
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 15 April 2022
Sec. Molecular Psychiatry
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.866052
This article is part of the Research Topic Unravelling the Role of CB2r in Neuropsychiatric Diseases View all 6 articles
 Alvaro Morcuende1
Alvaro Morcuende1 María Salud García-Gutiérrez1,2,3
María Salud García-Gutiérrez1,2,3 Simone Tambaro4
Simone Tambaro4 Elena Nieto1
Elena Nieto1 Jorge Manzanares1,2,3
Jorge Manzanares1,2,3 Teresa Femenia1,2*
Teresa Femenia1,2*
Emotional behavior, memory, and learning have been associated with alterations in the immune system in neuropsychiatric and neurodegenerative diseases. In recent years, several studies pointed out the involvement of the cannabinoid receptor 2 (CB2r) in the immune system and the regulation of inflammation. This receptor is widely distributed in different tissues and organs with higher expression in spleen and immune system cells. However, CB2r has also been detected in several brain areas and different brain cell types, such as neurons and glia. These findings suggest that CB2r may closely relate the immune system and the brain circuits regulating inflammation, mood, and cognitive functions. Therefore, we review the studies that may help elucidate the molecular bases of CB2r in regulating inflammation in different brain cells and its role in the pathophysiology of psychiatric and neurodegenerative disorders.
Psychiatric disorders are a heterogeneous group where alterations at the inflammatory level have been identified, relating in some cases to the onset or progression of psychiatric disorders.
Studies reported increased circulating inflammation in individuals with mood disorders characterized by cognitive dysfunction (1–9).
Inflammation is a process that helps repair tissue damage or resolve an infection, in which the rapid response of the innate immune system plays a significant role. Different mediators known as Damage Associated Molecular Patterns (DAMPS) are released into the extracellular space during tissue damage. These DAMPS are recognized by specific receptors, Pattern Recognition Receptors (PRR), composed of several families of receptors. The most important PRRs are the Toll-Like Receptors (TLRs) or NOD-Like Receptors (NLRs). These receptors recognize DAMPS and Pathogen-associated Molecular Patterns (PAMPS) or products associated with oxidative stress such as reactive oxygen species (ROS). Once these PRRs are activated, they initiate an intracellular signaling cascade triggering the release of inflammatory mediators through the Type I interferon and Nuclear Factor Kappa-B (NF-kB) signaling pathways, the most studied pathways of pro-inflammatory cytokine production (10, 11). In an inflammatory process, there is a resolution phase where anti-inflammatory mechanisms such as anti-inflammatory cytokines, maresins, and resolvins are stimulated (12). When this fails, the consequence is chronic and pathological inflammation, but still, the organism will try to compensate for this inflammation to reach homeostasis. In this context, a delicate balance between the levels of pro-inflammatory cytokines (e.g., L-1β, IL-6, and TNF-α) and anti-inflammatory (e.g., IL-4, IL-10, IL-11, and IL-13) is necessary for a proper resolution of the inflammation process. These signals are regulated upon the phenotype acquired of the immune cells and microglia at specific time windows (i.e., M1 pro-inflammatory phenotype or M2 anti-inflammatory phenotype) (13, 14).
Noteworthy, the release of DAMPS and the consequent release of pro-inflammatory cytokines produce a systemic sterile inflammation without the participation of pathogens. This systemic inflammation may promote disruption of the blood-brain barrier, making it more permeable to the passage of DAMPS, pro-inflammatory cytokines, or infiltration of leukocytes and macrophages associated with neuroinflammation (15–17). In this way, sterile inflammation has been linked to various psychiatric illnesses, especially to episodes of major depression or bipolar disorder (8, 10, 11). In this sense, it has been observed in different clinical studies that the administration of non-steroidal anti-inflammatory drugs (NSAIDs), mainly using selective COX-2 inhibitors, can improve the psychiatric pathology, evidencing the close correlation between these diseases and the immune system (18).
In this regard, during the last years, many findings suggested the participation of the endogenous cannabinoid system in the modulation of behavior and inflammation. Therefore, due to the possible importance of inflammation in the onset and development of psychiatric diseases, it is necessary to investigate further the precise mechanisms underlying this process to discover new therapeutic targets.
The endogenous cannabinoid system (ECS) is composed of several receptors, including the cannabinoid receptor 1 (CB1r) and the cannabinoid receptor 2 (CB2r), endogenous ligands, and enzymes responsible for their synthesis and degradation. In addition, it should be noted that there are other atypical receptors, not CB1/CB2, which include the GPR family receptors. Their effects could be mediated through the activation of different mechanisms, such as gene transcription mediated by peroxisome proliferator-activated receptors (PPARs), transient receptor potential channels (TRP), or vanilloid receptors like TRPV1 (19–21).
The main actions of CB1r and CB2r include inhibition of adenylate cyclase, receptors coupled to G proteins of the Go and Gi type. However, while CB1r and CB2r have a similar affinity for Gi type, CB2r has a lower affinity for Go than Gi (22); inhibition of voltage-dependent calcium channels; activation of MAP kinases; and modulation of potassium channels. Their activation leads to cell physiology changes as diverse as synaptic function, gene transcription, and cell migration (19, 21, 23, 24). However, cannabinoid receptors combine among themselves; Callen et al. (25) report the presence of CB1-CB2 receptor heteromers in different brain areas, showing a bidirectional cross-antagonism phenomenon between them.
Regarding the distribution of endocannabinoid receptors, CB1r is widely distributed in the central nervous system (CNS), in brain regions such as the cortex, basal ganglia, hippocampus, cerebellum, dorsal and ventral striatum, globus pallidus, and substantia nigra, among other areas. These receptors are found in neurons and glia and seem to play an important role in motivation and cognition, involved in GABAergic and glutamatergic pathways (19, 21, 23, 24). On the other hand, CB2r was initially discovered in the spleen, suggesting a role in immunity. It was believed to be the primary expression site for a long time and with no expression in the brain. Since the CB2R expression in the healthy brain is low and the available tools have important limitations (i.e., issues with antibodies specificity). It was classically thought to be expressed in microglial cells and vascular elements and to increase in large numbers in the presence of tissue damage or neuroinflammation (19, 21, 23, 24). However, it has also been found in basal conditions in neurons and brain areas highly involved in emotional, rewarding, and cognitive behaviors, such as the ventral tegmental area (VTA), nucleus accumbens, amygdala, or hippocampus. Therefore, it is suggested that it may have an essential role in several brain disorders related to these behaviors (24).
Among the endogenous cannabinoids (EC), the two most studied ones are the arachidonoylethanolamine or anandamide (AEA) and the 2-arachidonoylglycerol (2-AG) (23). Typically, the endogenous ligands are derived from membrane phospholipids and therefore are not stored in synaptic vesicles, in contrast to other neurotransmitters (19, 23). However, other EC ligands have been characterized, such as lysophosphatidylinositol (LlI), virodhamine, and noladin ether.
In the synthesis of EC, both AEA and 2-AG are derived from arachidonic acid, the primary source of arachidonic acid, the omega-6 polyunsaturated fatty acids (PUFAs). The AEA is produced from the precursor N-arachidonoyl-phosphatidyl-ethanol (NAPE). It is thought that AEA is obtained from this precursor and by four possible routes, being NAPE phospholipase D (NAPE-PLD), a direct and well-characterized route (19, 23). Although the synthesis of 2-AG appears to be simple, the majority comes from the sequential hydrolysis of arachidonoyl-phosphatidylinositol-bisphosphate by a phospholipase (PLCβ) followed by hydrolysis of the resulting diacylglycerol by the enzyme diacylglycerol lipase (DAGL) (19, 23). The importance of 2-AG as an intermediate metabolite in lipid synthesis must be emphasized. The primary source of arachidonic acid for the biosynthesis of prostaglandins (19, 23) stands out for their involvement as an inflammatory mediator. Two isoforms of DAGL, α, and β, have been characterized. In animal studies, it was proposed that DAGLα is a crucial enzyme for neuronal plasticity in adults.
The metabolization of AEA in the central nervous system is carried out mainly by the fatty acid aminohydrolase or FAAH, which is found in postsynaptic neurons and can also degrade various fatty acid derivatives. In addition, anandamide can be metabolized by cyclooxygenase-2 (COX-2) or N-acylethanolamine amidase (NAAA) (19, 21, 23). The degradation will be carried out mainly by the monoacylglycerol lipase (MAGL) located in presynaptic neurons. In turn, other enzymes that can hydrolyze 2-AG are ABHD6, usually located in dendrites and dendritic spines of excitatory neurons, and ABH12. Similarly, like AEA, 2-AG can also be metabolized by COX-2, and under certain conditions, FAAH could act in its degradation (19, 21, 23). Therefore, it could be hypothesized that COX-2 inhibitors may elevate basal levels of endocannabinoids, producing the beneficial effect observed.
On the other hand, the most popular phytocannabinoid is the delta-9-tetrahydrocannabinol (Δ9-THC), the main psychoactive component of Cannabis sativa cannabidiol (CBD), this one devoid of abuse and dependence, subject of intense medical research. CBD can bind to more than 65 targets and displays 100 times less affinity for CB1r than Δ9-THC (26, 27). In addition, one of the first synthetic treatments employed in humans that acts selectively to the EC system was the antagonist/inverse agonist of CB1r SR141716A (rimonabant). This compound was used to treat obesity, but its commercialization stopped due to a few cases of suicide reported in patients treated with this drug (19–21, 23, 24). This fact pointed out how the modulation of the endogenous cannabinoid system impacts psychiatric disorders.
Currently, CB2r are emerging as potential immunomodulatory agents with specific roles in cell-type specificity. Therefore, knowing these receptors will help understand the mechanisms by which CB2r could impact psychiatry due to the importance of inflammation in neuropsychiatric disorders. A large amount of studies have demonstrated the involvement of inflammatory mediators in mood disorders and in particular, IL-1β and TNF-α in behavioral alterations [For review see. Dantzer et al. (28), Remus and Dantzer (29), and Raison et al. (30)] For this reason, we will review in the following sections how the modulation of CB2 receptors shows promising results for the management of psychiatric diseases (Figure 1).
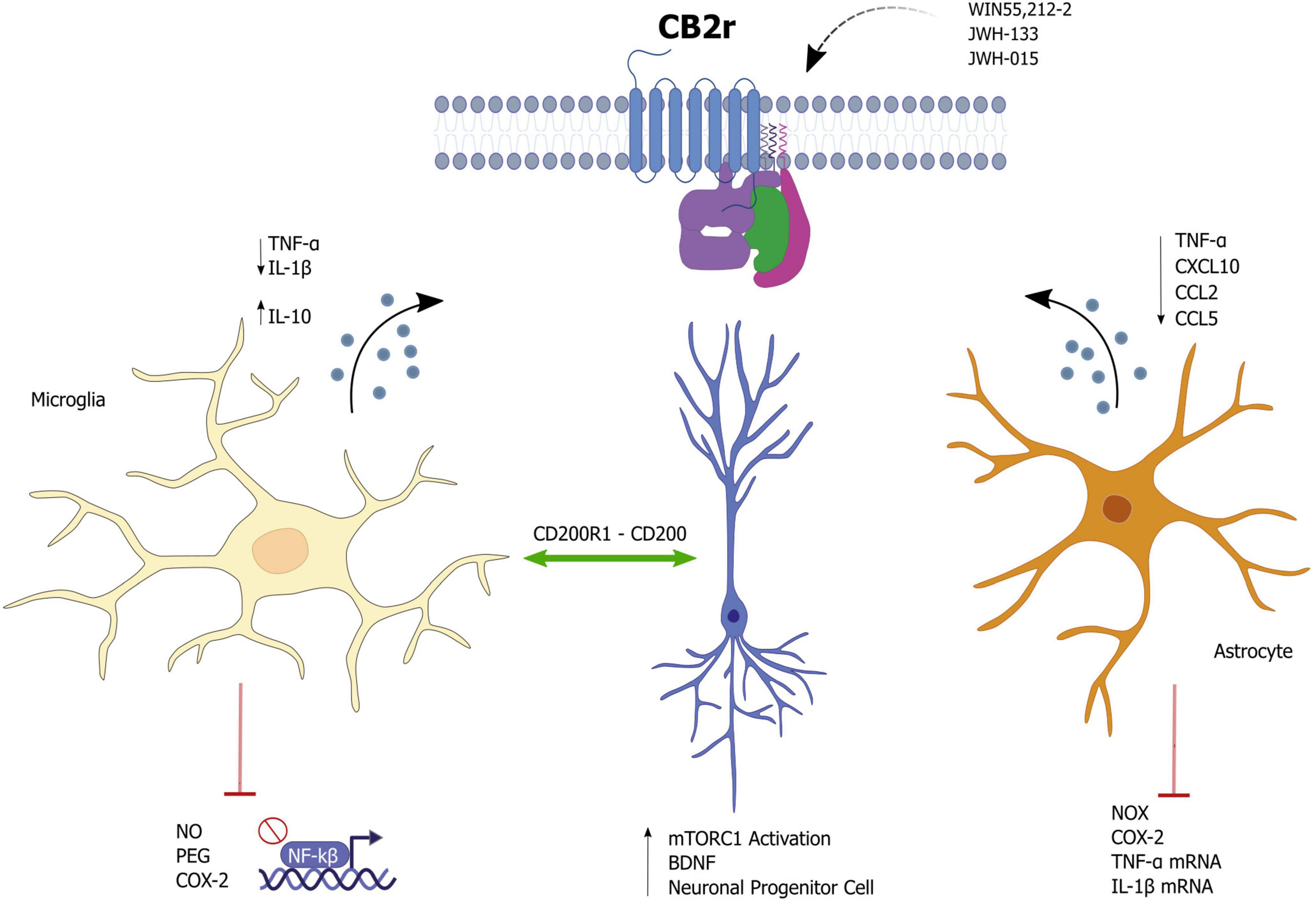
Figure 1. Effects of cannabinoid receptor 2 (CB2r) activation as a potential modulator of neuroinflammation.
