 Béla Kiss
Béla Kiss Balázs Krámos2
Balázs Krámos2- 1Pharmacological and Drug Safety Research, Gedeon Richter Plc., Budapest, Hungary
- 2Spectroscopic Research Department, Gedeon Richter Plc., Budapest, Hungary
- 3Global Medical Division, Gedeon Richter Plc., Budapest, Hungary
Dysfunctions of the dopaminergic system are believed to play a major role in the core symptoms of schizophrenia such as positive, negative, and cognitive symptoms. The first line of treatment of schizophrenia are antipsychotics, a class of medications that targets several neurotransmitter receptors in the brain, including dopaminergic, serotonergic, adrenergic and/or muscarinic receptors, depending on the given agent. Although the currently used antipsychotics display in vitro activity at several receptors, majority of them share the common property of having high/moderate in vitro affinity for dopamine D2 receptors (D2Rs) and D3 receptors (D3Rs). In terms of mode of action, these antipsychotics are either antagonist or partial agonist at the above-mentioned receptors. Although D2Rs and D3Rs possess high degree of homology in their molecular structure, have common signaling pathways and similar in vitro pharmacology, they have different in vivo pharmacology and therefore behavioral roles. The aim of this review, with summarizing preclinical and clinical evidence is to demonstrate that while currently used antipsychotics display substantial in vitro affinity for both D3Rs and D2Rs, only very few can significantly occupy D3Rs in vivo. The relative importance of the level of endogenous extracellular dopamine in the brain and the degree of in vitro D3Rs receptor affinity and selectivity as determinant factors for in vivo D3Rs occupancy by antipsychotics, are also discussed.
Introduction
It is widely accepted that dysfunction of the dopaminergic neurotransmitter system plays a major role in the pathophysiology of schizophrenia. The primary pharmacotherapy of schizophrenia involves the use of antipsychotics, a group of drugs representing great heterogeneity in their chemical structure, pharmacological and functional profile, as well as clinical efficacy. At present, all available antipsychotics display affinity for D2Rs, and it is widely accepted that D2R antagonism or partial agonism is essential for their antipsychotic efficacy. Currently used antipsychotics display medium-to-high in vitro affinity for D2R as well as D3R, and high correlation can be demonstrated between their affinities for these receptors. This is not surprising considering the high structural homology, and the in vitro functional and pharmacological similarities of the two receptors. On the other hand, significant differences have been demonstrated in their in vivo pharmacology and behavioral roles. All currently used antipsychotics, in agreement with their in vitro D2R affinity, show significant in vivo brain D2R occupancy at their antipsychotic effective doses. However, despite their substantial in vitro D3R affinity, not all antipsychotics demonstrated in vivo D3R occupancy in animals or in humans. Here, a review is given on the data available for the in vitro affinity for D2Rs and D3Rs and a hypothesis is provided as to why a group of antipsychotics do not show significant in vivo brain D3R occupancy despite their notable in vitro D3R affinity.
Schizophrenia
Schizophrenia is one of the most serious and debilitating psychiatric disorder affecting about 1% of the population disregarding economic, social, or cultural background of the society (1). Schizophrenia is characterized by positive symptoms (delusions, hallucinations) negative symptoms (social and emotional withdrawal, anhedonia, lack of motivation) and cognitive dysfunction, as well. All these symptoms may be mixed with aggressive behavior, depression, or anxiety (2–4).
The early, so called “dopamine hypothesis” stated that low prefrontal dopamine activity would cause “deficit symptoms” whereas enhanced activity in mesolimbic dopamine system would be in the background of the positive symptoms (5). In fact, the increased dopamine transmission has been demonstrated by positron emission tomography (PET) (6, 7). Further, presynaptically increased synthesis of dopamine in the basal ganglia has been found [(8, 9), see for review]. Loss of glutamatergic functions is also hypothesized and is thought to explain negative symptoms (9–11).
Antipsychotics
Recognition of the neuroleptic action of chlorpromazine in 1952 represented a breakthrough in the drug treatment of schizophrenia (12). Chlorpromazine was soon followed by introduction of several other “neuroleptics” such as haloperidol, fluphenazine, pimozide, sulpiride, thioridazine etc. (Interestingly enough, this group of drugs was named/categorized by their side effect profile).
At the time of their discovery, the main mechanism of action of the first-generation antipsychotics was believed to be mediated by their actions on the monoaminergic system. Carlsson and Lindquist demonstrated that haloperidol and chlorpromazine increased monoamine turnover in the rat brain and these changes were attributed to the monoamine receptor antagonism action of these compounds (13). Van Rossum was the first describing that antipsychotics exert their therapeutic effects through the blockade of dopamine receptors (14). For the history of antipsychotics’ discovery see the recent review by Seeman (15).
Some antipsychotics, such as clozapine, fluperlapine and melperone were found to produce weak catalepsy in rodents, with minimal extrapyramidal symptoms and serum prolactin elevation in humans, compared to the earlier typical antipsychotic drugs, such as haloperidol. Meltzer and Matsubara explored the basis of these differences by testing the affinity of 38 antipsychotics for the rat striatal dopamine D1 receptors (D1Rs), D2R and serotonin 5-HT2 receptors (5-HT2R). They found that the 5-HT2R/D2R affinity ratio was the most useful means of differentiation from the typical antipsychotics. They demonstrated that compounds displaying 5-HT2R/D2R affinity ratio of 1.12 or higher were the ones showing the atypical characteristics (16). These findings had significant impact on the antipsychotic drug research: the primary aim was to find antipsychotics possessing a significant serotonin 5-HT2A receptor (5-HT2AR) affinity that would be similar or higher than that for the D2R. The quest for compounds with D2R/5-HT2AR affinity led to discovery of risperidone, asenapine, olanzapine, quetiapine, ziprasidone, blonanserin and lurasidone, collectively classified as atypical or second-generation antipsychotics.
Atypical antipsychotics, like to the typical antipsychotics, are efficacious in the treatment of positive symptoms of schizophrenia but display relatively lower propensity to cause extrapyramidal side effects. However, it was claimed that the label of “atypical” is not fully justified as they are different from first-generation antipsychotics only in their side effect profile (e.g., weight gain, alteration in metabolic parameters, cardiovascular complications) (17–19). In fact, neither group represented major step forward in the treatment of other symptoms of schizophrenia, such as negative or cognitive symptoms.
Distinct category of second-generation antipsychotics with partial agonism at dopamine D2R, D3R and serotonin 5-HT1A receptors (5-HT1AR) as well as antagonism at serotonin 5-HT2AR and 5-HT2B receptors (5-HT2BRs) is represented by aripiprazole, cariprazine and brexpiprazole. Amongst these three partial agonist antipsychotics, aripiprazole and brexpiprazole display preferential binding affinity for dopamine D2R (20, 21), whereas cariprazine has higher affinity for dopamine D3R over D2R receptors (22). These dopamine receptor partial agonists may be referred to as third generation antipsychotics (23). These dopamine-serotonin partial agonists were originally approved for acute schizophrenia, schizophrenia maintenance, later, however, they were found to be useful in treatment of mania, bipolar disorder, and as adjunct in unipolar depression (24).
Dopamine Receptors
Effects of dopamine are mediated through five receptors subtypes, namely D1-, D2-, D3-, D4-, and D5-receptors. All dopamine receptors belong to G-protein coupled receptor (GPCR) family: D1 and D5 receptors (D1-receptor family) stimulate cAMP signaling pathway through a Gαs G-proteins, whereas D2-, D3- and D4-receptors (D2-receptor family) inhibit cAMP signaling through a Gαi/o G-proteins (25–29).
Expression of dopamine D1 receptors (D1R) is the highest in basal ganglia (caudate nucleus, putamen and globus pallidus), accumbens nuclei, substantia nigra, amygdala and the frontal cortex. The cortex, substantia nigra, hypothalamus and the hippocampus express low level of dopamine D5 receptors (D5Rs). High levels of D2Rs are found in the basal ganglia, while cortical regions express low level of these receptors. D2Rs are the primary drug targets in schizophrenia, Parkinson’s disease, restless leg syndrome and neuroendocrine tumors. Highest expression of dopamine D3Rs are found mainly in the limbic system (islands of Calleja, nucleus accumbens, ventral part of caudate nucleus), with minor/low levels of expression in cortical regions. Dopamine D4 receptors (D4Rs) are found with relatively low level of expression in the amygdala, hippocampus, hypothalamus, cortex and, in the substantia nigra (25–28, 30–34).
D2Rs as Key Targets for the Therapeutic Action of Aps
In vitro Affinity and Selectivity of Antipsychotics for Dopamine Receptors of D2R-Subtype
First and second-generation antipsychotics possess diverse structural, pharmacological (in vitro receptor profile, functional activity, e.g., antagonism, partial agonism, inverse agonism) and behavioral effects and side-effect profiles. However, their common property is that all display medium-to-high affinity for dopamine receptors of D2R-subtype (i.e., D2R, D3R, and/or D4R) under in vitro conditions (18, 35–40). The in vitro affinities of currently used antipsychotics for dopamine D2R-like (i.e., D2R, D3R, and D4R subtypes) and their degree of D3R selectivity are summarized in Table 1.
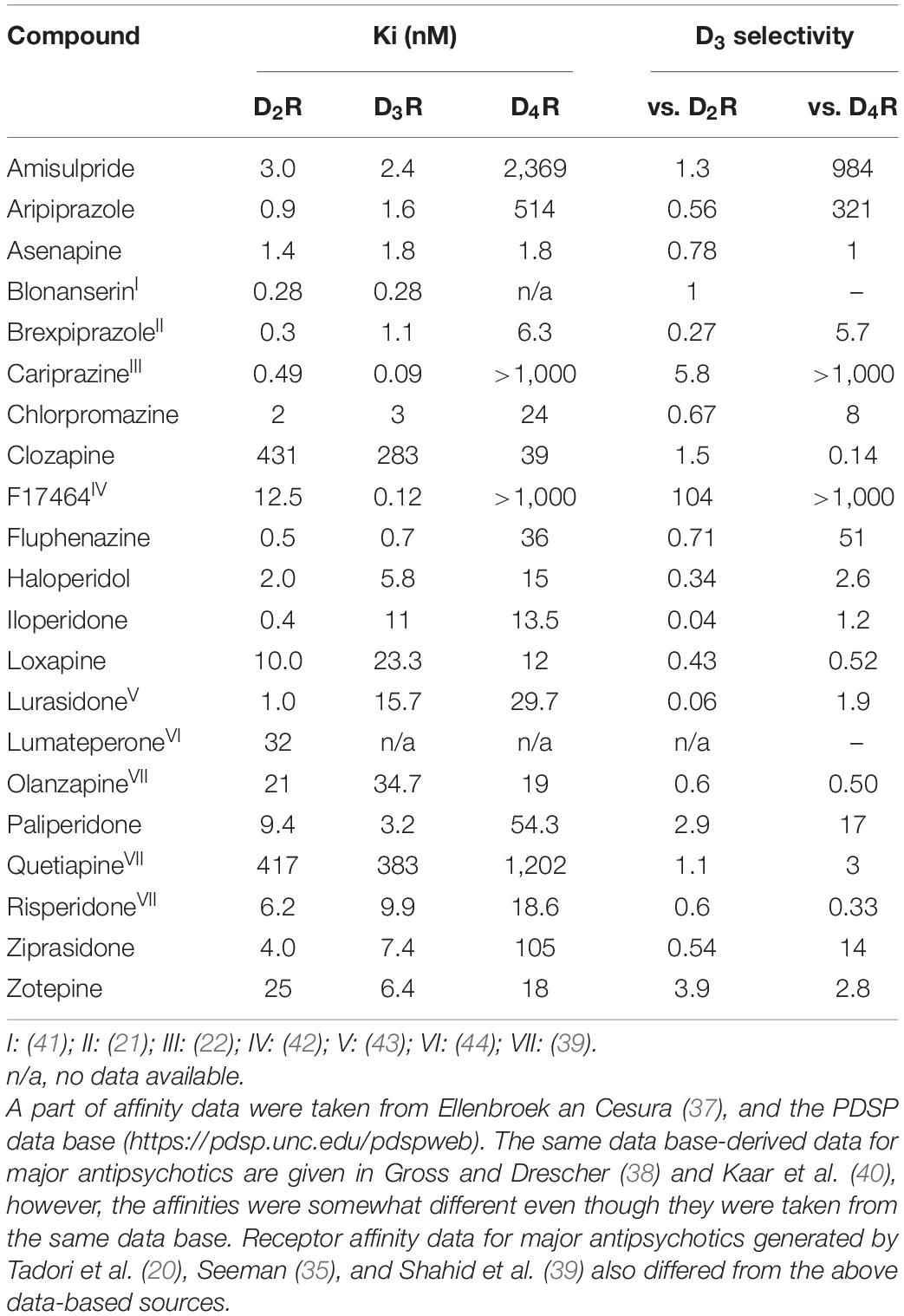
Table 1. In vitro affinity of major first-, second-, and third-generation antipsychotics at human dopamine receptors of D2R-type and their degree of their D3R selectivity.
Daily Dose and Plasma Levels of Antipsychotics Correlates With Their in vitro Affinity for Dopamine D2Rs
Seeman demonstrated a close correlation between the therapeutic doses of antipsychotics and their in vitro D2R receptor affinity, but no correlation was found with D1R affinity (45, 46). Correlation between D2R affinities, optimal occupancy of brain D2R for antipsychotic efficacy (i.e., 60–70%) and the free plasma levels of antipsychotics were also demonstrated (47).
Antipsychotics Occupy D2Rs in Brain
At present, it is broadly accepted that D2R affinity is the primary mechanism for antipsychotic efficacy (18, 36, 48, 49). Positron emission tomography (PET) studies demonstrated that for the clinical efficacy of D2R antagonist antipsychotics, a 60–75% occupancy of brain D2R is essential (50). In case of partial agonist antipsychotics, such as aripiprazole or cariprazine D2R occupancy can be as high as 95% at dose levels with established clinical efficacy (51–53), whereas brexpiprazole produced only 80% occupancy at the highest dose applied (54).
At present, despite the great efforts to develop non-dopamine antipsychotics, no such compounds are approved for the treatment of positive, negative, or cognitive symptoms of schizophrenia (55).
D3R, a Potential Novel Target in the Therapy of Central Nervous System Disorders: Comparison With D2R
Similarities and Differences of D2Rs and D3Rs
Structural
The D3R is a member of the largest phylogenetic class of GPCRs, known as class A, which contains the transmembrane domain without a large extracellular domain. Native ligands of aminergic GPCRs bind directly to the transmembrane domain, which is composed of seven transmembrane (TM) helices embedded in the cell membrane connected by three extracellular (EL) and three intracellular (IL) loops (56). The C-terminus of the protein is the eighth small α-helix (H8).
