
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 21 December 2022
Sec. Psychological Therapy and Psychosomatics
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.1046471
This article is part of the Research TopicReviews in Emotional and Behavioral Dyscontrol in Neurological DisordersView all 5 articles
 Anna Digiovanni1,2*†
Anna Digiovanni1,2*† Paola Ajdinaj1,2†
Paola Ajdinaj1,2† Mirella Russo1,2
Mirella Russo1,2 Stefano L. Sensi1,2
Stefano L. Sensi1,2 Marco Onofrj1,2
Marco Onofrj1,2 Astrid Thomas1
Astrid Thomas1Psychiatric symptoms frequently predate or complicate neurological disorders, such as neurodegenerative diseases. Symptoms of bipolar spectrum disorders (BSD), like mood, behavioral, and psychotic alterations, are known to occur – individually or as a syndromic cluster – in Parkinson’s disease and in the behavioral variant of frontotemporal dementia (FTD). Nonetheless, due to shared pathophysiological mechanisms, or genetic predisposition, several other neurological disorders show significant, yet neglected, clinical and biological overlaps with BSD like neuroinflammation, ion channel dysfunctions, neurotransmission imbalance, or neurodegeneration. BSD pathophysiology is still largely unclear, but large-scale network dysfunctions are known to participate in the onset of mood disorders and psychotic symptoms. Thus, functional alterations can unleash BSD symptoms years before the evidence of an organic disease of the central nervous system. The aim of our narrative review was to illustrate the numerous intersections between BSD and neurological disorders from a clinical-biological point of view and the underlying predisposing factors, to guide future diagnostic and therapeutical research in the field.
Bipolar spectrum disorders (BSD) are psychiatric conditions characterized by unstable mood and the presence of alternating manic/hypomanic and depressive episodes. BSD markedly impact the global quality of life of patients (1). Interestingly, the comorbidity of BSD with neurological diseases has been extensively described (2). Shared genetic predisposition and neurodevelopmental alterations are a common ground for developing different neuropsychiatric conditions.
According to the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) BSD are classified into three primary patterns: bipolar disorder (BD) I, II, and Cyclothymia (3). BD I is the classical “manic-depressive disorder” characterized by the occurrence of at least one manic episode, i.e., “a period of abnormally and persistently elevated, expansive, or irritable mood and abnormally and persistently increased goal-directed activity or energy” (3, 4). During the manic phase, BD patients frequently suffer from impulse control disorders (ICDs), like gambling, hypersexuality, shopping sprees, bulimia, aggressive driving, and hoarding (5). Most subjects with BD I also experience major depressive episodes in their lifetime, although its presence is not a required diagnostic criterium. BD II diagnosis requires at least one episode of major depression and at least one hypomanic episode that, differently from manic episodes, are not severe enough to cause severe impairment in social or occupational functioning or necessitate hospitalization (3). The Cyclothymic Disorder is diagnosed when, for at least 2 years, the subject has experienced hypomanic and depressive periods without fulfilling the criteria for a full-blown episode of mania, hypomania, or major depression (3).
Bipolar disorder categorization has mainly been conceptualized by American authors. However, French and German psychiatrists have the merit to have drawn attention to Cyclic Psychoses largely before the American studies (6–12). Also, European studies attempted to extend the DSM categorization from three to six clinical syndromic entities (13, 14). These six conditions range from hypomania, not interfering with adaptation to social life and career goals, to patterns associated with neurodegeneration. BD type VI, in particular, is characterized by cognitive decline and mood instability and may be interpreted as a late-onset BSD. In these patients, an accurate evaluation of premorbid personality and familiar history of BSD may help define the therapeutic strategy since mood stabilization may be more effective than antidepressants and acetylcholinesterase inhibitors (14).
In a MEDLINE (Medical Literature Analysis and Retrieval System Online) search, “Bipolar Disorders” AND “Dementia” keywords generate more than 1,400 results, “Bipolar Disorders” and “Parkinson” more than 700 papers, “Multiple Sclerosis” and “Bipolar Disorders” above 200 papers, and finally the combined search with Epilepsy finds about 1,000 papers.
The present review analyzes the literature on the co-occurrence of BSDs and Neurological Disorders to understand whether different comorbidity patterns indicate clusters, endophenotypes, or statistical overlaps (Figure 1). We omit from the review the rare genetic and metabolic disease, where coexistence of psychiatric and neurologic symptoms, together with signs of medical and laboratory dysfunctions, are core elements for the diagnosis, like Wilson’s disease (15).
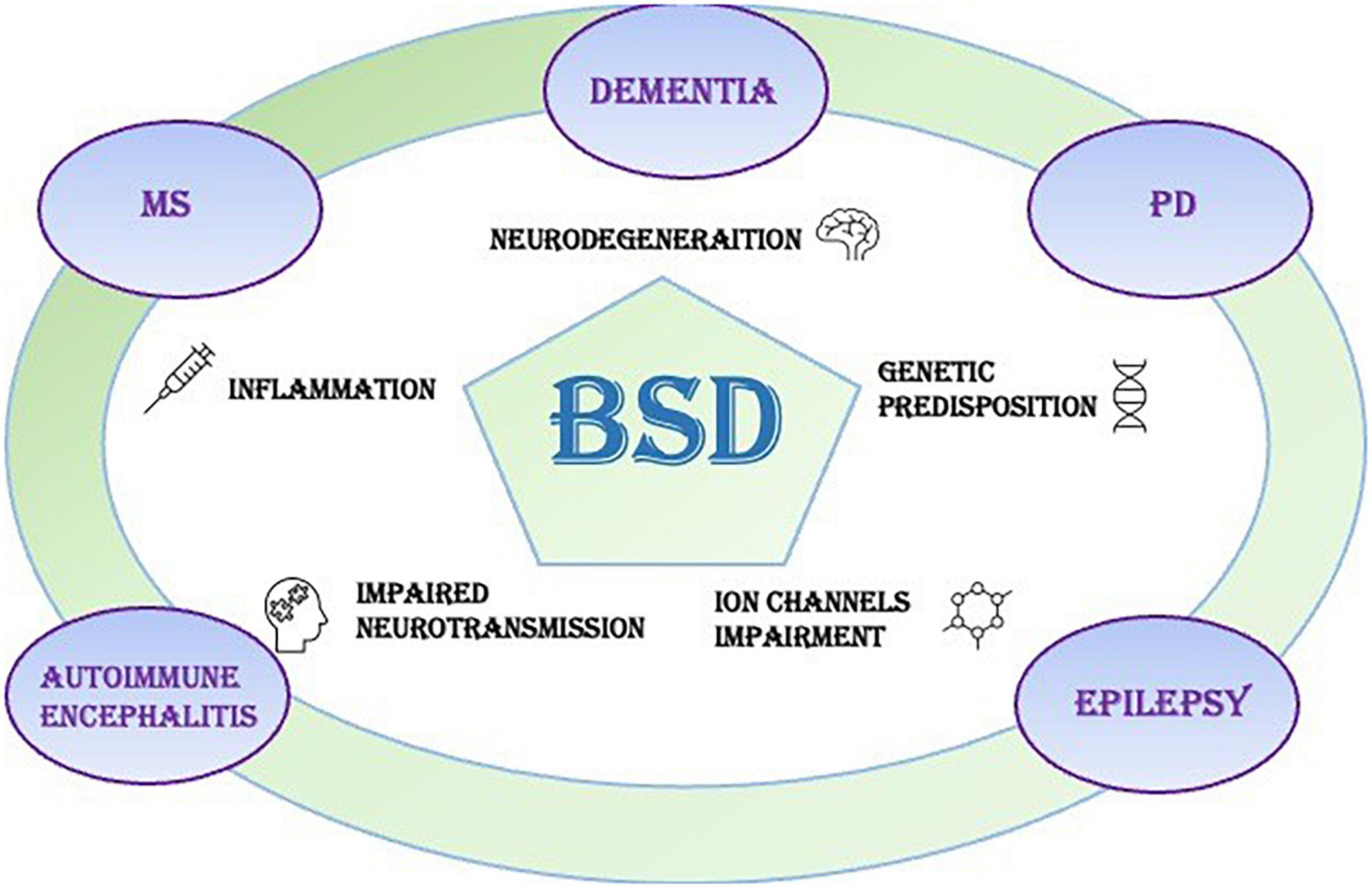
Figure 1. Bipolar spectrum disorders (BSDs) frequently occur in neurological disorders of the central nervous system, like dementias, Parkinson’s disease (PD), multiple sclerosis (MS), autoimmune encephalitis (AE), and epilepsy. The pathophysiological triggers of the neurological symptoms can play a role also in the onset of mood disorders like BSD.
Compared to other adult psychiatric disorders BSD, are associated with an increased risk of developing neurodegenerative disorders and cerebrovascular diseases (16). Several studies concur in showing that dementia risk in BSD patients is remarkably higher (20–25%) than in the general population (7%) (17). In some cases, BSD present a progressive course, with an increasing number of hospitalizations, poor response to treatment, and steady cognitive decline in the late phases of the disease (16). However, some other studies suggest that early alterations of cognitive functions are part of the BSD phenotype (17–19). A remarkable inter-subject heterogeneity is observed in terms of compromised cognitive domains (20), converging toward a more prominent impairment of verbal memory, attention, executive functions, and visual memory (20). Some data suggest that cognitive decline correlates with the clinical course of BSD, for instance, with the number of altered mood episodes – in particular the maniac ones –, the occurrence of hospitalizations, and the duration of illness (16, 21, 22). It has been hypothesized that mood swings trigger cumulative damage in neural cells and then feed a vicious circle causing more manic or depressive episodes (23). Inflammation, genetic predisposition, and neural vulnerability associated with the disease duration, and the number of mood episodes may favor accelerated brain aging in BSD (1). This mechanism may explain the coexistence of the two conditions, neuropathological findings in the brain of BSD patients and imaging signs of cortical atrophy associated with cognitive decline. Moreover, some dementia subtypes are characterized by early mood and behavioral alterations (16), as later detailed.
The frontotemporal dementia (FTD) spectrum encompasses several neurodegenerative entities that share prominent damage of the frontal and temporal lobes (16). According to the clinical presentation, FTD can be categorized into three subtypes: the behavioral variant (bvFTD) and three forms of Primary Progressive Aphasia (APP) (logopenic, semantic, and non-fluent/agrammatic) (24). Behavioral features appear consistently also in patients whose phenotype is showing prominent APP features, especially in the semantic variant (25). From a genetic standpoint, FTD is known to be highly inheritable and related with several genes, such as TAR DNA-binding protein (TDP), progranulin (GRN), chromosome 9 open reading frame 72 (C9orf72), fused in sarcoma (FUS) and some other rare disease genes (26). Patients with bvFTD may be diagnosed as suffering from psychiatric conditions (Major Depressive Disorder – MDD, BSD, Schizophrenia – SCZ) more often than dementia patients affected by Alzheimer’s disease (AD) or other FTD subtypes (16, 27). Due to symptoms like disinhibition or increased talkativeness and distractibility, early-onset FTD cases must be differentiated from late-onset BSD (28). From a neuropsychological standpoint, bvFTD is characterized by deficits in executive functions, verbal memory, and emotional processing (28). All these alterations may also be found in subsets of BSD patients, especially late in life (28). Psychosis is the most common psychiatric symptom in bvFTD patients, occurring in almost the 50% of cases (16), mainly as delusions and hallucinations. Visual hallucinations, which were initially considered a feature of dementia with Lewy bodies (DLB), are described in C9orf72 and GRN genetic variant carriers (25). Manic or hypomanic episodes, in the absence of cognitive alterations, may appear years before other symptoms of bvFTD and be a prodrome of dementia, possibly indicating an anterior temporal involvement in the disease (16). On the other hand, patients with late-onset bipolar disorder may exhibit a non-progressive “FTD Phenocopy Syndrome” (28), defined as a syndrome that initially meets the criteria for possible FTD but remains stable after 3 years and does not show neuroimaging abnormalities. Furthermore, a distinct subtype of post-BD dementia has been described, with clinical features similar to bvFTD (29).
