 Rana El Rawas
Rana El Rawas Inês M. Amaral
Inês M. Amaral Alex Hofer
Alex Hofer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Psychiatry, 30 May 2022
Sec. Schizophrenia
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.868244
This article is part of the Research TopicReviews in Psychiatry 2022: SchizophreniaView all 9 articles
Current pharmacological therapy has limited effects on the cognitive impairments and negative symptoms associated with schizophrenia. Therefore, understanding the molecular underpinnings of this disorder is essential for the development of effective treatments. It appears that a reduction in calcium/calmodulin-dependent protein kinase II (α-CaMKII) activity is a common mechanism underlying the abnormal social behavior and cognitive deficits associated with schizophrenia. In addition, in a previous study social interaction with a partner of the same sex and weight increased the activity of α-CaMKII in rats. Here, we propose that boosting of CaMKII signaling, in a manner that counteracts this neuropsychiatric disease without disrupting the normal brain function, might ameliorate the abnormalities in social cognition and the negative symptoms of schizophrenia.
Schizophrenia is a complex, chronic, and polygenic neuropsychiatric disorder that affects more than one percent of the world adult population. Typical clinical manifestations are positive symptoms (e.g., hallucinations, delusions, disordered thoughts, and speech), negative symptoms (e.g., deficits in social interaction, diminished expression and motivation, anhedonia, apathy), and deficits in both neurocognition (processing speed, attention/vigilance, working memory, verbal learning and memory, visual learning and memory, reasoning and problem solving, verbal comprehension, and verbal fluency) and social cognition (emotional processing, social perception and knowledge, theory of mind, and attributional bias). Notably, negative and cognitive symptoms have a larger impact on patients’ functioning than positive symptoms (1) and correlate with the degree of disability (2, 3).
Around 20–35% of the people affected by schizophrenia fail to respond to antipsychotics (4) and current pharmacological therapy has limited effects on cognitive impairments and negative symptoms (5). Moreover, existing treatments reduce the severity of symptoms rather than providing a cure. Therefore, understanding the molecular mechanisms underlying schizophrenia is essential for the development of effective treatments.
For many years, the essential role of dopamine in the pathogenesis of schizophrenia has been proposed for the reasons that all currently available antipsychotic agents target hyperdopaminergia in the brain via postsynaptic dopamine receptor blockade (6) and that in humans, dopamine-like agents such as amphetamine mimic the positive symptoms of schizophrenia (7). On the other hand, phencyclidine (PCP) and ketamine, both non-competitive N-methyl-D-aspartate (NMDA) receptor antagonists, induce schizophrenia-like psychosis (8), thereby supporting a “glutamatergic” implication in the pathophysiology of schizophrenia. Since then, many researchers have suggested that insufficient glutamate neurotransmission is involved in this disorder (9). Particularly, it has been proposed that dysfunction in calcium/calmodulin-dependent protein kinase II (CaMKII) expression and activity is a common mechanism underlying changes in glutamatergic structural and functional synaptic plasticity that may directly contribute to neuropsychiatric diseases (10).
Calcium/calmodulin-dependent protein kinase II is a serine/threonine kinase found throughout the brain (11, 12) and is activated upon Ca2+/calmodulin (CaM) binding. This kinase has a key role in synaptic signaling and consequently in learning and memory, not only due to its cellular and subcellular location, but also due to the time-course of its activity and autophosphorylation properties (10, 12, 13). In mammals, CaMKII subunits are encoded by closely related gene products—α, β, γ, and δ. Of note, CaMKII α and β isoforms are predominant in the brain (11).
Each CaMKII isoform comprises (1) an N-terminal catalytic domain that contains the ATP- and the substrate-binding (S) sites, (2) an auto-inhibitory regulatory domain that includes a pseudo-substrate segment and a threonine residue 286 (Thr286) segment (or Thr287, depending on the CaMKII isoform), and (3) a C-terminal association domain. The auto-inhibitory and catalytic domains form a gate that regulates activity in a way that when these domains bind to each other at the S site (binding to the pseudo-substrate region) and at a site known as T (binding to the region around Thr286) of the catalytic domain, the enzyme is inhibited and the gate is closed (12). The association domain is required for oligomerization (13, 14). This region is linked to the catalytic and regulatory domains by a variable region that is responsible for most of the structural differences between isoforms (12).
In the presence of Ca2+, the Ca2+/CaM complex can bind to CaMKII on a region that overlaps with the pseudo-substrate region, opening the gate and inducing a conformational change that will expose the catalytic domain and thus activate CaMKII (14). A site on the NMDA receptor NR2B subunit can bind to the T side, keeping the gate open and the enzyme active even after the dissociation of calmodulin (12). In the presence of Ca2+/CaM, the Thr286 residue on the auto-inhibitory domain of α-CaMKII (or Thr287 on βCaMKII) can become autophosphorylated by a neighboring, activated subunit (14). Even when intracellular Ca2+ levels decrease and CaM dissociates from its complex, the inter-subunit autophosphorylation prevents CaMKII from reverting back to its inactive state (12), acquiring autonomous and Ca2+-independent activity. De-phosphorylation returns the enzyme to an inactive state and is catalyzed by protein phosphatase types 1 and 2A (14).