Analysis of amino acid sequence of human and rat dopamine D2R and D3R exhibits a high level of general sequence identity which is increased in the transmembrane helices forming a highly conserved orthosteric binding site (OBS) (see Figures 1A–C). The most obvious differences in the sequences can be found in the intracellular loop region (ICL3) between transmembrane helices of the TM5 and TM6. However, this region is quite distant from the orthosteric binding site, and thus the differences in the ECL2 (between the TM4 and TM5) and in the secondary binding site (SBS) are more relevant for the discovery of selective D3R vs. D2R ligands (57, 58). Moreover, targeting SBS may be a tool for fine tuning functional activity and biased agonism (59, 60). The shape and the sequence of the ECL2 is highly different in D2R and D3R (see Figure 1D). The SBS is the most probable binding site for the tail group of several elongated D3R ligands, where for instance the amino acid at the position 1.39 [Ballesteros-Weinstein numbering; (61)] is leucine in the D2R and tyrosine in the D3R. The amino acids forming the OBS are identical, but comparison of D2R and D3R structures suggest a slightly different shape of OBS because of the slightly different TM6 orientation (62).
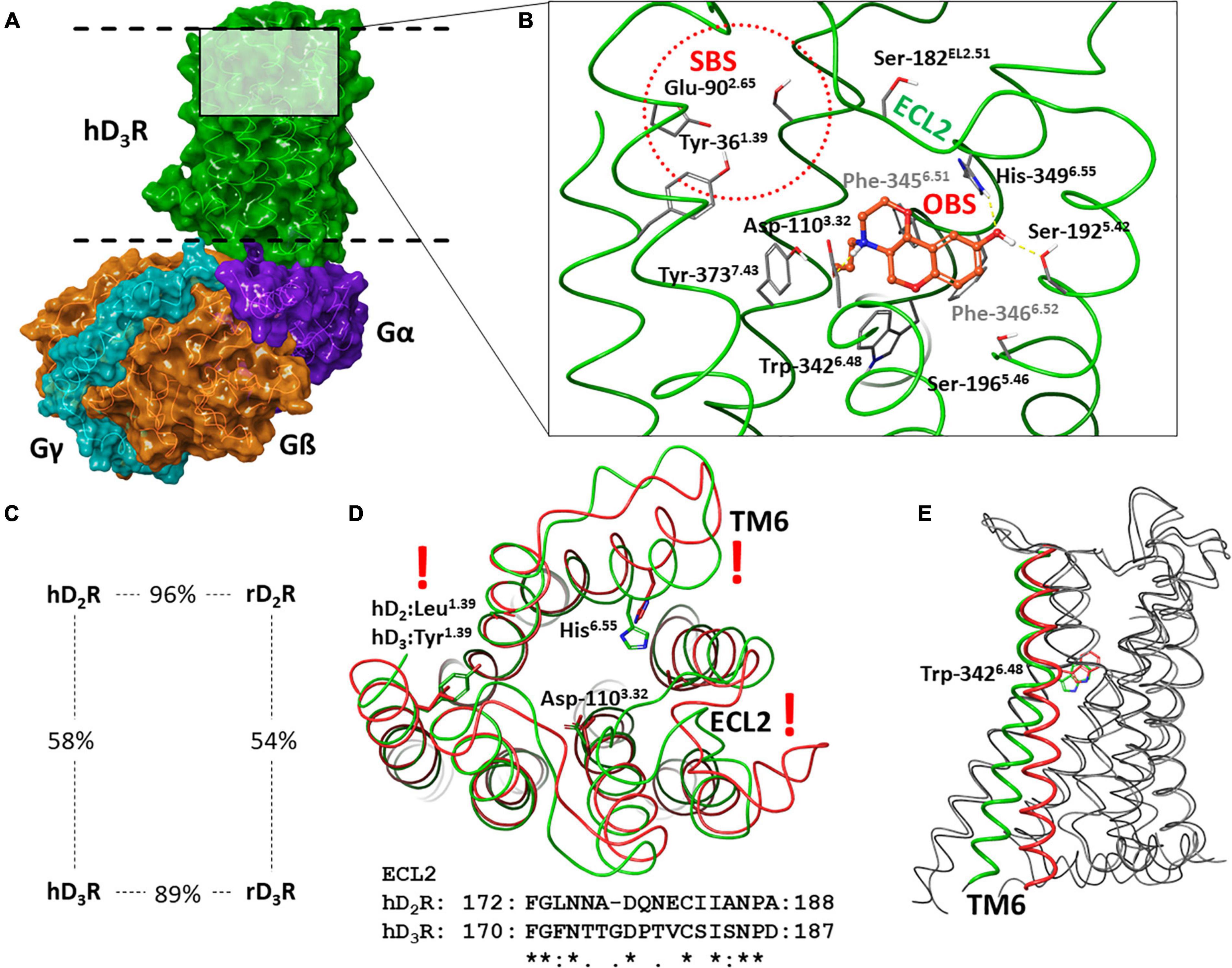
Figure 1. Structure of hD3R: (A) hD3R-Gi complex; (B) binding of the selective agonist PD128907 in the orthosteric binding site (Secondary binding site and ECL2 are also depicted); (C) percent identity of human and rat D2R and D3R sequences; (D) comparison of hD2R (red) and hD3R (green); (E) comparison of active (green) and inactive (red) conformation of hD3R*.
Recently published experimental structures of D2R and D3R (62–68) provide extensive information sources on ligand binding and receptor function. Like other GPCRs, the most conspicuous change during activation is the movement of the TM6, which enables the G-protein to connect to the receptor (see Figure 1E). The Trp in the position 6.48 may have a key role in the activation since it is close to the OBS and its position is related to the TM6 orientation (62).
Intracellular Signaling Pathways
All dopamine receptors belong to GPCR family: D1R and D5R receptors (D1-receptors family) stimulate cAMP signaling pathway through Gαs G-proteins whereas D2R, D3R, and D4R (D2R family) inhibit this pathway through Gαi/o G-proteins. There exists cAMP- independent pathways such as the recently recognized ß-arrestin pathway which is thought to be involved in several physiological functions and drugs’ effects (25–29).
Upon activation, both isoforms of D2R (i.e., D2Short and D2Long) and D3R inhibit the enzyme adenylyl cyclase (AC) through Gαi/o subtype of G-protein leading to inhibition of cAMP-PKA-pDARPP32-PPI pathway. However, differences may exist in the coupling efficiency of the two receptors and AC (or its subtypes).
In different cell lines, both D2R and D3R can activate ERK/MAPK signaling albeit with different mechanisms: D2Rs are coupled to and activate through α-subunit of Gi/o protein following agonist stimulation whereas, D3R functions through Go or Gß subunit depending on the signaling machinery of the given cell line. Both D2Rs and D3Rs are positively coupled to ß-arrestin-Akt-GSK3 pathway. GSK3ß is expressed in several brain regions and plays important role in neuronal development, neurovegetative and psychiatric diseases such as schizophrenia or bipolar disorder (26, 29, 70–79).
In vitro Pharmacological Profile of Dopaminergic Agents at D3Rs vs. D2Rs
It has been demonstrated that significant correlation exists between the in vitro affinities of various dopaminergic agents (agonists, antagonists, partial agonists) for D2Rs and D3Rs (80) (Figure 2).
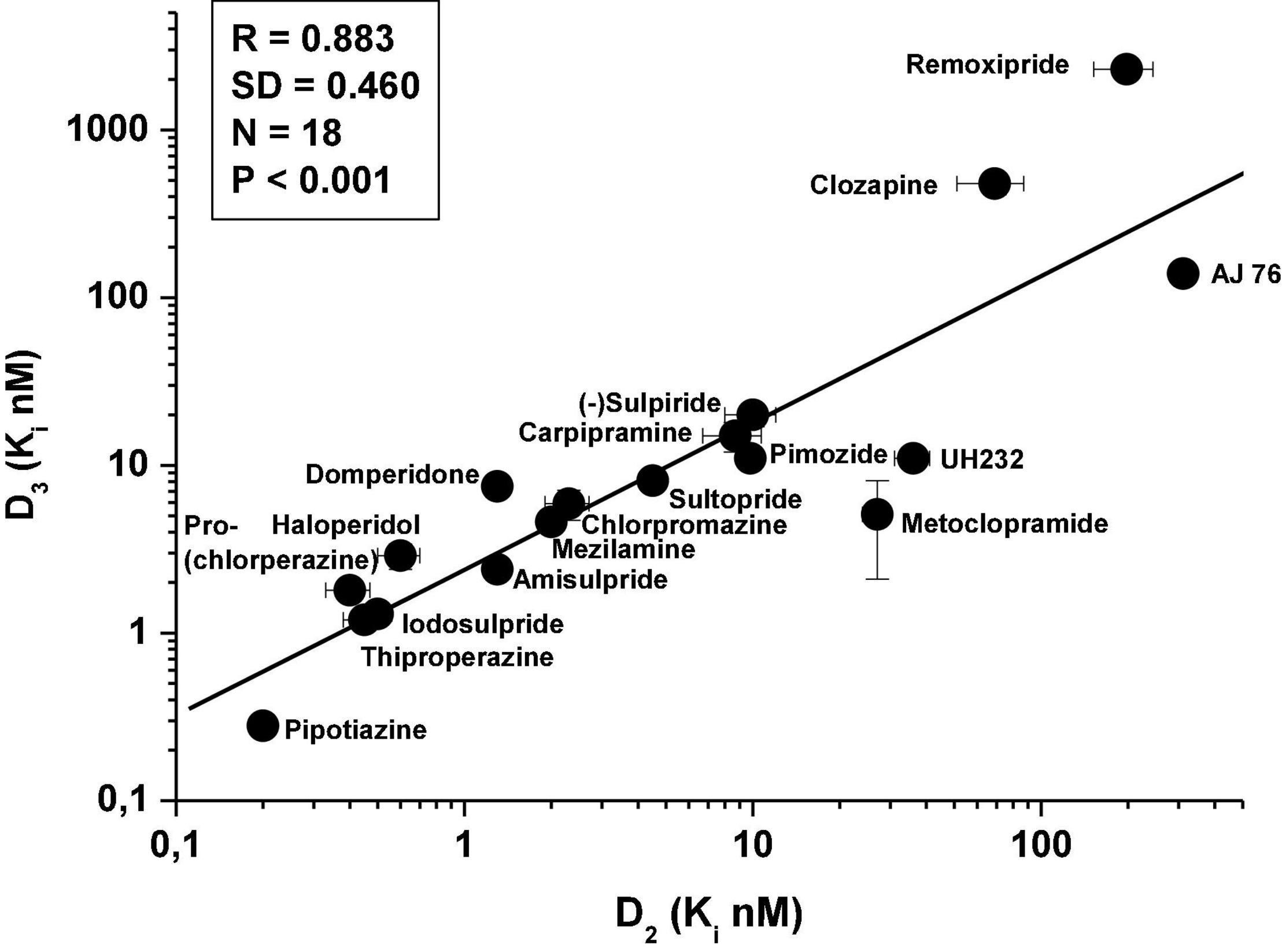
Figure 2. Correlation between in vitro affinity of various dopaminergic antagonists for human recombinant D3Rs and D2Rs [data taken from Sokoloff et al. (80)]; ligand: [125I]sulpiride.
Further results, using additional compounds, have confirmed earlier evidence showing close correlation between affinities of antipsychotics for human recombinant D2Rs and D3Rs (Figure 3A). However, no such correlation was found between D1R vs. D3R or D3R vs. D4R (data not shown). Similarly, high level of correlation was found between the affinity of antipsychotics for the rat D2R and D3Rs using [3H](+)-PHNO radioligand (81, 82) (Figure 3B).
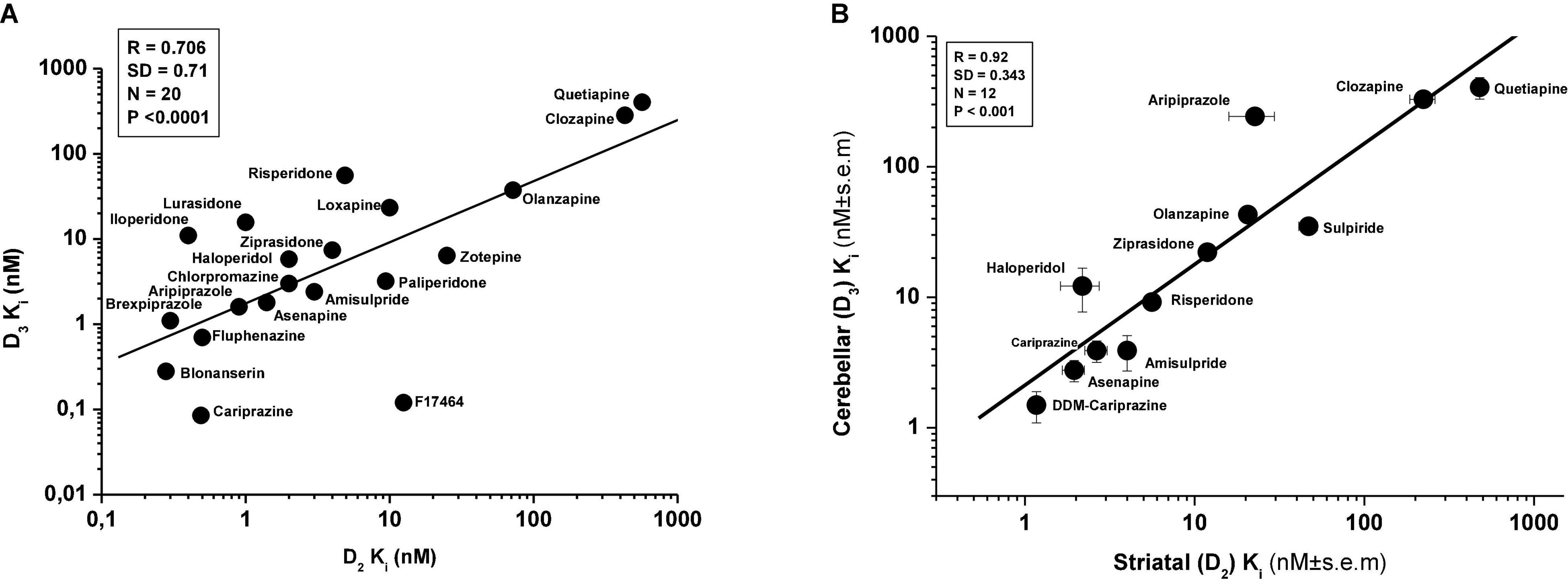
Figure 3. Correlation between in vitro affinity of antipsychotics for human D2Rs and D3Rs (A, data taken from Table 1; various radioligands) and for rat striatal D2Rs (striatal membrane) and cerebellar D3Rs (CB L9,10 membrane) (B); rat D2R and D3R affinity data derive from the extension of the study of Kiss et al. (81) and Kiss et al. (82). Determination of D2R and D3R affinity in membranes from CHO cells expressing human D3R or cerebellar L9,10 membranes (ligand: [3H]-(+)-PHNO) is described in Kiss et al. (81).
Based on recognition that D3Rs are mainly expressed in the limbic system (vide supra), the region is involved in schizophrenia pathology, and that significant correlation existed between the affinity of antipsychotics for D2Rs and D3Rs, it was thought that D3R affinity may play a role in the therapeutic efficacy of antipsychotics and led to propose development of selective D3R antagonists as novel antipsychotics (30, 80, 83–85).
Predicted Binding Mode of Antipsychotics in the D3R
One of the available experimental structure studies of D3R has been carried out with the antagonist eticlopride (63), and the other two with the agonists, pramipexole and PD128907 (62). All these agents bind to the orthosteric binding site (see Figure 1B). The most important interactions are the salt bridge with the Asp-1103.32 as well as the π-π interactions with the aromatic residues (e.g., Trp-3426.48, Phe-3456.51, Phe-3466.52), which form a lipophilic cavity. Hydrogen bond interaction with the serines in the 5.42 and 5.46 positions is typical for agonist binding state in D3R (62), and also in D2R structures (64, 65).
