 Melody J. Y. Kang
Melody J. Y. Kang Emily Hawken2,3
Emily Hawken2,3 Gustavo Hector Vazquez
Gustavo Hector Vazquez
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Psychiatry, 25 April 2022
Sec. Molecular Psychiatry
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.860882
This article is part of the Research TopicThe Glutamate Hypothesis of Mood Disorders: Neuroplasticity Processes, Clinical Features, Treatment PerspectivesView all 6 articles
The mechanism of action underlying ketamine’s rapid antidepressant effects in patients with depression, both suffering from major depressive disorder (MDD) and bipolar disorder (BD), including treatment resistant depression (TRD), remains unclear. Of the many speculated routes that ketamine may act through, restoring deficits in neuroplasticity may be the most parsimonious mechanism in both human patients and preclinical models of depression. Here, we conducted a literature search using PubMed for any reports of ketamine inducing neuroplasticity relevant to depression, to identify cellular and molecular events, relevant to neuroplasticity, immediately observed with rapid mood improvements in humans or antidepressant-like effects in animals. After screening reports using our inclusion/exclusion criteria, 139 publications with data from cell cultures, animal models, and patients with BD or MDD were included (registered on PROSPERO, ID: CRD42019123346). We found accumulating evidence to support that ketamine induces an increase in molecules involved in modulating neuroplasticity, and that these changes are paired with rapid antidepressant effects. Molecules or complexes of high interest include glutamate, AMPA receptors (AMPAR), mTOR, BDNF/TrkB, VGF, eEF2K, p70S6K, GSK-3, IGF2, Erk, and microRNAs. In summary, these studies suggest a robust relationship between improvements in mood, and ketamine-induced increases in molecular neuroplasticity, particularly regarding intracellular signaling molecules.
Depressive Disorders affect 280 million people globally, being the leading cause of disability worldwide (1). One widely accepted neurobiological theory for this highly prevalent mental disorder is the monoamine hypothesis of depression, developed over 50 years ago (2), suggesting that serotonin, norepinephrine, and dopamine deficiencies were responsible for the occurrence of depressive symptoms. Since then, all antidepressant drugs, such as selective serotonin reuptake inhibitors, monoamine oxidase inhibitors, and tricyclic antidepressants have targeted this system to provide relief. While effective for some, remission rates remain low with a delayed onset of clinical efficacy, and up to 46% of patients do not respond effectively to available treatments—resulting in treatment-resistant depression (TRD) (3). One accepted definition of TRD is when patients exhibit inadequate responses to two or more antidepressants after adequate dosing and duration (3). However, in practice, many patients with TRD have exhausted all available treatments, yet struggle to find symptom relief. This highlights the complexity of this disorder as well as the deficiency of evidence regarding its pathogenesis and methods of treatment.
The neuroplasticity hypothesis of depression is a recent neurobiological theory of major depressive disorder (MDD). Neuroplasticity is a phenomenon characterized by neuronal adaptation, i.e., the brain’s ability to reorganize itself when facing an internal or external stimulus (4–8). MDD is strongly linked to abnormalities in neuroplasticity shown through neuroimaging and pharmacological studies (9, 10). When referring to neuroplasticity, this term generally encompasses three large categories; structural, molecular, and functional neuroplasticity (11). Changes in volumes of brain regions, phenotypical changes in neuronal cell types, and differing levels of biomarkers involved in neuroplasticity signaling cascades are all observed in both patients with depressive symptoms and animal models of depression, compared to healthy controls (9, 10). Furthermore, traditional antidepressant medications that target the monoamine hypothesis of depression show downstream effects of improved neuroplasticity in patients, and rescued neuroplasticity in animal models of depression (11–13). Through these observations, the deficit of neuroplasticity has been elucidated as a potential target in developing novel therapeutics for the pathology of depression.
Ketamine, an N-methyl-D-aspartate receptor antagonist within the glutamatergic system, was first approved by the U.S. Food and Drug Administration (FDA) as an anesthetic in 1970. In recent years, many studies have explored its antidepressant properties and demonstrated its potential for patients with MDD and TRD. Following these studies, the FDA approved the isomer (s)-ketamine, also known as esketamine, as the first glutamatergic antidepressant in the form of an intranasal spray called Spravato, in 2019 (14). Unlike currently available pharmacological antidepressants, ketamine elicits its effects as fast as 1 h following administration and is sustained for up to 1–2 weeks (15, 16). It also shows high response and remission rates at approximately 60–70% and 30%, respectively (17–20). Additionally, ketamine also affects several domains of neuroplasticity including molecular biomarkers, cellular structures, and regional brain volumes, raising the question about the underlying mechanisms of action mediating ketamine’s rapid clinical antidepressant effects.
The exact relationship between the pathology of depression, deficits in neuroplasticity, and ketamine’s rapid antidepressant effect remain poorly understood. Current results of the literature seem to fall into two different categories of mechanisms that converge to enact ketamine’s antidepressant effect: molecular neuroplasticity for its immediate effects (minutes to hours), and structural neuroplasticity for its sustained effects (days). Molecular plasticity affected by ketamine include changes in receptors, primary and secondary effectors, proteins involved in signaling cascades, neurotransmitters, and microRNAs. These changes are observed immediately post infusion in both humans and animal models and seem to precede any other structural or functional plasticity markers observed. Here we have summarized the broad range of ketamine’s effects on neuroplasticity in the context of depression, with a specific focus on the immediate molecular cascades that may mediate ketamine’s rapid antidepressant effects. In conjunction, we postulate that deficits or alterations in molecular neuroplasticity may constitute pathological markers of depression and that ketamine might serve to restore plasticity to induce its rapid antidepressant effect.
Using Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines (21), the search for this systematic review was conducted through January 2022 and registered on PROSPERO (ID: CRD42019123346). The database PubMed was used to identify peer-reviewed literature regarding ketamine and neuroplasticity in both preclinical and clinical trials, with or without controls or randomization. Our search included open-label studies, as well as citations from reviews for additional papers to include.
Search terms were the following: “ketamine” and “neuroplasticity, dendritic spines, spinogenesis, synaptogenesis, dendritic arbor, synapse, neurogenesis, neuroregeneration, neurotransmission, BDNF, neurotrophic, neuronal plasticity, nerve growth factor, mTOR, synaptic plasticity, functional plasticity, glutamate, cognitive flexibility, and GSK-3.” The search strategy was reviewed by all authors and reports were reviewed by two investigators independently (M.JY.K and E.R.H) to verify correct filtering and sampling process. Screening selected titles and abstracts, as well as full article eligibility assessments were completed by the same two investigators independently, with continuation to full texts of reports. Inclusion criteria included reports that were peer-reviewed and published that evaluated ketamine’s neuroplastic nature either in vivo, in vitro, both in animals and humans, in an antidepressant context, and written in English. Exclusion criteria included studies regarding schizophrenia or any other disorder not relevant to bipolar disorder, MDD or TRD, chart reviews, and reports lacking data on significant changes. In addition, any studies regarding structural or functional (electrophysiology, connectivity, potentiation) neuroplastic changes post-ketamine were excluded as it was out of the scope of the current review. Data was sought for study design, sample size, route of administration or exposure to ketamine, measures of improvement, main results, and fundamental mechanism of neuroplasticity, conclusions, and limitations.
During the first search, 8,352 reports were identified, and after removal of duplicates, 5,176 papers were considered. Another 4,730 papers were excluded during the title and abstract screening process for lack of relevance, with a remainder of 446 reports for full text review. This process led to another exclusion of 305 reports for reasons including manuscripts only studying ketamine at anesthetic doses, results mainly surrounding ketamine’s metabolite hydroxynorketamine, results only surrounding ketamine-induced behavioral changes, main pathologies explored were not MDD or TRD, timelines explored were only long-term and chronic, and results were surrounding functional or structural plasticity which was out of scope for our current review which focuses on molecular plasticity, resulting in a final number of 141. The results from these reports are all included in this systematic analysis (Figure 1).
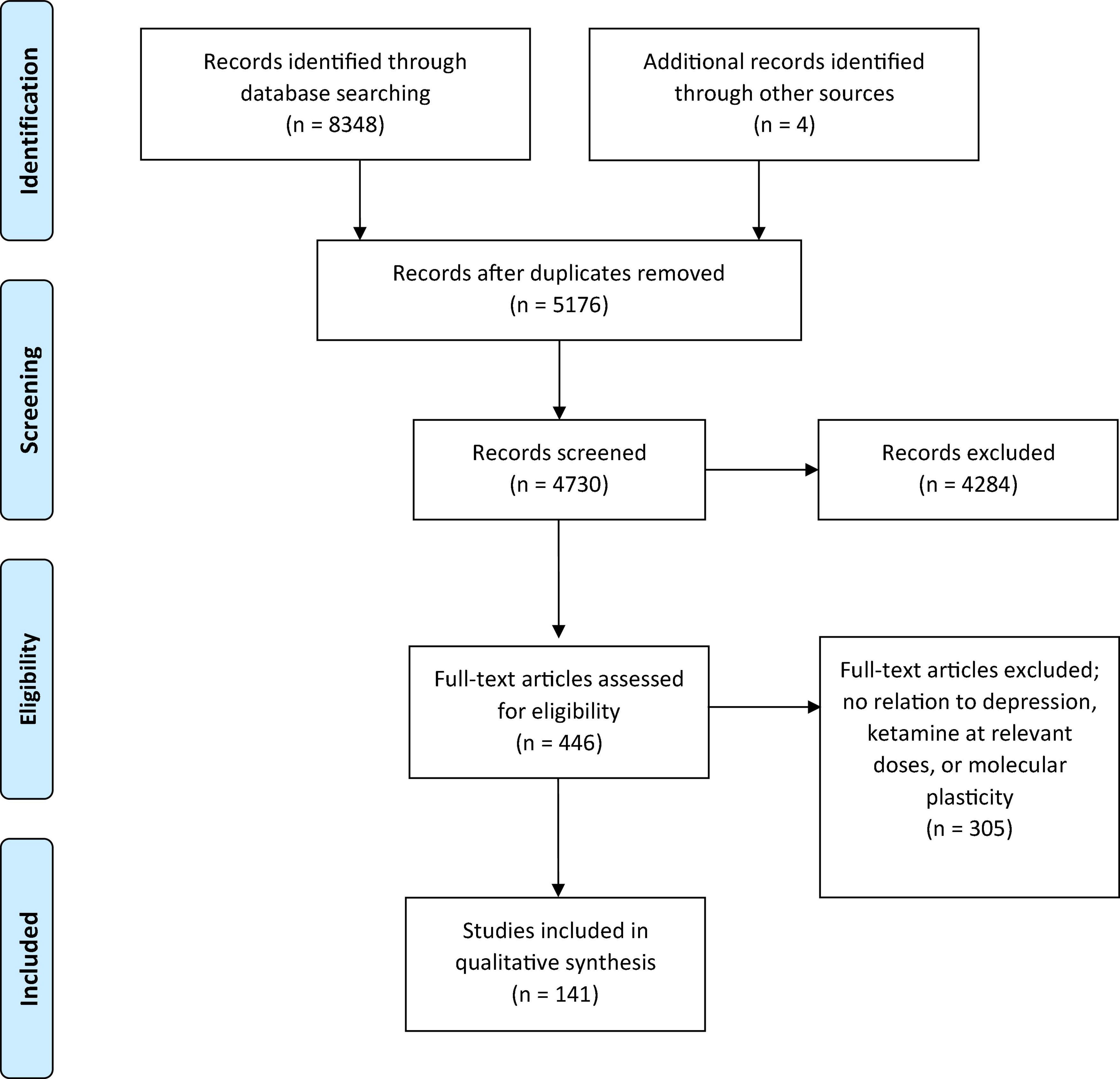
Figure 1. Schematic representation of PRISMA methods utilized for this systematic review.
Glutamate is the main excitatory neurotransmitter in the brain and has functionality in over half of the total neuronal synapses. Differing levels of glutamate has been reported in healthy controls vs. patients with depression (22), as well as in post-mortem studies of suicide victims (23). The transmission of glutamate, its precursor glutamine, as well as the main inhibitory neurotransmitter gamma-aminobutyric acid (GABA), has been explored as a part of ketamine’s mechanism of action.
