 Lei Cao-Lei
Lei Cao-Lei Daniel Saumier1
Daniel Saumier1 Justine Fortin
Justine Fortin Alain Brunet
Alain Brunet
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 07 November 2022
Sec. Psychopathology
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.857087
This article is part of the Research Topic Comprehensive Understanding of PTSD: From Pathogenesis to Intervention View all 9 articles
Epigenetic research in post-traumatic stress disorder (PTSD) is essential, given that environmental stressors and fear play such a crucial role in its development. As such, it may provide a framework for understanding individual differences in the prevalence of the disorder and in treatment response. This paper reviews the epigenetic markers associated with PTSD and its treatment, including candidate genes and epigenome-wide studies. Because the etiopathogenesis of PTSD rests heavily on learning and memory, we also draw upon animal neuroepigenetic research on the acquisition, update and erasure of fear memory, focusing on the mechanisms associated with memory reconsolidation. Reconsolidation blockade (or impairment) treatment in PTSD has been studied in clinical trials and, from a neurological perspective, may hold promise for identifying epigenetic markers of successful therapy. We conclude this paper by discussing several key considerations and challenges in epigenetic research on PTSD in humans.
Post-traumatic stress disorder (PTSD) is caused by the experience of a traumatic event and is associated with four clusters of symptoms, including intrusive phenomena, avoidance, changes in mood and cognition, as well as hyper-arousal (1). While most individuals have experienced at least one traumatic event during their lifetime (2), the prevalence of the disorder in the general population is approximately 9–12% (3). This raises the question of why some trauma-exposed individuals develop PTSD and others don’t. Genetic factors can confer an increased vulnerability to PTSD following trauma exposure (4, 5). While other risk factors that contribute to the development of the disorder (6), epigenetics may provide a framework for understanding how gene expression is influenced by traumatic experiences to produce individual differences in the prevalence of PTSD. Considering that PTSD treatment outcome may also be quite variable (7, 8), our knowledge of these mechanisms may potentially serve as biomarkers of treatment response in individuals diagnosed with the disorder. In this review, we introduce some general mechanisms of genomic transcription and provide an overview of studies that have established epigenetic associations with PTSD. Assuming that these associations may not only be relevant to the development of the disorder but also potentially contribute as mediators of treatment response in patients, we review human epigenetic findings pertaining to PTSD treatment. We also discuss some of the challenges and future directions for epigenetic research in this field.
Epigenetics refers to the chemical changes to chromatin that control genomic transcription. These changes in gene expression occur through alterations in chromatin structure rather than changes in DNA sequence per se (9). DNA methylation, histone modification and non-coding RNA modification are the three most common epigenetic mechanisms. Epigenetic patterns are responsible for cell-type specific gene expression patterns (10), which bestow cellular identity to DNA. They are formed during development by a highly regulated and orderly process.
DNA methylation has been the most frequently investigated epigenetic mechanism. The covalent methylation modification of the DNA (11), catalyzed by DNA methyltransferases (DNMT), at the 5′ position of the cytosine ring, occurs without changing the DNA sequence. DNA methylation is involved in gene regulation primarily by inhibiting promoter activity, either by interfering with binding of transcription factors or by recruiting methylated DNA binding proteins (MBD) that recruit histone deacetylases (HDACs) and histone methyltransferases (HMTase) that induce chromatin inactivation (12, 13).
Histone modification includes acetylation, methylation, phosphorylation, ubiquitination and sumoylation. The most studied histone modification is acetylation. Acetylation results from the effect of the enzyme histone acetyltransferases (HATs), causing a loss in the histone’s chromatin structure and promoting transcription (14). Histone methylation is a highly complex modification process. Depending on the position of methylation and the number of methyl groups transferred to the histone tail, it can stimulate or repress transcription. Histone phosphorylation is involved in transcriptional activation (15), but this modification process is less studied than the two others.
Published studies in the last decade have provided evidence that epigenetic patterns can be altered in response to environmental stimuli (16–18), leading to long-term alterations in gene activity by regulating gene expression, which further plays a critical role in disease susceptibility, etiology, and progression. The study of epigenetic mechanisms that emerge out of traumatic experiences is part of this rapidly growing scientific literature. A number of human-based epigenetic studies of PTSD have been published (19–22), with the majority of them focusing on candidate genes which are chosen based on animal models or genetic association findings, such as NR3C1 (23, 24), FKBP5 (25, 26), SLC6A3 (27), and BDNF (28). For instance, BDNF (brain-derived neurotrophic factor) has been associated with memory, stress, and neuropsychiatric disorders. In a predator stress model of PTSD in rodents (30) the methylation of Bdnf exon-IV was specifically modified in hippocampus, whereas no changes were found in the prefrontal cortex or amygdala (28). The related physiological systems involved in PTSD have included the hypothalamic-pituitary-adrenal (HPA) axis, immune function, serotonergic system, catecholaminergic system and others [see (19) for a comprehensive review]. Unbiased epigenome-wide association approaches have also been employed in PTSD research. We will briefly review main findings of candidate gene and epigenome-wide research as it pertains to PTSD.
Extensive animal and human studies have been conducted to determine how altered HPA axis functioning contributes to the development and maintenance of PTSD (31). In rodent studies, significant disruptions were observed in the functioning of the HPA axis, including reduced basal glucocorticoid levels and increased dexamethasone-induced inhibition of cortisol levels (29, 32, 33). Some (but not all) PTSD patients have been reported to have lower levels of the stress hormone cortisol (34–38)–a finding attributed to glucocorticoid receptor (GR) hypersensitivity, as well as an increased negative feedback inhibition of the HPA axis (39, 40). HPA axis-related genes have been widely studied in animals (31, 41–43) and significant epigenetic changes that occur in conjunction with PTSD have been reported in humans (23, 24). The HPA-axis related gene NR3C1, which encodes GR, is a candidate gene most extensively studied in stress and PTSD research (44–46). As shown in a recent review (47) a growing body of studies on early life adversity in both rodents and humans have reported increased methylation levels of the exon 1F promoter at the NR3C1 (analog of Nr3c1 17 in rodents). However, evidence from PTSD research suggests that there may be lower DNA methylation levels at this gene. For instance, Yehuda et al. (24) compared a cohort of combat veterans diagnosed with PTSD to a sample of trauma-exposed individuals without PTSD and found that the veterans with PTSD displayed lower DNA methylation levels of the NR3C1 exon 1F promoter from their peripheral blood samples (24). Likewise, Labonté et al. (23) reported lower T-cell levels of NR3C1 exon 1B and 1C and higher GR expression in civilians with PTSD compared to participants with current or remitted PTSD.
Although previous studies have targeted candidate genes in PTSD research, large-scale, unbiased epigenome-wide association approaches are increasingly used to search for methylation transformations across a variety of newly discovered genes. Epigenome-wide studies have focused primarily on DNA methylation. There has been a number of such studies investigating DNA methylation profiling in PTSD and reviews have summarized these findings (19, 20, 48). Uddin et al. (49) first identified methylation profiles underlying immune system changes associated with lifetime PTSD. This study not only revealed differences among PTSD-affected individuals with respect to the number of uniquely methylated genes, but also indicated that uniquely unmethylated genes were associated with a strong signature of immune system involvement.
Another group (50) working on immune system and DNA methylation reported differentially methylated CpGs in five genes (TPR, CLEC9A, APC5, ANXA2, and TLR8) associated with PTSD (50). Furthermore, in a PTSD study on epigenome-wide gene expression and DNA methylation profiles, eight odorant/olfactory receptor-associated genes, as well as genes related to immunological activation, the Gamma-Aminobutyric Acid A (GABAA) receptor, and vitamin D synthesis, were found to be up-regulated with PTSD (51). In a meta-analysis of PTSD epigenome-wide association studies in three trauma-exposed civilian cohorts, the NRG1 and HGS were identified as blood-based biomarkers associated with PTSD (52). Moreover, a DNA methylation study of PTSD in World Trade Center first-responders (53) identified a set of novel genes (ZDHHC11, CSMD2, COL9A3, PDCD6IP, TBC1D24, and FAM164A) associated with PTSD. Moreover, the gene BRSK1, LCN8, NFG, DOCK2 (54) and ZFP57, RNF39, HIST1H2APS2 (55) were found to be linked to the severity of PTSD symptoms.
