 Michael Carter1,2,3*
Michael Carter1,2,3* Sophie Casey1,4
Sophie Casey1,4 Gerard W. O'Keeffe1,4
Gerard W. O'Keeffe1,4 Louise Gibson1,2
Louise Gibson1,2 Louise Gallagher5,6
Louise Gallagher5,6 Deirdre M. Murray1,2*
Deirdre M. Murray1,2*- 1INFANT Research Centre, University College Cork, Cork, Ireland
- 2Department of Paediatrics and Child Health, University College Cork, Cork, Ireland
- 3National Children's Research Centre, Dublin, Ireland
- 4Department of Anatomy and Neuroscience, University College Cork, Cork, Ireland
- 5Department of Psychiatry, School of Medicine, Trinity College Dublin, Dublin, Ireland
- 6Trinity Translational Medicine Institute, St. James's Hospital, Dublin, Ireland
Autism spectrum disorder (ASD) is the commonest neurodevelopmental disability. It is a highly complex disorder with an increasing prevalence and an unclear etiology. Consensus indicates that ASD arises as a genetically modulated, and environmentally influenced condition. Although pathogenic rare genetic variants are detected in around 20% of cases of ASD, no single factor is responsible for the vast majority of ASD cases or that explains their characteristic clinical heterogeneity. However, a growing body of evidence suggests that ASD susceptibility involves an interplay between genetic factors and environmental exposures. One such environmental exposure which has received significant attention in this regard is maternal immune activation (MIA) resulting from bacterial or viral infection during pregnancy. Reproducible rodent models of ASD are well-established whereby induction of MIA in pregnant dams, leads to offspring displaying neuroanatomical, functional, and behavioral changes analogous to those seen in ASD. Blockade of specific inflammatory cytokines such as interleukin-17A during gestation remediates many of these observed behavioral effects, suggesting a causative or contributory role. Here, we review the growing body of animal and human-based evidence indicating that interleukin-17A may mediate the observed effects of MIA on neurodevelopmental outcomes in the offspring. This is particularly important given the current corona virus disease-2019 (COVID-19) pandemic as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection during pregnancy is a potent stimulator of the maternal immune response, however the long-term effects of maternal SARS-CoV-2 infection on neurodevelopmental outcomes is unclear. This underscores the importance of monitoring neurodevelopmental outcomes in children exposed to SARS-CoV-2-induced MIA during gestation.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by a spectrum of deficits in social interactions and communication combined with stereotypical and repetitive behaviors. Up to 50% of those affected can have intellectual disability (ID) and limited verbal communication (1–3). In recent decades, the prevalence of ASD has consistently increased from approximately 1 in 1,000 in the 1960s (4), to 1 in 44 today in the United States (5). Increasing prevalence may in part, be explained by changes in reporting practices, increased recognition of ASD symptoms, broadening of the ASD diagnosis (1), and improved accessibility to services (6, 7). A significant ratio of 4:1 from male to female still exists with markedly differing prevalence rates between the sexes, 1/38 in males and 1/151 among females (8). Although genetic susceptibilities are recognized, the mechanism of disease development is unknown and does not follow a clear pattern of inheritance (9, 10). This suggests possible mediation by additional unknown biological or environmental factors (11). Both common and rare genetic risk factors have been identified with more than 400 diverse genes now linked to ASD. Singly, these genetic factors each convey only a modest increase in ASD risk (~1%), however collectively they can contribute to a far greater risk (12, 13). Up to 20% of individuals with ASD may possess copy number variants (CNVs) and de novo loss of function single nucleotide variants (SNVs) that are individually rare but in combination, increase an individual's ASD risk (12). While newer methods of genetic analysis (such as whole genome sequencing) are uncovering new candidate genes with regularity (14), the heterogeneity of the clinical and phenotypic groups within ASD strongly suggest that in those with a genetic predisposition, environmental factors may act in concert to bring about a multisystem dysfunction leading to ASD. A well-characterized environmental factor known to impact early fetal brain development and increase ASD risk is maternal inflammation during pregnancy, which is commonly called maternal immune activation (MIA). Numerous epidemiological studies have linked gestational infections with elevated risk of ASD in offspring (15–17), and animal models of MIA have simulated gestational infection resulting in MIA-induced neural and behavioral abnormalities analogous to those seen in ASD (18–20).
Focused early intervention in young children with ASD has been shown to result in normalized patterns of brain activity, and is associated with improved functional outcomes and reduced morbidity (21, 22). Most children affected by ASD can have a reliable and stable ASD diagnosis from as early as 14 months of age (23), yet in spite of this, the average age of ASD diagnosis is closer to 5 years (24, 25). Numerous studies sought to identify blood-based biomarkers of ASD in affected adolescents and adults (26, 27) and have reported alterations of molecules involved in iron transport (28), inflammation (29, 30), brain development (31), and metabolism (32). None to date has identified and validated reliable mechanistic biomarkers with the ability to improve ASD detection in the crucial early developmental period. Multiple descriptive ASD biomarkers such as characteristic MRI brain findings, abnormalities of gaze preference on eye tracking or characteristic EEG findings in infants with ASD; show promise in terms of aiding earlier ASD detection. However, none is directly involved in the pathogenesis of ASD and arises of the condition rather than contributes to it. The infant brain doubles in volume over the first year coinciding with maximal neuroplasticity and synaptogenesis. Recognition of an early mechanistic biomarker gives us the best chance of implementing strategies during this critical early childhood window allowing ASD diagnosis and intervention at the earliest possible stage.
Here, we highlight recent research in this area, both from pre-clinical animal studies and epidemiological human studies, along with a proposed mechanistic pathway, that we can encourage other research groups with access to suitable maternal-child cohorts to examine this question. We encourage researchers to look at the prospective study of children born during the corona virus disease-2019 (COVID-19) era, when their gestations may have been complicated by mild or even asymptomatic severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Otherwise, the long-term effect, if any, of COVID-19 on the fetal brain could remain unknown for years to come.
Inflammation, Viral Infection, and ASD: What Are the Implications of the COVID-19 Pandemic?
There is growing scientific evidence that aberrant immune activation occurs in ASD (27, 33) based on studies of autistic children and young adults (34, 35). As early as 1971, Stella Chess reported ASD cases associated with the 1964 Rubella outbreak in the United States (36), and in a 1977 follow up study, Chess et al. quoted ASD prevalence rates of 8–13% in children of mothers who were infected during that outbreak (16). Large epidemiological studies indicate that conditions such as maternal autoimmune disorders and mid-trimester viral infections that trigger gestational pro-inflammatory states (i.e., MIA), are linked with elevated ASD, schizophrenia, and bipolar disorder risk in offspring (16, 17, 37, 38). More recently, a range of conditions associated with proinflammatory states in pregnancy such as obesity, psychosocial stress, and pre-eclampsia were associated with increased ASD risk in children (39, 40). Thus, gestational MIA appears to play a role in the pathogenesis of the ASD phenotype in exposed offspring.
Maternal Immune Activation and Neurodevelopmental Outcomes
We define MIA as a triggering of the maternal immune system by infectious or infectious-like stimuli resulting in an increase in measurable inflammatory markers during pregnancy (41, 42). Maternal immune activation has been most commonly simulated in preclinical rodent, murine and non-human primate (rhesus macaque) animal models by Poly (I:C) (polyinosinic-polycytidylic acid) or LPS (lipopolysaccharide) injection which, respectively, model viral and bacterial infection (18, 43, 44). Poly (I:C) is a synthetic analog of double stranded RNA, mimics the effects of viral infection (45). The triggered immune response results in offspring with behavioral, immunological, and neurological abnormalities that approximate to autistic symptoms observed in humans, notably, impaired sociability and repetitive behaviors (18, 46, 47). Offspring born to poly (I:C) treated dams have consistently, across all exposure categories [administration of varying doses of poly (I:C) and at varying gestations], shown impairment of social interaction, this is manifest as reduced communication in ultrasonic vocalizations (USV) which are usually triggered by separation from the dam in the first two postnatal weeks. Marble burying, a well-recognized behavioral paradigm to measure repetitive behaviors in rodents, again is consistently increased in murine offspring following poly (I:C) treatment (48). These offspring have proven useful in pre-clinical etiological studies as well as identification of therapeutic targets.
Cytokine dysregulation may play a causative role in observed neuronal dysfunction in pre-clinical models of MIA (20, 46, 49). In a recent study, Choi et al. convincingly demonstrated that simulated MIA in murine models leads to elevation in maternal IL-6, which in turn activates maternal Th17 cells. These maternal Th17 cells produce IL-17, which is thought to cross the placenta triggering increased expression of IL-17AR in the fetal brain and leading to cortical malformations and behavioral abnormalities (18, 50). These malformations parallel abnormalities found in brain development in children, adolescents and adults with ASD (51, 52). Poly (I:C) treatment also leads to raised IL-17A mRNA levels in placental tissue of these mice (18). Through inhibition of IL-6 and IL-17A signaling with antibody blockade of the IL-17A cytokine, Choi at al also determined that a sustained increase in IL-17A expression seemed to be pathogenic in ASD, as IL-17A blockade prevented the development of ASD-like phenotypes (18). Specific behaviors in mice which model core diagnostic features of ASD (including repetitive burying and increased neonatal USV) were normalized in the previously MIA-exposed offspring (53, 54).
Improved fetal resilience is associated with lower intensity of MIA. Autism spectrum disorder risk after prenatal exposure to maternal fever has been found to increase in a dose dependent manner (55, 56) and similar effects were identified in animal models of MIA (57). A balanced maternal diet seems to contribute to improved fetal resilience also (58–60). Exposure to relatively higher grades of immune activation via high intensity MIA (40), intrapartum infection (61, 62) and genetic risk factors lead to reduced fetal resilience, and increased likelihood of unfavorable developmental outcomes.