To model the pathophysiology of schizophrenia, many transgenic mouse lines have been generated. In addition, other animal models based on pharmacological manipulations of the glutamatergic or the dopaminergic system have been explored. It is perceived that reduced CaMKII function (Figure 1) could be a common mechanism for various symptoms observed in schizophrenia (10).
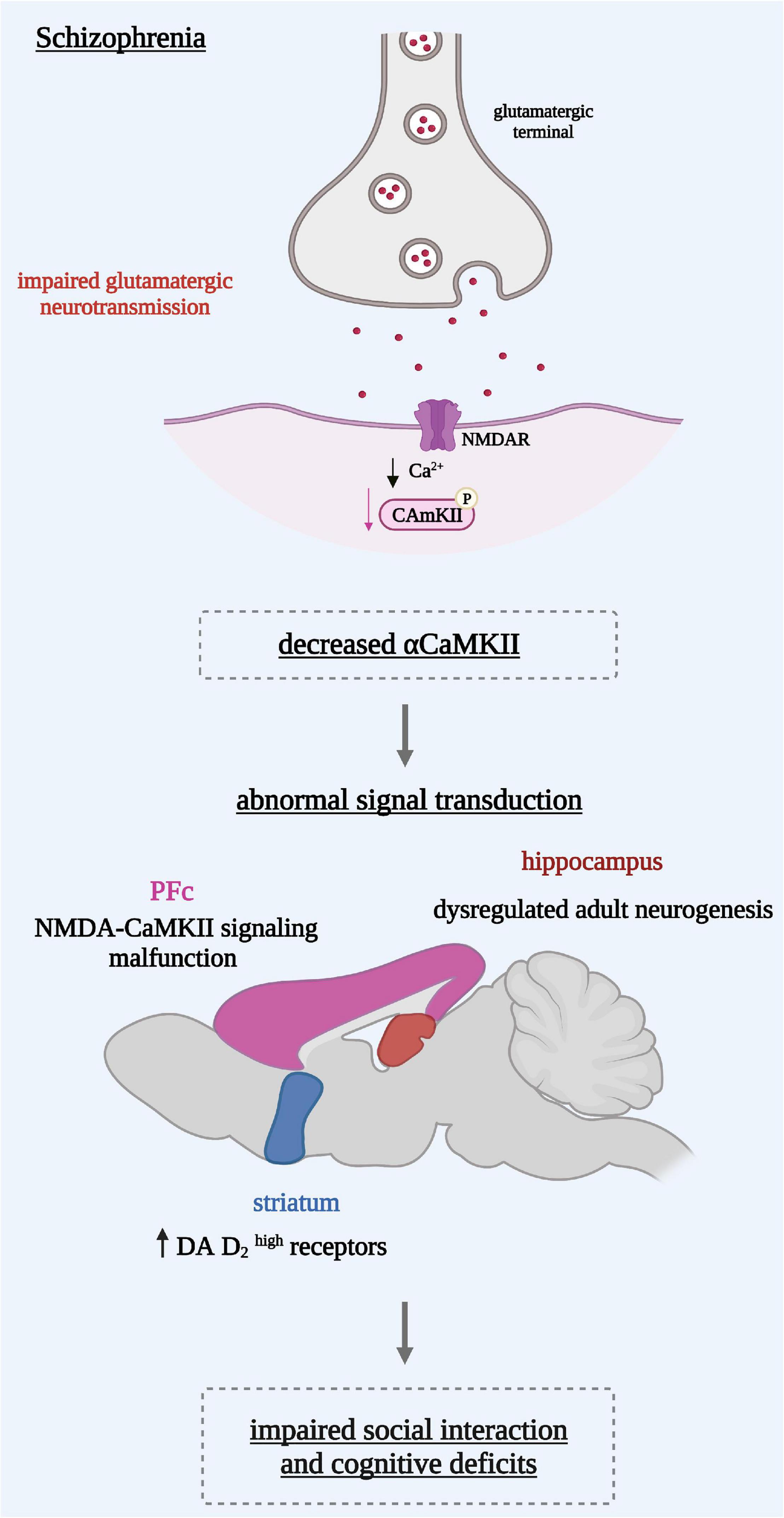
Figure 1. Reduced α-CaMKII induces on the cellular level an abnormal signal transduction reflected in different brain regions. In the hippocampus: a dysregulated adult neurogenesis leading to an immature dentate gyrus in the hippocampus. In the pre-frontal cortex (PFc): a malfunction of NMDA receptor signaling in the associated with dopaminergic hypo-function. In the striatum: dopamine (DA) D2 receptors in a state with a high affinity for DA leading to a hyperdopaminergic state. These mechanisms might underlie behavioral abnormalities such as the social interaction impairments and cognitive deficits seen in schizophrenia.
The most prominent behavioral phenotypes were those carrying a heterozygous null mutation for α-calcium/calmodulin kinase II or α-CaMKII +/− mice. These mice showed features analogous to the ones found in schizophrenia. Most notably, they showed increased locomotor activity, a severe working memory deficit, disrupted circadian activity, and social withdrawal in addition to high levels of aggression toward cage mates (15–17). Moreover, transcriptome analysis and comprehensive autoradiography studies indicated that the mice had marked abnormalities in gene expression and receptor binding in the hippocampus, specifically in the dentate gyrus (DG) (16) where adult neurogenesis partially occurs (18). Whereas the number of newborn neurons in the mutant DG mice was increased by more than 50%, the number of mature neurons was intensely decreased in a way that the DG neurons in the α-CaMKII +/− mice were mostly containing immature neurons leading to an “immature DG” (16).