Several comprehensive reviews describing epigenetic mechanisms and PTSD with the candidate gene and epigenome-wide approaches have been recently published (19, 20, 48). A detailed review of these studies is beyond the scope of the current paper.
Although research has identified epigenetic markers in PTSD, only a few studies have explored whether these markers affect treatment response (see Table 1).
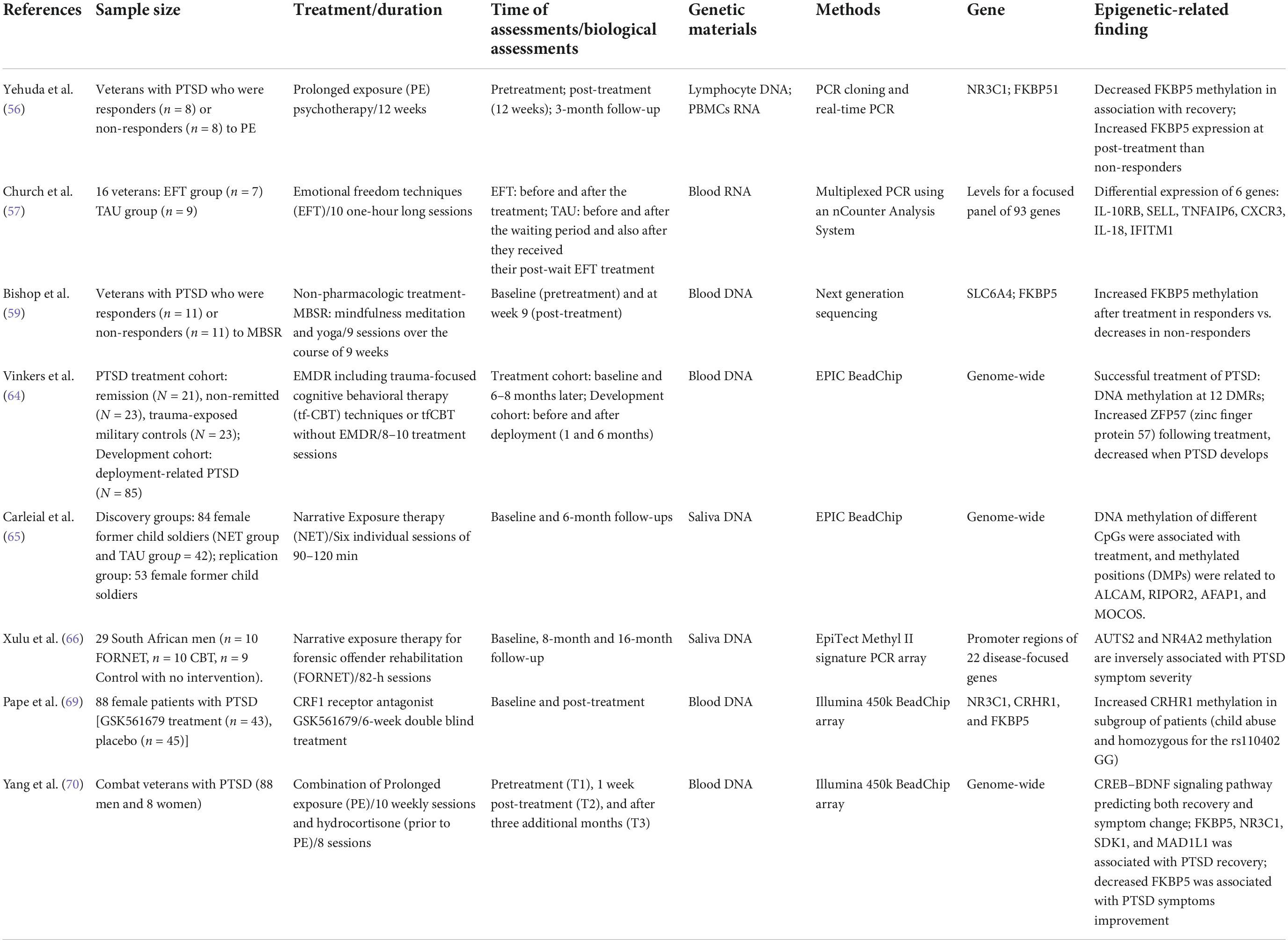
Table 1. Epigenetic studies in PTSD treatment.
Yehuda et al. (56) were the first to investigate the role of epigenetics in psychotherapy interventions. Their study involved a small sample of veterans with PTSD receiving a 12-week prolonged exposure (PE) psychotherapy intervention. Blood samples were taken at pre-treatment, after 12 weeks of psychotherapy and at a 3-month follow-up visit. Blood-based DNA methylation at the NR3C1 promoter region was found to predict positive treatment response, (higher DNA methylation assessed at pre-treatment was associated with lower PTSD symptoms at follow-up). In contrast, the FK506 binding protein 5 (FKBP5) promoter did not predict treatment response, while decreased DNA methylation was found to be associated with PTSD recovery. Moreover, higher gene expression of FKBP5 at post-treatment was observed in treatment responders. The authors suggested that these epigenetic markers may be associated with prognosis (NR3C1 methylation) and symptom severity (FKBP5 methylation). This study represents an important initial first step in establishing relevant molecular markers for PTSD therapies.
Church et al. (57) conducted a pilot randomized controlled trial in which emotional freedom techniques (EFT) were applied to treat veterans suffering from PTSD. The method incorporates elements exposure and cognitive therapies, as well as somatic stimulation, including acupressure (58). Participants received ten one-hour long sections of EFT and blood samples were drawn before and after treatment. The authors examined a panel of 92 gene expressions using blood RNA before and after treatment. The pre- vs. post-treatment comparisons revealed six significant differential gene expressions (IL-10RB, SELL, TNFAIP6, CXCR3, IL-18, and IFITM1) associated with treatment response to EFT.
Using mindfulness-based stress reduction (MBSR) techniques (i.e., meditation and yoga) to treat PTSD in veterans, Bishop et al. (59) used a case-control study design that involved psychoeducation, followed by 7 weekly 2.5 h group-therapy sessions and a 6.5 h retreat, for a total of nine sessions spanning a 9 week period. Methylation analyses were based on blood draws obtained at post-treatment. The methylation of two candidate genes, serotonin transporter (SLC6A4) and FKBP5, which are involved in monoamine and HPA axis function (60–63), were examined in treatment responders and non-responders. The authors reported a time-by-responder group interaction in which responders exhibited an increase in FKBP5 methylation following treatment, with non-responders showing a decrease in methylation after treatment.
Vinkers et al. (64) investigated epigenome-wide DNA methylation changes associated with symptomatic remission after psychosocial interventions that included trauma-focused cognitive behavioral therapy (tf-CBT) and eye movement desensitization and reprocessing (EMDR) within military participants with PTSD. Participants received 8–10 treatment sessions and underwent genotyping and methylation quantification 6–8 months after treatment. The findings revealed 12 differentially methylated regions (DMRs) in participants with reduced PTSD symptoms following treatment. Furthermore, the authors examined whether these DMRs were associated with the development of PTSD in an independent prospective cohort with deployment-related PTSD. Interestingly, the methylation of the Zinc finger protein 57 (ZFP57), a transcriptional regulator of genomic imprinting, was found to be related to PTSD symptom reduction in this cohort.