Typical imaging findings of full-blown FTD (cortical atrophy of the frontal and temporal lobes) may not be present in the disease’s early stages. Also, certain degrees of cortical atrophy can be found in the late course of BSD (28, 30, 31), even though less prominent than in FTD, thereby representing a further complication for the differential diagnosis. 18-Fluorodeoxyglucose-positron emission tomography (FDG-PET) is employed to confirm the diagnosis of bvFTD over BSD, since brain hypometabolism is an early marker of neurodegeneration. Still, several false positive and false negative cases have been described. Also, unspecific hypometabolism, especially in the dorsomedial prefrontal cortex and the superior temporal gyrus, can be found in BSD patients (28). Only follow-up visits can discriminate between the two conditions in these cases.
Both disorders present substantial degrees of inheritance, estimated to be 25% for FTD and 70% for BSD (28). Some genetic loci seem relevant in FTD and BSD (e.g., C9ORF72 and progranulin) (16, 28). For instance, a case of late-onset BSD presenting the C9ORF72 hexanucleotide expansion (32) has been described as well as cases presenting progranulin mutations (33). Moreover, the variability of the GNR gene seems to be related to the susceptibility to develop the BD-I subtype of BSD (34). Phenotypical similarities between late-onset BSD and early bvFTD, as well as the results of GWAS studies, prompted the hypothesis of common molecular mechanisms that cause a neuropsychiatric continuum between the two conditions. Inflammation, a well-known predisposing factor for BSD, has been studied when exploring the association between BSD and FTD. For instance, increased levels of interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and progranulin have been recognized as common inflammatory markers in BSD and FTD (16). Moreover, some human leukocytes antigen (HLA) haplotypes have been related to both BSD and FTD (16).
All these data, taken together, prompted the definition of a new framework in which psychiatric and neurologic conditions – so far considered compartmentalized nosological entities – are described as a continuum epiphenomenon of staged accumulations of structural and functional damage of the cerebral tissue (accelerated brain aging).
Although AD, in its early stages, may present with mood disorders, mainly depressive episodes, other clinical features of BSD are not typically present in AD. However, both conditions are characterized by increased agitation and aggression, affective lability, dysphoria, apathy, impaired self-regulation, and, in some cases, psychosis (35, 36). Of note, the frontal variant of AD (37) is featured by euphoria, disinhibition, and other FTD-like symptoms. Genetic, early-onset AD (EOAD) – e.g., related to PSEN1 mutation – can present with frontal dysfunction (38) and, since it affects subjects in their 30s–40s, may require a differential diagnosis from psychiatric disorders including BSD. Finally, some cases of BSD ultimately lead to cognitive and independence decline, as suggested by Akiskal’s classification (type VI) (13).
BSD and AD share common structural and functional alterations in the hippocampal area. In AD, the hippocampus shows the most prominent and early features of neurodegeneration (37, 39). Alterations in the functional activity of the hippocampus have also been associated with the presence of emotional and cognitive impairment in several mental disorders, including BSD (40). As expected, age-related structural changes in the hippocampus are found in BSD patients (41). Interestingly, the hippocampal volume is also reduced – compared to controls – in young BSD patients naïve to treatment. At the same time, it is relatively increased in lithium-treated BSD patients, thereby suggesting a potential neuroprotective role for the compound (42). Thus, long-term untreated BSD patients may exhibit structural neuroradiological findings that mimic hallmark AD-related neurodegeneration.
From a molecular point of view, BSD and AD share some epigenetic features. These include the hyper/hypo methylation of some promoter regions of the DNA that drives to impaired expression of crucial proteins (e.g., neurotrophic factors and metabolizing enzymes) (41). Accordingly, the expression of the Brain-Derived Neurotropic Factor (BDNF) is decreased in old BSD patients (43), as well as in AD (44). Moreover, BSD patients show increased levels of AD-related amyloid-β (Aβ) (45, 46), with a positive correlation with the number of affective episodes (46). In contrast, no significant changes in cerebrospinal fluid (CSF) levels of tau protein have been found (47). No AD-related CSF alterations have been observed in BSD patients, but some impairment of the Aβ clearance during affective episodes was found, suggesting an association between symptoms and brain amyloidosis in BSD (48). Moreover, dementia in BSD is not associated with the e4 allele of apolipoprotein E (APOE*4), a well-established risk factor for AD (48).
Bipolar spectrum disorders, Parkinson’s disease (PD), and DLB share several clinical features, such as the presence of ICD, and psychotic symptoms (49–52). For instance, hallucinations and delusions are typical of DLB and PD (49, 50) but also of the manic phases of BSD (51). While visual modality is the most common presentation of hallucinations in all three conditions (49–51), delusional content may differ, with paranoid delusions more commonly found in DLB and grandiose types in BSD (53). Impulsivity is a core feature of BSD that leads to engagement in risky behaviors, especially during the manic/hypomanic phases (3, 54) and is also present in parkinsonian patients (55). However, for decades, parkinsonian ICDs have been attributed to iatrogenic side effects of Dopamine Agonist therapies (56–64), before being recognized as a core symptom of the disease, appearing often before any exposition to drug treatments. This phenotype is described in carriers of PRKN and GBA mutations (65–67) leading to PD. Because of the assumed effect of drug treatments, the co-occurrence of BSD and PD has been largely neglected, until large meta-analysis and cohort studies have shown an unquestionable correlation (68–70). Bipolar patients carry a 3.4- to 6.78-fold higher risk of developing PD (68, 69). The risk seems related to the magnitude and frequency of mood shifts (68). The overlap between the two conditions shapes the disease course. Compared to “pure” PD patients, BSD-PD patients, for instance, have been reported to carry higher degrees of neuropsychiatric symptoms like ICD, delusions, or somatizations (5). Furthermore, cognitive impairment may occur earlier in these BSD-PD patients, who also typically exhibit worst outcomes after a Deep Brain Stimulation surgical procedure (71).
The link between BSD and PD has been challenged, by some authors, by calling into question the potential occurrence of iatrogenic parkinsonism in lithium (72) – or valproate (73) – treated patients. However, the high prevalence of dopaminergic transmission impairment at molecular neuroimaging (20%), in bipolar subjects (72), confirm underlying neurodegeneration in many cases. Furthermore, several fMRI studies have documented, in BSD patients, a lack of deactivation during tasks of cortical regions belonging to the default mode network (DMN) [namely the medial prefrontal cortex (mPFC) (74–76), and posterior cingulate cortex (PCC) (77)] along with a decreased activation of dorsolateral prefrontal cortex (DLPFC) (78). This condition, considered a robust indicator of BSD, may represent the hallmark of a deranged connectivity [possibly due to white matter damage (79)] between the DMN and the attentional networks (a condition also known as “corticolimbic dysfunction”) (78, 80, 81). As a consequence, the reality-checking failure in these patients is impaired. The same findings were described in patients with PD or DLB, consisting of disinhibition of PCC and decreased activation of DLPFC (82, 83). In DLB the activity of PCC is proposed as a diagnostic index, vs. other types of dementia, and is denominated Cingulate Island Sign (82, 83). Therefore, psychosis, in the two disorders, shares a common pathophysiological ground as evidenced by networks dysfunctions (84, 85).
The dysregulation of the dopaminergic circuitry – the leading cause of several parkinsonian symptoms – has also been recognized in BSD. In particular, the combined mood/motor fluctuations of PD (e.g., motor and psychic off states vs. dyskinesia and ICD) have been compared to BSD-related mood swings (69, 86). However, while mechanisms by which dopaminergic dysregulations and related neurodegeneration occur in PD are quite clear the BSD pathophysiological cascade is less understood. For instance, the neuropathology of BSD does not account for a unique, distinctive pattern. However, sporadic observations have confirmed the possible presence of Lewy body pathology in bipolar patients (87, 88). Moreover, a decreased parietal concentration of monomeric α-synuclein, possibly reflecting a synaptic impairment, has been observed in bipolar patients who had developed parkinsonism (89). Still, further studies are needed to assess the biochemical and pathological relationships between synucleinopathies and BSD.
Multiple sclerosis (MS) is a chronic autoimmune inflammatory demyelinating disorder of the central nervous system (CNS) with a variable and unpredictable course (90, 91). Mood disorders are commonly the most reported neuropsychiatric comorbidity in MS. These are also associated with lower treatment compliance, poor outcomes and functional status, and overall decreased quality of life (90, 92–94). An increased risk for psychiatric disorders is recognized even before a definite diagnosis of MS is made (OR = 1.4, IC 1.05–1.88) (95). The lifetime prevalence of BSD is higher in people with MS than in sex- and age-matched controls, and estimated to range from 0 to 16.2% in different population-based studies (91, 96–98). BSD also affect the pediatric MS population with a prevalence of 3.57% (99). In a recent meta-analysis on MS, a higher BSD prevalence was found in the Americas than in Europe (90, 98, 100).
White matter abnormalities (WMa) are a typical MS neuroradiological finding. In BSD patients, WMa are also common. They occur, predominantly in the right posterior temporoparietal and left cingulate regions, along with thinning of the cortical gray matter in the frontal, temporal, and parietal regions of both hemispheres (101, 102). MS lesions in brain areas, critical for the control of affective functions, could be a substantial contributing factor toward an increased diagnosis of BSD in this population (98, 103, 104).
The link between MS and BSD has not been fully explored. However, a possible genetic association involving the HLA region has been suggested in patients with MS and a family history of BSD (98, 105–109). Neuroinflammation is a shared trait between MS and – to a lesser extent – BSD (90). Neuroinflammation is involved in the pathogenesis of mood disorders, as indicated by the post-mortem detection of increased protein and mRNA levels of interleukin 1β (IL-1β), interleukin 1 receptor, myeloid differentiation factor 88 (MyD88), nuclear factor-kappa B (NFκB) subunits and astroglial and microglial markers (glial fibrillary acidic protein, GFAP; inducible nitric oxide synthase, iNOS; c-fos, and CD11b) in the frontal cortex of BSD patients (110, 111). In addition, mood symptoms are exacerbated by MS treatments like corticosteroids or baclofen (112). High-dose corticosteroids, in addition to anti-inflammatory effects, can modulate metabolism, cognitive performance, and emotional responses due to the interaction with several neurotransmitters and neuropeptide systems, as well as increasing adrenal responsiveness (113). Thus, while steroids’ capacity to trigger manic episodes is well understood, the mechanism through which baclofen – a myorelaxant – induces manic symptoms is not well identified but may partially rely on its effects on the dopaminergic or serotoninergic systems (114).