Cannabinoid receptor 2 in microglia appears to be involved in a variety of paradigms and diseases involving neuroinflammation, including Alzheimer’s disease (AD), Parkinson’s disease, multiple sclerosis (MS), as well as stress or addiction (8, 31, 32). Some studies showed that activation of CB2 receptors prevents the activation of microglia and the release of pro-inflammatory mediators (33–35). This anti-inflammatory action seems to be responsible for a neuroprotective effect in different animal models of AD, MS, stroke, amyotrophic lateral sclerosis (ALS), and other diseases involving inflammation (36). However, the molecular mechanisms associated with these anti-inflammatory effects remain unknown.
Evidence suggests that CB2r controls microglial activity by interfering with the NF-κB pathway (37) and with the mitogen-activated protein kinases (MAPK) pathways [c-Jun N-terminal Kinase (JNK), ERK, or p38] (38). These kinases are well-known targets for the modulation of inflammation by interfering in downstream signaling pathways of the innate immune response and the production of inflammatory mediators (39). However, the activation of CB2r coupling to MAPKs is dependent on cellular context. A wide range of activation and inhibition has been observed depending on the cell type, cell differentiation status, and co-modulators of MAPK cascades (36).
Early reports showed that CB2r agonists prevented neuronal injury during neuroinflammation by regulating mitogen-activated protein kinase phosphatase-1 (MKP-1), resulting in Erk1/2 inhibition (34). Similarly, Romero-Sandoval reported that activation of CB2r with JWH-015, a selective CB2r agonist, induced an anti-inflammatory phenotype in microglia consistent with a reduction in TNF-α expression and a decrease in microglial migration through inhibition of ERK1/2 phosphorylation and MKP induction (40). Another study also found a reduction of p-ERK and p-p38 133 with an anti-inflammatory effect upon CB2r activation using JWH-133 (41). Moreover, oleamide (ODA), an amide fatty acid with actions at CB1 and CB2 receptors (42), showed a protective effect by inhibiting Nitric Oxide (NO), Prostaglandin E, COX-2 production through inhibition of Akt, p38, and ERK phosphorylation when BV2 microglia were treated with Lipopolysaccharide (LPS). This TLR4 agonist triggers inflammation through NF-kB (43). Guo et al. (37) also reported reduced levels of pro-inflammatory cytokines, such as IL-1β, TNF-α, and IL-6 in BV2 microglia following hypoxic exposure and treatment with Trans-caryophyllene. These anti-inflammatory effects were mediated by inhibiting the NF-κB pathway (37).
Noteworthy, Viscomi et al. (44) found that in axotomized neurons, the JWH-015 agonist did not produce neuroprotection via the ERK1/2 and p38 pathways during remote cell death. Instead, the beneficial effect was seen through an increase in p-Akt and a decrease in p-JNK, a PI3K-dependent effect, suggesting a protective effect of JWH-015 through regulation of JNK via the PI3K/Akt pathway. Importantly, there is a constitutive control of CB2r-mediated basal JNK activity in the mouse brain; the selective CB2r agonist JWH133 reduced JNK phosphorylation, whereas acute administration of the antagonist AM630 would increase it. In contrast, chronic administration of AM630 markedly decreases basal JNK activation, revealing a reversal of the acute effect of the antagonist (45).
In an in vitro model of multiple sclerosis, anandamide showed an anti-inflammatory action by inhibiting LPS/IFNγ-induced NF-kB activation. In particular, anandamide strongly activated the phosphorylation of ERK1/2 and JNK pathways while reducing Akt phosphorylation, which increased IL-10 production in microglia (46).
In addition, several reports suggest that the anti-inflammatory effect of CB2r activation might involve other different pathways such as JAK (Janus kinase)/STAT1 (signal transducer and activator of transcription) (35) or activation of the pCREB-Bcl-2 pathway (47).
CB2r has been positioned as an essential target mechanism for phenotypic change of microglia (38, 48). One of the main ideas of how CB2r can counteract inflammation is shifting microglia toward an anti-inflammatory phenotype, M2, with a consequent increase in the release of anti-inflammatory cytokines such as IL-10. Essentially, the endogenous endocannabinoids 2-AG and AEA increase the expression of the anti-inflammatory phenotype by microglia while decreasing M1 inflammatory microglia markers (46, 49). Similarly, palmitoylethanolamide (PEA) blunted the increase of M1 pro-inflammatory markers induced by lipopolysaccharide (LPS), concomitantly increasing those M2 anti-inflammatory markers in N9 microglial cells partially through CB2r activation (50).
The mechanisms involved in the switch of microglia to an anti-inflammatory phenotype by CB2r are the (cyclic AMP)-dependent protein kinase-mediated pathway found in in vivo and in vitro models of brain injury (51), the PI3K/Akt/mTOR/NF-kB pathway in neuroprotection induced in an animal model of vascular dementia (VD) (52), and the protein kinase C (PKC) and PGC-1α, a transcription protein involved in the regulation of mitochondrial biogenesis (50, 53).
Interestingly, the expression of cannabinoid receptors, mainly CB2, has been related to the microglial phenotype (49). For instance, results from in vitro studies suggest that the deletion of microglial CB2r leads to suppressing inflammatory phenotypes. Also, primary microglia from CB2r deficient mice are less responsive to pro-inflammatory stimuli (54). On the other hand, microglia from CB2r deficient mice cannot polarize to an M2a phenotype (49). Altogether, these findings suggest that CB2rs play a role in microglia activation either to a pro- or anti-inflammatory phenotype. Nevertheless, these findings are still ambiguous, and further research will clarify the fine-tune mechanisms involved in regulating pro- or anti-inflammatory behaviors associated with CB2.
Altogether, these data highlight the critical immunomodulatory functions of CB2 receptors in microglia. Moreover, modulation of this receptor may be an excellent strategy to target neuroinflammation in psychiatric diseases.
It is well-known the role of astrocytes at the level of neuronal support and metabolism (55). Still, it has also been seen how they can intervene, after systemic inflammation, in regulating cognitive functions (56). In this aspect, cb2 receptors expressed in the astrocytes could be contributing to the modulation of inflammation and metabolism in astrocytes to support neuronal function.
The treatment with the CB2r agonist WIN55,212-2, in IL-1β-stimulated human astrocytes, inhibited iNOS together with reduced release of pro-inflammatory mediators such as TNF-α, CXCL10, CCL2, and CCL5. Therefore, the authors suggested that it was primarily mediated by CB2r receptors (57). On the other hand, Jia et al. (58) showed that CB2r could mediate anti-inflammatory actions in astrocytes. CB2 receptor expression is upregulated in astrocytes treated with 1-methyl-4-phenylpyridinium (MPP+), a toxic metabolite that activates glial cells to release inflammatory mediators. Furthermore, the authors showed that pretreatment with the CB2r agonist JWH133 inhibits MPP+-induced iNOS and COX-2 protein up-regulation and TNF-α and IL-1β mRNA up-regulation in astrocytes. Moreover, the CB2r agonist inhibited MPP+-induced iron influx into astrocytes. The authors concluded that all these effects depended on CB2r (58). Additional anti-inflammatory effects for a CB2r agonist have also been reported in the context of spinal glial activation and pain sensitization (59). These findings suggest a functional relationship between astrocytic CB2r signaling and inhibition of p38 phosphorylation, which is associated with an anti-inflammatory effect and suppression of IL-1-induced CX3CL1 upregulation (60, 61).
In addition, pretreatment with a CB2r agonist protected against increased blood-brain barrier permeability in a model of spinal cord ischemia-reperfusion. This action may be primarily mediated by reduced expression of MyD88/NF-κB level pathway and the astrocytic TLR4/Matrix metallopeptidase 9 (MMP-9), an enzyme involved in BBB leakage (62). Other studies show how the stimulation with JWH133 alleviated neuroinflammation and protected BBB permeability in rat models of intracerebral hemorrhage (63, 64). In addition, CB2r is expressed in cell types, different from astrocytes, that form the blood-brain barrier, such as endothelial cells (65), showing the implication of the CB2r in the maintenance of the brain-periphery homeostasis.
The CB2 receptors are involved in protecting neuronal damage caused by neuroinflammation. The most studied mechanisms of this protection were those involving microglia. However, CB2 receptors are also expressed in neurons. This protective effect could be due to neuronal intracellular mechanisms, although it is unknown whether neuronal CB2r can modulate immune functions.
Marchant et al. showed how chronic activation of CB1 and CB2 receptors by WIN-2 proven an anti-inflammatory cytokine profile in the hippocampus of aged rats. While the agonist actions of WIN-2 at CB1/2 receptors promoted neurogenesis in the dentate gyrus, the antagonist actions of WIN-1 at the TRPV1 receptor were responsible for the reduction in microglial activation (66); Furthermore, CB2 receptors have been shown to promote neural progenitor cell proliferation through the activation of mTORC1 signaling (67).
To date, few studies have investigated the differential signaling pathways triggered by CB2r in microglial and neuronal cells. However, neuronal and microglial CB2 receptors have been shown to play different roles in behavioral regulation (68). One of the possible mechanisms that have been observed is the modulation of the transcription factor NRF2 through CB2 receptors. NRF2 is a master regulator of inflammation and modulates microglial dynamics. It has been shown to counteract inflammation in several neurodegenerative diseases by interacting with the transcription factor NF-κB. However, these findings have not been reproduced at the neuronal level (69). Therefore, this raises the possibility that CB2r in neurons has other functions not involved in the regulation of inflammation.
Another neuroprotective mechanism of CB2r is the bidirectional interaction between neurons and glial cells through the release of soluble factors and the formation of ligand-receptor associations. It is thought that CB2r-mediated modulation of neuron-glia crosstalk would be highly relevant for neuronal survival when brain homeostasis is lost. Recent studies have shown that MAGL inhibition increases the risk of neuronal death in neuronal cultures but not in neuron/microglia co-cultures. The authors conclude that the neuroprotective effect of MAGL inhibition was due to CB2r in microglia (70).