Recently, Carleial et al. (65) investigated methylome-wide changes in saliva samples of 84 female former child soldiers from Eastern Congo having a DSM-5 diagnosis of PTSD related to experiences of abduction and other traumatic events. The participants received either Narrative Exposure Therapy (NET) or treatment-as-usual. The NET group received six individual sessions of 90–120 min each in length. The saliva samples were collected at baseline and at a 6-month follow-up visit. DNA methylation of different CpGs were associated with treatment response, and methylated positions (DMPs) were related to four genes (ALCAM, RIPOR2, AFAP1, and MOCOS). Treatment-related DMPs were also found to be involved in memory formation and in biochemical pathways that are typically affected by trauma-related disorders. Furthermore, the findings were partially replicated in a separate group of 53 female former child soldiers.
Xulu et al. (66) applied NET as a forensic offender rehabilitation (FORNET) intervention in order to investigate DNA methylation changes associated PTSD treatment response and appetitive aggression symptoms among South African men. Participants with chronic trauma exposure received one of three interventions: FORNET, CBT or no intervention (waitlist). The FORNET intervention involved 82-h sessions that comprised psychoeducation. Saliva samples were collected at baseline, 8- and 16-months following treatment. DNA methylation of promoter regions were analyzed for a disease-focused gene panel (22 genes). The study revealed that increased gene methylation involved in dopaminergic neurotransmission (NR4A2) and synaptic plasticity (AUTS2) was linked to a reduction in PTSD symptoms in the FORNET group.
As a pharmacological treatment, a novel CRF1 receptor antagonist GSK561679 has been shown to be effective in the treatment of PTSD (67, 68). Based on these findings, Pape et al. (69) tested the hypothesis that DNA methylation could serve as a potential marker for this treatment. The authors analyzed the association between blood-based DNA methylation levels of CRHR1, NR3C1, and FKBP5 with treatment response in a cohort of women with a history of childhood abuse, as well as in a subgroup of these participants who showed high CRF activity. The treatment protocol involved a 6-week double-blind treatment design and blood draws were taken for genotyping after 5 weeks of study treatment. The findings showed significant differences in CRHR1 methylation levels following GSK561679 treatment in the subgroup with high CRF activity and NR3C1 methylation at baseline interacted with child abuse status (none-to-mild abuse vs. moderate-to-severe abuse) in predicting PTSD symptom change after GSK561679 treatment.
More recently, Yehuda’s team (70) combined prolonged-exposure (PE) psychotherapy (10 weekly sessions) with a pharmacological drug (hydrocortisone) (taken 45 min prior to each PE session; 8 total) to explore epigenetic markers involved in the treatment of military deployment PTSD through epigenome-wide analyses in a longitudinal study. The CREB–BDNF signaling pathway, which is involved in fear learning and memory formation (71), was identified as a marker that predicted both recovery and symptom change, based on blood samples taken at 3 months post-treatment. FKBP5, NR3C1, SDK1, and MAD1L1 were also found to be associated with PTSD recovery, especially decreased FKBP5 was associated with PTSD symptom improvement.
Although our understanding of the pathophysiology of PTSD is limited, one model assumes that the disorder is associated with fear memory formation (72). Pavlovian fear conditioning has been postulated as a model of PTSD (73) and is assumed to drive the “re-experiencing” of PTSD symptoms (74). Pavlovian fear conditioning is putatively involved in the development of fear-related memory by generating the conditioned stimulus (CS)-unconditioned stimulus (US) associations. The process by which fear memories develop has also been conceptualized as involving memory consolidation (73). During this process, memory traces are integrated into long-term and stable memory (75). PTSD symptoms are usually induced in animals using physical, social, or psychological stressors which have been reported to reveal universal or distinct neuroadaptation patterns across all models (76). In the case of PTSD, stress hormones (such as cortisol) and neurotransmitters (such as norepinephrine) affect amygdala functioning by “hyper-consolidating” the traumatic memory trace as primarily sensory-emotional associations which are dissociated from declarative or episodic information that is normally registered via the hippocampal-cortical pathways (77). Once reactivated, the previously consolidated memories reconsolidate and may be potentiated by the stress hormones and neurotransmitters. Studies have shown that various amnesic agents may be effective at impairing reconsolidation in animals (78) and in patients (79, 80).
Animal studies have enhanced our understanding of the epigenetic mechanisms underlying fear memory [for a review see (81–83)]. These studies provided important information regarding the underlying epigenetic mechanisms for each stage of the memory process, with focus on the amygdala and hippocampus (84, 85). The amygdala is essential for the establishment of the CS -US link in fear conditioning, while the hippocampus is engaged in learning the contextual information (86). Diamond and Zoladz proposed that the amygdala has rather a hyperfunctional role than a disfunctional role in PTSD and considered that the amygdala functions to ensure a person’s survival in PTSD condition (87). This research group also reported that reactivation of a remote emotional memory can exert a powerful intrusive effect on new hippocampal memory processing in rats 1 year after the original traumatic experience (88).
Various intracellular signaling pathways and regulators of gene expression have been proposed in mediating synaptic plasticity in memory consolidation and reconsolidation (89). Reconsolidation, in particular, requires engaging transcription factors CEBP and Zif268 and the kinase ERK/MAPK (90) which, furthermore, control epigenetic mechanisms that regulate gene expression and result in a complex pattern of intracellular changes and long-term neuro-structural alterations. Two main epigenetic mechanisms (histone modifications and DNA methylation) have been associated with fear memory consolidation/reconsolidation. In rodents, administration of an HDAC inhibitor, which enhances histone acetylation, results in enhanced auditory fear memory and increased H3 acetylation in the amygdala (91–93). Furthermore, retrieval of contextually conditioned fear memories increase histone H3 phosphorylation and acetylation at specific gene promoters after inhibition of HDAC activity in the hippocampus (94). As a result, histone acetylation in the amygdala impacts auditory fear memory, whereas histone acetylation in the hippocampus modulates contextual fear.
DNA modification is also involved in the transcriptional regulation of gene expression during reconsolidation. A small number of animal studies have examined the role of DNA methylation involved in reconsolidation. There is evidence indicating the importance of transcriptional regulation through DNA methylation (82, 83). For instance, the inhibition of DNA methylation by the infusion of 5-aza-2-deoxycytidine (5-AZA) or RG108 during fear memory reconsolidation has been shown to reduce fear responses associated with the memory (93). DNA methylation inhibition is also linked to lower levels of H3 acetylation, suggesting an association between DNA methylation and histone acetylation.
Animal research on the consolidation and reconsolidation of fear memories suggests that there are epigenetic changes underlying such memories. From a therapeutic perspective, such changes can be targeted for reversal or inhibition through environmental or pharmacological interventions. While exposure-based methods have been most often employed in clinical practice, fear responses may often remit with exposure to reactivating fear stimuli or with the passage of time. Yet, epigenetic studies have shown that the blocking of hdac activity may promote extinction memory and contextual fear (95–98). Alternatively, fear memories may be weakened by blocking consolidation and reconsolidation processes, particularly those related to protein synthesis mechanisms involved in these processes. Reconsolidation blockade treatment of PTSD has been studied in clinical trials and from a neurological perspective, may hold promise for identifying epigenetic markers of therapy. Reconsolidation blockade using propranolol is supported by an animal model (99), by two meta-analyses involving healthy participants and patient populations (100, 101), and by imaging studies (102, 103). Single doses of sirolimus (rapamycin), administered immediately after trauma memory retrieval, and has been shown to reduce PTSD symptoms in male combat veterans (104). Propofol, which belongs to hypnotics or anesthetics’ class of drugs (105), has been shown to impair emotional episodic memory retrieval following memory reactivation in a non-clinical population (106). Because of the accumulating evidence that these interventions involve the blockade of memory reconsolidation, epigenetic studies involving reconsolidation blockade may allow researchers to develop more specific and methodologically sound hypotheses regarding epigenetic modifications associated with changes in traumatic memories. While there has been accumulating evidence in both animal and human research that has validated this treatment approach, no study has yet examined the epigenetic mechanisms that may underlie consolidation or reconsolidation blockade for weakening fear memories in humans.
