. doi: 10.1146/annurev-clinpsy-050718-095514
13. Meltzer, HY. An overview of the mechanism of action of clozapine. J Clin Psychiatry. (1994) 55:47–52.
14. Li, P, Snyder, GL, and Vanover, KE. Dopamine targeting drugs for the treatment of schizophrenia: past, present and future. Curr Top Med Chem. (2016) 16:3385–403. doi: 10.2174/1568026616666160608084834
15. Lahdelma, L, and Appelberg, B. Clozapine-induced agranulocytosis in Finland, 1982-2007: long-term monitoring of patients is still warranted. J Clin Psychiatry. (2012) 73:837–42. doi: 10.4088/JCP.11m07244
16. Idänpään-Heikkilä, J, Alhava, E, Olkinuora, M, Palva, IP, et al. Agranulocytosis during treatment with clozapine. Eur J Clin Pharmacol. (1977) 11:193–8. doi: 10.1007/BF00606409
17. Mijovic, A, and MacCabe, JH. Clozapine-induced agranulocytosis. Ann Hematol. (2020) 99:2477–82. doi: 10.1007/s00277-020-04215-y
18. Kane, J, Honigfeld, G, Singer, J, and Meltzer, H. Clozapine for the treatment-resistant schizophrenic: a double-blind comparison with chlorpromazine. Arch Gen Psychiatry. (1988) 45:789–96. doi: 10.1001/archpsyc.1988.01800330013001
19. Khan, AH, and Zaidi, S. Clozapine: improvement of negative symptoms of schizophrenia. Cureus. (2017) 9:e1973. doi: 10.7759/cureus.1973
20. Fabrazzo, M, Cipolla, S, Camerlengo, A, Perris, F, and Catapano, F. Second-generation antipsychotics’ effectiveness and tolerability: a review of real-world studies in patients with schizophrenia and related disorders. J Clin Med. (2022) 11:4530. doi: 10.3390/jcm11154530
21. Citrome, L, McEvoy, JP, Todtenkopf, MS, McDonnell, D, and Weiden, PJ. A commentary on the efficacy of olanzapine for the treatment of schizophrenia: the past, present, and future. Neuropsychiatr Dis Treat. (2019) 15:2559–69. doi: 10.2147/NDT.S209284
22. Sanger, TM, Lieberman, JA, Tohen, M, Grundy, S, Beasley, Jr, C, and Tollefson, GD. Olanzapine versus haloperidol treatment in first-episode psychosis. Am J Psychiatr. (1999) 156:79–87. doi: 10.1176/ajp.156.1.79
23. Szasz, M, Casoni, D, and Cimpoiu, C. Study on binding affinity profile, bioactivity, and lipophilicity of selected antipsychotic drugs. J Liq Chromatogr Relat Technol. (2023):1–6. doi: 10.1080/10826076.2023.2216780
24. Kikuchi, T, Maeda, K, Suzuki, M, Hirose, T, Futamura, T, and McQuade, RD. Discovery research and development history of the dopamine D2 receptor partial agonists, aripiprazole and brexpiprazole. Neuropsychopharmacol Rep. (2021) 41:134–43. doi: 10.1002/npr2.12180
25. Mailman, RB, and Murthy, V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. (2010) 16:488–501. doi: 10.2174/138161210790361461
26. Kim, DD, Barr, AM, Lian, L, Yuen, JWY, Fredrikson, D, Honer, WG, et al. Efficacy and tolerability of aripiprazole versus D2 antagonists in the early course of schizophrenia: a systematic review and meta-analysis. NPJ Schizophr. (2021) 7:29. doi: 10.1038/s41537-021-00158-z
27. Tandon, R, Lenderking, WR, Weiss, C, Shalhoub, H, Barbosa, CD, Chen, J, et al. The impact on functioning of second-generation antipsychotic medication side effects for patients with schizophrenia: a worldwide, cross-sectional, web-based survey. Ann General Psychiatry. (2020) 19:1–11. doi: 10.1186/s12991-020-00292-5