Alterations in Cytokine Expression in Human Studies
While many studies have examined the cytokine profiles of individuals with ASD, only a very limited number of studies to date have examined mid-gestation cytokine levels in mothers of children who subsequently develop ASD. Three studies retrospectively analyzed maternal blood sampled during pregnancy. A 2017 study by Jones et al., reported elevated mid-gestation cytokines and chemokines in mothers of children with ASD associated with ID, and particularly early onset ASD (as defined by the authors as early or sustained delays in language or social skills, and excluding those showing clear skill regression) (63). Dysregulation was noted in a number of cytokines including interleukins IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-8, and IL-17A between 15 and 19 weeks' gestation. An earlier study noted elevations in mid-gestation serum IL-4, IL-5, and IFN-gamma levels in mothers of ASD affected children (15). While, more recently, Irwin et al. demonstrated alterations in IL-4, MCP-1, and IL-10 levels in 28-week gestation serum of mothers who birthed ASD affected children (64). Other authors have examined amniotic fluid at mid-gestation and found elevated levels of IL-4, IL-10, TNF-α, and TNF-β in ASD patients vs. controls (65). Yet, amniotic fluid cytokine concentrations are more indicative of the fetal immune state rather than the maternal state (66, 67). In Table 1, we outline a number of the cytokines most frequently found to be dysregulated in the serum or cerebrospinal fluid (CSF) of ASD affected individuals, and gestational serum and amniotic fluid samples from mothers of ASD affected children.
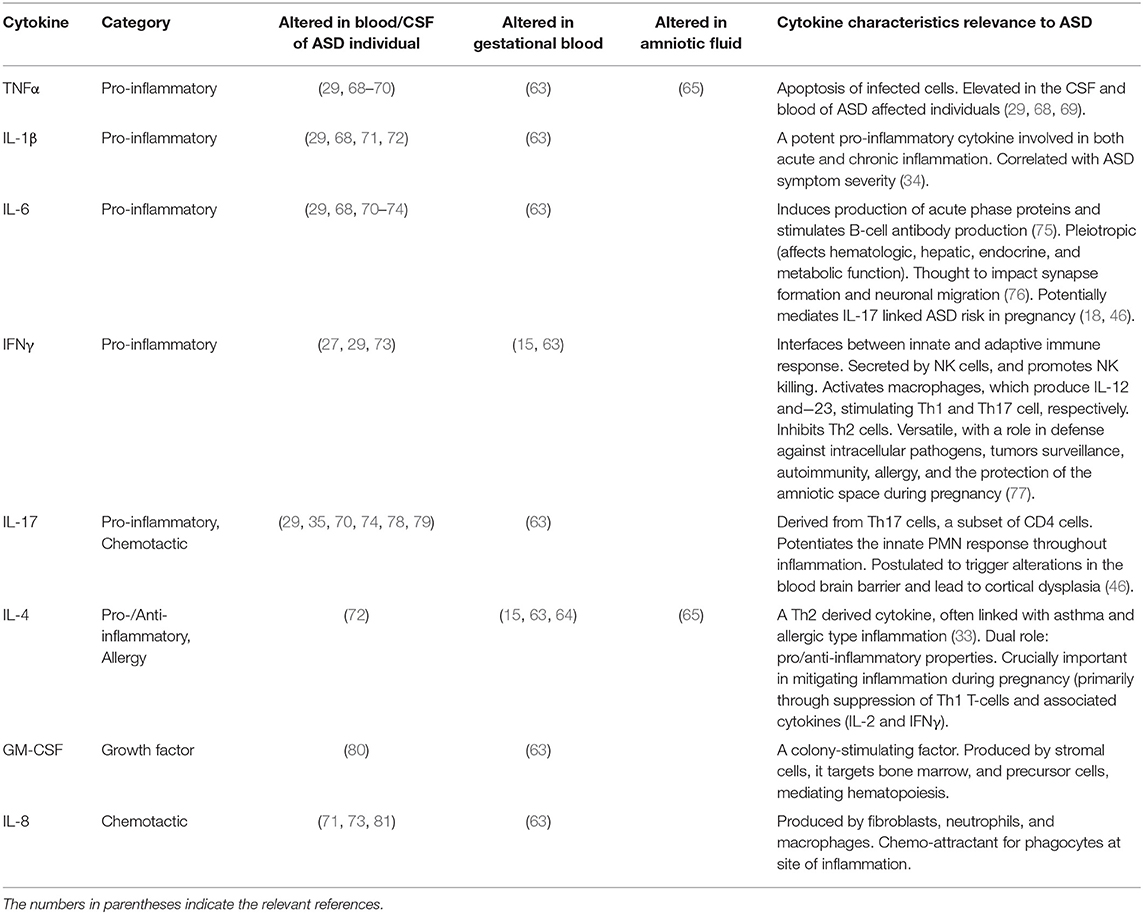
Table 1. Cytokine dysregulation in ASD affected individuals and in gestational serum and amniotic fluid samples of mothers with ASD affected offspring.
A growing body of evidence supports a role in ASD pathogenesis for Th17 cells and their product cytokine, IL-17A (Figure 1) (79, 82). The IL17A gene itself has been identified by a small genome-wide CNV study to have amplified CNVs in ASD affected cohorts (83). Elevated levels of IL-17A have been reported in the blood of ASD affected individuals, and these correlate positively with severity of ASD behavioral symptoms (35, 63, 79). Yet, others have found high concentrations of IL-17A in individuals affected by obesity or high BMI (84), both of which are more likely in ASD groups (85). This is a potential confounder for any retrospective cohort based study designs.
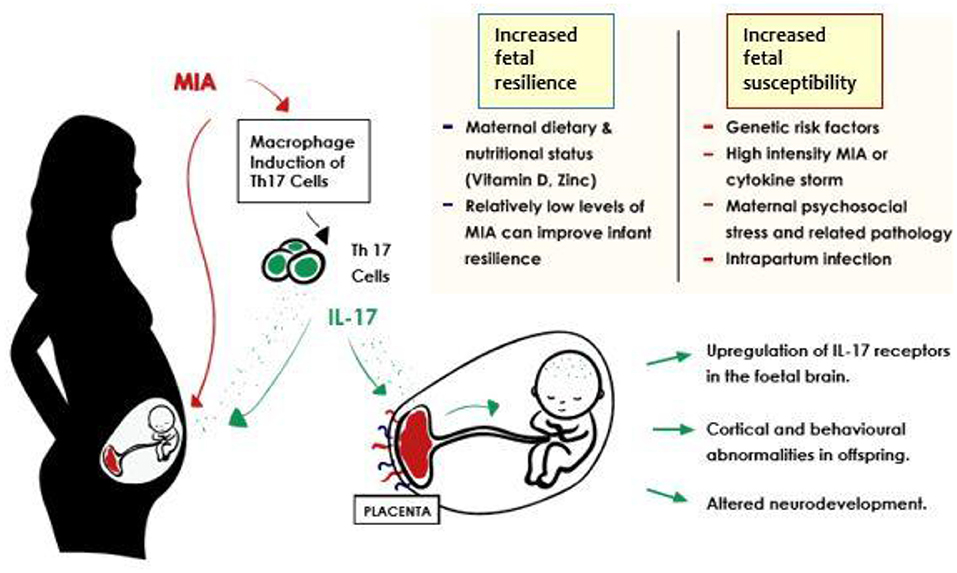
Figure 1. Potential outcomes in the inflammation-exposed fetus in the context of MIA related IL-17 induction. Improved fetal resilience is associated with lower intensity of maternal immune activation. Autism spectrum disorder risk after prenatal exposure to maternal fever has been found to increase in a dose dependent manner (55, 56) and similar effects were identified in animal models of MIA (57). A balanced maternal diet seems to contribute to improved fetal resilience also (58–60). Exposure to relatively higher grades of immune activation via high intensity MIA (40), intrapartum infection (61, 62), and genetic risk factors lead to reduced fetal resilience, and increased likelihood of unfavorable developmental outcomes.
STRING analysis (Figure 2) (86) indicates that IL-17A has proven or predicted interactions with IL-2, IL-6, IL-10, IL-13, IL-17F, IL-17RA, IL-17RC, CTLA4, STAT3, and STAT6. Each of these proteins have been previously reported to have altered expression in children with ASD, as outlined below. Of these, the most persistently described, and hence, potential key player is IL-17A, along with its receptor IL17RA and receptor complex, IL17RC.
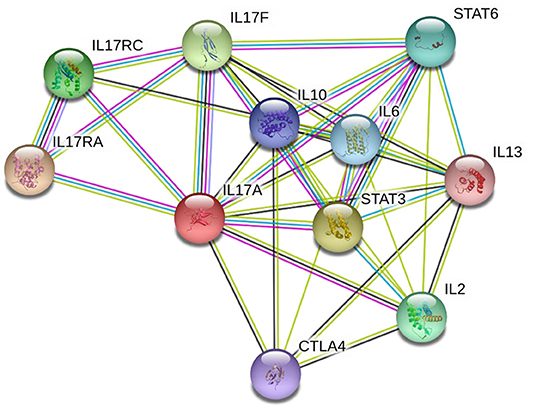
Figure 2. STRING diagram illustrating the known and predicted protein interactions for IL-17A. Network nodes represent proteins—each node represents all the proteins produced by a single, protein-coding gene locus. Edges (lines) represent protein-protein associations that are specific and meaningful, i.e., proteins jointly contribute to a shared function; this does not necessarily mean they are physically binding each other. Blue connecting lines indicate that protein interaction information was derived from curated databases, pink indicates the interaction was experimentally determined, yellow indicates the interaction was determined via text mining, black indicates protein co-expression, and lilac indicates protein homology. Analysis was performed on 28 July 2021 via the string-db.org domain.
Network nodes represent proteins—each node represents all the proteins produced by a single, protein-coding gene locus. Edges (lines) represent protein-protein associations that are specific and meaningful, i.e., proteins jointly contribute to a shared function; this does not necessarily mean they are physically binding each other. Blue connecting lines indicate that protein interaction information was derived from curated databases, pink indicates the interaction was experimentally determined, yellow indicates the interaction was determined via text mining, black indicates protein co-expression, and lilac indicates protein homology. Analysis was performed on 28 July 2021 via the string-db.org domain.
IL-17A Associated Pro-Inflammatory Mediators in ASD
Upregulation of pro-inflammatory pathways has been persistently associated with ASD. IL-6 is a versatile cytokine, with multiple functions throughout the body. It plays roles in immunity, inflammation, hematopoiesis, and oncogenesis. IL-6 works to promote pro-inflammatory Th17 cells (IL-17 producers) and to downregulate anti-inflammatory Treg cells (regulatory T-Helper cells) (87, 88). Th17 cells produce cytokines that cross the placental barrier (20). This transplacental effect has been well-characterized with IL-6, which was shown to alter offspring behavior and brain development (20, 89).
