 Stuti Chhabra
Stuti Chhabra Leonardo Nardi
Leonardo Nardi Petra Leukel
Petra Leukel Clemens J. Sommer
Clemens J. Sommer Michael J. Schmeisser
Michael J. Schmeisser
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Psychiatry , 08 March 2023
Sec. Molecular Psychiatry
Volume 14 - 2023 | https://doi.org/10.3389/fpsyt.2023.1110525
This article is part of the Research Topic Toward an Understanding Of Neurodevelopmental Disorders: Novel Insights From Molecular, Behavioural and Neuroinflammatory Mechanisms View all 5 articles
Autism spectrum disorder (ASD) comprises a wide range of neurodevelopmental phenotypes united by impaired social interaction and repetitive behavior. Environmental and genetic factors are associated with the pathogenesis of ASD, while other cases are classified as idiopathic. The dopaminergic system has a profound impact in the modulation of motor and reward-motivated behaviors, and defects in dopaminergic circuits are implicated in ASD. In our study, we compare three well-established mouse models of ASD, one idiopathic, the BTBR strain, and two syndromic, Fmr1 and Shank3 mutants. In these models, and in humans with ASD, alterations in dopaminergic metabolism and neurotransmission were highlighted. Still, accurate knowledge about the distribution of dopamine receptor densities in the basal ganglia is lacking. Using receptor autoradiography, we describe the neuroanatomical distribution of D1 and D2 receptors in dorsal and ventral striatum at late infancy and adulthood in the above-mentioned models. We show that D1 receptor binding density is different among the models irrespective of the region. A significant convergence in increased D2 receptor binding density in the ventral striatum at adulthood becomes apparent in BTBR and Shank3 lines, and a similar trend was observed in the Fmr1 line. Altogether, our results confirm the involvement of the dopaminergic system, showing defined alterations in dopamine receptor binding density in three well-established ASD lines, which may provide a plausible explanation to some of the prevalent traits of ASD. Moreover, our study provides a neuroanatomical framework to explain the utilization of D2-acting drugs such as Risperidone and Aripiprazole in ASD.
Autism spectrum disorder (ASD) is a range of neurodevelopmental conditions defined by impairment in social interaction and repetitive behavior. These core symptoms are accompanied by other comorbidities such as cognitive disabilities, anxiety, sleep disturbances, hyperactivity, and motor impairments. The broad span of ASD symptoms can affect each individual to a varying degree of severity (1). Global prevalence of ASD has risen in recent years, with 1 in 100 children being affected across all socioeconomic, racial, and ethnic groups (2). However, etiology of ASD is still poorly understood, with no single underlying cause, rather a complex interplay between genetic and environmental factors that alone or in combination are implicated in development of the disorder (1, 3). Heterogeneous presentation of the disease reveals that different brain regions and discrete perturbances in specific neural circuits contribute to the distinct features of ASD symptomatology (4).
The dopaminergic system has been characterized in controlling motor functions and reward-motivated behavior. Dopaminergic neurons are found mainly in the substantia nigra, pars compacta, and in the ventral tegmental area (VTA). The projections arising in the substantia nigra, pars compacta target mainly the dorsal striatum (DS), forming the nigrostriatal pathway, which is relevant in the control of voluntary movement (5). The axons from neurons, which reside in the VTA, project mainly to the Ventral striatum (VS) and prefrontal cortex, making up the mesocorticolimbic pathway, involved in reward, motivation and emotion (6). Dopamine (DA) receptors belong to the family of G protein–coupled receptors. According to pharmacological and binding studies, DA receptors are grouped into D1-like (D1 and D5) and D2-like (D2, D3, and D4) families, depending upon their ability to regulate positively or negatively intracellular concentration of cyclic adenosine monophosphate, respectively (7).
In recent years, a possible role of a dysfunctional dopaminergic system in ASD has gained a lot of attention. Multiple studies have highlighted the association between alterations in the dopaminergic system and ASD (8–10). However, the impact of these changes in contributing to ASD pathophysiology still needs to be further investigated. Additional evidence in support of this view comes from the fact that the only FDA approved drugs for ASD are the antipsychotics risperidone and aripiprazole, which act as D2 receptor antagonist and partial agonist, respectively (11, 12). Neuroanatomical alterations of the basal ganglia have been discovered in individuals with ASD. Significant changes in volume and neuronal density were reported in the DS (13). Also, a significantly smaller bilateral VS and larger cerebral ventricles were identified in individuals with a later diagnosis of ASD (14). In mice, it has also been observed that chemogenetic inhibition of dopaminergic neurons in the VTA leads to reduced exploration of novel social stimuli (15). As a result of the abundant evidence available, a DA hypothesis of ASD has been advanced. It has been postulated that variations in the nigrostriatal pathway may lead to motor impairments and stereotyped behaviors in subjects with ASD. Also, any alterations in the mesocorticolimbic pathway could lead to social deficits, reduced motivation, and changes in reward-related behavior (16–18).
Various mouse models recapitulating ASD-like phenotypes are widely utilized as preclinical tools to understand the causal role of genetic and environmental factors. Approximately 75% of ASD is idiopathic. Remaining cases show a specific genetic cause, such as fragile X syndrome, tuberous sclerosis and Phelan-McDermid syndrome (19). For this study, we chose three well-established mouse strains that recapitulate ASD features. Black and tan brachyury (BTBR) mice are an inbred strain characterized by the lack of corpus callosum and a severe reduction of hippocampal commissure (20). This line provides face validity for the majority of core symptoms of ASD, making it a suitable model for idiopathic ASD (21). Fragile X mental retardation 1 (Fmr1) knockout mice are the most investigated model for the human fragile X syndrome, the most common cause for intellectual disability and ASD in human. This gene codes for a mRNA binding protein (FMRP), whose activity is essential for proper synaptic plasticity and architecture (22). SH3 and multiple ankyrin repeat domain 3 (SHANK3) is a postsynaptic scaffold protein, which interacts with several postsynaptic receptors, signaling molecules, and cytoskeletal proteins (23). Mutations of SHANK3 are associated with the Phelan-McDermid syndrome and ASD (24). It has been inferred from previous studies that these three mouse lines display altered D1 and D2 receptor mediated neurotransmission and striatal pathway disruption, thus making the selected mouse strains a suitable choice for this study (25–28).
In an attempt to unravel the pathomechanistic underpinnings of a dysfunctional dopaminergic system in ASD, we investigated the alteration of DA receptor densities in the three above-mentioned ASD mouse lines in an age and region-dependent fashion.
BTBR (BTBR T+ Itprtf/J, stock #002282), C57BL6/J (stock #000664), Fmr1 (B6.129P2-Fmr1tm1Cgr/J, stock #003025), and Shank3b (B6.129-Shank3tm2Gfng/J, stock #017688) mice were purchased from Jackson laboratories and housed in a pathogen-free facility with 12 h light/dark cycle, food, and water available ad libitum. Fmr1−/y (Fmr1 KO) mice were generated by (29). Shank3b (B6.129-Shank3tm2Gfng/J) mice were generated by replacing exons 13–16 with a neomycin resistance cassette (26). BTBR, Fmr1−/y (Fmr1 KO), and Shank3b−/− (Shank3b KO) were used as test animals. C57BL6/J mice were used as controls for BTBR mice, wildtype littermates for Fmr1 KO and Shank3b KO. Breeding was approved by the local authorities. Only male mice were used in the experiments, seen the higher prevalence of ASD in male individuals (2). The number of animals tested in each experiment is reported in every figure legend. All the experiments were performed according to guidelines of the central animal facility institution (TARC, Mainz University Medical Center) representing those of the German Animal Welfare Act and the European Directive 2010/63/EU for the protection of animals used for scientific purposes. Reporting was carried out according to the ARRIVE guidelines for reporting in vivo experiments.
After decapitation, brains were rapidly removed, frozen in isopentane and stored at −80°C until cutting. The brains were serially cut (20 μm thickness) in the coronal plane with a cryostat microtome (Leica, Germany). Totally, 4 slices were collected between the bregma points 1.53 mm 0.97 mm according to Paxinos and Franklin (30), where both DS and VS can be properly visualized. The first two slices were used for histological staining, the successive two were incubated with the [H3]-labeled ligands for D1 and D2 receptors. The slices were stored at −80°C until further histological and autoradiographic experiments.