After exposure to ketamine, a glutamate burst in the synapse is observed, and this is thought to be caused by the binding of ketamine to inhibitory interneurons and thereby enhancing the actions of pyramidal excitatory neurons (24–28). Downstream signaling molecules of this process, such as neuregulin-1, has shown to be downregulated post ketamine administration in parvalbumin-expressing (PV) interneurons, resulting in cortical disinhibition (29). In addition, this glutamate burst is not observed with traditional antidepressants (24–28, 30), though it seems to be dependent on specific synapses and is not globally observed (31). Some studies suggest that this burst may also be sex-dependent (32).
Fluctuating levels of glutamate, glutamine, and GABA following ketamine depend on brain region, timing of measurement, and species. Clinical studies offer varied results. Acute increases in levels of glutamate, glutamine, and GABA have been found in the medial prefrontal cortex (mPFC) of patients with MDD post-ketamine, with GABA levels decreasing after 3–4 h (33, 34). This reduction is maintained for up to 24 h post-ketamine (35). Another study, published by the same group, found the opposite result in the ventro-mPFC, with glutamine and glutamate (Glx) levels decreasing post ketamine, with lower mean Glx levels associated with a better antidepressant response (36). They postulate the reason for their opposite result to be the existence of a placebo group in the latter trial (36). Glx cycling has also shown to contribute to ketamine’s mechanism in the mPFC (37) and the pregenual anterior cingulate cortex (pgACC) (38, 39). The pgACC of healthy controls and patients with MDD demonstrated no changes in glutamate levels, but a significant increase in the ratio of glutamine/glutamate, after 24 h (38, 39). Supporting this, ketamine induced an increase in prefrontal glutamate release in humans, measured by 13C-glutamine labeling of glutamine, which were more prominent than 13C-glutamate (40). This suggests an increased production of glutamine, but not glutamate post-ketamine. The above clinical studies employed magnetic resonance spectroscopy (MRS) and used the most observed subanesthetic dose of 0.5 mg/kg of ketamine, with the exception of the 13C-MRS study, which administered a 0.23 mg/kg bolus followed by a 0.58 mg/kg infusion.
Similar fluctuation patterns are observed in the prefrontal cortex (PFC) and hippocampus of rats (41–44), though one study showed decreased levels of Glx in the mouse hippocampus after 14 h, and increased GABA levels 72 h after ketamine exposure (45). Receptor subunit alterations are also observed. Hippocampal protein levels of GABAA receptor subunit α1 significantly decreases acutely after ketamine in mice (45). Levels of GAD67, a GABA synthetic enzyme, are restored with ketamine administration in stressed models of mice (46), though another found reduced levels of GAD67 post-ketamine in the rat PFC (47). One study found that ketamine’s mechanism may involve changes in G-protein signaling regulators, such as RGS4, in the glutamatergic synapse (48). The rapid antidepressant effects may result directly from the alterations or restorations of these glutamatergic neurotransmitters, neuromodulatory regulators, or from a trigger of receptor activated signaling cascades from the increase of glutamine and glutamate. The nature of these changes in glutamate, glutamine, or GABA levels is still not defined. Ketamine doses used in animal studies most commonly range from 10 to 30 mg/kg, a comparable subanesthetic dose used frequently in rodent and animal studies.
AMPA receptors (AMPAR), one of three glutamatergic receptors, mediate most of the synaptic transmission that occurs in the brain and is likely integral in ketamine’s mechanism of action. AMPARs are located on post-synaptic neurons, and are comprised of GluA1, GluA2, GluA3, and GluA4 subunits (49, 50). Under normal conditions, the consequences of AMPAR activation result in downstream effects including fast excitatory synaptic transmission and increased signaling cascades integral to synaptic plasticity (49, 51). AMPARs and their receptor subunits have been implicated as pivotal factors in the control of mood as well as the pathophysiology and treatment of depression (52). Observed increases in glutamate is presumed to enhance the activation of AMPARs, to carry out ketamine’s antidepressant effects.
Many studies support activity at AMPARs as significant contributions to ketamine’s antidepressant effect. For example, when ketamine is administered at a dose below the threshold of response, an AMPA agonist induces an antidepressant-like effect in a rat model of depression (53). Even when the AMPA agonist is administered alone, similar responses are observed in rats, followed by increased downstream molecular effects, such as increased mTOR activity and BDNF levels (54). Ketamine also upregulates the mRNA of AMPAR subunits 1.5–2-fold (55). When an AMPAR antagonist, NBQX, is administered, ketamine’s rapid behavioral antidepressant-like effects are blocked (54, 56–58). NBQX is shown to inhibit ketamine-induced glutamate and GABA release in the mPFC of mice (58). In addition, ketamine-induced increases in mTOR signaling and protein levels in the hippocampus and prefrontal cortex are blocked by AMPAR antagonists (54, 59, 60). The inverse is true as well—downstream signaling inhibition reduces AMPAR activation and prevents the effects of ketamine (61). As expected, ketamine administration also increases the AMPA to NMDA receptor ratio transiently, a marker of increased activity at AMPARs, in the hippocampus of rat models of anxiety (62). These observations are not limited to in vivo studies, as AMPA antagonists abolish all spontaneous activity of ketamine in cultured neurons of the dorsal raphe nucleus (61).
There is some debate on whether ketamine administration phosphorylates subunits of the AMPAR, as a method of enhancing AMPA activation. Ketamine administration has shown to induce phosphorylation of the AMPA GluA1 subunit on the post synaptic terminal (63, 64). In addition, animals with a knockout of this phosphorylation site were unable to produce ketamine’s electrophysiological and rapid antidepressant-like effects (63). This suggests that phosphorylation of GluA1 is a requirement for a therapeutic response. However, a separate study observed ketamine inducing significantly lower levels of rodent hippocampal phosphorylated GluA1 (57). This effect was inhibited by AMPA antagonists (56). Opposite to results observed in GluA1, a stress paradigm in a rat model of depression increased hippocampal GluA2 phosphorylation levels, which was partially reduced by acute ketamine administration (65), though some studies showed opposite results where ketamine upregulated GluA2 subunits in both the mPFC and hippocampus of rodents (45, 64). In in vitro studies of human induced pluripotent stem cell (iPSC)-derived dopaminergic neurons, GluA1 and GluA2 increases were observed (66), though this was specific to dendrites. Only GluA2 and not GluA1 upregulation was observed in the soma (66). Levels of subunit GluA3 was significantly reduced in the hippocampus of vehicle mice treated with s-ketamine (45). Selective upregulation of subunit expression specific to regions of the neuron and brain may contribute to ketamine’s mechanism.
Along with receptors for glutamate, glutamate transporters are affected by ketamine administration. Stressed mice show decreased levels of excitatory amino acid transporters (EAAT) 2 and 3 and increased extracellular concentrations of glutamate—exhibiting improper glutamate transmission (67). Ketamine administration show restoration of EAAT levels coupled with behavioral antidepressant-like effects (67). Ketamine as an augmentative compound administered with guanosine diminished depressive-like behavior in rodent models of depression, and normalized decreased levels of hippocampal levels of glutamate transporter-1 (GLT-1) (68–70). However, another study showed decreased levels of GLT-1 with ketamine exposure in the mPFC (64). This suggests differential mechanisms of ketamine in the hippocampus and mPFC of animal models and are summarized in Table 1, and illustrated in Figure 2.

Table 1. Molecular changes observed after ketamine administration in preclinical (cell cultures, animals), and clinical studies.
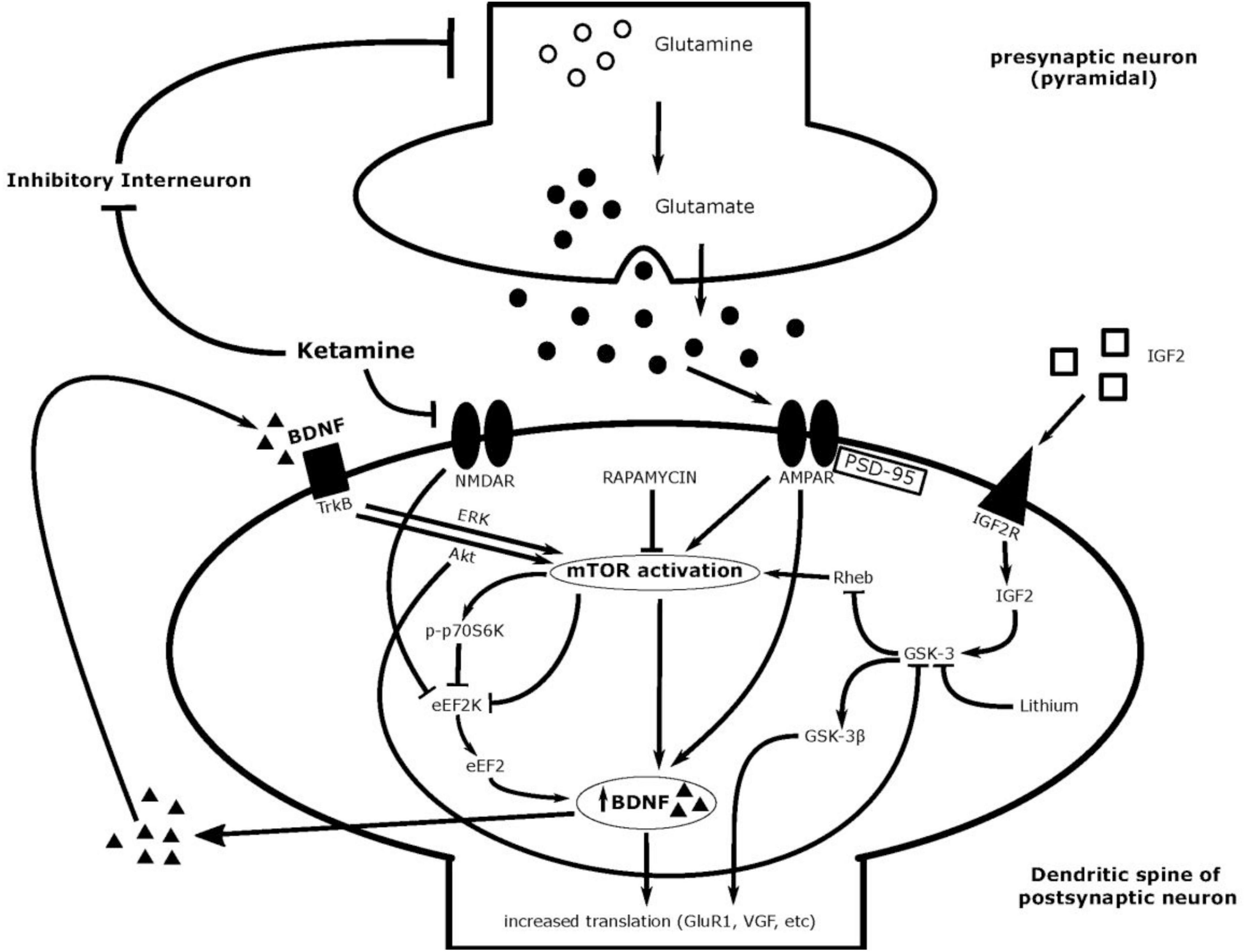
Figure 2. Ketamine’s postulated mechanism of action and associated molecules.
Recent studies outline the mechanistic target of rapamycin (mTOR) as one of the kinases involved in ketamine’s mechanism of action. mTOR is triggered by both the activation of AMPARs and the antagonism of NMDA receptors from ketamine binding. This protein kinase has many upstream and downstream effectors and is known as a master regulator of cell growth that responds to various physiological cues such as amino acids, stress, growth factors, and metabolism (71). mTOR combines with several different proteins to form a complex which function to execute cell survival, maturation, and maintenance (71). Knockdown of mTOR has previously evoked depressive-like states in mice (72) and rapamycin, an mTOR inhibitor, has allowed for the investigation of ketamine’s role in the mTOR signaling pathway.