In human epigenetic studies of PTSD, most of the published research has involved cross-sectional investigations. It is therefore difficult to ascertain whether gene methylation is a predisposing factor for the outcomes reported, or whether it is a consequence of developing the disorder. A small number of studies have implemented longitudinal approaches in PTSD epigenetic research (56, 59, 64, 70). While it is difficult to establish generalized conclusions from such studies, they allow for the dynamic tracking of epigenetic markers over time in relation to PTSD predictors and/or PTSD symptom change, which may allow for a more specific evaluation of treatment targets.
A candidate gene approach may aid in identifying specific gene targets that may be associated with a psychiatric disorders. The candidate gene approach examines the main effect of specific genes on the expression of a disorder and typically focuses on biological candidates that are selected according to existing biological evidence. The epigenome-wide study (49, 50, 53, 70), which employs an unbiased approach, is a theoretical and exploratory method that provides opportunities to inform the researcher of genetic patterns across the epigenome. This approach could result in the discovery of new epigenetic biomarkers and functional gene pathways, leading to new potential mechanistic insights into the biological basis of PTSD. In this regard, further research on PTSD epigenetics should not be limited to selective candidate genes, but rather use new approaches with the aim of discovering new biomarkers for PTSD research.
Epigenetic biomarkers can be used as disease biomarkers to explain differences between disease and non-disease states or to discriminate between different diagnostic conditions. While there are no such markers that have been adequately validated from a regulatory (e.g., Food and Drug Administration, European Medicines Agency) perspective (107), they may ultimately provide informational measures of therapeutic effectiveness in clinical trials. They may also represent targets for enhancing psychotherapeutic and pharmacological effects in therapeutic trials in PTSD. However, the relationship between epigenetic biomarkers of disorder and therapeutic outcomes needs to be established in PTSD research.
Our review of the empirical literature on the epigenetics of PTSD and PTSD treatment suggests that there may indeed be common epigenetic markers that reflect PTSD vulnerability and onset, but also correlate (directly or inversely) with disease progression and treatment outcome. For instance, the HPA-axis related genes NR3C1 and FKBP5 are not only associated with the diagnosis of PTSD (19, 23), but also involved in the treatment of PTSD (56, 69). Moreover, ZFP57 (64) has been associated with both the development and treatment of PTSD. However, there is considerable variability in the epigenetic markers (i.e., the differentially methylated CpG sites/genes) identified as clinically relevant to PTSD diagnosis across studies, and most of these do not appear to be associated to treatment-related changes in PTSD patients.
While more studies are clearly needed to further identify and validate epigenetic markers in PTSD and its treatment, several theoretical and methodological issues should be considered while designing these studies. First, epigenetic association studies in PTSD vary considerably in terms of inclusion criteria, types of stressors/traumas examined, as well as the epigenome platforms employed. Second, most epigenetic studies in the domain of PTSD have investigated either the development of PTSD or PTSD treatment effects, but not both. Thus, it is difficult to establish a causal relationship between trauma exposure, epigenetic regulation, and treatment outcome. On the other hand, prospective controlled studies may provide the possibility to establish in a more systematic manner the reciprocal interactions that may exist between these elements (108). Vinkers et al.’s (64) study, which prospectively examined epigenetic markers of PTSD onset and treatment outcome in the same sample of participants, represents this kind of needed research. On the other hand, such markers may not necessarily be implicated in explaining treatment change, particularly if the underlying mechanisms associated with symptom improvement (for example, emotion regulation processes associated with frontal lobe neural mechanisms) are distinct from those related to symptom onset (e.g., amygdala-related fear responses).
Another inherent issue in clinical epigenetic research in psychiatry concerns the heterogeneity of the treatment interventions examined (109). Considering that the PTSD studies reviewed here involve the use of very different clinical interventions (from PE to mindfulness), it remains unclear from a mechanistic viewpoint as to what is really going on during the therapy processes in these studies. The heterogeneity of epigenetic findings may reflect different underlying mechanisms of change, including self-regulation, extinction, cognitive restructuring, etc. Clinical research will need to systematically take into consideration specific components of psychotherapeutic interventions so as to pinpoint their specific underlying epigenetic mechanisms.
Several studies highlight the possible use of epigenetic markers in peripheral biological samples, such as the blood and saliva, as diagnostic markers in PTSD (23, 24, 110). Given that such sampling is cell- or tissue- specific (111), the findings may reflect a heterogeneous mixture of different types of cells and give rise to variability in the DNA methylation estimations. For instance, epigenetic research with blood samples reveals that cellular composition accounts for a large portion of the observed heterogeneity in DNA methylation (112–114). Although it would be ideal to isolate and employ a single cell type to analyze epigenetic state in future studies, there is growing evidence suggesting that correcting for cell type distribution using statistical methodologies would be a relevant strategy (115, 116). As a result, it may be critical to consider cell diversity when conducting epigenetic investigations using blood and other biological sources.
Neuroimaging research also provides a valuable non-invasive opportunity to explore the brain region functions in humans’ PTSD research (117, 118). In addition, comparing post-mortem human brain tissue from individuals with PTSD could provide valuable information about PTSD-related epigenetic markers. These methods could also be used to investigate potentially common epigenetic mechanisms across rodents and humans in analogous brain regions. The advantage of studying epigenetic markers in the brain is that these approaches could capture brain-specific epigenetic signals that may be distinct from those derived from peripheral tissues (21). Very few studies have been conducted in post-mortem human brain tissues of individuals with PTSD (119–122) and none have explored epigenetic mechanisms.
Another issue pertaining to the interpretation of epigenetic markers in PTSD research concerns sex differences in epigenetic mechanisms. Animal research has shown epigenetic influences in establishing brain sex differences in terms of fear response acquisition (123, 124). For instance, female mice that are resistant to fear extinction exhibit increased DNA methylation of Bdnf with a corresponding mRNA level decrease in the medial prefrontal cortex (123). In a study employing an animal model of PTSD, epigenetic changes of histone acetylation subsequent to maternal separation was found to correlate with BDNF-programmed synaptic changes with sex difference (124). It is worth noting that most animal PTSD studies have been conducted on male rodents, although females are twice as likely as males to develop PTSD in humans (125, 126).
Although there is thus far a limited number of human epigenetic studies, the growing literature suggests that DNA methylation alterations of many genes occur in response to environmental stress occur in a sex-specific manner (127). Similarly, a review paper (128) reported that sex-related factors could affect PTSD risk directly through epigenetic mechanisms. Interestingly, Maddox et al (129) focused on the role of HDAC4 regulation in predicting PTSD risk in women and suggested that estrogen levels, in part through their modulation of HDAC4, may enhance the risk of PTSD in some women. This finding was supported from the animal model results of estrogen- and stress-dependent regulation of Hdac4 within the amygdala (129). Thus, investigating epigenetic sex differences may help shed light on the sexually dimorphic risk and/or resilience to development of PTSD.
In this narrative review, we provided a brief overview of key concepts in epigenetic research as they pertain to the study of PTSD. While genetic predispositions may interact with traumatic experiences to account for individual differences stress response (21, 130), experience induced epigenetic changes may also result in brain modifications that give rise to behavioral manifestations of PTSD (20). These changes may represent susceptibility factors for the development of PTSD, arise from traumatic experiences themselves, or represent markers of untreated symptom change over time. Because of the widespread use of accessible peripheral tissues in human PTSD research, it will be important to identify peripheral epigenetic markers that can characterize individuals as being particularly susceptible or resistant to developing PTSD. However, alternative post-mortem and neuroimaging methods need to be given further consideration as complementary methods of investigation.