Non-selective ligands most probably bind to both the D2R and D3R in the same binding mode, forming a very similar interaction pattern. Thus, the D2R structural binding results obtained for non-selective D2R/D3R antagonists, such as risperidone, haloperidol or spiperone can be predictive of their binding mode at the D3R. It should be noted that distinct inactive conformations of D3R exists, and ligands may have different preferences which lead to different functional behaviors of antagonists (antagonism vs. inverse agonism, sensitivity for sodium ions) (86). These results are in line with the well-known highly dynamic nature of the GPCRs (87).
Based on the available experimental structural information supplemented by computational investigations (60, 88–90) the binding mode of antipsychotics at the D3R can be predicted at a reliable manner. In order to illustrate this, we docked several selected ligands into the D3R structures available in the Protein Data Bank (PDB ID: 7CMV (62) for dopamine and 3PBL (63) for the others) using the Glide, induced-fit-docking and the protein-ligand complex refinement protocols implemented in the Schrödinger software package (Schrödinger Release 2020-2) (Figure 4).
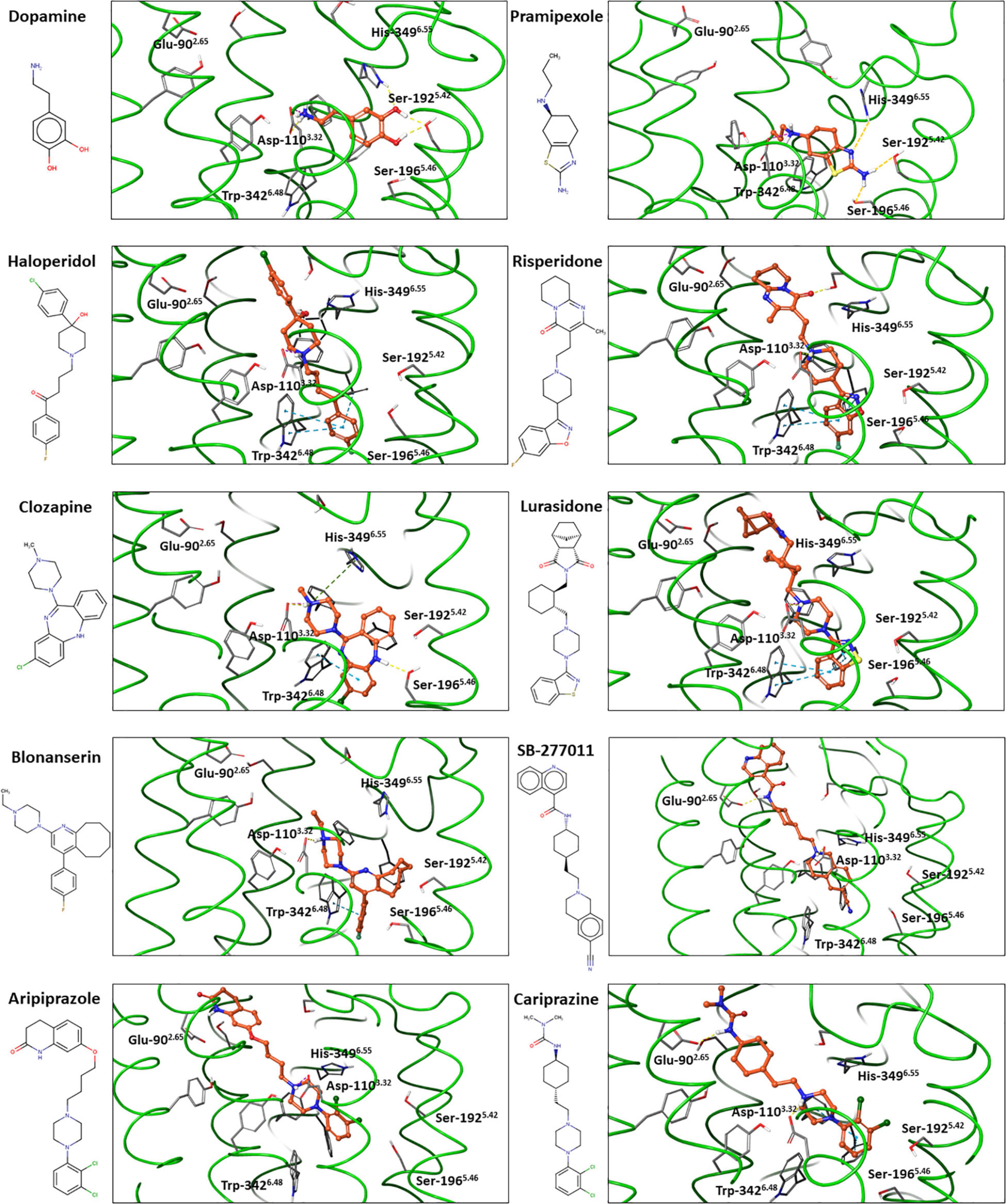
Figure 4. Dopamine, pramipexole and selected antipsychotics docked into experimental D3R structures*.
In vivo Roles and Behavioral Pharmacology of D3R and D2Rs Is Different
Despite the similarities in the in vitro properties of D3Rs and D2Rs described above, the in vivo roles and behavioral pharmacology of D3Rs compared to D2Rs are remarkably different. Animal data suggest opposite role of D2R vs. D3R in the control of locomotor activity, and cortical functions such as learning and memory (91, 92). On the other hand, both D2R and D3R receptor agonists were shown to impair certain social functions and cognitions (93–95). Enhanced expression of striatal dopamine D3R receptors impairs motivation (96). Antagonists of dopamine D2R receptors stimulate prolactin secretion (18), whereas D3R antagonism does not produce such effect either in rats or in human (97, 98). Majority of D2R antagonist antipsychotics (e.g., haloperidol, risperidone, and olanzapine) elicit catalepsy at higher doses (99). In contrast, D3R antagonists do not cause catalepsy (97), they rather inhibit haloperidol-induced catalepsy (100, 101).
Microdialysis studies demonstrated that D2R antagonist antipsychotics enhance, whereas selective D3R antagonists (such as SB-277011) (97, 102) or D3R-preferring D3R/D2R (such as S33138) antagonists (84, 103) exert no or minimal effects on cortical or striatal dopamine release (104).
Little is known on the functions of dopamine D3R receptors in humans although their involvement is assumed in central nervous system (CNS) diseases such as schizophrenia, Parkinson’s disease, addiction, anxiety, and depression or in the clinical effects of antipsychotics (26, 38, 70, 75).
Selective Agonists or Antagonists for D3R: the Challenge of Drug Research
The availability of drugs displaying high selectivity and affinity for D2R or D3R receptors are of great importance. Such compounds are useful tools in the exploration of neural mechanisms related to dopamine D3R receptors and may lead to novel agents for the treatment of various CNS disorders. Because of the close similarity in structure and signaling pathways of D2R and D3R, development of highly selective compounds for either subtype has been very challenging (34, 105).
Amongst agonists, the in vitro D3R affinity and selectivity of 7-OH-DPAT, PD128907 and pramipexole demonstrated great variability depending on the assay conditions used (105). Nevertheless, their degree of D3R vs. D2R selectivity seems adequate for use as tools for in vitro studies and their in vivo D3R selectivity may not be optimal, as they may also stimulate D2Rs within a narrow dose range (38, 106–109). For example, all three compounds produce biphasic behavioral effects in rats, some of which can be inhibited by either D3R and/or D2R selective antagonists, depending on the exposure levels of these agonists (95, 110–114).
The quest for high affinity, selective antagonists for D3R receptors (i.e., low-nanomolar Ki with D2R/D3R selectivity ≥100) began soon after the discovery of D3R. Several antagonists fulfilling the selectivity requirements such as SB-277011A (97), ABT-925 (115), GSK598809 (116), compound 74 in Micheli et al. (117) are currently available for experimental purposes. The pharmacological properties of the selective D3R antagonists have been reviewed by Gross et al. (84). L-741626 seems to be relatively selective for D2R reaching 100-fold higher D2R affinity vs. D3R, depending on the assay system used (118).
Selective D3R Antagonists as Antipsychotics?
Compounds with relatively high selectivity for dopamine D3Rs such as SB-277011A (97), S33084 (119), ABT-925 (115, 120), GSK598809 (116, 117), or the D3R-preferring D3R/D2R antagonist S33138 (103), or the D3R-preferring partial agonist BP-897 (121) demonstrated antipsychotic-like properties in animal models, however none of them reached therapeutic application. The high affinity D3R -preferring antagonist F17464 with partial agonism at serotonin 5-HT1AR and antagonism at dopamine D2R (42) showed promising preclinical profile as well as clinical efficacy in schizophrenia patients in a Phase II study. This compound is still under development and (122, 123). Propose the development of selective D3R antagonist for the treatment of negative symptoms of schizophrenia based on the available scientific evidence (84).
Imaging the D3Rs In Vivo
PHNO for Labeling D3Rs
Number of tracers have been tried to develop for selective imaging of D3Rs in the brain (124–126), however, the only radioligand currently available for labeling of D3R in occupancy studies suitable for separation of D3R and D2R signal is the [3H]- or [11C]-labeled (+)-4-propyl-9-hydroxynaphthoxazine [(+)-PHNO, naxagolide]. (+)-PHNO was originally described as an orally acting, potent dopamine receptor full agonist (127). (+)-PHNO was shown to possess 50-fold selectivity for human recombinant D3R (Ki: 0.16 nM) vs. D2R (Ki: 8.5 nM) (128).
[11C]-(+)-PHNO was synthesized by Wilson et al. (129) and it was shown that, in contrast with the antagonists such as [11C]raclopride, [18F]Fallypride, [11C]FLB-457 or the agonist [11C]N-methyl-norapomorphine, (+)-PHNO highly binds to regions rich in D3Rs. Using selective compounds such as the D3R antagonists SB-277011A, GSK598809 or the D2R antagonist SV-156, [11C]-PHNO proved to be useful for the separation of D3R and D2R binding signal and quantification of D3Rs in the brain, thus becoming an important tool for the investigation of the in vivo D3R occupancy by antipsychotics (116, 129–136).
D3R Occupancy of Antipsychotics—Animal Studies With [3H](+)PHNO
It was reported that after intravenous administration of [3H](+)-PHNO, D3Rs are labeled in the rat cerebellum L9,10 and D2R in the striatum. This is based on the finding that the selective D3R antagonist, SB-277011 inhibited [3H](+)-PHNO binding in CB L9,10 membranes but not in the striatum whereas, the opposite profile was obtained with the D2R selective antagonist, SV-156 (118) (compound 9); (81).
Using the above approach, olanzapine, risperidone, haloperidol, and clozapine given acutely or chronically, at doses corresponding to human doses, showed nearly full occupancy in the striatum and NAC (D2R rich regions) with significantly lower level or no occupancy in VP, ICj and substantia nigra (SN) (D3R rich regions). In contrast, in the in vitro autoradiography experiments all these antipsychotics inhibited [3H]-(+)-PHNO binding in the above regions except CB L9,10. It was concluded that under in vivo conditions the above-mentioned antipsychotics occupy dopamine D2R but not D3Rs despite their significant affinity for D3Rs in vitro (137, 138).
We extended this approach and compared the in vitro affinity of several dopamine D2R/D3R agonists, partial agonists, and antipsychotics using membranes prepared from rat striatum (D2R-rich) and cerebellar L9,10 region (D3R rich) to determine their in vivo D3R and D2R occupancy. The affinity data are given in Kiss et al. (82). We also compared the effects of systemic administration of selected full agonists, partial agonists and antipsychotics on the in vivo binding/uptake of intravenously given [3H](+)-PHNO binding/uptake in the rat striatum and cerebellar L,910 regions. The results are summarized in Table 2. Among the drugs with subnanomolar or low nanomolar Ki values for D3R, full agonists pramipexole and PHNO potently inhibited [3H](+)-PHNO binding of CB L,910 membranes with marked preference toward CB L9,10 D3Rs. Cariprazine, didesmethyl-cariprazine (DDCAR), asenapine, raclopride and amisulpride, produced dose-dependent inhibition of [3H](+)-PHNO binding/uptake both in the striatal and CB L9,10 regions. Raclopride and asenapine, however demonstrated high striatal vs. cerebellar selectivity (82). The antipsychotics, aripiprazole, olanzapine, risperidone, quetiapine, ziprasidone (all with high nanomolar Ki values) produced inhibition of [3H](+)-PHNO binding/uptake in the striatum and little or modest level of inhibition in the CB L9,10.
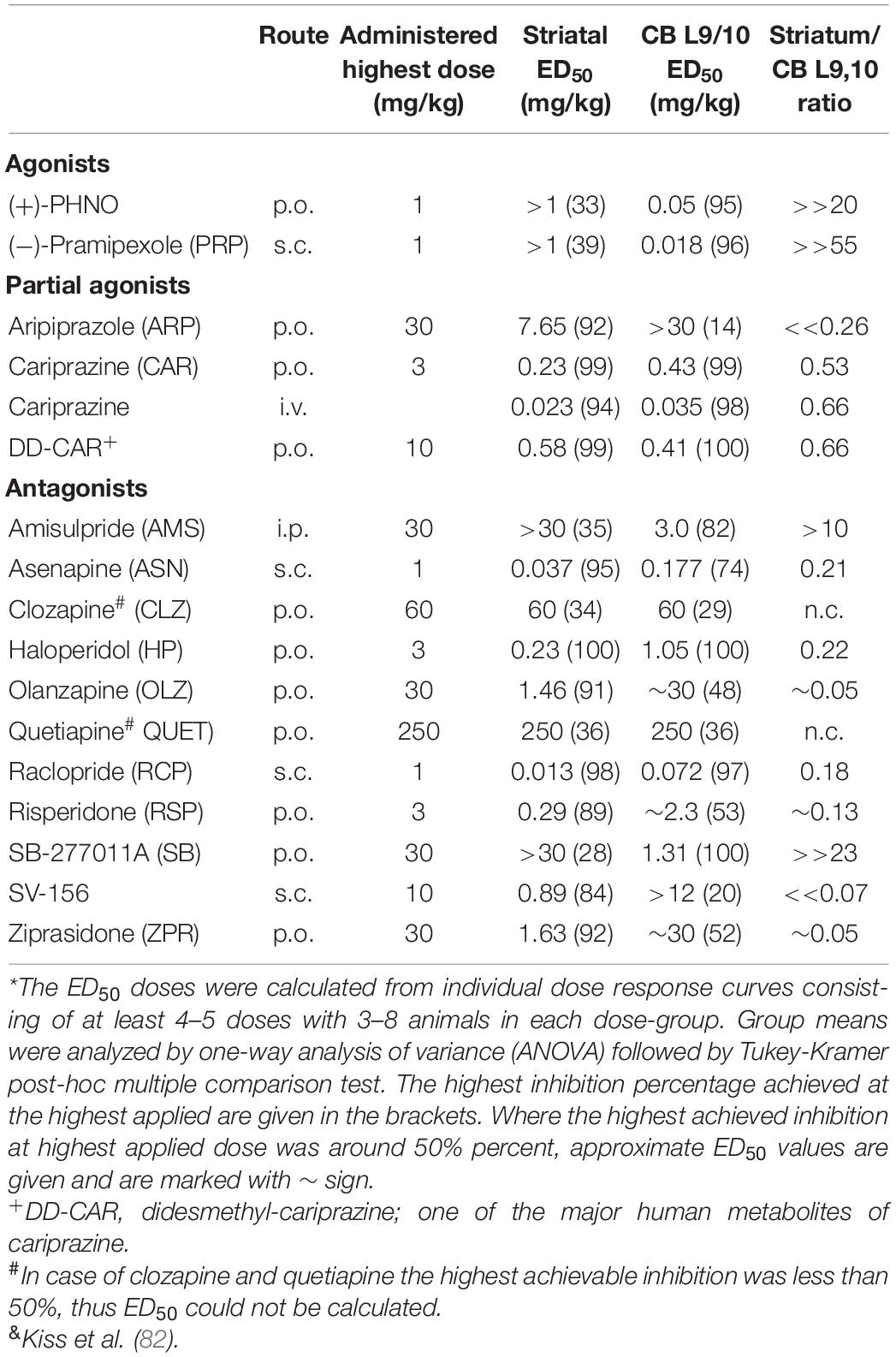
Table 2. Effects of selected antipsychotics, D3R agonists, antagonists, on the [3H](+)-PHNO uptake in rat striatum and cerebellum L9,10 region*,&.