Hematoxylin–eosin staining was performed to help spatially localize the DS and VS on the autoradiograms. Briefly, the frozen brain slices were acclimatized at room temperature for 10 min. The slices were then incubated in acetone for 5 min, briefly air dried, and dipped in hematoxylin (Thermo Fisher) for 1 min. After washing in running water for 10 min, the slices were put for 10 s in Eosin Y (Thermo Fisher). Then, the slices were dehydrated in increasing ethanol concentrations (96 and 100%) each for 2 min. Finally, the slices were placed for 3 min in xylol, and cover slipped with Cytoseal XYL (Thermo Fisher). Pictures were scanned at 4× magnification with a Leica microscope (Leica, Germany), digitized, and transferred to the MCID program.
The receptor binding density for D1 and D2 receptors was adapted from the protocols described in Sommer et al. (31) and Behuet et al.’s study (32). The tritiated ligands [3H]-SCH23390 and [3H]-Raclopride were purchased from PerkinElmer (Germany). SCH23390 is a potent D1 and D5 receptor antagonist. Raclopride is a D2 and D3 selective receptor antagonist. Both ligands bind to the receptors localized on the cell surface, without penetrating inside the cell membrane. In the first step, the pre-incubation, endogenous ligands were washed off. In the following main incubation, the tritiated ligands were incubated both in the presence of a competitor, in order to determine the unspecific binding, and without it, in order to assess the total binding. Finally, the slices were rinsed. The slices incubated with [3H]-Raclopride were additionally dried with a cold air stream. A detailed description of the protocols used is reported in Table 1.
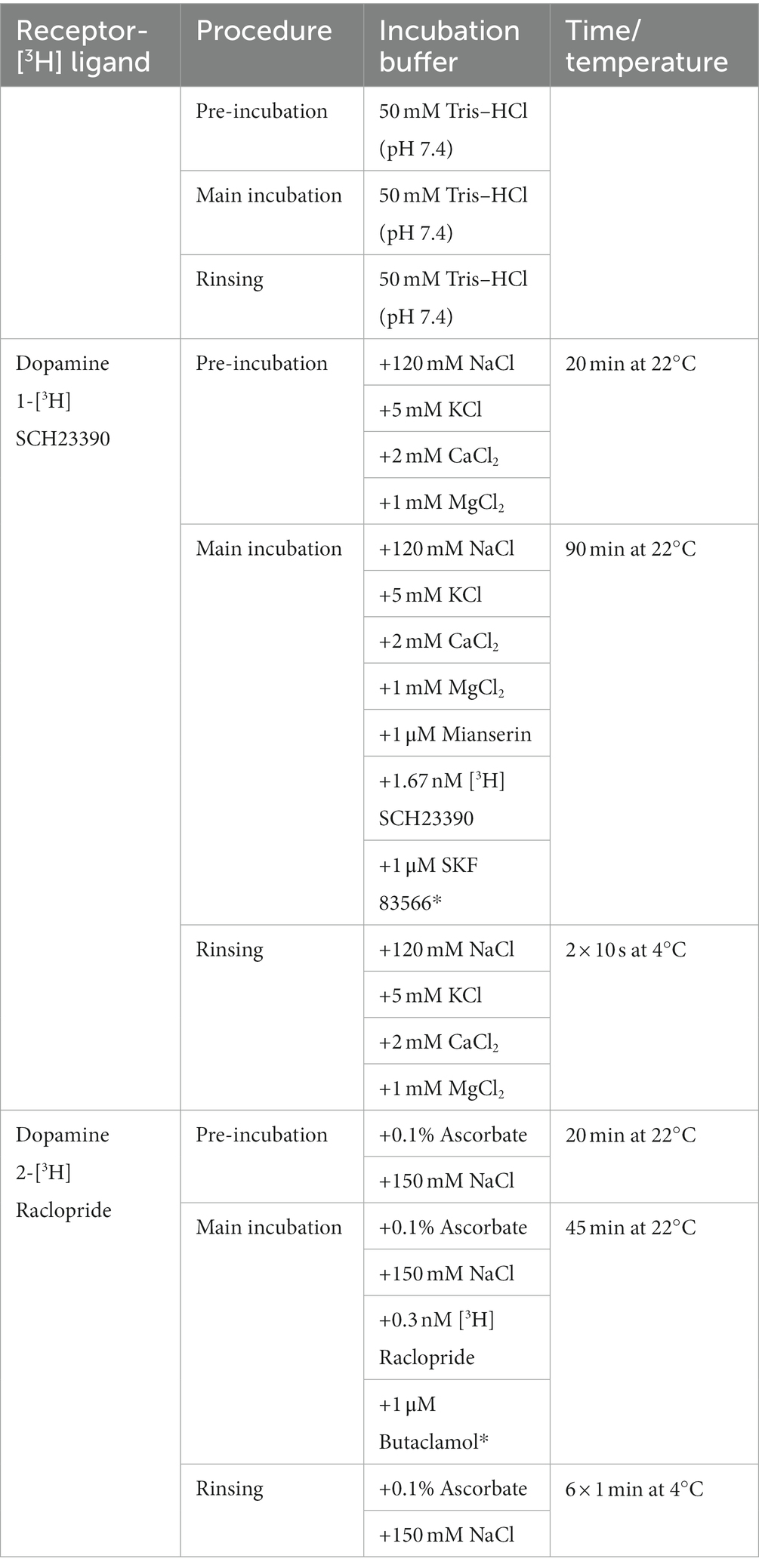
Table 1. Receptor binding protocols for the dopaminergic [3H] ligands with competitors (noted with *) and incubation conditions.
Image acquisition and analysis were performed as described in Mammele et al.’s study (33). [3H] plastic standards (Microscales®; Amersham, Freiburg, Germany) were exposed together with the tritium-labeled sections to a [3H]-sensitive film (Bio Max MR-1 Autoradiography Film, KODAKTM) for 7 ([3H]-SCH23390) and 10 weeks ([3H]-Raclopride). The autoradiograms and the standards were scanned in equal lighting conditions with the digital CoolSNAP camera (Roper Scientific, Photometrics CoolSNAPTM cf., Ottobrunn/Munich Germany) and digitized with the MCID image analysis system (Imaging Research Inc., St. Catharines, Ontario, Canada). The standards were used to calculate the relationship between the gray values of the autoradiograms and the concentration of radioactivity. Total binding was calculated on the autoradiograms on both hemispheres in DS and VS after tracing the boundary of the region of interest on the hematoxylin–eosin staining (Figure 1). The unspecific binding was consistently slightly above background signal or completely lacking. The value was then subtracted from the total binding.