Like IL-17A, IL-17F is also produced by Th17 cells (90). IL-17F is reported to be involved in the regulation of proinflammatory gene expression and responses (91). IL-17RA and IL-17RC are both members of the IL-17 receptor family. In order for IL-17A (or indeed IL-17F) to have biological effects on tissues, IL-17RA must be present (90). IL-17RA is expressed in immune cells, and some children affected by ASD appear to possess higher levels of this receptor compared to neuro-typical controls (92). IL-17RA blockade may reduce monocyte associated oxidative stress which may improve neuro-inflammation associated with ASD (92). IL-17RC is also essential for the formation of the IL-17 receptor complex (46). IL-17RC levels in neutrophils are raised in children with ASD compared to neuro-typical controls. In fact, expression of this receptor (mRNA and protein) was completely absent in a cohort of neuro-typical children. The presence of both IL-17A receptor subunits in ASD patients may magnify the effects of IL-17A resulting in an autistic phenotype (93).
The transcription factor STAT3 (signal transducer and activator of transcription 3) is a key player in the development of T helper cells and regulates the expression of the T helper cell specific transcriptional regulator—retinoic acid receptor related orphan receptor γ-t (RORγt) via IL-6 (94, 95). IL-6 is a potent driver of RORγt activity. RORγt is exclusively found in lymphoid cells such as Th17 cells (CD 4 helper cells), and is required for differentiation of Tregs to Th17 cells (95). STAT3 proteins occur at elevated levels in the peripheral blood mononuclear cells (PBMCs) of children affected by ASD (96). Inhibition of STAT3 mitigates MIA associated behavioral and immunological abnormalities seen in animal models (49), while RORγt KO models reverse outcomes in MIA exposed mouse pups (18).
Lastly, IL-13 is a cytokine derived from T cells, which has both inflammatory and anti-inflammatory properties. IL-13 inhibits the production of other inflammatory cytokines (IL-1α, IL-1β, IL-6) through its effects on inflammatory macrophages (97). IL-13 is recognized as a key driver in allergic and inflammatory airway disease, where its effects are potentiated by IL-17 (98). Raised IL-13 has been noted in the plasma and PMBCs of children affected by ASD (29, 99), particularly those with comorbid asthma (although IL-13 is known to be skewed in those with co-morbid atopic conditions) (35).
IL-17A Associated Anti-Inflammatory Mediators in ASD
Another member of the STAT family, STAT6, suppresses the IL-17A inflammatory response. In certain conditions, STAT6 signaling attenuates IL-17A producing T-cells, reducing their production of IL-17A (100). IL-4 mediated inhibition of Th17 cells and IL-17A production is STAT6 dependent (101). In human studies, children with ASD reportedly have reduced levels of STAT6-expressing CD45 cells (CD45+STAT6+) in their PBMC profile compared to neuro-typical controls (80). STAT6, as part of the IL-4 signaling cascade can enhance the expression of anti-inflammatory mediators. This pathway is critical for acceptance of the fetal graft, through reduction of Th17 cells and increase of both IL-4 and Tregs in the fetal environment (102, 103).
In addition to downregulation of the STAT6 mediated pathways, downregulation of other anti-inflammatory cytokines is also reported in autism. Anti-inflammatory cytokine IL-10 acts as a “master” immuno-regulator (104) and IL-10 concentrations are significantly lower in ASD children compared with neuro-typical controls (79, 105). Cytotoxic T-lymphocyte antigen 4 (CTLA4) is a glycoprotein located on T cells (106) and is induced following T cell activation. This anti-inflammatory molecule is expressed at lower levels in the PBMCs of children with ASD (107). Reductions in the levels of these anti-inflammatory and regulatory proteins may lead those with ASD to acquire a more pro-inflammatory state.
Linking Immunity and Genetics in ASD
Bioinformatics analysis of large CNV studies suggest strongly that innate immune processes are implicated in ASD risk (108), this may indicate that immune dysfunction in ASD may be genetically driven or influenced. Maternal immune activation downregulates expression of susceptibility genes known to be highly penetrant in ASD and heavily involved in neurogenesis, cell signaling, synaptogenesis, and axonal guidance in the early stages of fetal development (108, 109). When compared with curated ASD associated gene sets [e.g., via the SFARI Gene database (http://gene.sfari.org/)], MIA downregulated genes were substantially enriched. The strongest enrichment of MIA downregulated genes was observed in the ASD gene categories with the highest likelihood of a link to ASD i.e., SFARI “High Confidence” or “Syndromic” ASD gene sets. This suggests that MIA may bestow increased ASD risk through downregulating the expression of the same genes that are highly penetrant in ASD during the early stages of fetal development.
Loss of function mutations in TSC1 and TSC2 genes are linked to syndromic ASD, and these genes are critical upstream regulators of the mammalian target of rapamycin (mTor) pathway. mTor has important functions in innate immunity and metabolism in particular (52, 110, 111).
Maternal immune activation also has downstream effects, in some cases influencing the transcriptome rather the genes themselves. Fragile X mental retardation 1 gene (FMR1) and CHD8 are both highly penetrant genes for ASD, yet MIA does not seem to influence expression of these genes directly. Rather, it wields an influence on downstream gene targets such as FMRP (fragile X syndrome protein complex). This raises the possibility that MIA may act as an environmental factor disrupting crucial early developmental genomic pathways through influence on downstream gene targets (108). This might suggest that MIA could act both in a direct (genetic) and indirect fashion (epigenetic/regulatory) with the end effects converging on similar pathways.
As previously, mentioned, normal pregnancy is associated with suppression of immunity, allowing the fetus to develop inside the mother's innate immune system. Human leukocyte antigen G coding gene antigen recognition controls the placental immune response and allows acceptance of the fetal graft. Human leukocyte antigen G coding gene interacts with the CD8 cell surface antigen found on most cytotoxic T-lymphocytes that mediate efficient cell–cell interactions within the immune system (112). Higher rates of HLA-G mutations have been found in mothers of children with ASD (113). The Th17 pathway in particular has been identified as a likely effector of inflammatory changes on the developing fetal brain, with downstream effects on behavior and cognitive development (46, 114). We hypothesize that the physiological changes in maternal immunity during pregnancy are dysregulated in some mothers of children with ASD.
In summary, many of the inflammatory proteins reported to have altered expression in ASD are linked to pro-inflammatory Th-17 cells, their product IL-17A, and the IL-17 receptors and receptor complexes. It appears that IL-6 activation (regulated by STAT3 and STAT6 via RORγt activity) of IL-17 expression, and subsequent upregulation of IL-17 receptors and receptor complexes may have a key role in the pathogenesis of ASD. The majority of linked molecules identified above are pro-inflammatory and found in higher quantities in those with ASD, with a corresponding downregulation of anti-inflammatory proteins. Whether this dysregulation of IL-17 is an inherent or acquired state is unclear.
Circulating T cell and IL-17A levels are altered in a subset of children with ASD. Maternal immune activation (including IL-17A) seems to play a role in altering important developmental pathways through direct interaction with ASD susceptibility genes, and indirectly, through interaction with their gene products. Circulating levels of IL-17A are dysregulated during pregnancy in mothers of children who develop ASD and ID (63, 79, 83). Murine models support a causative role for IL-17A in the pathogenesis of ASD. We conclude from the existing evidence that IL-17A dysregulation in the mother or developing infant could play a causal role in the development of at least some subsets of ASD and may be the link between environmental exposure and genetic susceptibility. Understanding the role of IL-17A and its associated targets on neurodevelopmental outcomes is now becomin increasingly important.
What Is the Relevance of the Ongoing COVID-19 Pandemic to MIA-Induced ASD Risk?
Coronavirus disease 2019 (COVID-19), a disease caused by the novel coronavirus, SARS-CoV-2, has become a pandemic, affecting every corner of the globe. Although, the disease (COVID-19) affects primarily the respiratory systems of those affected, it has been found to affect and damage other organs, including the kidneys (115), liver (116), brain (117, 118), and heart (119, 120). Worldwide reported cases and COVID-19 related mortality are most likely an underestimate due to variability of public health capacities between countries, but as of August 2021, there have been almost 200 million confirmed cases of COVID-19, and over 4.2 million deaths reported to the WHO (121).
Our current knowledge of COVID-19 is based only on our limited experience with SARS-CoV-2 since December 2019 and analogously, through our experience of other coronaviruses (SARS CoV and MERS, Middle East Respiratory Syndrome). The long-term consequences of in-utero SARS-CoV-2 exposure and/or congenital infection are almost entirely unknown. There is clear evidence that prenatal exposure to viral infections increases the risk of adverse developmental, neurological, and psychiatric outcomes in later childhood and adult life (38, 44, 122). In this next section, we discuss the implications of the COVID-19 pandemic in the context of MIA-induced alterations in neurodevelopmental outcomes.
COVID-19 and Cytokine Storm
Preclinical work shows that MIA, which stimulates interleukin-17A release from Th17 cells, can establish sustained fetal-placental inflammatory responses. This inflammatory milieu can persist into childhood and affect the development of the young “primed” brain. Remarkably, in murine models, social difficulties in MIA-exposed offspring are remediable through a variety of mechanisms including IL-17 blockade (18, 46). Cytokine storm is a general term applied to maladaptive cytokine release in responses to infection and other stimuli (123). In the context of sepsis, cytokine storm is considered one of the major causes of acute respiratory distress syndrome (ARDS), systemic inflammatory response syndrome (SIRS), and multi-organ failure (124, 125). In COVID-19, cytokine storm seems to play a role in disease aggravation and correlates positively with severity of disease (126). IL-17A target IL-6 and C-reactive protein (CRP) specifically, have been shown to correlate positively with increased mortality (127). Elevated numbers of Th17 cells have been isolated in the blood of individuals with fatal COVID-19 infection (128), while many authors have demonstrated significantly elevated levels of IL-17A in those with both mild and severe COVID-19 (129–131). Coronavirus infection results in macrophage, and dendritic cell activation and IL-6 release (132). This instigates an amplification cascade (JAK–STAT1/3 pathway) that results in cis signaling (binding of cell membrane bound IL-6 receptors) in lymphocytes with downregulation of Tregs and increased differentiation of TH17 cells; as well as trans-signaling (binding of soluble IL-6 receptor) effects on many other cell types (endothelial cells). This widespread immune activation and cytokine production contributes to the pathophysiology of severe COVID-19 (133). Indeed, some authors have specifically suggested therapies intended to target both Th17 cells and IL-17A in COVID-19 disease (134, 135). We have already outlined how Th17 specific (T-helper 17 cell) pathways are initiated via activated macrophages that produce IL-6 and IL-1β. As outlined, IL-6 in particular, is a potent potentiator and trigger for IL-17A release (123, 134, 136). IL-17A therefore, may be a key player in the COVID-19 cytokine storm.