In addition, animal models of PTSD are hoped to shed light on the cellular and molecular pathways that underpin PTSD in humans. However, research has thus far revealed few common epigenetic markers across animal and human studies. There may obviously be potential constraints that influence the degree to which the results derived from animal models may be translated to humans. These include differences in the heterogeneity of the stressors (ex., conditioned stimuli, such as shocks, vs. the myriad of traumatic events that humans experience), complexity of the behavioral stress responses, temporality of symptom onset (ex., stress responses in rodent models normally occur relatively rapidly while PTSD symptoms in humans may be delayed), probability of developing stress reactions following a stressor or traumatic experience (ex., most humans do not develop PTSD following a traumatic event), as well as effects of life history. These differences may need to be given more consideration in animal research if the non-human models are to aid us in our understanding of the human cellular and molecular mechanisms of PTSD.
From a neurological perspective, investigating the underlying epigenetic mechanisms involved in PTSD treatment may hold promise for identifying epigenetic markers of therapy. Although epigenetic markers have been associated with treatment-related changes, it remains to be empirically determined whether there are common epigenetic mechanisms that underly PTSD susceptibility, diagnosis, and treatment change. Moreover, the clinical studies (56, 57, 59) discussed in this review indicate that there may be several epigenetic biomarkers involved in treatment response in patients suffering from PTSD. However, due to the varying methodologies employed, it is difficult to draw consistent conclusions from published literature regarding how epigenetic biomarkers relate to specific therapeutic (psychotherapy or pharmacotherapy) processes of change. We suggest that future clinical trials target specific components of interventions, such as exposure-based therapies, coping strategies, cognitive processing/restructuring (131), as well as reconsolidation-based techniques, to establish their underlying epigenetic mechanisms more firmly using either candidate gene or epigenome-wide approach.
LC-L and JF drafted an earlier version of the manuscript. LC-L and DS prepared the final version for submission. AB reviewed the final version. All authors listed on this manuscript has seen and approved this submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-5. Washington, DC: American Psychiatric Association (2013).
2. Benjet C, Bromet E, Karam EG, Kessler RC, McLaughlin KA, Ruscio AM, et al. The epidemiology of traumatic event exposure worldwide: results from the World Mental Health Survey Consortium. Psychol Med. (2016) 46:327–43. doi: 10.1017/s0033291715001981
3. Olff M. Sex and gender differences in post-traumatic stress disorder: an update. Eur J Psychotraumatol. (2017) 8(Supp 4):1351204.
4. Cornelis MC, Nugent NR, Amstadter AB, Koenen KC. Genetics of post-traumatic stress disorder: review and recommendations for genome-wide association studies. Curr Psychiatry Rep. (2010) 12:313–26. doi: 10.1007/s11920-010-0126-6
5. Nugent NR, Amstadter AB, Koenen KC. Genetics of post-traumatic stress disorder: informing clinical conceptualizations and promoting future research. Am J Med Genet C Semin Med Genet. (2008) 148c:127–32. doi: 10.1002/ajmg.c.30169
6. Sharma S, Ressler KJ. Genomic updates in understanding PTSD. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 90:197–203. doi: 10.1016/j.pnpbp.2018.11.010
7. Krediet E, Bostoen T, Breeksema J, van Schagen A, Passie T, Vermetten E. Reviewing the potential of psychedelics for the treatment of PTSD. Int J Neuropsychopharmacol. (2020) 23:385–400. doi: 10.1093/ijnp/pyaa018
8. Resick PA, LoSavio ST, Wachen JS, Dillon KH, Nason EE, Dondanville KA, et al. Predictors of treatment outcome in group or individual cognitive processing therapy for posttraumatic stress disorder among active duty military. Cogn Ther Res. (2020) 44:611–20.
9. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. (2007) 128:635–8.
10. Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity (Edinb). (2010) 105:4–13. doi: 10.1038/hdy.2010.54
11. Hotchkiss RD. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem. (1948) 175:315–32.
12. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. (2013) 38:23–38. doi: 10.1038/npp.2012.112
13. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. (1998) 393:386–9. doi: 10.1038/30764
14. Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin Epigenetics. (2016) 8:59. doi: 10.1186/s13148-016-0225-2
15. Grant PA. A tale of histone modifications. Genome Biol. (2001) 2:Reviews0003. doi: 10.1186/gb-2001-2-4-reviews0003
17. Marsit CJ. Influence of environmental exposure on human epigenetic regulation. J Exp Biol. (2015) 218(Pt 1):71–9. doi: 10.1242/jeb.106971
18. Martin EM, Fry RC. Environmental influences on the epigenome: exposure- associated DNA methylation in human populations. Annu Rev Public Health. (2018) 39:309–33. doi: 10.1146/annurev-publhealth-040617-014629
19. Kim GS, Smith AK, Nievergelt CM, Uddin M. Neuroepigenetics of post-traumatic stress disorder. Prog Mol Biol Transl Sci. (2018) 158:227–53. doi: 10.1016/bs.pmbts.2018.04.001
20. Howie H, Rijal CM, Ressler KJ. A review of epigenetic contributions to post-traumatic stress disorder?. Dialogues Clin Neurosci. (2019) 21:417–28. doi: 10.31887/DCNS.2019.21.4/kressler
21. Daskalakis NP, Rijal CM, King C, Huckins LM, Ressler KJ. Recent genetics and epigenetics approaches to PTSD. Curr Psychiatry Rep. (2018) 20:30. doi: 10.1007/s11920-018-0898-7
22. Bhattacharya S, Fontaine A, MacCallum PE, Drover J, Blundell J. Stress across generations: DNA methylation as a potential mechanism underlying intergenerational effects of stress in both post-traumatic stress disorder and pre-clinical predator stress rodent models. Front Behav Neurosci. (2019) 13:113. doi: 10.3389/fnbeh.2019.00113
23. Labonté B, Azoulay N, Yerko V, Turecki G, Brunet A. Epigenetic modulation of glucocorticoid receptors in posttraumatic stress disorder. Transl Psychiatry. (2014) 4:e368. doi: 10.1038/tp.2014.3
24. Yehuda R, Flory JD, Bierer LM, Henn-Haase C, Lehrner A, Desarnaud F, et al. Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatry. (2015) 77:356–64. doi: 10.1016/j.biopsych.2014.02.006
25. Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. (2008) 299:1291–305. doi: 10.1001/jama.299.11.1291
26. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. (2013) 16:33–41. doi: 10.1038/nn.3275
27. Chang SC, Koenen KC, Galea S, Aiello AE, Soliven R, Wildman DE, et al. Molecular variation at the SLC6A3 locus predicts lifetime risk of PTSD in the Detroit Neighborhood Health Study. PLoS One. (2012) 7:e39184. doi: 10.1371/journal.pone.0039184
28. Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. (2009) 65:760–9. doi: 10.1016/j.biopsych.2008.11.028
29. Zoladz PR, Fleshner M, Diamond DM. Psychosocial animal model of PTSD produces a long-lasting traumatic memory, an increase in general anxiety and PTSD-like glucocorticoid abnormalities. Psychoneuroendocrinology. (2012) 37:1531–45. doi: 10.1016/j.psyneuen.2012.02.007