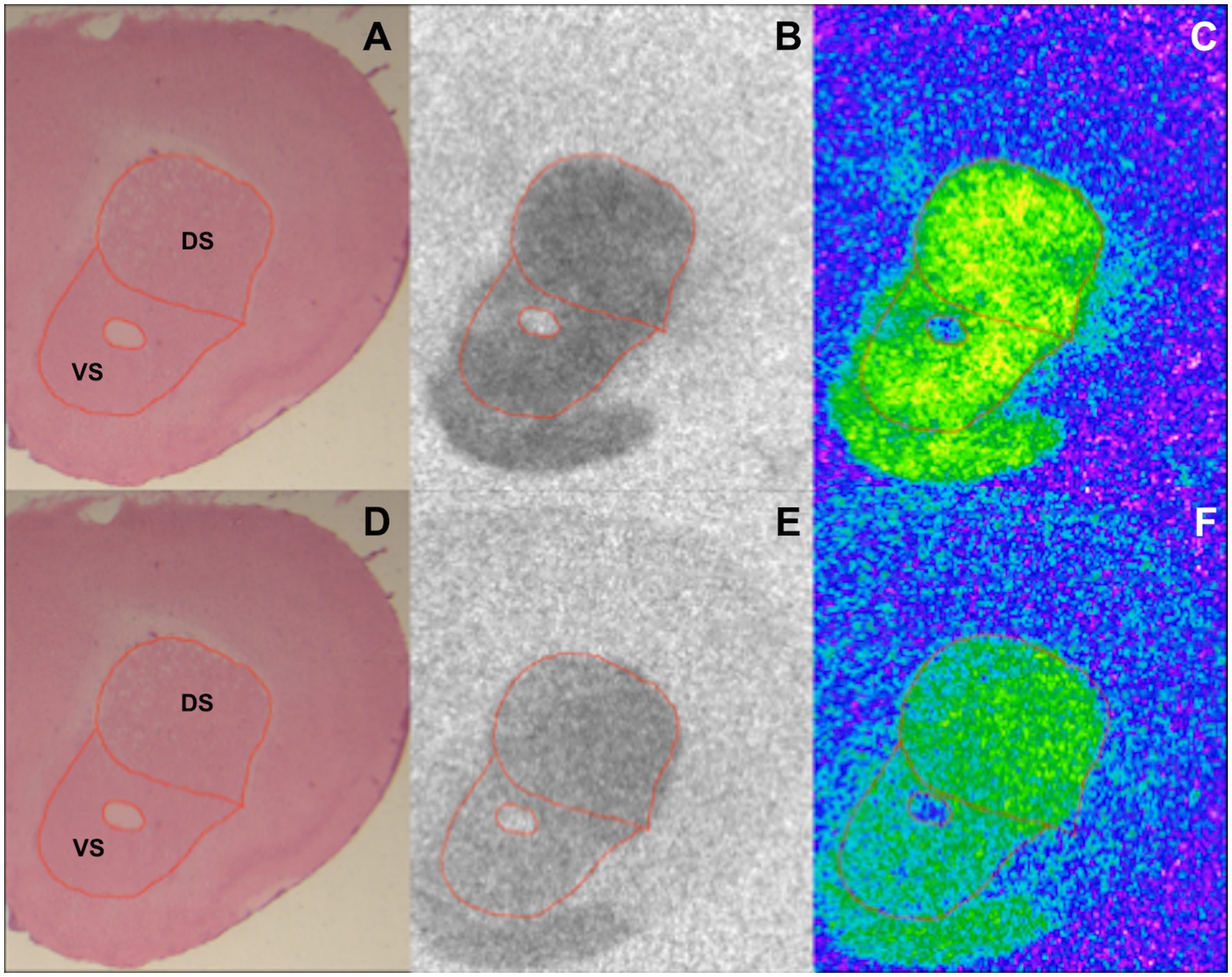
Figure 1. Exemplary section from a BTBR 12-week-old mouse showing DS and VS in an HE stained section (A,D), autoradiographic image of binding to D1 (B) and D2 receptors (E), and the corresponding color-coded picture of the same autoradiogram [(C,F) respectively].
Statistical analysis was carried out with Prism (GraphPad, Version 9). Student’s t-test was performed. p < 0.05 was taken as threshold for statistical significance and results are shown as the mean ± SEM. The autoradiography experiments were performed in a blinded manner. Data were expressed as percentage of controls.
HE-stained sections were used to trace DS and VS (Figures 1A,D). The boundaries of the regions of interest were then overlapped on the autoradiograms for D1 and D2 receptors prior to analysis (Figures 1B,C,E,F, respectively). The analysis of the binding density to D1 and D2 receptors revealed various alterations in DS and VS of both 4- and 12-week-old BTBR, Fmr1 KO, and Shank3b KO mice.
In 4-week-old BTBR mice, D1 receptor binding density was unaltered in both DS (p = 0.23) and VS (p = 0.45) (Figure 2A). At the same time point, no difference could be found in D2 receptor binding density in DS (p = 0.28), whereas it was increased in VS (p = 0.03) (Figure 3A). A similar pattern was observed also in 12-week-old BTBR mice. Binding density to D1 receptors was not changed both in DS (p = 0.22) and in VS (p = 0.8) (Figure 2B); D2 binding density remained unchanged in DS (p = 0.25) but was increased in VS (p = 0.014) (Figure 3B). Hence, VS showed at both time points increased D2 receptor binding density, while binding density to D1 receptors was not altered at either time point.
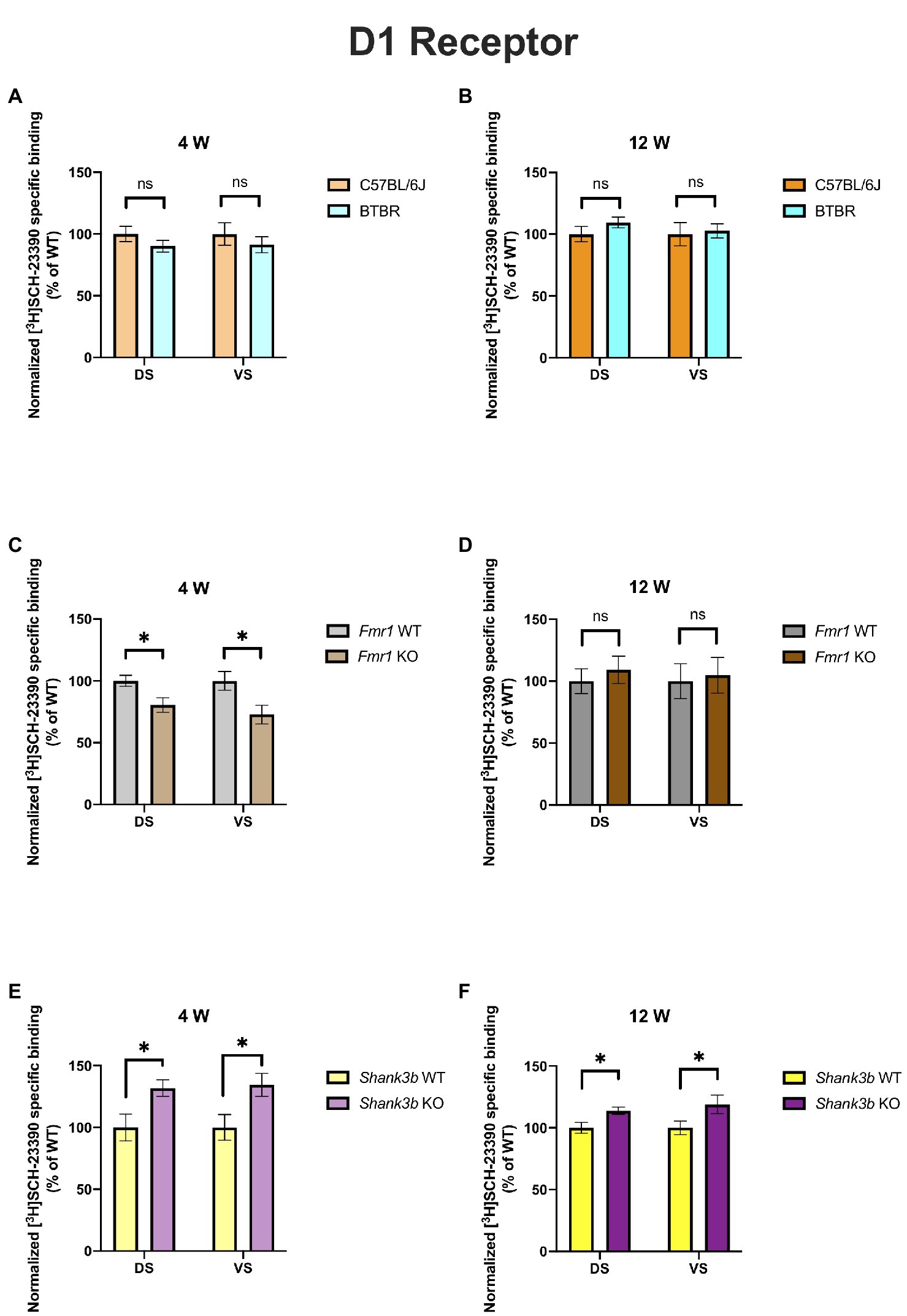
Figure 2. Bar charts representing mean and SEM of the receptor density for D1 receptors. DS: C57BL6/J (n = 8), BTBR (n = 8) and VS: C57BL6/J (n = 8), BTBR (n = 8) at 4 weeks of age (A); DS: C57BL6/J (n = 10), BTBR (n = 11) and VS: C57BL6/J (n = 10), BTBR (n = 11) at 12 weeks of age (B); DS: Fmr1 WT (n = 9), Fmr1 KO mice (n = 8) and VS: Fmr1 WT (n = 9) and Fmr1 KO mice (n = 7) at 4 weeks of age (C); DS: Fmr1 WT (n = 7), Fmr1 KO mice (n = 8) and VS: Fmr1 WT (n = 7) and Fmr1 KO mice (n = 8) at 12 weeks of age (D); DS: Shank3b WT (n = 5), Shank3b KO mice (n = 6) and VS: Shank3b WT (n = 5), Shank3b KO mice (n = 6) at 4 weeks of age (E); DS: Shank3b WT (n = 14), Shank3b KO mice (n = 12) and VS: Shank3b WT (n = 14) and Shank3b KO mice (n = 12) at 12 weeks of age (F). Significant differences are indicated with an asterisk (*p < 0.05). Changes are represented as percentage of the mean of C57BL6/J, Fmr1 WT, Shank3b WT mice, respectively.