Encephalitis is an inflammatory condition of the brain frequently caused by either viral or immune-mediated processes. The latter, namely called “autoimmune encephalitis” is due to the production of antibodies against intracellular neuronal proteins (onconeural proteins), or neuronal cell surface/synaptic proteins. The most common forms of AE are those caused by antibodies anti N-methyl-D-aspartate (NMDA) receptor, anti-leucine-rich glioma inactivated 1 (LGI1), anti-contactin-associated protein-like 2 (CASPR2), anti alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, anti gamma-aminobutyric acid A or B (GABA-A or GABA-B) receptor, anti IgLON family member 5 (IgLON5), anti dipeptidyl-peptidase-like protein-6 (DPPX), anti glycine receptor (GlyR), anti neurexin-3 alpha and anti myelin-oligodendrocyte glycoprotein (MOG) (115). Encephalitis – limbic variant above all – is characterized by neurologic dysfunction and psychiatric symptoms like irritability, mood disturbance, hallucinations, and personality disturbances, mostly present in anti-NMDAR, anti-GABA-A-R and anti-AMPAR encephalitis (115–118). BSD symptoms can be the only clinical manifestation of AE at the onset and can be attributed only to a psychiatric disorder, thereby leading to misdiagnosis (117, 118). Moreover, antibodies typical of different AE can be found in patients with BSD or other psychiatric disorders without neurological involvement (117, 118). Patients with bipolar disorder frequently present with antibodies against AMPAR, ATPA (H/K Adenosine Triphosphate), CASPR2, GAD65 (Glutamic Acid Decarboxylase), NMDAR and VGKC (Voltage Gated Potassium Channel) complex in serum (116, 119). Almost 67% of patients with anti-NMDAR encephalitis (ANMDARE) experience psychiatric symptoms and require psychiatric care (120–123). After a first “flu-like” phase of the disease, with subtle symptoms such as anxiety, agitation, and short-term memory loss, the following phase is characterized by 1–3 weeks of more severe and debilitating mood and behavioral disturbances, cognitive impairment, and psychosis. Eventually, the full-blown phase consists of dyskinesia, dystonic posture, seizures, autonomic instability, and decreased level of consciousness (121). A subset of patients affected by ANMDARE presents pure psychiatric symptoms during specific periods of their illness, with no neurological symptoms (121). BSD should also be considered in the differential diagnosis of catatonia, which is present in many other medical and neurologic conditions that require prompt recognition and intervention. Catatonia could be the next step of the delirious mania, characterized by an acute onset of manic signs and symptoms, accompanied by a waxing and waning pattern of consciousness (124) and is also a feature of AE dependent on different autoantibodies than the anti-NMDAR, e.g., anti-GABA (115). The case of a patient with a late-onset manic syndrome, followed by a worsening of the level of consciousness, and catatonic features, has been described (124). While, in the initial phase, the symptoms were attributed to BSD, further investigations were consistent with a diagnosis of Non-Convulsive Status Epilepticus and the patient was treated with benefits with immunotherapy, suggesting the probable diagnosis of AE (124). Moreover, also anti-AMPAR encephalitis can be characterized by psychiatric symptoms (125). In a case series of 22 patients with AMPA receptor antibodies psychosis with bipolar features has been described in one patient (125). In literature it has been also reported the case of a 20-year-old woman with Turner Syndrome who presented bipolar mood disorder, psychotic and other psychiatric symptoms, and anti-AMPA receptor encephalitis (126).
In a cohort of 571 patients with ANMDARE, it has been found that the levels of auto-Abs decreased across the manic episode, suggesting a possible role of anti-NMDAR auto-Abs as a biomarker of acute episode (120, 121). Moreover, anti-NMDA receptor Abs have also been detected in serum from ∼10% of healthy controls as well as in patients with pure psychotic symptoms suggesting an autoimmune process as a pathogenetic mechanism in a subgroup of bipolar patients who present increased levels of anti-NMDAR auto-Abs.
The imbalance of glutamatergic system is involved in the pathophysiology of neuropsychiatric symptoms, including BSD. Evidence derived from genetic, post-mortem, biochemical, and imaging studies suggest that BSD is characterized by altered levels of glutamate, abnormalities in the NMDAR gene expression, concentration, and functioning (111, 127). ANMDARE is triggered by IgG auto-Abs against the NR1 subunit of the NMDAR. It has been supposed that the early phase of the ANMDARE is related to initial intrathecal penetration and antibody diffusion into cortical and hippocampal gray matter with resulting disinhibition and psychiatric symptoms. In the late phase, a secondary immunologic expansion within the intrathecal compartment could determine the further imbalance of the glutamatergic neurotransmission with an increase of neurologic symptoms (121). The involvement of anti-AMPA receptor antibodies confirm the implication of the glutamatergic system in the pathogenesis of both BSD and AE, since glutamate-mediated excitotoxicity takes place in many types of acute and chronic CNS diseases (126). Activation of microglia and presence of specific inflammatory proteins such as IL-6 and interleukin-8 (IL-8) in the CSF of psychiatric patients with schizophrenia or affective disorders support the role of CNS inflammation in these disorders (116).
Bipolar spectrum disorders has been frequently described as rare in patients with epilepsy, due to anecdotical evidence rather than a standardized evaluation of the actual psychopathology (128). Nonetheless, growing evidence indicate an increasing prevalence of BSD and bipolar symptoms in people with epilepsy compared with that in the general population (128, 129). Moreover, psychogenic non-epileptic seizures (PNES), a subgroup of conversion disorders (CD) characterized by paroxysmal motor, non-motor, or behavioral alterations that resemble epileptic seizures without EEG correlates, are a common comorbidity of epilepsy and may be present in several psychiatric conditions, including BSD (130). A US survey reports manic symptoms to be present in 12.2% of patients with epilepsy, which is twice as common as in people with asthma and 7 times as common as in the healthy comparison group (128, 129, 131). A European study describes similar percentages (128, 132, 133). However, potential confounding variables should be considered. In most cases, bipolar symptoms occur around the ictal episode. Behavioral manifestations may precede (preictal) or follow (postictal) the ictal event, or they may represent the expression of the seizure activity (ictal) (128, 134, 135). An interictal dysphoric disorder (IDD) that may represent a phenotype copy of BSD has also been described (128, 132, 136). Manic symptoms may also be rare side effects of antiepileptic drugs (AEDs) (137, 138). AED-related manic symptoms may be due to the toxic effects of the drug or the forced normalization phenomenon (129). Currently, the link between the two conditions is still tenuous. However, considering the substantial burden of psychiatric disorders in epileptic patients, bipolar symptoms should be appropriately recognized and treated (128).
Bipolar spectrum disorders and epilepsy have a similar clinical course, characterized by acute and paroxysmal episodes (128, 129, 139) treated with AEDs like carbamazepine, valproate, and lamotrigine (128, 137). It has been hypothesized that the kindling model represents the underlying pathophysiology of seizures and BSD. Moreover, the efficacy of several AEDs in BSD could be explained by similar modifications in neurotransmitters or the functioning of voltage-opened ion channels (129). Different studies suggest altered GABAergic-related mechanisms in BSD and the involvement of glutamate in stress-related neurotoxicity. The same neurochemical imbalance has been described as a relevant pathogenetic driver of epilepsy, possibly playing a similar role in both conditions (140). In patients with acute mania and BSD, an excess of the inward sodium current known to correlate with neuronal hyperexcitability and seizure facilitation has been described (128).
Bipolar spectrum disorders show a high degree of heritability (60–80% in twins studies) (141), while no mendelian inheritance pattern has been found. GWAS studies have identified more than 40 SNPs associated with an increased risk of development of BSD (16, 142). A recent GWAS analysis has found 87 enriched pathways that are relevant for BSD. Most are involved in cellular processes, signal transduction, metabolic processes, neuronal activities, immune system, and inflammation-related processes (143). Six of these metabolic pathways (drug metabolism, retinol metabolism, pentose and glucuronate interconversions, porphyrin and chlorophyll metabolism, starch and sucrose metabolism, ascorbate, and aldarate metabolism) were connected to dementia-related mechanisms, thereby suggesting links between BSD and the risk of dementia (143).
Calcium dysregulation has long been implicated in BSD pathophysiology, since intracellular calcium signaling is increased in BSD (144). Transient receptor potential (TRP) is a family of non-selective cation permeable channels, most of which show a low selectivity for Ca2+ ions. The receptor has been implicated in a wide array of CNS disorders, such as AD, PD, stroke, epilepsy, depression, bipolar disorder, and migraine, all associated with disturbances in Ca2+ homeostasis (145, 146). In BSD, TRP channels play an important role in disease development and progression. Genetic studies on BSD have shown that a mutation of TRPM2 gene reduces the dephosphorylation of GSK-3β, a crucial pathway involved in Ca2+ homeostasis (146, 147). Thus, TRPM2, as well as other TRP channels, may play a role in neurodegenerative disorders by interfering with Ca2+ homeostasis and mitochondrial activity, therefore activating the apoptosis pathways (146). TRP family is also involved in the etiology of epilepsy and in the generation of seizures (146). A knockout (KO) TRPM2 murine model showed reduced levels of neuroinflammation and neurodegeneration and increased epilepsy-induced psychological disorders, suggesting that TRPM2 favors the development and evolution of epilepsy-related brain injury (148).
Sodium–potassium ATPase (Na+/K+-ATPase) is an integral protein complex, responsible for ions transfer across the plasma membrane. In the healthy brain, almost 50% of the ATP consumption is due to the activity of this complex. Its structural or functional alterations impair neuronal excitability (149) and promote intracellular Ca2+ overloads (149, 150). It has been suggested that mood state-related reductions in Na+/K+-ATPase plays a central role in the pathophysiology of BSD (150).
In particular, allelic association between BD and a Na+, K+-ATPase α subunit gene (ATP1A3) has been reported (151). The decreased Na+/K+-ATPase activity in peripheral blood cells has been described during the acute phases of BSD (150, 151). Several studies have reported that intracerebroventricular injection of ouabain (a Na+/K+-ATPase inhibitor) induces hyperactive behavior in rats. Moreover, Li+ increases the Na+/K+-ATPase activity by reducing the formation of myoinositol and, as a consequence, attenuates the second messenger response (150). Several protein aggregates implicated in the pathophysiology of neurodegenerative disorders (amyloid-β, α-synuclein, and protein tau) directly interact with the Na+/K+-ATPase, in particular with its α3-subunit, and its activity may be reduced by this pathological conditions (149), leading to the development of manic disorders and, at the same time, to intracellular Ca2+ overload, that enhances the neurodegenerative process.
