
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Psychiatry, 10 November 2022
Sec. Perinatal Psychiatry
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.945343
 Rezan Nehir Mavioglu1*
Rezan Nehir Mavioglu1* Laura Ramo-Fernandez1
Laura Ramo-Fernandez1 Anja M. Gumpp1
Anja M. Gumpp1 Iris-Tatjana Kolassa1
Iris-Tatjana Kolassa1 Alexander Karabatsiakis1,2
Alexander Karabatsiakis1,2Childhood maltreatment (CM) is associated with alterations in DNA methylation (DNAm) especially in stress response genes. Due to the higher risk of overall health complications of individuals with a parental history of CM, intergenerational transmission of CM-associated DNAm changes has been investigated but remains unclear. In this study, we investigated if different severities of CM have any influence on the DNAm of DNA methyltransferase 1 (DNMT1), an important enzyme of the DNAm machinery, in immune and buccal cells of mother-newborn dyads. DNAm was assessed by mass spectrometry using immune cell DNA from mothers (N = 117) and their newborns (N = 113), and buccal cell DNA of mother-newborn dyads (N = 68 each). Mothers with a history of CM had lower mean methylation of DNMT1 in immune cells compared to the mothers without a CM history. CM status only influenced maternal DNMT1 gene expression when at least moderate CM was reported. Buccal cell DNAm was not associated with CM status. Maternal history of CM was not linked to any alterations in DNMT1 mean DNAm in any of the cell types studied in newborns. We conclude that the CM-associated alterations in DNMT1 DNAm might point to allostatic load and can be physiologically relevant, especially in individuals with more severe CM experiences, resulting in an activated DNA methylation machinery that might influence stress response genes. Our lack of significant findings in buccal cells shows the tissue-specific effects of CM on DNAm. In our sample with low to moderate maternal CM history, there was no intergenerational transmission of DNMT1 DNAm in newborns.
Childhood maltreatment (CM) is a severe form of early life stressor and a widespread problem around the world (1) that may lead to physical and mental health problems throughout one’s lifespan (2, 3). One way CM induces such health effects a long time after its exposure might be through epigenetic alterations. Epigenetic alterations are tissue specific and can alter gene expression. One of the epigenetic mechanisms, DNA methylation (DNAm), is known to “shut down” genes when present in promoter regions, by preventing transcription factor binding and reducing gene expression (4). Psychoneuroimmunology research provided evidence that DNAm is affected by CM (5, 6). Epigenetic alterations were mainly reported in genes of the neuroendocrine stress-response such as NR3C1 and FKBP5, indicating mostly a positive CM-DNAm association in NR3C1 and a negative one in FKBP5 (5–7). These studies almost exclusively focused on a single tissue, and comparisons between the most two prominently studied tissues, peripheral blood mononuclear cells (PBMC) and buccal cells, are not available. Moreover, CM associated DNAm alterations were also observed in epigenome-wide association studies and studies concerning global DNAm (5, 6), hinting toward the involvement of fundamental regulators of DNAm in the relationship between CM and DNAm outcomes. How DNAm regulators moderate the effects of CM on DNAm outcomes might be through stress-response elements themselves. It has been proposed that glucocorticoid receptor (encoded by NR3C1)—glucocorticoid complexes can (1) bind to glucocorticoid response elements (GREs) within genes coding DNA methyltransferases (DNMTs) and change their expression (8, 9), or (2) activate specific genes that impact the expression of DNMT enzymes (10). The link between DNMTs and stress-response elements is possibly bidirectional since DNMTs can regulate methylation of the genes coding for these elements.
DNA methyltransferase 1 (DNMT1) is one of the five DNMT enzymes that are responsible for the transfer of methyl groups, along with DNMT2, DNMT3A, DNMT3B, and DNMT3L. DNMT1, DNMT3A, and DNMT3B are known to be canonical cytosine 5-DNMTs that catalyze the addition of methyl (–CH3) groups to cytosine of CpG dinucleotides (CpG sites) in both strands of genomic DNA (11). DNMT1 is both a maintenance and de novo methyltransferase meaning that it is involved in the methylation of hemimethylated CpG sites and contributes to the methylation of new CpG sites (12). Because of its crucial role in DNAm regulation, DNMT1 has been studied in the context of early life adversity and psychopathology in both animals and humans. In rats, increased DNMT1 mRNA levels in fetal cortex after prenatal stress was reported (13). Similarly, in male mice, increased DNMT1 protein levels were found in the frontal cortex after prenatal restraint stress (14). Another study showed the long-term effect of a maternal separation paradigm in male mice, reporting increased DNMT1 mRNA in the prefrontal cortex (PFC) in adult animals but not in 15 days-old animals (15). However, Blaze and Roth (16) reported less DNMT1 mRNA in PFC of adult male rats after experimental CM exposure. Overall, the results of these animal studies indicate that DNAm machinery is affected by early-life adversity on the levels of gene and protein expression. There have been no studies so far concerning the influence of early life adversity or CM on DNMT1 DNAm or gene expression in humans. Instead, the scientific focus was mostly directed on lifetime trauma and psychopathology. A longitudinal study reported increased levels of DNAm of one CpG site in DNMT1 in the venous blood of individuals with PTSD after trauma exposure, compared to their pre-trauma DNAm levels (17). However, there were no significant differences in DNAm after trauma in PTSD cases versus controls. Another study with mother-newborn dyads found a positive association between one methylation cluster of DNMT1 and genome-wide mean DNAm only in maternal venous blood but not in newborn cord blood (18). In addition, a negative association was found between war trauma and maternal genome-wide mean DNAm in their cohort with a history of stressful life events. The association between war trauma and DNMT1 DNAm was not directly tested (18) but results from these two studies hint toward an adulthood trauma-related regulation of DNMT1 DNAm in blood. DNMT1 gene expression levels were also not reported in these two studies (17, 18), limiting the evaluation of possible physiological effects of altered DNMT1 methylation. As for studies investigating the role of DNMT1 in psychopathology, a postmortem design with individuals with schizophrenia and bipolar disorder reported increased DNMT1 expression and enzymatic activity patients’ brains, compared to controls (19). They reported increased DNMT1 binding to GABAergic and glutamatergic promoters that led to the downregulation of these genes. This downregulation did not necessarily correlate with methylation of GABAergic and glutamatergic promoters, hinting toward other crucial roles of the DNMT1 enzyme beyond DNAm (19, 20). In line with this finding, antidepressant medications might reduce DNMT1 activity, which might be one of the mechanisms how these medications work in treating individuals with various mental health disorders (21, 22). This view was supported by a conditional forebrain DNMT1 knockout mice model showing less depressive-like behavior compared to wild-type mice (23). These studies show the importance of DNMT1 in the context of lifetime adversity and psychopathology.
As a cumulative stress factor, maternal history of CM might also influence next generations’ health. The offspring of individuals with a history of CM were shown to have a higher risk of developing physical as well as mental disorders (24, 25). One of the pathways that can explain this observation could be the intergenerational transmission of epigenetic stress profiles. There are two main ways how intergenerational transmission of trauma might occur in DNAm context. Firstly, intergenerational transmission of CM-related DNAm changes might refer to a direct transmission of DNAm signatures from parental gametes. The majority of methyl marks on the DNA are thought to be removed and not passed to the further generations, thus intergenerational transmission of DNAm implies incomplete removal of the methyl groups. There is no evidence so far that this form of intergenerational transmission of trauma exists in the DNAm of humans but it has been observed in some animals, limited to only a certain number of generations (26). However, the cross-species comparability of methylation regulation processes remains under debate. Another form of intergenerational transmission of trauma is through alterations in maternal physiology and increased prenatal stress that can affect the intrauterine development and the epigenetic makeup of the fetus (27). In extreme prenatal stress conditions, some studies conducted with animals (28, 29) as well as humans (30, 31) reported findings indicating intergenerational transmission of DNAm markers. Intergenerational transmission studies that are conducted later on in human life have the limitation that they can be confounded by certain environmental factors such as parenting style, shared lifestyle factors and other psychosocial influences. Although parenting behavior can be affected by a history of traumatic events, they can only qualify as “indirect” intergenerational transmission, since the impact occurs after birth and does not directly affect the gestational biology (27). Therefore, it is essentially important to study the intergenerational transmission of DNAm directly after birth. In our cohort of mother-newborn dyads, we did not find evidence for a transmission of maternal CM-induced DNAm in stress-response related genes in immune cells collected after birth of the newborn (7).
In this study, we aim to understand if a history of CM experiences can influence the DNAm profiles and gene expression of an important regulator of DNAm, DNMT1. To achieve this, we examined the role of different severities of CM on DNMT1 DNAm in PBMC and buccal cells of mothers who recently gave birth. DNMT1 relative gene expression levels were determined to assess the possible physiological implications of alterations in DNAm. To test if stress-response elements have any bidirectional relationships with DNMTs as proposed before (8–10), the associations between our previously published NR3C1 exon 1F DNAm and relative gene expression results from the same cohort (7), and DNMT1 DNAm and relative gene expression were explored in maternal PBMC. Lastly, we examined maternal CM related intergenerational DNMT1 DNAm transmission in newborn PBMC from umbilical cord blood and buccal cells both collected also shortly after parturition.
As part of the “My Childhood—Your Childhood” study, N = 548 clinically healthy mothers were recruited between October 2013 and December 2015, within the first week after giving birth (M = 2.71 days) at the Department of Obstetrics and Gynecology at Ulm University Hospital. Mothers were asked to provide written informed consent forms and answer a set of sociodemographic and psychological questionnaires. Peripheral blood of consenting mothers, umbilical cord blood of their newborns, and cheek swabs of mother-newborn dyads were collected. Out of 153 mothers that provided blood, only 58 mothers had a history of at least low CM. For the epigenetic analyses of PBMC, these mothers were matched with N = 59 mothers without a history of CM, resulting in a cohort of 117 mother-newborn dyads. Four newborn samples were excluded from the analyses (N = 3 twins, N = 1 sample loss due to technical difficulties during the cell isolation procedure), leaving N = 117 mothers and N = 113 newborns in the immune cell cohort [please see Ramo-Fernández et al. (7) for details about the selection procedure]. From these participants, a subsample of 68 mothers and infants was defined according to maternal CM status and availability of at least 1 μg DNA extracted from buccal cells for measurements of buccal cell epigenetics (buccal cell cohort). The study protocol was in accordance with the Declaration of Helsinki, which was approved by the ethics committee of Ulm University.