Fmr1 KO mice showed a reduced receptor binding density to D1 receptors both in DS (p = 0.01) and in VS (p = 0.04) (Figure 2C) but no changes for D2 receptor binding density (DS: p = 0.58; VS p = 0.72) at 4 weeks (Figure 3C). At 12 weeks no alteration was found in the binding density to D1 receptors (DS: p = 0.56; VS: p = 0.82) (Figure 2D). The binding profile to D2 receptors was instead significantly increased in DS (p = 0.02) and showed a strong tendency in the same direction also in VS (p = 0.08) (Figure 3D). For Fmr1 KO mice, it is hence possible to observe both in DS and VS at 4 weeks reduction of D1 binding density. Interestingly, this change was not observed at 12 weeks, implicating a possible role of developmental compensatory changes. Interestingly, the D2 binding density had an opposite dynamic in DS, being unmodified at 4 weeks, but increased at 12 weeks. The changes described at 12 weeks hence mirrors what we observed in the BTBR line.
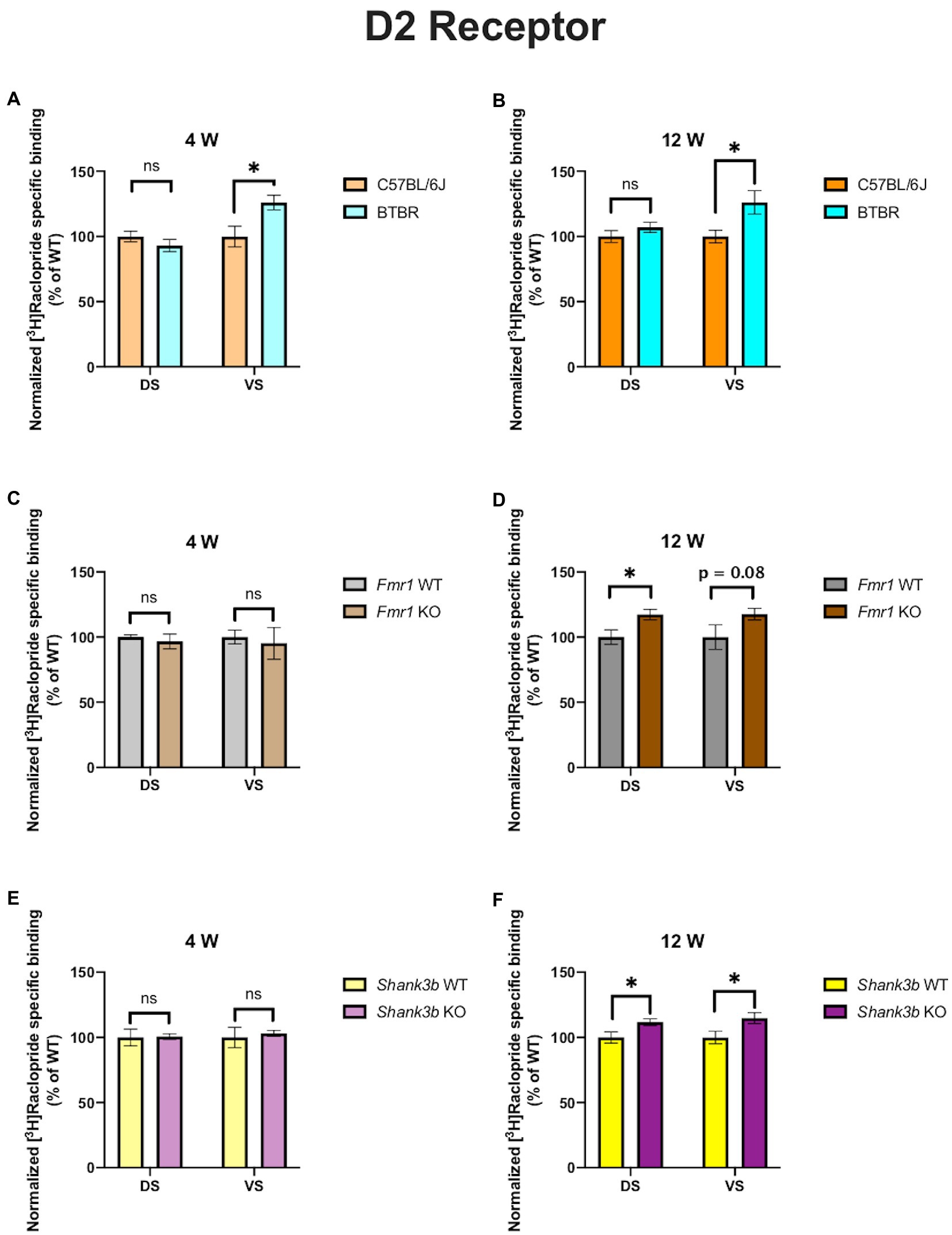
Figure 3. Bar charts representing mean and SEM of the receptor density for D2 receptors. DS: C57BL6/J (n = 9), BTBR (n = 8) and VS: C57BL6/J (n = 9), BTBR (n = 6) at 4 weeks of age (A); DS: C57BL6/J (n = 10), BTBR (n = 9) and VS: C57BL6/J (n = 10), BTBR (n = 7) at 12 weeks of age (B); DS: Fmr1 WT (n = 8), Fmr1 KO mice (n = 8) and VS: Fmr1 WT (n = 8) and Fmr1 KO mice (n = 8) at 4 weeks of age (C); DS: Fmr1 WT (n = 5), Fmr1 KO mice (n = 8) and VS: Fmr1 WT (n = 5) and Fmr1 KO mice (n = 8) at 12 weeks of age (D); DS: Shank3b WT (n = 6), Shank3b KO mice (n = 6) and VS: Shank3b WT (n = 6), Shank3b KO mice (n = 6) at 4 weeks of age (E); DS: Shank3b WT (n = 12), Shank3b KO mice (n = 12) and VS: Shank3b WT (n = 12) and Shank3b KO mice (n = 11) at 12 weeks of age (F). Significant differences are indicated with an asterisk (*p < 0.05). Changes are represented as percentage of the mean of C57BL6/J, Fmr1 WT, Shank3b WT mice, respectively.
Shank3b KO showed an increase of D1 receptor binding density in both DS and VS at 4 (DS: p = 0.02; VS: p = 0.03) (Figure 2E) and 12 weeks (DS: p = 0.01; VS: p = 0.04) (Figure 2F). With respect to D2 receptors, no change was found at 4 weeks (DS: p = 0.92; VS: p = 0.7) (Figure 3E), whereas an increased binding density was registered at 12 weeks in both regions (DS: p = 0.02; VS: p = 0.03) (Figure 3F). Taken together, we show that diverse ASD mouse models have a convergent increase in the D2 binding density in the VS at adulthood, implicating that the mesolimbic circuitry might be a common denominator in ASD pathophysiology. This brain region could therefore be acknowledged as a point of commonality among various ASD models in the context of dopamine signaling, thus making it an interesting candidate for further exploration. We also observed increased D2 binding in the DS of Fmr1 KO and Shank3b KO at adulthood. D1 binding density, on the contrary, showed no convergent changes among mouse lines at both time points, but an individual phenotypic profile.