A possible protective mechanism involved in CB2r-mediated microglia-neuron interaction is the potentiation of the CD200-CD200R interaction. CD200R is expressed on microglia, whereas CD200 is expressed on neurons, and their interaction plays a crucial role in neuronal protection in inflammation-mediated neurodegeneration. Furthermore, CD200R1-CD200 interaction was associated with decreased production of the pro-inflammatory cytokines IL-1b and IL-6 but increased IL-10 in activated microglia after anandamide treatment (32). Another possible mechanism related to microglia-neuron interaction and CB2r involves the up-regulation of microglial CX3CR1. This effect promotes a neurotrophic phenotype of microglia, associated with increased release of BDNF by microglia and increased proliferation of neuronal progenitor cells (71).
Altogether, these data highlight an essential role for brain CB2r in modulating inflammatory responses. CB2r can modulate immune function by suppressing inflammation, shifting microglia to an M2 anti-inflammatory phenotype, supporting neuron-glia coupling, promoting neuronal proliferation, and neuroprotection, which makes them potential modulators for psychiatric conditions, as they present immune alterations. Importantly, data show an anti-inflammatory effect in multiple inflammatory diseases. Data regarding the molecular mechanisms are still uncertain since studies are performed in different conditions. However, these data reveal that CB2r can impact central routes involved in microglial functions (JNK, p38, ERK, Akt, mTOR). These pathways are also involved in neuropsychiatric disorders and inflammatory conditions. CB2r is emerging as a potential therapeutic target. Further studies are needed to reveal how CB2r mediates anti-inflammatory and neuroprotective actions in neuropsychiatric disorders.
Approximately 16% of the world population presents a depressive disorder, the third leading cause of disability and the fourth leading cause of disease burden (72). Its clinical features include a great variety of symptoms such as sleep disturbances, sadness, apathy, anhedonia, and reduced social interest. Depressive disorders are associated with a detriment in social relationships, including family breakdown, absence from work, and reduced productivity in the workplace. The prevalence of depressive disorders varies depending on age and sex, with the highest rates in women (73–76). Interestingly, the severity of depressive disorders is more significant at older ages (77).
Despite the significant number of antidepressants available, the clinical response rate is low, reflecting that only one-third of patients achieve complete remission (78, 79). The main reason for this clinical reality lies in the incomplete knowledge of the biological basis underlying depressive disorders, especially considering gender and age influence. Thus, it is necessary to invest more efforts and resources in characterizing in more detail the underlying mechanisms with the final goal of identifying new and effective critical targets for treating these disorders. In this respect, cumulative data pointed out CB2r as a promising therapeutic option for several neuropsychiatric conditions, including depressive disorders. The first evidence came from animal models of depression showing alterations of CB2r in different brain regions, such as the hippocampus (HIPP), amygdala (AMY), and frontal cortex (80–83). A postmortem study from the Manzanares group performed in the brain of suicide victims showed a reduction of CB2r gene expression in the dorsolateral prefrontal cortex and AMY, critical regions in decision making and impulsivity emotional reactivity (84). Besides, the research team led by Onaivi has shown a high incidence of the Q63R polymorphism of CB2r in Japanese depressive patients (85). The R allele of this functional polymorphism was also associated with increased sensitivity for childhood trauma, possibly due to a loss in the inhibition of inflammation and overactivation of the hypothalamic-pituitary-adrenal (HPA) axis (86).
In the same way, genetic and pharmacological studies performed in rodents further supported the role of CB2r in emotional reactivity, including anxiety and depressive-like behaviors. On the one hand, mice overexpressing CB2r in the central nervous system (neurons and glia) (CB2xP) presented decreased depressive-like behaviors in the tail suspension and novelty suppressed feeding tests (81). On the other hand, mice lacking CB2r (CB2KO) showed just the opposite since their vulnerability was higher in the evaluated paradigms (87). Overexpression of CB2r was associated with an endophenotype resistant to stimuli promoting a depressive state in the unpredictable chronic mild stress paradigm (UCMS), a reliable animal model to study the pathophysiological mechanisms underlying depression as decreased neurogenesis, HPA axis alterations and neuroinflammation (88–92).
Likewise, pharmacological studies chronically administering the cannabinoid CB2r-antagonist AM630 (1 mg/kg/12 h) revealed a reduction of depressive behaviors induced by CUMS (81). In contrast to these data, it was previously shown that administration of AM630 (3 mg/kg/24 h) produced no effect on sucrose consumption in CUMS-exposed mice (80). These discrepancies could be due to notable differences between the two studies, such as (1) the rodent strains employed [BALB/c (80), ICR (81)], and (2) the dose and administration patterns evaluated [3 mg/kg/24 h (80), 1 mg/kg/12 h (81)].
Based on the neuroinflammation hypothesis of depression (93–95), administration of the LPS, which induces sickness and depressive-like behaviors and cognitive impairments, is used to clarify the underlying mechanism of depression (96–98). A significant up-regulation of CB2r was identified in activated microglia using this model (99). Additional evidence about the role of CB2r in modulating neuroinflammation and depressive-like behaviors came from pharmacological studies testing CB2r-agonists. Acute and chronic administration (7 days) of the CB2r-agonist 1-phenylisatin (PI) significantly improved LPS-behavioral and neuroinflammation, reducing depressive-like behaviors, oxidative stress and TNF-α levels, and preventing the decline of antioxidant enzymes as GSH and catalase (100). Interestingly, Youssef et al. (101) observed that the CB2r-agonist beta-caryophyllene (BCP) alleviates insulin resistance, oxidative stress, neuroinflammation, and psychological changes, including depressive-like behaviors and memory deficits induced by high fat/fructose diet (HFFD) in male Wistar rats. More in detail, this study indicated that CB2r, together with PPAR-γ, is involved in the anti-inflammatory, anxiolytic and antioxidant effects of BCP. Further data supported anxiolytic- and antidepressant-like effects of BCP in different paradigms (102, 103). Interestingly, pre-administration of the CB2r-antagonist AM630 completely blocked BCP effects, supporting the involvement of CB2r in such anxiolytic and antidepressant properties (104). Curiously, blockade of CB2r by the antagonist SR144528 attenuated the effects of the synthetic cannabinoid HU210 (CB1r and CB2r agonist) on reducing the increase of cytokines concentrations induced by LPS in the brain (105).
Complementary studies revealed the involvement of CB2r in the antidepressant properties of different compounds. Such is the case of a very recent study showing that CB2r, along with CB1r and PPAR- γ, are involved in the antidepressant-like effects of the rosmarinic acid in the LPS-induced neuroinflammatory model (106). Besides, the compound terpineol, monoterpenoid alcohol, showed immunomodulatory, neuroprotective, and antidepressant-like effects mainly through CB1r and CB2r (107).
Interestingly, previous studies demonstrated the regulatory role of CB2r in stress-induced excitotoxicity and neuroinflammation in mice. JWH133 prevented the increase in pro-inflammatory cytokines and the cellular oxidative and nitrosative damage (lipid peroxidation) induced by immobilization and acoustic stress (2 h/day for 4 days). Similarly, CB2xP exhibited the same anti-inflammatory and neuroprotective actions as those observed in mice pretreated with JWH133. Conversely, CB2KO mice showed intensified stress-induced neuroinflammatory responses (108).
Additional studies also revealed a closed association between CB2r and classical monoaminergic systems involved in depression, such as the dopaminergic and the serotoninergic systems (109–111). More importantly, CB2r also influences neuroplasticity. In this respect, genetic mice models revealed alterations in BDNF, one of the main neurotrophic factors involved in neurogenesis, which plays an essential role in modulating the plasticity of adult neurons and glia cells (112). Overexpression of CB2r (Cb2xP) increased BDNF levels in the HIPP, which was not reduced even after 8 weeks of CUMS (81). In contrast, CB2KO presented low BDNF, MAP2, NF200 and SYN-immunoreactive fibers and a reduced number of synapses in the HIPP, supporting abnormalities in the dendritic reorganization and the synaptic connections (113). Indeed, chronic administration of AM630 significantly increases CB2r and BDNF gene expression in the HIPP of mice exposed to the CUMS, being proposed as a mechanism underlying its antidepressant properties in this mice model (81). These results strongly suggest that CB2r may play an essential role in regulating BDNF gene expression. Recent pieces of evidence supported how microglia can modulate neuronal plasticity potentially through BDNF. Thus, it is necessary to study further if modulation of microglia by CB2r may be the mechanism underlying the increase of BDNF observed in CB2xP mice.
Furthermore, the lack of CB2r (CB2KO) reduced the glucocorticoid receptor (GR) gene expression in the HIPP (113). Increased phosphorylation of proteins involved in the mTOR signaling pathway, p70S6K and AKT, was found in CB2KO, revealing potential alterations in the translational process that controls the protein synthesis underlying synaptic neuronal plasticity and memory (113–116). Considering the role of stress and GR in regulating inflammatory processes and BDNF gene expression (117, 118), it is tempting to speculate that the reduction of GR gene expression may cause changes in BDNF gene expression, neuronal plasticity and consequently alter the neurogenesis processes.
More recently, the neuroprotective effects of CB2r agonists were associated with the suppression of microglial activation through the inhibition of neurotoxic factors and by decreasing neuronal cell damage (119). During CNS inflammation, activation of CB2r inhibited ERK-1/2 signaling in microglia, reducing iNOS production (34). Studies carried out with the BCP demonstrated that this CB2r-agonist presents a modulatory effect on the PGC-1α/BDNF pathway (103). Additional pharmacological studies using CB2r-agonists, such as JWH133 and AM1241, showed that activation of CB2r significantly upregulated BDNF while reducing a variety of neuroinflammatory markers (50, 120). Altogether revealed that CB2r, through the regulation of microglial activity, modulates BDNF gene expression and, consequently, neuroplasticity.
It is important to note that cumulative evidence highlight that the success of antidepressant treatments lies in their ability to reduce inflammatory processes and improve trophic factors (121, 122). Moreover, recent studies have demonstrated that antidepressants promote microglial phenotype switching to M2-like cells, which can secrete anti-inflammatory cytokines, an effect that is also found by activation of CB2r on these cells (123, 124). Interestingly, microglia show sex- and age-differential characteristics. Aging female microglia selectively upregulates M1 markers with a poor response to noxious and stress stimuli (125). These results indicate the CB2r as a promising target for treating depressive disorders that deserve further exploration, especially considering sex and age. For summarized data see Table 1.