28. Chadda, RK, Ramshankar, P, Deb, KS, and Sood, M. Metabolic syndrome in schizophrenia: differences between antipsychotic-naïve and treated patients. J Pharmacol Pharmacother. (2013) 4:176–86. doi: 10.4103/0976-500X.114596
29. Yuen, JW, Kim, DD, Procyshyn, RM, Panenka, WJ, Honer, WG, and Barr, AM. A focused review of the metabolic side-effects of clozapine. Front Endocrinol. (2021) 12:609240. doi: 10.3389/fendo.2021.609240
30. Huang, PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. (2009) 2:231–7. doi: 10.1242/dmm.001180
31. Papanastasiou, E. The prevalence and mechanisms of metabolic syndrome in schizophrenia: a review. Therap Adv Psychopharmacol. (2013) 3:33–51. doi: 10.1177/2045125312464385
32. Hasnain, M, Fredrickson, SK, Vieweg, WVR, and Pandurangi, AK. Metabolic syndrome associated with schizophrenia and atypical antipsychotics. Curr Diab Rep. (2010) 10:209–16. doi: 10.1007/s11892-010-0112-8
33. Correll, CU, Solmi, M, Veronese, N, Bortolato, B, Rosson, S, Santonastaso, P, et al. Prevalence, incidence and mortality from cardiovascular disease in patients with pooled and specific severe mental illness: a large-scale meta-analysis of 3,211,768 patients and 113,383,368 controls. World Psychiatry. (2018) 17:120. doi: 10.1002/wps.20503
34. Calabrese, J, Al Khalili, Y, and Shaheen, K. Psychosis (nursing) In: StatPearls [Internet] Treasure Island (FL): StatPearls Publishing (2022) Available from: https://www.ncbi.nlm.nih.gov/books/NBK568796/ (Accessed May 1, 2023).
35. Taipale, H, Tanskanen, A, Mehtälä, J, Vattulainen, P, Correll, CU, and Tiihonen, J. 20-year follow-up study of physical morbidity and mortality in relationship to antipsychotic treatment in a nationwide cohort of 62,250 patients with schizophrenia (FIN20). World Psychiatry. (2020) 19:61–8. doi: 10.1002/wps.20699
36. Pillinger, T, McCutcheon, RA, Vano, L, Mizuno, Y, Arumuham, A, Hindley, G, et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: a systematic review and network meta-analysis. Lancet Psychiatry. (2020) 7:64–77. doi: 10.1016/S2215-0366(19)30416-X
37. Citrome, L. A review of aripiprazole in the treatment of patients with schizophrenia or bipolar I disorder. Neuropsychiatr Dis Treat. (2006) 2:427–43. doi: 10.2147/nedt.2006.2.4.427
38. San-Millán, I. The key role of mitochondrial function in health and disease. Antioxidants. (2023) 12:782. doi: 10.3390/antiox12040782
39. Bhatti, JS, Bhatti, GK, and Reddy, PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders—a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1066–77. doi: 10.1016/j.bbadis.2016.11.010
40. Bugger, H, and Dale Abel, E. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci. (2008) 114:195–210. doi: 10.1042/CS20070166
41. Gonzalez-Franquesa, A, and Patti, M-E. Insulin resistance and mitochondrial dysfunction. Mitochon Dynam Cardiovasc Med. (2017) 982:465–520. doi: 10.1007/978-3-319-55330-6_25
42. Scaini, G, Quevedo, J, Velligan, D, Roberts, DL, Raventos, H, and Walss-Bass, C. Second generation antipsychotic-induced mitochondrial alterations: implications for increased risk of metabolic syndrome in patients with schizophrenia. Eur Neuropsychopharmacol. (2018) 28:369–80. doi: 10.1016/j.euroneuro.2018.01.004