It has been suggested that adult neurogenesis has a potential role in psychiatric disorders including schizophrenia (19). Indeed, dysregulated adult neurogenesis has been associated with neurocognitive impairments in forms of learning and memory (20), and abnormal hippocampal function (17). In line with these facts, α-CaMKII +/− mice exhibited specific learning impairments, in particular in regards of spatial learning (21).
On the other hand, the levels of dopamine (DA) D2 receptors in a state with a high affinity for DA (D2 high receptors) were found to be elevated in the striatum of α-CaMKII +/− mice, thereby reflecting the hyperdopaminergic state seen in patients with schizophrenia. This high affinity state of DA D2 receptors might possibly be a consequence of elevated β-CaMKII mRNA levels observed in the striatum of these mice with reduced α-CaMKII expression (22), which is probably a compensatory effect. Elevated β-CaMKII subunit mRNA expression in rats’ striatum was also found in the amphetamine sensitization animal model of psychosis (23). In addition, these hyperactive animals show elevated levels of D2 high receptors (22). Remarkably, the CaMKII inhibitor, KN-93, markedly reduced the D2 high states in the rat striatum (22), suggesting that β-CaMKII may increase the D2 high receptors state in the striatum of animals and possibly in schizophrenia (22).
Phencyclidine, a non-competitive NMDA antagonist, reproduces a schizophrenia-like psychosis including positive and negative symptoms as well as cognitive deficits. PCP treated mice have been shown to exhibit hyperlocomotion as an index of positive symptoms, negative symptoms reflected by an enhanced immobility in a forced swimming test, and reduced social interaction and cognitive deficits revealed by impairments of latent learning in a water finding test and recognition memory (24–26). In these mice, α-CaMKII phosphorylation (Thr286) was reduced in the prefrontal cortex (PFc) in comparison to control mice (24, 25, 27, 28). As behavioral impairments and abnormal intracellular signaling were alleviated after potentiation of NMDA receptor function, it has been suggested that repeated PCP treatment induces dysfunction of NMDA-CaMKII signaling in the PFc (24, 28). Moreover, the animals treated repeatedly with PCP failed to release DA in response to high potassium stimulation or a challenge of PCP in the PFc (28). Thus, it is possible that repeated PCP treatment induces a malfunction of NMDA-CaMKII signaling in the PFc, which is associated with dopaminergic hypo-function (28).
This uniquely well situated substrate, predominantly located in the postsynaptic density of excitatory glutamatergic neurons (29), appears to be a common actor in schizophrenia. In other animal models of this disorder, in particular ketamine-treated mice were shown to exhibit deficiencies in sociability and social novelty behavior associated with a significant decrease in hippocampal α-CaMKII expression (30). Previous studies indicate that post-pubertal neonatal ventral hippocampal lesioned rats exhibit impairments in prepulse inhibition (PPI), spontaneous locomotion, social interaction behavior, and working memory (31, 32). In these animals, CaMKII autophosphorylation is significantly reduced, especially in the medial PFc, the striatum, and the hippocampal CA1 region relative to control animals (31, 32). In a model of early life stress, α-CaMKII was found to be downregulated in the PFc (33). Furthermore, late adolescent stress in combination with disrupted-in-Schizophrenia 1 (DISC1) genetic risk impaired activation of NMDA-Ca2+/calmodulin kinase II signaling in the PFc (34), resulting in impaired social interaction and novelty preference for object recognition memory (34). In dysbindin-1-deficient mice, reduced levels of CaMKII were reported in the medial PFc (35). Notably, dysbindin-1 in the PFc has also been shown to be reduced in schizophrenia patients (36, 37). This reduction is thought to promote NMDA receptor hypo-function, thereby leading to the cognitive deficits observed in schizophrenia (37).
Altogether, these findings indicate that the α-CaMKII +/− mouse and others like it may provide genetic biomarkers that can be used to improve treatments for schizophrenia (10). In Table 1, we summarize the findings reporting that dysregulated CaMKII signaling causes impaired social interaction and cognitive deficits.