Ketamine administration rapidly activates the mTOR pathway in animal models, and consequently increases levels of associated signaling proteins dose-dependently, such as phospho- eukaryotic initiation factor 4E binding protein 1 (p4E-BP), p70S6 kinase (p70S6K), extracellular signal-regulated kinase (ERK), and Akt (also named protein kinase B, PKB), an effect that is sustained for at least 72 h (73). Levels of phosphorylated mTOR increase in rat hippocampus and dorsal raphe nucleus post ketamine exposure (61, 74). Blocking the mTOR pathway with rapamycin results in inhibition of ketamine’s behavioral and molecular effects (73, 75) including increases in synaptic proteins such as postsynaptic density protein 95 (PSD-95), and GluA1 in the PFC (73, 75) and hippocampus (76). Ketamine-induced increases in PSD-95 were also observed in the somatosensory cortex of mice (77). These molecular changes may be very specific to brain region, as the ventral hippocampus of mice exhibit greater upregulations of proteins in the mTOR pathway, than the dorsal hippocampus (78). Pre-treatment with ketamine also prevents reductions of PSD-95, GluA1, and synapsin induced by models of depression (79–81). Ketamine also reverses stressed-induced changes in levels of GluA1 in the amygdala (82), hippocampus (76), and levels of GluA4 in the dentate gyrus (83).
In vivo deletion of GluN2B (NR2B), a subunit of the glutamatergic NMDA receptor increases mTOR signaling and mimics ketamine’s behavioral antidepressant-like effects in mice (84, 85), though one study found increases in GluN2B in the rat mPFC after ketamine exposure (64). Interestingly, deletion of another NMDAR subunit, GluN2C, fully preserves ketamine’s antidepressant-like effects in mice (86), suggesting only selective NMDA isomers are required in this mechanism.
An upstream G-protein, Ras homolog enriched in brain (Rheb), is involved in the regulation mTOR activity (87). Rheb, when bound to glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and therefore sequestered, inhibits mTOR activation. Conversely, it is implied that when Rheb-GAPDH binding is blocked, mTOR activation will increase and potential antidepressant mechanisms can ensue (88). In the presence of ketamine, levels of Rheb are significantly increased in cell culture, which is presumed to be closely associated with the increased levels of mTOR activity (88). When NMDA receptors are concomitantly activated, Rheb levels decrease, supporting NMDAR antagonism as part of ketamine’s mechanism of action (88).
The mTOR pathway largely initiates mRNA protein translation through eukaryotic initiation factor 4E-binding proteins (4E-BP), and the mTOR-4E-BP pathway has shown to be pivotal for synaptic plasticity (89, 90). Downstream signaling proteins 4E-BP1 and 4E-BP2 are required for antidepressant-like effects of ketamine and its metabolite, (2R,6R)-HNK, in rodents (89). Additionally, another regulator called extracellular signal-related kinases (ERK) has shown involvement in ketamine’s mechanism, which has previously demonstrated increased levels of phosphorylation in response to traditional antidepressants such as escitalopram, paroxetine, and tranylcypromine in rat hippocampal cultures (91). Ketamine reversed stress-induced decreases in levels of ERK 44 and 42 in the rat amygdala, reflective of increased mTOR activation (82). The mTOR and ERK pathway may only be specific to the mechanism of (S)-ketamine, as (R)-ketamine did not show changes in levels of phosphorylation in the mouse PFC (92).
A neurotrophin largely responsible for neuroplasticity in the brain is brain-derived neurotrophic factor (BDNF). BDNF is a marker of neuronal survival and growth and has been heavily associated with mood and antidepressant therapy (93, 94). BDNF is produced by mTOR activation, released into the synapse by the neuron, and stimulates its receptor, tropomyosin receptor kinase B (TrkB) on the same postsynaptic neuron (95, 96). This further stimulates mTOR activation and functions as a positive feedback loop (96).
Infusion of a BDNF neutralizing antibody, BDNF-knockout, and TrkB-knockout resulted in abolishment of ketamine’s behavioral antidepressant-like effects in animal models (90, 97–100). Similar results are observed with a TrkB antagonist (81, 101). Stress-induced decreases in levels of BDNF in the mouse hippocampus and the ventromedial prefrontal cortex is effectively restored with ketamine administration (102–105). Ketamine also acutely increases BDNF levels in the hippocampus and amygdala (106–111), dentate gyrus (109), and serum (112) of rodents. Administration of a TrkB inhibitor to the hippocampus blocks ketamine’s behavioral and molecular effects in a rat model of depression (81, 113). In vitro studies show similar effects. Primary neuronal cultures incubated with ketamine show increased release of BDNF at 15 min (60). Neuronal cultures isolated from the hippocampus of rat models of depression show increased levels of BDNF when the rats were treated with ketamine, as opposed to those that were not (114).
Interestingly, ketamine’s metabolite, (2R,6R)-HNK also upregulated BDNF levels in the rat ventrolateral periaqueductal gray, hippocampus, and mPFC (115). Infusion of (2R,6R)-HNK into the intramedial-mPFC or systemic administration induced antidepressant-like effects in mice (97). In vitro studies show that ketamine and (2R,6R)-HNK disrupts TrkB receptor interaction with a protein complex crucial for endocytosis, adaptor protein complex 2 (AP2M) (116). This suggests differential regulation mechanisms.
One study also showed differential effects of the R- and S- isomer requiring TrkB receptor activation involved in ketamine’s mechanism (92). Behavioral effects of R-ketamine required TrkB receptors, while S-ketamine did not (92). Though further research is required, these studies robustly support involvement of BDNF-TrkB signaling in ketamine’s mechanism.
Specific phases of upstream or downstream signaling of BDNF are also involved. One study showed ketamine-induced restoration of hippocampal dendritic trafficking of BDNF mRNA that was impaired by chronic mild stress in rats, though global levels of BDNF mRNA were not rescued (117). Significantly increased levels of BDNF promotor IV’s transcriptional activity has also been found post-ketamine (118). Ketamine decreases levels of a transcriptional regulator, histone deacetylase (HDAC), in the mouse striata, hippocampus, and PFC (109). However, knocking out histone deacetylase 5 (HDAC5) resulted in a dysregulated increase of BDNF, which abolished ketamine-induced increases (118).
ProBDNF, a BDNF precursor, is cleaved to produce mature BDNF (mBDNF) and this cleavage is assumed to be integral in producing antidepressant effects (119). The administration of ketamine increases mBDNF levels and decreases the ratio of proBDNF to mBDNF in stressed mice (119). When this cleavage is inhibited with a tissue plasminogen activator (tPA), mice show increased depressive-like symptoms (119). Additionally, the release of mBDNF post S-ketamine in the mouse mPFC was shown to be dependent on the expression of stress-responsive glucocorticoid receptor co-chaperone FKBP51, and FKBP51-knockout mice did not show antidepressant-like effects of ketamine (120). This suggests that this cleavage, and the expression of mBDNF, is integral in ketamine’s mechanism of action.
While most researchers agree on the importance of BDNF in ketamine’s rapid therapeutic effects, the exact role of BDNF is still contested. Measurement of BDNF protein levels induced by ketamine seems to vary as a function of time. Rats who were sacrificed immediately after administration had higher BDNF levels in the prefrontal cortex, amygdala, and hippocampus than those sacrificed 1 and 6 h post-administration (121). Acute administration of ketamine results in a spike of BDNF levels in the hippocampus (122), though chronic administration for 14 days results in BDNF hippocampal levels being unaffected (123, 124). BDNF protein levels seem to increase acutely, and decrease slowly, suggesting a trigger in homeostatic mechanisms to clear increased protein levels (121). However, increased BDNF is seemingly unnecessary for the sustained antidepressant effect. Though protein levels returned to baseline after 24 h, behavioral antidepressant-like responses were still intact in mice (90). In one study, ketamine was still able to manifest antidepressant-like effects in BDNF knockout mice unable to produce BDNF, suggesting that ketamine elicits its clinical effects via additional mechanisms (125).
Results of BDNF levels in humans show variability, and all clinical studies employed the common subanesthetic dose of 0.5 mg/kg, with the exception of two studies which included a range of 0.2–0.5 mg/kg. Ketamine-induced increases in BDNF are observed in serum after 1 week (126) and plasma after 230–240 min (127, 128). Depressive symptoms of patients correlated with BDNF plasma levels for up to 72 h (128), while one found no correlation with mood responses (129). Other studies have found no differences in BDNF levels in plasma at the same time points despite significant improvements in mood (129–133). In a genome-wide association study, single nucleotide polymorphisms as well as whole genes involved in BDNF-TrkB signaling were shown to be associated with rapid and sustained antidepressant effects in patients with TRD (134). Specific polymorphisms associated with BDNF, namely those with the Val/Val BDNF allele at rs65, were more likely to exhibit an antidepressant response (135) and an anti-suicidal response (136) to intravenous ketamine than those with the Val66Met polymorphism.
One critical limitation of using BDNF as a biomarker arises from the fact that quantification of BDNF is often conducted in animal models through brain tissue, and peripheral blood in humans. Robust animal data may not translate to humans in practice. For example, a robust increase in plasma BDNF from ketamine was shown to have no effect on brain BDNF in rats, and no correlation was seen between peripheral and central BDNF levels (137). Studies like this contribute to the challenge of interpreting BDNF’s exact role in ketamine’s rapid antidepressant effects.
BDNF acts to modulate several downstream processes essential to ketamine’s treatment efficacy. A protein that is regulated by and downstream to BDNF, is neuropeptide VGF (non-acronym). VGF is an exercise regulated protein that is downregulated by stress and models of depression, and upregulated with exercise (138, 139). VGF levels increase with ketamine administration in the ventromedial prefrontal cortex in mice (140). Inhibition of VGF in the prefrontal cortex decreases ketamine-induced mTOR signaling and inhibits rapid antidepressant-like effects in mice (141). When VGF is artificially overexpressed, behavioral deficits typically caused by chronic restraint stress is prevented (140). When VGF is knocked out, ketamine exposed mice were more susceptible to chronic stress. Thus, it is likely that VGF is one of many proteins utilized by ketamine, via BDNF, to execute its rapid clinical effects.
Beyond BDNF but still within the mTOR signaling pathway is eukaryotic elongation factor 2 kinase (eEF2K), a downstream kinase of mTOR. This kinase is paired with the protein eEF2, an important factor of ribosome translocation of protein synthesis (142). When NMDAR is at its resting state, eEF2K phosphorylates eEF2, which pauses translation of proteins. Thus, NMDAR antagonism stops eEF2 phosphorylation, which promotes translation of transcripts. Administration of ketamine led to rapid decreases in p-eEF2 in the hippocampus, and artificially inhibiting eEF2K led to increased BDNF protein expression (90). In addition, inhibiting eEF2K in BDNF knockout mice had no antidepressant-like effect, further supporting BDNF’s involvement in ketamine’s actions (90). Ketamine administration was shown to increase levels of phosphorylated eEF2K in the prefrontal cortex (143), yet the significance of this result is yet to be clarified, as the location of phosphorylation determines increased or decreased activity and impacts on eEF2 (144).
P70 ribosomal S6 kinase (p70S6K) is another downstream substrate of mTOR that works to promote protein synthesis, and has previously shown decreased levels in depressed subjects compared to healthy controls (145). Ketamine significantly increased levels of p70S6K in primary neuronal cultures, in the mPFC of rats, and NAc, ventral tegmental area, substantia nigra, hippocampus, and basolateral amygdala of mice, with rapamycin abolishing these effects (59, 60, 88, 146, 147). Ketamine also restores levels of p70S6K in mice models of depression. Interestingly, this ketamine-induced increase in p70S6K does not occur in dopamine 3 receptor (D3R) knockout mice (146). This suggests that ketamine’s mechanism of action depends on viable D3Rs (146).
Glycogen synthase kinase 3, or GSK-3, is one of the enzymes in the mTOR signaling pathway responsible for regulating many aspects of the cell cycle such as proliferation and apoptosis (148). This serine/threonine kinase has previously been studied in the context of bipolar disorder, diabetes, and Alzheimer’s disease. In the context of depression, inhibition of this enzyme is required for ketamine’s behavioral antidepressant effect in numerous animal models of depression (149, 150). Constitutively active GSK-3 is associated with resistance to ketamine’s antidepressant effect, and the usage of a GSK-3 inhibitor, lithium, mimics ketamine’s effects (149). Ketamine has shown to decrease levels of GSK-3 in the mouse hippocampus, striata, dentate gyrus, and PFC (109), and may also act through increasing levels of phosphorylated GSK-3β, the inactive form, in rat prefrontal cortex (151, 152). GSK-3 inhibitors act to augment antidepressant-like effects of ketamine when the dose of ketamine is at a sub-threshold value (152).