During the last decades, growing evidence has supported the role of inflammation in the development of mood symptoms and BSD. The observation of the so-called “sickness behavior,” i.e., the behavioral changes observed in inflammatory conditions, characterized by changes in mood, social behavior, and cognition, has suggested a role of the immune system in the development of mood disorders (111, 152). Even the SARS-CoV-2 infection has been related with the development of manic episodes (153). Moreover, a certain degree of association between BSD and autoimmune diseases is known (107) and a prior hospitalization due to an autoimmune disease or infection has been recognized as a risk factor for developing a major mood disorder (111). Following this observational evidence, many studies have investigated the inflammatory state of patients with BSD, highlighting the presence of increased plasma levels of some pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) in BSD patients, compared to healthy controls (152), mainly during manic episodes. BSD also show higher levels of CRP compared to healthy controls in any stage of the disease. These levels increase more during the manic phase than during depression or euthymia (152, 154). Autoimmune pathologies (such as MS and systemic autoimmune diseases) and infections can trigger a condition of chronic low-grade inflammation. The mechanisms that can explain the neuropsychiatric alteration following infectious events are molecular mimicry, stress-induced microglial activation, disruption of the BBB, and alterations of epigenetic pathways (111). Chronic inflammation can lead to neurotoxicity and neurodegeneration as pro-inflammatory cytokines increase oxidative stress and promote microglial hyperactivation, thereby impairing neurotransmission and triggering neuronal loss (155). Post-mortem evaluations have revealed significant increases in excitotoxicity and neuroinflammatory markers in the prefrontal cortex of BSD patients, confirming the hyperactivation of the IL-1R receptor cascade and increases in NMDA receptor activity (155). The overlap between neuroinflammatory and psychiatric conditions is becoming so evident that some authors have conceptualized the notion of “autoimmune psychosis.” From this point of view, besides the clear autoimmune conditions defined in the spectrum of AE, it is possible to define a large number of psychiatric disorders as the expression of an overactivation of immune response that increases the risk of developing psychotic symptoms (111). Therefore, these authors have proposed a diagnostic algorithm to better identify immunologic alterations in patients with new-onset neuropsychiatric symptoms that involves screening for blood, CSF, and imaging markers (111).
Excessive dopamine neurotransmission is involved in the development of manic and psychotic symptoms (156). Reduced dopamine transporter functioning, i.e., the impaired regulation of dopamine levels at synapses, recapitulates many aspects of BSD-related manic manifestations in preclinical models (157). Reduced plasma levels of dopamine-β-hydroxylase (DβH), an enzyme converting dopamine to norepinephrine, relate to the severity of BSD symptoms (158), and, compared to controls, striatal dopamine synthesis is increased in BSD patients with psychosis (159). The glutamatergic system has also been implicated in the pathophysiology of BSD (160, 161). The hyperactivation of these pathways can synergistically promote neuronal and non-neuronal stress, which leads to functional impairment and cell depletion. Dopamine has a high redox potential, reacts with nitrogen species, and increases oxidative stress (162). GABAergic transmission is decreased in BSD (140), thereby promoting glutamate/GABA imbalance with increased excitotoxicity (163) (164). Serotonin (5-HT) has been extensively associated with several psychiatric disorders, including BSD and ICD (165–167). Selective serotonin reuptake inhibitors (SSRIs) are the most used drugs for depressive mood disorders (168). Some evidence indicate a close interaction between the serotoninergic system and neurotrophic factors. For instance, BDNF promotes the development and optimal activity of serotoninergic neurons. At the same time, 5-HT controls and enhances BDNF expression (169), providing another functional crosslink between mood disorders and neurodegeneration.
Our review outlines the outstanding co-occurrence of Neurologic Disorders and BSDs (Figures 1–3).
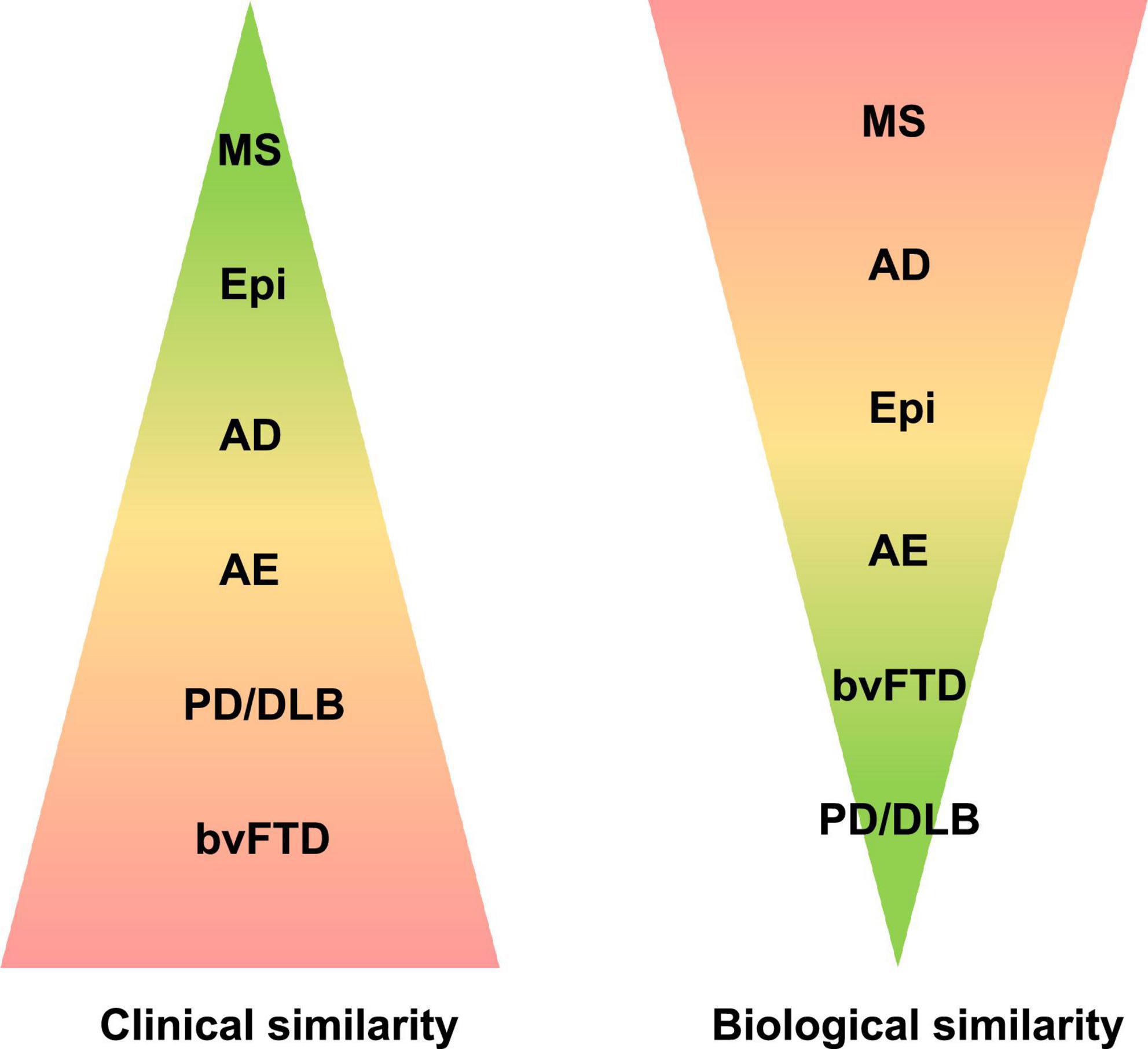
Figure 2. Neurological disorders of the central nervous system are here classified according to clinical and biological similarity to bipolar spectrum disorders (BSDs). Color coding and the corresponding area indicate the size of similarity. AD, Alzheimer’s disease; AE, autoimmune encephalitis; bvFTD, behavioral variant of frontotemporal dementia; Epi, epilepsy; MS, multiple sclerosis; PD/DLB, Parkinson’s disease, dementia with Lewy bodies.
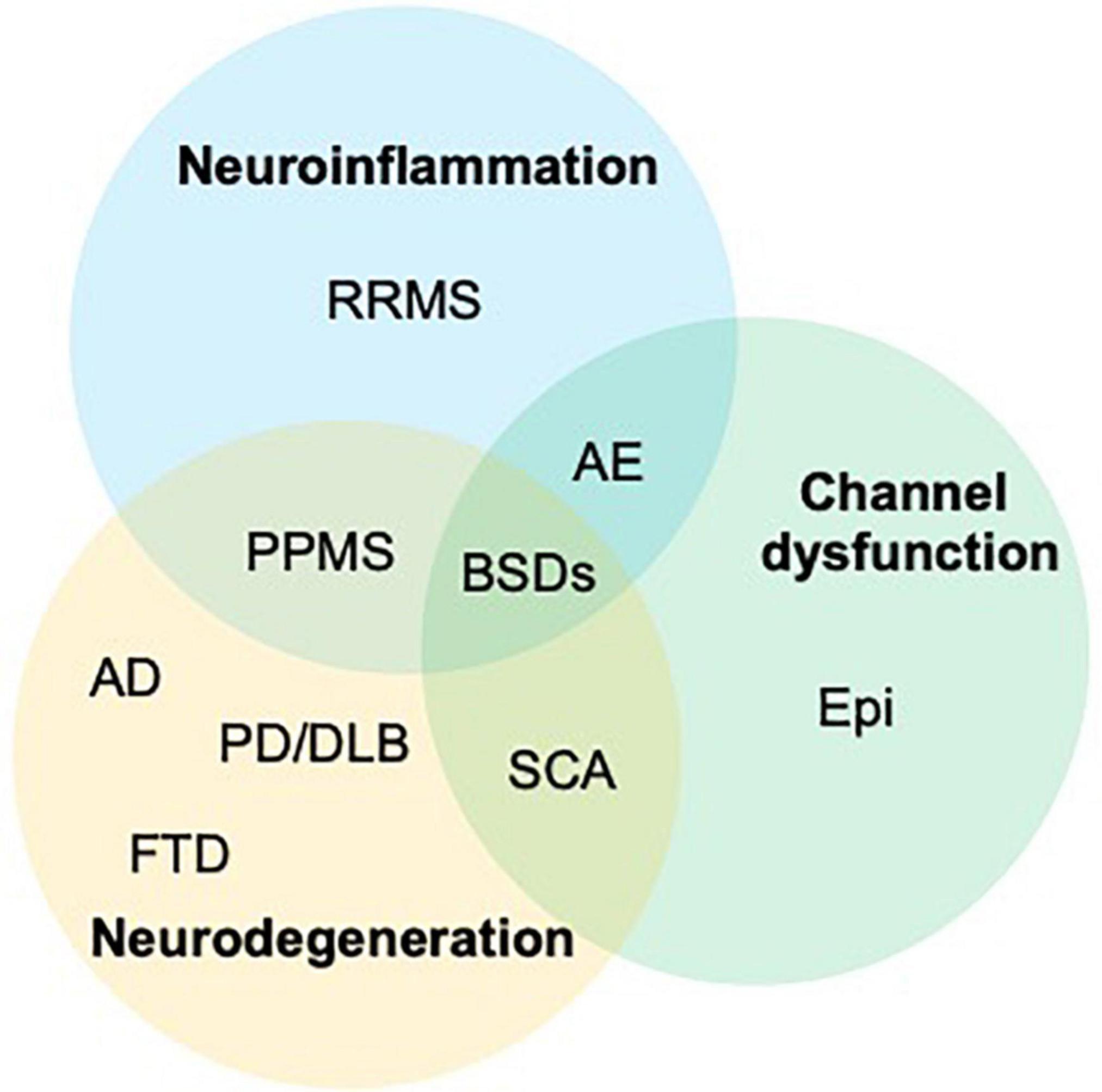
Figure 3. The graph outlines the intersections of three main pathological changes affecting the central nervous system and shows how all three are involved in the onset of bipolar spectrum disorders (BSDs). AD, Alzheimer’s disease; AE, autoimmune encephalitis; bvFTD, behavioral variant of frontotemporal dementia; Epi, epilepsy; PD/DLB, Parkinson’s disease, dementia with Lewy bodies; PPMS, primary progressive MS; RRMS, relapsing remitting multiple sclerosis; SCA, spinocerebellar ataxias.