Mothers provided sociodemographic and health information, such as age, newborns’ sex, smoking, and medical history. Please find the information about the relevant variables in Table 1.
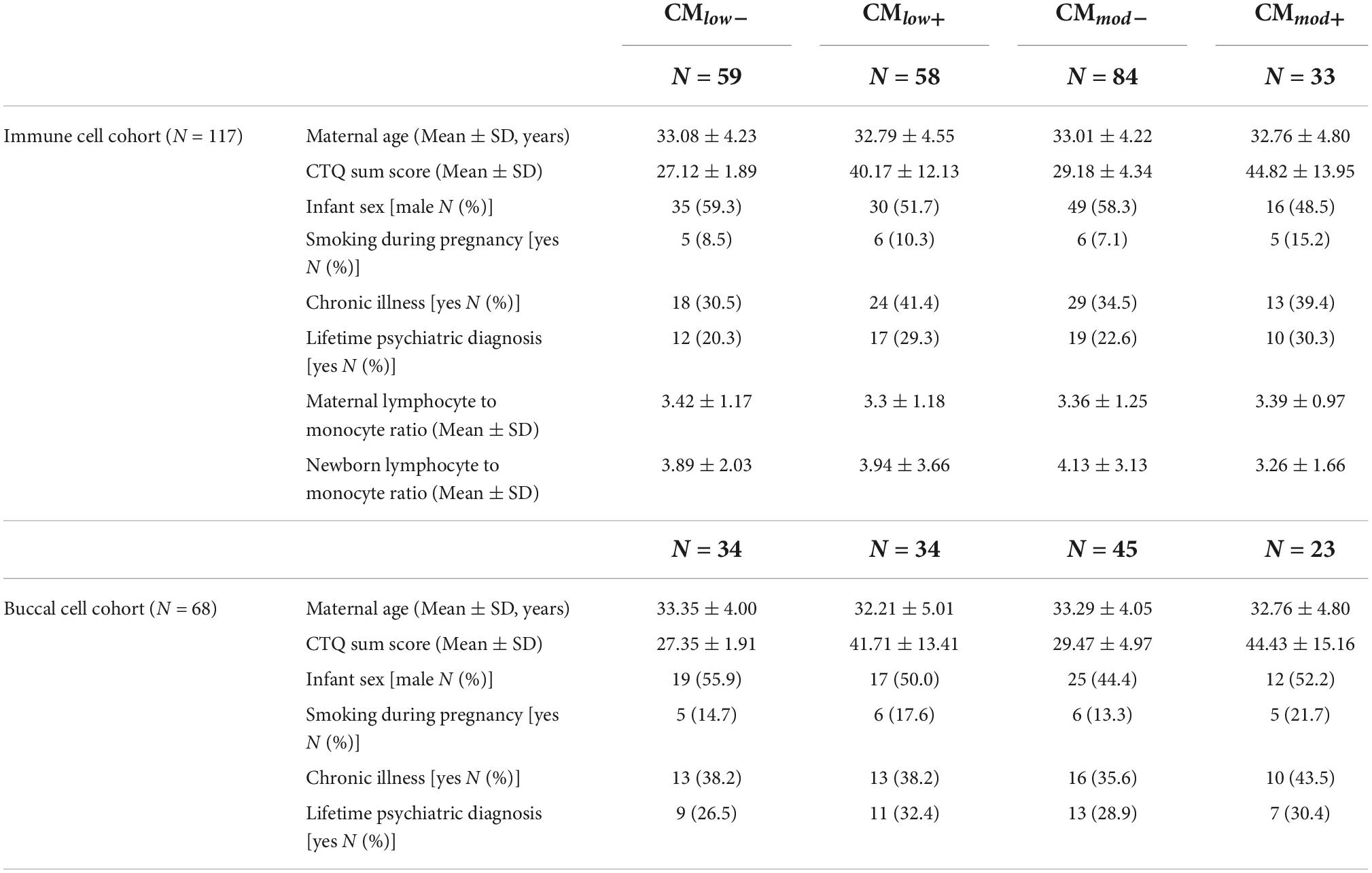
Table 1. Sociodemographic and health information in mother-newborn dyads in immune and buccal cell cohorts, categorized according to low and moderate childhood maltreatment (CM) severity groups.
To assess the history of CM, the German short version of the Childhood Trauma Questionnaire (CTQ) (32, 33) was filled in by the mothers. The CTQ is a Likert-type questionnaire with five subscales (emotional abuse, physical abuse, sexual abuse, emotional neglect, and physical neglect), scores of each can range from 5 to 25, with higher scores indicating a more severe history of CM. The CTQ sum score is calculated by adding each subscale score together, and the total score can range from 25 to 125 to measure CM load. CTQ was used to dichotomize the mothers according to their low and moderate CM experiences. To achieve this, mothers were categorized as being experienced none to mild, low, moderate or severe maltreatment in each of the five subscales, according to the CTQ manual (33). For low dichotomization, mothers who experienced low to severe maltreatment according to at least one subscale were classified as CMlow+. The mothers who did not fulfill this criterion were classified as CMlow–. Similarly, for moderate dichotomization, mothers who experienced moderate to severe maltreatment according to at least one subscale were classified as CMmod+. The mothers who did not fulfill this criterion were classified as CMmod–.
Peripheral blood mononuclear cells (PBMC) and umbilical cord blood mononuclear cells (UBMC) were isolated by Ficoll-Hypaque density gradient centrifugation (GE Healthcare, Chalfont St Giles, UK) according to the manufacturer’s protocol, from maternal peripheral blood and newborn umbilical cord blood that were collected into CPDA-buffered blood collection tubes (Sarstedt S-Monovette, Nürmbrecht, Germany). Cells were cryopreserved in medium (1:10 dimethyl sulphoxide: fetal calf serum: Sigma-Aldrich, St. Louis, MO, USA) and stored at –80°C until DNA isolation. Additionally, a K3EDTA-buffered collection tube was used for samples from mothers and newborns and handed to the Department of Clinical Chemistry and Central Laboratory of Ulm University for standard hemogram analysis. Buccal cells (BC) were collected from mothers and newborns with buccal swabs (Isohelix, Harrietsham, Kent, United Kingdom), and the swabs were placed at –80°C until DNA isolation.
DNA was purified from PBMC, UBMC and BC using an automated MagNAPure 96 platform (Roche, Basel, Switzerland). DNA concentrations were measured with a Qubit fluorometer (Life Technologies, Carlsbad, CA, USA) and DNA eluates were set to 40 ng/μl. DNA samples were stored at –20°C and sent to Varionostic GmbH (Ulm, Germany) for DNAm analyses.
Thawed DNA samples (1 μg minimum) were treated with bisulfite, amplified with two primer pairs resulting in two amplicons by polymerase chain reaction (PCR), reverse transcribed and treated with RNase A, before performing EpiTYPER mass spectrometry (Agena Bioscience, San Diego, CA, USA). RNase A cleaves the reverse transcribed PCR products at the uracil residues, therefore the resulting products consist of CpG units that can contain multiple CpG sites. Products with too high or too low mass cannot be successfully read, as a limitation of the technique (34). The DNA methylation percentage of a CpG unit represents the average DNA methylation percentage of the CpG sites in that unit. The primers were chosen to span the regulatory regions in the first intron of the DNMT1 gene [Chr19:10,194,327-10,194,571 (GRCh38/hg38), primer sequences PF1: 5′-AGGAAGAGAGGATGTATAGTTTTGGGGGAAAGGTA-3′, PR1: 5′-CAGTAATACGACTCACTATAGGGAGAAGGCTAAAAATTCTCTTTTAATCCCCAAAT-3′], and the beginning of the first exon including the gene promoter [chr19:10,194,533-10,195,072 (GRCh38/hg38), primer sequences PF2: 5′-AGGAAGAGAGTTAAAGTTTGTTGTATTTGGGGATT-3′, PR2: 5′-CAGTAATACGACTCACTATAGGGAGAAGGCTTTCCATCCTTCTACACAAAATATC-3′]. The chosen sequence had an overlapping length of 746 bp with a total of 73 CpG sites, and 69% GC content. Please see Supplementary Figure 1 for a detailed depiction of the sequence, as well as regulatory regions, and the CpG sites. Along with the promoter, the sequence contains enhancers, microRNA targets, and transcription start sites (TSS). Please see Supplementary Table 1 for the genomic and regulatory regions in the sequence, their positions, and the corresponding CpG units.
NR3C1 exon 1F methylation assessment was performed as a part of a previous study (7). Briefly, primers were chosen to span NR3C1 exon 1F, to assess the sequence Chr5:143,413,957-143,414,659 (GRCh38/hg38). The chosen sequence had a length of 703 bp with a total of 79 CpG sites. Please see Ramo-Fernández et al. (7) for details.
Gene expression analyses were performed in N = 72 mothers due to the limited amount of remaining PBMC. Qiagen RNeasy Kit (QIAGEN, Hilden, Germany) was used for RNA isolation. After quantification with a Qubit spectrophotometer (Life Technologies, Carlsbad, CA, USA), RNA was transcribed to cDNA using a high-capacity cDNA reverse transcription kit (Thermo Fischer Scientific, Waltham, MA, USA). Please check Ramo-Fernández et al. (7) for details about RNA purification and cDNA conversion. Gene expression assays (real-time quantitative PCR) were performed using QuantStudio 6 (Applied Biosystems, Waltham, MA, USA) with TaqMan Gene Expression assays (Thermo Fischer Scientific, Waltham, MA, USA) for DNMT1 as the target gene (Hs00154749_m1; covers exon boundary 1-2) and IPO8 as the housekeeping gene (Hs00183533_m1). Each cDNA sample (20 ng) was measured in triplicates and an interplate calibrator which was prepared as a mixture of the sample cDNA was used to control for interplate differences in a total of six plates. Relative DNMT1 gene expression of the samples was calculated using the Livak method, with the interplate calibrator as the reference sample [2–ΔΔCt, Livak and Schmittgen (35)].