GSK-3’s activity is a prerequisite for biomolecular changes observed in animal models after ketamine administration. Specifically, ketamine increases membrane levels of the AMPAR subunit, GluA1 (147, 153). However, when GSK-3 is knocked out, ketamine’s effects on GluA1 are abolished, and previously observed increases in BDNF are inhibited (153). Similar results occur when rapamycin is administrated (147). This mechanism may be monitored through postsynaptic density protein 95 (PSD-95), a substrate for GSK-3 that also regulates AMPAR trafficking (153). To add further, ketamine decreases hippocampal membrane levels of phosphorylated-PSD-95, the target of GSK-3 that would promote AMPA-GluA1 internalization (153). Decreased p-PSD-95 would mean more AMPARs would be available on the membrane to be activated. Ketamine also increased PSD-95 in the rat hippocampus (108, 154) and normalized levels of transcription factor Egr-1 (154).
Ketamine’s modulatory actions also converge on to pathways downstream of NMDAR. In a poststroke depression model of rats, ketamine administration significantly improved antidepressant-like symptoms (155). Along with this, there was an upregulation of subtype NMDAR2-β and downregulation of subtype NMDAR2-α, as well as their downstream signaling proteins, β-CaMKII and α-phosphorylation, respectively, in the dentate gyrus (155). Interestingly, no effects on NR1 and NR3 subtypes were seen, suggesting that ketamine is not dependent on all subunits of NMDARs (155).
Decreased levels of a protein upstream of GSK-3, called insulin-like growth factor 2 (IGF2), has previously been implicated in rodent models of depression, with artificial induction of IGF2 expression having protective effects against depressive-like behaviors (156). IGF2 is upregulated with ketamine in vivo and in vitro (150, 157)—and this requires the inhibition of GSK-3 as well (150). Knocking out IGF2 reduces ketamine’s behavioral antidepressant effect, and mice resilient to induced depression show higher amounts of IGF2 than mice that were susceptible to depression paradigms (150).
There are several limitations to all mTOR results discussed so far. The first limitation is that there seem to be sex-dependent differences in mTOR signaling. Ketamine treatment increased levels of BDNF mRNA in female, not male mice (158), suggesting sex-dependent mechanisms that require both protein translation and transcription. While female rats display greater sensitivity to ketamine at lower doses than male rats, such as at 2.5 mg/kg, this rapid behavioral antidepressant effect was not paired with an increased phosphorylation level of mTOR (159). A similar result was observed in female mice, where there was actually a decrease of mTOR phosphorylation in the prefrontal cortex (32). In addition, ketamine-induced reductions in levels of eEF2K activation in male rats were in agreement with previous results; however, this was not seen in female rats (159). In fact, the increased sensitivity to ketamine was not seen when female rats were ovariectomized—and restored when artificial estrogen and progesterone were administered, suggesting integral roles of gonadal hormones in ketamine’s mechanism of action (159). This gender-specific response to ketamine is not isolated to this one study, as a similar result is seen with acetylation of α-tubulin—another marker of neuroplasticity. Increased acetylation of α-tubulin was observed only in females 24 h post-ketamine administration, suggesting increased stabilization of microtubules (160). These differences may point to possible variations of the mechanism of action or metabolism for ketamine depending on the sex.
The second limitation arises from the fact that these results stem from a combination of different rodent models of depression. Each model may manifest through various phenotypes of depression, and therefore cannot be considered as one consistent valid method of inducing depression. In fact, when a model of resistant depression was utilized, mTOR levels were significantly reduced in the prefrontal cortex, despite a behavioral antidepressant response (161). This suggests that an increase in mTOR levels do not necessarily reflect a behavioral antidepressant response in all cases.
Lastly, the third limitation is observed from a recent study that showed oral rapamycin in humans with depression prolonging ketamine’s antidepressant effect at 2 weeks (162). However, the addition of rapamycin did not affect ketamine’s acute antidepressant effects (162). This study puts preclinical results into perspective and generates questions about the function of mTOR in sustained antidepressant effects. Taken together, while the current field of literature supports mTOR involvement in the mechanism of action for ketamine, these are limitations to consider.
MAPK/ErK signaling pathway is another important pathway involved in neural plasticity, carrying signals from the cell surface to the nucleus. The MAPK pathway regulates CREB, an integral modulator of cell survival. Inhibition of this pathway shows further potentiation of ketamine’s effects in the rat brain. Ketamine increased levels of p-TrkB and pCREB in cultured neurons and the prefrontal cortex, and decreased pCREB in the hippocampus (163, 164). A MAPK inhibitor alone showed similar results and augmented ketamine’s effects when administered together, suggesting that MAPK signaling inhibition may be involved in ketamine’s mechanism (163).
One of the upstream extracellular signaling growth factors that stimulate the MAPK/Erk pathway, among many others, is vascular endothelial growth factor (VEGF), which has previously been discussed as a potential target for treating depression (165). It is an important mitogen that functions in the survival of endothelial cells, neurons, as well as maintenance of synaptic transmission (165), and has previously shown to mimic actions of antidepressants in behavioral models of rodents (166). Neuron-specific deletion of VEGF or its receptor, or the usage of a VEGF neutralizing antibody resulted in ketamine’s behavioral and synaptogenic effects being abolished in rodents (167, 168). Additionally, infusion of VEGF alone into the mPFC produced rapid antidepressant-like effects in these animal models (167, 168). However, in the rat hippocampus, ketamine administration decreased levels of VEGF after 2 h (169). Data from clinical studies show contradictory results. One found no effects in plasma VEGF levels after treatment with ketamine (131), while another found increased plasma levels of VEGFA mRNA and VEGFA protein (170). Ratio of the mRNA of VEGF to pigment epithelial-derived factor (PEDF), a signaling molecule closely linked to VEGF, was increased with ketamine as well (170). This may suggest VEGF as a part of ketamine’s mechanism.
MicroRNAs (or miRNAs) are non-coding small molecules that are integral in synthesizing proteins, and modulators of protein expression. Members of the MiR-29 family have shown to regulate neuropathological processes relevant to neuroplasticity. Metabotropic glutamate receptor 4 (GRM4), which is predicted to be a target of MiR-29, regulates many major neurotransmitters such as dopamine and glutamate. Inducing depression in rat models was paired with downregulation of miR-29b-3p and increased levels of GRM4 in the prefrontal cortex, which were restored by ketamine administration (171). In fact, pre-treatment of ketamine prevented upregulation of GRM4 in the rat prefrontal cortex, and miR-29b-3p overexpression was followed by a relief of behavioral depressive symptoms (171). An miRNA from a different family, miR-206, was down-regulated in ketamine-treated rodents, which was determined to be a critical regulator of BDNF protein expression (172). In addition, overexpression of miR-206 in primary cultured hippocampal pyramidal neurons impaired ketamine-induced upregulation of neuronal BDNF protein expression (172). Rats vulnerable to chronic mild stress exhibited reduced levels of another microRNA, miR-9-5p, in the hippocampus, which was reversed by ketamine administration in 24 h (173). An in vitro model of stress also showed restored levels of miR-9-5p with ketamine (173). These results suggest that the pathway involving microRNAs may significantly be involved in ketamine’s antidepressant mechanism of action.
Ketamine’s rapid antidepressant potential in unipolar depression have initiated the exploration of its treatment effects in bipolar disorder (BD) as well (174–180). However, evidence supporting ketamine’s effects for BD is limited, and the number of randomized controlled trials is significantly less than those exploring unipolar depression. In addition, extra caution is required due to relevant ketamine’s psychomimetic and dissociative effects (181). Still, current evidence seems to support acute ketamine’s rapid antidepressant and anti-suicidal effects in bipolar depression (182–185), though the data that measures the sustainability of this effect is limited. Studies that differentiate BD-I and BD-II is even fewer, though Zarate et al. (178) suggests that results may be similar in both subtypes.
According to our literature search, there are very few studies that investigate ketamine’s mechanism of action for bipolar disorder specifically. Assumptions can be made from mechanistic actions of lithium, a common treatment option for BD, as both ketamine and lithium inhibit GSK-3 (153, 186) in animal models. This suggests glutamatergic modulation as one of the mechanisms employed by ketamine. A clinical study, Lally et al. (187) found that ketamine improved anhedonia, independent from mood, in patients with bipolar depression, and this effect was significantly associated with increased glucose metabolism in the dorsal anterior cingulate cortex and putamen. Interestingly, patients who were taking concomitant lithium, but not valproate, had greater hedonic improvements (187). BDNF as a molecular marker has been robustly linked with bipolar episodes in patients, with low levels of BDNF associated with depressive and manic episodes, as well as their severity (188–190). BDNF upregulation after ketamine administration may partially explain ketamine’s mechanism in BD (191). Patients with euthymic BD have reported decreased expression of HDAC genes compared to control (192), and ketamine has shown to alter levels of HDAC in animal models as well (109, 193), suggesting the involvement of epigenetic mechanisms of ketamine.
The present systematic review summarized the wide-ranging scope of current literature regarding molecular neuroplastic changes that occur rapidly after exposure to ketamine. We found that these changes in molecular neuroplasticity revolving glutamate, AMPA receptors (AMPAR), mTOR, BDNF/TrkB, VGF, eEF2K, p70S6K, GSK-3, IGF2, Erk, and microRNAs may likely be responsible in mediating and producing ketamine’s rapid antidepressant effects in MDD and bipolar disorder depression. We found studies that report similar findings post ketamine, such as increased glutamate/glutamine cycling, enhanced actions of AMPA receptors, increased activation of mTOR, BDNF/TrkB, and associated signaling molecules. However, no matter how robust a field of evidence is, such as mTOR’s involvement pathway, potential inconsistencies still exist, such as sex-dependent results and differences in sources of tissue in animal models vs. clinical studies. In addition, these studies employ a range of methodologies in various lab environments, models of depression, and doses of ketamine. It is most probable that many different pathways contribute to ketamine’s actions, and it is currently difficult to narrow down which mechanisms are most dominant over others to produce this effect, due to the widespread nature of ongoing research. Further studies should focus on reproducing currently available data with different animal models of depression, larger sample sizes, and long-term studies exploring mechanisms of maintenance and sustained effects to strengthen the evidence.
The original results presented in the study are included in the article, and further inquiries can be directed to the corresponding author/s.
MJYK: acquisition, analysis, interpretation of data for the writing of original manuscript, and writing original manuscript. EH: substantial contributions to the conception and design, and revising it critically for important intellectual content. GV: substantial contributions to the conception or design and final approval of the version to be published. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This manuscript is supported in part by a 2019 Innovative Research Grant from Providence Care Hospital.
2. Bunney WE. Norepinephrine in depressive reactions: a review. Arch Gen Psychiatry. (1965) 13:483. doi: 10.1001/archpsyc.1965.01730060001001
3. Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am. (1996) 19:179–200. doi: 10.1016/s0193-953x(05)70283-5
4. Cramer SC, Sur M, Dobkin BH, O’Brien C, Sanger TD, Trojanowski JQ, et al. Harnessing neuroplasticity for clinical applications. Brain. (2011) 134:1591–609.
5. Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. (2012) 338:68–72. doi: 10.1126/science.1222939
6. Pascual-Leone A, Amedi A, Fregni F, Merabet LB. The plastic human brain cortex. Annu Rev Neurosci. (2005) 28:377–401. doi: 10.1146/annurev.neuro.27.070203.144216
7. Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. (2008) 33:88–109. doi: 10.1038/sj.npp.1301574
8. Price RB, Duman R. Neuroplasticity in cognitive and psychological mechanisms of depression: an integrative model. Mol Psychiatry. (2020) 25:530–43. doi: 10.1038/s41380-019-0615-x
9. Duman RS, Malberg J, Nakagawa S, D’Sa C. Neuronal plasticity and survival in mood disorders. Biol Psychiatry. (2000) 48:732–9. doi: 10.1016/s0006-3223(00)00935-5
10. Koolschijn PC, van Haren NE, Lensvelt-Mulders GJ, Hulshoff Pol HE, Kahn RS. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum Brain Mapp. (2009) 30:3719–35. doi: 10.1002/hbm.20801
11. D’Sa C, Duman RS. Antidepressants and neuroplasticity. Bipolar Disord. (2002) 4:183–94. doi: 10.1034/j.1399-5618.2002.01203.x
12. Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, et al. Hippocampal neurogenesis induced by antidepressant drugs: an epiphenomenon in their mood-improving actions. Mol Psychiatry. (2009) 14:739. doi: 10.1038/mp.2009.75
13. Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. (2016) 22:238–49. doi: 10.1038/nm.4050
14. U.S. Food and Drug Administration [USFDA]. Spravato NDA211243/S-000 Approval Letter. Silver Spring, MD: U.S. Food and Drug Administration (2019).
15. Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. (2000) 47:351–4. doi: 10.1016/s0006-3223(99)00230-9
16. Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A Randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. (2006) 63:856. doi: 10.1001/archpsyc.63.8.856
17. Aan Het Rot M, Zarate CA, Charney DS, Mathew SJ. Ketamine for depression: where do we go from here? Biol Psychiatry. (2012) 72:537–47. doi: 10.1016/j.biopsych.2012.05.003
18. Murrough JW. Ketamine as a novel antidepressant: from synapse to behavior. Clin Pharmacol Ther. (2012) 91:303–9. doi: 10.1038/clpt.2011.244
19. Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, Aan Het Rot M, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. (2013) 74:250–6. doi: 10.1016/j.biopsych.2012.06.022
20. Phillips JL, Norris S, Talbot J, Birmingham M, Hatchard T, Ortiz A, et al. Single, repeated, and maintenance ketamine infusions for treatment-resistant depression: a randomized controlled trial. Am J Psychiatry. (2019) 176:401–9. doi: 10.1176/appi.ajp.2018.18070834
21. Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. (2009) 6:e1000100. doi: 10.1371/journal.pmed.1000100
22. Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. (2012) 72:1313–33. doi: 10.2165/11633130-000000000-00000
23. Nowak G, Ordway GA, Paul IA. Alterations in the N-methyl-d-asparatate (n.d.) receptor complex in the frontal cortex of suicide victims. Brain Res. (1995) 675:157–64. doi: 10.1016/0006-8993(95)00057-w
24. Ali F, Gerhard DM, Sweasy K, Pothula S, Pittenger C, Duman RS, et al. Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines. Nat Commun. (2020) 11:72.
25. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. (2007) 27:11496–500. doi: 10.1523/JNEUROSCI.2213-07.2007
26. Miller OH, Moran JT, Hall BJ. Two cellular hypotheses explaining the initiation of ketamine’s antidepressant actions: direct inhibition and disinhibition. Neuropharmacology. (2016) 100:17–26. doi: 10.1016/j.neuropharm.2015.07.028
27. Widman AJ, McMahon LL. Disinhibition of CA1 pyramidal cells by low-dose ketamine and other antagonists with rapid antidepressant efficacy. Proc Natl Acad Sci U S A. (2018) 115:E3007–16. doi: 10.1073/pnas.1718883115
28. Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. (2018) 23:801–11. doi: 10.1038/mp.2017.255
29. Grieco SF, Qiao X, Johnston KG, Chen L, Nelson RR, Lai C, et al. Neuregulin signaling mediates the acute and sustained antidepressant effects of subanesthetic ketamine. Transl Psychiatry. (2021) 11:144. doi: 10.1038/s41398-021-01255-4
30. Zhang B, Yang X, Ye L, Liu R, Ye B, Du W, et al. Ketamine activated glutamatergic neurotransmission by GABAergic disinhibition in the medial prefrontal cortex. Neuropharmacology. (2021) 194:108382. doi: 10.1016/j.neuropharm.2020.108382
31. Lazarevic V, Yang Y, Flais I, Svenningsson P. Ketamine decreases neuronally released glutamate via retrograde stimulation of presynaptic adenosine A1 receptors. Mol Psychiatry. (2021) 26:7425–35. doi: 10.1038/s41380-021-01246-3
32. Thelen C, Flaherty E, Saurine J, Sens J, Mohamed S, Pitychoutis PM. Sex differences in the temporal neuromolecular and synaptogenic effects of the rapid-acting antidepressant drug ketamine in the mouse brain. Neuroscience. (2019) 398:182–92. doi: 10.1016/j.neuroscience.2018.11.053
33. Milak MS, Proper CJ, Mulhern ST, Parter AL, Kegeles LS, Ogden RT, et al. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol Psychiatry. (2016) 21:320–7. doi: 10.1038/mp.2015.83
34. Scheidegger M, Fuchs A, Lehmann M, Grimm S, Boeker H, Seifritz E, et al. Effects of an Antidepressant Dose of Ketamine on Prefrontal Aspartate, Glutamine and GABA Levels in Healthy Subjects: Assessing the Post-Infusion Interval with 1H-MRS. Red Hook, NY: Curran Associates, Inc (2013).
35. Njau S, Vasavada M, Leaver A, Wade B, Kubicki A, Congdon E, et al. F151. GABAergic neurotransmission modulates therapeutic response to ketamine infusion in major depression. Biol Psychiatry. (2018) 83:S296–7.
36. Milak MS, Rashid R, Dong Z, Kegeles LS, Grunebaum MF, Ogden RT, et al. Assessment of relationship of ketamine dose with magnetic resonance spectroscopy of Glx and GABA responses in adults with major depression: a randomized clinical trial. JAMA Netw Open. (2020) 3:e2013211. doi: 10.1001/jamanetworkopen.2020.13211
37. Abdallah CG, Jackowski A, Salas R, Gupta S, Sato JR, Mao X, et al. The nucleus accumbens and ketamine treatment in major depressive disorder. Neuropsychopharmacology. (2017) 42:1739–46. doi: 10.1038/npp.2017.49
38. Evans JW, Lally N, An L, Li N, Nugent AC, Banerjee D, et al. 7T 1H-MRS in major depressive disorder: a ketamine treatment study. Neuropsychopharmacology. (2018) 43:1908–14. doi: 10.1038/s41386-018-0057-1
39. Li M, Woelfer M, Colic L, Safron A, Chang C, Heinze H-J, et al. Default mode network connectivity change corresponds to ketamine’s delayed glutamatergic effects. Eur Arch Psychiatry Clin Neurosci. (2020) 270:207–16. doi: 10.1007/s00406-018-0942-y
40. Abdallah CG, De Feyter HM, Averill LA, Jiang L, Averill CL, Chowdhury GMI, et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology. (2018) 43:2154–60. doi: 10.1038/s41386-018-0136-3
41. Chowdhury GMI, Zhang J, Thomas M, Banasr M, Ma X, Pittman B, et al. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol Psychiatry. (2017) 22:120–6. doi: 10.1038/mp.2016.34
42. Pham TH, Defaix C, Xu X, Deng S-X, Fabresse N, Alvarez J-C, et al. Common Neurotransmission Recruited in (R,S)-Ketamine and (2R,6R)-hydroxynorketamine–induced sustained antidepressant-like effects. Biol Psychiatry. (2018) 84:e3–6. doi: 10.1016/j.biopsych.2017.10.020
43. Wang N, Zhang G-F, Liu X-Y, Sun H-L, Wang X-M, Qiu L-L, et al. Downregulation of neuregulin 1-ErbB4 signaling in parvalbumin interneurons in the rat brain may contribute to the antidepressant properties of ketamine. J Mol Neurosci. (2014) 54:211–8. doi: 10.1007/s12031-014-0277-8
44. Wu M, Minkowicz S, Dumrongprechachan V, Hamilton P, Kozorovitskiy Y. Ketamine rapidly enhances glutamate-evoked dendritic spinogenesis in medial prefrontal cortex through dopaminergic mechanisms. Biol Psychiatry. (2021) 89:1096–105. doi: 10.1016/j.biopsych.2020.12.022
45. Weckmann K, Deery MJ, Howard JA, Feret R, Asara JM, Dethloff F, et al. Ketamine’s effects on the Glutamatergic and GABAergic systems: a proteomics and metabolomics study in mice. Mol Neuropsychiatry. (2019) 5:42–51. doi: 10.1159/000493425
46. Ghosal S, Duman CH, Liu R-J, Wu M, Terwilliger R, Girgenti MJ, et al. Ketamine rapidly reverses stress-induced impairments in GABAergic transmission in the prefrontal cortex in male rodents. Neurobiol Dis. (2020) 134:104669. doi: 10.1016/j.nbd.2019.104669
47. Zhou Z, Zhang G, Li X, Liu X, Wang N, Qiu L, et al. Loss of phenotype of parvalbumin interneurons in rat prefrontal cortex is involved in antidepressant- and propsychotic-like behaviors following acute and repeated ketamine administration. Mol Neurobiol. (2015) 51:808–19. doi: 10.1007/s12035-014-8798-2
48. Lur G, Fariborzi M, Higley MJ. Ketamine disrupts neuromodulatory control of glutamatergic synaptic transmission. PLoS One. (2019) 14:e0213721. doi: 10.1371/journal.pone.0213721
49. Hollmann M, O’Shea-Greenfield A, Rogers SW, Heinemann S. Cloning by functional expression of a member of the glutamate receptor family. Nature. (1989) 342:643–8. doi: 10.1038/342643a0
50. Nakanishi N, Shneider NA, Axel R. A family of glutamate receptor genes: evidence for the formation of heteromultimeric receptors with distinct channel properties. Neuron. (1990) 5:569–81. doi: 10.1016/0896-6273(90)90212-x
51. Raymond LA, Blackstone CD, Huganir RL. Phosphorylation of amino acid neurotransmitter receptors in synaptic plasticity. Trends Neurosci. (1993) 16:147–53. doi: 10.1016/0166-2236(93)90123-4
52. Alt A, Nisenbaum ES, Bleakman D, Witkin JM. A role for AMPA receptors in mood disorders. Biochem Pharmacol. (2006) 71:1273–88. doi: 10.1016/j.bcp.2005.12.022
53. Akinfiresoye L, Tizabi Y. Antidepressant effects of AMPA and ketamine combination: role of hippocampal BDNF, synapsin, and mTOR. Psychopharmacology. (2013) 230:291–8. doi: 10.1007/s00213-013-3153-2
54. Zhou W, Wang N, Yang C, Li X-M, Zhou Z-Q, Yang J-J. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatr. (2014) 29:419–23. doi: 10.1016/j.eurpsy.2013.10.005
55. Ho M-F, Correia C, Ingle JN, Kaddurah-Daouk R, Wang L, Kaufmann SH, et al. Ketamine and ketamine metabolites as novel estrogen receptor ligands: induction of cytochrome P450 and AMPA glutamate receptor gene expression. Biochem Pharmacol. (2018) 152:279–92. doi: 10.1016/j.bcp.2018.03.032
56. Du J, Machado-Vieira R, Maeng S, Martinowich K, Manji HK, Zarate CA. Enhancing AMPA to NMDA throughput as a convergent mechanism for antidepressant action. Drug Discov Today Ther Strateg. (2006) 3:519–26. doi: 10.1016/j.ddstr.2006.11.012
57. Maeng S, Zarate CA, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of α-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic acid receptors. Biol Psychiatry. (2008) 63:349–52. doi: 10.1016/j.biopsych.2007.05.028
58. Pham TH, Defaix C, Nguyen TML, Mendez-David I, Tritschler L, David DJ, et al. Cortical and raphe GABAA, AMPA receptors and glial GLT-1 glutamate transporter contribute to the sustained antidepressant activity of ketamine. Pharmacol Biochem Behav. (2020) 192:172913. doi: 10.1016/j.pbb.2020.172913
59. Girgenti MJ, Ghosal S, LoPresto D, Taylor JR, Duman RS. Ketamine accelerates fear extinction via mTORC1 signaling. Neurobiol Dis. (2017) 100:1–8. doi: 10.1016/j.nbd.2016.12.026
60. Lepack AE, Bang E, Lee B, Dwyer JM, Duman RS. Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology. (2016) 111:242–52. doi: 10.1016/j.neuropharm.2016.09.011
61. Llamosas N, Perez-Caballero L, Berrocoso E, Bruzos-Cidon C, Ugedo L, Torrecilla M. Ketamine promotes rapid and transient activation of AMPA receptor-mediated synaptic transmission in the dorsal raphe nucleus. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 88:243–52. doi: 10.1016/j.pnpbp.2018.07.022
62. Tizabi Y, Bhatti BH, Manaye KF, Das JR, Akinfiresoye L. Antidepressant-like effects of low ketamine dose is associated with increased hippocampal AMPA/NMDA receptor density ratio in female Wistar–Kyoto rats. Neuroscience. (2012) 213:72–80. doi: 10.1016/j.neuroscience.2012.03.052
63. Zhang K, Xu T, Yuan Z, Wei Z, Yamaki VN, Huang M, et al. Essential roles of AMPA receptor GluA1 phosphorylation and presynaptic HCN channels in fast-acting antidepressant responses of ketamine. Sci Signal. (2016) 9:ra123. doi: 10.1126/scisignal.aai7884
64. Piva A, Caffino L, Mottarlini F, Pintori N, Castillo Díaz F, Fumagalli F, et al. Metaplastic effects of ketamine and MK-801 on glutamate receptors expression in rat medial prefrontal cortex and hippocampus. Mol Neurobiol. (2021) 58:3443–56. doi: 10.1007/s12035-021-02352-7
65. Elhussiny MEA, Carini G, Mingardi J, Tornese P, Sala N, Bono F, et al. Modulation by chronic stress and ketamine of ionotropic AMPA/NMDA and metabotropic glutamate receptors in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. (2021) 104:110033. doi: 10.1016/j.pnpbp.2020.110033
66. Collo G, Cavalleri L, Chiamulera C, Merlo Pich E. Ketamine increases the expression of GluR1 and GluR2 α-amino-3-hydroxy-5-methy-4-isoxazole propionate receptor subunits in human dopaminergic neurons differentiated from induced pluripotent stem cells. Neuroreport. (2019) 30:207–12. doi: 10.1097/WNR.0000000000001185