Among the different studies, attention was addressed to possible common mechanism related to synaptic malfunctions (e.g., epilepsy), network disruption (e.g., MS), neurotransmitters imbalance (PD and dementias). Some studies attempted an investigation of possible phenotypes of Dementia associated with BSD.
For FTD and AE the occurrence of BSD symptoms was interpreted as a misdiagnosis of BSD, and this interpretation may be deemed acceptable, due to the prominent occurrence of psychosis in FTD (16, 27, 28) and to the short disease course in Encephalitis.
As this interpretation appears untenable for other Neurologic Disorders, the co-occurrence of BSD and Neurologic Disorders was, mostly, explained as a dependent on epidemiological overlaps, due to the prevalence of the disorders in general populations.
Bipolar spectrum disorders has a prevalence of around 5%, when considering plainly the three variants categorized by DSM-5 (3). PD has a prevalence of 2% in populations aged above 65, Dementias, also, have age-dependent prevalences, ranging from 5 to 20% or more according to age. The population prevalence of Epilepsy in its severe/refractory forms and of MS is around 0.05%. Therefore, in a gross estimate, 5% of patients with neurologic disorders would be expected to present with a history of BSD. Given the high prevalence of BSD, PD, and Dementia the epidemiological overlaps would result in a considerable number of patients sharing common features.
Therefore, in Neurologic Disorders, the co-occurrence was interpreted as a statistical effect (27, 70, 91), or random finding, until recent powerful epidemiology and meta-analysis studies outlined the coexistence of disorders, and the predictive value for the development of symptoms and response to treatments (16, 98, 129).
This statement is definitely valid for PD and Dementia (5, 20, 27, 87, 170–172), needs validation for Epilepsy and MS.
The evidence of an undeniable statistical correlation, suggests the necessity for different considerations, addressing practical and theoretical issues.
The robust evidence of a phenotype including BSD symptoms for Neurologic Diseases, with emergence of manic conditions, and its corollaries, including ICDs, somatic symptoms disorders and addiction behaviors, suggests that some assumptions related to drug effects should be reconsidered with caution.
An outstanding example is provided by the history of dopamine agonist (DAs) treatments in PD.
Dopamine agonist induce psychosis and ICDs in a variable percentage of PD patients, ranging from 8 to 30%. ICD, as described in these patients, consisted of gambling, hypersexuality, paraphilias, shopping sprees, aggressive driving, and hoarding, addictive consumption of drugs, compulsory activities often termed “punding,” with a denomination borrowed from definitions of effects of cocaine addiction (173–177).
This effect of Das was described since the early introduction of ergolinic Das, like bromocriptine, cabergoline, or pergolide (174, 178–181), and was also described in patients receiving only L-dopa based treatments (182–185). It must be remembered also, that J Updike, in his “Rabbit Tetralogy” (186) described, masterly, the mood shift induced by L-dopa, with hypersexuality, at a time when L-dopa was still a new, “experimental” treatment. It was, however, with the introduction of non-ergolinic Das (pramipexole, ropinirole, and rotigotine, which, at difference with ergolinic Das, were not causing organ fibrosis and cardiac vegetations) that ICD induction reached a worldwide mediatic coverage (58, 187–191), with presentation of class actions, successfully claiming compensations from pharmaceutical companies and prescribers (medical doctors).
These actions ended definitively the research on Das, including apomorphine, a DA with wider receptor interactions than ergolinic and non-ergolinic Das (192).
Despite the grossly evident similarity of these Das effects with the behavioral (manic) shifts induced by antidepressants in BSD patients, the pharmacological mechanisms were not investigated, therefore there is no understanding of the reason why drugs scarcely, or not, acting on serotoninergic pathways should induce similar, or same, effects as drugs acting on serotonine reuptake.
This blurring by mediatic clamors, left many other questions unanswered, in example:
(1) On the precise pharmacological effects of Das, i.e., the question is whether the known presynaptic effect of Das (193–195), as Das do not act only at a post-synaptic level but also on the release of catecholamine quantums from presynaptic vesicles (193), is effected at a general presynaptic level on catecholamines, including serotonine, or it acts on the single neurotransmitter dopamine.
(2) On the effect, and dose requirements, of DAs in patients with severe depression, as a mood shift induction should be properly investigated, in order to provide new treatment options in other mental disorders (196–198).
(3) On the mechanism of DAs withdrawal syndrome (DAWS) (199), which consists of severe dysphoria with dysautonomia, observed in a minority of PD after the interruption of DAs treatments, and which is again similar to severe rebounds of depression in patients experiencing withdrawal of antidepressant drugs.
Moreover, there are no studies on the follow-up of patients who, putatively, experienced ICD because of DAs exposition, and it is not known, therefore, whether the effect was unique or linked to more complex predispositions.
The example of PD shows the disruptive effect of mediatic clamor on scientific inquiry, with its burden of unanswered questions, but other unanswered questions emerge when considering, among the other conditions, the co-occurrence of BSD and Dementia or Epilepsy.
Properly designed studies should address questions on antidepressant drugs effects and mechanisms in the subphenotypes expressing the association of disorders, in order to attempt the tailored approach to treatments.
The coexistence of different neurologic disorders and bipolar disorder suggests that the behavioral patterns which are deemed proper of BSD are the result of predispositions to unstable control system acting at the network or at the synaptic level, giving origin to the cascade of symptoms as a final malfunctioning pathway. Similar malfunctions of the DMN are observed in psychotic episodes of FTD, PD, Dementias, some Epilepsies, and manic episodes with psychosis of BSD patients (78, 80, 81). The core malfunction results in disinhibition of PCC, which is released from the control of Frontal Cortex, possibly resulting in the activation of short distance connections (“small world” networks, as opposed to “large world” connections including the Fronto-Parietal Control network) (200, 201) and the emergence of resilient hallucinations, delusions and subcortical behaviors, like the impulsive ones linked to frontal lobes alterations. The hypothesis could be pushed forward, to hypothesize that BSD, in its most severe features, could represent the final phenotype of dysfunctions taking origins from different causative factors, but converging to a common eventual pathway. In this context, the current debate on the psychiatric classification system highlights the importance of adopting the “medical model” of disease categorization, which assumes that a disease is characterized by three components: an etiological agent, a pathological process, and symptoms/signs (202). This approach is deemed essential to strengthen the “scientific credibility” of psychiatry. Nowadays, the accepted systematization of psychiatric disorders is mainly based on the clinical presentation [DSM-5, (3)] and systematically leaves out the etiologic question. On the other hand, a growing amount of evidence from the neurobiological correlates of psychiatric conditions is setting the foundations for a more biological, and thus “valid” and “reliable,” approach to the field (203). In the future, the identification of a definite pathophysiological background will strengthen the nosographic understanding of BSD as a continuum and mitigate the common problem, in psychiatry, of low diagnostic validity, as juxtaposed to good diagnostic reliability (204, 205). Thus, future neurobiological and neuroimaging studies should focus on discovering a “BSD fingerprint,” which could help disentangle complex and overlapping clinical pictures and guide the formulation of future diagnostic criteria. As discussed in a recent paper (206), the Frontal lobe, due to its evolutional characteristics, is the weak point of human brain, as the size increment of the structure was not paralleled by size increments of brainstem structures providing the neurotransmitters supply (5, 206). With this hypothesis, the need for complex recategorization of BSD, such as proposed by Akiskal and Pinto, who provided examples of several BSD prototypes (13), finds a robust support, and opens new approaches to research.
AD, PA, MO, and AT: conceptualization. AD, PA, and MO: methodology. AD, PA, MR, MO, and AT: writing—original draft preparation. AD, PA, MR, SS, MO, and AT: writing—review and editing. SS, MO, and AT: supervision. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AD, Alzheimer’s disease; AE, autoimmune encephalitis; AEDs, antiepileptic drugs; ANMDARE, anti-NMDAR encephalitis; APP, primary progressive aphasia; BSD, bipolar spectrum disorder; CD, conversion disorders; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies; DLPFC, dorsolateral prefrontal cortex; DMN, default mode network; EOAD, early-onset Alzheimer’s disease; FTD, frontotemporal dementia; bvFTD, behavioral variant frontotemporal dementia; ICDs, impulse control disorders; IDD, interictal dysphoric disorder; MDD, major depressive disorder; mPFC, medial prefrontal cortex; MS, multiple sclerosis; PCC, posterior cingulate cortex; PD, Parkinson’s disease; PNES, psychogenic non-epileptic seizures; SCA, spinocerebellar ataxias; SCZ, schizophrenia; Wma, white matter abnormalities.
1. Bonnín CDM, Reinares M, Martínez-Arán A, Jiménez E, Sánchez-Moreno J, Solé B, et al. Improving functioning, quality of life, and well-being in patients with bipolar disorder. Int J Neuropsychopharmacol. (2019) 22:467–77. doi: 10.1093/ijnp/pyz018
2. Sinha A, Shariq A, Said K, Sharma A, Jeffrey Newport D, Salloum IM. Medical comorbidities in bipolar disorder. Curr Psychiatry Rep. (2018) 20:36. doi: 10.1007/s11920-018-0897-8
3. American Psychiatric Association. DSM-5 Task Force., Diagnostic and Statistical Manual of Mental Disorders : DSM-5. 5th ed. Washington, DC: American Psychiatric Association (2013). 947 p.
4. Muzina DJ. Bipolar spectrum disorder: differential diagnosis and treatment. Prim Care. (2007) 34:521–50. doi: 10.1016/j.pop.2007.06.001
5. Onofrj M, Di Iorio A, Carrarini C, Russo M, Franciotti R, Espay A, et al. Preexisting bipolar disorder influences the subsequent phenotype of Parkinson’s disease. Mov Disord. (2021) 36:2840–52. doi: 10.1002/mds.28745
6. Pichot P. The birth of the bipolar disorder. Eur Psychiatry. (1995) 10:1–10. doi: 10.1016/0767-399X(96)80069-1
7. Leonhard K. [Significance of twin birth for the appearance of cyclic psychoses]. Psychiatr Neurol Med Psychol. (1976) 28:89–98.