Blonanserin, an antipsychotic marketed in Japan, was originally described as D2R and serotonin 5-HT2R antagonist (139). It has recently been found that blonanserin displayed high affinity in vitro for human D2R and D3Rs (Ki: 0.28 nM). Using the in vivo [3H](+)-PHNO method it caused dose-dependent, high occupancy of striatal D2R and D3R in the rat CB L9,10. In agreement with our data (see above) risperidone, olanzapine and aripiprazole demonstrated high occupancy only in the striatum and moderate or no occupancy was noted in the CB L9,10 region (41).
D3R Occupancy of Antipsychotics—Human PET Studies
In patients suffering from schizophrenia, occupancy of D2Rs and D3Rs following long-term treatment with risperidone, clozapine or olanzapine was examined using [11C]raclopride or [11C](+)-PHNO PET. This study demonstrated that these antipsychotics caused high D2R occupancy in the D2R-rich dorsal striatum, using either [11C]raclopride or [11C](+)-PHNO. However, they failed to show binding signal in the D3R-rich globus pallidus using [11C](+)-PHNO (140). Similar results with [11C](+)-PHNO PET were reported by Mizrahi et al. demonstrating that in drug-naive, first episode schizophrenia patients, olanzapine and risperidone resulted in high occupancy in the D2R-rich regions but not in the globus pallidus where even “negative occupancy” was noted (141). On the other hand, blonanserin, (hD2R Ki: 0.284 nM; hD3R Ki: 0.277 nM), in agreement with data obtained in rats, achieved significant D3R occupancy in healthy volunteers (142).
PET studies in healthy volunteers using [11C]raclopride (51) as well as in patients with schizophrenia using [18F]Fallypride (52) aripiprazole with D2R preference showed dose-dependent occupancy in the D2R-rich striatum without causing extrapyramidal side effects. A subsequent study with D3R preferring PET ligand, [11C](+)-PHNO confirmed the D2R occupancy of aripiprazole however, minor levels of D3R occupancy was detected (143).
Cariprazine, a D3R preferring D3R/D2R partial agonist antipsychotic (hD2R Ki: 0.49 nM; hD3R Ki: 0.09 nM) (22) dose-dependently inhibited [11C](+)-PHNO binding in brain regions with varying D2R and D3R expression. It showed significant occupancy of both D2R and D3R, albeit with approximately 3–6-fold selectivity for D3R (53, 143).
Brexpiprazole is also a partial agonist antipsychotic with D2R preference (hD2R Ki: 0.3 nM; D3R Ki: 1.1 nM) (21). Occupancy study in healthy volunteers showed that in the therapeutic dose range (1 and 4 mg/d) it produced only very low levels (i.e., 2–13%) of D3R occupancy whereas it achieved 36 and 59% D2R occupancy, respectively, in the applied dose range (54).
F17464 with remarkable affinity for D3Rs (D3R Ki: 0.16 nM; D2R Ki: 12 nM) demonstrated antipsychotic-like activity in animal experiments (42, 144). It was reported that in a double blind, multicenter Phase II study, F17464 (20 mg/bd) improved schizophrenia symptoms (122). In a phase I study, F17464 resulted in 69–95% occupancy of D3Rs whereas only a 20% occupancy of D2Rs were noted (145).
Potential Explanation for Why Significant In Vitro Affinity May Not Guarantee Substantial D3 Occupancy In Vivo
Role of Endogenous Dopamine
Affinity of Dopamine for D3R
The dopamine displays considerably higher in vitro affinities for D3Rs (Ki values vary from 30 nM to 100 nM) compared with D2R (Ki values vary from 200 nM to 1000 nM)1 ; (70). The in vitro Ki values greatly depend on several in vitro binding conditions such as the receptor source, radioligands used for binding assays, and assay methodology.
As to the dopamine Ki values for D3Rs the picture is further complicated since like D2R, D3R may also exist in low and high affinity state. Sokoloff et al. did not find differences between affinity of dopamine for D3R in the absence or presence of Gpp(NH)p (24 vs. 27 nM) (30, 80). However, Gross and Drescher (38) and Seeman et al. (146) reported remarkable difference between the low and high affinity states of D3R. D3Rs are prone to dimerization and to form heteromers with D1Rs or D2Rs, or with non-dopaminergic receptors (147). Affinity of dopamine (and the signalization pathway) as well as that of other dopaminergics (including antipsychotics) toward D3R di- or heteromers may also change.
Endogenous Dopamine Concentrations
As determined by in vivo microdialysis in rodents, under physiological conditions the extracellular, (i.e., resting or steady state) dopamine concentrations are in the low nanomolar range in various brain regions, including n. accumbens (∼1.5–4.5 nM), striatum (∼2–5 nM), hippocampus (∼1 nM) and in subnanomolar range in the prefrontal cortex (∼0.3–0.6 nM) (104, 148–154).
Little is known about the endogenous dopamine concentration in the human brain. Using [11C]-(+)-PHNO PET Caravaggio et al. estimated that the Kd (dissociation constant) of dopamine is 22–24 nM and they reported that concentration of dopamine is between 8 and 9 nM in the ventral striatum, caudate and putamen and 2.8 nM in the globus pallidus (155).
Endogenous Dopamine May Compete With Antipsychotics for Occupying D3Rs
Using ex vivo autoradiography Schotte et al. (156) demonstrated that endogenous dopamine had greater ability to occupy D3Rs as compared to D2Rs and concluded that D3Rs are preferably occupied by endogenous dopamine which “limits the binding of antipsychotic drugs to D3 receptors in the rat brain.”
The alkylating agent, EEDQ (1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline) concentration dependently reduced the in vitro [3H]7-OH-DPAT and [3H]spiperone binding in membranes from rat ventral striatum. In vivo treatment of rats with EEDQ resulted in reduction of the ex vivo [3H]spiperone binding in striatal membranes but did not alter [3H]7-OH-DPAT binding in membranes from ventral striatum. The author concluded [3H]7-OH-DPAT binding sites (i.e., mostly D3R) seem to be resistant to EEDQ-induced inactivation in vivo sites (157).
In agreement with these results, Zang et al. using autoradiography, demonstrated that treatment of rats with EEDQ or NIPS (N- p-iso-thiocyanatophenethyl-spiperone) did not alkylate D3Rs receptors in n. accumbens and in the island of Calleja at doses that resulted in blockade of D2Rs receptors in caudate and n. accumbens. On the other hand, under in vitro conditions when slices from the above regions were incubated with EEDQ or NIPS, both inhibited dopamine D2Rs as well as D3Rs and inhibition at D3R sites were prevented by dopamine in nanomolar concentration range whereas only millimolar concentration of dopamine was able to protect D2Rs. The authors concluded that their results “are consistent with the view that alkylation of D3 receptors in vivo is prevented by its high affinity for even minor concentrations of endogenous dopamine” (158).
Modulation of Extracellular Dopamine by D2R Antipsychotic Treatment
Microdialysis Studies
The partial agonists antipsychotics such as aripiprazole (159, 160), brexpiprazole (21) and cariprazine (153, 154) caused only moderate or no change of the extracellular dopamine concentration in the rat prefrontal cortex, hippocampus, n. accumbens and in the striatum. It is interesting to note that the high affinity D3R-preferring antagonist antipsychotic, F17464 (Ki for D3R: 0.16 nM; Ki for D2R: 12.1 nM) also did not significantly change extracellular dopamine concentration in the medial prefrontal cortex (42).
Both the typical antipsychotic haloperidol and the atypicals such as such as asenapine, blonanserin, clozapine, risperidone, olanzapine, lurasidone, and ziprasidone dose-dependently and remarkably (by 2- to 4-fold) elevated the extracellular dopamine concentrations in the rat prefrontal cortex, hippocampus, n. accumbens and in the striatum (104, 148–152, 161, 162). It should be mentioned that the above antipsychotics, beside their D2R affinity, display high affinity for adrenergic alpha, dopamine D3, D4, serotonin 5-HT2A, 5-HT1A, 5-HT2B, 5-HT6, and 5-HT7, muscarinic, and histaminergic receptors (37) which may influence the extracellular dopamine levels evoked via D2R antagonism. In fact, among atypical antipsychotics risperidone, asenapine, increased extracellular concentration of serotonin in the prefrontal cortex (151, 161), while olanzapine (162), lurasidone (152) blonanserin (137), and clozapine (138) resulted in modest or no effect. Olanzapine, blonanserin, asenapine and haloperidol significantly increased extracellular norepinephrine levels, as well (137, 148, 163).
D2R Antagonists Directly Inhibit Dopamine Transporter
Former studies showed that D2R antagonists can inhibit dopamine uptake via D2Rs (164). Amato et al. have recently proposed that beside D2R antagonism/occupancy, the direct blockade of DAT by antipsychotics, i.e., the modulation of extracellular dopamine, is a likely important factor in the antipsychotic efficacy (165–167).
The involvement of D3Rs in the regulation of DAT or the effects of antipsychotics via D3Rs on the DAT is much less known. Zapata et al. found that D3R upregulate DAT (168), whereas Luis-Ravelo et al. demonstrated that the regulation appears to be biphasic, i.e., acute D3R activation increased DAT expression whereas prolonged activations reduced dopamine uptake (169).
Turnover Studies
Early studies found greatly increased dopamine turnover rate in the rat or cat brain after antipsychotic treatment (170–172).
We compared the effects of selected antipsychotics, D3R or D2R antagonists and D3R preferring dopamine agonists on the dopamine turnover index in the rat striatum (and in olfactory tubercle and n. accumbens, data not shown) with D3R occupancy ED50 doses (i.e., doses causing 50% inhibition of [3H](+)PHNO uptake/occupancy, Table 2) in the striatum and in CB L9,10.
At cerebellar (i.e., CB L9,10 D3R) occupancy ED50 doses, the agonists (+)-PHNO and (-)-pramipexole reduced the striatal dopamine turnover index by about 50%, whereas antipsychotics such as asenapine, haloperidol, olanzapine, risperidone, and ziprasidone and the D2R preferring antagonist SV-156 greatly enhanced (by about 3–4-fold) striatal dopamine turnover index (Figure 5A). Blonanserin was not involved in this study, but it is reported that it caused 3–4-fold increase of striatal, frontal and limbic (i.e., olfactory tubercle and n. accumbens) DOPAC and HVA, which are all clearly indicate turnover increasing effect of blonanserin (139). The partial agonist cariprazine, the cariprazine metabolite, DD-CAR did not significantly change the striatal dopamine turnover index as was noted with amisulpride and the D3R antagonist SB-277011A. Interestingly enough, the D2R partial agonist aripiprazole produced effects more like to those seen with D2R antagonist antipsychotics.
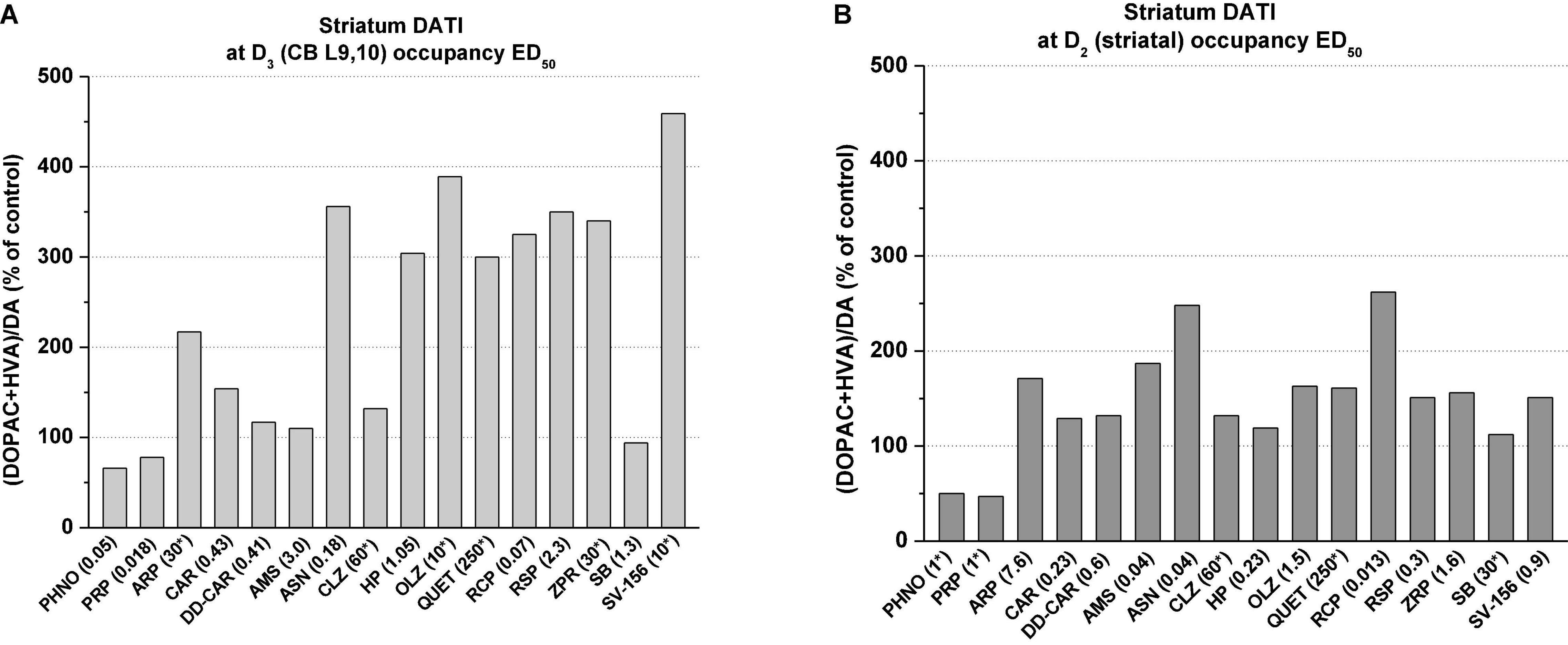
Figure 5. Graphical presentation of percent changes in striatal dopamine turnover indices (DATI) at D3R (i.e., cerebellar L9, 10) occupancy ED50 doses (A) and at D2R (i.e., striatal) occupancy ED50 doses (B) in vivo*,#. *Doses in brackets denote the occupancy ED50 doses (or close to ED50) taken from Table 2. Dopamine turnover index (DATI) was estimated from turnover dose-response curves (consisting of at least 4–5 doses, with five rats in each dose-group) for individual compounds listed in this figure. In cases where the occupancy ED50 values could not be exactly calculated (see Table 2) the turnover indices were determined at doses denoted with asterisks. Dopamine turnover index was defined as DA/(DOPAC+HVA). Determination of tissue dopamine, DOPAC and HVA was carried out exactly as described in Kiss et al. (22) (for abbreviations of drugs’ names, see Table 2). #Dopamine turnover data for cariprazine and DD-CAR were published in Kiss et al. (173). Turnover results of other compounds are unpublished and are on file at G. Richter. Plc.