67. Zhu X, Ye G, Wang Z, Luo J, Hao X. Sub-anesthetic doses of ketamine exert antidepressant-like effects and upregulate the expression of glutamate transporters in the hippocampus of rats. Neurosci Lett. (2017) 639:132–7. doi: 10.1016/j.neulet.2016.12.070
68. Camargo A, Dalmagro AP, M Rosa J, B Zeni AL, P Kaster M, Tasca CI, et al. Subthreshold doses of guanosine plus ketamine elicit antidepressant-like effect in a mouse model of depression induced by corticosterone: role of GR/NF-κB/IDO-1 signaling. Neurochem Int. (2020) 139:104797. doi: 10.1016/j.neuint.2020.104797
69. Camargo A, Dalmagro AP, Zeni ALB, Rodrigues ALS. Guanosine potentiates the antidepressant-like effect of subthreshold doses of ketamine: possible role of pro-synaptogenic signaling pathway. J Affect Disord. (2020) 271:100–8. doi: 10.1016/j.jad.2020.03.186
70. Camargo A, Pazini FL, Rosa JM, Wolin IAV, Moretti M, Rosa PB, et al. Augmentation effect of ketamine by guanosine in the novelty-suppressed feeding test is dependent on mTOR signaling pathway. J Psychiatr Res. (2019) 115:103–12. doi: 10.1016/j.jpsychires.2019.05.017
71. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
72. Garro-Martínez E, Fullana MN, Florensa-Zanuy E, Senserrich J, Paz V, Ruiz-Bronchal E, et al. mTOR knockdown in the infralimbic cortex evokes a depressive-like state in mouse. Int J Mol Sci. (2021) 22:8671. doi: 10.3390/ijms22168671
73. Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. (2010) 329:959–64. doi: 10.1126/science.1190287
74. Johnston JN, Thacker JS, Desjardins C, Kulyk BD, Romay-Tallon R, Kalynchuk LE, et al. Ketamine rescues hippocampal reelin expression and synaptic markers in the repeated-corticosterone chronic stress paradigm. Front Pharmacol. (2020) 11:559627. doi: 10.3389/fphar.2020.559627
75. Li N, Liu R-J, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. (2011) 69:754–61. doi: 10.1016/j.biopsych.2010.12.015
76. Wen G, Yao H, Li Y, Ding R, Ren X, Tan Y, et al. Regulation of tau protein on the antidepressant effects of ketamine in the chronic unpredictable mild stress model. Front Psychiatry. (2019) 10:287. doi: 10.3389/fpsyt.2019.00287
77. Pryazhnikov E, Mugantseva E, Casarotto P, Kolikova J, Fred SM, Toptunov D, et al. Longitudinal two-photon imaging in somatosensory cortex of behaving mice reveals dendritic spine formation enhancement by subchronic administration of low-dose ketamine. Sci Rep. (2018) 8:6464. doi: 10.1038/s41598-018-24933-8
78. Yamada J, Jinno S. Potential link between antidepressant-like effects of ketamine and promotion of adult neurogenesis in the ventral hippocampus of mice. Neuropharmacology. (2019) 158:107710. doi: 10.1016/j.neuropharm.2019.107710
79. Camargo A, Dalmagro AP, de Souza MM, Zeni ALB, Rodrigues ALS. Ketamine, but not guanosine, as a prophylactic agent against corticosterone-induced depressive-like behavior: possible role of long-lasting pro-synaptogenic signaling pathway. Exp Neurol. (2020) 334:113459. doi: 10.1016/j.expneurol.2020.113459
80. Camargo A, Dalmagro AP, Wolin IAV, Siteneski A, Zeni ALB, Rodrigues ALS. A low-dose combination of ketamine and guanosine counteracts corticosterone-induced depressive-like behavior and hippocampal synaptic impairments via mTORC1 signaling. Prog Neuropsychopharmacol Biol Psychiatry. (2021) 111:110371. doi: 10.1016/j.pnpbp.2021.110371
81. Qu Y, Shan J, Wang S, Chang L, Pu Y, Wang X, et al. Rapid-acting and long-lasting antidepressant-like action of (R)-ketamine in Nrf2 knock-out mice: a role of TrkB signaling. Eur Arch Psychiatry Clin Neurosci. (2021) 271:439–46. doi: 10.1007/s00406-020-01208-w
82. Lee C-W, Chen Y-J, Wu H-F, Chung Y-J, Lee Y-C, Li C-T, et al. Ketamine ameliorates severe traumatic event-induced antidepressant-resistant depression in a rat model through ERK activation. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 93:102–13. doi: 10.1016/j.pnpbp.2019.03.015
83. Michaëlsson H, Andersson M, Svensson J, Karlsson L, Ehn J, Culley G, et al. The novel antidepressant ketamine enhances dentate gyrus proliferation with no effects on synaptic plasticity or hippocampal function in depressive-like rats. Acta Physiol (Oxf). (2019) 225:e13211. doi: 10.1111/apha.13211
84. Gerhard DM, Pothula S, Liu R-J, Wu M, Li X-Y, Girgenti MJ, et al. GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. J Clin Invest. (2020) 130:1336–49. doi: 10.1172/JCI130808
85. Miller OH, Yang L, Wang C-C, Hargroder EA, Zhang Y, Delpire E, et al. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife. (2014) 3:e03581. doi: 10.7554/eLife.03581
86. Tarrés-Gatius M, Miquel-Rio L, Campa L, Artigas F, Castañé A. Involvement of NMDA receptors containing the GluN2C subunit in the psychotomimetic and antidepressant-like effects of ketamine. Transl Psychiatry. (2020) 10:427. doi: 10.1038/s41398-020-01110-y
87. Lee MN, Ha SH, Kim J, Koh A, Lee CS, Kim JH, et al. Glycolytic flux signals to mTOR through glyceraldehyde-3-phosphate dehydrogenase-mediated regulation of Rheb. Mol Cell Biol. (2009) 29:3991–4001. doi: 10.1128/MCB.00165-09
88. Harraz MM, Tyagi R, Cortés P, Snyder SH. Antidepressant action of ketamine via mTOR is mediated by inhibition of nitrergic Rheb degradation. Mol Psychiatry. (2016) 21:313–9. doi: 10.1038/mp.2015.211
89. Aguilar-Valles A, De Gregorio D, Matta-Camacho E, Eslamizade MJ, Khlaifia A, Skaleka A, et al. Antidepressant actions of ketamine engage cell-specific translation via eIF4E. Nature. (2021) 590:315–9. doi: 10.1038/s41586-020-03047-0
90. Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. (2011) 475:91–5. doi: 10.1038/nature10130
91. Park SW, Lee JG, Seo MK, Lee CH, Cho HY, Lee BJ, et al. Differential effects of antidepressant drugs on mTOR signalling in rat hippocampal neurons. Int J Neuropsychopharmacology. (2014) 17:1831–46. doi: 10.1017/S1461145714000534
92. Rafało-Ulińska A, Pałucha-Poniewiera A. The effectiveness of (R)-ketamine and its mechanism of action differ from those of (S)-ketamine in a chronic unpredictable mild stress model of depression in C57BL/6J mice. Behav Brain Res. (2022) 418:113633. doi: 10.1016/j.bbr.2021.113633
93. Castren E. Neurotrophic effects of antidepressant drugs. Curr Opin Pharmacol. (2004) 4:58–64. doi: 10.1016/j.coph.2003.10.004
94. Chen B, Dowlatshahi D, MacQueen GM, Wang J-F, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. (2001) 50:260–5. doi: 10.1016/s0006-3223(01)01083-6
95. Cavalleri L, Merlo Pich E, Millan MJ, Chiamulera C, Kunath T, Spano PF, et al. Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Mol Psychiatry. (2018) 23:812–23. doi: 10.1038/mp.2017.241
96. Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. (2010) 3:1. doi: 10.3389/neuro.02.001.2010
97. Fukumoto K, Fogaça MV, Liu R-J, Duman C, Kato T, Li X-Y, et al. Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc Natl Acad Sci U S A. (2019) 116:297–302. doi: 10.1073/pnas.1814709116
98. Lepack AE, Fuchikami M, Dwyer JM, Banasr M, Duman RS. BDNF release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol. (2015) 18:pyu033. doi: 10.1093/ijnp/pyu033
99. Lin P-Y, Ma ZZ, Mahgoub M, Kavalali ET, Monteggia LM. A synaptic locus for TrkB signaling underlying ketamine rapid antidepressant action. Cell Rep. (2021) 36:109513. doi: 10.1016/j.celrep.2021.109513
100. Ma Z, Zang T, Birnbaum SG, Wang Z, Johnson JE, Zhang C-L, et al. TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nat Commun. (2017) 8:1668. doi: 10.1038/s41467-017-01709-8
101. Tan Y, Fujita Y, Qu Y, Chang L, Pu Y, Wang S, et al. Phencyclidine-induced cognitive deficits in mice are ameliorated by subsequent repeated intermittent administration of (R)-ketamine, but not (S)-ketamine: role of BDNF-TrkB signaling. Pharmacol Biochem Behav. (2020) 188:172839. doi: 10.1016/j.pbb.2019.172839
102. Asim M, Hao B, Yang Y-H, Fan B-F, Xue L, Shi Y-W, et al. Ketamine alleviates fear generalization through GluN2B-BDNF signaling in mice. Neurosci Bull. (2020) 36:153–64. doi: 10.1007/s12264-019-00422-4
103. Silva Pereira V, Elfving B, Joca SRL, Wegener G. Ketamine and aminoguanidine differentially affect Bdnf and Mtor gene expression in the prefrontal cortex of adult male rats. Eur J Pharmacol. (2017) 815:304–11. doi: 10.1016/j.ejphar.2017.09.029
104. Tang X-H, Zhang G-F, Xu N, Duan G-F, Jia M, Liu R, et al. Extrasynaptic CaMKIIα is involved in the antidepressant effects of ketamine by downregulating GluN2B receptors in an LPS-induced depression model. J Neuroinflammation. (2020) 17:181. doi: 10.1186/s12974-020-01843-z
105. Yang C, Hu Y-M, Zhou Z-Q, Zhang G-F, Yang J-J. Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Ups J Med Sci. (2013) 118:3–8. doi: 10.3109/03009734.2012.724118
106. Fan J-F, Tang Z-H, Wang S-Y, Lei S, Zhang B, Tian S-W. Ketamine enhances novel object recognition memory reconsolidation via the BDNF/TrkB pathway in mice. Physiol Behav. (2021) 242:113626. doi: 10.1016/j.physbeh.2021.113626
107. Kim J-W, Monteggia LM. Increasing doses of ketamine curtail antidepressant responses and suppress associated synaptic signaling pathways. Behav Brain Res. (2020) 380:112378. doi: 10.1016/j.bbr.2019.112378
108. Pazini FL, Rosa JM, Camargo A, Fraga DB, Moretti M, Siteneski A, et al. mTORC1-dependent signaling pathway underlies the rapid effect of creatine and ketamine in the novelty-suppressed feeding test. Chem Biol Interact. (2020) 332:109281. doi: 10.1016/j.cbi.2020.109281
109. Viana GSB, Vale EM, Araujo ARA, Coelho NC, Andrade SM, Costa RO, et al. Rapid and long-lasting antidepressant-like effects of ketamine and their relationship with the expression of brain enzymes, BDNF, and astrocytes. Braz J Med Biol Res. (2020) 54:e10107. doi: 10.1590/1414-431X202010107
110. Xu SX, Zhou ZQ, Li XM, Ji MH, Zhang GF, Yang JJ. The activation of adenosine monophosphate-activated protein kinase in rat hippocampus contributes to the rapid antidepressant effect of ketamine. Behav Brain Res. (2013) 253:305–9. doi: 10.1016/j.bbr.2013.07.032
111. Zhang M, Radford KD, Driscoll M, Purnomo S, Kim J, Choi KH. Effects of subanesthetic intravenous ketamine infusion on neuroplasticity-related proteins in the prefrontal cortex, amygdala, and hippocampus of Sprague-Dawley rats. IBRO Rep. (2019) 6:87–94. doi: 10.1016/j.ibror.2019.01.006
112. Ardalan M, Elfving B, Rafati AH, Mansouri M, Zarate CA, Mathe AA, et al. Rapid effects of S-ketamine on the morphology of hippocampal astrocytes and BDNF serum levels in a sex-dependent manner. Eur Neuropsychopharmacol. (2020) 32:94–103. doi: 10.1016/j.euroneuro.2020.01.001
113. Liu W-X, Wang J, Xie Z-M, Xu N, Zhang G-F, Jia M, et al. Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a role in the anti-apoptotic and antidepressant effects of ketamine in chronic unpredictable stress model of depression. Psychopharmacology. (2016) 233:405–15. doi: 10.1007/s00213-015-4128-2
114. Chang D, Zhao J, Zhang X, Lian H, Du X, Yuan R, et al. Effect of ketamine combined with DHA on lipopolysaccharide-induced depression-like behavior in rats. Int Immunopharmacol. (2019) 75:105788. doi: 10.1016/j.intimp.2019.105788
115. Chou D. Brain-derived neurotrophic factor in the ventrolateral periaqueductal gray contributes to (2R,6R)-hydroxynorketamine-mediated actions. Neuropharmacology. (2020) 170:108068. doi: 10.1016/j.neuropharm.2020.108068