8. Groth M, Lundin T, Stål K, Tjelldén G. [Cyclic psychoses – a typical case]. Lakartidningen. (1977) 74:4221–2.
10. Kendler KS, Engstrom EJ. Kahlbaum, Hecker, and Kraepelin and the transition from psychiatric symptom complexes to empirical disease forms. Am J Psychiatry. (2017) 174:102–9. doi: 10.1176/appi.ajp.2016.16030375
11. Wolman BB. New ideas on mental disorders. Am J Psychother. (1977) 31:546–60. doi: 10.1176/appi.psychotherapy.1977.31.4.546
12. Leonhard K. Cycloid psychoses–endogenous psychoses which are neither schizophrenic nor manic-depressive. J Ment Sci. (1961) 107:633–48. doi: 10.1192/bjp.107.449.633
13. Akiskal HS, Pinto O. The evolving bipolar spectrum. Prototypes I, II, III, and IV. Psychiatr Clin North Am. (1999) 22:517–34. doi: 10.1016/s0193-953x(05)70093-9
14. Ng B, Camacho A, Lara DR, Brunstein MG, Pinto OC, Akiskal HS. A case series on the hypothesized connection between dementia and bipolar spectrum disorders: bipolar type VI? J Affect Disord. (2008) 107:307–15. doi: 10.1016/j.jad.2007.08.018
15. Kasztelan-Szczerbinska B, Cichoz-Lach H. Wilson’s disease: an update on the diagnostic workup and management. J Clin Med. (2021) 10:5097. doi: 10.3390/jcm10215097
16. Nascimento C, Nunes V, Diehl RR, Takada L, Suemoto C, Grinberg L, et al. A review on shared clinical and molecular mechanisms between bipolar disorder and frontotemporal dementia. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 93:269–83. doi: 10.1016/j.pnpbp.2019.04.008
17. Callahan BL, McLaren-Gradinaru M, Burles F, Iaria G. How does dementia begin to manifest in bipolar disorder? A description of prodromal clinical and cognitive changes. J Alzheimers Dis. (2021) 82:737–48. doi: 10.3233/JAD-201240
18. Lee RS, Hermens D, Scott J, Redoblado-Hodge M, Naismith S, Lagopoulos J, et al. A meta-analysis of neuropsychological functioning in first-episode bipolar disorders. J Psychiatr Res. (2014) 57:1–11. doi: 10.1016/j.jpsychires.2014.06.019
19. Daglas R, Yücel M, Cotton S, Allott K, Hetrick S, Berk M. Cognitive impairment in first-episode mania: a systematic review of the evidence in the acute and remission phases of the illness. Int J Bipolar Disord. (2015) 3:9. doi: 10.1186/s40345-015-0024-2
20. Lopes R, Fernandes L. Bipolar disorder: clinical perspectives and implications with cognitive dysfunction and dementia. Depress Res Treat. (2012) 2012:275957. doi: 10.1155/2012/275957
21. Cardoso T, Bauer IE, Meyer TD, Kapczinski F, Soares JC. Neuroprogression and cognitive functioning in bipolar disorder: a systematic review. Curr Psychiatry Rep. (2015) 17:75. doi: 10.1007/s11920-015-0605-x
22. Cao B, Passos I, Mwangi B, Bauer I, Zunta-Soares G, Kapczinski F, et al. Hippocampal volume and verbal memory performance in late-stage bipolar disorder. J Psychiatr Res. (2016) 73:102–7. doi: 10.1016/j.jpsychires.2015.12.012
23. Vieta E, Popovic D, Rosa A, Solé B, Grande I, Frey B, et al. The clinical implications of cognitive impairment and allostatic load in bipolar disorder. Eur Psychiatry. (2013) 28:21–9. doi: 10.1016/j.eurpsy.2011.11.007
24. Gorno-Tempini ML, Hillis A, Weintraub S, Kertesz A, Mendez M, Cappa S, et al. Classification of primary progressive aphasia and its variants. Neurology. (2011) 76:1006–14. doi: 10.1212/WNL.0b013e31821103e6
25. Perry DC, Brown J, Possin K, Datta S, Trujillo A, Radke A, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. (2017) 140:3329–45. doi: 10.1093/brain/awx254
26. Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. (2016) 138 Suppl 1:32–53. doi: 10.1111/jnc.13622
27. Woolley JD, Khan BK, Murthy NK, Miller BL, Rankin KP. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. J Clin Psychiatry. (2011) 72:126–33. doi: 10.4088/JCP.10m06382oli
28. Maia da Silva MN, Porto F, Lopes P, Sodré de Castro Prado C, Frota N, Alves C, et al. Frontotemporal dementia and late-onset bipolar disorder: the many directions of a busy road. Front Psychiatry. (2021) 12:768722. doi: 10.3389/fpsyt.2021.768722
29. Lebert F, Lys H, Haëm E, Pasquier F. [Dementia following bipolar disorder]. Encephale. (2008) 34:606–10. doi: 10.1016/j.encep.2007.12.007
30. Coyle TR, Kochunov P, Patel R, Nery F, Lancaster J, Mangin J, et al. Cortical sulci and bipolar disorder. Neuroreport. (2006) 17:1739–42. doi: 10.1097/01.wnr.0000239957.53072.f0
31. Lyoo IK, Sung Y, Dager S, Friedman S, Lee J, Kim S, et al. Regional cerebral cortical thinning in bipolar disorder. Bipolar Disord. (2006) 8:65–74. doi: 10.1111/j.1399-5618.2006.00284.x
32. Floris G, Borghero G, Cannas A, Stefano F, Murru M, Corongiu D, et al. Bipolar affective disorder preceding frontotemporal dementia in a patient with C9ORF72 mutation: is there a genetic link between these two disorders? J Neurol. (2013) 260:1155–7. doi: 10.1007/s00415-013-6833-2
33. Rubino E, Vacca A, Gallone S, Govone F, Zucca M, Gai A, et al. Late onset bipolar disorder and frontotemporal dementia with mutation in progranulin gene: a case report. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:624–6. doi: 10.1080/21678421.2017.1339716
34. Galimberti D, Prunas C, Paoli R, Dell’Osso B, Fenoglio C, Villa C, et al. Progranulin gene variability influences the risk for bipolar I disorder, but not bipolar II disorder. Bipolar Disord. (2014) 16:769–72. doi: 10.1111/bdi.12180
35. Besga A, Gonzalez I, Echeburua E, Savio A, Ayerdi B, Chyzhyk D, et al. Discrimination between Alzheimer’s disease and late onset bipolar disorder using multivariate analysis. Front Aging Neurosci. (2015) 7:231. doi: 10.3389/fnagi.2015.00231
36. Albert MS, Blacker D. Mild cognitive impairment and dementia. Annu Rev Clin Psychol. (2006) 2:379–88. doi: 10.1146/annurev.clinpsy.1.102803.144039
37. Dubois B, Feldman H, Jacova C, Hampel H, Molinuevo J, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. (2014) 13:614–29. doi: 10.1016/S1474-4422(14)70090-0
38. Riudavets MA, Bartoloni L, Troncoso J, Pletnikova O, St George-Hyslop P, Schultz M, et al. Familial dementia with frontotemporal features associated with M146V presenilin-1 mutation. Brain Pathol. (2013) 23:595–600. doi: 10.1111/bpa.12051
39. Frisoni GB, Winblad B, O’Brien JT. Revised NIA-AA criteria for the diagnosis of Alzheimer’s disease: a step forward but not yet ready for widespread clinical use. Int Psychogeriatr. (2011) 23:1191–6. doi: 10.1017/S1041610211001220
40. Femenía T, ómez-Galán MG, Lindskog M, Magara S. Dysfunctional hippocampal activity affects emotion and cognition in mood disorders. Brain Res. (2012) 1476:58–70. doi: 10.1016/j.brainres.2012.03.053
41. Corrêa-Velloso JC, Gonçalves MC, Naaldijk Y, Oliveira-Giacomelli Á, Pillat MM, Ulrich H. Pathophysiology in the comorbidity of bipolar disorder and Alzheimer’s disease: pharmacological and stem cell approaches. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 80 (Pt A):34–53. doi: 10.1016/j.pnpbp.2017.04.033
42. Bearden CE, Thompson P, Dutton R, Frey B, Peluso M, Nicoletti M, et al. Three-dimensional mapping of hippocampal anatomy in unmedicated and lithium-treated patients with bipolar disorder. Neuropsychopharmacology. (2008) 33:1229–38. doi: 10.1038/sj.npp.1301507
43. Soares AT, Andreazza A, Rej S, Rajji T, Gildengers A, Lafer B, et al. Decreased brain-derived neurotrophic factor in older adults with bipolar disorder. Am J Geriatr Psychiatry. (2016) 24:596–601. doi: 10.1016/j.jagp.2016.02.052
44. O’Bryant SE, Hobson V, Hall JR, Waring SC, Chan W, Massman P, et al. Brain-derived neurotrophic factor levels in Alzheimer’s disease. J Alzheimers Dis. (2009) 17:337–41. doi: 10.3233/JAD-2009-1051
45. Jakobsson J, Zetterberg H, Blennow K, Johan Ekman C, Johansson AG, Landén M. Altered concentrations of amyloid precursor protein metabolites in the cerebrospinal fluid of patients with bipolar disorder. Neuropsychopharmacology. (2013) 38:664–72. doi: 10.1038/npp.2012.231
46. Piccinni A, Origlia N, Veltri A, Vizzaccaro C, Marazziti D, Catena-Dell’osso M, et al. Plasma β-amyloid peptides levels: a pilot study in bipolar depressed patients. J Affect Disord. (2012) 138:160–4. doi: 10.1016/j.jad.2011.12.042
47. Forlenza OV, Aprahamian I, Radanovic M, Talib L, Camargo M, Stella F, et al. Cognitive impairment in late-life bipolar disorder is not associated with Alzheimer’s disease pathological signature in the cerebrospinal fluid. Bipolar Disord. (2016) 18:63–70. doi: 10.1111/bdi.12360
48. Kerr DS, Stella F, Radanovic M, Aprahamian I, Bertollucci PH, Forlenza OV. Apolipoprotein E genotype is not associated with cognitive impairment in older adults with bipolar disorder. Bipolar Disord. (2016) 18:71–7. doi: 10.1111/bdi.12367
49. McKeith IG, Boeve B, Dickson D, Halliday G, Taylor J, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB consortium. Neurology. (2017) 89:88–100. doi: 10.1212/WNL.0000000000004058
50. Onofrj M, Espay AJ, Bonanni L, Delli Pizzi S, Sensi SL. Hallucinations, somatic-functional disorders of PD-DLB as expressions of thalamic dysfunction. Mov Disord. (2019) 34:1100–11. doi: 10.1002/mds.27781
51. Tillman R, Geller B, Klages T, Corrigan M, Bolhofner K, Zimerman B. Psychotic phenomena in 257 young children and adolescents with bipolar I disorder: delusions and hallucinations (benign and pathological). Bipolar Disord. (2008) 10:45–55. doi: 10.1111/j.1399-5618.2008.00480.x