To our knowledge, we provide the first thorough neuroanatomical characterization of D1 and D2 receptor distribution by the means of receptor autoradiography in the DS and VS of several ASD mouse models at different developmental stages. For our analysis, we chose two time points that correspond to late infancy (4 weeks) and adulthood (12 weeks), a time frame in which several synaptic proteins are highly modulated (34). Although ASD can be reliably diagnosed by 3 years of age (35), ASD is a lifelong condition that deserves to be investigated with a dynamic outlook. It is hence fundamental to study ASD brains at several developmental time points, in order to optimize diagnostic and therapeutical interventions. Developmental changes of D1- and D2-family receptors have been demonstrated in the human brain (36). Recent PET imaging studies in humans with ASD investigated the D1 (37) and D2 binding (38) in DS. These two studies can only within limits be compared with our observations because (a) only adult subjects were considered; (b) no distinction between idiopathic and syndromic forms of ASD was made; (c) VS was not analyzed as an entity per se, even though it has already been considered in other human studies as potential therapeutical target in case of repetitive behavior (39, 40); and d) functional assays with a focus on differences in responses to tasks (38) or correlations to behavioral data (37) were performed.
The DA hypothesis of ASD is supported by alterations in the dopaminergic metabolism and transmission in several different mouse models such as Df(h22q11)/+ mice (model for the human 22q11.2 microdeletion syndrome) (41), Cntnap4 KO (KO model of a protein highly associated to ASD and schizophrenia) (42), Ube3a KO (model for Angelman syndrome) (43), Chd8+/− mice (model for the CHD8-related syndrome) (44), and VPA (model for the prenatal exposure to valproic acid) (45).
The models investigated in our study have also been analyzed by other groups regarding alterations in DA metabolism and neurotransmission. BTBR mice have no altered mRNA expression of D1 and D2 receptors in DS but reduced D2 postsynaptic signaling at adulthood (27). We also found no alterations in the receptor binding density for D1 in the DS, whereas D2 binding density was incremented (Figures 2A,B and 3A,B). Pharmacological interventions affecting DA signaling have proved effective in the BTBR line. The administration of p-cresol, a metabolite found elevated in children with ASD, leads to increased DA turnover in DS and VS of BTBR mice. Moreover, it aggravates repetitive behavior and social interaction deficits (46). Multi-targeting ligands such as ST-713 and ST-2223, both histamine H3- and DA D2-receptor antagonists, proved effective in reducing repetitive behavior and social deficits (47, 48). This evidence points again at an involvement of the dopaminergic systemic in BTBR mice, albeit the interplay with other neurotransmitter systems might complicate the general understanding. Moreover, it was recently shown that levels of tyrosine hydroxylase, the enzyme which catalyzes the first reaction in the biosynthesis of DA, are reduced in the DS and VTA but not in the VS of BTBR mice. Intranasal injection of DA could restore the levels of tyrosine hydroxylase and restore the observed social approach deficit. Interestingly, in the same study, Fmr1 KO mice were also investigated. Contrary to the BTBR mice, no changes were found in tyrosine hydroxylase levels neither in the DS nor in the VS of Fmr1 KO animals, despite an improvement of social preference upon administration of intranasal DA (25). This might seem to point at the fact, that different ASD models might have profoundly divergent molecular etiologies. In our study too, the receptor binding profiles to D1 receptor show no convergency in the BTBR and Fmr1 models (Figures 2A,C). Nonetheless, at 12 weeks, we observed increased D2 receptor binding density in the VS of BTBR (Figure 3B), and Shank3b KO mice (Figure 3F). This is an interesting convergency concerning both an idiopathic and a genetic mouse model of ASD, which deserves to be more profoundly explored in future studies. A similar tendency was also observed in the Fmr1 KO mice (Figure 3D).
Fragile X mental retardation protein, the RNA-binding protein encoded by the gene FMR1, has been proven as a crucial mediator of the integration between the D1- and D2-signaling and glutamatergic neurotransmission (49). In striatal cultures from Fmr1 KO mice, reduced coupling of the D1 receptor with the Gs protein was detected upon stimulation with a D1 agonist. Moreover, an increased phosphorylation at serine sites was also registered (50). We also found alterations in the D1 binding density both in DS and VS at 4 but not 12 weeks in the Fmr1 KO mice (Figures 2C,D). Such observations, however, pinpoint the fact that mapping the receptor binding density or the quantity of the receptor itself might not be enough to comprehend in depth the entity of the alterations. In a later study, DA release was measured in striatal slices at different time points. At 12 and 15 weeks, but not at 10 weeks, a significant decrease in the DA release was observed (51). In the Fmr1 KO model hence, an increased D1 receptor phosphorylation is accompanied by a time-dependent fashion reduction of the DA release. A subsequent study confirmed reduced protein kinase A as a consequence of the reduced D1 signaling (52). Interestingly, protein kinase A is part of the cyclic adenosine monophosphate signaling, on which both D1 and D2 receptor families converge, either stimulating or inhibiting adenylyl cyclase (7). Furthermore, ultrastructural and electrophysiological alterations were found in the VS of Fmr1 KO mice (53). We detected at 4 weeks a reduction of the D1 receptor binding both in DS and in VS. At 12 weeks of age, this was not observed (Figures 2C,D).
Although Shank3 mutant mice are among the most deeply investigated monogenic models for ASD, studies regarding dopamine transmission remain unclear. This is even more surprising considering that the SHANK3 protein is enriched in the DS (54) and that available evidence in human patients with Phelan-McDermid syndrome points at major alterations in the DS (55, 56). Silencing SHANK3 in the VTA leads to reduced dopaminergic activity and impaired social preference, which can be reversed by modulation of glutamatergic transmission. Effects on the downstream targets of the VTA such as VS or DS were not investigated though (57). Deletion of SHANK3 in the DS using Dlx5/6 and Drd2-Cre mice leads to increased perseverative explorative behavior. Moreover, deletion of SHANK3 in neurons expressing D1 or D2 leads to increased excitability in the DS (58). Altered glutamatergic transmission was however highlighted in the Shank3b model (59, 60). Although these two studies did not directly approach dopaminergic transmission, inhibition of protein kinase A was able to ameliorate the behavioral deficits of the Shank3b KO mice. Morphological and electrophysiological alterations of D2-expressing medium spiny neurons were also reported in 6–8 weeks old Shank3b KO mice (28). We also found increased binding to the D2 receptor both in DS and in VS at 12, but not 4 weeks of age (Figures 3E,F). In the same study, moreover, it was highlighted that voltage-gated calcium channels Cav1.3 are impaired in D2-expressing medium spiny neurons. As previously demonstrated (61, 62), SHANK3 mediates the regulation of D2 receptors on the Cav1.3 ones which, on turn, enable glutamatergic signaling. A recent in vitro study on brains from individuals with ASD also highlighted increased D2 mRNA in the caudate and putamen but unchanged D2 receptor binding (63). Interestingly, we found in the DS of Fmr1 KO and Shank3b KO increased D2 binding density at adulthood (Figures 3D,F). The differences observed between that study and ours might be due to the different [3H] ligands used and to different analytical approaches. However, other mouse lines associated to ASD showed alterations in the D2 signaling. The mouse model of 16p11.2 deletion syndrome displays elevated number of D2 expressing neurons both in DS and VS (64). In the above-mentioned Cntnap4 KO model, administration of haloperidol, a D2-antagonist, led to reduced repetitive behavior (42).