43. Newcomer, JW, Haupt, DW, Fucetola, R, Melson, AK, Schweiger, JA, Cooper, BP, et al. Abnormalities in glucose regulation during antipsychotic treatment of schizophrenia. Arch Gen Psychiatry. (2002) 59:337–45. doi: 10.1001/archpsyc.59.4.337
44. Schmidt, RH, Jokinen, JD, Massey, VL, Falkner, KC, Shi, X, Yin, X, et al. Olanzapine activates hepatic mammalian target of rapamycin: new mechanistic insight into metabolic dysregulation with atypical antipsychotic drugs. J Pharmacol Exp Ther. (2013) 347:126–35. doi: 10.1124/jpet.113.207621
45. Klingerman, CM, Stipanovic, ME, Bader, M, and Lynch, CJ. Second-generation antipsychotics cause a rapid switch to fat oxidation that is required for survival in C57BL/6J mice. Schizophr Bull. (2014) 40:327–40. doi: 10.1093/schbul/sbs196
46. Albaugh, VL, Vary, TC, Ilkayeva, O, Wenner, BR, Maresca, KP, Joyal, JL, et al. Atypical antipsychotics rapidly and inappropriately switch peripheral fuel utilization to lipids, impairing metabolic flexibility in rodents. Schizophr Bull. (2012) 38:153–66. doi: 10.1093/schbul/sbq053
47. Leverve, X., Batandier, C., and Fontaine, E. (2006). “Choosing the right substrate” in Sepsis: New insights, new therapies: Novartis Foundation symposium 280. Wiley Online Library.
48. Oh, K-J, Park, J, Lee, SY, Hwang, I, Kim, JB, Park, TS, et al. Atypical antipsychotic drugs perturb AMPK-dependent regulation of hepatic lipid metabolism. Am J Physiol Endocrinol Metabol. (2011) 300:E624–32. doi: 10.1152/ajpendo.00502.2010
49. He, M, Zhang, Q, Deng, C, Wang, H, Lian, J, and Huang, XF. Hypothalamic histamine H1 receptor-AMPK signaling time-dependently mediates olanzapine-induced hyperphagia and weight gain in female rats. Psychoneuroendocrinology. (2014) 42:153–64. doi: 10.1016/j.psyneuen.2014.01.018
50. Ikegami, M, Ikeda, H, Ishikawa, Y, Ohsawa, M, Ohashi, T, Kai, M, et al. Olanzapine induces glucose intolerance through the activation of AMPK in the mouse hypothalamus. Eur J Pharmacol. (2013) 718:376–82. doi: 10.1016/j.ejphar.2013.08.006
51. Kim, MK, Kim, SH, Yu, HS, Park, HG, Kang, UG, Ahn, YM, et al. The effect of clozapine on the AMPK-ACC-CPT1 pathway in the rat frontal cortex. Int J Neuropsychopharmacol. (2012) 15:907–17. doi: 10.1017/S1461145711000976
52. Okada, M, Fukuyama, K, and Motomura, E. Dose-dependent biphasic action of quetiapine on ampk signalling via 5-ht7 receptor: exploring pathophysiology of clinical and adverse effects of quetiapine. Int J Mol Sci. (2022) 23:9103. doi: 10.3390/ijms23169103
53. Long, YC, and Zierath, JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. (2006) 116:1776–83. doi: 10.1172/JCI29044
54. Carli, M, Kolachalam, S, Longoni, B, Pintaudi, A, Baldini, M, Aringhieri, S, et al. Atypical antipsychotics and metabolic syndrome: from molecular mechanisms to clinical differences. Pharmaceuticals. (2021) 14:238. doi: 10.3390/ph14030238
55. Stapel, B, Kotsiari, A, Scherr, M, Hilfiker-Kleiner, D, Bleich, S, Frieling, H, et al. Olanzapine and aripiprazole differentially affect glucose uptake and energy metabolism in human mononuclear blood cells. J Psychiatr Res. (2017) 88:18–27. doi: 10.1016/j.jpsychires.2016.12.012