On the other hand, at the D2R occupancy ED50 doses (i.e., doses causing 50% inhibition of striatal [3H](+)-PHNO uptake) which were much lower than that of necessary for 50% occupancy of CB L9, 10 D3Rs, all antipsychotics (i.e., asenapine, haloperidol, olanzapine, risperidone, and ziprasidone and the D2R preferring antagonist SV-156) caused much less increase in dopamine turnover index (Figure 5B). The effects of the partial agonist cariprazine, DD-CAR and the SB-277011A, at their D2R occupancy doses, produced modest turnover changes in the striatum as was seen at their D3R ED50 occupancy doses.
The results of dopamine turnover studies, in agreement with microdialysis results, indicate that D2R antagonist antipsychotics greatly increase the dynamics of dopamine metabolism including the increase of extracellular dopamine at doses sufficient to achieve occupancy of D3Rs. Opposite effects were seen with dopamine D3R-preferring agonists (-)-pramipexole and (+)-PHNO (which is probably due to the D2R agonist effects manifested under in vivo conditions). At pharmacological doses, neither cariprazine nor its one of the major metabolite, DD-CAR did not seem to alter significant alteration in dopamine metabolism in rat striatum.
Affinity and/or Selectivity of Compounds for D3Rs in vitro vs. D3R Occupancy in vivo
In Table 3, a summary is given on the D2R and D3R affinity and selectivity of some D3R selective antagonist, agonists, and antipsychotics along with their D3R occupancy determined in rats or in human.
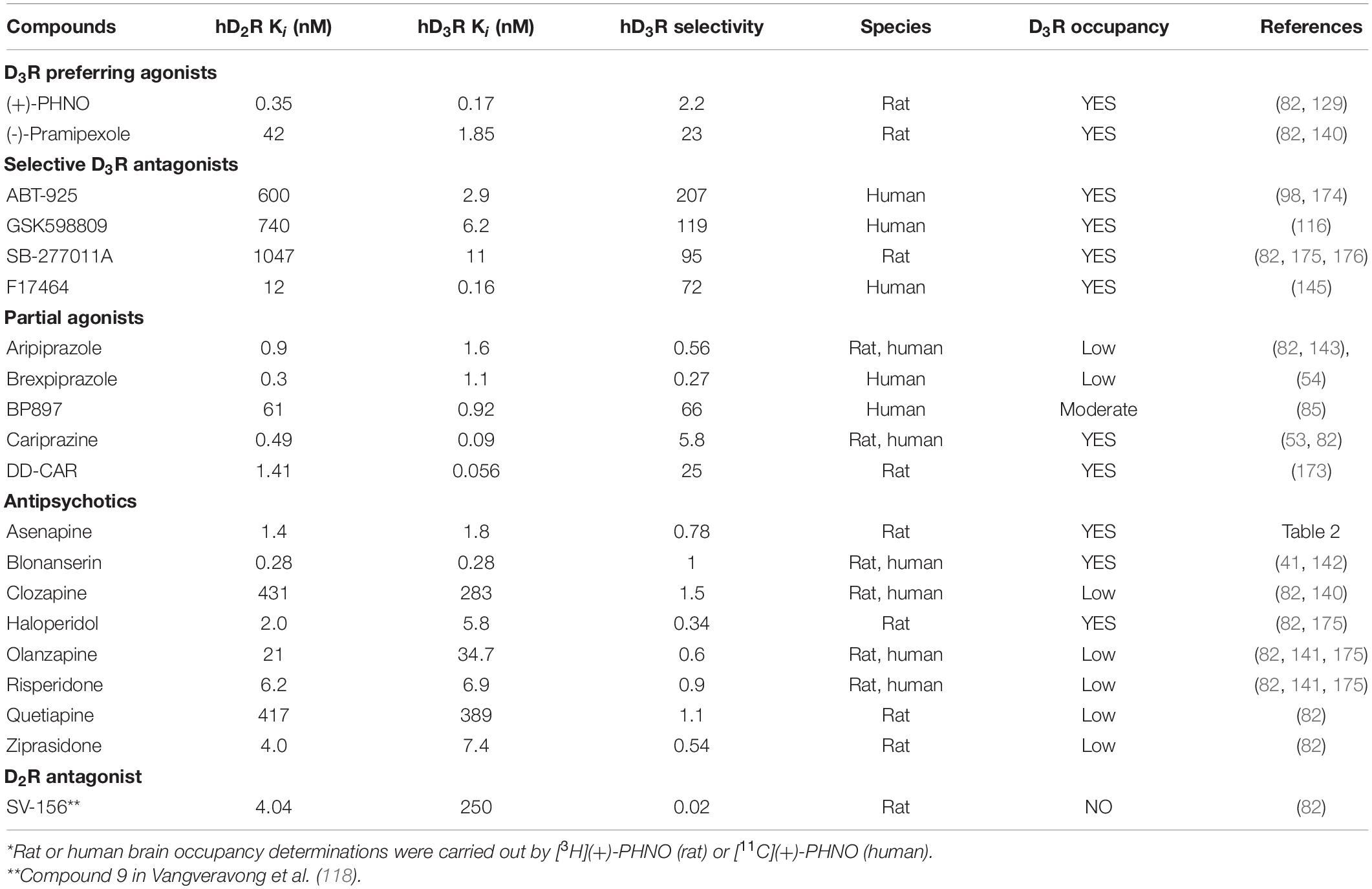
Table 3. Summary of in vitro human D3R affinity, D3R selectivity and occupancy* of some antipsychotics, partial agonists, highly preferring/selective D3R agonists, antagonists.
Based solely on the in vitro affinity data one may expect compounds with low-or sub-nanomolar affinities for both receptor subtypes, would show D2R as well as D3R occupancy in vivo. However, the preclinical and human occupancy studies summarized above do not necessarily support such a correlation.
Both D3R-preferring agonist, (+)-PHNO and pramipexole as well as the antagonists (ABT-925, GSK598890, SB-277011A and the antipsychotic candidate F17464) all display low- or sub-nanomolar D3R affinity and high selectivity for D3Rs in vitro. These compounds produced D3R occupancy in rat or human studies. The same (i.e., high D3R affinity, selectivity in vitro and high D3R occupancy) is applicable for the partial agonists, cariprazine and its metabolite, DD-CAR. Although aripiprazole and brexpiprazole displayed low nanomolar in vitro D3R affinity, their D3R selectivity was below 1, which could explain their lack of D3R occupancy in vivo. Among the currently used antipsychotics, only the D2R/D3R antagonist blonanserin, which has low- or sub-nanomolar affinity for these receptors has been shown to have significant in vivo occupancy for both receptors in rats. Second generation antipsychotics (i.e., risperidone, quetiapine, clozapine) with low D3R affinity (Ki: >3–10 nM) and selectivity resulted in negligible D3R occupancy.
Limitations
Our knowledge about the occupancy of D3Rs in the rat or human brain comes from the use of [3H](+)-PHNO or the [11C](+)-PHNO radiotracers. Their use represented a great advance in the in vivo imaging of D3Rs and determination of occupancy of brain D3Rs by antipsychotics.
[3H](+)-PHNO or the [11C](+)-PHNO however, are not ideal ligands/tracers for several reasons. They may not be sensitive enough for more detailed mapping of D3Rs in regions having low D3R expression e.g., cerebral cortex. Although both display higher affinity than dopamine for D3R, they are still sensitive to endogenous dopamine (155, 177).
Furthermore, both D2Rs and D3Rs may exist in high- or low-affinity states and they are prone to di- or heteromerization (147, 178, 179). It was reported that recombinant human or rat D3R, like D2R, may exist in low- and high-affinity state and the affinity of PHNO shows significant difference for these states (30, 81, 128, 146, 180) which may have implication in drugs’ imaging studies (140, 155).
These conditions (i.e., the high/low affinity state and di- or heteromerization, if they exist) may greatly change the affinity of the two receptors toward the agonist tracer and the affinity of drugs to be examined and their occupancy. Thus, the quest for better ligands (agonist or antagonist?) for the demonstration of brain D3Rs occupancy in vivo by therapeutically useful compounds (e.g., antipsychotics among others) continues (109, 125, 126, 177, 181).
Moreover, in contrast with the known therapeutically optimal occupancy of antipsychotics at D2Rs (i.e., 65–75%) there is no reliable information on the optimal level of D3R occupancy for manifestation of therapeutic effect.
Summary and Conclusion
All currently used antipsychotics display high-to-medium affinity for both D2R and D3Rs in vitro. In agreement with the in vitro D2R affinity they show significant D2R occupancy in the rat and human brain at their antipsychotic-effective doses. However, as revealed by animal and human occupancy studies, despite the considerable in vitro D3R affinity, not all antipsychotics demonstrated brain D3R occupancy in vivo.
There may exist several possibilities for this dichotomy, as outlined in the following:
First, dopamine displays much higher affinity for D3Rs than for D2Rs and thus endogenous dopamine might, at least partly, keeps D3Rs occupied even under basal conditions.
Second, animal microdialysis and turnover studies revealed that acute treatment with dopamine agonists such as (-)-pramipexole and (+)-PHNO reduced dopamine turnover, i.e., they decrease extracellular dopamine and increase D3R availability. Administration of antipsychotics (e.g., risperidone, olanzapine, haloperidol, ziprasidone, clozapine, quetiapine), due to antagonism of presynaptic and biosynthesis and release regulating D2Rs, leads to several-fold increase of extracellular dopamine. Further, Amato et al. demonstrated that antipsychotics initially suppress dopamine transporter (DAT) activity leading to increase of dopamine in synaptic cleft, a mechanism which represents a further possible alternative way to modulate extracellular dopamine (166). Thus, the increase of extracellular dopamine following antipsychotics with D2R antagonism seems to be a likely important factor in the lack or low levels of in vivo D3R occupancy; given the higher affinity of dopamine for D3R vs. D2R. Thus, D2R antagonist antipsychotics inhibit their own binding at D3Rs by increasing extracellular dopamine.
Third, beside the effects on the endogenous dopamine levels, the D3R affinity and selectivity appear to be further factors of importance. All three selective D3R antagonists (D3R vs. D2R selectivity ≥100) such as ABT-925, GSK595809 and SB-277011 (with the in vitro low nanomolar D3R) affinity produced high D3R occupancy in animal or human studies, indicating primary importance of selectivity to achieve D3R occupancy in vivo.
Example of antipsychotics such as the D3R/D2R partial agonist cariprazine and the D2R/D3R antagonist blonanserin shows that, in the presence of relatively high affinity for D2Rs, subnanomolar affinity for D3Rs appears to be necessary for D3R occupancy in vivo. Further, cariprazine and the F17464 (subnanomolar affinity for D3R with 75-fold D3R vs. D2R), do not increase extracellular dopamine and hence are able to compete for D3Rs vs. extracellular dopamine.
The case of D2R/D3R partial agonist antipsychotics, aripiprazole and brexpiprazole is somewhat controversial. Both demonstrated low nanomolar affinity for D2Rs and D3Rs (with D2R preference) in vitro, with negligible effects on extracellular dopamine in vivo. However, both produced no or very low occupancy of D3Rs for which the likely explanation is the D2R preference.
In conclusion, data reviewed and discussed here regarding the current antipsychotics’ in vitro D2R/D3R affinity vs. their brain D3R occupancy in vivo, indicate that levels of extracellular dopamine (or its change) in different brain regions is a key factor regarding D3R occupancy. On the other hand, the compounds’ high (i.e., subnanomolar) D3R affinity and/or high D3R vs. D2R selectivity are also important determining factors to achieve significant D3R occupancy in the brain.
Author Contributions
BKi and BKr drafted the manuscript with several inputs from IL. All authors were participating in the final editing and critical revision of the article and approved the final version to be published.
Conflict of Interest
BKi, BKr, and IL were employees of Gedeon Richter Plc.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Dr. Nika Adham of AbbVie Inc. for her critical reading, suggestions, and valuable comments on the manuscript.
Footnotes
References
1. Perälä J, Suvisaari J, Saarni SI, Kuoppasalmi K, Isometsä E, Pirkola S, et al. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch Gen Psychiatry. (2007) 64:19–28. doi: 10.1001/archpsyc.64.1.19
2. Millan MJ, Agid Y, Brüne M, Bullmore ET, Carter CS, Clayton NS, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. (2012) 11:141–68. doi: 10.1038/nrd3628
3. Howes OD, Murray RM. Schizophrenia: an integrated sociodevelopmental-cognitive model. Lancet. (2014) 383:1677–87. doi: 10.1016/s0140-6736(13)62036-x
4. Millan MJ, Fone K, Steckler T, Horan WP. Negative symptoms of schizophrenia: clinical characteristics, pathophysiological substrates, experimental models and prospects for improved treatment. Eur J Neuropsychopharmacol. (2014) 24:645–92. doi: 10.1016/j.euroneuro.2014.03.008
5. Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. (1991) 148:1474–86. doi: 10.1176/ajp.148.11.1474
6. Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. (1999) 6:56–72. doi: 10.1016/s0006-3223(99)00067-0
7. Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce J, Gil R, Kegeles LS, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA. (2000) 97:8104–9. doi: 10.1073/pnas.97.14.8104
8. Fusar-Poli P, Meyer-Lindenberg A. Striatal presynaptic dopamine in schizophrenia, part II: meta-analysis of [(18)F/(11)C]-DOPA PET studies. Schizophr Bull. (2013) 39:33–42. doi: 10.1093/schbul/sbr180
9. McCutcheon RA, Krystal JH, Howes OD. Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World Psychiatry. (2020) 19:15–33. doi: 10.1002/wps.20693
10. Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol. (2015) 29:97–115. doi: 10.1177/0269881114563634
11. Coyle JT, Ruzicka WB, Balu DT. Fifty years of research on schizophrenia: the ascendance of the glutamatergic synapse. Am J Psychiatry. (2020) 177:1119–28. doi: 10.1176/appi.ajp.2020.20101481
12. Delay J, Deniker P, Harl J. Traitment des etats d’excitation et d’agitation par une methode medicamentense derivee de l’hibernotherapie. Ann Med Psychol. (1952) 110:267–73.
13. Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol. (1963) 20:140–4. doi: 10.1111/j.1600-0773.1963.tb01730.x
14. van Rossum J. The significance of dopamine-receptor blockade for the action of neuroleptic drugs. In: H Brill, J Cole, P Deniker, H Hippies, P Bradley editors. Neuropsychopharmacology, Proceedings of the 5th Collegium Internationale Neuropsychopharmacologicum. Amsterdam: Excerpt Medica (1967). p. 321–9.
15. Seeman MV. History of the dopamine hypothesis of antipsychotic action. World J Psychiatry. (2021) 11:355–64. doi: 10.5498/wjp.v11.i7.355
16. Meltzer HY, Matsubara S, Lee J-C. Classification of typical and atypical antipsychotic drugs based on dopamine D-1, D-2 and serotonin2 pKi values. J Pharmacol Exp Ther. (1989) 251:238–46.
17. Gründer G, Hippius H, Carlsson A. The “atypicality” of antipsychotics: a concept re-examined and re-defined. Nat Rev Drug Discov. (2009) 8:197–202. doi: 10.1038/nrd2806
18. Stahl SM. Antipsychotic Agents in: Stahl’s Essential Psychopharmacology, Neuroscientific Basis and Practical Applications. 4th ed. New York: Cambridge University Press (2013). p. 129–237.
19. Lieberman JA, Stroup TS. The NIMH-CATIE schizophrenia study: what did we learn? Am J Psychiatry. (2011) 168:770–5. doi: 10.1176/appi.ajp.2011.11010039
20. Tadori Y, Forbes RA, McQuade RD, Kikuchi T. In vitro pharmacology of aripiprazole, its metabolite and experimental dopamine partial agonists at human dopamine D2 and D3 receptors. Eur J Pharmacol. (2011) 668:355–65. doi: 10.1016/j.ejphar.2011.07.020
21. Maeda K, Sugino H, Akazawa H, Amada N, Shimada J, Futamura T, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. (2014) 350:589–604. doi: 10.1124/jpet.114.213793
22. Kiss B, Horváth A, Némethy Z, Schmidt É, Laszlovszky I, Bugovics G, et al. Cariprazine (RGH-188), a dopamine D3 receptor-preferring, D3/D2 dopamine receptor antagonist–partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. (2010) 333:328–40. doi: 10.1124/jpet.109.160432
23. Citrome L. Aripiprazole, brexpiprazole, and cariprazine: not all the same. Understanding the key differences among these agents can help inform treatment decisions. Curr Psychiatry. (2018) 17:35–44.
24. Cookson J, Pimm J. Partial agonists of dopamine receptors: mechanisms and clinical effects of aripiprazole, brexpiprazole and cariprazine. BJPsych Adv. (2021):1–6. doi: 10.1192/bja.2021.49
25. Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. (1998) 78:189–225. doi: 10.1152/physrev.1998.78.1.189
26. Beaulieu J-M, Espinoza S, Gainetdinov RR. Dopamine receptors – IUPHAR Review 13. Brit J Pharmacol. (2015) 172:1–23. doi: 10.1111/bph.12906
27. Prieto GA. Abnormalities of dopamine D3 receptor signaling in the diseased brain. J Central Nervous Syst Dis. (2017) 9:117957351772633. doi: 10.1177/1179573517726335
28. Klein MO, Battagello DS, Cardoso AR, Hauser DN, Bittencourt JC, Correa RG. Dopamine: functions, signaling, and association with neurological diseases. Cell Mol Neurobiol. (2019) 39:31–59. doi: 10.1007/s10571-018-0632-3
29. Rangel-Barajas C, Coronel I, Florán B. Dopamine receptors and neurodegeneration. Aging Dis. (2015) 6:349. doi: 10.14336/ad.2015.0330
30. Sokoloff P, Giros B, Martres M-P, Bouthenet M-L, Schwartz J-C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. (1990) 347:146–51. doi: 10.1038/347146a0
31. Bouthenet M-L, Souil E, Martres M-P, Sokoloff P, Giros B, Schwartz J-C. Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: comparison with dopamine D2 receptor mRNA. Brain Res. (1991) 564:203–19. doi: 10.1016/0006-8993(91)91456-b
32. Meador-Woodruff J. Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology. (1996) 15:17–29. doi: 10.1016/0893-133x(95)00150-c
33. Gurevich E. Distribution of dopamine D3 receptor expressing neurons in the human forebrain comparison with D2 receptor expressing neurons. Neuropsychopharmacology. (1999) 20:60–80. doi: 10.1016/s0893-133x(98)00066-9
34. Moritz AE, Free RB, Sibley DR. Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cell Signal. (2018) 41:75–81. doi: 10.1016/j.cellsig.2017.07.003
35. Seeman P. Antipsychotic drugs, dopamine receptors, and schizophrenia. Clin Neurosci Res. (2001) 1:53–60. doi: 10.1016/s1566-2772(00)00007-4
36. Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Progr Neuro Psychopharmacol Biol Psychiatry. (2003) 27:1081–90. doi: 10.1016/j.pnpbp.2003.09.004
37. Ellenbroek BA, Cesura AM. Antipsychotics and the dopamine–serotonin connection. In: S Celanire, S Poli editors. Small Molecule Therapeutics for Schizophrenia. Topics in Medicinal Chemistry. (Vol. 13), Cham: Springer (2014). doi: 10.1007/7355_2014_51
38. Gross G, Drescher K. The role of dopamine D3 receptors in antipsychotic activity and cognitive functions. Handb Exp Pharmacol. (2012) 213:167–210. doi: 10.1007/978-3-642-25758-2_7
39. Shahid M, Walker G, Zorn S, Wong E. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol. (2008) 23:65–73. doi: 10.1177/0269881107082944
40. Kaar SJ, Natesan S, McCutcheon R, Howes OD. Antipsychotics: mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology. (2020) 172:107704. doi: 10.1016/j.neuropharm.2019.107704
41. Baba S, Enomoto T, Horisawa T, Hashimoto T, Ono M. Blonanserin extensively occupies rat dopamine D3 receptors at antipsychotic dose range. J Pharmacol Sci. (2015) 127:326–31. doi: 10.1016/j.jphs.2015.01.007
42. Cosi C, Martel J-C, Auclair AL, Collo G, Cavalleri L, Heusler P, et al. Pharmacology profile of F17464, a dopamine D3 receptor preferential antagonist. Eur J Pharmacol. (2021) 890:173635. doi: 10.1016/j.ejphar.2020.173635
43. Ishiyama T, Loebel A, Cucciaro J, Horisawa T, Tokuda K, Ogasa M, et al. Comparative rereceptor binding profile of lurasidone and other first and second generation antipsychotics. Poster NR6-40 (2010) APA, May 22–26, New Orleans, LA.
44. Snyder GL, Vanover KE, Davis RE, Li P, Fienberg A, Mates S. A review of the pharmacology and clinical profile of lumateperone for the treatment of schizophrenia. Adv Pharmacol. (2021) 90:253–76. doi: 10.1016/bs.apha.2020.09.001
45. Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science (1975) 188:1217–9. doi: 10.1126/science.1145194
47. Seeman P. Targeting the dopamine D2 receptor in schizophrenia. Exp Opin Ther Targets. (2006) 10:515–31. doi: 10.1517/14728222.10.4.515
48. Ginovart N, Kapur S. Role of dopamine D(2) receptors for antipsychotic activity. Handb Exp Pharmacol. (2012) 212:27–52. doi: 10.1007/978-3-642-25761-2_2
49. Seeman P. Schizophrenia and dopamine receptors. Eur Neuropsychopharmacol. (2013) 23:999–1009. doi: 10.1016/j.euroneuro.2013.06.005
50. Nord M, Farde L. Antipsychotic occupancy of dopamine receptors in schizophrenia. CNS Neurosci Ther. (2010) 17:97–103. doi: 10.1111/j.1755-5949.2010.00222.x
51. Yokoi F, Gründer G, Biziere K, Stephane M, Dogan AS, Dannals RF, et al. Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]raclopride. Neuropsychopharmacology. (2002) 27:248–59. doi: 10.1016/s0893-133x(02)00304-4
52. Kegeles LS, Slifstein M, Frankle WG, Xu X, Hackett E, Bae S-A, et al. Dose–occupancy study of striatal and extrastriatal dopamine D2 receptors by aripiprazole in schizophrenia with PET and [18F]Fallypride. Neuropsychopharmacology. (2008) 33:3111–25. doi: 10.1038/npp.2008.33
53. Girgis RR, Slifstein M, D’Souza D, Lee Y, Periclou A, Ghahramani P, et al. Preferential binding to dopamine D3 over D2 receptors by cariprazine in patients with schizophrenia using PET with the D3/D2 receptor ligand [11C](+)-PHNO. Psychopharmacology. (2016) 233:3503–12. doi: 10.1007/s00213-016-4382-y
54. Girgis RR, Forbes A, Abi-Dargham A, Slifstein M. A positron emission tomography occupancy study of brexpiprazole at dopamine D2 and D3 and serotonin 5-HT1A and 5-HT2A receptors, and serotonin reuptake transporters in subjects with schizophrenia. Neuropsychopharmacology. (2019) 45:786–92. doi: 10.1038/s41386-019-0590-6
55. Girgis RR, Zoghbi AW, Javitt DC, Lieberman JA. The past and future of novel, non-dopamine-2 receptor therapeutics for schizophrenia: a critical and comprehensive review. J Psychiatric Res. (2019) 108:57–83. doi: 10.1016/j.jpsychires.2018.07.006
56. Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature (2009) 459:356–63. doi: 10.1038/nature08144
57. Michino M, Beuming T, Donthamsetti P, Newman AH, Javitch JA, Shi L. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol Rev. (2014) 67:198–213. doi: 10.1124/pr.114.009944
58. Maramai S, Gemma S, Brogi S, Campiani G, Butini S, Stark H, et al. Dopamine D3 receptor antagonists as potential therapeutics for the treatment of neurological diseases. Front Neurosci. (2016) 10:451. doi: 10.3389/fnins.2016.00451
59. Egyed A, Domány-Kovács K, Koványi B, Horti F, Kurkó D, Kiss DJ, et al. Controlling receptor function from the extracellular vestibule of G-protein coupled receptors. Chem Commun. (2020) 56:14167–70. doi: 10.1039/d0cc05532h
60. Klein Herenbrink C, Verma R, Lim HD, Kopinathan A, Keen A, Shonberg J, et al. Molecular determinants of the intrinsic efficacy of the antipsychotic aripiprazole. ACS Chem Biol. (2019) 14:1780–92. doi: 10.1021/acschembio.9b00342
61. Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. (1995) 25:366–428. doi: 10.1016/s1043-9471(05)80049-7
62. Xu P, Huang S, Mao C, Krumm BE, Zhou XE, Tan Y, et al. Structures of the human dopamine D3 receptor-Gi complexes. Mol Cell. (2021) 81:1147–59.e4. doi: 10.1016/j.molcel.2021.01.003
63. Chien EYT, Liu W, Zhao Q, Katritch V, Won Han G, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. (2010) 330:1091–5. doi: 10.1126/science.1197410
64. Yin J, Chen KM, Clark MJ, Hijazi M, Kumari P, Bai XC, et al. Structure of a D2 dopamine receptor–G-protein complex in a lipid membrane. Nature. (2020) 584:125–9. doi: 10.1038/s41586-020-2379-5
65. Zhuang Y, Xu P, Mao C, Wang L, Krumm B, Zhou XE, et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell. (2021) 184:931–42.e18. doi: 10.1016/j.cell.2021.01.027
66. Im D, Inoue A, Fujiwara T, Nakane T, Yamanaka Y, Uemura T, et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat Commun. (2020) 11:6442. doi: 10.1038/s41467-020-20221-0
67. Fan L, Tan L, Chen Z, Qi J, Nie F, Luo Z, et al. Haloperidol bound D2 dopamine receptor structure inspired the discovery of subtype selective ligands. Nat Commun. (2020) 11:1074. doi: 10.1038/s41467-020-14884-y
68. Wang S, Che T, Levit A, Shoicet BK, Wacker D, Roth BL. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature. (2018) 555:269–73. doi: 10.1038/nature25758
69. Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. (2019) 47:W636–41. doi: 10.1093/nar/gkz268