NR3C1 relative gene expression was performed for another study (7). Briefly, with the same QuantStudio 6 (Applied Biosystems, Waltham, MA, USA), a real-time quantitative PCR was performed using TaqMan Gene Expression assays (Thermo Fischer Scientific, Waltham, MA, USA) for NR3C1 as the target gene (Hs00353740_m1; covers exon boundary 4-5) and SDHA (Hs00188166_m1) and IPO8 (Hs00183533_m1) as housekeeping genes. Please see Ramo-Fernández et al. (7) for details.
Raw methylation data with the mean percentages of each CpG unit were pre-processed to verify high data quality. In summary, CpG units unable to be analyzed in more than 30% of the whole sample, as well as samples that had more than 50% of missing values overall, were excluded from the analysis. 32 CpG units (57 CpG sites), samples from N = 109 mothers and N = 110 newborns for analyses with PBMC (N = 102 dyads with both mother and infant data), and N = 62 mothers and N = 65 newborns for analyses with BC (N = 59 dyads with both mother and infant data) remained after this process. The CpG units with the same calculated molecular weight (due to their shared nucleotide content) were grouped together, in the cases of unsuccessful discrimination of the CpG units by mass spectrometry as a limitation of the technique. Mean percentage methylation scores were created by calculating the mean of the available CpG unit methylation percentages. As for NR3C1 exon 1F mean DNAm, data from N = 113 mothers was used and 17 CpG units (29 CpG sites) remained after pre-processing [please see Ramo-Fernández et al. (7) for details].
Due to non-normal distributions and unequal variances, non-parametric tests were performed. Spearman’s rank-order correlation was used to determine the bidirectional relationships between the variables. Wilcoxon signed-rank test was performed for within-subjects analyses, to compare immune and buccal cell DNAm of the same individuals. To compare the DNAm and relative gene expression values of CM− and CM+ groups, Brunner Munzel test (36) with t-approximation was used using rankFD package (37) in R 4.1.3. Briefly, Brunner Munzel test is a non-parametric rank test that can be used when two populations have unequal variances, unlike Wilcoxon tests. Its test statistic is T. It measures stochastic equality of two populations, meaning the frequency of larger values is equal among populations. The relative estimate (P) is between 0 and 1, and if it is around 0.5 stochastic equality is given. If it is significantly smaller than 0.5, the first group has a higher frequency of larger values compared to the second group and if it is larger than 0.5, the second group has a higher frequency of larger values (36). Relative estimates were reported with 95% confidence intervals (CI) in brackets such as [Lower CI, Upper CI]. Calculations were performed with CMlow– or CMmod– as the first group. False discovery rate (FDR) correction due to multiple testing was performed according to Benjamini-Hochberg procedure (38). In order to test the effects of possible covariates in the relationship between CM and DNAm or relative gene expression, robust regressions were performed using MASS package (39) in R 4.1.3. These covariates were maternal age, number of days between parturition and blood sampling, history of psychiatric diagnosis, medication use, folic acid intake during pregnancy (N = 8 reported), smoking, presence of chronic illness, and lymphocyte to monocyte ratio (for PBMC only) for analyses with mothers. For analyses with newborns, the covariates were gestational age, birth weight, sex, lymphocyte to monocyte ratio (for UBMC only), and the maternal covariates mentioned above. The covariates were tested one by one in separate models. Mean and standard deviations were reported as descriptives for comparability reasons. All tests were performed two-tailed, and alpha was set to 0.05.
Please see Table 1 for descriptives for sociodemographic and health variables, categorized by CM status in immune cell and buccal cell cohorts. The comparisons of the variables within CMlow and CMmod groups did not reveal any differences (ps > 0.05) except for the CTQ sum scores which differed between CMlow– and CMlow+ (T = 36.496, P = 0.970, p < 0.001), and CMmod– and CMmod+ (T = 16.621, P = 0.924, p < 0.001) in immune cell cohort, meaning higher CTQ scores in groups with at least low and at least moderate CM history. Analyses with the buccal cell cohort yielded similar results.
CMlow+ mothers (4.15 ± 1.53%) had lower mean percentage DNMT1 DNAm than CMlow– mothers (4.54 ± 1.25%, T = –2.527, P = 0.363 [0.255, 0.471], p = 0.013, Figure 1A). Similarly, CMmod+ mothers (3.91 ± 1.16%) had lower mean percentage DNMT1 methylation than CMmod– mothers (4.52 ± 1.48%, T = –2.108, P = 0.373 [0.253, 0.494], p = 0.040, Figure 1B). These findings did not change when tested with the covariates (see Section “Data processing and statistical analyses”). Mean percentage DNMT1 methylation had a trend level negative association with CTQ sum score (rs = –0.167, p = 0.083, Figure 1C). Analyses with single CpG units revealed significant differences depending on CM status with overlaps in low and moderate cut-offs that survived FDR correction (see Supplementary Tables 2, 3). Twelve out of 32 CpG units differed in terms of mean percentage DNAm of CMlow– and CMlow+ mothers (Supplementary Table 2), whereas, only two out of 32 CpG units were differentially methylated in terms of CMmod status of mothers (Supplementary Table 3). For the CMlow categorization, differentially methylated CpG units were predicted to be located at enhancers, exon, and TSS (see Supplementary Table 1 for locations of regulatory regions and corresponding CpG units). Except for 2 CpG units, CMlow+ mothers had lower mean percentage DNAm than CMlow– mothers. Mean percentage DNAm differences ranged from 1.0 to 4.9% (Supplementary Table 2). Two CpG units were detected to be lower in terms of mean percentage DNAm in CMmod+ compared to CMmod– mothers. These units were predicted to be located in the enhancer and TSS. Mean percentage DNAm differences were 1.8 (enhancer) and 7.3% (TSS; Supplementary Table 3).
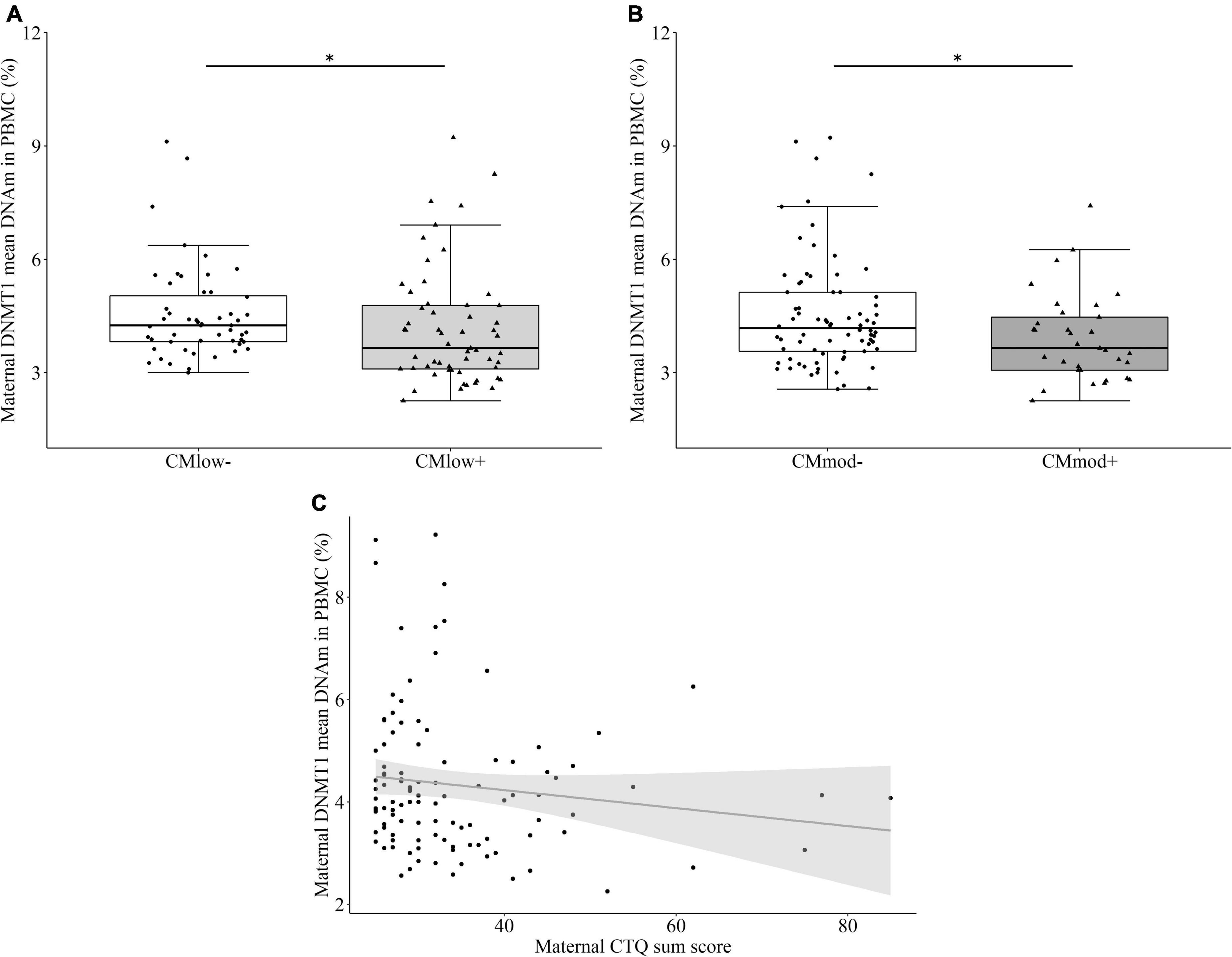
Figure 1. DNA methyltransferase 1 (DNMT1) mean DNA methylation (DNAm) and relative gene expression in peripheral blood mononuclear cells (PBMC) of the mothers. (A) Mothers with none to mild CM experiences (CMlow–) had higher DNMT1 mean DNAm compared to mothers with at least low CM experiences (CMlow+; p = 0.013). (B) Mothers with none to low CM experiences (CMmod–) had higher DNMT1 mean DNAm compared to mothers with at least moderate CM experiences (CMmod+; p = 0.040). (C) DNMT1 mean DNAm had a negative trend level correlation with Childhood Trauma Questionnaire (CTQ) sum score (rs = –0.167, p = 0.083). *p < 0.05.