56. Li, R, Zhu, W, Huang, P, Yang, Y, Luo, F, Dai, W, et al. Olanzapine leads to nonalcoholic fatty liver disease through the apolipoprotein A5 pathway. Biomed Pharmacother. (2021) 141:111803. doi: 10.1016/j.biopha.2021.111803
57. Walss-Bass, C, Weintraub, ST, Hatch, J, Mintz, J, and Chaudhuri, AR. Clozapine causes oxidation of proteins involved in energy metabolism: a possible mechanism for antipsychotic-induced metabolic alterations. Int J Neuropsychopharmacol. (2008) 11:1097–104. doi: 10.1017/S1461145708008882
58. Casademont, J, Garrabou, G, Miró, Ò, López, S, Pons, A, Bernardo, M, et al. Neuroleptic treatment effect on mitochondrial electron transport chain: peripheral blood mononuclear cells analysis in psychotic patients. J Clin Psychopharmacol. (2007) 27:284–8. doi: 10.1097/JCP.0b013e318054753e
59. Fatemi, SH, Reutiman, TJ, Folsom, TD, Bell, C, Nos, L, Fried, P, et al. Chronic olanzapine treatment causes differential expression of genes in frontal cortex of rats as revealed by DNA microarray technique. Neuropsychopharmacology. (2006) 31:1888–99. doi: 10.1038/sj.npp.1301002
60. Contreras-Shannon, V, Heart, DL, Paredes, RM, Navaira, E, Catano, G, Maffi, SK, et al. Clozapine-induced mitochondria alterations and inflammation in brain and insulin-responsive cells. PLoS One. (2013) 8:e59012. doi: 10.1371/journal.pone.0059012
61. Eftekhari, A, Azarmi, Y, Parvizpur, A, and Eghbal, MA. Involvement of oxidative stress and mitochondrial/lysosomal cross-talk in olanzapine cytotoxicity in freshly isolated rat hepatocytes. Xenobiotica. (2016) 46:369–78. doi: 10.3109/00498254.2015.1078522
62. Sipos, I, Tretter, L, and Adam-Vizi, V. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem. (2003) 84:112–8. doi: 10.1046/j.1471-4159.2003.01513.x
63. Guo, C, Sun, L, Chen, X, and Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. (2013) 8:2003–14. doi: 10.3969/j.issn.1673-5374.2013.21.009
64. Zorova, LD, Popkov, VA, Plotnikov, EY, Silachev, DN, Pevzner, IB, Jankauskas, SS, et al. Mitochondrial membrane potential. Anal Biochem. (2018) 552:50–9. doi: 10.1016/j.ab.2017.07.009
65. Cikánková, T, Fišar, Z, Bakhouche, Y, Ľupták, M, and Hroudová, J. In vitro effects of antipsychotics on mitochondrial respiration. Naunyn Schmiedeberg’s Arch Pharmacol. (2019) 392:1209–23. doi: 10.1007/s00210-019-01665-8
66. Burkhardt, C, Kelly, JP, Lim, YH, Filley, CM, and Parker, WD Jr. Neuroleptic medications inhibit complex I of the electron transport chain. Ann Neurol. (1993) 33:512–7. doi: 10.1002/ana.410330516
Keywords: antipsychotics, psychosis, mitochondria, energy metabolism, metabolic syndrome
Citation: Mortimer KRH, Katshu MZUH and Chakrabarti L (2023) Second-generation antipsychotics and metabolic syndrome: a role for mitochondria. Front. Psychiatry. 14:1257460. doi: 10.3389/fpsyt.2023.1257460
Edited by:
Anna Rita Atti, University of Bologna, ItalyReviewed by:
Magdalena Sowa-Kucma, University of Rzeszow, PolandCopyright © 2023 Mortimer, Katshu and Chakrabarti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Chakrabarti, TGlzYS5DaGFrcmFiYXJ0aUBub3R0aW5naGFtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.