Coronavirus (SARS-CoV-2) Neurotropism and Neurological Effects
Coronaviruses have a demonstrated specific neuro-tropism that allows them access to, and to proliferate in, the host's CNS (137, 138). Cell entry seems to occur through the angiotensin-converting enzyme-2 (ACE-2) and transmembrane protease serine 2 (TMP S2) receptors, both of which are widely expressed in the placenta and at the feto-maternal interface. While trans-placental infection of the fetus is, yet to be proven conclusively, vertical transmission is certainly plausible and may lead directly to inflammatory processes in the fetal brain, in addition to indirect effects via the host/maternal immune response. The neurological sequelae of COVID-19 are wide-ranging and relatively common. The majority of neurological presentations so far have fallen into five categories, (i) Encephalopathy (including delirium and impaired consciousness), (ii) Inflammatory CNS disorders [including encephalitis and Acute Disseminating Encephalomyelitis (ADEM)], (iii) Cerebrovascular accident (CVA)/stroke, (iv) PNS disorders [including Guillain-Barré Syndrome (GBS) and cranial nerve palsies], (v) “Miscellaneous” central neurological disorders (such as raised intracranial pressure, seizures, and myelitis) (139). Hyposmia/Anosmia and hypogeusia (140) are recognized as two important hallmarks of acute SARS-CoV-2 infection, while more severe neurological complications have included CVAs, encephalitis, encephalopathy, and neuropsychiatric disorders (118, 141). Protein-protein network analysis for GBS and COVID-19 revealed that the combined gene set showed an increased connectivity as compared to COVID-19 or GBS alone, this was particularly true of genes related to Th17 cell differentiation. Transcriptome analysis of PBMC from patients with COVID-19 and GBS demonstrated the activation of interleukin-17 signaling in both conditions (142). Viral RNA has been isolated in clinical CSF samples in those with COVID-19 and neurological symptoms (143), and post-mortem examination of brain tissue has identified both viral RNA and neutrophilic infiltrates suggestive of aberrant immune response (144).
Recent pluripotent stem cell derived organoid models have been used to model SARS-CoV-2 infection in a wide range of tissues including gut, lung, liver, kidney, and brain (117, 145). These models demonstrate the virus' ability to infiltrate and proliferate in a variety of different cell/tissue types. Within the brain, the areas with the highest avidity for SARS-CoV-2 are the choroid plexus and the hippocampus (117). This is an interesting finding, as the choroid plexuses themselves represent the interface between CSFand blood compartments (in a similar fashion to the blood-brain barrier). They are located in each of the four ventricles, and are intimately related with immediately adjacent CSF, capillary blood supply, and neural tissue. Angiotensin-converting enzyme-2 receptors also appear to be highly expressed in the choroid plexus (146). In this sense, they provide a comprehensive roadmap upon which SARS-CoV-2 can potentially travel. The neurological features on COVID-19 infection are diverse and wide-ranging. Most studies to date have focused on symptomology in adult patients, but novel models of SARS-CoV-2 infection in a variety of human and animal tissues is casting new light on the mechanisms underlying COVID's infectivity and its ill-effects. There appears to be a variety of mechanisms underlying COVID's pathogenicity, not limited to direct viral effects on tissue, but also collateral effects via immune and thrombotic processes (147). Although there is little research on the effects of COVID on fetuses in early pregnancy, the same processes of direct viral effects and secondary immune and inflammatory effects are likely to be at play.
Maternal COVID-19 Infection and Perinatal Exposure
Pregnant women are not thought to be more susceptible to contracting coronavirus than the general population (148), but given alterations in the pregnant immune state (103), they may be more susceptible to more severe infection (149, 150). Studies from previous pandemics, H1N1 influenza (2009), SARS (2003), and MERS (2012), suggest the possibility of significant maternal and neonatal morbidity and mortality (151, 152). Indeed, both MERS and SARS resulted in maternal death in a significant number of cases, but the specific risk factors for a fatal outcome during pregnancy are not clear. Our experience with these previous coronaviruses indicates higher risk of adverse outcomes for the fetus and infant including fetal growth restriction (FGR), and preterm delivery, both of which have previously been linked to increased ASD incidence (153) as well as NICU admission, spontaneous abortion, and perinatal death. As with other Coronaviruses, maternal SARS-CoV-2 infection has been associated with negative perinatal outcomes. Preterm delivery, fetal distress, stillbirth, and perinatal death have been widely reported (150, 154–156). Figures from China show that while up to 3% of pregnant women infected with COVID-19 required admission to intensive care (157, 158), a UK study showed 1% of pregnant women admitted with SARS-CoV-2 required ECMO (Extra-corporeal membrane oxygenation) and 10% Intensive Care Unit (ICU) management (159).
Cesarean section (CS) has been implicated as a risk factor for the development of ASD in offspring. The mechanisms underlying this are unclear, yet the risk of ASD is increased by approximately 33% in both elective and emergency CS procedures (160). In a systematic review of perinatal and maternal outcomes during the pandemic, CS rates were reported at extremely high levels, up to 90% in some centers (range from approximately 50–90%) (161). For comparison in work published in 2020, Turner at al noted an all-cause national CS rate in Ireland of approximately 26% (162). These higher rates were observed in most centers in spite of recommendations from the Royal College of Obstetrics and Gynecology (RCOG) and the International Federation of Gynecology and Obstetrics (IFGO) against decisions for CS being influenced by maternal SARS-CoV-2 status.
More specifically to neonatal outcomes, the WHO quotes worldwide preterm delivery rates of approximately 10% (163). Two large review studies reported preterm delivery rates of 20–25% in SARS-CoV-2 affected pregnancies (164, 165). Women with SARS-CoV-2 seemed to be more likely to endure a preterm delivery (165). The majority of these deliveries were iatrogenic, but in some reviews, up to half were attributable to either fetal or maternal compromise (166).
Maternal and neonatal ICU admission rates were also higher in the SARS-CoV-2 affected cohorts. Maternal ICU admission and mechanical ventilation rates were high vs. age matched non-pregnant women (165). While rates of stillbirth and neonatal death appear similar to uninfected fetuses, NICU admission rates were notably higher in COVID affected pregnancies (159), commonly as a precautionary step in the care of the neonate. Neonatal morbidity was higher in the SARS-CoV-2 affected groups and was associated with preterm delivery in mothers with more severe COVID-19 primary infection. Hypoxemia and respiratory difficulties in mothers had knock on effects of reduced placenta perfusion, pre-placental hypoxemia, fetal distress, and preterm delivery (167).
Given our knowledge of the potential developmental effects of Th17 activation in pregnancy, children in-utero during this pandemic may have significant inflammatory exposures if maternal infection occurs. There remain unanswered questions about the impact that asymptomatic and mild maternal infection has on the fetal brain in early pregnancy. Prospective follow up studies will need to follow infants whose mothers were infected as well as health unaffected controls. There is enormous potential to leverage archived serological samples from pregnancy and neonatal cohorts to study the relationships (or associations) between markers of maternal inflammation and later neurodevelopmental outcomes in offspring born during the pandemic. While in general, the likelihood of intrauterine maternal-fetal transmission of coronaviruses is low—there have been no documented cases of vertical transmission occurring with either SARS or MERS. There are current reports of possible vertical transmission of SARS-CoV-2 in several cases of third trimester maternal infection (168–170). Little to no information exists about children exposed in the first and second trimesters yet. While generally placental seeding does not seem common, some cases have reported strong evidence of placental infection with the demonstration of high viral load and immuno-histological evidence of SARS-CoV-2 in placental tissue (168). Currently, we can only surmise what the true effect (if any) of gestational COVID-19 on the incidence of ASD will be, but already some have concerns that the incidence may increase (171, 172). No studies have yet been reported on neurodevelopmental outcomes, as the oldest offspring are still in early childhood. Still, the evidence we have outlined within this review from MIA studies examining IL-17A and its pathway members provides a strong basis to build upon our current hypothesis and ask the question; could COVID-19 induced MIA act via IL-17A signaling to increase the risk of ASD-like phenotypes in vulnerable offspring?
Discussion: Improving Outcomes for ASD Affected Individuals and Families
We believe that in spite of the tragedy of the COVID-19 emergency, we are presented with a serendipitous opportunity to progress scientific knowledge regarding prenatal exposures and ASD risk. During the COVID-19 pandemic, we have witnessed a novel infection, affect an immunologically naïve population within an extremely well-defined period of exposure. COVID-19 is now a notifiable illness, and has been characterized and monitored more than any illness in history. Many countries have developed stringent mandatory testing protocols, and track and trace programmes. Within all this, exists an opportunity to study the longitudinal effects of this infection on offspring of those affected by gestational COVID. Further investigation of mid-gestational cytokine profiles (IL-17A in particular) and their potential for genetic interplay could be a crucial cog in the development of actionable and cost-effective improvements in the current models of ASD care. Identification of pathways of immune dysregulation during pregnancy could lead to the identification of a risk marker of ASD that could be characterized in broader ASD cohorts. This would facilitate the identification of a predictive marker of ASD allowing earlier dedicated ASD screening in at risk children. Coupled with these potential biochemical markers, known early clinical signs of ASD exist. Crystallization of the ASD diagnosis can be as early as 14 months old according to some authors, and there are clinically detectable signs of ASD from a younger age still (23, 173, 174). The first children born of this pandemic are now reaching their toddler years, and they may represent a group with increased risk of ASD or other developmental conditions. Taken together, a postulated early biochemical marker and established early clinical markers could allow targeted early ASD screening, which would lead to earlier intervention, and improved outcomes. Therapies instituted in this age group have the potential to significantly improve clinical outcomes in ASD affected children. The timing of therapy is important with the most dramatic symptomatic and developmental improvements in those detected at an earlier age of diagnosis (175, 176).