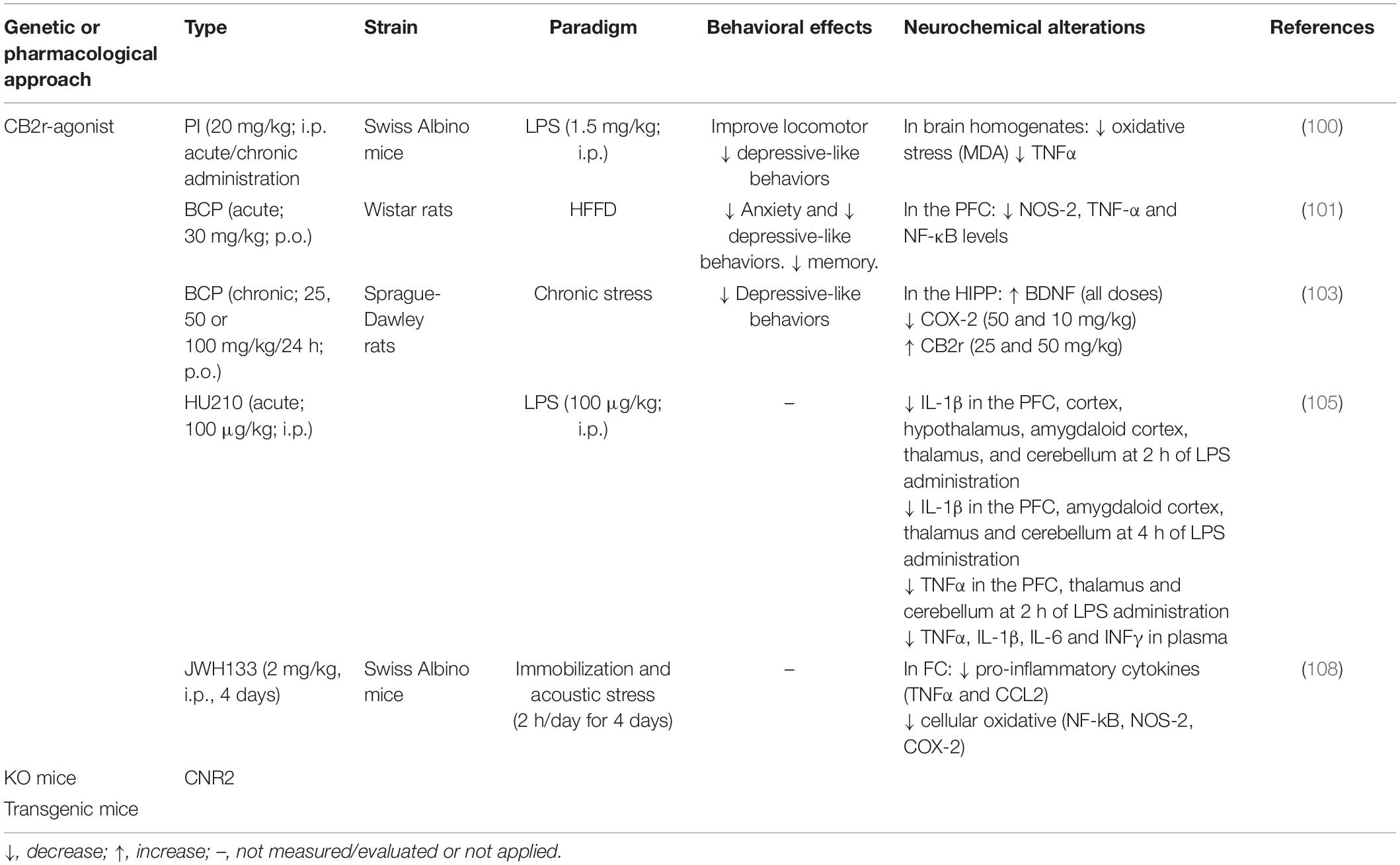
Table 1. Summary of the modulatory effects of cannabinoid receptor 2 (CB2r) on neuroinflammation in depressive disorders.
Anxiety disorders are the most common mental diseases, affecting nearly 30% of adults at some point in their lives (126). They present high comorbidity with other psychiatric disorders, among which depressive disorders stand out (73, 127, 128). Different factors have increased the likelihood of developing anxiety, such as genetic, environmental, psychological, and developmental causes (129–135). The treatment of anxiety disorders includes psychotherapy and pharmacotherapy such as anxiolytic and antidepressant drugs. Among anxiolytics, benzodiazepines (BZP) are the most common, generally prescribed only for short periods. Selective serotonin reuptake inhibitor (SSRI) and serotonin and norepinephrine reuptake inhibitor (SNRI) antidepressant classes are the first-line medication treatments (136, 137). Despite all the therapeutic options available, their limited efficacy and side effects (138–143) prompted further research to elucidate the underlying mechanisms. Therefore, identifying new targets and drugs alone with current pharmacological options would improve clinical effectiveness and safety outcomes.
The identification of CB2r in brain regions is closely related to the response to stress, as the HIPP and AMY (81, 144, 145) accelerated the development of studies to clarify its role in modulating anxiety. In rodents, exposure to different types of stressful stimuli induced significant changes in the expression of CB2r in the brain. Such is the case of maternal deprivation (MD), a model used for evaluating long-lasting effects of early life stress, including anxiety- and depressive-like behaviors, cognitive impairments and neuroendocrine alterations (146–148). In rats, MD significantly increased the expression of CB2r in the HIPP (149). Similarly, additional studies revealed sex-dependent differences, observing an increase of CB2r gene expression in the frontal cortex, ventral and dorsal striatum, dorsal HIPP and AMY in MD-male rats. In females, such an increase was only observed in the HIPP, suggesting that consequences of early life stress depend on sex and brain region (83). Moreover, a rapid increase in CB2r gene expression has been observed in the HIPP of mice exposed to social defeat (150).
Studies using mice modified genetically demonstrated that CB2xP showed resistance to anxiogenic-like stimuli in the light-dark box and elevated plus maze tests (151). On the contrary, CB2KO mice presented anxious behaviors (87). More interestingly, genetic manipulation studies allowed us to go more deeply into the cell-specific active involvement of CB2r in the HIPP, dissecting the effects of CB2r gene expression disruption or overexpression in hippocampal neurons or microglia in the regulation of anxiety behavior and cognition (68). The results revealed that overexpression of CB2r in CA1 pyramidal neurons significantly reduced anxiety levels. In the case of microglia, the elevation of CB2r increased contextual fear memory, whereas the absence of CB2r induced a reduction. Moreover, the deletion of CB2r in VTA dopaminergic neurons caused a significant anxiolytic-like effect (152). Altogether, these results revealed that CB2r plays distinct roles in regulating anxiety and memory depending on the type of cells expressed.
Pharmacological studies also revealed the role of this cannabinoid receptor in stress response and anxiety. Acute treatment with the CB2r-agonist JWH015 reduced anxiety-like behaviors in stressed mice; however, its chronic administration induced opposite effects (153). In line with these results, acute activation of CB2r by BCP caused an anxiolytic-like effect entirely abolished by the CB2r-antagonist AM630, supporting the role of CB2r on BCP effects (102, 104). Alternatively, it has been demonstrated that chronic administration of the CB2r-antagonist AM630 reduced the anxious behaviors induced by the CUMS (81). Similarly, chronic intracerebroventricular administration of the CB2r antisense oligonucleotide induced anxiolytic-like effects (154). Although additional studies are needed, the data collected to the date indicated potential differences between acute and chronic administration effects of compounds acting on CB2r. The CB2r agonists appear to be more helpful in producing an acute anxiolytic effect, whereas CB2r antagonists would be more appropriate for chronic anxiolytic treatments. CB2r has been closely related to crucial targets in response to stress and anxiety, including the HPA axis (151), the GABAergic system (151, 155), and more recently, neuroinflammation. The modulation of neuroinflammatory elements was associated with the anxiolytic-like effects of the non-selective CB2r-agonist WIN55,212-2 in mice exposed to repeat social deficits. This cannabinoid compound decreased IL-1β gene expression in microglia/macrophages and the accumulation of peripheral inflammatory monocytes (156). Interestingly, in animal models of traumatic brain injury, the modulation of neuroinflammation by CB2r agonist reduced the development of anxiety and depressive-like behaviors in rodents (157–159). Thus, modulation neuroinflammation appears to be a mechanism by which CB2r modulates emotional responses deserving an in-depth exploration (Table 2).

Table 2. Summary of the modulatory effects of cannabinoid receptor 2 (CB2r) on neuroinflammation in anxiety disorders.
The role of CB2r in the onset of Alzheimer’s disease (AD) has been intensively investigated in the last decades. Inflammation has been shown to prompt cognitive dysfunction and dementia later in life (160). Several studies associate depression with a high risk of developing dementia, and often those diseases are concomitant (161).
Unraveling its pathophysiological role in AD can provide a new therapeutical strategy. CB2r are poorly expressed in the neuronal brain cells in healthy conditions and are primarily detected in glial cells. However, CB2r levels drastically increase in neurodegenerative disorders, playing an essential role in modulating pro-inflammatory mediators such as AD (162, 163). High levels of CB2r have been found in senile plaques surrounding microglia and astrocytes in both cortex and hippocampus in postmortem human AD patients (164, 165). Similarly, in a preclinical murine AD model, a high expression of CB2r was also found in astrocytes and microglia surrounding the amyloid plaques but not neuronal cells (166). Interestingly, expression levels of CB2r correlated with two relevant AD molecular markers, Aβ42 levels and senile plaque score. Both human and rodents’ findings support a potential role of CB2r in the inflammatory response generated by plaque deposition.
Although most studies found a correlation of CB2r expression and Aβ42 levels, Tau overexpression appears to be involved in modulating CB2r levels in both mouse and human brains (167). This study showed a TAU-dependent increase of CB2r expression on a neuronal level, and most importantly, the deletion of CB2r in TAU mice was associated with a cognitive improvement. However, it was ineffective to reverse neuroinflammation supporting a neuronal CB2 effect in TAU mice rather than glial, as reported in APP Tg AD mouse models. To further investigate the role of CB2r, Aso et al. (168) produced an AD(APP/PS1)/CB2 KO transgenic mice and evaluated the specific contribution of CB2r in the AD pathology. The deletion of the CB2r increased cortical Aβ deposition and Aβ40 soluble levels on the APP/PS1 mice. However, no effect was reported on tau pathology. Similar results were also previously observed in the mice model J20APP/CB2 KO (169). In addition, in this study, the deletion of CB2r reduced total Tau expression, supporting a divergent effect of CB2r between the Aβ and Tau pathology. In other studies, in contradiction with Aso’s et al. (168) findings, CB2r deletion in APP/PS1 mice was associated with improved cognitive and learning deficits. These findings were accompanied by reduced neuronal loss, decreased plaque levels, and a reduction of activated microglia (170).
CB2r has been a target of several in vitro and in vivo tests in preclinical studies, sometimes contradictory results. In primary rat hippocampal neuronal cultures, it has been shown that the CB2r agonist JWH133 treatment reduces the Aβ42–induced neuron apoptosis (171), reverted effect by the selective CB2r antagonist AM630. CB2r activation with the selective CB2r agonist JWH-015 suppressed IFN-γ-induced microglia activation, TNF-α, and nitric oxide production (35). In APP/PS1 mice, the same compound enhanced the novel object recognition memory deficiency but was inefficient for hippocampus-dependent spatial cognitive dysfunction in the Morris water maze test. Moreover, activation of CB2r did not affect plaque deposition. Pretreatment with MDA7, a selective CB2r agonist, in an Aβ42 induced AD-Murine model, was associated with a reduction in microglia activation and pro-inflammatory IL-1β production. An improvement in memory impairment was found in the Morris water maze test (172). Recently, similar results were reported after treating APP/PS1 mice with anandamide analog (NITyr). NITyr treatment improved motor coordination spatial memory and reduced Aβ40 and Aβ42 without affecting the APP expression (173).
Administration of CB2r agonists may affect microglia-dependent neuroinflammation. In addition, CB2r activation over CB1r compounds results in a more appealing therapeutic strategy since CB2r activation is not linked to the psychoactive effects linked to CB1r activation. Taken together (Table 3), these findings, and considering the high levels of CB2r in microglia observed in preclinical and clinical studies, CB2r can be regarded as a novel target for AD therapy.
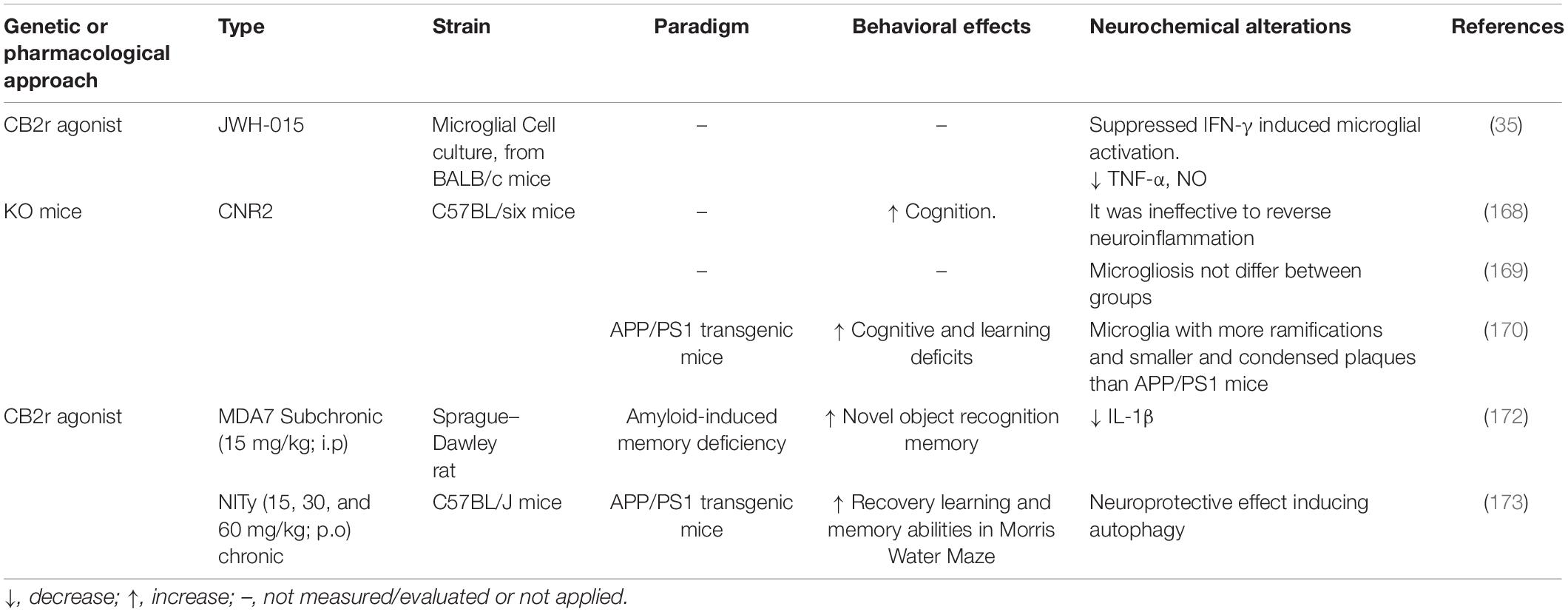
Table 3. Summary of the modulatory effects of cannabinoid receptor 2 (CB2r) on neuroinflammation in cognitive dysfunction and neurodegeneration.