There was no significant difference in the relative DNMT1 gene expression between CMlow– (1.16 ± 1.93) and CMlow+ mothers (1.17 ± 1.44, T = 0.151, P = 0.511 [0.366, 0.656], p = 0.881, Figure 2A). However CMmod+ mothers (1.23 ± 1.03) had higher relative DNMT1 gene expression than CMmod– mothers (1.14 ± 1.86, T = 2.487, P = 0.664 [0.532, 0.795], p = 0.015, Figure 2B). Therefore, relative gene expression of DNMT1 differed depending on the severity of CM categorization. The results did not change when certain variables were accounted for as covariates (see Section “Data processing and statistical analyses”). Relative DNMT1 gene expression did not have an association with CTQ sum score (rs = 0.034, p = 0.782, Figure 2C). No association between mean percentage DNMT1 DNAm and relative DNMT1 gene expression was found (rs = –0.012, p = 0.921, Figure 2D).
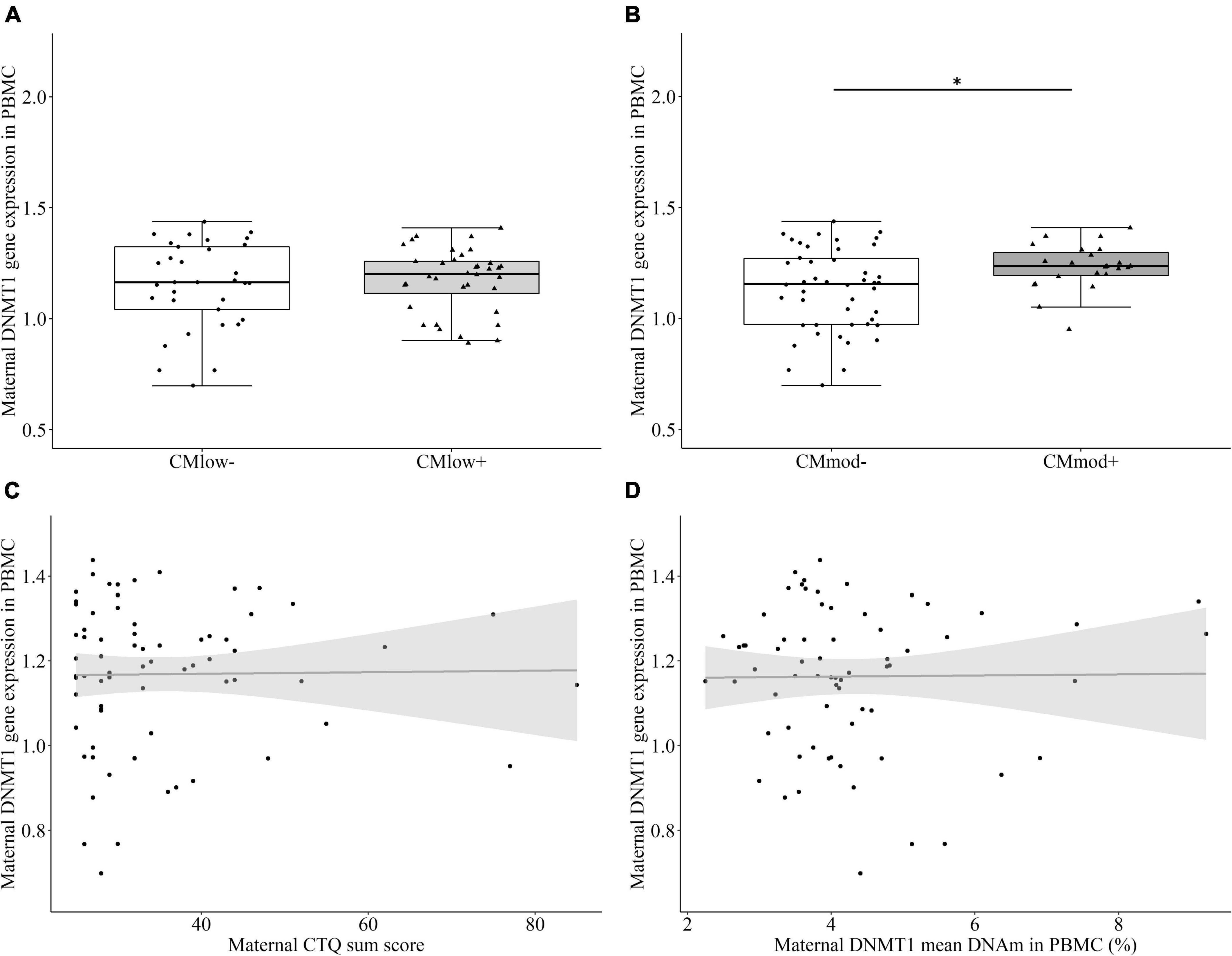
Figure 2. DNA methyltransferase 1 (DNMT1) relative gene expression in peripheral blood mononuclear cells (PBMC) of the mothers. (A) There was no significant difference between CMlow– and CMlow+ mothers in terms of their DNMT1 relative gene expression levels. (B) CMmod– mothers had lower DNMT1 relative gene expression compared to CMmod+ mothers (p = 0.015). (C) DNMT1 relative gene expression did not have a significant correlation with CTQ sum score (rs = 0.034, p = 0.782). (D) DNMT1 relative gene expression did not have a significant correlation with DNMT1 mean DNAm (rs = –0.012, p = 0.921). *p < 0.05.
There was no significant correlation between DNMT1 and NR3C1 exon 1F mean DNAm (rs = 0.031, p = 0.752, Figure 3A), but a trend toward significance between DNMT1 mean DNAm and NR3C1 relative gene expression (rs = 0.215, p = 0.086). However, there was a significant positive correlation between relative DNMT1 gene expression and NR3C1 exon 1F mean DNAm (rs = 0.444, p < 0.001, Figure 3B). No association was present between DNMT1 and NR3C1 relative gene expression levels (rs = –0.189, p = 0.156).
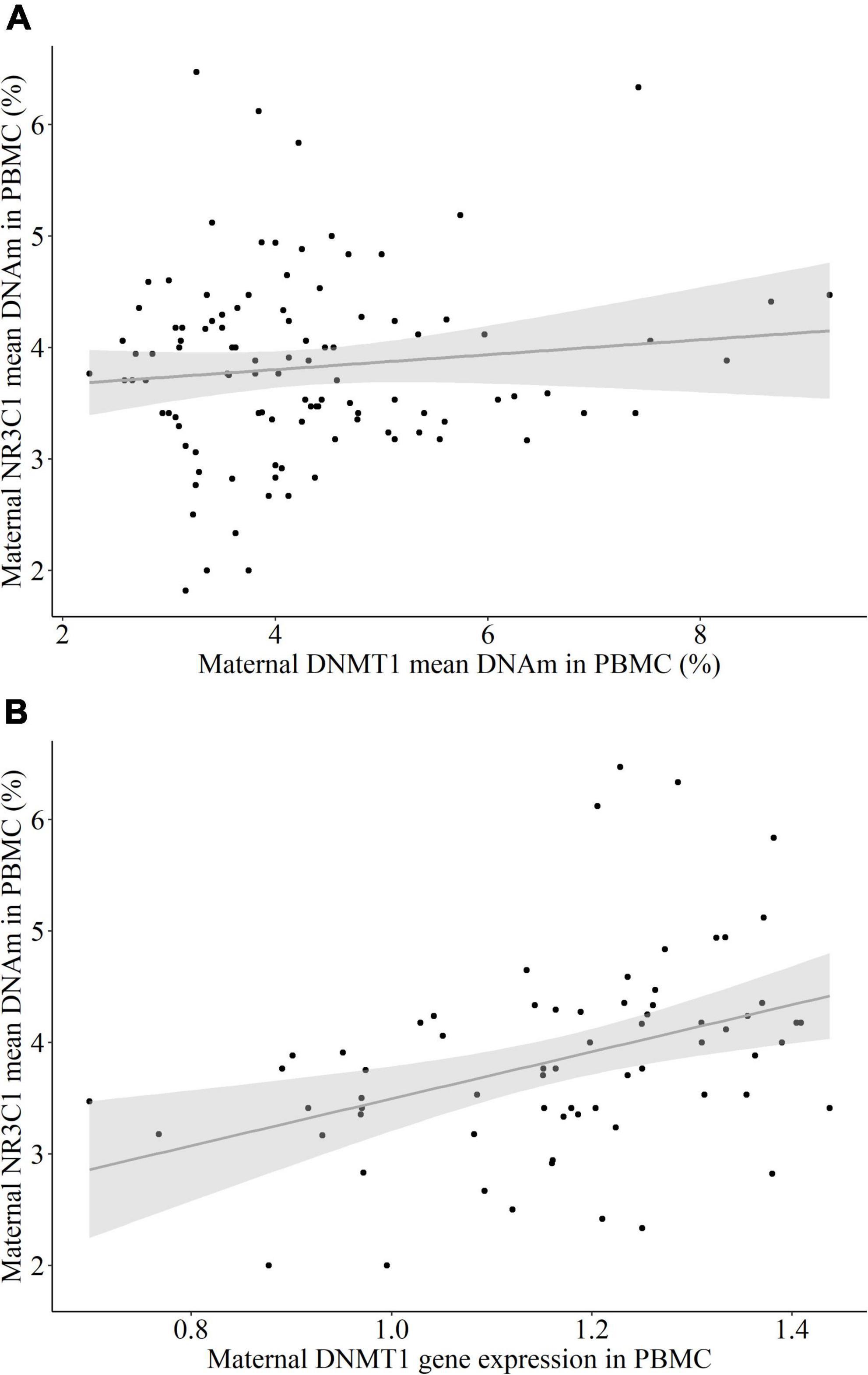
Figure 3. The association between NR3C1 mean DNA methylation (DNAm) and DNMT1 DNAm and relative gene expression in peripheral blood mononuclear cells (PBMC) of the mothers. (A) DNMT1 mean DNAm did not have a significant correlation with NR3C1 exon 1F mean DNAm (rs = 0.031, p = 0.752). (B) DNMT1 relative gene expression had a positive correlation with NR3C1 exon 1F mean DNAm (rs = 0.444, p < 0.001).