We believe that it is the obligation of the scientific community to glean what benefit we can from this pandemic. In spite of social distancing measures, systematic national “lockdowns,” and working from home, there has been unprecedented scientific collaboration to try to counter the scourge of COVID. This has led to some outstanding success, not least in the development of two highly effective mRNA vaccines. In order to facilitate international research, the development of an international gestational COVID-19 consortium and registry would be an important step in coordinating research activities and aims. Isolation of relevant clinical bio-samples and prospective identification of patients will have already begun in some centers, and should be facilitated by the public health infrastructures that have been built up around the pandemic. Multidisciplinary collaborative follow up programmes should be established to identify, assess, and treat children with potential negative post-COVID outcomes.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author Contributions
MC wrote the manuscript, reviewed the literature, and synthesized the hypothesis. SC, LGi, and LGa commented on the manuscript at all stages. GO'K commented on the manuscript and aided with literature review. DM commented on the manuscript, helped to synthesize the hypothesis, review the literature, and was the key supervisor. All authors have read and approved the final manuscript.
Funding
Funding was provided to MC by the National Children's Research Center (NCRC) (grant D/19/1), Our Lady's Children's Hospital, Crumlin, Dublin 12, Ireland.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACE-2, angiotensin-converting enzyme-2; ADHD, Attention Deficit Hyperactivity Disorder; ARDS, acute respiratory distress syndrome; ASD, autism spectrum disorder; CS, cesarean section; CD8 cell, cluster of differentiation 8, cytotoxic T-lymphocytes; CHD8, chromodomain helicase DNA binding protein 8 gene; CNV, copy number variant; COVID-19, corona virus disease-2019; FMR1, fragile X mental retardation 1 gene; GWAS, genome-wide association study; HLA-G gene, human leukocyte antigen G coding gene; ID, intellectual disability; IL, interleukin; IL17A gene, interleukin 17A gene; LPS, lipopolysaccharide; MERS, Middle Eastern Respiratory Syndrome; MIA, maternal immune activation; mTor, mammalian target of rapamycin; Poly (I:C), polyinosinic:polycytidylic acid; PNS, peripheral nervous system; RORγt, retinoid-related orphan receptor gamma t; SARS-CoV-2, severe acute respiratory syndrome-coronavirus 2; SNV, single nucleotide variant; Th17, T helper 17 cell; TSC1/TSC 2, Tuberous sclerosis complex ½.
References
1. American Psychiatric Association. DSM-V. 5th ed. Washington, DC: American Psychiatric Association (2013).
2. Birtwell KB. Social, cognitive, and behavioral development of children and adolescents with autism spectrum disorder. In: McDougle C, editor. Autism Spectrum Disorder. Section 1, Chapter 2. Oxford: UK: Oxford Press (2016). p. 19–30. doi: 10.1093/med/9780199349722.003.0002
3. Magiati I, Ong C, Lim XY, Tan JW, Ong AY, Patrycia F, et al. Anxiety symptoms in young people with autism spectrum disorder attending special schools: associations with gender, adaptive functioning and autism symptomatology. Autism. (2016) 20:306–20. doi: 10.1177/1362361315577519
4. Treffert DA. Epidemiology of infantile autism. Arch Gen Psychiatry. (1970) 22:431–8. doi: 10.1001/archpsyc.1970.01740290047006
5. Maenner MJ, Shaw KA, Bakian AV, Bilder DA, Durkin MS, Esler A, et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MWR Surveill Summ. (2021) 70:1–16. doi: 10.15585/mmwr.ss7011a1
6. Lundstrom S, Reichenberg A, Anckarsater H, Lichtenstein P, Gillberg C. Autism phenotype versus registered diagnosis in Swedish children: prevalence trends over 10 years in general population samples. BMJ. (2015) 350:h1961. doi: 10.1136/bmj.h1961
7. Hansen SN, Schendel DE, Parner ET. Explaining the increase in the prevalence of autism spectrum disorders: the proportion attributable to changes in reporting practices. JAMA Pediatr. (2015) 169:56–62. doi: 10.1001/jamapediatrics.2014.1893
8. Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MWR Surveill Summ. (2018) 67:1–23. doi: 10.15585/mmwr.mm6745a7
9. de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat Med. (2016). 22:345–61. doi: 10.1038/nm.4071
10. Fernandes IR, Cruz ACP, Ferrasa A, Phan D, Herai RH, Muotri AR. Genetic variations on SETD5 underlying autistic conditions. Dev Neurobiol. (2018) 78:500–18. doi: 10.1002/dneu.22584
11. Palmer N, Beam A, Agniel D, Eran A, Manrai A, Spettell C, et al. Association of sex with recurrence of autism spectrum disorder among siblings. JAMA Pediatr. (2017) 171:1107–12. doi: 10.1001/jamapediatrics.2017.2832
12. Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. (2014) 46:881–5. doi: 10.1038/ng.3039
13. Vorstman JAS, Parr JR, Moreno-De-Luca D, Anney RJL, Nurnberger JI Jr, Hallmayer JF. Autism genetics: opportunities and challenges for clinical translation Nature reviews. Genetics. (2017) 18:362–76. doi: 10.1038/nrg.2017.4
14. F NG, Gallagher L, Lopez LM. Autism spectrum disorder genomics: the progress and potential of genomic technologies. Genomics. (2020). 112:5136–42. doi: 10.1016/j.ygeno.2020.09.022
15. Goines PE, Croen LA, Braunschweig D, Yoshida CK, Grether J, Hansen R, et al. Increased midgestational IFN-gamma, IL-4 and IL-5 in women bearing a child with autism: a case-control study. Mol Autism. (2011) 2:13. doi: 10.1186/2040-2392-2-13
16. Chess S. Follow-up report on autism in congenital rubella. J Autism Child Schizophr. (1977) 7:69–81. doi: 10.1007/BF01531116
17. Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. (2010) 40:1423–30. doi: 10.1007/s10803-010-1006-y
18. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. (2016) 351:933–9. doi: 10.1126/science.aad0314
19. Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. (2013) 31:54–68. doi: 10.1016/j.bbi.2012.07.008
20. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
21. Dawson G. Early intensive behavioral intervention appears beneficial for young children with autism spectrum disorders. J Pediatr. (2013) 162:1080–1. doi: 10.1016/j.jpeds.2013.02.049
22. Estes A, Munson J, Rogers SJ, Greenson J, Winter J, Dawson G. Long-term outcomes of early intervention in 6-year-old children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. (2015) 54:580–7. doi: 10.1016/j.jaac.2015.04.005
23. Pierce K, Gazestani VH, Bacon E, Barnes CC, Cha D, Nalabolu S, et al. Evaluation of the diagnostic stability of the early autism spectrum disorder phenotype in the general population starting at 12 months. JAMA Pediatr. (2019) 173:578–87. doi: 10.1001/jamapediatrics.2019.0624
24. Landa RJ. Diagnosis of autism spectrum disorders in the first 3 years of life. Nat Clin Pract Neurol. (2008) 4:138–47. doi: 10.1038/ncpneuro0731
25. Rogers SJ, Estes A, Lord C, Vismara L, Winter J, Fitzpatrick A, et al. Effects of a brief Early Start Denver model (ESDM)-based parent intervention on toddlers at risk for autism spectrum disorders: a randomized controlled trial. J Am Acad Child Adolesc Psychiatry. (2012) 51:1052–65. doi: 10.1016/j.jaac.2012.08.003
26. Broek JA, Brombacher E, Stelzhammer V, Guest PC, Rahmoune H, Bahn S. The need for a comprehensive molecular characterization of autism spectrum disorders. Int J Neuropsychopharmacol. (2014) 17:651–73. doi: 10.1017/S146114571300117X
27. Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. Mol Psychiatry. (2015) 20:440–6. doi: 10.1038/mp.2014.59
28. Gunes S, Ekinci O, Celik T. Iron deficiency parameters in autism spectrum disorder: clinical correlates and associated factors. Ital J Pediatr. (2017) 43:86. doi: 10.1186/s13052-017-0407-3
29. Suzuki K, Matsuzaki H, Iwata K, Kameno Y, Shimmura C, Kawai S, et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS ONE. (2011) 6:e20470. doi: 10.1371/journal.pone.0020470
30. Ashwood P, Wills S, Van de Water J. The immune response in autism: a new frontier for autism research. J Leukoc Biol. (2006) 80:1–15. doi: 10.1189/jlb.1205707
31. Morato Torres CA, Wassouf Z, Zafar F, Sastre D, Outeiro TF, Schüle B. The role of alpha-synuclein and other Parkinson's genes in neurodevelopmental and neurodegenerative disorders. Int J Mol Sci. (2020). 21:5724. doi: 10.3390/ijms21165724
32. Zou M, Li D, Wang L, Li L, Xie S, Liu Y, et al. Identification of amino acid dysregulation as a potential biomarker for autism spectrum disorder in China. Neurotox Res. (2020) 38:992–1000. doi: 10.1007/s12640-020-00242-9
33. Masi A, Glozier N, Dale R, Guastella AJ. The immune system, cytokines, and biomarkers in autism spectrum disorder. Neurosci Bull. (2017) 33:194–204. doi: 10.1007/s12264-017-0103-8
34. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Van de Water J. Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. J Neuroimmunol. (2011) 232:196–9. doi: 10.1016/j.jneuroim.2010.10.025
35. Akintunde ME, Rose M, Krakowiak P, Heuer L, Ashwood P, Hansen R, et al. Increased production of IL-17 in children with autism spectrum disorders and co-morbid asthma. J Neuroimmunol. (2015) 286:33–41. doi: 10.1016/j.jneuroim.2015.07.003
36. Chess S. Autism in children with congenital rubella. J Autism Child Schizophr. (1971) 1:33–47. doi: 10.1007/BF01537741
37. Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX, et al. Maternal infection during pregnancy and risk of autism spectrum disorders: a systematic review and meta-analysis. Brain Behav Immun. (2016) 58:165–72. doi: 10.1016/j.bbi.2016.06.005
38. Conway F, Brown AS. Maternal immune activation and related factors in the risk of offspring psychiatric disorders. Front Psychiatry. (2019) 10:430. doi: 10.3389/fpsyt.2019.00430
39. Curran EA, O'Keeffe GW, Looney AM, Moloney G, Hegarty SV, Murray DM, et al. Exposure to hypertensive disorders of pregnancy increases the risk of autism spectrum disorder in affected offspring. Mol Neurobiol. (2018) 55:5557–64. doi: 10.1007/s12035-017-0794-x
40. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol. (2014) 10:643–60. doi: 10.1038/nrneurol.2014.187
41. Boulanger-Bertolus J, Pancaro C, Mashour GA. Increasing role of maternal immune activation in neurodevelopmental disorders. Front Behav Neurosci. (2018). 12:230. doi: 10.3389/fnbeh.2018.00230
42. Minakova E, Warner BB. Maternal immune activation, central nervous system development and behavioral phenotypes. Birth Defec Res. (2018) 110:1539–50. doi: 10.1002/bdr2.1416
43. Bauman MD, Iosif AM, Smith SE, Bregere C, Amaral DG, Patterson PH. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol Psychiatry. (2014) 75:332–41. doi: 10.1016/j.biopsych.2013.06.025
44. Careaga M, Murai T, Bauman MD. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry. (2017) 81:391–401. doi: 10.1016/j.biopsych.2016.10.020
45. Meyer U, Feldon J. To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology. (2012) 62:1308–21. doi: 10.1016/j.neuropharm.2011.01.009
46. Wong H, Hoeffer C. Maternal IL-17A in autism. Exp Neurol. (2018). 299(Pt A):228–40. doi: 10.1016/j.expneurol.2017.04.010
47. Xuan ICY, Hampson DR. Gender-dependent effects of maternal immune activation on the behavior of mouse offspring. PLoS ONE. (2014) 9:e104433. doi: 10.1371/journal.pone.0104433
48. Haddad FL, Patel SV, Schmid S. Maternal immune activation by poly I:c as a preclinical model for neurodevelopmental disorders: a focus on autism and schizophrenia. Neurosci Biobehav Rev. (2020) 113:546–67. doi: 10.1016/j.neubiorev.2020.04.012
49. Parker-Athill EC, Tan J. Maternal immune activation and autism spectrum disorder: interleukin-6 signaling as a key mechanistic pathway. Neurosignals. (2010) 18:113–28. doi: 10.1159/000319828
50. Estes ML, McAllister AK. IMMUNOLOGY. Maternal TH17 cells take a toll on baby's brain. Science. (2016) 351:919–20. doi: 10.1126/science.aaf2850
51. Casanova MF, El-Baz AS, Kamat SS, Dombroski BA, Khalifa F, Elnakib A, et al. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol Commun. (2013) 1:67. doi: 10.1186/2051-5960-1-67
52. Varghese M, Keshav N, Jacot-Descombes S, Warda T, Wicinski B, Dickstein DL, et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol. (2017) 134:537–66. doi: 10.1007/s00401-017-1736-4
53. Kugelberg E. Neuroimmunology: IL-17A mediates a path to autism. Nat Rev Immunol. (2016) 16:205. doi: 10.1038/nri.2016.35
54. Chang YC, Cole TB, Costa LG. Behavioral phenotyping for autism spectrum disorders in mice. Curr Protoc Toxicol. (2017). 72:11.22.1–21. doi: 10.1002/cptx.19
55. Hornig M, Bresnahan MA, Che X, Schultz AF, Ukaigwe JE, Eddy ML, et al. Prenatal fever and autism risk. Mol Psychiatry. (2018) 23:759–66. doi: 10.1038/mp.2017.119
56. Atladóttir HÓ, Henriksen TB, Schendel DE, Parner ET. Autism after infection, febrile episodes, and antibiotic use during pregnancy: an exploratory study. Pediatrics. (2012) 130:e1447. doi: 10.1542/peds.2012-1107
57. Mueller FS, Polesel M, Richetto J, Meyer U, Weber-Stadlbauer U. Mouse models of maternal immune activation: Mind your caging system! Brain Behav Immunity (2018) 73:643–60. doi: 10.1016/j.bbi.2018.07.014
58. Li J, Robinson M, Malacova E, Jacoby P, Foster J, van Eekelen A. Maternal life stress events in pregnancy link to children's school achievement at age 10 years. J Pediatr. (2013) 162:483–9. doi: 10.1016/j.jpeds.2012.09.007
59. Chua JSC, Cowley CJ, Manavis J, Rofe AM, Coyle P. Prenatal exposure to lipopolysaccharide results in neurodevelopmental damage that is ameliorated by zinc in mice. Brain Behav Immun. (2012) 26:326–36. doi: 10.1016/j.bbi.2011.10.002
60. Luan W, Hammond LA, Vuillermot S, Meyer U, Eyles DW. Maternal vitamin D prevents abnormal dopaminergic development and function in a mouse model of prenatal immune activation. Sci Rep. (2018) 8:9741. doi: 10.1038/s41598-018-28090-w
61. Rovira N, Alarcon A, Iriondo M, Ibañez M, Poo P, Cusi V, et al. Impact of histological chorioamnionitis, funisitis and clinical chorioamnionitis on neurodevelopmental outcome of preterm infants. Early Hum Dev. (2011) 87:253–7. doi: 10.1016/j.earlhumdev.2011.01.024
62. Lee I, Neil JJ, Huettner PC, Smyser CD, Rogers CE, Shimony JS, et al. The impact of prenatal and neonatal infection on neurodevelopmental outcomes in very preterm infants. J Perinatol. (2014) 34:741–7. doi: 10.1038/jp.2014.79
63. Jones KL, Croen LA, Yoshida CK, Heuer L, Hansen R, Zerbo O, et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol Psychiatry. (2017) 22:273–9. doi: 10.1038/mp.2016.77
64. Irwin JL, Yeates AJ, Mulhern MS, McSorley EM, Strain JJ, Watson GE, et al. Maternal gestational immune response and autism spectrum disorder phenotypes at 7 years of age in the seychelles child development study. Mol Neurobiol. (2019) 56:5000–8. doi: 10.1007/s12035-018-1424-y
65. Abdallah MW, Larsen N, Grove J, Norgaard-Pedersen B, Thorsen P, Mortensen EL, et al. Amniotic fluid inflammatory cytokines: potential markers of immunologic dysfunction in autism spectrum disorders. World J Biol Psychiatry. (2013) 14:528–38. doi: 10.3109/15622975.2011.639803
66. Shobokshi A, Shaarawy M. Maternal serum and amniotic fluid cytokines in patients with preterm premature rupture of membranes with and without intrauterine infection. Int J Gynaecol Obstet. (2002) 79:209–15. doi: 10.1016/S0020-7292(02)00238-2
67. Rounioja S, Räsänen J, Glumoff V, Ojaniemi M, Mäkikallio K, Hallman M. Intra-amniotic lipopolysaccharide leads to fetal cardiac dysfunction. A mouse model for fetal inflammatory response. Cardiovasc Res. (2003) 60:156–64. doi: 10.1016/S0008-6363(03)00338-9
68. Ricci S, Businaro R, Ippoliti F, Lo Vasco VR, Massoni F, Onofri E, et al. Altered cytokine and BDNF levels in autism spectrum disorder. Neurotox Res. (2013) 24:491–501. doi: 10.1007/s12640-013-9393-4
69. Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol. (2007) 36:361–5. doi: 10.1016/j.pediatrneurol.2007.01.012
70. Eftekharian MM, Ghafouri-Fard S, Noroozi R, Omrani MD, Arsang-Jang S, Ganji M, et al. Cytokine profile in autistic patients. Cytokine. (2018) 108:120–6. doi: 10.1016/j.cyto.2018.03.034
71. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. (2011) 25:40–5. doi: 10.1016/j.bbi.2010.08.003
72. Kordulewska NK, Kostyra E, Piskorz-Ogórek K, Moszyńska M, Cieślińska A, Fiedorowicz E, et al. Serum cytokine levels in children with spectrum autism disorder: Differences in pro- and anti-inflammatory balance. J Neuroimmunol. (2019) 337:577066. doi: 10.1016/j.jneuroim.2019.577066
73. Heuer LS, Croen LA, Jones KL, Yoshida CK, Hansen RL, Yolken R, et al. An exploratory examination of neonatal cytokines and chemokines as predictors of autism risk: the early markers for autism study. Biol Psychiatry. (2019) 86:255–64. doi: 10.1016/j.biopsych.2019.04.037
74. Kutuk MO, Tufan E, Gokcen C, Kilicaslan F, Karadag M, Mutluer T, et al. Cytokine expression profiles in autism spectrum disorder: a multi-center study from Turkey. Cytokine. (2020) 133:155152. doi: 10.1016/j.cyto.2020.155152
75. Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. (2006) 2:619–26. doi: 10.1038/ncprheum0338
76. Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT, et al. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation. (2011) 8:52. doi: 10.1186/1742-2094-8-52
77. Murphy SP, Tayade C, Ashkar AA, Hatta K, Zhang J, Croy BA. Interferon gamma in successful pregnancies. Biol Reprod. (2009) 80:848–59. doi: 10.1095/biolreprod.108.073353
78. Al-Ayadhi LY, Mostafa GA. Elevated serum levels of interleukin-17A in children with autism. J Neuroinflammation. (2012) 9:158. doi: 10.1186/1742-2094-9-158
79. Moaaz M, Youssry S, Elfatatry A, El Rahman MA. Th17/Treg cells imbalance and their related cytokines (IL-17, IL-10 and TGF-β) in children with autism spectrum disorder. J Neuroimmunol. (2019) 337:577071. doi: 10.1016/j.jneuroim.2019.577071
80. Ahmad SF, Ansari MA, Nadeem A, Bakheet SA, Al-Ayadhi LY, Alasmari AF, et al. Involvement of CD45 cells in the development of autism spectrum disorder through dysregulation of granulocyte-macrophage colony-stimulating factor, key inflammatory cytokines, and transcription factors. Int Immunopharmacol. (2020) 83:106466. doi: 10.1016/j.intimp.2020.106466
81. Bryn V, Aass HC, Skjeldal OH, Isaksen J, Saugstad OD, Ormstad H. Cytokine profile in autism spectrum disorders in children. J Mol Neurosci. (2017) 61:1–7. doi: 10.1007/s12031-016-0847-z
82. Casey S, Carter M, Looney AM, Livingstone V, Moloney G, O'Keeffe GW, et al. Maternal mid-gestation cytokine dysregulation in mothers of children with autism spectrum disorder. J Autism Dev Disord. (2021). doi: 10.1007/s10803-021-05271-7 [Epub ahead of print].