Studies developed to the date indicated that CB2r is involved in emotional response and cognition. Its pharmacological modulation could be an exciting tool to treat different neuropsychiatric disorders. Despite the correlation between the immune system and the brain, the implication of CB2r needs to be further elucidated. CB2r expression in cells of immune systems points out the possible involvement of this system in psychiatry and neurology with the implication of CB2r. Here, different actions have been related to CB2r and its pharmacological modulation, including changes in inflammatory pathways and microglial functions associated with NF-κB, JAK, MAPK, and AKT signaling pathways. Remarkably, a neuroprotective action has been proposed for this receptor, possibly by producing a shift of microglia toward an anti-inflammatory phenotype. In addition, CB2r modulation also changes the response of astrocytes and neurons on inflammation, protecting from BBB leakage or promoting neural plasticity. Consequently, the CB2r modulation may be a promising target to improve neuropsychiatric diseases associated with neuroinflammation.
Several human clinical trials are ongoing to look at the potential modulation of the cannabinoid system in human disease. However, no studies are currently underway with selective modulation of the CB2r to treat neuropsychiatric illnesses. Among the following strategies, we find the use of FAAH inhibitors, for instance, using the compound PF-04457845 in Tourette syndrome (NCT02134080) or cannabis use disorder (NCT03386487). The potential use of URB597 for the treatment of schizophrenia has been shown to protect against NLRP3 inflammasome activation (97). However, no studies with MAGL inhibitors are currently underway. Furthermore, the potential use of CBD, directly modulating the EC system, to treat anxiety (NCT02548559), psychosis (NCT03883360), as well as the anxiety and agitation symptoms in mild to moderate Alzheimer Disease (NCT04075435), are currently being evaluated. Besides, the use of nabilone, an analog of THC, is also being studied for non-motor symptoms in patients with Parkinson’s Disease (PD) (NCT03773796).
It is unclear whether the therapeutic effect could be due to the improvement of inflammation through modulation of the CB2r or other critical targets of the ECS. Therefore, the specific role of the CB2r in inflammation associated with psychiatric disorders requires further study.
AM and TF designed, coordinated, and reviewed the sections and contents of the review manuscript. TF oversaw the organization to distribute the writing tasks among the authors and the article writing. AM, MSG-G, ST, and EN performed the literature searches. AM, MSG-G, ST, JM, and TF participated in the manuscript writing. All authors critically reviewed and approved the final version of the manuscript.
The preparation of the manuscript was supported by Instituto de Salud Carlos III (RD. PI18/00576), (RD21/0009/0008 Red de Investigación en Atención Primaria de Adicciones), and Delegación del Gobierno para el Plan Nacional Sobre Drogas (PNSD, 2019I012) from the Spanish Ministry of Health to JM and Ministerio de Ciencia e Innovación Programa Ramón y Cajal (RYC201722666) to TF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Sergio Javaloy for his excellent assistance in the graphical design.
1. Osimo EF, Cardinal RN, Jones PB, Khandaker GM. Prevalence and correlates of low-grade systemic inflammation in adult psychiatric inpatients: an electronic health record-based study. Psychoneuroendocrinology. (2018) 91:226–34. doi: 10.1016/j.psyneuen.2018.02.031
2. Vogelzangs N, Beekman AT, de Jonge P, Penninx BW. Anxiety disorders and inflammation in a large adult cohort. Transl Psychiatry. (2013) 3:e249. doi: 10.1038/tp.2013.27
3. Hoge EA, Brandstetter K, Moshier S, Pollack MH, Wong KK, Simon NM. Broad spectrum of cytokine abnormalities in panic disorder and posttraumatic stress disorder. Depress Anxiety. (2009) 26:447–55. doi: 10.1002/da.20564
4. Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. (2010) 67:446–57. doi: 10.1016/j.biopsych.2009.09.033
5. Ortiz-Dominguez A, Hernandez ME, Berlanga C, Gutierrez-Mora D, Moreno J, Heinze G, et al. Immune variations in bipolar disorder: phasic differences. Bipolar Disord. (2007) 9:596–602. doi: 10.1111/j.1399-5618.2007.00493.x
6. Misiak B, Leszek J, Kiejna A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease–the emerging role of systemic low-grade inflammation and adiposity. Brain Res Bull. (2012) 89:144–9. doi: 10.1016/j.brainresbull.2012.08.003
7. Menard C, Pfau ML, Hodes GE, Russo SJ. Immune and Neuroendocrine Mechanisms of Stress Vulnerability and Resilience. Neuropsychopharmacology. (2017) 42:62–80. doi: 10.1038/npp.2016.90
8. Bauer ME, Teixeira AL. Inflammation in psychiatric disorders: what comes first? Ann NY Acad Sci. (2019) 1437:57–67. doi: 10.1111/nyas.13712
9. Hodes GE, Kana V, Menard C, Merad M, Russo SJ. Neuroimmune mechanisms of depression. Nat Neurosci. (2015) 18:1386–93. doi: 10.1038/nn.4113
10. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
11. Franklin TC, Xu C, Duman RS. Depression and sterile inflammation: essential role of danger associated molecular patterns. Brain Behav Immun. (2018) 72:2–13. doi: 10.1016/j.bbi.2017.10.025
12. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. (2018) 128:2657–69. doi: 10.1172/JCI97943
13. Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. (2016) 53:1181–94. doi: 10.1007/s12035-014-9070-5
14. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13.
15. Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. (2017) 60:1–12. doi: 10.1016/j.bbi.2016.03.010
16. Niklasson F, Agren H. Brain energy metabolism and blood-brain barrier permeability in depressive patients: analyses of creatine, creatinine, urate, and albumin in CSF and blood. Biol Psychiatry. (1984) 19:1183–206.
17. Sumner JA, Chen Q, Roberts AL, Winning A, Rimm EB, Gilsanz P, et al. Posttraumatic stress disorder onset and inflammatory and endothelial function biomarkers in women. Brain Behav Immun. (2018) 69:203–9. doi: 10.1016/j.bbi.2017.11.013
18. Muller N. COX-2 Inhibitors, Aspirin, and Other Potential Anti-Inflammatory Treatments for Psychiatric Disorders. Front Psychiatry. (2019) 10:375. doi: 10.3389/fpsyt.2019.00375
19. Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol. (2013) 64:21–47.
20. Sharir H, Abood ME. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol Ther. (2010) 126:301–13. doi: 10.1016/j.pharmthera.2010.02.004
21. Marco EM, Garcia-Gutierrez MS, Bermudez-Silva FJ, Moreira FA, Guimaraes F, Manzanares J, et al. Endocannabinoid system and psychiatry: in search of a neurobiological basis for detrimental and potential therapeutic effects. Front Behav Neurosci. (2011) 5:63. doi: 10.3389/fnbeh.2011.00063
22. Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB(1) and CB(2) receptors. Mol Pharmacol. (1999) 56:1362–9. doi: 10.1124/mol.56.6.1362
23. Lu HC, Mackie K. An Introduction to the Endogenous Cannabinoid System. Biol Psychiatry. (2016) 79:516–25. doi: 10.1016/j.biopsych.2015.07.028
24. Manzanares J, Cabanero D, Puente N, Garcia-Gutierrez MS, Grandes P, Maldonado R. Role of the endocannabinoid system in drug addiction. Biochem Pharmacol. (2018) 157:108–21. doi: 10.1016/j.bcp.2018.09.013
25. Callen L, Moreno E, Barroso-Chinea P, Moreno-Delgado D, Cortes A, Mallol J, et al. Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J Biol Chem. (2012) 287:20851–65. doi: 10.1074/jbc.M111.335273
26. Garcia-Gutierrez MS, Navarrete F, Gasparyan A, Austrich-Olivares A, Sala F, Manzanares J. Cannabidiol: a potential new alternative for the treatment of anxiety, depression, and psychotic disorders. Biomolecules (2020) 10:1575. doi: 10.3390/biom10111575
27. Zlebnik NE, Cheer JF. Beyond the CB1 receptor: is cannabidiol the answer for disorders of motivation? Annu Rev Neurosci. (2016) 39:1–17. doi: 10.1146/annurev-neuro-070815-014038
28. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. (2008) 9:46–56. doi: 10.1038/nrn2297
29. Remus JL, Dantzer R. Inflammation models of depression in rodents: relevance to psychotropic drug discovery. Int J Neuropsychopharmacol. (2016) 19:yw028. doi: 10.1093/ijnp/pyw028
30. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. (2006) 27:24–31. doi: 10.1016/j.it.2005.11.006
31. Benito C, Tolon RM, Pazos MR, Nunez E, Castillo AI, Romero J. Cannabinoid CB2 receptors in human brain inflammation. Br J Pharmacol. (2008) 153:277–85. doi: 10.1038/sj.bjp.0707505
32. Hernangomez M, Mestre L, Correa FG, Loria F, Mecha M, Inigo PM, et al. CD200-CD200R1 interaction contributes to neuroprotective effects of anandamide on experimentally induced inflammation. Glia. (2012) 60:1437–50. doi: 10.1002/glia.22366
33. Klegeris A, Bissonnette CJ, McGeer PL. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br J Pharmacol. (2003) 139:775–86. doi: 10.1038/sj.bjp.0705304
34. Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. (2006) 49:67–79. doi: 10.1016/j.neuron.2005.11.027
35. Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, et al. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflamm. (2005) 2:29. doi: 10.1186/1742-2094-2-29
36. Rom S, Persidsky Y. Cannabinoid receptor 2: potential role in immunomodulation and neuroinflammation. J Neuroimmune Pharmacol. (2013) 8:608–20. doi: 10.1007/s11481-013-9445-9