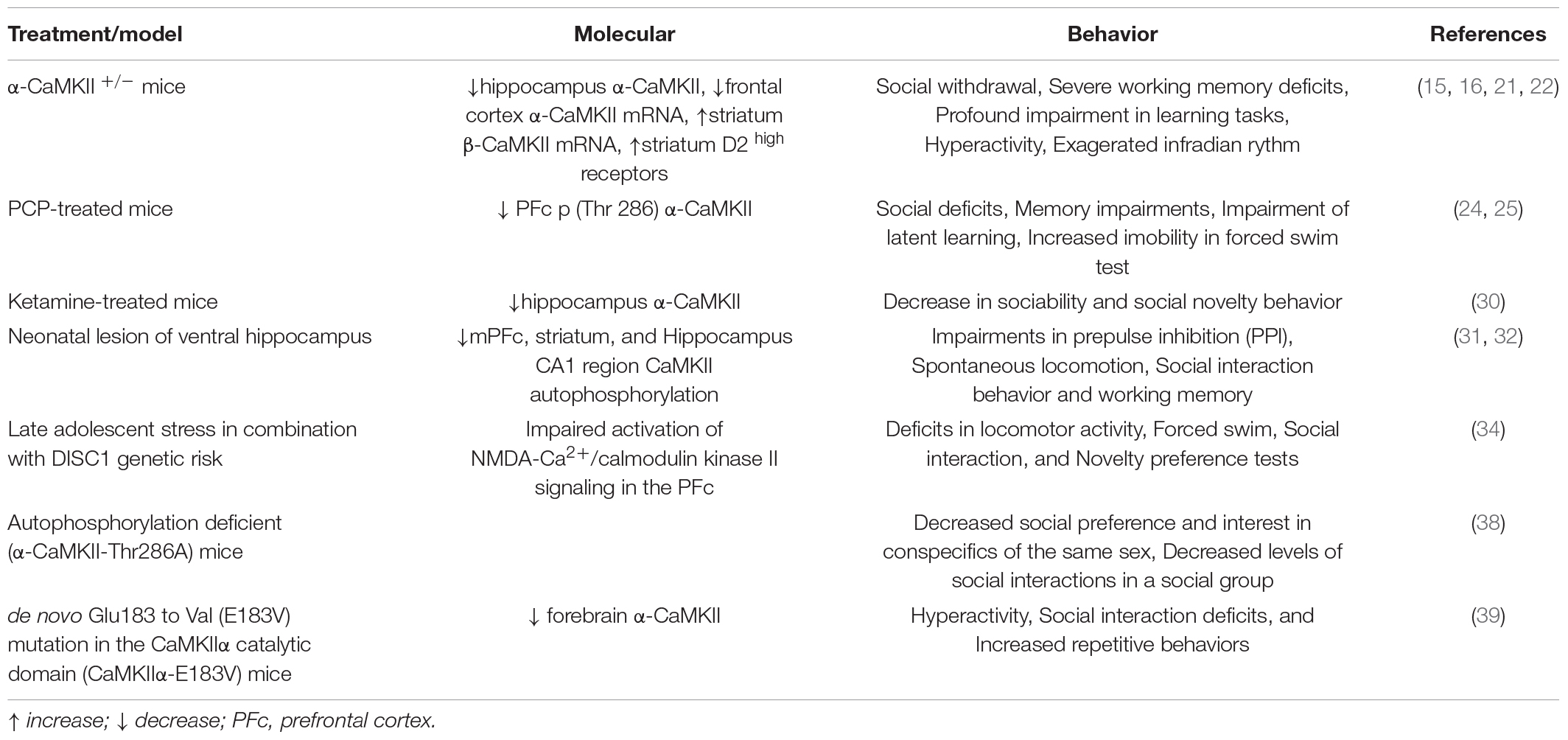
Table 1. Summary of the findings reporting that dysregulated CaMKII signaling causes impaired social interaction and cognitive deficits.
As is the case with many psychiatric disorders and as mentioned above, schizophrenia is characterized by different symptoms: positive symptoms, negative symptoms, and cognitive impairments. Evidently, some of these symptoms are uniquely human and impossible to model in an animal (17). Importantly, the vast majority of the animal models commonly share the profile of impaired social interaction. This behavioral abnormality seems to be tightly linked to CaMKII. Indeed, autophosphorylation-deficient (α-CaMKII-Thr286A) mutant female mice show abnormal social behaviors characterized by decreased social preference and interest in conspecifics of the same sex, as compared to controls (38). Moreover, these mutant mice show decreased levels of social interactions in a social group, as compared to control mice (38). Whereas, control mice increase the frequency of close social interactions during a learning task, α-CaMKII-T286A mutant mice do not (38). In line with these findings, mice with a mutation in the CaMKII-α catalytic domain having lower total forebrain CaMKII-α levels, display aberrant behavioral phenotypes, in particular social interaction deficits (39).
In a recent study, a conditioned place preference (CPP) to a social interaction partner of the same sex, age, and weight was shown to increase α-CaMKII activity in the nucleus accumbens (NAc) (40). In the CPP paradigm, the animal learns to associate a stimulus with a specific context during conditioning, and if this stimulus is appetitive, the animal will prefer to spend more time in the context associated with this stimulus when the choice to “prefer” between a stimulus or a neutral-associated context is given. This study also demonstrated that inhibition of CaMKII in the NAc shell decreases the preference for social interaction (40). These results suggest that social interaction reward is associated with an increased α-CaMKII phosphorylation in this region (Figure 2).