70. Levant B. The D3 dopamine receptor: neurobiology and potential clinical relevance. Pharmacol Rev. (1997) 49:231–52.
71. Kim K-M, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. Differential regulation of the dopamine D2and D3 receptors by g protein-coupled receptor kinases and β-arrestins. J Biol Chem. (2001) 276:37409–14. doi: 10.1074/jbc.m106728200
72. Ahlgren-Beckendorf JA, Levant B. Signaling mechanisms of the D3 dopamine receptor. J Recept Signal Transduct Res. (2004) 24:117–30. doi: 10.1081/rrs-200029953
73. Beaulieu J-M, Tirotta E, Sotnikova TD, Masri B, Salahpour A, Gainetdinov RR, et al. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J Neurosci. (2007) 27:881–5. doi: 10.1523/jneurosci.5074-06.2007
74. Beom S, Cheong D, Torres G, Caron MG, Kim K-M. comparative studies of molecular mechanisms of dopamine D2 and D3 receptors for the activation of extracellular signal-regulated kinase. J Biol Chem. (2004) 279:28304–14. doi: 10.1074/jbc.m403899200
75. Leggio GM, Bucolo C, Platania CBM, Salomone S, Drago F. Current drug treatments targeting dopamine D3 receptor. Pharmacol Ther. (2016) 165:164–77. doi: 10.1016/j.pharmthera.2016.06.007
76. Jin M, Min C, Zheng M, Cho D-I, Cheong S-J, Kurose H, et al. Multiple signaling routes involved in the regulation of adenylyl cyclase and extracellular regulated kinase by dopamine D2 and D3 receptors. Pharmacol Res. (2013) 67:31–41. doi: 10.1016/j.phrs.2012.09.012
77. Peineau S, Bradley C, Taghibiglou C, Doherty A, Bortolotto ZA, Wang YT, et al. The role of GSK-3 in synaptic plasticity. Br J Pharmacol. (2008) 153:S428–37. doi: 10.1038/bjp.2008.2
78. Mannoury la Cour C, Salles M-J, Pasteau V, Millan MJ. Signaling pathways leading to phosphorylation of akt and GSK-3β by activation of cloned human and rat cerebral D2 and D3 receptors. Mol Pharmacol. (2011) 79:91–105. doi: 10.1124/mol.110.065409
79. Collo G, Zanetti S, Missale C, Spano P. Dopamine D3 receptor-preferring agonists increase dendrite arborization of mesencephalic dopaminergic neurons via extracellular signal-regulated kinase phosphorylation. Eur J Neurosci. (2008) 28:1231–40. doi: 10.1111/j.1460-9568.2008.06423.x
80. Sokoloff P, Andrieux M, Besançon R, Pilon C, Martres M-P, Giros B, et al. Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: comparison with D2 receptor. Eur J Pharmacol Mol Pharmacol. (1992) 225:331–7. doi: 10.1016/0922-4106(92)90107-7
81. Kiss B, Horti F, Bobok A. In vitro and in vivo comparison of [3H](+)-PHNO and [3H]raclopride binding to rat striatum and lobes 9 and 10 of the cerebellum: a method to distinguish dopamine D3 from D2 receptor sites: a method to distinguish dopamine D3 from D2 receptor sites. Synapse. (2011) 65:467–78. doi: 10.1002/syn.20867
82. Kiss B, Horti F, Bobok A. Poster #16 cariprazine, a D3/D2 dopamine receptor partial agonist antipsychotic, displays greater D3 receptor occupancy in vivo compared with other antipsychotics. Schizophr Res. (2012) 136:S190. doi: 10.1016/s0920-9964(12)70588-1
83. Leriche L, Bezard E, Gross C, Guillin O, Foll B, Diaz J, et al. The dopamine D3 receptor: a therapeutic target for the treatment of neuropsychiatric disorders. CNS Neurol Disord Drug Targets. (2006) 5:25–43. doi: 10.2174/187152706784111551
84. Gross G, Wicke K, Drescher KU. Dopamine D3 receptor antagonism—still a therapeutic option for the treatment of schizophrenia. Naunyn Schmiedeberg’s Arch Pharmacol. (2012) 386:155–66. doi: 10.1007/s00210-012-0806-3
85. Sokoloff P, Le Foll B. The dopamine D3 receptor, a quarter century later. Eur J Neurosci. (2016) 45:2–19. doi: 10.1111/ejn.13390
86. Lane JR, Abramyan AM, Adhikari P, Keen AC, Lee K-H, Sanchez J, et al. Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism. eLife. (2020) 9:e52189. doi: 10.7554/eLife.52189
87. Latorraca NR, Venkatakrishnan AJ, Dror RO. GPCR dynamics: structures in motion. Chem Rev. (2017) 117:139–55. doi: 10.1021/acs.chemrev.6b00177
88. Frank A, Kiss DJ, Keserű GM, Stark H. Binding kinetics of cariprazine and aripiprazole at the dopamine D3 receptor. Sci Rep. (2018) 8:12509. doi: 10.1038/s41598-018-30794-y
89. Ferruz N, Doerr S, Vanase-Frawley MA, Zou Y, Chen X, Marr ES, et al. Dopamine D3 receptor antagonist reveals a cryptic pocket in aminergic GPCRs. Sci Rep. (2018) 8:897. doi: 10.1038/s41598-018-19345-7
90. Michino M, Boateng CA, Donthamsetti P, Yano H, Bakare OM, Bonifazi A, et al. Toward understanding the structural basis of partial agonism at the dopamine D3 receptor. J Med Chem. (2017) 60:580–93. doi: 10.1021/acs.jmedchem.6b01148
91. Svensson K, Carlsson A, Huff RM, Kling-Petersen T, Waters N. Behavioral and neurochemical data suggest functional differences between dopamine D2 and D3 receptors. Eur J Pharmacol. (1994) 263:235–43. doi: 10.1016/0014-2999(94)90718-8
92. Sigala S, Missale C, Spano P. Opposite effects of dopamine D2 and D3 receptors on learning and memory in the rat. Eur J Pharmacol. (1997) 336:107–12. doi: 10.1016/s0014-2999(97)01235-1
93. Santesso DL, Evins AE, Frank MJ, Schetter EC, Bogdan R, Pizzagalli DA. Single dose of a dopamine agonist impairs reinforcement learning in humans: evidence from event-related potentials and computational modeling of striatal-cortical function. Hum Brain Mapp. (2009) 30:1963–76. doi: 10.1002/hbm.20642
94. Watson DJ, Loiseau F, Ingallinesi M, Millan MJ, Marsden CA, Fone KC. Selective blockade of dopamine D3 receptors enhances while D2 receptor antagonism impairs social novelty discrimination and novel object recognition in rats: a key role for the prefrontal cortex. Neuropsychopharmacology. (2011) 37:770–86. doi: 10.1038/npp.2011.254
95. Kagaya T, Yonaga M, Furuya Y, Hashimoto T, Kuroki J, Nishizawa Y. Dopamine D3 agonists disrupt social behavior in rats. Brain Res. (1996) 721:229–32. doi: 10.1016/0006-8993(96)00288-0
96. Simpson EH, Winiger V, Biezonski DK, Haq I, Kandel ER, Kellendonk C. Selective overexpression of dopamine D3 receptors in the striatum disrupts motivation but not cognition. Biol Psychiatry. (2014) 76:823–31. doi: 10.1016/j.biopsych.2013.11.023
97. Reavill C, Taylor SG, Wood MD, Ashmeade T, Austin NE, Avenell KY, et al. Pharmacological actions of a novel, high-affinity, and selective human dopamine D(3) receptor antagonist, SB-277011-A. J Pharmacol Exp Ther. (2000) 294:1154–65.
98. Redden L, Rendenbach-Mueller B, Abi-Saab WM, Katz DA, Goenjian A, Robieson WZ, et al. A double-blind, randomized, placebo-controlled study of the dopamine D3 receptor antagonist ABT-925 in patients with acute schizophrenia. J Clin Psychopharmacol. (2011) 31:221–5. doi: 10.1097/jcp.0b013e31820e4818
99. Bardin L, Kleven MS, Barret-Grévoz C, Depoortère R, Newman-Tancredi A. Antipsychotic-like vs cataleptogenic actions in mice of novel antipsychotics having D2 Antagonist and 5-HT1A agonist properties. Neuropsychopharmacology. (2005) 31:1869–79. doi: 10.1038/sj.npp.1300940
100. Millan MJ, Gressier H, Brocco M. The dopamine D3 receptor antagonist, (+)-S14297, blocks the cataleptic properties of haloperidol in rats. Eur J Pharmacol. (1997) 321:R7–9. doi: 10.1016/s0014-2999(97)00049-6
101. Gyertyán I, Sághy K. The selective dopamine D3 receptor antagonists, SB 277011-A and S 33084 block haloperidol-induced catalepsy in rats. Eur J Pharmacol. (2007) 572:171–4. doi: 10.1016/j.ejphar.2007.06.035
102. Lacroix LP, Hows MEP, Shah AJ, Hagan JJ, Heidbreder CA. Selective antagonism at dopamine D3 receptors enhances monoaminergic and cholinergic neurotransmission in the rat anterior cingulate cortex. Neuropsychopharmacology. (2002) 28:839–49. doi: 10.1038/sj.npp.1300114
103. Millan MJ, Svenningsson P, Ashby CR Jr., Hill M, Egeland M, Dekeyne A, et al. S33138 [N-[4-[2-[(3aS,9bR)-8-cyano-1,3a,4,9b-tetrahydro[1]-benzopyrano[3,4-c]pyrrol-2(3 H)-yl)-ethyl]phenylacetamide], a preferential dopamine D3 versus D2 receptor antagonist and potential antipsychotic agent. II. A neurochemical, electrophysiological and behavioral characterization in vivo. J Pharmacol Exp Ther. (2008) 324:600–11.
104. Koch S, Perry KW, Bymaster FP. Brain region and dose effects of an olanzapine/fluoxetine combination on extracellular monoamine concentrations in rat. Neuropharmacology. (2004) 46:232–42. doi: 10.1016/j.neuropharm.2003.09.001
105. Kiss B, Laszlovszky I, Krámos B, Visegrády A, Bobok A, Lévay G, et al. Neuronal dopamine D3 receptors: translational implications for preclinical research and CNS disorders. Biomolecules. (2021) 11:104. doi: 10.3390/biom11010104
106. Levesque D, Diaz J, Pilon C, Martres MP, Giros B, Souil E, et al. Identification, characterization, and localization of the dopamine D3 receptor in rat brain using 7-[3H]hydroxy-N,N-di-n-propyl-2-aminotetralin. Proc Nat Acad Sci USA. (1992) 89:8155–9. doi: 10.1073/pnas.89.17.8155
107. Pugsley TA, Davis MD, Akunne HC, MacKenzie RG, Shih YH, Damsma G, et al. Neurochemical and functional characterization of the preferentially selective dopamine D3 agonist PD 128907. J Pharmacol Exp Ther. (1995) 275:1355–66.
108. Mierau J, Schneider FJ, Ensinger HA, Chio CL, Lajiness ME, Huff RM. Pramipexole binding and activation of cloned and expressed dopamine D2, D3 and D4 receptors. Eur J Pharmacol Mol Pharmacol. (1995) 290:29–36. doi: 10.1016/0922-4106(95)90013-6
109. Kassel S, Schwed JS, Stark H. Dopamine D3 receptor agonists as pharmacological tools. Eur Neuropsychopharmacol. (2015) 25:1480–99. doi: 10.1016/j.euroneuro.2014.11.005
110. Mogilnicka E, Klimek V. Drugs affecting dopamine neurons and yawning behavior. Pharmacol Biochem Behav. (1977) 7:303–5. doi: 10.1016/0091-3057(77)90224-6
111. Kurashima M, Yamada K, Nagashima M, Shirakawa K, Furukawa T. Effects of putative dopamine D3 receptor agonists, 7-OH-DPAT, and quinpirole, on yawning, stereotypy, and body temperature in rats. Pharmacol Biochem Behav. (1995) 52:503–8. doi: 10.1016/0091-3057(95)00103-4
112. Bristow LJ, Kramer MS, Kulagowski J, Patel S, Ragan CI, Seabrook GR. Schizophrenia and L-745,870, a novel dopamine D4 receptor antagonist. Trends Pharmacol Sci. (1997) 18:186–8. doi: 10.1016/s0165-6147(97)01066-3
113. Collins GT, Witkin JM, Newman AH, Svensson KA, Grundt P, Cao J, et al. Dopamine agonist-induced yawning in rats: a dopamine D3 receptor-mediated behavior. J Pharmacol Exp Ther. (2005) 314:310–9. doi: 10.1124/jpet.105.085472
114. Collins GT, Newman AH, Grundt P, Rice KC, Husbands SM, Chauvignac C, et al. Yawning and hypothermia in rats: effects of dopamine D3 and D2 agonists and antagonists. Psychopharmacology. (2007) 193:159–70. doi: 10.1007/s00213-007-0766-3
115. Geneste H, Amberg W, Backfisch G, Beyerbach A, Braje WM, Delzer J, et al. Synthesis and SAR of highly potent and selective dopamine D3-receptor antagonists: variations on the 1H-pyrimidin-2-one theme. Bioorg Med Chem Lett. (2006) 16:1934–7. doi: 10.1016/j.bmcl.2005.12.079
116. Searle G, Beaver JD, Comley RA, Bani M, Tziortzi A, Slifstein M, et al. Imaging dopamine D3 receptors in the human brain with positron emission tomography, [11C]PHNO, and a selective D3 receptor antagonist. Biol Psychiatry. (2010) 68:392–9. doi: 10.1016/j.biopsych.2010.04.038
117. Micheli F, Arista L, Bonanomi G, Blaney FE, Braggio S, Capelli AM, et al. 1,2,4-Triazolyl azabicyclo[3.1.0]hexanes: a new series of potent and selective dopamine D3 receptor antagonists. J Med Chem. (2009) 53:374–91. doi: 10.1021/jm901319p
118. Vangveravong S, McElveen E, Taylor M, Xu J, Tu Z, Luedtke RR, et al. Synthesis and characterization of selective dopamine D2 receptor antagonists. Bioorg Med Chem. (2006) 14:815–25. doi: 10.1016/j.bmc.2005.09.008
119. Millan MJ, Dekeyne A, Rivet J-M, Dubuffet T, Lavielle G, Brocco M. S33084, a novel, potent, selective and competitive antagonist at dopamine D3 receptors: II. Functional and behavioral profile compared with GR218,231 and L741,626. J Pharmacol Exp Ther. (2000) 293:1063–73.
120. Gross G, Bialojan S, Drescher K, Freeman AS, Garcia-Ladona FJ, Höger T, et al. Evaluation of D3 receptor antagonists. Eur Neuropsychopharmacol. (1997) 1002:S120.
121. Pilla M, Perachon S, Sautel F, Garrido F, Mann A, Wermuth CG, et al. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature (1999) 400:371–5. doi: 10.1038/22560
122. Bitter I, Groc M, Delsol C, Fabre C, Fagard M, Barthe L, et al. Efficacy of F17464, a new preferential D3 antagonist in a placebo-controlled phase 2 study of patients with an acute exacerbation of schizophrenia. Eur Psychiatry (2017) 41:S387. doi: 10.1016/j.eurpsy.2017.02.428
123. Bitter I, Lieberman JA, Gaudoux F, Sokoloff P, Groc M, Chavda R, et al. Randomized, double-blind, placebo-controlled study of F17464, a preferential D3 antagonist, in the treatment of acute exacerbation of schizophrenia. Neuropsychopharmacology. (2019) 44:1917–24. doi: 10.1038/s41386-019-0355-2
124. Finnema SJ, Bang-Andersen B, Wikström HV, Halldin C. Current state of agonist radioligands for imaging of brain dopamine D2/D3 receptors in vivo with positron emission tomography. Curr Top Med Chem. (2010) 10:1477–98. doi: 10.2174/156802610793176837
125. Mach RH, Luedtke RR. Challenges in the development of dopamine D2- and D3-selective radiotracers for PET imaging studies. J Labelled Comp Radiopharm. (2017) 61:291–8. doi: 10.1002/jlcr.3558
126. Doot RK, Dubroff JG, Labban KJ, Mach RH. Selectivity of probes for PET imaging of dopamine D3 receptors. Neurosci Lett. (2019) 691:18–25. doi: 10.1016/j.neulet.2018.03.006
127. Martin GE, Williams M, Pettibone DJ, Zrada MM, Lotti VJ, Taylor DA, et al. Selectivity of (1)-4-propyl-9-ydroxynaphthoxazine (+)-PHNO for dopamine receptors in vitro and in vivo. J Pharmacol Exp Ther. (1985) 233:395–401.
128. Freedman SB, Patel S, Marwood R, Emms F, Seabrook GR, Knowles MR, et al. Expression and pharmacological chacterization of the human D3 dopamine receptor. J Pharmacol Exp Ther. (1994) 268:417–26.