52. Onofrj M, Russo M, Carrarini C, Delli PS, Thomas A, Bonanni L, et al. Functional neurological disorder and somatic symptom disorder in Parkinson’s disease. J Neurol Sci. (2022) 433:120017. doi: 10.1016/j.jns.2021.120017
53. Naasan G, Shdo S, Rodriguez E, Spina S, Grinberg L, Lopez L, et al. Psychosis in neurodegenerative disease: differential patterns of hallucination and delusion symptoms. Brain. (2021) 144:999–1012. doi: 10.1093/brain/awaa413
54. Ekinci O, Albayrak Y, Ekinci AE, Caykoylu A. Relationship of trait impulsivity with clinical presentation in euthymic bipolar disorder patients. Psychiatry Res. (2011) 190:259–64. doi: 10.1016/j.psychres.2011.06.010
55. Izzo VA, Donati MA, Torre E, Ramat S, Primi C. Impulse control disorders in Parkinson’s disease versus in healthy controls: a different predictive model. J Neuropsychol. (2020) 14:318–32. doi: 10.1111/jnp.12193
56. Weintraub D, Potenza MN. Impulse control disorders in Parkinson’s disease. Curr Neurol Neurosci Rep. (2006) 6:302–6. doi: 10.1007/s11910-006-0022-y
57. Moore SF, Barker RA. Predictors of Parkinson’s disease dementia: towards targeted therapies for a heterogeneous disease. Parkinsonism Relat Disord. (2014) 20(Suppl. 1):S104–7. doi: 10.1016/S1353-8020(13)70026-9
58. Garcia-Ruiz PJ, Martinez Castrillo J, Alonso-Canovas A, Herranz Barcenas A, Vela L, Sanchez Alonso P, et al. Impulse control disorder in patients with Parkinson’s disease under dopamine agonist therapy: a multicentre study. J Neurol Neurosurg Psychiatry. (2014) 85:840–4. doi: 10.1136/jnnp-2013-306787
59. Weintraub D, Claassen DO. Impulse control and related disorders in Parkinson’s disease. Int Rev Neurobiol. (2017) 133:679–717. doi: 10.1016/bs.irn.2017.04.006
60. Fenu S, Wardas J, Morelli M. Impulse control disorders and dopamine dysregulation syndrome associated with dopamine agonist therapy in Parkinson’s disease. Behav Pharmacol. (2009) 20:363–79. doi: 10.1097/FBP.0b013e32833109a0
61. Alonso Cánovas A, Luquin Piudo R, García Ruiz-Espiga P, Burguera J, Campos Arillo V, Castro A, et al. Dopaminergic agonists in Parkinson’s disease. Neurologia. (2014) 29:230–41. doi: 10.1016/j.nrl.2011.04.012
62. Grall-Bronnec M, Victorri-Vigneau C, Donnio Y, Leboucher J, Rousselet M, Thiabaud E, et al. Dopamine agonists and impulse control disorders: a complex association. Drug Saf. (2018) 41:19–75. doi: 10.1007/s40264-017-0590-6
63. Marques A, Durif F, Fernagut PO. Impulse control disorders in Parkinson’s disease. J Neural Transm. (2018) 125:1299–312. doi: 10.1007/s00702-018-1870-8
64. Vargas AP, Vaz LS, Reuter A, Couto CM, Costa Cardoso FE. Impulse control symptoms in patients with Parkinson’s disease: the influence of dopaminergic agonist. Parkinsonism Relat Disord. (2019) 68:17–21. doi: 10.1016/j.parkreldis.2019.06.019
65. Morgante F, Fasano A, Ginevrino M, Petrucci S, Ricciardi L, Bove F, et al. Impulsive-compulsive behaviors in parkin-associated Parkinson disease. Neurology. (2016) 87:1436–41. doi: 10.1212/WNL.0000000000003177
66. Bandres-Ciga S, Blauwendraat C, Singleton AB. Assessment of genetic association between Parkinson disease and bipolar disorder. JAMA Neurol. (2020) 77:1034–5. doi: 10.1001/jamaneurol.2020.0248
67. Petrucci S, Ginevrino M, Trezzi I, Monfrini E, Ricciardi L, Albanese A, et al. GBA-related Parkinson’s disease: dissection of genotype-phenotype correlates in a large italian cohort. Mov Disord. (2020) 35:2106–11. doi: 10.1002/mds.28195
68. Huang MH, Cheng C, Huang K, Hsu J, Bai Y, Su T, et al. Bipolar disorder and risk of Parkinson disease: a nationwide longitudinal study. Neurology. (2019) 92:e2735–42. doi: 10.1212/WNL.0000000000007649
69. Faustino PR, Duarte G, Chendo I, Castro CA, Reimão S, Fernandes R, et al. Risk of developing Parkinson disease in bipolar disorder: a systematic review and meta-analysis. JAMA Neurol. (2020) 77:192–8. doi: 10.1001/jamaneurol.2019.3446
70. Harrison PJ, Luciano S. Incidence of Parkinson’s disease, dementia, cerebrovascular disease and stroke in bipolar disorder compared to other psychiatric disorders: an electronic health records network study of 66 million people. Bipolar Disord. (2021) 23:454–62. doi: 10.1111/bdi.13022
71. Bacciardi S, Elefante C, Brancati G, Mazzucchi S, Del PE, Frosini D, et al. Bipolar spectrum disorders in Parkinson’s disease: a systematic evaluation. CNS Spectr. (2022) 27:355–61. doi: 10.1017/S1092852920002126
72. Lei P, Ayton S, Appukuttan A, Moon S, Duce J, Volitakis I, et al. Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol Psychiatry. (2017) 22:396–406. doi: 10.1038/mp.2016.96
73. Brugger F, Bhatia KP, Besag FM. Valproate-associated parkinsonism: a critical review of the literature. CNS Drugs. (2016) 30:527–40. doi: 10.1007/s40263-016-0341-8
74. Pomarol-Clotet E, Alonso-Lana S, Moro N, Sarró S, Bonnin M, Goikolea J, et al. Brain functional changes across the different phases of bipolar disorder. Br J Psychiatry. (2015) 206:136–44. doi: 10.1192/bjp.bp.114.152033
75. Rodríguez-Cano E, Alonso-Lana S, Sarró S, Fernández-Corcuera P, Goikolea J, Vieta E, et al. Differential failure to deactivate the default mode network in unipolar and bipolar depression. Bipolar Disord. (2017) 19:386–95. doi: 10.1111/bdi.12517
76. Ongür D, Lundy M, Greenhouse I, Shinn A, Menon V, Cohen B, et al. Default mode network abnormalities in bipolar disorder and schizophrenia. Psychiatry Res. (2010) 183:59–68. doi: 10.1016/j.pscychresns.2010.04.008
77. Costafreda SG, Fu C, Picchioni M, Toulopoulou T, McDonald C, Kravariti E, et al. Pattern of neural responses to verbal fluency shows diagnostic specificity for schizophrenia and bipolar disorder. BMC Psychiatry. (2011) 11:18. doi: 10.1186/1471-244X-11-18
78. Fernández-Corcuera P, Salvador R, Monté G, Salvador SS, Goikolea J, Amann B, et al. Bipolar depressed patients show both failure to activate and failure to de-activate during performance of a working memory task. J Affect Disord. (2013) 148:170–8. doi: 10.1016/j.jad.2012.04.009
79. Adler CM, DelBello MP, Strakowski SM. Brain network dysfunction in bipolar disorder. CNS Spectr. (2006) 11:312–20. doi: 10.1017/s1092852900020800
80. Nabulsi L, McPhilemy G, Kilmartin L, Whittaker J, Martyn F, Hallahan B, et al. Frontolimbic, frontoparietal, and default mode involvement in functional dysconnectivity in psychotic bipolar disorder. Biol Psychiatry Cogn Neurosci Neuroimaging. (2020) 5:140–51. doi: 10.1016/j.bpsc.2019.10.015
81. Lois G, Linke J, Wessa M. Altered functional connectivity between emotional and cognitive resting state networks in euthymic bipolar I disorder patients. PLoS One. (2014) 9:e107829. doi: 10.1371/journal.pone.0107829
82. Patterson L, Firbank MJ, Colloby SJ, Attems J, Thomas AJ, Morris CM. Neuropathological changes in dementia with lewy bodies and the cingulate Island sign. J Neuropathol Exp Neurol. (2019) 78:717–24. doi: 10.1093/jnen/nlz047
83. Iizuka T, Iizuka R, Kameyama M. Cingulate island sign temporally changes in dementia with Lewy bodies. Sci Rep. (2017) 7:14745. doi: 10.1038/s41598-017-15263-2
84. Delli Pizzi S, Maruotti V, Taylor J, Franciotti R, Caulo M, Tartaro A, et al. Relevance of subcortical visual pathways disruption to visual symptoms in dementia with Lewy bodies. Cortex. (2014) 59:12–21. doi: 10.1016/j.cortex.2014.07.003
85. Franciotti R, Delli Pizzi S, Perfetti B, Tartaro A, Bonanni L, Thomas A, et al. Default mode network links to visual hallucinations: a comparison between Parkinson’s disease and multiple system atrophy. Mov Disord. (2015) 30:1237–47. doi: 10.1002/mds.26285
86. Berk M, Dodd S, Kauer-Sant’anna M, Malhi GS, Bourin M, Kapczinski F, et al. Dopamine dysregulation syndrome: implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr Scand Suppl. (2007) 116:41–9. doi: 10.1111/j.1600-0447.2007.01058.x
87. Shioya A, Saito Y, Arima K, Kakuta Y, Yuzuriha T, Tanaka N, et al. Neurodegenerative changes in patients with clinical history of bipolar disorders. Neuropathology. (2015) 35:245–53. doi: 10.1111/neup.12191
88. Jellinger KA. Lewy body/alpha-synucleinopathy in schizophrenia and depression: a preliminary neuropathological study. Acta Neuropathol. (2009) 117:423–7. doi: 10.1007/s00401-009-0492-5
89. Gray LJ, Dean B, Kronsbein HC, Robinson PJ, Scarr E. Region and diagnosis-specific changes in synaptic proteins in schizophrenia and bipolar I disorder. Psychiatry Res. (2010) 178:374–80. doi: 10.1016/j.psychres.2008.07.012
90. Jun-O’Connell AH, Butala A, Morales I, Henninger N, Deligiannidis K, Byatt N, et al. The prevalence of bipolar disorders and association with quality of life in a cohort of patients with multiple sclerosis. J Neuropsychiatry Clin Neurosci. (2017) 29:45–51. doi: 10.1176/appi.neuropsych.15120403
91. Marrie RA, Reingold S, Cohen J, Stuve O, Trojano M, Sorensen P, et al. The incidence and prevalence of psychiatric disorders in multiple sclerosis: a systematic review. Mult Scler. (2015) 21:305–17. doi: 10.1177/1352458514564487
92. Marrie RA, Horwitz R, Cutter G, Tyry T. Cumulative impact of comorbidity on quality of life in MS. Acta Neurol Scand. (2012) 125:180–6. doi: 10.1111/j.1600-0404.2011.01526.x
93. Turner AP, Alschuler K, Hughes A, Beier M, Haselkorn J, Sloan A, et al. Mental health comorbidity in MS: depression, anxiety, and bipolar disorder. Curr Neurol Neurosci Rep. (2016) 16:106. doi: 10.1007/s11910-016-0706-x