37. Guo K, Mou X, Huang J, Xiong N, Li H. Trans-caryophyllene suppresses hypoxia-induced neuroinflammatory responses by inhibiting NF-kappaB activation in microglia. J Mol Neurosci. (2014) 54:41–8. doi: 10.1007/s12031-014-0243-5
38. Komorowska-Muller JA, Schmole AC. CB2 Receptor in Microglia: the Guardian of Self-Control. Int J Mol Sci. (2020) 22:19. doi: 10.3390/ijms22010019
39. Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy–from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. (2005) 1754:253–62. doi: 10.1016/j.bbapap.2005.08.017
40. Romero-Sandoval EA, Horvath R, Landry RP, DeLeo JA. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol Pain. (2009) 5:25. doi: 10.1186/1744-8069-5-25
41. Tang J, Tao Y, Tan L, Yang L, Niu Y, Chen Q, et al. Cannabinoid receptor 2 attenuates microglial accumulation and brain injury following germinal matrix hemorrhage via ERK dephosphorylation in vivo and in vitro. Neuropharmacology. (2015) 95:424–33. doi: 10.1016/j.neuropharm.2015.04.028
42. Maya-Lopez M, Rubio-Lopez LC, Rodriguez-Alvarez IV, Orduno-Piceno J, Flores-Valdivia Y, Colonnello A, et al. A cannabinoid receptor-mediated mechanism participates in the neuroprotective effects of oleamide against excitotoxic damage in rat brain synaptosomes and cortical slices. Neurotox Res. (2020) 37:126–35. doi: 10.1007/s12640-019-00083-1
43. Oh YT, Lee JY, Lee J, Lee JH, Kim JE, Ha J, et al. Oleamide suppresses lipopolysaccharide-induced expression of iNOS and COX-2 through inhibition of NF-kappaB activation in BV2 murine microglial cells. Neurosci Lett. (2010) 474:148–53. doi: 10.1016/j.neulet.2010.03.026
44. Viscomi MT, Oddi S, Latini L, Pasquariello N, Florenzano F, Bernardi G, et al. Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J Neurosci. (2009) 29:4564–70. doi: 10.1523/JNEUROSCI.0786-09.2009
45. Salort G, Alvaro-Bartolome M, Garcia-Sevilla JA. Regulation of cannabinoid CB2 receptor constitutive activity in vivo: repeated treatments with inverse agonists reverse the acute activation of JNK and associated apoptotic signaling in mouse brain. Psychopharmacology. (2017) 234:925–41. doi: 10.1007/s00213-017-4537-5
46. Correa F, Hernangomez M, Mestre L, Loria F, Spagnolo A, Docagne F, et al. Anandamide enhances IL-10 production in activated microglia by targeting CB(2) receptors: roles of ERK1/2, JNK, and NF-kappaB. Glia. (2010) 58:135–47. doi: 10.1002/glia.20907
47. Fujii M, Sherchan P, Soejima Y, Hasegawa Y, Flores J, Doycheva D, et al. Cannabinoid receptor type 2 agonist attenuates apoptosis by activation of phosphorylated CREB-Bcl-2 pathway after subarachnoid hemorrhage in rats. Exp Neurol. (2014) 261:396–403. doi: 10.1016/j.expneurol.2014.07.005
48. Mecha M, Carrillo-Salinas FJ, Feliu A, Mestre L, Guaza C. Microglia activation states and cannabinoid system: therapeutic implications. Pharmacol Ther. (2016) 166:40–55. doi: 10.1016/j.pharmthera.2016.06.011
49. Mecha M, Feliu A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutierrez S, de Sola RG, et al. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immun. (2015) 49:233–45. doi: 10.1016/j.bbi.2015.06.002
50. Ma L, Jia J, Liu X, Bai F, Wang Q, Xiong L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem Biophys Res Commun. (2015) 458:92–7. doi: 10.1016/j.bbrc.2015.01.073
51. Tao Y, Li L, Jiang B, Feng Z, Yang L, Tang J, et al. Cannabinoid receptor-2 stimulation suppresses neuroinflammation by regulating microglial M1/M2 polarization through the cAMP/PKA pathway in an experimental GMH rat model. Brain Behav Immun. (2016) 58:118–29. doi: 10.1016/j.bbi.2016.05.020
52. Luo XQ, Li A, Yang X, Xiao X, Hu R, Wang TW, et al. Paeoniflorin exerts neuroprotective effects by modulating the M1/M2 subset polarization of microglia/macrophages in the hippocampal CA1 region of vascular dementia rats via cannabinoid receptor 2. Chin Med. (2018) 13:14. doi: 10.1186/s13020-018-0173-1
53. Ma L, Niu W, Lv J, Jia J, Zhu M, Yang S. PGC-1alpha-mediated mitochondrial biogenesis is involved in cannabinoid receptor 2 agonist AM1241-induced microglial phenotype amelioration. Cell Mol Neurobiol. (2018) 38:1529–37. doi: 10.1007/s10571-018-0628-z
54. Schmole AC, Lundt R, Ternes S, Albayram O, Ulas T, Schultze JL, et al. Cannabinoid receptor 2 deficiency results in reduced neuroinflammation in an Alzheimer’s disease mouse model. Neurobiol Aging. (2015) 36:710–9. doi: 10.1016/j.neurobiolaging.2014.09.019
55. Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. (2011) 14:724–38. doi: 10.1016/j.cmet.2011.08.016
56. Femenia T, Gimenez-Cassina A, Codeluppi S, Fernandez-Zafra T, Katsu-Jimenez Y, Terrando N, et al. Disrupted Neuroglial Metabolic Coupling after Peripheral Surgery. J Neurosci. (2018) 38:452–64. doi: 10.1523/JNEUROSCI.1797-17.2017
57. Sheng WS, Hu S, Min X, Cabral GA, Lokensgard JR, Peterson PK. Synthetic cannabinoid WIN55,212-2 inhibits generation of inflammatory mediators by IL-1beta-stimulated human astrocytes. Glia. (2005) 49:211–9. doi: 10.1002/glia.20108
58. Jia Y, Deng H, Qin Q, Ma Z. JWH133 inhibits MPP(+)-induced inflammatory response and iron influx in astrocytes. Neurosci Lett. (2020) 720:134779. doi: 10.1016/j.neulet.2020.134779
59. Tumati S, Largent-Milnes TM, Keresztes A, Ren J, Roeske WR, Vanderah TW, et al. Repeated morphine treatment-mediated hyperalgesia, allodynia and spinal glial activation are blocked by co-administration of a selective cannabinoid receptor type-2 agonist. J Neuroimmunol. (2012) 244:23–31. doi: 10.1016/j.jneuroim.2011.12.021
60. Alkaitis MS, Solorzano C, Landry RP, Piomelli D, DeLeo JA, Romero-Sandoval EA. Evidence for a role of endocannabinoids, astrocytes and p38 phosphorylation in the resolution of postoperative pain. PLoS One. (2010) 5:e10891. doi: 10.1371/journal.pone.0010891
61. Sheng WS, Hu S, Ni HT, Rock RB, Peterson PK. WIN55,212-2 inhibits production of CX3CL1 by human astrocytes: involvement of p38 MAP kinase. J Neuroimmune Pharmacol. (2009) 4:244–8. doi: 10.1007/s11481-009-9147-5
62. Jing N, Fang B, Li Z, Tian A. Exogenous activation of cannabinoid-2 receptor modulates TLR4/MMP9 expression in a spinal cord ischemia reperfusion rat model. J Neuroinflamm. (2020) 17:101. doi: 10.1186/s12974-020-01784-7
63. Wang Z, Li Y, Cai S, Li R, Cao G. Cannabinoid receptor 2 agonist attenuates bloodbrain barrier damage in a rat model of intracerebral hemorrhage by activating the Rac1 pathway. Int J Mol Med. (2018) 42:2914–22. doi: 10.3892/ijmm.2018.3834
64. Li L, Yun D, Zhang Y, Tao Y, Tan Q, Qiao F, et al. A cannabinoid receptor 2 agonist reduces blood-brain barrier damage via induction of MKP-1 after intracerebral hemorrhage in rats. Brain Res. (2018) 1697:113–23. doi: 10.1016/j.brainres.2018.06.006
65. Rajesh M, Mukhopadhyay P, Batkai S, Hasko G, Liaudet L, Huffman JW, et al. CB2-receptor stimulation attenuates TNF-alpha-induced human endothelial cell activation, transendothelial migration of monocytes, and monocyte-endothelial adhesion. Am J Physiol Heart Circ Physiol. (2007) 293:H2210–8. doi: 10.1152/ajpheart.00688.2007
66. Marchalant Y, Brothers HM, Norman GJ, Karelina K, DeVries AC, Wenk GL. Cannabinoids attenuate the effects of aging upon neuroinflammation and neurogenesis. Neurobiol Dis. (2009) 34:300–7. doi: 10.1016/j.nbd.2009.01.014
67. Palazuelos J, Ortega Z, Diaz-Alonso J, Guzman M, Galve-Roperh I. CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J Biol Chem. (2012) 287:1198–209. doi: 10.1074/jbc.M111.291294
68. Li Y, Kim J. Distinct roles of neuronal and microglial CB2 cannabinoid receptors in the mouse hippocampus. Neuroscience. (2017) 363:11–25. doi: 10.1016/j.neuroscience.2017.08.053
69. Galan-Ganga M, Del Rio R, Jimenez-Moreno N, Diaz-Guerra M, Lastres-Becker I. Cannabinoid CB2 Receptor Modulation by the Transcription Factor NRF2 is Specific in Microglial Cells. Cell Mol Neurobiol. (2020) 40:167–77. doi: 10.1007/s10571-019-00719-y
70. Rojo-Bustamante E, Inigo-Marco I, Abellanas MA, Vinueza-Gavilanes R, Baltanas A, Luquin E, et al. CB2 Receptors and Neuron-Glia Interactions Modulate Neurotoxicity Generated by MAGL Inhibition. Biomolecules. (2020) 10:1198. doi: 10.3390/biom10081198
71. Chung T, Creswell KG, Bachrach R, Clark DB, Martin CS. Adolescent binge drinking. Alcohol Res. (2018) 39:5–15.
73. Kessler RC, Gruber M, Hettema JM, Hwang I, Sampson N, Yonkers KA. Co-morbid major depression and generalized anxiety disorders in the National Comorbidity Survey follow-up. Psychol Med. (2008) 38:365–74. doi: 10.1017/S0033291707002012
74. Deshpande SS, Kalmegh B, Patil PN, Ghate MR, Sarmukaddam S, Paralikar VP. Stresses and Disability in Depression across Gender. Depress Res Treat. (2014) 2014:735307. doi: 10.1155/2014/735307
75. Vieta E, Alonso J, Perez-Sola V, Roca M, Hernando T, Sicras-Mainar A, et al. Epidemiology and costs of depressive disorder in Spain: the EPICO study. Eur Neuropsychopharmacol. (2021) 50:93–103. doi: 10.1016/j.euroneuro.2021.04.022
76. Terlizzi EP, Villarroel MA. Symptoms of depression among adults: United States, 2019. NCHS Data Brief. (2020) 2020:1–8. doi: 10.1080/1028415x.2020.1794422
77. Arias de la Torre J, Vilagut G, Ronaldson A, Dregan A, Ricci-Cabello I, Hatch SL, et al. Prevalence and age patterns of depression in the United Kingdom. A population-based study. J Affect Disord. (2021) 279:164–72. doi: 10.1016/j.jad.2020.09.129
78. Tranter R, O’Donovan C, Chandarana P, Kennedy S. Prevalence and outcome of partial remission in depression. J Psychiatry Neurosci. (2002) 27:241–7.