In conclusion, several lines of evidence show that the balance between D1 and D2 activation in the DS and VS is warranted (18). A disbalance toward higher activation of D1 signaling might lead, for example, to enhance stereotyped behavior (65, 66). Its modulation, on the opposite, either through optogenetics or pharmacologically, can reduce the repetitiveness (67–69). Trying to undermine the role of dopaminergic system in ASD is even more difficult considering that DA receptors D1 and D2 might form in DS and VS (and also in the hippocampus, cingulate cortex, and frontal cortex) heteromers with other G-coupled receptors such as metabotropic glutamate receptors, oxytocine-, serotonin-, and cannabinoid-receptors (70). Notably, both Aripiprazole and Risperidone, the only drugs approved so far from the FDA for ASD, act, even if with different mechanisms, pleiotropically on D2 and serotonin receptors. Both drugs were reported as effective against irritability and stereotypical behavior in idiopathic autism (71–73) and also proved effective in patients with Phelan-McDermid syndrome (74, 75) and Fragile-X syndrome (76, 77). As our results highlight, we found a significant increase of D2 receptor density in the VS at adulthood in the BTBR and Shank3b lines. A similar tendency was also observed in the Fmr1 line. Our results hence appear to corroborate in a translational fashion, on the neuroanatomical level, evidence derived from pharmacological studies. Future experiments should be designed in order to tackle the intertwined molecular interplay between dopaminergic and other neurotransmitter systems. Targeted pharmacological intervention aiming at restoring the intracellular molecular cascades might prove effective, but further in-vivo investigations are needed.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethical review and approval was not required for the animal study because organ removal from mice for scientific purpose (the brain in this study) does not require approval by an ethics committee in Germany.
SC, LN, PL, CJS, and MJS planned the autoradiographic experiments. SC, LN, and PL conducted the autoradiographic experiments and analyzed the data. SC and LN drafted the manuscript. PL, CJS, and MJS critically revised and edited the manuscript. All authors contributed to the article and approved the submitted version.
LN was supported by an internal grant of the University Medical Center, Mainz (Stufe I), MJS by the German Research Foundation (DFG, Collaborative Research Center 1080, Project B10) and the Werner Reichenberger Foundation.
The authors would like to thank Magdeleine Herkt, Christine Oswald and Alexander Wenzel for supreme technical assistance and Dilja Krueger-Burg for sparkling scientific discussion. We are particularly grateful to Martin Michel for helping with statistics and to Guilherme Horta for his guidance on mouse behavior.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
[3H], tritium; ASD, autism spectrum disorder; BTBR, black and tan brachyury; DA, dopamine; DS, dorsal striatum; FMR1, fragile X mental retardation 1; KO, knockout; SEM, standard error of the mean; SHANK, SH3 and multiple ankyrin repeat domains protein 3; VS, ventral striatum; VTA, ventral tegmental area; WT, wildtype.
1. Lord, C, Brugha, TS, Charman, T, Cusack, J, Dumas, G, Frazier, T, et al. Autism spectrum disorder. Nat Rev Dis Primers. (2020) 6:5. doi: 10.1038/s41572-019-0138-4
2. Zeidan, J, Fombonne, E, Scorah, J, Ibrahim, A, Durkin, MS, Saxena, S, et al. Global prevalence of autism: a systematic review update. Autism Res. (2022) 15:778–90. doi: 10.1002/aur.2696
3. Won, H, Mah, W, and Kim, E. Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front Mol Neurosci. (2013) 6:19–9. doi: 10.3389/fnmol.2013.00019
4. Masi, A, DeMayo, MM, Glozier, N, and Guastella, AJ. An overview of autism Spectrum disorder, heterogeneity and treatment options. Neurosci Bull. (2017) 33:183–93. doi: 10.1007/s12264-017-0100-y
5. Wise, RA. Dopamine, learning and motivation. Nat Rev Neurosci. (2004) 5:483–94. doi: 10.1038/nrn1406
6. Arias-Carrion, O, Stamelou, M, Murillo-Rodriguez, E, Menendez-Gonzalez, M, and Poppel, E. Dopaminergic reward system: a short integrative review. Int Arch Med. (2010) 3:24. doi: 10.1186/1755-7682-3-24
7. Missale, C, Nash, SR, Robinson, SW, Jaber, M, and Caron, MG. Dopamine receptors: from structure to function. Physiol Rev. (1998) 78:189–225. doi: 10.1152/physrev.1998.78.1.189
8. Hamilton, PJ, Campbell, NG, Sharma, S, Erreger, K, Herborg Hansen, F, Saunders, C, et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry. (2013) 18:1315–23. doi: 10.1038/mp.2013.102
9. Mariggiò, MA, Palumbi, R, Vinella, A, Laterza, R, Petruzzelli, MG, Peschechera, A, et al. DRD1 and DRD2 receptor polymorphisms: genetic neuromodulation of the dopaminergic system as a risk factor for ASD, ADHD and ASD/ADHD overlap. Front Neurosci. (2021) 15:705890. doi: 10.3389/fnins.2021.705890
10. Padmakumar, M, Van Raes, E, Van Geet, C, and Freson, K. Blood platelet research in autism spectrum disorders: in search of biomarkers. Res Pract Thromb Haemost. (2019) 3:566–77. doi: 10.1002/rth2.12239
11. Eissa, N, Al-Houqani, M, Sadeq, A, Ojha, SK, Sasse, A, and Sadek, B. Current enlightenment about etiology and pharmacological treatment of autism Spectrum disorder. Front Neurosci. (2018) 12:304. doi: 10.3389/fnins.2018.00304
12. Lamy, M, Pedapati, EV, Dominick, KL, Wink, LK, and Erickson, CA. Recent advances in the pharmacological Management of Behavioral Disturbances Associated with autism Spectrum disorder in children and adolescents. Paediatr Drugs. (2020) 22:473–83. doi: 10.1007/s40272-020-00408-0
13. Wegiel, J, Flory, M, Kuchna, I, Nowicki, K, Ma, SY, Imaki, H, et al. Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol Commun. (2014) 2:141. doi: 10.1186/s40478-014-0141-7
14. Shiohama, T, Ortug, A, Warren, JLA, Valli, B, Levman, J, Faja, SK, et al. Small nucleus Accumbens and large cerebral ventricles in infants and toddlers prior to receiving diagnoses of autism Spectrum disorder. Cereb Cortex. (2022) 32:1200–11. doi: 10.1093/cercor/bhab283
15. Bariselli, S, Hornberg, H, Prevost-Solie, C, Musardo, S, Hatstatt-Burkle, L, Scheiffele, P, et al. Role of VTA dopamine neurons and neuroligin 3 in sociability traits related to nonfamiliar conspecific interaction. Nat Commun. (2018) 9:3173. doi: 10.1038/s41467-018-05382-3
16. Kosillo, P, and Bateup, HS. Dopaminergic dysregulation in syndromic autism Spectrum disorders: insights from genetic mouse models. Front Neural Circuits. (2021) 15:700968. doi: 10.3389/fncir.2021.700968
17. Paval, D. A dopamine hypothesis of autism Spectrum disorder. Dev Neurosci. (2017) 39:355–60. doi: 10.1159/000478725
18. Paval, D, and Miclutia, IV. The dopamine hypothesis of autism Spectrum disorder revisited: current status and future prospects. Dev Neurosci. (2021) 43:73–83. doi: 10.1159/000515751
19. Fernandez, BA, and Scherer, SW. Syndromic autism spectrum disorders: moving from a clinically defined to a molecularly defined approach. Dialogues Clin Neurosci. (2017) 19:353–71. doi: 10.31887/DCNS.2017.19.4/sscherer
20. Wahlsten, D, Metten, P, and Crabbe, JC. Survey of 21 inbred mouse strains in two laboratories reveals that BTBR T/+ tf/tf has severely reduced hippocampal commissure and absent corpus callosum. Brain Res. (2003) 971:47–54. doi: 10.1016/s0006-8993(03)02354-0
21. Meyza, KZ, and Blanchard, DC. The BTBR mouse model of idiopathic autism - current view on mechanisms. Neurosci Biobehav Rev. (2017) 76:99–110. doi: 10.1016/j.neubiorev.2016.12.037
22. Richter, JD, and Zhao, X. The molecular biology of FMRP: new insights into fragile X syndrome. Nat Rev Neurosci. (2021) 22:209–22. doi: 10.1038/s41583-021-00432-0
23. Durand, CM, Betancur, C, Boeckers, TM, Bockmann, J, Chaste, P, Fauchereau, F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. (2007) 39:25–7. doi: 10.1038/ng1933
24. Vyas, Y, Cheyne, JE, Lee, K, Jung, Y, Cheung, PY, and Montgomery, JM. Shankopathies in the developing brain in autism Spectrum disorders. Front Neurosci. (2021) 15:775431. doi: 10.3389/fnins.2021.775431
25. Chao, OY, Pathak, SS, Zhang, H, Dunaway, N, Li, JS, Mattern, C, et al. Altered dopaminergic pathways and therapeutic effects of intranasal dopamine in two distinct mouse models of autism. Mol Brain. (2020) 13:111. doi: 10.1186/s13041-020-00649-7
26. Peca, J, Feliciano, C, Ting, JT, Wang, W, Wells, MF, Venkatraman, TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. (2011) 472:437–42. doi: 10.1038/nature09965
27. Squillace, M, Dodero, L, Federici, M, Migliarini, S, Errico, F, Napolitano, F, et al. Dysfunctional dopaminergic neurotransmission in asocial BTBR mice. Transl Psychiatry. (2014) 4:e427. doi: 10.1038/tp.2014.69
28. Wang, W, Li, C, Chen, Q, van der Goes, MS, Hawrot, J, Yao, AY, et al. Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J Clin Invest. (2017) 127:1978–90. doi: 10.1172/JCI87997
29. Bakker, CE, Verheij, C, Willemsen, R, Vanderhelm, R, Oerlemans, F, Vermey, M, et al. Fmr1 knockout mice - a model to study fragile-X mental-retardation. Cells. (1994) 78:23–33.