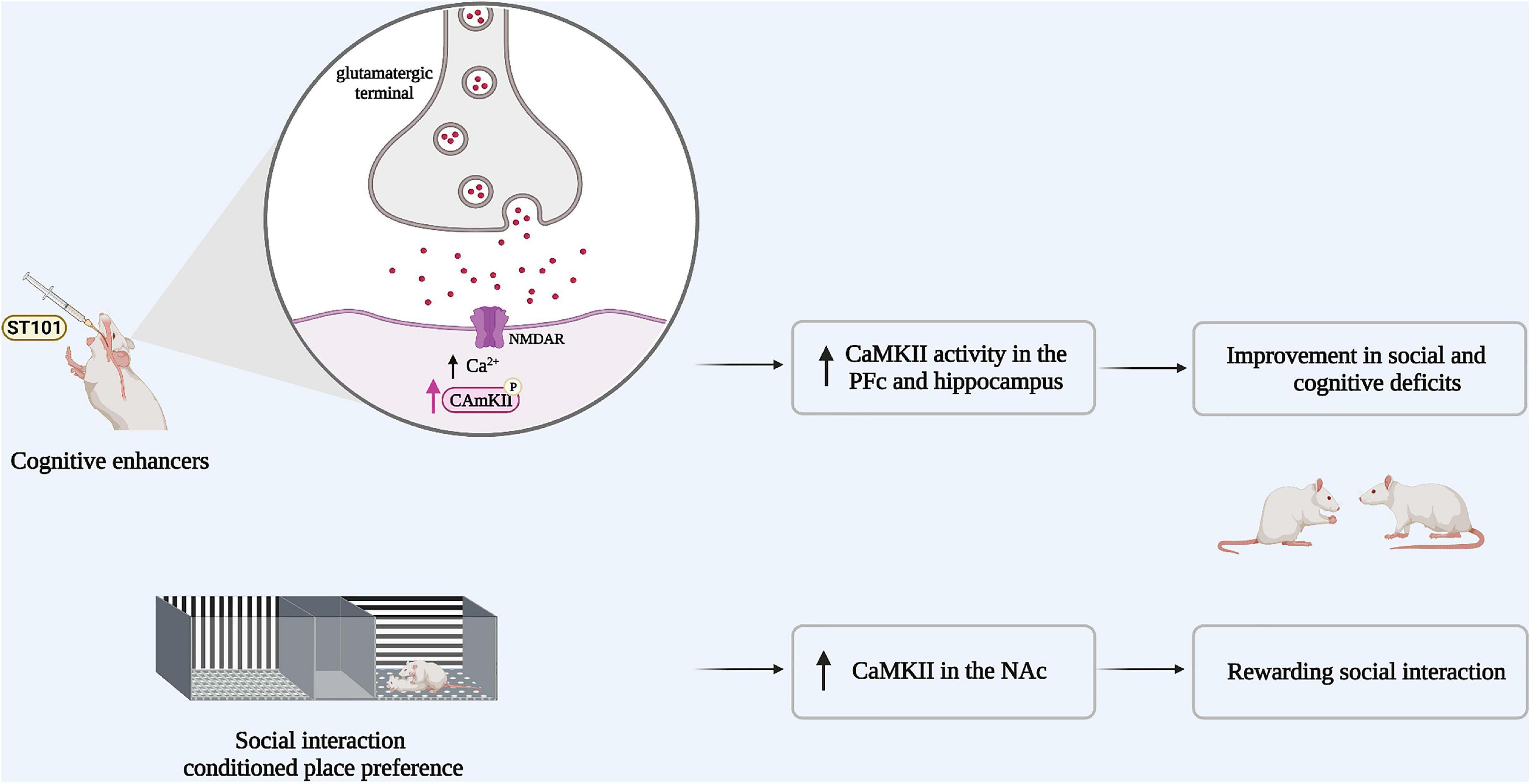
Figure 2. Boosting CaMKII signaling could improve both social and cognitive deficits in schizophrenia. Animal models of schizophrenia share the same behavioral profile, in particular social withdrawal. Impaired social interaction is associated with reduced α-CaMKII activity. If CaMKII activity was potentiated via the administration of cognitive enhancers such as (ST101), CaMKII activity in the pre-frontal cortex (PFc) and the hippocampus is increased and the social impairment is rescued. In parallel, social interaction reward increases α-CaMKII activity in the nucleus accumbens (NAc).
Rats with a neonatal ventral hippocampus lesion exhibit impaired social interaction and reduced CaMKII signaling in memory-related brain regions, resistant to second generation antipsychotics such as risperidone (32, 41). Notably, the administration of the cognitive enhancer spiro[imi-dazo[1,2-a]pyridine-3,2-indan]-2(3H)-one (ST101), an enhancer of T-type calcium channels (42, 43), stimulates CaMKII activity in the hippocampus and the medial PFc and significantly improves deficits in social interaction and cognitive function in these rats (43). It has therefore been proposed that ST101 may improve social interaction and cognitive deficits in neonatal ventral hippocampal lesioned rats by indirectly restoring CaMKII signaling (43) at the opposite of the specific ways to potentially enhance CaMKII activity through gain of function-mutations (44).
Since a reduction in CaMKII activity may underly abnormal social behavior and the cognitive deficits associated with schizophrenia, we hypothesize that an the enhancement of CaMKII signaling could improve both social cognition and negative symptomatology in those living with this disorder (43) (Figure 2).