30. Zoladz PR, Diamond DM. Predator-based psychosocial stress animal model of PTSD: Preclinical assessment of traumatic stress at cognitive, hormonal, pharmacological, cardiovascular and epigenetic levels of analysis. Exp Neurol. (2016) 284(Pt B):211–9. doi: 10.1016/j.expneurol.2016.06.003
31. Dunlop BW, Wong A. The hypothalamic-pituitary-adrenal axis in PTSD: Pathophysiology and treatment interventions. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 89:361–79. doi: 10.1016/j.pnpbp.2018.10.010
32. Chester JA, Kirchhoff AM, Barrenha GD. Relation between corticosterone and fear-related behavior in mice selectively bred for high or low alcohol preference. Addict Biol. (2014) 19:663–75. doi: 10.1111/adb.12034
33. Starcevic A, Petricevic S, Radojicic Z, Djulejic V, Ilankovic A, Starcevic B, et al. Glucocorticoid levels after exposure to predator odor and chronic psychosocial stress with dexamethasone application in rats. Kaohsiung J Med Sci. (2016) 32:235–40. doi: 10.1016/j.kjms.2016.04.011
34. Meewisse ML, Reitsma JB, de Vries GJ, Gersons BP, Olff M. Cortisol and post-traumatic stress disorder in adults: systematic review and meta-analysis. Br J Psychiatry. (2007) 191:387–92. doi: 10.1192/bjp.bp.106.024877
35. Mason JW, Giller EL, Kosten TR, Ostroff RB, Podd L. Urinary free-cortisol levels in posttraumatic stress disorder patients. J Nerv Ment Dis. (1986) 174:145–9. doi: 10.1097/00005053-198603000-00003
36. Yehuda R, Southwick SM, Nussbaum G, Wahby V, Giller EL Jr., Mason JW. Low urinary cortisol excretion in patients with posttraumatic stress disorder. J Nerv Ment Dis. (1990) 178:366–9. doi: 10.1097/00005053-199006000-00004
37. Pan X, Wang Z, Wu X, Wen SW, Liu A. Salivary cortisol in post-traumatic stress disorder: a systematic review and meta-analysis. BMC Psychiatry. (2018) 18:324. doi: 10.1186/s12888-018-1910-9
38. Wahbeh H, Oken BS. Salivary cortisol lower in posttraumatic stress disorder. J Trauma Stress. (2013) 26:241–8. doi: 10.1002/jts.21798
39. Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann N Y Acad Sci. (2009) 1179:56–69. doi: 10.1111/j.1749-6632.2009.04979.x
40. Mehta D, Binder EB. Gene × environment vulnerability factors for PTSD: the HPA-axis. Neuropharmacology. (2012) 62:654–62. doi: 10.1016/j.neuropharm.2011.03.009
41. Plank AC, Frey S, Basedow LA, Solati J, Canneva F, von Hörsten S, et al. Prenatally traumatized mice reveal hippocampal methylation and expression changes of the stress-related genes Crhr1 and Fkbp5. Transl Psychiatry. (2021) 11:183. doi: 10.1038/s41398-021-01293-y
42. Richter-Levin G, Stork O, Schmidt MV. Animal models of PTSD: a challenge to be met. Mol Psychiatry. (2019) 24:1135–56. doi: 10.1038/s41380-018-0272-5
43. Daskalakis NP, Yehuda R, Diamond DM. Animal models in translational studies of PTSD. Psychoneuroendocrinology. (2013) 38:1895–911. doi: 10.1016/j.psyneuen.2013.06.006
44. Argentieri MA, Nagarajan S, Seddighzadeh B, Baccarelli AA, Shields AE. Epigenetic pathways in human disease: The impact of DNA methylation on stress-related pathogenesis and current challenges in biomarker development. EBioMedicine. (2017) 18:327–50. doi: 10.1016/j.ebiom.2017.03.044
45. Watkeys OJ, Kremerskothen K, Quidé Y, Fullerton JM, Green MJ. Glucocorticoid receptor gene (NR3C1) DNA methylation in association with trauma, psychopathology, transcript expression, or genotypic variation: A systematic review. Neurosci Biobehav Rev. (2018) 95:85–122. doi: 10.1016/j.neubiorev.2018.08.017
46. Palma-Gudiel H, Córdova-Palomera A, Leza JC, Fañanás L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: A critical review. Neurosci Biobehav Rev. (2015) 55:520–35. doi: 10.1016/j.neubiorev.2015.05.016
47. Cao-Lei L, de Rooij SR, King S, Matthews SG, Metz GAS, Roseboom TJ, et al. Prenatal stress and epigenetics. Neurosci Biobehav Rev. (2020) 117:198–210. doi: 10.1016/j.neubiorev.2017.05.016
48. Blacker CJ, Frye MA, Morava E, Kozicz T, Veldic M. A Review of epigenetics of PTSD in comorbid psychiatric conditions. Genes (Basel). (2019) 10:140. doi: 10.3390/genes10020140
49. Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, et al. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci USA. (2010) 107:9470–5. doi: 10.1073/pnas.0910794107
50. Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B, et al. Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B Neuropsychiatr Genet. (2011) 156b:700–8. doi: 10.1002/ajmg.b.31212
51. Chen Y, Li X, Kobayashi I, Tsao D, Mellman TA. Expression and methylation in posttraumatic stress disorder and resilience; evidence of a role for odorant receptors. Psychiatry Res. (2016) 245:36–44. doi: 10.1016/j.psychres.2016.07.045
52. Uddin M, Ratanatharathorn A, Armstrong D, Kuan PF, Aiello AE, Bromet EJ, et al. Epigenetic meta-analysis across three civilian cohorts identifies NRG1 and HGS as blood-based biomarkers for post-traumatic stress disorder. Epigenomics. (2018) 10:1585–601. doi: 10.2217/epi-2018-0049
53. Kuan PF, Waszczuk MA, Kotov R, Marsit CJ, Guffanti G, Gonzalez A, et al. An epigenome-wide DNA methylation study of PTSD and depression in world trade center responders. Transl Psychiatry. (2017) 7:e1158. doi: 10.1038/tp.2017.130
54. Mehta D, Bruenig D, Carrillo-Roa T, Lawford B, Harvey W, Morris CP, et al. Genomewide DNA methylation analysis in combat veterans reveals a novel locus for PTSD. Acta Psychiatr Scand. (2017) 136:493–505. doi: 10.1111/acps.12778
55. Rutten BPF, Vermetten E, Vinkers CH, Ursini G, Daskalakis NP, Pishva E, et al. Longitudinal analyses of the DNA methylome in deployed military servicemen identify susceptibility loci for post-traumatic stress disorder. Mol Psychiatry. (2018) 23:1145–56. doi: 10.1038/mp.2017.120
56. Yehuda R, Daskalakis NP, Desarnaud F, Makotkine I, Lehrner AL, Koch E, et al. Epigenetic biomarkers as predictors and correlates of symptom improvement following psychotherapy in combat veterans with PTSD. Front Psychiatry. (2013) 4:118. doi: 10.3389/fpsyt.2013.00118
57. Church D, Yount G, Rachlin K, Fox L, Nelms J. Epigenetic effects of PTSD remediation in veterans using clinical emotional freedom techniques: a randomized controlled pilot study. Am J Health Promot. (2018) 32:112–22. doi: 10.1177/0890117116661154
58. Church D. Clinical EFT as an evidence-based practice for the treatment of psychological and physiological conditions. Psychology. (2013) 4:645. doi: 10.3390/healthcare6040146
59. Bishop JR, Lee AM, Mills LJ, Thuras PD, Eum S, Clancy D, et al. Methylation of FKBP5 and SLC6A4 in relation to treatment response to mindfulness based stress reduction for posttraumatic stress disorder. Front Psychiatry. (2018) 9:418. doi: 10.3389/fpsyt.2018.00418
60. Smoller JW. The genetics of stress-related disorders: PTSD, depression, and anxiety disorders. Neuropsychopharmacology. (2016) 41:297–319.
61. Zannas AS, Provençal N, Binder EB. Epigenetics of posttraumatic stress disorder: current evidence, challenges, and future directions. Biol Psychiatry. (2015) 78:327–35. doi: 10.1016/j.biopsych.2015.04.003
62. Domschke K, Tidow N, Schwarte K, Deckert J, Lesch K-P, Arolt V, et al. Serotonin transporter gene hypomethylation predicts impaired antidepressant treatment response. Int J Neuropsychopharmacol. (2014) 17:1167–76.
63. Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry. (2010) 15:473–500.