No significant difference in the mean percentage of buccal cell DNMT1 DNAm between CMlow– (7.84 ± 2.78%) and CMlow+ mothers (8.05 ± 2.54%, T = 0.328, P = 0.525 [0.375, 0.674], p = 0.744, Figure 4A), nor CMmod– (7.80 ± 2.75%) and CMmod+ mothers (8.24 ± 2.47%, T = 0.829, P = 0.566 [0.405, 0.727], p = 0.412, Figure 4B) was found. There was no association between mean percentage buccal cell DNMT1 DNAm and CTQ sum score (rs = –0.086, p = 0.504, Figure 4C). These results did not change when covariates were introduced in robust regressions (see Section “Data processing and statistical analyses”). No relationship was present between the mean percentage DNMT1 methylation levels of immune and buccal cells (rs = –0.006, p = 0.965, Figure 4D). Buccal cell DNA (7.93 ± 2.65%) was significantly more methylated than PBMC DNA (4.34 ± 1.41%, W = 1853.0, p < 0.001). Analyses with single CpG units revealed no CM-status dependent difference in DNAm percentages after FDR correction (see Supplementary Tables 4, 5).
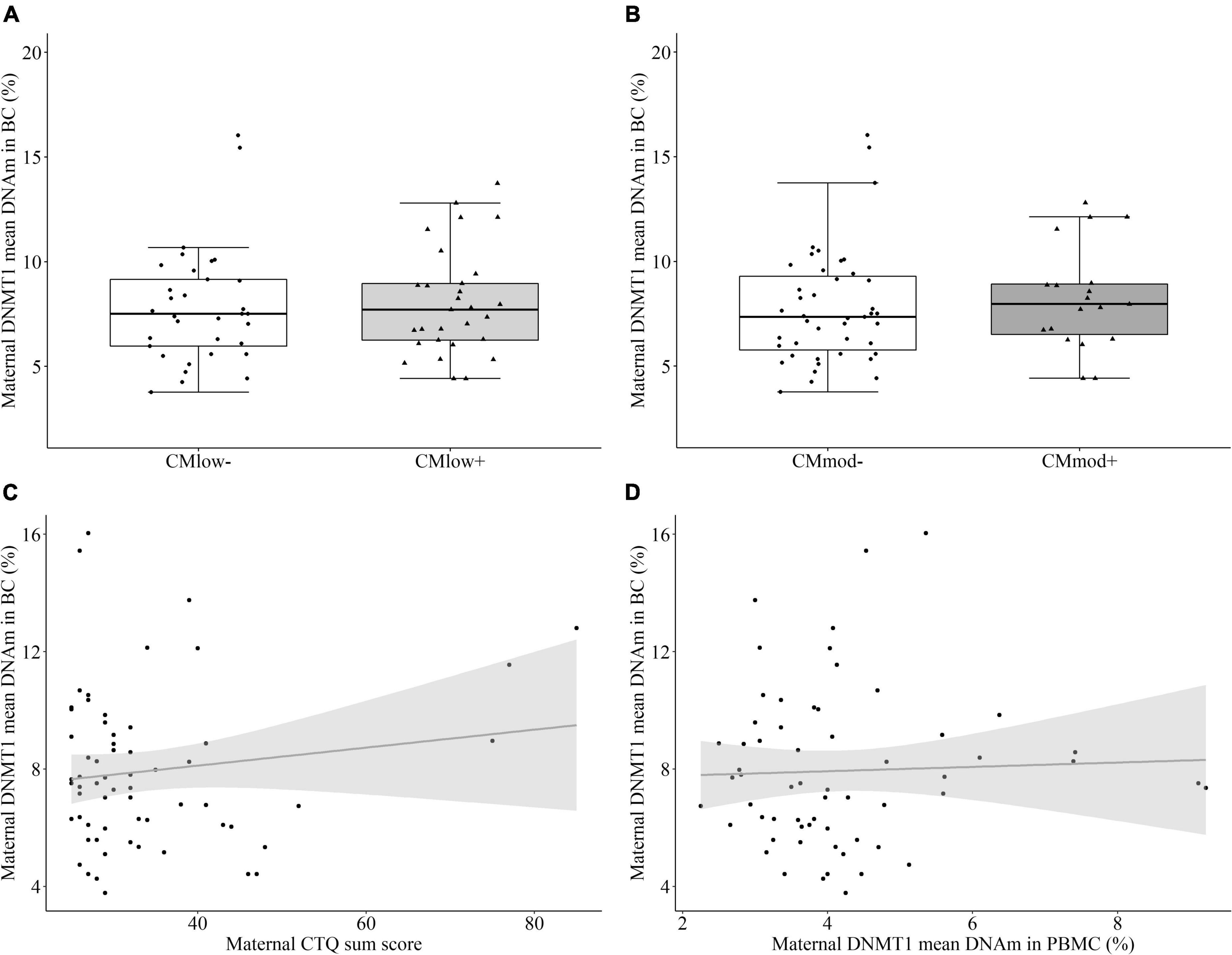
Figure 4. DNA methyltransferase 1 (DNMT1) mean DNA methylation (DNAm) in buccal cells (BC) of the mothers. (A,B) DNMT1 mean DNA methylation did not differ in mothers categorized with their childhood maltreatment (CM) experiences depending on the low cut-off (CMlow– versus CMlow+) or moderate cut-off (CMmod– versus CMmod+). (C) DNMT1 mean DNAm did not have a significant correlation with CTQ sum score (rs = –0.086, p = 0.504). (D) There was no significant relationship between DNMT1 mean DNAm in immune and buccal cells (rs = –0.006, p = 0.965).
In newborns, maternal CM status did not predict differential mean percentage DNMT1 methylation in UBMC according to neither the low CM cut-off [CMlow– (4.19 ± 0.71%) versus CMlow+ (4.31 ± 1.45%), T = –0.801, P = 0.455 [0.343, 0.567], p = 0.425, Figure 5A] nor the moderate cut-off [CMmod– (4.32 ± 1.18%) versus CMmod+ (4.07 ± 0.93%), T = –1.083, P = 0.430 [0.299, 0.560], p = 0.285, Figure 5B]. There was no association between newborn mean DNMT1 methylation and maternal CTQ sum score (rs = –0.042, p = 0.662, Figure 5C). The results did not change when tested for the effects of covariates (see Section “Data processing and statistical analyses”). There was no difference in female (4.40 ± 1.34%) and male (4.07 ± 0.76%) newborns in terms of mean DNMT1 DNAm (T = –1.161, P = 0.436 [0.323, 0.544], p = 0.240). Single CpG units were not differentially methylated according to maternal CM status (Supplementary Tables 6, 7). Maternal and newborn mean DNMT1 methylation also did not have a significant relationship (rs = 0.146, p = 0.143, Figure 5D). However, DNAm percentage of nine single CpG units were correlated between mother-newborn dyads, independent of the CM status (Supplementary Table 8).
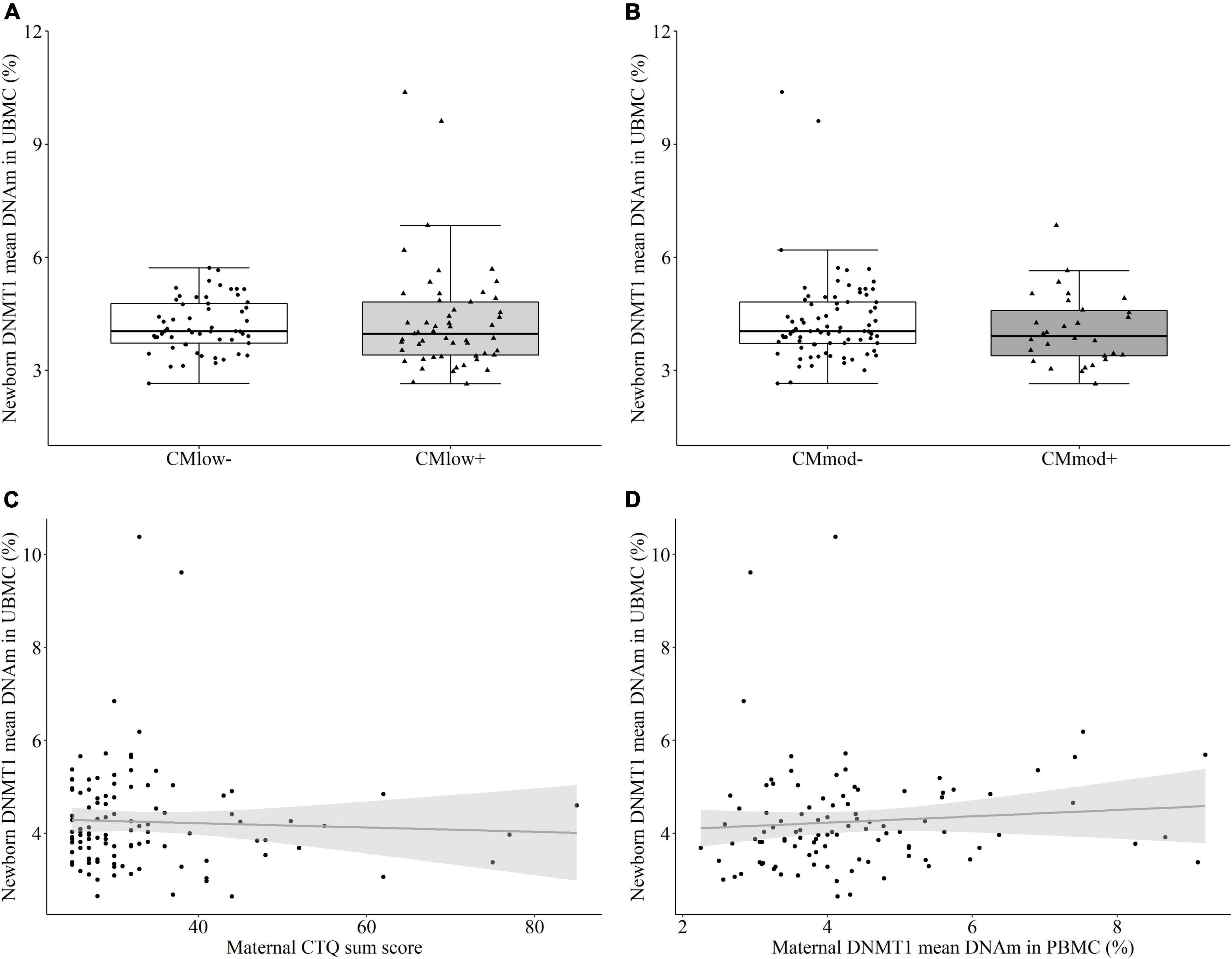
Figure 5. DNA methyltransferase 1 (DNMT1) mean DNA methylation (DNAm) in umbilical cord blood mononuclear cell (UBMC) of the newborns. (A,B) DNMT1 mean DNA methylation did not differ in newborns with a maternal history of childhood maltreatment (CM) experiences depending on the low cut-off (CMlow– versus CMlow+) or moderate cut-off (CMmod– versus CMmod+). (C) DNMT1 mean DNAm did not have a significant correlation with maternal CTQ sum score (rs = –0.042, p = 0.662). (D) There was no significant correlation between DNMT1 mean DNAm of mothers and newborns (rs = 0.146, p = 0.143).