Studies performed in patients with schizophrenia focused on the expression of CaMKII in post-mortem cerebral frontal cortex. Whereas α-and β-CaMKII protein expression were reported to be significantly reduced in this brain region (45), the expression of β-CaMKII mRNA has been shown to be significantly elevated (46). Additionally, it was reported that the prefrontal cortical expression of α-CaMKII mRNA is comparable in patients with schizophrenia and healthy control subjects (47). Recently, six mutations were found in the α-isoform of CaMKII in patients suffering from schizophrenia (48). Of these mutations, two CaMKII variants show impaired biochemical functions (48). Thus, CaMKII mutations causing impairments in CaMKII function can be a driver for schizophrenia in humans (48). In line with these findings and given that CaMKII is essential for learning and memory formation, several studies reported about new variants in the CaMKII genes that are linked to intellectual disability [for review: (44)]. Specifically, one de novo missense mutation in α-CaMKII was found in patients with autism (44), a disorder comprising social interaction and communication deficits (39). Interestingly, mice carrying the same mutation in α-CaMKII display reduced α-CaMKII protein forebrain levels and deficits in social interactions (39).
These assumptions are in agreement with studies performed in rodents. Indeed, the heterozygous CaMKII knockout, the neonatal ventral hippocampus lesion, and the NMDA-antagonism based models of schizophrenia show decreased CaMKII activity associated to a schizophrenia-like profile, thereby suggesting CaMKII as a potential therapeutic target in schizophrenia. Moreover, preclinical rodent studies enhancing CaMKII activity have demonstrated a potential for the treatment of social and cognitive impairments in schizophrenia (43), previously showing resistance to antipsychotics. This resistance to antipsychotics might be due to the fact that repeated treatment with antipsychotics decreases α-CaMKII protein levels in the striatum (49). Therefore, it is plausible that boosting CaMKII activity in a manner that counteracts this neuropsychiatric disease without disrupting the normal functioning of the brain, might restore this unmet need in the treatment of schizophrenia-like symptoms.
RE, IA, and AH wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
The experiments from our group reported in this mini-review were funded by The Austrian Science Fund (FWF) (Grant Number: T758-BBL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Figures created with Biorender.com.
1. Kharawala S, Hastedt C, Podhorna J, Shukla H, Kappelhoff B, Harvey PD. The relationship between cognition and functioning in schizophrenia: a semi-systematic review. Schizophr Res Cogn. (2022) 27:100217. doi: 10.1016/j.scog.2021.100217
2. McCleery A, Green MF, Hellemann GS, Baade LE, Gold JM, Keefe RSE, et al. Latent structure of cognition in schizophrenia: a confirmatory factor analysis of the MATRICS Consensus Cognitive Battery (MCCB). Psychol Med. (2015) 45:2657–66. doi: 10.1017/S0033291715000641
3. Strauss GP, Horan WP, Kirkpatrick B, Fischer BA, Keller WR, Miski P, et al. Deconstructing negative symptoms of schizophrenia: avolition-apathy and diminished expression clusters predict clinical presentation and functional outcome. J Psychiatr Res. (2013) 47:783–90. doi: 10.1016/j.jpsychires.2013.01.015
4. Mørup MF, Kymes SM, Åström DO. A modelling approach to estimate the prevalence of treatment-resistant schizophrenia in the United States. PLoS One. (2020) 15:234121. doi: 10.1371/journal.pone.0234121
5. Correll CU, Schooler NR. Negative symptoms in schizophrenia: a review and clinical guide for recognition, assessment, and treatment. Neuropsychiatr Dis Treat. (2020) 16:519–34. doi: 10.2147/NDT.S225643
6. Bruijnzeel D, Suryadevara U, Tandon R. Antipsychotic treatment of schizophrenia: an update. Asian J Psychiatr. (2014) 11:3–7. doi: 10.1016/j.ajp.2014.08.002
7. Bramness JG, Gundersen ØH, Guterstam J, Rognli EB, Konstenius M, Løberg EM, et al. Amphetamine-induced psychosis - a separate diagnostic entity or primary psychosis triggered in the vulnerable? BMC Psychiatry. (2012) 12:221. doi: 10.1186/1471-244X-12-221
8. Ellison G. The N-methyl-d-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias. Brain Res Rev. (1995) 20:250–67. doi: 10.1016/0165-0173(94)00014-G
9. Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. (2014) 511:421–7. doi: 10.1038/nature13595