79. Thase ME. Update on partial response in depression. J Clin Psychiatry. (2009) 70(Suppl 6):4–9. doi: 10.4088/JCP.8133su1c.01
80. Onaivi ES, Ishiguro H, Gong JP, Patel S, Meozzi PA, Myers L, et al. Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann NY Acad Sci. (2008) 1139:434–49. doi: 10.1196/annals.1432.036
81. Garcia-Gutierrez MS, Perez-Ortiz JM, Gutierrez-Adan A, Manzanares J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB(2) receptors. Br J Pharmacol. (2010) 160:1773–84. doi: 10.1111/j.1476-5381.2010.00819.x
82. Smaga I, Jastrzebska J, Zaniewska M, Bystrowska B, Gawlinski D, Faron-Gorecka A, et al. Changes in the brain endocannabinoid system in rat models of depression. Neurotox Res. (2017) 31:421–35. doi: 10.1007/s12640-017-9708-y
83. Marco EM, Echeverry-Alzate V, Lopez-Moreno JA, Gine E, Penasco S, Viveros MP. Consequences of early life stress on the expression of endocannabinoid-related genes in the rat brain. Behavioural Pharmacol. (2014) 25:547–56. doi: 10.1097/FBP.0000000000000068
84. Garcia-Gutierrez MS, Navarrete F, Navarro G, Reyes-Resina I, Franco R, Lanciego JL, et al. Alterations in gene and protein expression of cannabinoid cb2 and gpr55 receptors in the dorsolateral prefrontal cortex of suicide victims. Neurotherapeutics. (2018) 15:796–806. doi: 10.1007/s13311-018-0610-y
85. Onaivi ES, Ishiguro H, Gong JP, Patel S, Meozzi PA, Myers L, et al. Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: from mice to human subjects. PLoS One. (2008) 3:e1640. doi: 10.1371/journal.pone.0001640
86. Lazary J, Eszlari N, Juhasz G, Bagdy G. A functional variant of CB2 receptor gene interacts with childhood trauma and FAAH gene on anxious and depressive phenotypes. J Affect Disord. (2019) 257:716–22. doi: 10.1016/j.jad.2019.07.083
87. Ortega-Alvaro A, Aracil-Fernandez A, Garcia-Gutierrez MS, Navarrete F, Manzanares J. Deletion of CB2 cannabinoid receptor induces schizophrenia-related behaviors in mice. Neuropsychopharmacology. (2011) 36:1489–504. doi: 10.1038/npp.2011.34
88. Cai L, Mu YR, Liu MM, Tang WJ, Li R. Antidepressant-like effects of penta-acetyl geniposide in chronic unpredictable mild stress-induced depression rat model: involvement of inhibiting neuroinflammation in prefrontal cortex and regulating hypothalamic-pituitaryadrenal axis. Int Immunopharmacol. (2020) 80:106182. doi: 10.1016/j.intimp.2019.106182
89. Liu B, Xu C, Wu X, Liu F, Du Y, Sun J, et al. Icariin exerts an antidepressant effect in an unpredictable chronic mild stress model of depression in rats and is associated with the regulation of hippocampal neuroinflammation. Neuroscience. (2015) 294:193–205. doi: 10.1016/j.neuroscience.2015.02.053
90. Farooq RK, Isingrini E, Tanti A, Le Guisquet AM, Arlicot N, Minier F, et al. Is unpredictable chronic mild stress (UCMS) a reliable model to study depression-induced neuroinflammation? Behav Brain Res. (2012) 231:130–7. doi: 10.1016/j.bbr.2012.03.020
91. Willner P. Validity, reliability and utility of the chronic mild stress model of depression: a 10-year review and evaluation. Psychopharmacology. (1997) 134:319–29. doi: 10.1007/s002130050456
92. Willner P. The chronic mild stress (CMS) model of depression: history, evaluation and usage. Neurobiol Stress. (2017) 6:78–93. doi: 10.1016/j.ynstr.2016.08.002
93. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. (2016) 16:22–34. doi: 10.1038/nri.2015.5
94. Roohi E, Jaafari N, Hashemian F. On inflammatory hypothesis of depression: what is the role of IL-6 in the middle of the chaos? J Neuroinflamm. (2021) 18:45. doi: 10.1186/s12974-021-02100-7
95. Wu Z, Xiao L, Wang H, Wang G. Neurogenic hypothesis of positive psychology in stress-induced depression: adult hippocampal neurogenesis, neuroinflammation, and stress resilience. Int Immunopharmacol. (2021) 97:107653. doi: 10.1016/j.intimp.2021.107653
96. Ming Z, Sawicki G, Bekar LK. Acute systemic LPS-mediated inflammation induces lasting changes in mouse cortical neuromodulation and behavior. Neurosci Lett. (2015) 590:96–100. doi: 10.1016/j.neulet.2015.01.081
97. Casaril AM, Domingues M, Fronza M, Vieira B, Begnini K, Lenardao EJ, et al. Antidepressant-like effect of a new selenium-containing compound is accompanied by a reduction of neuroinflammation and oxidative stress in lipopolysaccharide-challenged mice. J Psychopharmacol. (2017) 31:1263–73. doi: 10.1177/0269881117711713
98. O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. (2009) 14:511–22. doi: 10.1038/sj.mp.4002148
99. Carlisle SJ, Marciano-Cabral F, Staab A, Ludwick C, Cabral GA. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int Immunopharmacol. (2002) 2:69–82. doi: 10.1016/s1567-5769(01)00147-3
100. Sahu P, Mudgal J, Arora D, Kinra M, Mallik SB, Rao CM, et al. Cannabinoid receptor 2 activation mitigates lipopolysaccharide-induced neuroinflammation and sickness behavior in mice. Psychopharmacology. (2019) 236:1829–38. doi: 10.1007/s00213-019-5166-y
101. Youssef DA, El-Fayoumi HM, Mahmoud MF. Beta-caryophyllene alleviates diet-induced neurobehavioral changes in rats: the role of CB2 and PPAR-gamma receptors. Biomed Pharmacother. (2019) 110:145–54. doi: 10.1016/j.biopha.2018.11.039
102. Bahi A, Al Mansouri S, Al Memari E, Al Ameri M, Nurulain SM, Ojha S. beta-Caryophyllene, a CB2 receptor agonist produces multiple behavioral changes relevant to anxiety and depression in mice. Physiol Behav. (2014) 135:119–24. doi: 10.1016/j.physbeh.2014.06.003
103. Hwang ES, Kim HB, Lee S, Kim MJ, Kim KJ, Han G, et al. Antidepressant-like effects of beta-caryophyllene on restraint plus stress-induced depression. Behav Brain Res. (2020) 380:112439. doi: 10.1016/j.bbr.2019.112439
104. Galdino PM, Nascimento MV, Florentino IF, Lino RC, Fajemiroye JO, Chaibub BA, et al. The anxiolytic-like effect of an essential oil derived from Spiranthera odoratissima A. St. Hil. leaves and its major component, beta-caryophyllene, in male mice. Prog Neuropsychopharmacol Biol Psychiatry. (2012) 38:276–84. doi: 10.1016/j.pnpbp.2012.04.012
105. Roche M, Diamond M, Kelly JP, Finn DP. In vivo modulation of LPS-induced alterations in brain and peripheral cytokines and HPA axis activity by cannabinoids. J Neuroimmunol. (2006) 181:57–67. doi: 10.1016/j.jneuroim.2006.08.001
106. Lataliza AAB, de Assis PM, da Rocha Laurindo L, Goncalves ECD, Raposo NRB, Dutra RC. Antidepressant-like effect of rosmarinic acid during LPS-induced neuroinflammatory model: the potential role of cannabinoid receptors/PPAR-gamma signaling pathway. Phytother Res. (2021) 35:6974–89. doi: 10.1002/ptr.7318
107. Vieira G, Cavalli J, Goncalves ECD, Braga SFP, Ferreira RS, Santos ARS, et al. Antidepressant-like effect of terpineol in an inflammatory model of depression: involvement of the cannabinoid system and D2 dopamine receptor. Biomolecules. (2020) 10:792. doi: 10.3390/biom10050792
108. Zoppi S, Madrigal JL, Caso JR, Garcia-Gutierrez MS, Manzanares J, Leza JC, et al. Regulatory role of the cannabinoid CB2 receptor in stress-induced neuroinflammation in mice. Br J Pharmacol. (2014) 171:2814–26. doi: 10.1111/bph.12607
109. Franklin JM, Vasiljevik T, Prisinzano TE, Carrasco GA. Cannabinoid 2 receptor- and beta Arrestin 2-dependent upregulation of serotonin 2A receptors. Eur Neuropsychopharmacol. (2013) 23:760–7. doi: 10.1016/j.euroneuro.2012.06.012
110. Franklin JM, Carrasco GA. Cannabinoid-induced enhanced interaction and protein levels of serotonin 5-HT(2A) and dopamine D(2) receptors in rat prefrontal cortex. J Psychopharmacol. (2012) 26:1333–47. doi: 10.1177/0269881112450786
111. Franklin JM, Carrasco GA. Cannabinoid receptor agonists upregulate and enhance serotonin 2A (5-HT(2A)) receptor activity via ERK1/2 signaling. Synapse. (2013) 67:145–59. doi: 10.1002/syn.21626
112. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. (2001) 24:677–736. doi: 10.1146/annurev.neuro.24.1.677
113. Garcia-Gutierrez MS, Ortega-Alvaro A, Busquets-Garcia A, Perez-Ortiz JM, Caltana L, Ricatti MJ, et al. Synaptic plasticity alterations associated with memory impairment induced by deletion of CB2 cannabinoid receptors. Neuropharmacology. (2013) 73:388–96. doi: 10.1016/j.neuropharm.2013.05.034
114. Lehman JA, Calvo V, Gomez-Cambronero J. Mechanism of ribosomal p70S6 kinase activation by granulocyte macrophage colony-stimulating factor in neutrophils: cooperation of a MEK-related, THR421/SER424 kinase and a rapamycin-sensitive, m-TOR-related THR389 kinase. J Biol Chem. (2003) 278:28130–8. doi: 10.1074/jbc.M300376200
115. Richter JD, Klann E. Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. (2009) 23:1–11. doi: 10.1101/gad.1735809
116. Zhou X, Lin DS, Zheng F, Sutton MA, Wang H. Intracellular calcium and calmodulin link brain-derived neurotrophic factor to p70S6 kinase phosphorylation and dendritic protein synthesis. J Neurosci Res. (2010) 88:1420–32. doi: 10.1002/jnr.22321
117. Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. (1995) 15(3 Pt 1):1768–77. doi: 10.1523/JNEUROSCI.15-03-01768.1995
118. Cazakoff BN, Howland JG. Acute stress disrupts paired pulse facilitation and long-term potentiation in rat dorsal hippocampus through activation of glucocorticoid receptors. Hippocampus. (2010) 20:1327–31. doi: 10.1002/hipo.20738
119. Rivest S. Cannabinoids in microglia: a new trick for immune surveillance and neuroprotection. Neuron. (2006) 49:4–8. doi: 10.1016/j.neuron.2005.12.004
120. Tang J, Miao H, Jiang B, Chen Q, Tan L, Tao Y, et al. A selective CB2R agonist (JWH133) restores neuronal circuit after Germinal Matrix Hemorrhage in the preterm via CX3CR1(+) microglia. Neuropharmacology. (2017) 119:157–69. doi: 10.1016/j.neuropharm.2017.01.027
121. Hayley S, Litteljohn D. Neuroplasticity and the next wave of antidepressant strategies. Front Cell Neurosci. (2013) 7:218. doi: 10.3389/fncel.2013.00218
122. Prowse N, Hayley S. Microglia and BDNF at the crossroads of stressor related disorders: towards a unique trophic phenotype. Neurosci Biobehav Rev. (2021) 131:135–63. doi: 10.1016/j.neubiorev.2021.09.018
123. Boorman E, Zajkowska Z, Ahmed R, Pariante CM, Zunszain PA. Crosstalk between endocannabinoid and immune systems: a potential dysregulation in depression? Psychopharmacology. (2016) 233:1591–604. doi: 10.1007/s00213-015-4105-9
124. Su F, Yi H, Xu L, Zhang Z. Fluoxetine and S-citalopram inhibit M1 activation and promote M2 activation of microglia in vitro. Neuroscience. (2015) 294:60–8. doi: 10.1016/j.neuroscience.2015.02.028
125. Han J, Fan Y, Zhou K, Blomgren K, Harris RA. Uncovering sex differences of rodent microglia. J Neuroinflamm. (2021) 18:74. doi: 10.1186/s12974-021-02124-z
126. American Psychological Association. What are anxiety disorders?. Washington, D.C: American Psychological Association (2021).