Similar to UBMC, there was no maternal CM-status dependent change in the mean percentage of DNMT1 methylation in buccal cells according to neither the low CM cut-off [CMlow– (7.69 ± 2.84%) versus CMlow+ (7.73 ± 2.98%), T = 0.052, P = 0.504 [0.356, 0.651], p = 0.959, Figure 6A] nor the moderate cut-off [CMmod– (7.95 ± 3.09%) versus CMmod+ (7.16 ± 2.32%), T = –0.609, P = 0.454 [0.304, 0.605], p = 0.545, Figure 6B]. Maternal CTQ sum score was not associated with newborn buccal cell mean DNMT1 methylation (rs = 0.84, p = 0.505, Figure 6C). The results were similar when tested for the effects of covariates (see Section “Data processing and statistical analyses”). DNAm percentage of single CpG units did not differ according to the maternal CM status (Supplementary Tables 9, 10). In buccal cells, maternal and newborn mean DNMT1 methylation (rs = 0.061, p = 0.647, Figure 6D) as well as DNAm percentage of single CpG units (Supplementary Table 4) also did not have an association. Similar to mothers, no relationship was found between newborn buccal and immune cell mean DNMT1 methylation (rs = –0.058, p = 0.655), and buccal cell mean methylation (7.71 ± 2.88%) was higher than immune cell mean DNAm (4.25 ± 1.12%, W = 1868.0, p < 0.001).
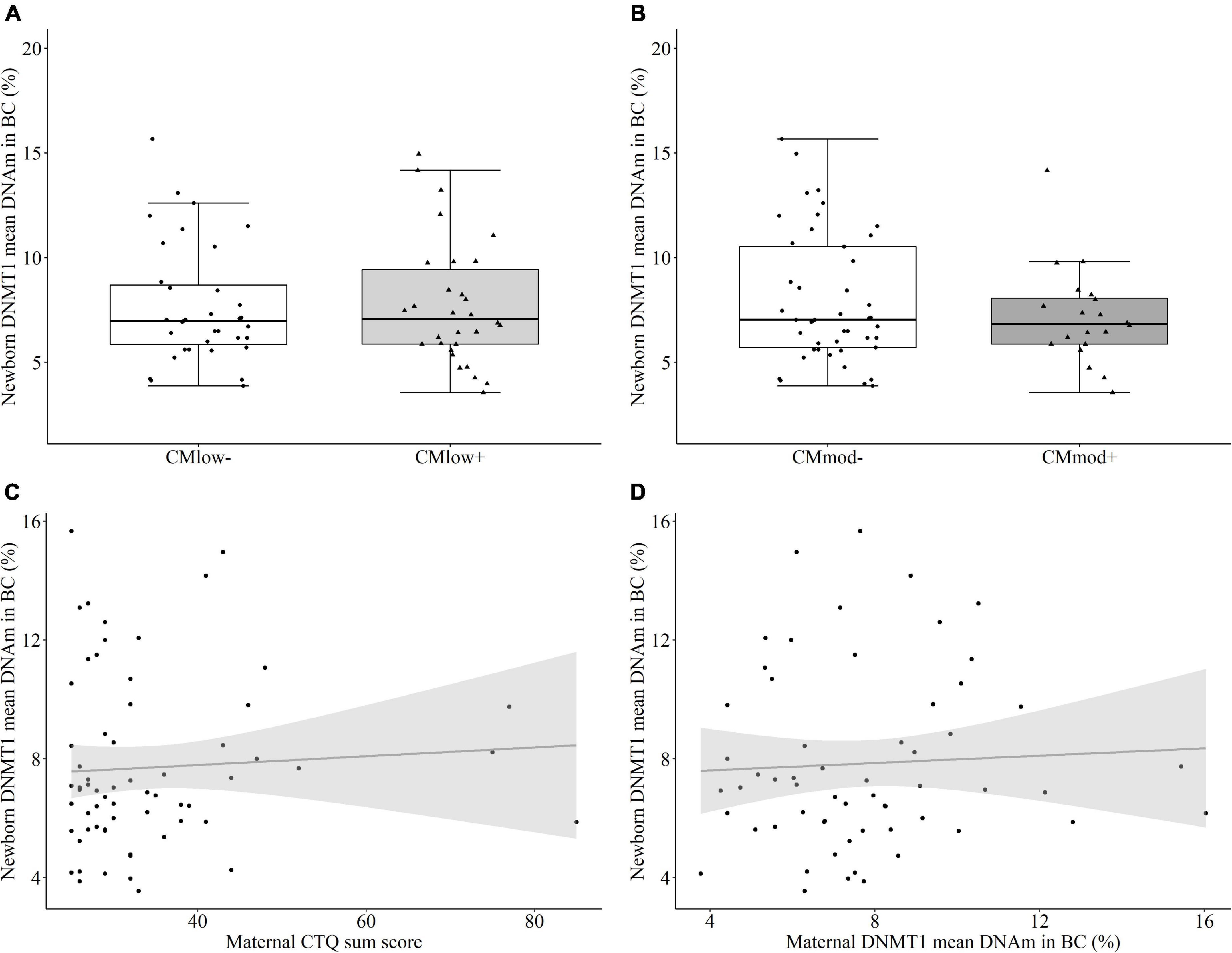
Figure 6. DNA methyltransferase 1 (DNMT1) mean DNA methylation (DNAm) in buccal cells (BC) of the newborns. (A,B) DNMT1 mean DNA methylation did not differ in newborns with a maternal history of childhood maltreatment (CM) experiences depending on the low cut-off (CMlow– versus CMlow+) or moderate cut-off (CMmod– versus CMmod+). (C) DNMT1 mean DNAm did not have a significant correlation with maternal CTQ sum score (rs = 0.84, p = 0.505). (D) There was no significant correlation between DNMT1 mean DNAm of mothers and newborns (rs = 0.061, p = 0.647).
This study aimed to explore the effects of different severities of maternal CM history on DNMT1 methylation in immune and buccal cells of mother-newborn dyads. The results revealed associations between maternal DNMT1 methylation and low as well as moderate CM in immune cells, such that mothers with a history of CM had significantly lower DNMT1 mean DNAm than mothers without CM. CM load measured by CTQ sum score was associated with DNMT1 mean DNAm in a trend level (Figure 1C). CM status was also linked to the maternal DNMT1 gene expression in PBMC, depending on the severity of CM: Mothers with a history of moderate CM (CMmod+) had higher relative DNMT1 expression than mothers without a moderate CM history (CMmod–). However, no such difference was found between CMlow– and CMlow+ mothers. CM history was not related to DNMT1 DNAm in buccal cells. As for intergenerational analyses, maternal CM status was not found to be relevant in newborns’ DNMT1 methylation levels neither in UBMC nor in buccal cells. There was also no association between maternal and infant mean DNMT1 DNAm levels in immune or buccal cells. These results indicate no intergenerational transmission of low or moderate history of maternal CM in terms of DNMT1 DNAm, both in terms of mean methylation and single CpG units.
The direction of the results for maternal PBMC supports most animal research and might explain some variance in the differential methylation profiles due to CM. Following diverse early life adversity paradigms, animal studies reported increased DNMT1 mRNA and protein levels in the brain (13–15). We have shown for the first time in humans, that CM is associated with decreased DNMT1 DNAm in immune cells of women who recently gave birth. A history of moderate but not low CM was linked to higher relative DNMT1 expression, compared to no or mild CM. Higher gene expression might mean higher enzymatic activity of DNMT1, meaning more methylation of hemimethylated strands as well as de novo CpG sites (12), depending on CM status. Adopting the traditional view of the relationship between DNA methylation and gene expression, the difference in the results according to CM severity might mean that the impact of CM on DNMT1 DNAm is physiologically meaningful only for individuals with a moderate but not low CM status. However, apart from its influence on gene expression, DNAm might still impact the function of regulatory elements, other epigenetic mechanisms such as microRNA production and histone modification, as well as translational mechanisms such as isoform production (40–42). We have reported no association between mean DNMT1 DNAm and relative DNMT1 expression. Although DNAm of two CpG units did negatively correlate with relative expression (data not shown), new literature challenges the expected negative relationship between DNAm and gene expression, reporting null and even positive associations (43, 44). Additionally, the difference in mean DNMT1 DNAm between groups with and without CM is very low, with a difference of 0.4% in low CM and 0.6% in moderate CM groups. The physiological relevance of this small difference might be challenged and should be treated with caution (45). Nevertheless, CpG sites around the promoter region are known to be rarely methylated (41), therefore a big difference in methylation depending on CM should not be expected in this highly conserved gene.
In complex phenotypes (i.e., psychopathology) that might be influenced by certain environmental factors such as early life adversity, DNAm was proposed to create subtle differences between individuals with and without a certain environmental exposure which might explain some of the variance of the phenotype (42). Sipahi et al. reported 0.8% difference in DNMT1 DNAm in PTSD cases before and after trauma (17), which is close to our findings in a cohort with mostly low CM experiences. Their reported mean DNAm values were around 3–4%, which were also close to our results. They reported a change opposite to the direction of our results, with an increased DNMT1 DNAm after trauma. This contradiction might be due to the time course of the trauma, adulthood versus childhood, which might be associated with differing pathologies (46), thus physiological correlates of these two might differ.