83. van der Zwaag B, Franke L, Poot M, Hochstenbach R, Spierenburg HA, Vorstman JA, et al. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PLoS ONE. (2009) 4:e5324. doi: 10.1371/journal.pone.0005324
84. Chehimi M, Vidal H, Eljaafari A. Pathogenic role of IL-17-producing immune cells in obesity, and related inflammatory diseases. J Clin Med. (2017). 6:68. doi: 10.3390/jcm6070068
85. Hill AP, Zuckerman KE, Fombonne E. Obesity and autism. Pediatrics. (2015) 136:1051–61. doi: 10.1542/peds.2015-1437
86. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. (2019) 47:D607–13. doi: 10.1093/nar/gky1131
87. Kimura A, Kishimoto T. IL-6: Regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
88. Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. (2009) 64:61–78. doi: 10.1016/j.neuron.2009.09.002
89. Wu WL, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun. (2017) 62:11–23. doi: 10.1016/j.bbi.2016.11.007
90. Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, Whitters MJ, et al. The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J Immunol. (2008) 181:2799. doi: 10.4049/jimmunol.181.4.2799
91. Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. (2008) 205:1063–75. doi: 10.1084/jem.20071978
92. Nadeem A, Ahmad SF, Attia SM, Bakheet SA, Al-Harbi NO, Al-Ayadhi LY. Activation of IL-17 receptor leads to increased oxidative inflammation in peripheral monocytes of autistic children. Brain Behav Immun. (2018) 67:335–44. doi: 10.1016/j.bbi.2017.09.010
93. Nadeem A, Ahmad SF, Attia SM, Al-Ayadhi LY, Bakheet SA, Al-Harbi NO. Oxidative and inflammatory mediators are upregulated in neutrophils of autistic children: role of IL-17A receptor signaling. Prog Neuro Psychopharmacol Biol Psychiatry. (2019) 90:204–11. doi: 10.1016/j.pnpbp.2018.12.002
94. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
95. Ivanov McKenzie BS II, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006). 126:1121–33. doi: 10.1016/j.cell.2006.07.035
96. Ahmad SF, Zoheir KMA, Ansari MA, Nadeem A, Bakheet SA, Al-Ayadhi LY, et al. Dysregulation of Th1, Th2, Th17, and T regulatory cell-related transcription factor signaling in children with autism. Mol Neurobiol. (2017) 54:4390–400. doi: 10.1007/s12035-016-9977-0
97. Zhu C, Zhang A, Huang S, Ding G, Pan X, Chen R. Interleukin-13 inhibits cytokines synthesis by blocking nuclear factor-κB and c-Jun N-terminal kinase in human mesangial cells. J Biomed Res. (2010) 24:308–16. doi: 10.1016/S1674-8301(10)60043-7
98. Hall SL, Baker T, Lajoie S, Richgels PK, Yang Y, McAlees JW, et al. IL-17A enhances IL-13 activity by enhancing IL-13-induced signal transducer and activator of transcription 6 activation. J Allergy Clin Immunol. (2017). 139(2):462.e14–71.e14. doi: 10.1016/j.jaci.2016.04.037
99. Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P, et al. Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol. (2006) 172:198–205. doi: 10.1016/j.jneuroim.2005.11.007
100. Bloodworth MH, Newcomb DC, Dulek DE, Stier MT, Cephus JY, Zhang J, et al. STAT6 signaling attenuates interleukin-17-producing γδ T cells during acute Klebsiella pneumoniae infection. Infect Immun. (2016) 84:1548–55. doi: 10.1128/IAI.00646-15
101. Cooney LA, Towery K, Endres J, Fox DA. Sensitivity and resistance to regulation by IL-4 during Th17 maturation. J Immunol. (2011) 187:4440–50. doi: 10.4049/jimmunol.1002860
102. Chatterjee P, Chiasson VL, Bounds KR, Mitchell BM. Regulation of the anti-inflammatory cytokines interleukin-4 and interleukin-10 during pregnancy. Front Immunol. (2014) 5:253. doi: 10.3389/fimmu.2014.00253
103. Jonakait GM. The effects of maternal inflammation on neuronal development: possible mechanisms. Int J Dev Neurosci. (2007) 25:415–25. doi: 10.1016/j.ijdevneu.2007.08.017
104. Couper KN, Blount DG, Riley EM. IL-10: The master regulator of immunity to infection. J Immunol. (2008) 180:5771. doi: 10.4049/jimmunol.180.9.5771
105. Abdallah MW, Larsen N, Mortensen EL, Atladóttir HÓ, Nørgaard-Pedersen B, Bonefeld-Jørgensen EC, et al. Neonatal levels of cytokines and risk of autism spectrum disorders: an exploratory register-based historic birth cohort study utilizing the Danish Newborn Screening Biobank. J Neuroimmunol. (2012) 252:75–82. doi: 10.1016/j.jneuroim.2012.07.013
106. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. (1994) 1:405–13. doi: 10.1016/1074-7613(94)90071-X
107. Ahmad SF, Nadeem A, Ansari MA, Bakheet SA, Attia SM, Zoheir KMA, et al. Imbalance between the anti- and pro-inflammatory milieu in blood leukocytes of autistic children. Mol Immunol. (2017) 82:57–65. doi: 10.1016/j.molimm.2016.12.019
108. Lombardo MV, Moon HM, Su J, Palmer TD, Courchesne E, Pramparo T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol Psychiatry. (2018) 23:1001–13. doi: 10.1038/mp.2017.15
109. Courchesne E, Pramparo T, Gazestani VH, Lombardo MV, Pierce K, Lewis NE. The ASD living biology: from cell proliferation to clinical phenotype. Mol Psychiatry. (2019) 24:88–107. doi: 10.1038/s41380-018-0056-y
110. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. (2015) 15:599–614. doi: 10.1038/nri3901
111. Petrasek T, Vojtechova I, Klovrza O, Tuckova K, Vejmola C, Rak J, et al. mTOR inhibitor improves autistic-like behaviors related to Tsc2 haploinsufficiency but not following developmental status epilepticus. J Neurodev Disord. (2021). 13:14. doi: 10.1186/s11689-021-09357-2
112. Sanders SK, Giblin PA, Kavathas P. Cell-cell adhesion mediated by CD8 and human histocompatibility leukocyte antigen G, a nonclassical major histocompatibility complex class 1 molecule on cytotrophoblasts. J Exp Med. (1991) 174:737–40. doi: 10.1084/jem.174.3.737
113. Guerini FR, Bolognesi E, Chiappedi M, Ripamonti E, Ghezzo A, Zanette M, et al. HLA-G coding region polymorphism is skewed in autistic spectrum disorders. Brain Behav Immun. (2018) 67:308–13. doi: 10.1016/j.bbi.2017.09.007
114. Shin Yim Y, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, et al. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature. (2017) 549:482–7. doi: 10.1038/nature23909
115. Watchorn J, Huang DY, Joslin J, Bramham K, Hutchings SD. Critically ill COVID-19 patients with acute kidney injury have reduced renal blood flow and perfusion despite preserved cardiac function. A case-control study using contrast enhanced ultrasound. Shock. (2020) 55:479–87. doi: 10.2139/ssrn.3627340
116. Kumar A, Kumar P, Dungdung A, Kumar Gupta A, Anurag A, Kumar A. Pattern of liver function and clinical profile in COVID-19: a cross-sectional study of 91 patients. Diabetes Metab Syndr. (2020) 14:1951–4. doi: 10.1016/j.dsx.2020.10.001
117. Jacob F, Pather SR, Huang WK, Zhang F, Wong SZH, Zhou H, et al. Human pluripotent stem cell-derived neural cells and brain organoids reveal SARS-CoV-2 neurotropism predominates in choroid plexus epithelium. Cell Stem Cell. (2020) 27:937–50.e9. doi: 10.1101/2020.07.28.225151
118. Rifino N, Censori B, Agazzi E, Alimonti D, Bonito V, Camera G, et al. Neurologic manifestations in 1760 COVID-19 patients admitted to Papa Giovanni XXIII Hospital, Bergamo, Italy. J Neurol. (2020) 268:2331–8. doi: 10.1007/s00415-020-10251-5
119. Peltzer B, Manocha KK, Ying X, Kirzner J, Ip JE, Thomas G, et al. Outcomes and mortality associated with atrial arrhythmias among patients hospitalized with COVID-19. J Cardiovasc Electrophysiol. (2020) 31:3077–85. doi: 10.1111/jce.14770
120. Nakamura Y, Shimizu M, Yamaki T, Kushimoto K, Yamashita A, Hayase K, et al. Myocardial injury in a patient with severe coronavirus disease: a case report. J Infect Chemother. (2020) 27:364–8. doi: 10.1016/j.jiac.2020.09.023
121. Organisation WH. WHO Coronavirus Disease (COVID-19) Dashboard. (2020). Available online at: https://covid19.who.int/ (accessed August 30, 2021).
122. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science. (2016) 353:772–7. doi: 10.1126/science.aag3194
123. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine Storm' in COVID-19. J Infect. (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
124. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. (2017) 39:517–28. doi: 10.1007/s00281-017-0639-8
125. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. Jama. (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
126. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
127. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intens Care Med. (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
128. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
129. Ghazavi A, Ganji A, Keshavarzian N, Rabiemajd S, Mosayebi G. Cytokine profile and disease severity in patients with COVID-19. Cytokine. (2021) 137:155323. doi: 10.1016/j.cyto.2020.155323
130. Qi D, Yan X, Tang X, Peng J, Yu Q, Feng L, et al. Epidemiological and clinical features of 2019-nCoV acute respiratory disease cases in Chongqing municipality, China: a retrospective, descriptive, multiple-center study. medRxiv. (2020) 2020.03.01.20029397. doi: 10.1101/2020.03.01.20029397
131. Ouyang Y, Yin J, Wang W, Shi H, Shi Y, Xu B, et al. Downregulated gene expression spectrum and immune responses changed during the disease progression in patients with COVID-19. Clin Infect Dis. (2020) 71:2052–60. doi: 10.1093/cid/ciaa462
132. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. (2020) 108:17–41. doi: 10.1002/JLB.3COVR0520-272R
133. Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. (2020) 368:473–4. doi: 10.1126/science.abb8925
134. Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. (2020) 53:368–70. doi: 10.1016/j.jmii.2020.03.005
135. Bulat V, Situm M, Azdajic MD, Likic R. Potential role of IL-17 blocking agents in the treatment of severe COVID-19? Br J Clin Pharmacol. (2021) 87:1578–81. doi: 10.1111/bcp.14437
136. Chen L, Liu HG, Liu W, Liu J, Liu K, Shang J, et al. [Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi. (2020) 43:E005. doi: 10.3760/cma.j.issn.1001-0939.2020.0005
137. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. (2005) 202:415–24. doi: 10.1084/jem.20050828
138. Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol. (2008) 82:7264–75. doi: 10.1128/JVI.00737-08
139. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. (2020) 143:3104–20. doi: 10.1093/brain/awaa240
140. Finsterer J, Stollberger C. Causes of hypogeusia/hyposmia in SARS-CoV2 infected patients. J Med Virol. (2020) 92:1793–4. doi: 10.1002/jmv.25903
141. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. (2020) 77:1–9. doi: 10.1001/jamaneurol.2020.1127
142. Li Z, Huang Z, Li X, Huang C, Shen J, Li S, et al. Bioinformatic analyses hinted at augmented T helper 17 cell differentiation and cytokine response as the central mechanism of COVID-19-associated Guillain-Barré syndrome. Cell Prolif. (2021). 54:e13024. doi: 10.1111/cpr.13024
143. Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. (2020) 383:590–2. doi: 10.1056/NEJMc2011400
144. Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. (2020) 1: E290–9. doi: 10.1016/S2666-5247(20)30144-0
145. Ramani A, Müller L, Ostermann PN, Gabriel E, Abida-Islam P, Müller-Schiffmann A, et al. SARS-CoV-2 targets cortical neurons of 3D human brain organoids and shows neurodegeneration-like effects. BioRxiv. (2020) 2020.05.20.106575. doi: 10.15252/embj.2020106230
146. Chen R, Wang K, Yu J, Chen Z, Wen C, Xu Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain. BioRxiv. (2020) 2020.04.07.030650. doi: 10.1101/2020.04.07.030650
147. Wool GD, Miller JL. The impact of COVID-19 disease on platelets and coagulation. Pathobiology. (2020) 88:15-27. doi: 10.1159/000512007
148. Chen Y, Li Z, Zhang Y-Y, Zhao W-H, Yu Z-Y. Maternal health care management during the outbreak of coronavirus disease 2019. J Med Virol. (2020) 92:731–9. doi: 10.1002/jmv.25787
149. Favre G, Pomar L, Musso D, Baud D. 2019-nCoV epidemic: what about pregnancies? Lancet. (2020) 395:e40. doi: 10.1016/S0140-6736(20)30311-1
150. Wastnedge EAN, Reynolds RM, Boeckel SRv, Stock SJ, Denison FC, Maybin JA, et al. Pregnancy and COVID-19. Physiol Rev. (2021) 101:303–18. doi: 10.1152/physrev.00024.2020
151. Alfaraj SH, Al-Tawfiq JA, Memish ZA. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) infection during pregnancy: report of two cases & review of the literature. J Microbiol Immunol Infect. (2019) 52:501–3. doi: 10.1016/j.jmii.2018.04.005
152. Siston AM, Rasmussen SA, Honein MA, Fry AM, Seib K, Callaghan WM, et al. Pandemic 2009 influenza A(H1N1) virus illness among pregnant women in the United States. Jama. (2010) 303:1517–25. doi: 10.1001/jama.2010.479
153. Lampi KM, Lehtonen L, Tran PL, Suominen A, Lehti V, Banerjee PN, et al. Risk of autism spectrum disorders in low birth weight and small for gestational age infants. J Pediatr. (2012) 161:830–6. doi: 10.1016/j.jpeds.2012.04.058
154. Fan C, Lei D, Fang C, Li C, Wang M, Liu Y, et al. Perinatal transmission of COVID-19 associated SARS-CoV-2: should we worry? Clin Infect Dis. (2020) 72:862–4. doi: 10.1093/cid/ciaa226
155. Chen H, Guo J, Wang C, Luo F, Yu X, Zhang W, et al. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: a retrospective review of medical records. Lancet. (2020) 395:809–15. doi: 10.1016/S0140-6736(20)30360-3
156. Salem D, Katranji F, Bakdash T. COVID-19 infection in pregnant women: review of maternal and fetal outcomes. Int J Gynaecol Obstet. (2021) 152:291–8. doi: 10.1002/ijgo.13533
157. Liu Y, Chen H, Tang K, Guo Y. Clinical manifestations and outcome of SARS-CoV-2 infection during pregnancy. J Infect. (2020) 2020:S0163-4453(20)30109-2. doi: 10.1016/j.jinf.2020.02.028
158. Wang X, Zhou Z, Zhang J, Zhu F, Tang Y, Shen X, et al. A case of 2019 novel coronavirus in a pregnant woman with preterm delivery. Clin Infect Dis. (2020) 71:844–6. doi: 10.1093/cid/ciaa200
159. Knight M, Bunch K, Vousden N, Morris E, Simpson N, Gale C, et al. Characteristics and outcomes of pregnant women admitted to hospital with confirmed SARS-CoV-2 infection in UK: national population based cohort study. BMJ. (2020) 369:m2107. doi: 10.1136/bmj.m2107
160. Zhang T, Sidorchuk A, Sevilla-Cermeño L, Vilaplana-Pérez A, Chang Z, Larsson H, et al. Association of cesarean delivery with risk of neurodevelopmental and psychiatric disorders in the offspring: a systematic review and meta-analysis. JAMA Network Open. (2019). 2:e1910236-e. doi: 10.1001/jamanetworkopen.2019.10236
161. Papapanou M, Papaioannou M, Petta A, Routsi E, Farmaki M, Vlahos N, et al. Maternal and neonatal characteristics and outcomes of COVID-19 in pregnancy: an overview of systematic reviews. Int J Environ Res Public Health. (2021). 18:596. doi: 10.3390/ijerph18020596
162. Turner MJ, Reynolds CME, McMahon LE, O'Malley EG, O'Connell MP, Sheehan SR. Caesarean section rates in women in the Republic of Ireland who chose to attend their obstetrician privately: a retrospective observational study. BMC Pregnancy Childbirth. (2020) 20:548. doi: 10.1186/s12884-020-03199-x
163. World Health O. Born Too Soon: The Global Action Report on Preterm Birth. Geneva: World Health Organization (2012).
164. Dhir SK, Kumar J, Meena J, Kumar P. Clinical features and outcome of SARS-CoV-2 infection in neonates: a systematic review. J Trop Pediatr. (2021). 67:fmaa059. doi: 10.1093/tropej/fmaa059
165. Allotey J, Stallings E, Bonet M, Yap M, Chatterjee S, Kew T, et al. Clinical manifestations, risk factors, and maternal and perinatal outcomes of coronavirus disease 2019 in pregnancy: living systematic review and meta-analysis. BMJ. (2020) 370:m3320. doi: 10.1136/bmj.m3320
166. Turan O, Hakim A, Dashraath P, Jeslyn WJL, Wright A, Abdul-Kadir R. Clinical characteristics, prognostic factors, and maternal and neonatal outcomes of SARS-CoV-2 infection among hospitalized pregnant women: a systematic review. Int J Gynaecol Obstet. (2020) 151:7–16. doi: 10.1002/ijgo.13329
167. Yoon SH, Kang JM, Ahn JG. Clinical outcomes of 201 neonates born to mothers with COVID-19: a systematic review. Eur Rev Med Pharmacol Sci. (2020) 24:7804–15. doi: 10.26355/eurrev_202007_22285
168. Vivanti AJ, Vauloup-Fellous C, Prevot S, Zupan V, Suffee C, Do Cao J, et al. Transplacental transmission of SARS-CoV-2 infection. Nat Commun. (2020) 11:3572. doi: 10.1038/s41467-020-17436-6
169. Kirtsman M, Diambomba Y, Poutanen SM, Malinowski AK, Vlachodimitropoulou E, Parks WT, et al. Probable congenital SARS-CoV-2 infection in a neonate born to a woman with active SARS-CoV-2 infection. CMAJ. (2020) 192:E647–50. doi: 10.1503/cmaj.200821
170. Egloff C, Vauloup-Fellous C, Picone O, Mandelbrot L, Roques P. Evidence and possible mechanisms of rare maternal-fetal transmission of SARS-CoV-2. J Clin Virol. (2020) 128:104447. doi: 10.1016/j.jcv.2020.104447
171. Steinman G. COVID-19 and autism. Med Hypotheses. (2020) 142:109797. doi: 10.1016/j.mehy.2020.109797
172. Shuid AN, Jayusman PA, Shuid N, Ismail J, Kamal Nor N, Mohamed IN. Association between viral infections and risk of autistic disorder: an overview. Int J Environ Res Public Health. (2021) 18:2817. doi: 10.3390/ijerph18062817
173. Libertus K, Sheperd KA, Ross SW, Landa RJ. Limited fine motor and grasping skills in 6-month-old infants at high risk for autism. Child Dev. (2014) 85:2218–31. doi: 10.1111/cdev.12262
174. Ozonoff S, Macari S, Young GS, Goldring S, Thompson M, Rogers SJ. Atypical object exploration at 12 months of age is associated with autism in a prospective sample. Autism. (2008) 12:457–72. doi: 10.1177/1362361308096402
175. Oono IP, Honey EJ, McConachie H. Parent-mediated early intervention for young children with autism spectrum disorders (ASD). Cochrane Database Syst Rev. (2013) 4:Cd009774. doi: 10.1002/14651858.CD009774.pub2
Keywords: ASD, autism, cytokine, maternal immune activation, MIA, interleukin-17A (IL-17A), COVID-19
Citation: Carter M, Casey S, O'Keeffe GW, Gibson L, Gallagher L and Murray DM (2022) Maternal Immune Activation and Interleukin 17A in the Pathogenesis of Autistic Spectrum Disorder and Why It Matters in the COVID-19 Era. Front. Psychiatry 13:823096. doi: 10.3389/fpsyt.2022.823096
Received: 26 November 2021; Accepted: 21 January 2022;
Published: 17 February 2022.
Edited by:
Hideo Matsuzaki, University of Fukui, JapanReviewed by:
Kohji Fukunaga, Tohoku University, JapanToru Takumi, RIKEN Brain Science Institute (BSI), Japan
Copyright © 2022 Carter, Casey, O'Keeffe, Gibson, Gallagher and Murray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Carter, bWljaGFlbC5jYXJ0ZXJAdWNjLmll; Deirdre M. Murray, ZC5tdXJyYXlAdWNjLmll