127. Grant BF, Hasin DS, Stinson FS, Dawson DA, June Ruan W, Goldstein RB, et al. Prevalence, correlates, co-morbidity, and comparative disability of DSM-IV generalized anxiety disorder in the USA: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Psychol Med. (2005) 35:1747–59. doi: 10.1017/S0033291705006069
128. Konac D, Young KS, Lau J, Barker. Comorbidity Between Depression and Anxiety in Adolescents: bridge Symptoms and Relevance of Risk and Protective Factors. J Psychopathol Behav Assess. (2021) 43:583–96. doi: 10.1007/s10862-021-09880-5
129. Struijs SY, de Jong PJ, Jeronimus BF, van der Does W, Riese H, Spinhoven P. Psychological risk factors and the course of depression and anxiety disorders: a review of 15 years NESDA research. J Affect Disord. (2021) 295:1347–59. doi: 10.1016/j.jad.2021.08.086
130. Tiguman GMB, Silva MT, Galvao TF. Prevalence of depressive and anxiety symptoms and their relationship with life-threatening events, tobacco dependence and hazardous alcohol drinking: a population-based study in the Brazilian Amazon. J Affect Disord. (2022) 298(Pt A):224–31. doi: 10.1016/j.jad.2021.10.059
131. Martin J, Asjadi K, Hubbard L, Kendall K, Pardinas AF, Jermy B, et al. Examining sex differences in neurodevelopmental and psychiatric genetic risk in anxiety and depression. PLoS One. (2021) 16:e0248254. doi: 10.1371/journal.pone.0248254
132. Hajj A, Hachem R, Khoury R, Hallit S, El JB, Nasr F, et al. Clinical and genetic factors associated with anxiety and depression in breast cancer patients: a cross-sectional study. BMC Cancer. (2021) 21:872. doi: 10.1186/s12885-021-08615-9
133. Hettema JM, Prescott CA, Myers JM, Neale MC, Kendler KS. The structure of genetic and environmental risk factors for anxiety disorders in men and women. Arch Gen Psychiatry. (2005) 62:182–9. doi: 10.1001/archpsyc.62.2.182
134. Scattolin MAA, Resegue RM, Rosario MCD. The impact of the environment on neurodevelopmental disorders in early childhood. J Pediatr. (2021). [Epub ahead of print]. doi: 10.1016/j.jped.2021.11.002
135. Berg N, Kiviruusu O, Grundstrom J, Huurre T, Marttunen M. Stress, development and mental health study, the follow-up study of Finnish TAM cohort from adolescence to midlife: cohort profile. BMJ Open. (2021) 11:e046654. doi: 10.1136/bmjopen-2020-046654
136. Bighelli I, Trespidi C, Castellazzi M, Cipriani A, Furukawa TA, Girlanda F, et al. Antidepressants and benzodiazepines for panic disorder in adults. Cochrane Database Syst Rev. (2016) 9:CD011567. doi: 10.1002/14651858.CD011567.pub2
137. Skapinakis P, Caldwell DM, Hollingworth W, Bryden P, Fineberg NA, Salkovskis P, et al. Pharmacological and psychotherapeutic interventions for management of obsessive-compulsive disorder in adults: a systematic review and network meta-analysis. Lancet Psychiatry. (2016) 3:730–9. doi: 10.1016/S2215-0366(16)30069-4
138. Patterson B, Van Ameringen M. Augmentation Strategies for Treatment-Resistant Anxiety Disorders: a Systematic Review and Meta-Analysis. Depress Anxiety. (2016) 33:728–36. doi: 10.1002/da.22525
139. Ammar G, Naja WJ, Pelissolo A. [Treatment-resistant anxiety disorders: a literature review of drug therapy strategies]. Encephale. (2015) 41:260–5. doi: 10.1016/j.encep.2013.11.002
140. Chapoutot M, Peter-Derex L, Bastuji H, Leslie W, Schoendorff B, Heinzer R, et al. Cognitive behavioral therapy and acceptance and commitment therapy for the discontinuation of long-term benzodiazepine use in insomnia and anxiety disorders. Int J Environ Res Public Health. (2021) 18:10222. doi: 10.3390/ijerph181910222
142. Schanzer B, Rivas-Grajales AM, Khan A, Mathew SJ. Novel investigational therapeutics for generalized anxiety disorder (GAD). Expert Opin Investig Drugs. (2019) 28:1003–12. doi: 10.1080/13543784.2019.1680638
143. Bruggeman C, O’Day CS. Selective Serotonin Reuptake Inhibitor Toxicity. Treasure Island, FL: StatPearls (2021).
144. Brusco A, Tagliaferro PA, Saez T, Onaivi ES. Ultrastructural localization of neuronal brain CB2 cannabinoid receptors. Ann NY Acad Sci. (2008) 1139:450–7. doi: 10.1196/annals.1432.037
145. Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. (2006) 1071:10–23. doi: 10.1016/j.brainres.2005.11.035
146. Nishi M. Effects of Early-Life Stress on the Brain and Behaviors: implications of Early Maternal Separation in Rodents. Int J Mol Sci. (2020) 21:7212. doi: 10.3390/ijms21197212
147. Rincel M, Darnaudery M. Maternal separation in rodents: a journey from gut to brain and nutritional perspectives. Proc Nutr Soc. (2020) 79:113–32. doi: 10.1017/S0029665119000958
148. Vetulani J. Early maternal separation: a rodent model of depression and a prevailing human condition. Pharmacol Rep. (2013) 65:1451–61. doi: 10.1016/s1734-1140(13)71505-6
149. Llorente-Berzal A, Assis MA, Rubino T, Zamberletti E, Marco EM, Parolaro D, et al. Sex-dependent changes in brain CB1R expression and functionality and immune CB2R expression as a consequence of maternal deprivation and adolescent cocaine exposure. Pharmacol Res. (2013) 74:23–33. doi: 10.1016/j.phrs.2013.05.001
150. Robertson JM, Achua JK, Smith JP, Prince MA, Staton CD, Ronan PJ, et al. Anxious behavior induces elevated hippocampal Cb2 receptor gene expression. Neuroscience. (2017) 352:273–84. doi: 10.1016/j.neuroscience.2017.03.061
151. Garcia-Gutierrez MS, Manzanares J. Overexpression of CB2 cannabinoid receptors decreased vulnerability to anxiety and impaired anxiolytic action of alprazolam in mice. J Psychopharmacol. (2011) 25:111–20. doi: 10.1177/0269881110379507
152. Liu QR, Canseco-Alba A, Zhang HY, Tagliaferro P, Chung M, Dennis E, et al. Cannabinoid type 2 receptors in dopamine neurons inhibits psychomotor behaviors, alters anxiety, depression and alcohol preference. Sci Rep. (2017) 7:17410. doi: 10.1038/s41598-017-17796-y
153. Onaivi ES, Carpio O, Ishiguro H, Schanz N, Uhl GR, Benno R. Behavioral effects of CB2 cannabinoid receptor activation and its influence on food and alcohol consumption. Ann NY Acad Sci. (2008) 1139:426–33. doi: 10.1196/annals.1432.035
154. Onaivi ES, Ishiguro H, Sejal P, Meozzi PA, Myers L, Tagliaferro P, et al. Methods to study the behavioral effects and expression of CB2 cannabinoid receptor and its gene transcripts in the chronic mild stress model of depression. Methods Mol Med. (2006) 123:291–8. doi: 10.1385/1-59259-999-0:291
155. Garcia-Gutierrez MS, Garcia-Bueno B, Zoppi S, Leza JC, Manzanares J. Chronic blockade of cannabinoid CB2 receptors induces anxiolytic-like actions associated with alterations in GABA(A) receptors. Br J Pharmacol. (2012) 165:951–64. doi: 10.1111/j.1476-5381.2011.01625.x
156. Lisboa SF, Niraula A, Resstel LB, Guimaraes FS, Godbout JP, Sheridan JF. Repeated social defeat-induced neuroinflammation, anxiety-like behavior and resistance to fear extinction were attenuated by the cannabinoid receptor agonist WIN55,212-2. Neuropsychopharmacology. (2018) 43:1924–33. doi: 10.1038/s41386-018-0064-2
157. Kasatkina LA, Rittchen S, Sturm EM. Neuroprotective and Immunomodulatory Action of the Endocannabinoid System under Neuroinflammation. Int J Mol Sci. (2021) 22:5431. doi: 10.3390/ijms22115431
158. Tanaka M, Sackett S, Zhang Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front Neurol. (2020) 11:87. doi: 10.3389/fneur.2020.00087
159. Amenta PS, Jallo JI, Tuma RF, Elliott MB. A cannabinoid type 2 receptor agonist attenuates blood-brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. J Neurosci Res. (2012) 90:2293–305. doi: 10.1002/jnr.23114
160. Sartori AC, Vance DE, Slater LZ, Crowe M. The impact of inflammation on cognitive function in older adults: implications for healthcare practice and research. J Neurosci Nurs. (2012) 44:206–17. doi: 10.1097/JNN.0b013e3182527690
161. Byers AL, Yaffe K. Depression and risk of developing dementia. Nat Rev Neurol. (2011) 7:323–31. doi: 10.1038/nrneurol.2011.60
162. Cassano T, Calcagnini S, Pace L, De Marco F, Romano A, Gaetani S. Cannabinoid Receptor 2 Signaling in Neurodegenerative Disorders: from Pathogenesis to a Promising Therapeutic Target. Front Neurosci. (2017) 11:30. doi: 10.3389/fnins.2017.0003
163. Abate G, Uberti D, Tambaro S. Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology. (2021) 10:542. doi: 10.3390/biology10060542
164. Benito C, Nunez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, et al. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J Neurosci. (2003) 23:11136–41. doi: 10.1523/JNEUROSCI.23-35-11136.2003
165. Solas M, Francis PT, Franco R, Ramirez MJ. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol Aging. (2013) 34:805–8. doi: 10.1016/j.neurobiolaging.2012.06.005
166. Savonenko AV, Melnikova T, Wang Y, Ravert H, Gao Y, Koppel J, et al. Cannabinoid CB2 Receptors in a Mouse Model of Abeta Amyloidosis: immunohistochemical Analysis and Suitability as a PET Biomarker of Neuroinflammation. PLoS One. (2015) 10:e0129618. doi: 10.1371/journal.pone.0129618
167. Galan-Ganga M, Rodriguez-Cueto C, Merchan-Rubira J, Hernandez F, Avila J, Posada-Ayala M, et al. Cannabinoid receptor CB2 ablation protects against TAU induced neurodegeneration. Acta Neuropathol Commun. (2021) 9:90. doi: 10.1186/s40478-021-01196-5
168. Aso E, Andres-Benito P, Carmona M, Maldonado R, Ferrer I. Cannabinoid Receptor 2 Participates in Amyloid-beta Processing in a Mouse Model of Alzheimer’s Disease but Plays a Minor Role in the Therapeutic Properties of a Cannabis-Based Medicine. J Alzheimers Dis. (2016) 51:489–500. doi: 10.3233/JAD-150913
169. Koppel J, Vingtdeux V, Marambaud P, d’Abramo C, Jimenez H, Stauber M, et al. CB2 receptor deficiency increases amyloid pathology and alters tau processing in a transgenic mouse model of Alzheimer’s disease. Mol Med. (2014) 20:29–36. doi: 10.2119/molmed.2013.00140.revised
170. Schmole AC, Lundt R, Toporowski G, Hansen JN, Beins E, Halle A, et al. Cannabinoid Receptor 2-Deficiency Ameliorates Disease Symptoms in a Mouse Model with Alzheimer’s Disease-Like Pathology. J Alzheimers Dis. (2018) 64:379–92. doi: 10.3233/JAD-180230
171. Zhao J, Wang M, Liu W, Ma Z, Wu J. Activation of cannabinoid receptor 2 protects rat hippocampal neurons against Abeta-induced neuronal toxicity. Neurosci Lett. (2020) 735:135207. doi: 10.1016/j.neulet.2020.135207
172. Wu J, Bie B, Yang H, Xu JJ, Brown DL, Naguib M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol Aging. (2013) 34:791–804. doi: 10.1016/j.neurobiolaging.2012.06.011
Keywords: cannabinoid receptor 2, psychiatry, anxiety, depression, Alzheimer’s disease, inflammation, immunomodulation
Citation: Morcuende A, García-Gutiérrez MS, Tambaro S, Nieto E, Manzanares J and Femenia T (2022) Immunomodulatory Role of CB2 Receptors in Emotional and Cognitive Disorders. Front. Psychiatry 13:866052. doi: 10.3389/fpsyt.2022.866052
Received: 30 January 2022; Accepted: 07 March 2022;
Published: 15 April 2022.
Edited by:
Stefania Schiavone, University of Foggia, ItalyReviewed by:
Caroline Menard, Laval University, CanadaCopyright © 2022 Morcuende, García-Gutiérrez, Tambaro, Nieto, Manzanares and Femenia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa Femenia, dGZlbWVuaWFAdW1oLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.