Apart from mean DNAm, the effects of single CpG unit methylation, which differ up to 7% in a unit that is located in a predicted TSS between groups with different CMmod status (Supplementary Tables 2, 3), might also be important since they can influence gene regulation and transcription factor binding. CpG units that seemed to be methylated less in CM+ groups are mostly in regulatory regions in both low and moderate CM categorizations. Considering the decreasing DNAm levels with increased CM severity, our results seem to be a sign of allostatic load for mothers with a history low and moderate CM, and they might also be physiologically relevant at least for mothers with moderate CM, and explain some findings in the literature regarding the CM-dependent differential DNAm. In line with this interpretation, psychopathological conditions with allostatic overload (47) have been previously associated with DNMT1 overactivation (19), and can be treated with antidepressants that have reported to reduce DNMT1 activity (21, 22).
We have reported a positive association between DNMT1 relative gene expression and NR3C1 exon 1F mean DNAm. It hints toward a link between mechanisms of stress-response regulation and DNA methylation, as previously suggested (8–10). As NR3C1 exon 1F DNAm has been repeatedly linked with early life adversity (5), it is possible that with increased levels of stress and allostatic load, glucocorticoid receptor–glucocorticoid complexes impact transcription of DNMT1, which in turn contributes to de novo methylation of the glucocorticoid receptor gene. Nevertheless, the meaning of this association and the direction of a causal relationship is not exactly clear and cannot be properly interpreted using our cross-sectional design. The relationship between the regulators of DNAm machinery and the stress-response systems should be investigated in longitudinal designs, including the remaining elements of these processes.
In line with our previous results (7) concerning stress-response genes, we did not find any evidence concerning an intergenerational transmission of maternal CM-associated DNAm changes. There was no differential methylation due to the maternal history of CM in DNMT1 DNAm in newborn UBMC of neither single CpG units nor mean percentage DNAm of these units. Our sample consisted of mostly healthy women who recently gave birth and their newborns, which enabled us to eliminate the effects of important confounders such as familial environment and parenting behavior that can be also influenced by parental history of CM (27). Our results suggest no intergenerational transmission, at least not as a result of incomplete erosion of CM-related changes in epigenetic signatures in parental gametes. Our results also do not suggest a maternal CM-related influence in fetus development or gestational biology in DNMT1 DNAm. Results might differ in a cohort with a maternal history of severe CM, and high prenatal stress associated with CM. Furthermore, maternal and newborn mean DNMT1 DNAm in immune cells were not associated, but there were associations between methylation levels in newborns and mothers at a single CpG unit level independently of CM status (Supplementary Table 8). Therefore, there might be transmission, or rather inheritance, of some methylation markers independent of any maternal history of childhood trauma. This “transmission” process might be explained by regular physiological processes, such as these regions being protected from demethylation during zygote formation, or being reestablished during fetal development (48). Lastly, different results might be obtained with newborn PBMC compared to UBMC, due to differences in cell type and composition in these cell populations (49).
Our results show the importance of tissue selection in DNAm studies. In contrast to our results with maternal PBMC, we did not find any CM related changes in DNMT1 DNAm in either single CpG units or mean percentage in the buccal cells of mothers. The levels of the methylation signatures in immune versus buccal cells were not correlated either. Studying buccal cells have its advantages compared to immune cells, such as they are more accessible, less invasive, and cheaper to collect. Although they also have a few subtypes (50), buccal cells might be more uniform in terms of cell composition which can give them a more homogeneous DNAm signature, compared to PBMCs which include several subtypes of lymphocytes, monocytes and natural killer cells. However, epigenetic modifications can be tissue and cell type-specific, and one should be mindful about studying the impact of a certain trait or environmental factor on DNAm in a certain tissue. In this study, we hypothesized that CM-related physiological changes such as alterations in immune and neuroendocrine systems (3, 51) might lead to alterations in DNAm of an important regulator of DNAm, DNMT1. These alterations should be relevant to immune cells but not necessarily to buccal cells. To our knowledge, no study so far investigated cross-tissue comparisons of DNAm profiles of stress-related genes or DNMT1 due to a psychological stressor, but one genome-wide DNAm study reported an association between buccal cell and blood DNAm levels of one CpG site linked to CM-related cortisol stress reactivity (52), and another study reported neural correlates of NR3C1 buccal cell methylation (53). Future studies should focus on cross-tissue associations and factors related to differential methylation in tissues.
This study has several limitations. Firstly, although immune cells might be more relevant targets compared to buccal cells, epigenetic changes might be cell type-specific, therefore changing cell compositions between individuals might lead to questionable results since it is not clear whether the results are due to epigenetic modifications or cell-type-specific proliferation. Our models including lymphocyte to monocyte ratios did not change our main results but we could not control for subtypes of these cells as the data was not available. Future studies should investigate how different immune cell subpopulations may impact difference in DNAm levels. One factor that can lead to changes in cell composition is certainly the postpartum state of the sample, which is known to have unique immunological alterations due to parturition (54) that might also compromise the generalizability of our results. However, this period is the best to study intergenerational transmission effects in humans, thereby it was chosen. Days since parturition was used in the robust regression models as a predictor in order to control for changing immune system within the postpartum period. Breastfeeding status, another postpartum variable that can influence immune system and overall metabolism (55, 56) thus our results, could not be assessed in this cohort. We also cannot generalize these results to a male cohort or an older cohort. Additionally, a retrospective self-report measure (CTQ) was used to assess CM. These measures were shown to be only in modest agreement with prospective measures, and report a smaller number of traumatic events compared to prospective measures (57), therefore results of our study might differ with a prospective measure of CM. This sample was limited in terms of CM severity, with mothers with CM having experienced mostly low CM. Nevertheless, even in this healthy population with low CM, we have observed CM-related DNMT1 DNAm changes in immune cells.
We conclude that CM-associated alterations in DNMT1 DNAm do exist and might have physiological relevance, especially in individuals with a more severe CM load. Increased DNMT1 expression due to demethylation linked to CM experiences with at least moderate severity might explain differential methylation profiles due to CM to some extent, especially in stress response genes as shown in NR3C1 DNAm. Our results might demonstrate the effect of stress-response elements on DNMT1 expression thus might reflect allostatic load in individuals with CM. The direction of the PBMC results supports most animal research about DNMT1 levels following adversity, and the pathophysiological conditions that have been characterized with allostatic overload. Furthermore, the CM-related differential methylation in single CpG units might impact the function of regulatory elements and translational mechanisms. Our lack of significant findings in buccal cells shows the tissue-specific effects of CM on DNMT1 DNAm. Importantly, no evidence for intergenerational transmission of DNMT1 DNAm was found, which complements previous findings reporting against transmission of DNAm signatures in cohorts with similar maternal CM experiences.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ulm University. Written informed consent to participate in this study was provided by the participants themselves or the participants’ legal guardians.
AK and ITK conceptualized the study. ITK acquired the funding and organized and supervised the data collection with her team. LRF and AK designed the methylation targets. LRF, AMG, and RNM contributed to biological analyses. RNM performed the statistical analyses, interpreted the findings, and wrote the first draft of the manuscript. All authors provided critical input to the manuscript and revised and approved the final version.
This study was funded by a grant from the Federal Ministry of Education and Research of Germany (BMBF funding number: 01KR1304A). AMG was supported by a scholarship of the Konrad Adenauer Foundation.
We thank Alexander Behnke for his suggestions in statistical analyses and the “My Childhood—Your Childhood” project team for their contributions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2022.945343/full#supplementary-material
1. Moody G, Cannings-John R, Hood K, Kemp A, Robling M. Establishing the international prevalence of self-reported child maltreatment: a systematic review by maltreatment type and gender. BMC Public Health. (2018). 18:1164. doi: 10.1186/s12889-018-6044-y
2. Springer KW, Sheridan J, Kuo D, Carnes M. Long-term physical and mental health consequences of childhood physical abuse: Results from a large population-based sample of men and women. Child Abuse Negl. (2007) 31:517–30. doi: 10.1016/j.chiabu.2007.01.003
3. Berens AE, Jensen SKG, Nelson CA. Biological embedding of childhood adversity: from physiological mechanisms to clinical implications. BMC Med. (2017) 15:135. doi: 10.1186/s12916-017-0895-4
4. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. (2013) 14:204–20. doi: 10.1038/nrg3354
5. Cecil CAM, Zhang Y, Nolte T. Childhood maltreatment and DNA methylation: A systematic review. Neurosci Biobehav Rev. (2020) 112:392–409. doi: 10.1016/j.neubiorev.2020.02.019