10. Robison AJ. Emerging role of CaMKII in neuropsychiatric disease. Trends Neurosci. (2014) 37:653–62. doi: 10.1016/j.tins.2014.07.001
11. Liu X-B, Murray KD. Neuronal excitability and calcium/calmodulin-dependent protein kinase type II: location, location, location. Epilepsia. (2012) 53:45–52. doi: 10.1111/j.1528-1167.2012.03474.x
12. Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. (2002) 3:175–90. doi: 10.1038/nrn753
13. Shioda N, Fukunaga K. Physiological and pathological roles of CaMKII-PP1 signaling in the brain. Int J Mol Sci. (2018) 19:19010020. doi: 10.3390/ijms19010020
14. Zhang X, Connelly J, Levitan ES, Sun D, Wang JQ. Calcium/calmodulin–dependent protein kinase II in cerebrovascular diseases. Transl Stroke Res. (2021) 12:513–29. doi: 10.1007/s12975-021-00901-9
15. Shin R, Kobayashi K, Hagihara H, Kogan JH, Miyake S, Tajinda K, et al. The immature dentate gyrus represents a shared phenotype of mouse models of epilepsy and psychiatric disease. Bipolar Disord. (2013) 15:405–21. doi: 10.1111/bdi.12064
16. Yamasaki N, Maekawa M, Kobayashi K, Kajii Y, Maeda J, Soma M, et al. Alpha-CaMKII deficiency causes immature dentate gyrus, a novel candidate endophenotype of psychiatric disorders. Mol Brain. (2008) 1:6. doi: 10.1186/1756-6606-1-6
17. Frankland PW, Sakaguchi M, Arruda-Carvalho M. Starting at the endophenotype: a role for alpha-CaMKII in schizophrenia? Mol Brain. (2008) 1:5. doi: 10.1186/1756-6606-1-5
18. Abbott LC, Nigussie F. Adult neurogenesis in the mammalian dentate gyrus. J Vet Med Ser C Anat Histol Embryol. (2020) 49:3–16. doi: 10.1111/ahe.12496
19. Schoenfeld TJ, Cameron HA. Adult neurogenesis and mental illness. Neuropsychopharmacology. (2015) 40:113–28. doi: 10.1038/npp.2014.230
20. Kang E, Wen Z, Song H, Christian KM, Ming GL. Adult neurogenesis and psychiatric disorders. Cold Spring Harb Perspect Biol. (2016) 8:a019026. doi: 10.1101/cshperspect.a019026
21. Silva AJ, Rosahl TW, Chapman PF, Marowitz Z, Friedman E, Frankland PW, et al. Impaired learning in mice with abnormal short-lived plasticity. Curr Biol. (1996) 6:1509–18. doi: 10.1016/S0960-9822(96)00756-7
22. Novak G, Seeman P. Hyperactive mice show elevated D2High receptors, A model for schizophrenia: calcium/calmodulin-dependent kinase II alpha knockouts. Synapse. (2010) 64:794–800. doi: 10.1002/syn.20786
23. Greenstein R, Novak G, Seeman P. Amphetamine sensitization elevates CaMKIIβ mRNA. Synapse. (2007) 61:827–34. doi: 10.1002/syn.20429
24. Nabeshima T, Mouri A, Murai R, Noda Y. Animal model of schizophrenia: dysfunction of NMDA receptor-signaling in mice following withdrawal from repeated administration of phencyclidine. Ann NY Acad Sci. (2006) 1086:160–8. doi: 10.1196/annals.1377.003
25. Aoyama Y, Mouri A, Toriumi K, Koseki T, Narusawa S, Ikawa N, et al. Clozapine ameliorates epigenetic and behavioral abnormalities induced by phencyclidine through activation of dopamine D1 receptor. Int J Neuropsychopharmacol. (2014) 17:723–37. doi: 10.1017/S1461145713001466
26. Mouri A, Noda Y, Enomoto T, Nabeshima T. Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem Int. (2007) 51:173–84. doi: 10.1016/j.neuint.2007.06.019
27. Molteni R, Pasini M, Moraschi S, Gennarelli M, Drago F, Racagni G, et al. Reduced activation of intracellular signaling pathways in rat prefrontal cortex after chronic phencyclidine administration. Pharmacol Res. (2008) 57:296–302. doi: 10.1016/j.phrs.2008.02.007
28. Mouri A, Noda Y, Noda A, Nakamura T, Tokura T, Yura Y, et al. Involvement of a dysfunctional dopamine-D1/N-methyl-D-aspartate-NR1 and Ca2+/calmodulin-dependent protein kinase II pathway in the impairment of latent learning in a model of schizophrenia induced by phencyclidine. Mol Pharmacol. (2007) 71:1598–609. doi: 10.1124/mol.106.032961
29. Fukunaga K, Goto S, Miyamoto E. Immunohistochemical localization of Ca2+/calmodulin-dependent protein kinase II in rat brain and various tissues. J Neurochem. (1988) 51:1070–8. doi: 10.1111/j.1471-4159.1988.tb03070.x
30. Ogundele OM, Lee CC. CaMKIIα expression in a mouse model of NMDAR hypofunction schizophrenia: putative roles for IGF-1R and TLR4. Brain Res Bull. (2018) 137:53–70. doi: 10.1016/j.brainresbull.2017.11.007
31. Yabuki Y, Nakagawasai O, Tadano T, Fukunaga K. Imaging monitoring method of CaMKII Activity by immunohistochemical analysis in schizophrenic model rats. Yakugaku Zasshi. (2013) 133:501–6. doi: 10.1248/yakushi.12-00278-3
32. Yabuki Y, Nakagawasai O, Moriguchi S, Shioda N, Onogi H, Tan-No K, et al. Decreased CaMKII and PKC activities in specific brain regions are associated with cognitive impairment in neonatal ventral hippocampus-lesioned rats. Neuroscience. (2013) 234:103–15. doi: 10.1016/j.neuroscience.2012.12.048