64. Vinkers CH, Geuze E, van Rooij SJH, Kennis M, Schür RR, Nispeling DM, et al. Successful treatment of post-traumatic stress disorder reverses DNA methylation marks. Mol Psychiatry. (2021) 26:1264–71. doi: 10.1038/s41380-019-0549-3
65. Carleial S, Nätt D, Unternährer E, Elbert T, Robjant K, Wilker S, et al. DNA methylation changes following narrative exposure therapy in a randomized controlled trial with female former child soldiers. Sci Rep. (2021) 11:18493. doi: 10.1038/s41598-021-98067-9
66. Xulu KR, Womersley JS, Sommer J, Hinsberger M, Elbert T, Weierstall R, et al. DNA methylation and psychotherapy response in trauma-exposed men with appetitive aggression. Psychiatry Res. (2021) 295:113608. doi: 10.1016/j.psychres.2020.113608
67. Dunlop BW, Rothbaum BO, Binder EB, Duncan E, Harvey PD, Jovanovic T, et al. Evaluation of a corticotropin releasing hormone type 1 receptor antagonist in women with posttraumatic stress disorder: study protocol for a randomized controlled trial. Trials. (2014) 15:240. doi: 10.1186/1745-6215-15-240
68. Dunlop BW, Binder EB, Iosifescu D, Mathew SJ, Neylan TC, Pape JC, et al. Corticotropin-Releasing factor receptor 1 antagonism is ineffective for women with posttraumatic stress disorder. Biol Psychiatry. (2017) 82:866–74. doi: 10.1016/j.biopsych.2017.06.024
69. Pape JC, Carrillo-Roa T, Rothbaum BO, Nemeroff CB, Czamara D, Zannas AS, et al. DNA methylation levels are associated with CRF(1) receptor antagonist treatment outcome in women with post-traumatic stress disorder. Clin Epigenetics. (2018) 10:136. doi: 10.1186/s13148-018-0569-x
70. Yang R, Xu C, Bierer LM, Flory JD, Gautam A, Bader HN, et al. Longitudinal genome-wide methylation study of PTSD treatment using prolonged exposure and hydrocortisone. Transl Psychiatry. (2021) 11:398. doi: 10.1038/s41398-021-01513-5
71. Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. (2010) 3:1. doi: 10.3389/neuro.02.001.2010
72. Desmedt A, Marighetto A, Piazza PV. Abnormal fear memory as a model for posttraumatic stress disorder. Biol Psychiatry. (2015) 78:290–7. doi: 10.1016/j.biopsych.2015.06.017
73. Mahan AL, Ressler KJ. Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci. (2012) 35:24–35. doi: 10.1016/j.tins.2011.06.007
74. Sauerhöfer E, Pamplona FA, Bedenk B, Moll GH, Dawirs RR, von Hörsten S, et al. Generalization of contextual fear depends on associative rather than non-associative memory components. Behav Brain Res. (2012) 233:483–93. doi: 10.1016/j.bbr.2012.05.016
75. McGaugh JL. Memory–a century of consolidation. Science. (2000) 287:248–51. doi: 10.1126/science.287.5451.248
76. Aykac A, Kalkan R. Epigenetic approach to PTSD: In the aspects of rat models. Glob Med Genet. (2022) 9:7–13. doi: 10.1055/s-0041-1736633
77. Eichenbaum H. A cortical-hippocampal system for declarative memory. Nat Rev Neurosci. (2000) 1:41–50. doi: 10.1038/35036213
78. Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. (2000) 406:722–6. doi: 10.1038/35021052
79. Brunet A, Orr SP, Tremblay J, Robertson K, Nader K, Pitman RK. Effect of post-retrieval propranolol on psychophysiologic responding during subsequent script-driven traumatic imagery in post-traumatic stress disorder. J Psychiatr Res. (2008) 42:503–6. doi: 10.1016/j.jpsychires.2007.05.006
80. Brunet A, Saumier D, Liu A, Streiner DL, Tremblay J, Pitman RK. Reduction of PTSD symptoms with pre-reactivation propranolol therapy: a randomized controlled trial. Am J Psychiatry. (2018) 175:427–33. doi: 10.1176/appi.ajp.2017.17050481
81. Lattal KM, Wood MA. Epigenetics and persistent memory: implications for reconsolidation and silent extinction beyond the zero. Nat Neurosci. (2013) 16:124–9. doi: 10.1038/nn.3302
82. Kwapis JL, Wood MA. Epigenetic mechanisms in fear conditioning: implications for treating post-traumatic stress disorder. Trends Neurosci. (2014) 37:706–20. doi: 10.1016/j.tins.2014.08.005
83. Jarome TJ, Lubin FD. Epigenetic mechanisms of memory formation and reconsolidation. Neurobiol Learn Mem. (2014) 115:116–27. doi: 10.1016/j.nlm.2014.08.002
84. Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. (2006) 1071:67–79.
85. Morey RA, Haswell CC, Hooper SR, De Bellis MD. Amygdala, hippocampus, and ventral medial prefrontal cortex volumes differ in maltreated youth with and without chronic posttraumatic stress disorder. Neuropsychopharmacology. (2016) 41:791–801. doi: 10.1038/npp.2015.205
86. Kim WB, Cho JH. Encoding of contextual fear memory in hippocampal-amygdala circuit. Nat Commun. (2020) 11:1382. doi: 10.1038/s41467-020-15121-2
87. Diamond DM, Zoladz PR. Dysfunctional or hyperfunctional? The amygdala in posttraumatic stress disorder is the bull in the evolutionary China shop. J Neurosci Res. (2016) 94:437–44. doi: 10.1002/jnr.23684
88. Zoladz PR, Woodson JC, Haynes VF, Diamond DM. Activation of a remote (1-year old) emotional memory interferes with the retrieval of a newly formed hippocampus-dependent memory in rats. Stress. (2010) 13:36–52. doi: 10.3109/10253890902853123
89. Alberini CM, Ledoux JE. Memory reconsolidation. Curr Biol. (2013) 23:R746–50. doi: 10.1016/j.cub.2013.06.046
90. Cestari V, Rossi-Arnaud C, Saraulli D, Costanzi M. The MAP(K) of fear: from memory consolidation to memory extinction. Brain Res Bull. (2014) 105:8–16. doi: 10.1016/j.brainresbull.2013.09.007
91. Maddox SA, Watts CS, Schafe GE. p300/CBP histone acetyltransferase activity is required for newly acquired and reactivated fear memories in the lateral amygdala. Learn Mem. (2013) 20:109–19. doi: 10.1101/lm.029157.112
92. Maddox SA, Watts CS, Doyère V, Schafe GE. A naturally-occurring histone acetyltransferase inhibitor derived from Garcinia indica impairs newly acquired and reactivated fear memories. PLoS One. (2013) 8:e54463. doi: 10.1371/journal.pone.0054463
93. Maddox SA, Schafe GE. Epigenetic alterations in the lateral amygdala are required for reconsolidation of a Pavlovian fear memory. Learn Mem. (2011) 18:579–93. doi: 10.1101/lm.2243411
94. Lubin FD, Sweatt JD. The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. (2007) 55:942–57. doi: 10.1016/j.neuron.2007.07.039
95. Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem. (2008) 15:39–45. doi: 10.1101/lm.801108
96. Lattal KM, Barrett RM, Wood MA. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci. (2007) 121:1125–31. doi: 10.1037/0735-7044.121.5.1125
97. Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM. Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol Psychiatry. (2012) 72:25–33. doi: 10.1016/j.biopsych.2011.12.012
98. Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. (2007) 14:268–76. doi: 10.1101/lm.500907
99. Debiec J, Ledoux JE. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience. (2004) 129:267–72. doi: 10.1016/j.neuroscience.2004.08.018
100. Lonergan MH, Olivera-Figueroa LA, Pitman RK, Brunet A. Propranolol’s effects on the consolidation and reconsolidation of long-term emotional memory in healthy participants: a meta-analysis. J Psychiatry Neurosci. (2013) 38:222–31. doi: 10.1503/jpn.120111
101. Young C, Butcher R. Propranolol for Post-Traumatic Stress Disorder: A Review of Clinical Effectiveness. Ottawa: CADTH (2020).