30. Paxinos, G, and Franklin, KBJ. Paxinos and Franklin’s The Mouse Brain in Stereotaxic Coordinates, 5th Edition, San Diego. Elsevier Academic Press. (2019).
31. Sommer, C, Fahrner, A, and Kiessling, M. Postischemic neuroprotection in the ischemia-tolerant state gerbil hippocampus is associated with increased ligand binding to inhibitory GABA(a) receptors. Acta Neuropathol. (2003) 105:197–202. doi: 10.1007/s00401-002-0632-7
32. Behuet, S, Cremer, JN, Cremer, M, Palomero-Gallagher, N, Zilles, K, and Amunts, K. Developmental changes of glutamate and GABA receptor densities in Wistar rats. Front Neuroanat, (2019).13–100. doi: 10.3389/fnana.2019.00100
33. Mammele, S, Frauenknecht, K, Sevimli, S, Diederich, K, Bauer, H, Grimm, C, et al. Prevention of an increase in cortical ligand binding to AMPA receptors may represent a novel mechanism of endogenous brain protection by G-CSF after ischemic stroke. Restor Neurol Neurosci. (2016) 34:665–75. doi: 10.3233/RNN-150543
34. Gonzalez-Lozano, MA, Klemmer, P, Gebuis, T, Hassan, C, van Nierop, P, van Kesteren, RE, et al. Dynamics of the mouse brain cortical synaptic proteome during postnatal brain development. Sci Rep. (2016) 6:35456. doi: 10.1038/srep35456
35. Hyman, SL, Levy, SE, Myers, SM, Council On Children With Disabilities, S.O.D, and Behavioral, P. Identification, evaluation, and management of children with autism spectrum disorder. Pediatrics. (2020) 145:e20193447. doi: 10.1542/peds.2019-3447
36. Rothmond, DA, Weickert, CS, and Webster, MJ. Developmental changes in human dopamine neurotransmission: cortical receptors and terminators. BMC Neurosci. (2012) 13:18. doi: 10.1186/1471-2202-13-18
37. Kubota, M, Fujino, J, Tei, S, Takahata, K, Matsuoka, K, Tagai, K, et al. Binding of dopamine D1 receptor and noradrenaline transporter in individuals with autism Spectrum disorder: a PET study. Cereb Cortex. (2020) 30:6458–68. doi: 10.1093/cercor/bhaa211
38. Zurcher, NR, Walsh, EC, Phillips, RD, Cernasov, PM, Tseng, CJ, Dharanikota, A, et al. A simultaneous [(11)C]raclopride positron emission tomography and functional magnetic resonance imaging investigation of striatal dopamine binding in autism. Transl Psychiatry. (2021) 11:33. doi: 10.1038/s41398-020-01170-0
39. Park, HR, Kim, IH, Kang, H, Lee, DS, Kim, BN, Kim, DG, et al. Nucleus accumbens deep brain stimulation for a patient with self-injurious behavior and autism spectrum disorder: functional and structural changes of the brain: report of a case and review of literature. Acta Neurochir. (2017) 159:137–43. doi: 10.1007/s00701-016-3002-2
40. Yan, H, Siegel, L, Breitbart, S, Gorodetsky, C, Fasano, A, Rahim, A, et al. An open-label prospective pilot trial of nucleus accumbens deep brain stimulation for children with autism spectrum disorder and severe, refractory self-injurious behavior: study protocol. Pilot Feasibility Stud. (2022) 8:24. doi: 10.1186/s40814-022-00988-3
41. Didriksen, M, Fejgin, K, Nilsson, SR, Birknow, MR, Grayton, HM, Larsen, PH, et al. Persistent gating deficit and increased sensitivity to NMDA receptor antagonism after puberty in a new mouse model of the human 22q11.2 microdeletion syndrome: a study in male mice. J Psychiatry Neurosci. (2017) 42:48–58. doi: 10.1503/jpn.150381
42. Karayannis, T, Au, E, Patel, JC, Kruglikov, I, Markx, S, Delorme, R, et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature. (2014) 511:236–40. doi: 10.1038/nature13248
43. Riday, TT, Dankoski, EC, Krouse, MC, Fish, EW, Walsh, PL, Han, JE, et al. Pathway-specific dopaminergic deficits in a mouse model of Angelman syndrome. J Clin Invest. (2012) 122:4544–54. doi: 10.1172/JCI61888
44. Platt, RJ, Zhou, Y, Slaymaker, IM, Shetty, AS, Weisbach, NR, Kim, JA, et al. Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep. (2017) 19:335–50. doi: 10.1016/j.celrep.2017.03.052
45. Kuo, HY, and Liu, FC. Valproic acid induces aberrant development of striatal compartments and corticostriatal pathways in a mouse model of autism spectrum disorder. FASEB J. (2017) 31:4458–71. doi: 10.1096/fj.201700054R
46. Pascucci, T, Colamartino, M, Fiori, E, Sacco, R, Coviello, A, Ventura, R, et al. P-cresol alters brain dopamine metabolism and exacerbates autism-like behaviors in the BTBR mouse. Brain Sci. (2020) 10:233. doi: 10.3390/brainsci10040233
47. Eissa, N, Venkatachalam, K, Jayaprakash, P, Falkenstein, M, Dubiel, M, Frank, A, et al. The multi-targeting ligand ST-2223 with histamine H3 receptor and dopamine D2/D3 receptor antagonist properties mitigates autism-like repetitive behaviors and brain oxid.ative stress in mice. Int J Mol Sci. (2021) 22:1947. doi: 10.3390/ijms22041947
48. Venkatachalam, K, Eissa, N, Awad, MA, Jayaprakash, P, Zhong, S, Stolting, F, et al. The histamine H3R and dopamine D2R/D3R antagonist ST-713 ameliorates autism-like behavioral features in BTBR T+tf/J mice by multiple actions. Biomed Pharmacother. (2021) 138:111517. doi: 10.1016/j.biopha.2021.111517
49. Khlghatyan, J, and Beaulieu, JM. Are FXR family proteins integrators of dopamine signaling and glutamatergic neurotransmission in mental illnesses? Front Synaptic Neurosci. (2018) 10:22. doi: 10.3389/fnsyn.2018.00022
50. Wang, H, Wu, LJ, Kim, SS, Lee, FJ, Gong, B, Toyoda, H, et al. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron. (2008) 59:634–47. doi: 10.1016/j.neuron.2008.06.027
51. Fulks, JL, O'Bryhim, BE, Wenzel, SK, Fowler, SC, Vorontsova, E, Pinkston, JW, et al. Dopamine release and uptake impairments and Behavioral alterations observed in mice that model fragile X mental retardation syndrome. ACS Chem Neurosci. (2010) 1:679–90. doi: 10.1021/cn100032f
52. Jiang, A, Wang, L, Lu, JYD, Freeman, A, Campbell, C, Su, P, et al. Sex differences in dopamine receptor signaling in Fmr1 knockout mice: a pilot study. Brain Sci. (2021) 11:1398. doi: 10.3390/brainsci11111398
53. Neuhofer, D, Henstridge, CM, Dudok, B, Sepers, M, Lassalle, O, Katona, I, et al. Functional and structural deficits at accumbens synapses in a mouse model of fragile X. Front Cell Neurosci. (2015) 9:100. doi: 10.3389/fncel.2015.00100
54. Schmeisser, MJ, Ey, E, Wegener, S, Bockmann, J, Stempel, AV, Kuebler, A, et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. (2012) 486:256–60. doi: 10.1038/nature11015