33. Györffy BA, Gulyássy P, Gellén B, Völgyi K, Madarasi D, Kis V, et al. Widespread alterations in the synaptic proteome of the adolescent cerebral cortex following prenatal immune activation in rats. Brain Behav Immun. (2016) 56:289–309. doi: 10.1016/j.bbi.2016.04.002
34. Matsumoto Y, Niwa M, Mouri A, Noda Y, Fukushima T, Ozaki N, et al. Adolescent stress leads to glutamatergic disturbance through dopaminergic abnormalities in the prefrontal cortex of genetically vulnerable mice. Psychopharmacology. (2017) 234:3055–74. doi: 10.1007/s00213-017-4704-8
35. Papaleo F, Yang F, Garcia S, Chen J, Lu B, Crawley JN, et al. Dysbindin-1 modulates prefrontal cortical activity and schizophrenia-like behaviors via dopamine/D2 pathways. Mol Psychiatry. (2012) 17:85–98. doi: 10.1038/mp.2010.106
36. Weickert CS, Straub RE, McClintock BW, Matsumoto M, Hashimoto R, Hyde TM, et al. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry. (2004) 61:544–55. doi: 10.1001/archpsyc.61.6.544
37. Tang J, LeGros RP, Louneva N, Yeh L, Cohen JW, Hahn CG, et al. Dysbindin-1 in dorsolateral prefrontal cortex of schizophrenia cases is reduced in an isoform-specific manner unrelated to dysbindin-1 mRNA expression. Hum Mol Genet. (2009) 18:3851–63. doi: 10.1093/hmg/ddp329
38. Harda Z, Dzik JM, Nalberczak-Skóra M, Meyza K, Łukasiewicz K, Łęski S, et al. Autophosphorylation of αCaMKII affects social interactions in mice. Genes, Brain Behav. (2018) 17:12457. doi: 10.1111/gbb.12457
39. Stephenson JR, Wang X, Perfitt TL, Parrish WP, Shonesy BC, Marks CR, et al. A novel human CAMK2a mutation disrupts dendritic morphology and synaptic transmission, and causes ASD-related behaviors. J Neurosci. (2017) 37:2216–33. doi: 10.1523/JNEUROSCI.2068-16.2017
40. Amaral IM, Scheffauer L, Langeder AB, Hofer A, El Rawas R. Rewarding social interaction in rats increases CaMKII in the nucleus accumbens. Biomedicines. (2021) 9:1886. doi: 10.3390/biomedicines9121886
41. Rueter LE, Ballard ME, Gallagher KB, Basso AM, Curzon P, Kohlhaas KL. Chronic low dose risperidone and clozapine alleviate positive but not negative symptoms in the rat neonatal ventral hippocampal lesion model of schizophrenia. Psychopharmacology. (2004) 176:312–9. doi: 10.1007/s00213-004-1897-4
42. Yabuki Y, Matsuo K, Izumi H, Haga H, Yoshida T, Wakamori M, et al. Pharmacological properties of SAK3, a novel T-type voltage-gated Ca2+ channel enhancer. Neuropharmacology. (2017) 117:1–13. doi: 10.1016/j.neuropharm.2017.01.011
43. Yabuki Y, Wu L, Fukunaga K. Cognitive enhancer ST101 improves schizophrenia-like behaviors in neonatal ventral hippocampus-lesioned rats in association with improved CaMKII/PKC pathway. J Pharmacol Sci. (2019) 140:263–72. doi: 10.1016/j.jphs.2019.07.015
44. Proietti Onori M, van Woerden GM. Role of calcium/calmodulin-dependent kinase 2 in neurodevelopmental disorders. Brain Res Bull. (2021) 171:209–20. doi: 10.1016/j.brainresbull.2021.03.014
45. Matas E, William DJF, Toro CT. Abnormal expression of post-synaptic proteins in prefrontal cortex of patients with schizophrenia. Neurosci Lett. (2021) 745:135629. doi: 10.1016/j.neulet.2021.135629
46. Novak G, Seeman P, Tallerico T. Increased expression of calcium/calmodulin-dependent protein kinase IIβ in frontal cortex in schizophrenia and depression. Synapse. (2006) 59:61–8. doi: 10.1002/syn.20211
47. Xing G, Russell S, Hough C, O’Grady J, Zhang L, Yang S, et al. Decreased prefrontal CaMKII α mRNA in bipolar illness. Neuroreport. (2002) 13:501–5. doi: 10.1097/00001756-200203250-00029
48. Brown CN, Cook SG, Allen HF, Crosby KC, Singh T, Coultrap SJ, et al. Characterization of six CaMKIIα variants found in patients with schizophrenia. iScience. (2021) 24:103184. doi: 10.1016/j.isci.2021.103184
Keywords: schizophrenia, CaMKII, social interaction, negative symptoms, biomarker
Citation: El Rawas R, Amaral IM and Hofer A (2022) The Anti-social Brain in Schizophrenia: A Role of CaMKII? Front. Psychiatry 13:868244. doi: 10.3389/fpsyt.2022.868244
Received: 02 February 2022; Accepted: 01 March 2022;
Published: 30 May 2022.
Edited by:
Neeltje E. M. Van Haren, Sophia Children’s Hospital, NetherlandsReviewed by:
Peter K. Giese, King’s College London, United KingdomCopyright © 2022 El Rawas, Amaral and Hofer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rana El Rawas, cmFuYS5lbC1yYXdhc0BpLW1lZC5hYy5hdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.