116. Fred SM, Laukkanen L, Brunello CA, Vesa L, Göös H, Cardon I, et al. Pharmacologically diverse antidepressants facilitate TRKB receptor activation by disrupting its interaction with the endocytic adaptor complex AP-2. J Biol Chem. (2019) 294:18150–61. doi: 10.1074/jbc.RA119.008837
117. Tornese P, Sala N, Bonini D, Bonifacino T, La Via L, Milanese M, et al. Chronic mild stress induces anhedonic behavior and changes in glutamate release, BDNF trafficking and dendrite morphology only in stress vulnerable rats. The rapid restorative action of ketamine. Neurobiol Stress. (2019) 10:100160. doi: 10.1016/j.ynstr.2019.100160
118. Choi M, Lee SH, Park MH, Kim Y-S, Son H. Ketamine induces brain-derived neurotrophic factor expression via phosphorylation of histone deacetylase 5 in rats. Biochem Biophys Res Commun. (2017) 489:420–5. doi: 10.1016/j.bbrc.2017.05.157
119. Zhang F, Luo J, Zhu X. Ketamine ameliorates depressive-like behaviors by tPA-mediated conversion of proBDNF to mBDNF in the hippocampus of stressed rats. Psychiatry Res. (2018) 269:646–51. doi: 10.1016/j.psychres.2018.08.075
120. Anderzhanova E, Hafner K, Genewsky AJ, Soliman A, Pöhlmann ML, Schmidt MV, et al. The stress susceptibility factor FKBP51 controls S-ketamine-evoked release of mBDNF in the prefrontal cortex of mice. Neurobiol Stress. (2020) 13:100239. doi: 10.1016/j.ynstr.2020.100239
121. Fraga DB, Réus GZ, Abelaira HM, De Luca RD, Canever L, Pfaffenseller B, et al. Ketamine alters behavior and decreases BDNF levels in the rat brain as a function of time after drug administration. Rev Bras Psiquiatr. (2013) 35:262–6. doi: 10.1590/1516-4446-2012-0858
122. Garcia LSB, Comim CM, Valvassori SS, Réus GZ, Barbosa LM, Andreazza AC, et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. (2008) 32:140–4. doi: 10.1016/j.pnpbp.2007.07.027
123. Garcia LS, Comim CM, Valvassori SS, Réus GZ, Andreazza AC, Stertz L, et al. Chronic administration of ketamine elicits antidepressant-like effects in rats without affecting hippocampal brain-derived neurotrophic factor protein levels. Basic Clin Pharmacol Toxicol. (2008) 103:502–6. doi: 10.1111/j.1742-7843.2008.00210.x
124. Garcia LSB, Comim CM, Valvassori SS, Réus GZ, Stertz L, Kapczinski F, et al. Ketamine treatment reverses behavioral and physiological alterations induced by chronic mild stress in rats. Prog Neuropsychopharmacol Biol Psychiatry. (2009) 33:450–5. doi: 10.1016/j.pnpbp.2009.01.004
125. Lindholm JSO, Autio H, Vesa L, Antila H, Lindemann L, Hoener MC, et al. The antidepressant-like effects of glutamatergic drugs ketamine and AMPA receptor potentiator LY 451646 are preserved in bdnf+/- heterozygous null mice. Neuropharmacology. (2012) 62:391–7. doi: 10.1016/j.neuropharm.2011.08.015
126. Allen AP, Naughton M, Dowling J, Walsh A, Ismail F, Shorten G, et al. Serum BDNF as a peripheral biomarker of treatment-resistant depression and the rapid antidepressant response: a comparison of ketamine and ECT. J Affect Disord. (2015) 186:306–11. doi: 10.1016/j.jad.2015.06.033
127. Duncan WC, Sarasso S, Ferrarelli F, Selter J, Riedner BA, Hejazi NS, et al. Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int J Neuropsychopharmacol. (2013) 16:301–11. doi: 10.1017/S1461145712000545
128. Haile CN, Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Foulkes A, et al. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int J Neuropsychopharm. (2014) 17:331–6. doi: 10.1017/S1461145713001119
129. Caliman-Fontes AT, Leal GC, Correia-Melo FS, Paixão CS, Carvalho MS, Jesus-Nunes AP, et al. Brain-derived neurotrophic factor serum levels following ketamine and esketamine intervention for treatment-resistant depression: secondary analysis from a randomized trial. Trends Psychiatry Psychother. (2021). [Epub ahead of print]. doi: 10.47626/2237-6089-2021-0298
130. Machado-Vieira R, Yuan P, Brutsche N, DiazGranados N, Luckenbaugh D, Manji HK, et al. Brain-derived neurotrophic factor and initial antidepressant response to an N -Methyl- D -aspartate antagonist. J Clin Psychiatry. (2009) 70:1662–6. doi: 10.4088/JCP.08m04659
131. Medeiros GC, Greenstein D, Kadriu B, Yuan P, Park LT, Gould TD, et al. Treatment of depression with ketamine does not change plasma levels of brain-derived neurotrophic factor or vascular endothelial growth factor. J Affect Disord. (2021) 280:136–9. doi: 10.1016/j.jad.2020.11.011
132. Woelfer M, Li M, Colic L, Liebe T, Di X, Biswal B, et al. Ketamine-induced changes in plasma brain-derived neurotrophic factor (BDNF) levels are associated with the resting-state functional connectivity of the prefrontal cortex. World J Biol Psychiatry. (2020) 21:696–710. doi: 10.1080/15622975.2019.1679391
133. Zheng W, Zhou Y-L, Wang C-Y, Lan X-F, Zhang B, Zhou S-M, et al. Association between plasma levels of BDNF and the antisuicidal effects of repeated ketamine infusions in depression with suicidal ideation. Ther Adv Psychopharmacol. (2020) 10:2045125320973794. doi: 10.1177/2045125320973794
134. Chen M-H, Kao C-F, Tsai S-J, Li C-T, Lin W-C, Hong C-J, et al. Treatment response to low-dose ketamine infusion for treatment-resistant depression: a gene-based genome-wide association study. Genomics. (2021) 113:507–14. doi: 10.1016/j.ygeno.2020.12.030
135. Laje G, Lally N, Mathews D, Brutsche N, Chemerinski A, Akula N, et al. Brain-derived neurotrophic factor val66met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol Psychiatry. (2012) 72:e27–8. doi: 10.1016/j.biopsych.2012.05.031
136. Chen M-H, Lin W-C, Wu H-J, Cheng C-M, Li C-T, Hong C-J, et al. Antisuicidal effect, BDNF Val66Met polymorphism, and low-dose ketamine infusion: reanalysis of adjunctive ketamine study of Taiwanese patients with treatment-resistant depression (AKSTP-TRD). J Affect Disord. (2019) 251:162–9. doi: 10.1016/j.jad.2019.03.075
137. Le Nedelec M, Glue P, Winter H, Goulton C, Broughton L, Medlicott N. Acute low-dose ketamine produces a rapid and robust increase in plasma BDNF without altering brain BDNF concentrations. Drug Deliv and Transl Res. (2018) 8:780–6. doi: 10.1007/s13346-017-0476-2
138. Hunsberger JG, Newton SS, Bennett AH, Duman CH, Russell DS, Salton SR, et al. Antidepressant actions of the exercise-regulated gene VGF. Nat Med. (2007) 13:1476–82. doi: 10.1038/nm1669
139. Jiang C, Lin W-J, Sadahiro M, Labonté B, Menard C, Pfau ML, et al. VGF function in depression and antidepressant efficacy. Mol Psychiatry. (2018) 23:1632–42. doi: 10.1038/mp.2017.233
140. Jiang C, Lin W-J, Labonté B, Tamminga CA, Turecki G, Nestler EJ, et al. VGF and its C-terminal peptide TLQP-62 in ventromedial prefrontal cortex regulate depression-related behaviors and the response to ketamine. Neuropsychopharmacology. (2019) 44:971–81. doi: 10.1038/s41386-018-0277-4
141. Shen M, Lv D, Liu X, Li S, Chen Y, Zhang Y, et al. Essential roles of neuropeptide VGF regulated TrkB/mTOR/BICC1 signaling and phosphorylation of AMPA receptor subunit GluA1 in the rapid antidepressant-like actions of ketamine in mice. Brain Res Bull. (2018) 143:58–65. doi: 10.1016/j.brainresbull.2018.10.004
142. Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron. (2007) 55:648–61. doi: 10.1016/j.neuron.2007.07.030
143. Abelaira HM, Réus GZ, Ignácio ZM, dos Santos MAB, de Moura AB, Matos D, et al. Effects of ketamine administration on mTOR and reticulum stress signaling pathways in the brain after the infusion of rapamycin into prefrontal cortex. J Psychiatric Res. (2017) 87:81–7. doi: 10.1016/j.jpsychires.2016.12.002
144. Monteggia LM, Gideons E, Kavalali ET. The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biol Psychiatry. (2013) 73:1199–203. doi: 10.1016/j.biopsych.2012.09.006
145. Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2011) 35:1774–9. doi: 10.1016/j.pnpbp.2011.05.010
146. Chiamulera C, di Chio M, Cavalleri L, Venniro M, Padovani L, Collo G. Ketamine effects on mammalian target of rapamycin signaling in the mouse limbic system depend on functional dopamine D3 receptors. Neuroreport. (2018) 29:615–20. doi: 10.1097/WNR.0000000000001008
147. Duman RS, Li N, Liu R-J, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. (2012) 62:35–41. doi: 10.1016/j.neuropharm.2011.08.044
148. Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. (2001) 359:1–16. doi: 10.1042/0264-6021:3590001
149. Beurel E, Song L, Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. (2011) 16:1068–70. doi: 10.1038/mp.2011.47
150. Grieco SF, Cheng Y, Eldar-Finkelman H, Jope RS, Beurel E. Up-regulation of insulin-like growth factor 2 by ketamine requires glycogen synthase kinase-3 inhibition. Prog Neuropsychopharmacol Biol Psychiatry. (2017) 72:49–54. doi: 10.1016/j.pnpbp.2016.08.008
151. Zhou W, Dong L, Wang N, Shi J, Yang J, Zuo Z, et al. Akt Mediates GSK-3β phosphorylation in the rat prefrontal cortex during the process of ketamine exerting rapid antidepressant actions. Neuroimmunomodulation. (2014) 21:183–8. doi: 10.1159/000356517
152. Liu R-J, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. (2013) 38:2268–77. doi: 10.1038/npp.2013.128
153. Beurel E, Grieco SF, Amadei C, Downey K, Jope RS. Ketamine-induced inhibition of glycogen synthase kinase-3 contributes to the augmentation of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor signaling. Bipolar Disord. (2016) 18:473–80. doi: 10.1111/bdi.12436
154. Zhang W, Wang H, Lv Y, Liu C, Sun W, Tian L. Downregulation of Egr-1 expression level via GluN2B underlies the antidepressant effects of ketamine in a chronic unpredictable stress animal model of depression. Neuroscience. (2018) 372:38–45. doi: 10.1016/j.neuroscience.2017.12.045
155. Abdoulaye IA, Wu S-S, Chibaatar E, Yu D-F, Le K, Cao X-J, et al. Ketamine induces lasting antidepressant effects by modulating the NMDAR/CaMKII-mediated synaptic plasticity of the hippocampal dentate gyrus in depressive stroke model. Neural Plast. (2021) 2021:6635084. doi: 10.1155/2021/6635084
156. Luo Y-W, Xu Y, Cao W-Y, Zhong X-L, Duan J, Wang X-Q, et al. Insulin-like growth factor 2 mitigates depressive behavior in a rat model of chronic stress. Neuropharmacology. (2015) 89:318–24. doi: 10.1016/j.neuropharm.2014.10.011
157. Grossert A, Mehrjardi NZ, Bailey SJ, Lindsay MA, Hescheler J, Šariæ T, et al. Ketamine increases proliferation of human iPSC-derived neuronal progenitor cells via insulin-like growth factor 2 and independent of the NMDA Receptor. Cells. (2019) 8:E1139. doi: 10.3390/cells8101139
158. Herzog DP, Mellema RM, Remmers F, Lutz B, Müller MB, Treccani G. Sexually dimorphic behavioral profile in a transgenic model enabling targeted recombination in active neurons in response to ketamine and (2R,6R)-hydroxynorketamine administration. Int J Mol Sci. (2020) 21:E2142. doi: 10.3390/ijms21062142
159. Carrier N, Kabbaj M. Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology. (2013) 70:27–34. doi: 10.1016/j.neuropharm.2012.12.009
160. Colic L, McDonnell C, Li M, Woelfer M, Liebe T, Kretzschmar M, et al. Neuronal glutamatergic changes and peripheral markers of cytoskeleton dynamics change synchronically 24 h after sub-anaesthetic dose of ketamine in healthy subjects. Behav Brain Res. (2019) 359:312–9. doi: 10.1016/j.bbr.2018.10.021
161. Kosel M, Tournier B, Ginovart N. Acute ketamine treatment normalizes depression-like behaviors in depression-prone RLA rats: involvement of the AMPA receptor system. (2014) Biol Psychiatry 75:S275–401.