102. Mahabir M, Tucholka A, Shin LM, Etienne P, Brunet A. Emotional face processing in post-traumatic stress disorder after reconsolidation impairment using propranolol: A pilot fMRI study. J Anxiety Disord. (2015) 36:127–33. doi: 10.1016/j.janxdis.2015.10.004
103. Schwabe L, Nader K, Wolf OT, Beaudry T, Pruessner JC. Neural signature of reconsolidation impairments by propranolol in humans. Biol Psychiatry. (2012) 71:380–6. doi: 10.1016/j.biopsych.2011.10.028
104. Surís A, Smith J, Powell C, North CS. Interfering with the reconsolidation of traumatic memory: sirolimus as a novel agent for treating veterans with posttraumatic stress disorder. Ann Clin Psychiatry. (2013) 25:33–40.
105. Bryson HM, Fulton BR, Faulds D. Propofol. An update of its use in anaesthesia and conscious sedation. Drugs. (1995) 50:513–59.
106. Vallejo AG, Kroes MCW, Rey E, Acedo MV, Moratti S, Fernández G, et al. Propofol-induced deep sedation reduces emotional episodic memory reconsolidation in humans. Sci Adv. (2019) 5:eaav3801. doi: 10.1126/sciadv.aav3801
107. García-Giménez JL, Seco-Cervera M, Tollefsbol TO, Romá-Mateo C, Peiró-Chova L, Lapunzina P, et al. Epigenetic biomarkers: Current strategies and future challenges for their use in the clinical laboratory. Crit Rev Clin Lab Sci. (2017) 54:529–50. doi: 10.1080/10408363.2017.1410520
108. Provenzi L, Brambilla M, Borgatti R, Montirosso R. Methodological challenges in developmental human behavioral epigenetics: insights into study design. Front Behav Neurosci. (2018) 12:286. doi: 10.3389/fnbeh.2018.00286
109. Kumsta R. The role of epigenetics for understanding mental health difficulties and its implications for psychotherapy research. Psychol Psychother. (2019) 92:190–207. doi: 10.1111/papt.12227
110. Lehrner A, Yehuda R. Biomarkers of PTSD: military applications and considerations. Eur J Psychotraumatol. (2014) 14:5. doi: 10.3402/ejpt.v5.23797
111. Razin A, Szyf M. DNA methylation patterns. Formation and function. Biochim Biophys Acta. (1984) 782:331–42. doi: 10.1016/0167-4781(84)90043-5
112. Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir G, et al. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS One. (2012) 7:e46705. doi: 10.1371/journal.pone.0046705
113. Qi L, Teschendorff AE. Cell-type heterogeneity: Why we should adjust for it in epigenome and biomarker studies. Clin Epigenetics. (2022) 14:31. doi: 10.1186/s13148-022-01253-3
114. Gervin K, Page CM, Aass HC, Jansen MA, Fjeldstad HE, Andreassen BK, et al. Cell type specific DNA methylation in cord blood: A 450K-reference data set and cell count-based validation of estimated cell type composition. Epigenetics. (2016) 11:690–8. doi: 10.1080/15592294.2016.1214782
115. Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. (2012) 13:86. doi: 10.1186/1471-2105-13-86
116. Lam LL, Emberly E, Fraser HB, Neumann SM, Chen E, Miller GE, et al. Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci USA. (2012) 109(Suppl 2):17253–60. doi: 10.1073/pnas.1121249109
117. Morey RA, Garrett ME, Stevens JS, Clarke EK, Haswell CC, van Rooij SJH, et al. Genetic predictors of hippocampal subfield volume in PTSD cases and trauma-exposed controls. Eur J Psychotraumatol. (2020) 11:1785994. doi: 10.1080/20008198.2020.1785994
118. Nisar S, Bhat AA, Hashem S, Syed N, Yadav SK, Uddin S, et al. Genetic and neuroimaging approaches to understanding post-traumatic stress disorder. Int J Mol Sci. (2020) 21:4503. doi: 10.3390/ijms21124503
119. Bracha HS, Garcia-Rill E, Mrak RE, Skinner R. Postmortem locus coeruleus neuron count in three American veterans with probable or possible war-related PTSD. J Neuropsychiatry Clin Neurosci. (2005) 17:503–9. doi: 10.1176/jnp.17.4.503
120. Licznerski P, Duric V, Banasr M, Alavian KN, Ota KT, Kang HJ, et al. Decreased SGK1 expression and function contributes to behavioral deficits induced by traumatic stress. PLoS Biol. (2015) 13:e1002282. doi: 10.1371/journal.pbio.1002282
121. Su YA, Wu J, Zhang L, Zhang Q, Su DM, He P, et al. Dysregulated mitochondrial genes and networks with drug targets in postmortem brain of patients with posttraumatic stress disorder (PTSD) revealed by human mitochondria-focused cDNA microarrays. Int J Biol Sci. (2008) 4:223–35. doi: 10.7150/ijbs.4.223
122. Young KA, Thompson PM, Cruz DA, Williamson DE, Selemon LD. BA11 FKBP5 expression levels correlate with dendritic spine density in postmortem PTSD and controls. Neurobiol Stress. (2015) 2:67–72. doi: 10.1016/j.ynstr.2015.07.002
123. Baker-Andresen D, Flavell CR, Li X, Bredy TW. Activation of BDNF signaling prevents the return of fear in female mice. Learn Mem. (2013) 20:237–40. doi: 10.1101/lm.029520.112
124. Sun H, Zhang X, Kong Y, Gou L, Lian B, Wang Y, et al. Maternal separation-induced histone acetylation correlates with BDNF-Programmed synaptic changes in an animal model of PTSD with sex differences. Mol Neurobiol. (2021) 58:1738–54. doi: 10.1007/s12035-020-02224-6
125. Breslau N, Kessler RC, Chilcoat HD, Schultz LR, Davis GC, Andreski P. Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Arch Gen Psychiatry. (1998) 55:626–32. doi: 10.1001/archpsyc.55.7.626
126. Olff M, Langeland W, Draijer N, Gersons BP. Gender differences in posttraumatic stress disorder. Psychol Bull. (2007) 133:183–204. doi: 10.1037/0033-2909.133.2.183
127. Uddin M, Sipahi L, Li J, Koenen KC. Sex differences in DNA methylation may contribute to risk of PTSD and depression: a review of existing evidence. Depress Anxiety. (2013) 30:1151–60. doi: 10.1002/da.22167
128. Christiansen DM, Berke ET. Gender- and sex-based contributors to sex differences in PTSD. Curr Psychiatry Rep. (2020) 22:19. doi: 10.1007/s11920-020-1140-y
129. Maddox SA, Kilaru V, Shin J, Jovanovic T, Almli LM, Dias BG, et al. Estrogen-dependent association of HDAC4 with fear in female mice and women with PTSD. Mol Psychiatry. (2018) 23:658–65. doi: 10.1038/mp.2016.250
130. Banerjee SB, Morrison FG, Ressler KJ. Genetic approaches for the study of PTSD: Advances and challenges. Neurosci Lett. (2017) 649:139–46. doi: 10.1016/j.neulet.2017.02.058
Keywords: PTSD, treatment, epigenetic markers, DNA methylation, intervention
Citation: Cao-Lei L, Saumier D, Fortin J and Brunet A (2022) A narrative review of the epigenetics of post-traumatic stress disorder and post-traumatic stress disorder treatment. Front. Psychiatry 13:857087. doi: 10.3389/fpsyt.2022.857087
Received: 18 January 2022; Accepted: 28 September 2022;
Published: 07 November 2022.
Edited by:
Antoine Bechara, University of Southern California, United StatesReviewed by:
David M. Diamond, University of South Florida, United StatesCopyright © 2022 Cao-Lei, Saumier, Fortin and Brunet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alain Brunet, YWxhaW4uYnJ1bmV0QG1jZ2lsbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.