55. Liu, C, Li, D, Yang, H, Li, H, Xu, Q, Zhou, B, et al. Altered striatum centered brain structures in SHANK3 deficient Chinese children with genotype and phenotype profiling. Prog Neurobiol. (2021) 200:101985. doi: 10.1016/j.pneurobio.2020.101985
56. Srivastava, S, Scherrer, B, Prohl, AK, Filip-Dhima, R, Kapur, K, Kolevzon, A, et al. Volumetric analysis of the basal ganglia and cerebellar structures in patients with Phelan-McDermid syndrome. Pediatr Neurol. (2019) 90:37–43. doi: 10.1016/j.pediatrneurol.2018.09.008
57. Bariselli, S, Tzanoulinou, S, Glangetas, C, Prevost-Solie, C, Pucci, L, Viguie, J, et al. SHANK3 controls maturation of social reward circuits in the VTA. Nat Neurosci. (2016) 19:926–34. doi: 10.1038/nn.4319
58. Bey, AL, Wang, X, Yan, H, Kim, N, Passman, RL, Yang, Y, et al. Brain region-specific disruption of Shank3 in mice reveals a dissociation for cortical and striatal circuits in autism-related behaviors. Transl Psychiatry. (2018) 8:94. doi: 10.1038/s41398-018-0142-6
59. Peixoto, RT, Chantranupong, L, Hakim, R, Levasseur, J, Wang, W, Merchant, T, et al. Abnormal striatal development underlies the early onset of Behavioral deficits in Shank3B(−/−) mice. Cell Rep. (2019) 29:2016–2027.e4. doi: 10.1016/j.celrep.2019.10.021
60. Peixoto, RT, Wang, W, Croney, DM, Kozorovitskiy, Y, and Sabatini, BL. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(−/−) mice. Nat Neurosci. (2016) 19:716–24. doi: 10.1038/nn.4260
61. Azdad, K, Gall, D, Woods, AS, Ledent, C, Ferre, S, and Schiffmann, SN. Dopamine D2 and adenosine A2A receptors regulate NMDA-mediated excitation in accumbens neurons through A2A-D2 receptor heteromerization. Neuropsychopharmacology. (2009) 34:972–86. doi: 10.1038/npp.2008.144
62. Olson, PA, Tkatch, T, Hernandez-Lopez, S, Ulrich, S, Ilijic, E, Mugnaini, E, et al. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci. (2005) 25:1050–62. doi: 10.1523/JNEUROSCI.3327-04.2005
63. Brandenburg, C, Soghomonian, JJ, Zhang, K, Sulkaj, I, Randolph, B, Kachadoorian, M, et al. Increased dopamine type 2 gene expression in the dorsal striatum in individuals with autism Spectrum disorder suggests alterations in indirect pathway signaling and circuitry. Front Cell Neurosci. (2020) 14:577858. doi: 10.3389/fncel.2020.577858
64. Portmann, T, Yang, M, Mao, R, Panagiotakos, G, Ellegood, J, Dolen, G, et al. Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep. (2014) 7:1077–92. doi: 10.1016/j.celrep.2014.03.036
65. Bouchekioua, Y, Tsutsui-Kimura, I, Sano, H, Koizumi, M, Tanaka, KF, Yoshida, K, et al. Striatonigral direct pathway activation is sufficient to induce repetitive behaviors. Neurosci Res. (2018) 132:53–7. doi: 10.1016/j.neures.2017.09.007
66. Lee, Y, Kim, H, Kim, JE, Park, JY, Choi, J, Lee, JE, et al. Excessive D1 dopamine receptor activation in the dorsal striatum promotes autistic-like behaviors. Mol Neurobiol. (2018) 55:5658–71. doi: 10.1007/s12035-017-0770-5
67. Gunaydin, LA, Grosenick, L, Finkelstein, JC, Kauvar, IV, Fenno, LE, Adhikari, A, et al. Natural neural projection dynamics underlying social behavior. Cells. (2014) 157:1535–51. doi: 10.1016/j.cell.2014.05.017
68. Muehlmann, AM, Maletz, S, King, MA, and Lewis, MH. Pharmacological targeting of striatal indirect pathway neurons improves subthalamic nucleus dysfunction and reduces repetitive behaviors in C58 mice. Behav Brain Res. (2020) 391:112708. doi: 10.1016/j.bbr.2020.112708
69. Presti, MF, Mikes, HM, and Lewis, MH. Selective blockade of spontaneous motor stereotypy via intrastriatal pharmacological manipulation. Pharmacol Biochem Behav. (2003) 74:833–9. doi: 10.1016/s0091-3057(02)01081-x
70. DelaCuesta-Barrutia, J, Penagarikano, O, and Erdozain, AM. G protein-coupled receptor Heteromers as putative Pharmacotherapeutic targets in autism. Front Cell Neurosci. (2020) 14:588662. doi: 10.3389/fncel.2020.588662
71. Aman, M, Rettiganti, M, Nagaraja, HN, Hollway, JA, McCracken, J, McDougle, CJ, et al. Tolerability, safety, and benefits of risperidone in children and adolescents with autism: 21-month follow-up after 8-week placebo-controlled trial. J Child Adolesc Psychopharmacol. (2015) 25:482–93. doi: 10.1089/cap.2015.0005
72. Hirsch, LE, and Pringsheim, T. Aripiprazole for autism spectrum disorders (ASD). Cochrane Database Syst Rev. (2016) 2016:CD009043. doi: 10.1002/14651858.CD009043.pub3
73. Persico, AM, Ricciardello, A, Lamberti, M, Turriziani, L, Cucinotta, F, Brogna, C, et al. The pediatric psychopharmacology of autism spectrum disorder: a systematic review - part I: the past and the present. Prog Neuro-Psychopharmacol Biol Psychiatry. (2021) 110:110326. doi: 10.1016/j.pnpbp.2021.110326
74. Cammarata-Scalisi, F, Callea, M, Martinelli, D, Willoughby, CE, Tadich, AC, Araya Castillo, M, et al. Clinical and genetic aspects of Phelan-McDermid syndrome: an interdisciplinary approach to management. Genes (Basel). (2022) 13:504. doi: 10.3390/genes13030504
75. Kolevzon, A, Delaby, E, Berry-Kravis, E, Buxbaum, JD, and Betancur, C. Neuropsychiatric decompensation in adolescents and adults with Phelan-McDermid syndrome: a systematic review of the literature. Mol Autism. (2019) 10:50. doi: 10.1186/s13229-019-0291-3
76. Eckert, EM, Dominick, KC, Pedapati, EV, Wink, LK, Shaffer, RC, Andrews, H, et al. Pharmacologic interventions for irritability, aggression, agitation and self-injurious behavior in fragile X syndrome: an initial cross-sectional analysis. J Autism Dev Disord. (2019) 49:4595–602. doi: 10.1007/s10803-019-04173-z
Keywords: ASD, autism (ASD), dopamine receptor (d1r and d2r), dorsal striatum, ventral striatum, receptor autoradiography
Citation: Chhabra S, Nardi L, Leukel P, Sommer CJ and Schmeisser MJ (2023) Striatal increase of dopamine receptor 2 density in idiopathic and syndromic mouse models of autism spectrum disorder. Front. Psychiatry. 14:1110525. doi: 10.3389/fpsyt.2023.1110525
Edited by:
Luca Pangrazzi, Institut Ludwig Pour la Recherche sur le Cancer (CHUV), SwitzerlandReviewed by:
Paola Bonsi, Santa Lucia Foundation (IRCCS), ItalyCopyright © 2023 Chhabra, Nardi, Leukel, Sommer and Schmeisser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Schmeisser, bXNjaG1laXNzZXJAdW5pLW1haW56LmRl
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.