162. Abdallah C, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Rapamycin, an Immunosuppressant and mTORC1 inhibitor, triples the antidepressant response rate of ketamine at 2 weeks following treatment: a double-blind, placebo-controlled, cross-over, randomized clinical trial. bioRxiv. [Preprint]. (2018). doi: 10.1101/500959
163. Réus GZ, Abaleira HM, Titus SE, Arent CO, Michels M, da Luz JR, et al. Effects of ketamine administration on the phosphorylation levels of CREB and TrKB and on oxidative damage after infusion of MEK inhibitor. Pharmacol Rep. (2016) 68:177–84. doi: 10.1016/j.pharep.2015.08.010
164. Wray NH, Schappi JM, Singh H, Senese NB, Rasenick MM. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol Psychiatry. (2019) 24:1833–43. doi: 10.1038/s41380-018-0083-8
165. Warner-Schmidt JL, Duman RS. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. (2008) 8:14–9. doi: 10.1016/j.coph.2007.10.013
166. Warner-Schmidt JL, Duman RS. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci U S A. (2007) 104:4647–52. doi: 10.1073/pnas.0610282104
167. Deyama S, Bang E, Kato T, Li X-Y, Duman RS. Neurotrophic and antidepressant actions of brain-derived neurotrophic factor require vascular endothelial growth factor. Biol Psychiatry. (2019) 86:143–52. doi: 10.1016/j.biopsych.2018.12.014
168. Deyama S, Bang E, Wohleb ES, Li X-Y, Kato T, Gerhard DM, et al. Role of neuronal VEGF signaling in the prefrontal cortex in the rapid antidepressant effects of ketamine. Am J Psychiatry. (2019) 176:388–400. doi: 10.1176/appi.ajp.2018.17121368
169. du Jardin KG, Müller HK, Sanchez C, Wegener G, Elfving B. A single dose of vortioxetine, but not ketamine or fluoxetine, increases plasticity-related gene expression in the rat frontal cortex. Eur J Pharmacol. (2016) 786:29–35. doi: 10.1016/j.ejphar.2016.05.029
170. McGrory CL, Ryan KM, Gallagher B, McLoughlin DM. Vascular endothelial growth factor and pigment epithelial-derived factor in the peripheral response to ketamine. J Affect Disord. (2020) 273:380–3. doi: 10.1016/j.jad.2020.04.013
171. Wan Y-Q, Feng J-G, Li M, Wang M-Z, Liu L, Liu X, et al. Prefrontal cortex miR-29b-3p plays a key role in the antidepressant-like effect of ketamine in rats. Exp Mol Med. (2018) 50:1–14. doi: 10.1038/s12276-018-0164-4
172. Yang X, Yang Q, Wang X, Luo C, Wan Y, Li J, et al. MicroRNA expression profile and functional analysis reveal that miR-206 is a critical novel gene for the expression of BDNF induced by ketamine. Neuromol Med. (2014) 16:594–605. doi: 10.1007/s12017-014-8312-z
173. Mingardi J, La Via L, Tornese P, Carini G, Trontti K, Seguini M, et al. miR-9-5p is involved in the rescue of stress-dependent dendritic shortening of hippocampal pyramidal neurons induced by acute antidepressant treatment with ketamine. Neurobiol Stress. (2021) 15:100381. doi: 10.1016/j.ynstr.2021.100381
174. Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. (2010) 67:793–802. doi: 10.1001/archgenpsychiatry.2010.90
175. Joseph B, Parsaik AK, Ahmed AT, Erwin PJ, Singh B. A systematic review on the efficacy of intravenous racemic ketamine for bipolar depression. J Clin Psychopharmacol. (2021) 41:71–5. doi: 10.1097/JCP.0000000000001317
176. Kantrowitz JT, Halberstam B, Gangwisch J. Single-dose ketamine followed by daily D-Cycloserine in treatment-resistant bipolar depression. J Clin Psychiatry. (2015) 76:737–8. doi: 10.4088/JCP.14l09527
177. Rybakowski JK, Permoda-Osip A, Bartkowska-Sniatkowska A. Ketamine augmentation rapidly improves depression scores in inpatients with treatment-resistant bipolar depression. Int J Psychiatry Clin Pract. (2017) 21:99–103. doi: 10.1080/13651501.2017.1297834
178. Zarate CA, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A, et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. (2012) 71:939–46. doi: 10.1016/j.biopsych.2011.12.010
179. Grunebaum MF, Ellis SP, Keilp JG, Moitra VK, Cooper TB, Marver JE, et al. Ketamine versus midazolam in bipolar depression with suicidal thoughts: a pilot midazolam-controlled randomized clinical trial. Bipolar Disord. (2017) 19:176–83. doi: 10.1111/bdi.12487
180. Zheng W, Zhou Y-L, Liu W-J, Wang C-Y, Zhan Y-N, Li H-Q, et al. Rapid and longer-term antidepressant effects of repeated-dose intravenous ketamine for patients with unipolar and bipolar depression. J Psychiatr Res. (2018) 106:61–8. doi: 10.1016/j.jpsychires.2018.09.013
181. Carta MG, Kalcev G, Fornaro M, Nardi AE. Novel experimental and early investigational drugs for the treatment of bipolar disorder. Expert Opin Investig Drugs. (2021) 30:1081–7. doi: 10.1080/13543784.2021.2000965
182. Dean RL, Marquardt T, Hurducas C, Spyridi S, Barnes A, Smith R, et al. Ketamine and other glutamate receptor modulators for depression in adults with bipolar disorder. Cochrane Database Syst Rev. (2021) 10:CD011611.
183. Xiong J, Lipsitz O, Chen-Li D, Rosenblat JD, Rodrigues NB, Carvalho I, et al. The acute antisuicidal effects of single-dose intravenous ketamine and intranasal esketamine in individuals with major depression and bipolar disorders: a systematic review and meta-analysis. J Psychiatr Res. (2021) 134:57–68. doi: 10.1016/j.jpsychires.2020.12.038
184. Lener MS, Kadriu B, Zarate CA. Ketamine and beyond: investigations into the potential of glutamatergic agents to treat depression. Drugs. (2017) 77:381–401. doi: 10.1007/s40265-017-0702-8
185. Bahji A, Vazquez GH, Zarate CA. Comparative efficacy of racemic ketamine and esketamine for depression: a systematic review and meta-analysis. J Affect Disord. (2021) 278:542–55. doi: 10.1016/j.jad.2020.09.071
186. Zarate CA, Machado-Vieira R. GSK-3: a key regulatory target for ketamine’s rapid antidepressant effects mediated by enhanced AMPA to NMDA throughput. Bipolar Disord. (2016) 18:702–5. doi: 10.1111/bdi.12452
187. Lally N, Nugent AC, Luckenbaugh DA, Ameli R, Roiser JP, Zarate CA. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Transl Psychiatry. (2014) 4:e469. doi: 10.1038/tp.2014.105
188. Grande I, Fries GR, Kunz M, Kapczinski F. The role of BDNF as a mediator of neuroplasticity in bipolar disorder. Psychiatry Investig. (2010) 7:243–50. doi: 10.4306/pi.2010.7.4.243
189. Cunha ABM, Frey BN, Andreazza AC, Goi JD, Rosa AR, Gonçalves CA, et al. Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neurosci Lett. (2006) 398:215–9. doi: 10.1016/j.neulet.2005.12.085
190. Machado-Vieira R, Dietrich MO, Leke R, Cereser VH, Zanatto V, Kapczinski F, et al. Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol Psychiatry. (2007) 61:142–4. doi: 10.1016/j.biopsych.2006.03.070
191. Wilkowska A, Szałach Ł, Cubała WJ. Ketamine in bipolar disorder: a review. Neuropsychiatr Dis Treat. (2020) 16:2707–17. doi: 10.2147/ndt.s282208
192. Hobara T, Uchida S, Otsuki K, Matsubara T, Funato H, Matsuo K, et al. Altered gene expression of histone deacetylases in mood disorder patients. J Psychiatr Res. (2010) 44:263–70. doi: 10.1016/j.jpsychires.2009.08.015
193. Réus GZ, Abelaira HM, dos Santos MAB, Carlessi AS, Tomaz DB, Neotti MV, et al. Ketamine and imipramine in the nucleus accumbens regulate histone deacetylation induced by maternal deprivation and are critical for associated behaviors. Behav Brain Res. (2013) 256:451–6. doi: 10.1016/j.bbr.2013.08.041
Keywords: bipolar disorder, ketamine, major depressive disorder, mechanism of action, neuroplasticity, treatment-resistant depression, rapid antidepressant effects
Citation: Kang MJY, Hawken E and Vazquez GH (2022) The Mechanisms Behind Rapid Antidepressant Effects of Ketamine: A Systematic Review With a Focus on Molecular Neuroplasticity. Front. Psychiatry 13:860882. doi: 10.3389/fpsyt.2022.860882
Received: 23 January 2022; Accepted: 18 March 2022;
Published: 25 April 2022.
Edited by:
Riccardo Guglielmo, Catholic University Medical School, ItalyReviewed by:
Laura Musazzi, University of Milano Bicocca, ItalyCopyright © 2022 Kang, Hawken and Vazquez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gustavo Hector Vazquez, Zy52YXpxdWV6QHF1ZWVuc3UuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.