6. Parade SH, Huffhines L, Daniels TE, Stroud LR, Nugent NR, Tyrka AR. A systematic review of childhood maltreatment and DNA methylation: candidate gene and epigenome-wide approaches. Transl Psychiatry. (2021) 11:134. doi: 10.1038/s41398-021-01207-y
7. Ramo-Fernández L, Boeck C, Koenig AM, Schury K, Binder EB, Gündel H, et al. The effects of childhood maltreatment on epigenetic regulation of stress-response associated genes: an intergenerational approach. Sci Rep. (2019) 9:983. doi: 10.1038/s41598-018-36689-2
8. Mourtzi N, Sertedaki A, Charmandari E. Glucocorticoid signaling and epigenetic alterations in stress-related disorders. Int J Mol Sci. (2021) 22:5964. doi: 10.3390/ijms22115964
9. Hong S, Zheng G, Wiley JW. Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology. (2015) 148:148–57. doi: 10.1053/j.gastro.2014.09.032
10. Duan Z, Lu J. DNA methyltransferases in depression: An update. Front Psychiatry. (2020) 11:538683. doi: 10.3389/fpsyt.2020.538683
11. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. (2018) 19:81–92. doi: 10.1038/nrg.2017.80
12. Jeltsch A, Jurkowska RZ. New concepts in DNA methylation. Trends Biochem Sci. (2014) 39:310–8. doi: 10.1016/j.tibs.2014.05.002
13. Jensen Peña C, Monk C, Champagne FA. Epigenetic effects of prenatal stress on 11β-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One. (2012) 7:e39791. doi: 10.1371/journal.pone.0039791
14. Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, et al. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. (2013) 68:184–94. doi: 10.1016/j.neuropharm.2012.04.013
15. Urb M, Anier K, Matsalu T, Aonurm-Helm A, Tasa G, Koppel I, et al. Glucocorticoid receptor stimulation resulting from early life stress affects expression of DNA methyltransferases in rat prefrontal cortex. J Mol Neurosci. (2019) 68:99–110. doi: 10.1007/s12031-019-01286-z
16. Blaze J, Roth TL. Exposure to caregiver maltreatment alters expression levels of epigenetic regulators in the medial prefrontal cortex. Int J Dev Neurosci. (2013) 31:804–10. doi: 10.1016/j.ijdevneu.2013.10.001
17. Sipahi L, Wildman DE, Aiello AE, Koenen KC, Galea S, Abbas A, et al. Longitudinal epigenetic variation of DNA methyltransferase genes is associated with vulnerability to post-traumatic stress disorder. Psychol Med. (2014) 44:3165–79. doi: 10.1017/S0033291714000968
18. Clukay CJ, Hughes DA, Rodney NC, Kertes DA, Mulligan CJ. DNA methylation of methylation complex genes in relation to stress and genome-wide methylation in mother-newborn dyads. Am J Phys Anthropol. (2018) 165:173–82. doi: 10.1002/ajpa.23341
19. Dong E, Ruzicka WB, Grayson DR, Guidotti A. DNA-methyltransferase1 (DNMT1) binding to CpG rich GABAergic and BDNF promoters is increased in the brain of schizophrenia and bipolar disorder patients. Schizophr Res. (2015) 167:35–41. doi: 10.1016/j.schres.2014.10.030
20. Mohan KN. DNMT1: catalytic and non-catalytic roles in different biological processes. Epigenomics. (2022) 14:629–43. doi: 10.2217/epi-2022-0035
21. Perisic T, Zimmermann N, Kirmeier T, Asmus M, Tuorto F, Uhr M, et al. Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology. (2010) 35:792–805. doi: 10.1038/npp.2009.188
22. Zimmermann N, Zschocke J, Perisic T, Yu S, Holsboer F, Rein T. Antidepressants inhibit DNA methyltransferase 1 through reducing G9a levels. Biochem J. (2012) 448:93–102. doi: 10.1042/BJ20120674
23. Morris MJ, Na ES, Autry AE, Monteggia LM. Impact of DNMT1 and DNMT3a forebrain knockout on depressive- and anxiety like behavior in mice. Neurobiol Learn Mem. (2016) 135:139–45. doi: 10.1016/j.nlm.2016.08.012
24. Negriff S, Palmer Molina A, Hackman DA. Parental exposure to childhood maltreatment and offspring’s mental health: investigating pathways through parental adversity and offspring exposure to maltreatment. Child Maltreat. (2020) 25:422–32. doi: 10.1177/1077559520912342
25. Jawaid A, Jehle K-L, Mansuy IM. Impact of parental exposure on offspring health in humans. Trends Genet. (2021) 37:373–88. doi: 10.1016/j.tig.2020.10.006
26. Perez MF, Lehner B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat Cell Biol. (2019) 21:143–51. doi: 10.1038/s41556-018-0242-9
27. Moog NK, Heim CM, Entringer S, Simhan HN, Wadhwa PD, Buss C. Transmission of the adverse consequences of childhood maltreatment across generations: Focus on gestational biology. Pharmacol Biochem Behav. (2022) 215:173372. doi: 10.1016/j.pbb.2022.173372
28. Dong E, Dzitoyeva SG, Matrisciano F, Tueting P, Grayson DR, Guidotti A. Brain-derived neurotrophic factor epigenetic modifications associated with schizophrenia-like phenotype induced by prenatal stress in mice. Biol Psychiatry. (2015) 77:589–96. doi: 10.1016/j.biopsych.2014.08.012
29. Gapp K, Bohacek J, Grossmann J, Brunner AM, Manuella F, Nanni P, et al. Potential of environmental enrichment to prevent transgenerational effects of paternal trauma. Neuropsychopharmacology. (2016) 41:2749–58. doi: 10.1038/npp.2016.87
30. Perroud N, Rutembesa E, Paoloni-Giacobino A, Mutabaruka J, Mutesa L, Stenz L, et al. The Tutsi genocide and transgenerational transmission of maternal stress: epigenetics and biology of the HPA axis. World J Biol Psychiatry. (2014) 15:334–45. doi: 10.3109/15622975.2013.866693
31. Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. (2014) 5:5592. doi: 10.1038/ncomms6592
32. Bader K, Hänny C, Schäfer V, Neuckel A, Kuhl C. Childhood trauma questionnaire — psychometrische eigenschaften einer deutschsprachigen version. Z Für Klin Psychol Psychother. (2009) 38:223–30. doi: 10.1026/1616-3443.38.4.223
33. Bernstein DP, Fink L. Childhood trauma questionnaire: A retrospective self-report manual. San Antonio, TX: Harcourt Brace & Co (1998). 68 p.
34. Suchiman HED, Slieker RC, Kremer D, Slagboom PE, Heijmans BT, Tobi EW. Design, measurement and processing of region-specific DNA methylation assays: the mass spectrometry-based method EpiTYPER. Front Genet. (2015) 6:287. doi: 10.3389/fgene.2015.00287
35. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
36. Brunner E, Munzel U. The nonparametric behrens-fisher problem: Asymptotic theory and a small-sample approximation. Biom J. (2000) 42:17–25.
37. Brunner E, Bathke AC, Konietschke F. Rank and pseudo-rank procedures for independent observations in factorial designs: Using R and SAS. Cham: Springer International Publishing (2018). doi: 10.1007/978-3-030-02914-2
38. Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
39. Venables WN, Ripley BD. Modern Applied Statistics with S. 4th ed. New York, NY: Springer (2002).
40. Chen Y-C, Elnitski L. Aberrant DNA methylation defines isoform usage in cancer, with functional implications. PLoS Comput Biol. (2019) 15:e1007095. doi: 10.1371/journal.pcbi.1007095
41. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. (2013) 38:23–38. doi: 10.1038/npp.2012.112
42. Leenen FAD, Muller CP, Turner JD. DNA methylation: conducting the orchestra from exposure to phenotype? Clin Epigenetics. (2016) 8:92. doi: 10.1186/s13148-016-0256-8
43. Taylor DL, Jackson AU, Narisu N, Hemani G, Erdos MR, Chines PS, et al. Integrative analysis of gene expression, DNA methylation, physiological traits, and genetic variation in human skeletal muscle. Proc Natl Acad Sci USA. (2019) 116:10883–8. doi: 10.1073/pnas.1814263116
44. Spainhour JC, Lim HS, Yi SV, Qiu P. Correlation patterns between DNA methylation and gene expression in the cancer genome atlas. Cancer Inform. (2019) 18:117693511982877. doi: 10.1177/1176935119828776
45. Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. (2013) 10:949–55. doi: 10.1038/nmeth.2632
46. Zlotnick C, Johnson J, Kohn R, Vicente B, Rioseco P, Saldivia S. Childhood trauma, trauma in adulthood, and psychiatric diagnoses: results from a community sample. Compr Psychiatry. (2008) 49:163–9. doi: 10.1016/j.comppsych.2007.08.007
47. Guidi J, Lucente M, Sonino N, Fava GA. Allostatic load and its impact on health: A systematic review. Psychother Psychosom. (2021) 90:11–27. doi: 10.1159/000510696
48. Seisenberger S, Peat JR, Hore TA, Santos F, Dean W, Reik W. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos Trans R Soc B Biol Sci. (2013) 368:20110330. doi: 10.1098/rstb.2011.0330
49. Paloczi K. Immunophenotypic and functional characterization of human umbilical cord blood mononuclear cells. Leukemia. (1999) 13:S87–9. doi: 10.1038/sj.leu.2401318
50. Theda C, Hwang SH, Czajko A, Loke YJ, Leong P, Craig JM. Quantitation of the cellular content of saliva and buccal swab samples. Sci Rep. (2018) 8:6944. doi: 10.1038/s41598-018-25311-0
51. do Prado CH, Grassi-Oliveira R, Daruy-Filho L, Wieck A, Bauer ME. Evidence for immune activation and resistance to glucocorticoids following childhood maltreatment in adolescents without psychopathology. Neuropsychopharmacology. (2017) 42:2272–82. doi: 10.1038/npp.2017.137
52. Houtepen LC, Vinkers CH, Carrillo-Roa T, Hiemstra M, van Lier PA, Meeus W, et al. Genome-wide DNA methylation levels and altered cortisol stress reactivity following childhood trauma in humans. Nat Commun. (2016) 7:10967. doi: 10.1038/ncomms10967
53. Di Sante J, Ismaylova E, Nemoda Z, Gouin J-P, Yu W-J, Caldwell W, et al. Peripheral DNA methylation of HPA axis-related genes in humans: Cross-tissue convergence, two-year stability and behavioural and neural correlates. Psychoneuroendocrinology. (2018) 97:196–205. doi: 10.1016/j.psyneuen.2018.07.019
54. Christiaens I, Zaragoza DB, Guilbert L, Robertson SA, Mitchell BF, Olson DM. Inflammatory processes in preterm and term parturition. J Reprod Immunol. (2008) 79:50–7. doi: 10.1016/j.jri.2008.04.002
55. Heinig MJ, Dewey KG. Health effects of breast feeding for mothers: a critical review. Nutr Res Rev. (1997) 10:35–56. doi: 10.1079/NRR19970004
56. Krol KM, Grossmann T. Psychological effects of breastfeeding on children and mothers. Bundesgesundheitsblatt – Gesundheitsforschung – Gesundheitsschutz. (2018) 61:977–85. doi: 10.1007/s00103-018-2769-0
Keywords: childhood maltreatment, early life adversity, DNA methylation, intergenerational transmission, DNMT1
Citation: Mavioglu RN, Ramo-Fernandez L, Gumpp AM, Kolassa IT and Karabatsiakis A (2022) A history of childhood maltreatment is associated with altered DNA methylation levels of DNA methyltransferase 1 in maternal but not neonatal mononuclear immune cells. Front. Psychiatry 13:945343. doi: 10.3389/fpsyt.2022.945343
Received: 16 May 2022; Accepted: 24 October 2022;
Published: 10 November 2022.
Edited by:
Jacqueline Samantha Womersley, Stellenbosch University, South AfricaReviewed by:
Annabel Katherine Short, University of California, Irvine, United StatesCopyright © 2022 Mavioglu, Ramo-Fernandez, Gumpp, Kolassa and Karabatsiakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rezan Nehir Mavioglu, bmVoaXIubWF2aW9nbHVAdW5pLXVsbS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.