129. Wilson AA, McCormick P, Kapur S, Willeit M, Garcia A, Hussey D, et al. Radiosynthesis and Evaluation of [11C]-(+)-4-Propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9-ol as a potential radiotracer for in vivo imaging of the dopamine D2 high-affinity state with positron emission tomography. J Med Chem. (2005) 48:4153–60. doi: 10.1021/jm050155n
130. Narendran R, Slifstein M, Guillin O, Hwang Y, Hwang D-R, Scher E, et al. Dopamine (D2/3) receptor agonist positron emission tomography radiotracer [11C]-(+)-PHNO is a D3 receptor preferring agonist in vivo. Synapse. (2006) 60:485–95. doi: 10.1002/syn.20325
131. Ginovart N, Galineau L, Willeit M, Mizrahi R, Bloomfield PM, Seeman P, et al. Binding characteristics and sensitivity to endogenous dopamine of [11C]-(+)-PHNO, a new agonist radiotracer for imaging the high-affinity state of D2 receptors in vivo using positron emission tomography. J Neurochem. (2006) 97:1089–103. doi: 10.1111/j.1471-4159.2006.03840.x
132. Ginovart N, Willeit M, Rusjan P, Graff A, Bloomfield PM, Houle S, et al. positron emission tomography quantification of [11C]-(+)-PHNO binding in the human brain. J Cereb Blood Flow Metab. (2006) 27:857–71. doi: 10.1038/sj.jcbfm.9600411
133. Rabiner EA, Slifstein M, Nobrega J, Plisson C, Huiban M, Raymond R, et al. In vivo quantification of regional dopamine-D3 receptor binding potential of (+)-PHNO: studies in non-human primates and transgenic mice. Synapse. (2009) 63:782–93. doi: 10.1002/syn.20658
134. Tziortzi AC, Searle GE, Tzimopoulou S, Salinas C, Beaver JD, Jenkinson M, et al. Imaging dopamine receptors in humans with [11C]-(+)-PHNO: dissection of D3 signal and anatomy. Neuroimage. (2011) 54:264–77. doi: 10.1016/j.neuroimage.2010.06.044
135. Gallezot J-D, Beaver JD, Gunn RN, Nabulsi N, Weinzimmer D, Singhal T, et al. Affinity and selectivity of [11C]-(+)-PHNO for the D3 and D2 receptors in the rhesus monkey brain in vivo. Synapse. (2012) 66:489–500. doi: 10.1002/syn.21535
136. Le Foll B, Collo G, Rabiner EA, Boileau I, Merlo Pich E, Sokoloff P. Dopamine D3 receptor ligands for drug addiction treatment. Prog Brain Res. (2014) 211:255–75. doi: 10.1016/B978-0-444-63425-2.00011-8
137. Huang M, Kwon S, Oyamada Y, Rajagopal L, Miyauchi M, Meltzer HY. Dopamine D3 antagonism contributes to blonanserin-indiced cortical and acetylcholine efflux and cognitive improvement. Pharmacol Biochem Behav. (2015) 138:49–57. doi: 10.1016/j.pbb.2015.09.011
138. Ichikawa J, Kuroki T, Dai J, Meltzer HY. Effect of antipsychotic drugs on extracellular serotonin levels in rat medial prefrontal cortex and nucleus accumbens. Eur J Pharmacol. (1998) 351:163–71. doi: 10.1016/s0014-2999(98)00308-2
139. Oka M, Noda Y, Ochi Y, Furukawa K, Une T, Kurumiya S, et al. Pharmacological profile of AD-5423, a novel antipsychotic with both potent dopamine-D2 and serotonin-S2 antagonist properties. J Pharmacol Exp Ther. (1993) 264:158–65.
140. Graff-Guerrero A, Mamo D, Shammi CM, Mizrahi R, Marcon H, Barsoum P, et al. The effect of antipsychotics on the high-affinity state of D2 and D3 receptors. Arch Gen Psychiatry. (2009) 66:606–15. doi: 10.1001/archgenpsychiatry.2009.43
141. Mizrahi R, Agid O, Borlido C, Suridjan I, Rusjan P, Houle S, et al. Effects of antipsychotics on D3 receptors: a clinical PET study in first episode antipsychotic naive patients with schizophrenia using [11C]-(+)-PHNO. Schizophr Res. (2011) 131:63–8. doi: 10.1016/j.schres.2011.05.005
142. Tateno A, Sakayori T, Kim W, Honjo K, Nakayama H, Arakawa R, et al. Comparison of dopamine D3 and D2 receptor occupancies by a single dose of blonanserin in healthy subjects: a positron emission tomography study with [11C]-(+)-PHNO. Int J Neuropsychopharmacol. (2018) 21:522–7. doi: 10.1093/ijnp/pyy004
143. Girgis R, Abi-Dargham A, Slifstein M, Chen L, Periclou A, Adham N, et al. In vivo dopamine D3 and D2 receptor occupancy profile of cariprazine versus aripiprazole: a PET study. Neuropsychopharmacology. (2017) 43:S595–6.
144. Heusler P, Martel JC, Gatti-McArthur S. In vitro profile of the new antipsychotic, F17464, at recombinant human neurotransmitter receptors. Eur Neuropsychopharmacol. (2016) 26:S490–1. doi: 10.1016/s0924-977x(16)31502-4
145. Slifstein M, Abi-Dargham A, Girgis RR, Suckow RF, Cooper TB, Divgi CR, et al. Binding of the D3-preferring antipsychotic candidate F17464 to dopamine D3 and D2 receptors: a PET study in healthy subjects with [11C]-(+)-PHNO. Psychopharmacology. (2019) 237:519–27. doi: 10.1007/s00213-019-05387-w
146. Seeman P, Ko F, Willeit M, McCormick P, Ginovart N. Antiparkinson concentrations of pramipexole and PHNO occupy dopamine D2high and D3high receptors. Synapse. (2005) 58:122–8. doi: 10.1002/syn.20193
147. Bono F, Mutti V, Fiorentini C, Missale C. Dopamine D3 receptor heteromerization: implications for neuroplasticity and neuroprotection. Biomolecules. (2020) 10:1016. doi: 10.3390/biom10071016
148. Li X-M, Perry KW, Wong DT, Bymaster FP. Olanzapine increases in vivo dopamine and norepinephrine release in rat prefrontal cortex, nucleus accumbens and striatum. Psychopharmacology. (1998) 136:153–61. doi: 10.1007/s002130050551
149. Kuroki T, Meltzer HY, Ichikawa J. Effects of antipsychotic drugs on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens. J Pharmacol Exp Ther. (1999) 288:774–81.
150. Westerink BHC, Kawahara Y, De Boer P, Geels C, De Vries JB, Wikström HV, et al. Antipsychotic drugs classified by their effects on the release of dopamine and noradrenaline in the prefrontal cortex and striatum. Eur J Pharmacol. (2001) 412:127–38. doi: 10.1016/s0014-2999(00)00935-3
151. Frånberg O, Marcus MM, Ivanov V, Schilström B, Shahid M, Svensson TH. Asenapine elevates cortical dopamine, noradrenaline and serotonin release. Evidence for activation of cortical and subcortical dopamine systems by different mechanisms. Psychopharmacology. (2009) 204:251–64. doi: 10.1007/s00213-008-1456-5
152. Huang M, Panos JJ, Kwon S, Oyamada Y, Rajagopal L, Meltzer HY. Comparative effect of lurasidone and blonanserin on cortical glutamate, dopamine, and acetylcholine efflux: role of relative serotonin (5-HT)2A and DA D2 antagonism and 5-HT1A partial agonism. J Neurochem. (2013) 128:938–49. doi: 10.1111/jnc.12512
153. Huang M, He W, Kiss B, Farkas B, Adham N, Meltzer HY. The role of dopamine D3 receptor partial agonism in cariprazine-induced neurotransmitter efflux in rat hippocampus and nucleus accumbens. J Pharmacol Exp Ther. (2019) 371:517–25. doi: 10.1124/jpet.119.259879
154. Kehr J, Yoshitake T, Ichinose F, Yoshitake S, Kiss B, Gyertyán I, et al. Effects of cariprazine on extracellular levels of glutamate, GABA, dopamine, noradrenaline and serotonin in the medial prefrontal cortex in the rat phencyclidine model of schizophrenia studied by microdialysis and simultaneous recordings of locomotor activity. Psychopharmacology. (2018) 235:1593–607. doi: 10.1007/s00213-018-4874-z
155. Caravaggio F, Kegeles LS, Wilson AA, Remington G, Borlido C, Mamo DC, et al. Estimating the effect of endogenous dopamine on baseline [11C]-(+)-PHNO binding in the human brain. Synapse. (2016) 70:453–60. doi: 10.1002/syn.21920
156. Schotte A, Janssen PFM, Gommeren W, Luyten WHLM, Leysen JE. Autoradiographic evidence for the occlusion of rat brain dopamine D3 receptors in vivo. Eur J Pharmacol. (1992) 218:373–5. doi: 10.1016/0014-2999(92)90196-b
157. Levant B. Differential sensitivity of [3H]7-OH-DPAT-labeled binding sites in rat brain to inactivation by N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline. Brain Res. (1995) 698:146–54. doi: 10.1016/0006-8993(95)00879-u
158. Zhang K, Weiss NT, Tarazi FI, Kula NS, Balessarini RJ. Effects of alkylating agent on dopamine D3 receptors in rat brain: selective protection by dopamine. Brain Res. (1999) 847:32–7. doi: 10.1016/s0006-8993(99)02024-7
159. Jordan S, Koprivica V, Dunn R, Tottori K, Kikuchi T, Altar CA. In vivo effects of aripiprazole on cortical and striatal dopaminergic and serotonergic function. Eur J Pharmacol. (2004) 483:45–53. doi: 10.1016/j.ejphar.2003.10.025
160. Bortolozzi A, Díaz-Mataix L, Toth M, Celada P, Artigas F. In vivo actions of aripiprazole on serotonergic and dopaminergic systems in rodent brain. Psychopharmacology. (2007) 191:745–58. doi: 10.1007/s00213-007-0698-y
161. Hertel P, Nomikos GG, Iurlo M, Svensson TH. Risperidone: regional effects in vivo on release and metabolism of dopamine and serotonin in the rat brain. Psychopharmacology. (1996) 124:74–86. doi: 10.1007/bf02245607
162. Koch S, Perry KW, Bymaster FP. Brain region and dose effects of an olanzapine/fluoxetine combination on extracellular monoamine concentrations in rat. Neuropharmacology. (2004) 46:232–42. doi: 10.1016/j.neuropharm.2003.09.001
163. Huang M, Li Z, Dai J, Shahid M, Wong EHF, Meltzer HY. Asenapine increases dopamine, norepinephrine, and acetylcholine efflux in the rat medial cortex and hippocampus. Neupropsychopharmacology. (2008) 33:2934–45. doi: 10.1038/npp.2008.20
164. Benoit-Marand M, Ballion B, Borrelli E, Boraud T, Gonon F. Inhibition of dopamine uptake by D2 antagonists: an in vivo study. J Neurochem. (2011) 116:449–58. doi: 10.1016/0304-3940(94)90096-5
165. Amato D, Vernon AC, Papaleo F. Dopamine, the antipsychotic molecule: a perspective on mechanimss underlying antipsychotc response. Neurosci Biobehav Rev. (2018) 85:146–59. doi: 10.1016/j.neubiorev.2017.09.027
166. Amato D, Kruyer A, Samaha A-N, Heinz A. Hypofunctional dopamine uptake and antipsychotic treatment-resistant schizophrenia. Front Psychiatry. (2019) 10:314. doi: 10.3389/fpsyt.2019.00314
167. Amato D, Canneva F, Cumming P, Maschaure S, Groos D, Dahlmanns JK, et al. A dopaminergic mechanism of antipsychotic drugs efficacy, failure, and failure reversal: the role of the dopamine transporter. Mol Pschiatry. (2020) 25:2101–18. doi: 10.1038/s41380-018-0114-5
168. Zapata A, Kivell B, Han Y, Javitch JA, Bolan A, Kuraguntla D, et al. Regulation of dopamine transporter function and cell surface expression by D3 dopamine receptor. J Biol Chem. (2007) 282:35842–54. doi: 10.1074/jbc.M611758200
169. Luis-Ravelo D, Fumagallo-Reading F, Castro-Hernandez J, Barroso-Chimea P, Alfonso-Oramas D, Febles-Casquero A, et al. Prolonged dopamine D3 receptor stimulation promotes dopamine transporter ubiquitination and degradation through a PKC-dependent mechanism. Pharmacol Res. (2021) 165:105464. doi: 10.1016/j.phrs.2021.105434
170. Andén N-E, Stock G. Effect of clozapine on the turnover of dopamine in the corpus striatum and in the limbic system. J Pharm Pharmacol. (1973) 25:346–8. doi: 10.1111/j.2042-7158.1973.tb10025.x
171. Bartholini G. Differential effect of neuroleptic drugs on dopamine turnover in the extrapyramidal and limbic system. J Pharm Pharmacol. (1976) 28:429–33. doi: 10.1111/j.2042-7158.1976.tb04648.x
172. Leysen JE, Janssen PM, Gommeren W, Wynants J, Pauwels PJ, Janssen PA. In vitro and in vivo receptor binding and effects on monoamine turnover in rat brain regions of the novel antipsychotics risperidone and ocaperidone. Mol Pharmacol. (1992) 41:494–508.
173. Kiss B, Némethy Z, Fazekas K, Kurkó D, Gyertyán I, Sághy K, et al. Preclinical pharmacodynamic and pharmacokinetic characterization of the major metabolites of cariprazine. Drug Des Dev Ther. (2019) 13:3229–48. doi: 10.2147/dddt.s188760
174. Mugnaini M, Iavarone L, Cavallini P, Griffante C, Oliosi B, Savoia C, et al. Occupancy of brain dopamine D3 receptors and drug craving: a translational approach. Neuropsychopharmacology. (2012) 38:302–12. doi: 10.1038/npp.2012.171
175. McCormick PN, Kapur S, Graff-Guerrero A, Raymond R, Nobrega JN, Wilson AA. the antipsychotics olanzapine, risperidone, clozapine, and haloperidol are D2-selective ex vivo but not in vitro. Neuropsychopharmacology. (2010) 35:1826–35. doi: 10.1038/npp.2010.50
176. McCormick PN, Wilson VS, Wilson AA, Remington GJ. Acutely administered antipsychotic drugs are highly selective for dopamine D2 over D3 receptors. Pharmacol Res. (2013) 70:66–71. doi: 10.1016/j.phrs.2013.01.002
177. Mach RH, Tu Z, Xu J, Li S, Jones LA, Taylor M, et al. Endogenous dopamine (DA) competes with the binding of a radiolabeled D3 receptor partial agonist in vivo: a positron emission tomography study. Synapse. (2011) 65:724–32. doi: 10.1002/syn.20891
178. Fiorentini C, Busi C, Gorruso E, Gotti C, Spano P, Missale C. Reciprocal regulation of dopamine d1 and D3 receptor function and trafficking by heterodimerization. Mol Pharmacol. (2008) 74:59–69. doi: 10.1124/mol.107.043885
179. Maggio R, Scarselli M, Capannolo M, Millan MJ. Novel dimensions of D3 receptor function: focus on heterodimerisation, transactivation and allosteric modulation. Eur Neuropsychopharmacol. (2015) 25:1470–9. doi: 10.1016/j.euroneuro.2014.09.016
180. van Wieringen J-P, Booij J, Shalgunov V, Elsinga P, Michel MC. Agonist high- and low-affinity states of dopamine D2 receptors: methods of detection and clinical implications. Naunyn Schmiedebergs Arch Pharmacol. (2012) 386:135–54. doi: 10.1007/s00210-012-0817-0
Keywords: schizophrenia, antipsychotics, D3 receptor, D2 receptor, dopamine, brain occupancy
Citation: Kiss B, Krámos B and Laszlovszky I (2022) Potential Mechanisms for Why Not All Antipsychotics Are Able to Occupy Dopamine D3 Receptors in the Brain in vivo. Front. Psychiatry 13:785592. doi: 10.3389/fpsyt.2022.785592
Received: 29 September 2021; Accepted: 25 February 2022;
Published: 24 March 2022.
Edited by:
Peter Falkai, LMU Munich University Hospital, GermanyReviewed by:
Davide Amato, Medical University of South Carolina, United StatesXenia Gonda, Semmelweis University, Hungary
Copyright © 2022 Kiss, Krámos and Laszlovszky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Béla Kiss, Yi5raXNzQHJpY2h0ZXIuaHU=; Ymtpc3M0NkBnbWFpbC5jb20=