94. Carta MG, Moro M, Lorefice L, Picardi A, Trincas G, Fenu G, et al. Multiple sclerosis and bipolar disorders: the burden of comorbidity and its consequences on quality of life. J Affect Disord. (2014) 167:192–7. doi: 10.1016/j.jad.2014.05.024
95. Hoang H, Laursen B, Stenager EN, Stenager E. Psychiatric co-morbidity in multiple sclerosis: the risk of depression and anxiety before and after MS diagnosis. Mult Scler. (2016) 22:347–53. doi: 10.1177/1352458515588973
96. Carta MG, Moro M, Lorefice L, Trincas G, Cocco E, Del GE, et al. The risk of bipolar disorders in multiple sclerosis. J Affect Disord. (2014) 155:255–60. doi: 10.1016/j.jad.2013.11.008
97. Marrie RA, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. The burden of mental comorbidity in multiple sclerosis: frequent, underdiagnosed, and undertreated. Mult Scler. (2009) 15:385–92. doi: 10.1177/1352458508099477
98. Joseph B, Nandakumar A, Ahmed A, Gopal N, Murad M, Frye M, et al. Prevalence of bipolar disorder in multiple sclerosis: a systematic review and meta-analysis. Evid Based Ment Health. (2021) 24:88–94. doi: 10.1136/ebmental-2020-300207
99. Goretti B, Ghezzi A, Portaccio E, Lori S, Zipoli V, Razzolini L, et al. Psychosocial issue in children and adolescents with multiple sclerosis. Neurol Sci. (2010) 31:467–70. doi: 10.1007/s10072-010-0281-x
100. Lorefice L, Fenu G, Trincas G, Moro M, Frau J, Coghe G, et al. Progressive multiple sclerosis and mood disorders. Neurol Sci. (2015) 36:1625–31. doi: 10.1007/s10072-015-2220-3
101. Lorefice L, Fenu G, Carta E, Frau J, Coghe G, Contu F, et al. Bipolar disorders and deep grey matter in multiple sclerosis: a preliminary quantitative MRI study. Mult Scler Relat Disord. (2020) 46:102564. doi: 10.1016/j.msard.2020.102564
102. Piaggio N, Schiavi S, Martino M, Bommarito G, Inglese M, Magioncalda P. Exploring mania-associated white matter injury by comparison with multiple sclerosis: a diffusion tensor imaging study. Psychiatry Res Neuroimaging. (2018) 281:78–84. doi: 10.1016/j.pscychresns.2018.09.005
103. Hibar DP, Westlye L, Doan N, Jahanshad N, Cheung J, Ching C, et al. Cortical abnormalities in bipolar disorder: an MRI analysis of 6503 individuals from the ENIGMA bipolar disorder working group. Mol Psychiatry. (2018) 23:932–42. doi: 10.1038/mp.2017.73
104. Nortje G, Stein DJ, Radua J, Mataix-Cols D, Horn N. Systematic review and voxel-based meta-analysis of diffusion tensor imaging studies in bipolar disorder. J Affect Disord. (2013) 150:192–200. doi: 10.1016/j.jad.2013.05.034
105. Andreassen OA, Harbo H, Wang Y, Thompson W, Schork A, Mattingsdal M, et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol Psychiatry. (2015) 20:207–14. doi: 10.1038/mp.2013.195
106. Tamouza R, Oliveira J, Etain B, Bengoufa D, Hamdani N, Manier C, et al. HLA genetics in bipolar disorder. Acta Psychiatr Scand. (2018) 138:464–71. doi: 10.1111/acps.12912
107. Rosenblat JD, McIntyre RS. Bipolar disorder and immune dysfunction: epidemiological findings, proposed pathophysiology and clinical implications. Brain Sci. (2017) 7:144. doi: 10.3390/brainsci7110144
108. Bozikas VP, Anagnostouli M, Petrikis P, Sitzoglou C, Phokas C, Tsakanikas C, et al. Familial bipolar disorder and multiple sclerosis: a three-generation HLA family study. Prog Neuropsychopharmacol Biol Psychiatry. (2003) 27:835–9. doi: 10.1016/S0278-5846(03)00116-7
109. Iacovides A, Andreoulakis E. Bipolar disorder and resembling special psychopathological manifestations in multiple sclerosis: a review. Curr Opin Psychiatry. (2011) 24:336–40. doi: 10.1097/YCO.0b013e328347341d
110. Rao JS, Harry GJ, Rapoport SI, Kim HW. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry. (2010) 15:384–92. doi: 10.1038/mp.2009.47
111. Pape K, Tamouza R, Leboyer M, Zipp F. Immunoneuropsychiatry - novel perspectives on brain disorders. Nat Rev Neurol. (2019) 15:317–28. doi: 10.1038/s41582-019-0174-4
112. Silveira C, Guedes R, Maia D, Curral R, Coelho R. Neuropsychiatric symptoms of multiple sclerosis: State of the art. Psychiatry Invest. (2019) 16:877–88. doi: 10.30773/pi.2019.0106
113. Lorefice L, Fenu G, Fois M, Frau J, Coghe G, Marrosu M, et al. Pulse steroid therapy in multiple sclerosis and mood changes: an exploratory prospective study. Mult Scler Relat Disord. (2018) 20:104–8. doi: 10.1016/j.msard.2018.01.012
114. Geoffroy PA, Auffret M, Deheul S, Bordet R, Cottencin O, Rolland B. Baclofen-induced manic symptoms: case report and systematic review. Psychosomatics. (2014) 55:326–32. doi: 10.1016/j.psym.2014.02.003
115. Uy CE, Binks S, Irani SR. Autoimmune encephalitis: clinical spectrum and management. Pract Neurol. (2021) 21:412–23. doi: 10.1136/practneurol-2020-002567
116. Hansen N, Lipp M, Vogelgsang J, Vukovich R, Zindler T, Luedecke D, et al. Autoantibody-associated psychiatric symptoms and syndromes in adults: a narrative review and proposed diagnostic approach. Brain Behav Immun Health. (2020) 9:100154. doi: 10.1016/j.bbih.2020.100154
117. Marrie RA, Walld R, Bolton J, Sareen J, Walker J, Patten S, et al. Rising incidence of psychiatric disorders before diagnosis of immune-mediated inflammatory disease. Epidemiol Psychiatr Sci. (2019) 28:333–42. doi: 10.1017/S2045796017000579
118. Marrie RA, Walld R, Bolton J, Sareen J, Walker J, Patten S, et al. Increased incidence of psychiatric disorders in immune-mediated inflammatory disease. J Psychosom Res. (2017) 101:17–23. doi: 10.1016/j.jpsychores.2017.07.015
119. Schou M, Sæther S, Borowski K, Teegen B, Kondziella D, Stoecker W, et al. Prevalence of serum anti-neuronal autoantibodies in patients admitted to acute psychiatric care. Psychol Med. (2016) 46:3303–13. doi: 10.1017/S0033291716002038
120. Choe CU, Karamatskos E, Schattling B, Leypoldt F, Liuzzi G, Gerloff C, et al. A clinical and neurobiological case of IgM NMDA receptor antibody associated encephalitis mimicking bipolar disorder. Psychiatry Res. (2013) 208:194–6. doi: 10.1016/j.psychres.2012.09.035
121. León-Caballero J, Pacchiarotti I, Murru A, Valentí M, Colom F, Benach B, et al. Bipolar disorder and antibodies against the N-methyl-d-aspartate receptor: a gate to the involvement of autoimmunity in the pathophysiology of bipolar illness. Neurosci Biobehav Rev. (2015) 55:403–12. doi: 10.1016/j.neubiorev.2015.05.012
122. Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti–N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. (2013) 70:1133–9. doi: 10.1001/jamaneurol.2013.3216
123. Guasp M, Giné-Servén E, Maudes E, Rosa-Justicia M, Martínez-Hernández E, Boix-Quintana E, et al. Clinical, neuroimmunologic, and CSF investigations in first episode psychosis. Neurology. (2021) 97:e61–75. doi: 10.1212/WNL.0000000000012191
124. Baqir H, Cosmo C, Benevenuto D, Morar D, Rizvi SA, Batista LM. “Ictal catatonia in autoimmune encephalitis,” (in eng). R I Med J. (2013) 103:55–8.
125. Höftberger R, van Sonderen A, Leypoldt F, Houghton D, Geschwind M, Gelfand J, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology. (2015) 84:2403–12. doi: 10.1212/WNL.0000000000001682
126. Quaranta G, Maremmani AG, Perugi G. Anti-AMPA-receptor encephalitis presenting as a rapid-cycling bipolar disorder in a young woman with turner syndrome. Case Rep Psychiatry. (2015) 2015:273192. doi: 10.1155/2015/273192
127. Rege S, Hodgkinson SJ. Immune dysregulation and autoimmunity in bipolar disorder: synthesis of the evidence and its clinical application. Aust N Z J Psychiatry. (2013) 47:1136–51. doi: 10.1177/0004867413499077
128. Mula M. The clinical spectrum of bipolar symptoms in epilepsy: a critical reappraisal. Postgrad Med. (2010) 122:17–23. doi: 10.3810/pgm.2010.07.2171
129. Mula M, Marotta AE, Monaco F. Epilepsy and bipolar disorders. Expert Rev Neurother. (2010) 10:13–23. doi: 10.1586/ern.09.139
130. Anzellotti F, Dono F, Evangelista G, Di Pietro M, Carrarini C, Russo M, et al. Psychogenic non-epileptic seizures and pseudo-refractory epilepsy, a management challenge. Front Neurol. (2020) 11:461. doi: 10.3389/fneur.2020.00461
131. Ettinger AB, Reed ML, Goldberg JF, Hirschfeld RM. Prevalence of bipolar symptoms in epilepsy vs other chronic health disorders. Neurology. (2005) 65:535–40. doi: 10.1212/01.wnl.0000172917.70752.05
132. Mula M, Jauch R, Cavanna A, Collimedaglia L, Barbagli D, Gaus V, et al. Clinical and psychopathological definition of the interictal dysphoric disorder of epilepsy. Epilepsia. (2008) 49:650–6. doi: 10.1111/j.1528-1167.2007.01434.x
133. Mula M, Schmitz B, Jauch R, Cavanna A, Cantello R, Monaco F, et al. On the prevalence of bipolar disorder in epilepsy. Epilepsy Behav. (2008) 13:658–61. doi: 10.1016/j.yebeh.2008.08.002
134. Scaramelli A, Braga P, Avellanal A, Bogacz A, Camejo C, Rega I, et al. Prodromal symptoms in epileptic patients: clinical characterization of the pre-ictal phase. Seizure. (2009) 18:246–50. doi: 10.1016/j.seizure.2008.10.007
135. Kanner AM, Soto A, Gross-Kanner H. Prevalence and clinical characteristics of postictal psychiatric symptoms in partial epilepsy. Neurology. (2004) 62:708–13. doi: 10.1212/01.wnl.0000113763.11862.26
136. Mula M, Jauch R, Cavanna A, Gaus V, Kretz R, Collimedaglia L, et al. Interictal dysphoric disorder and periictal dysphoric symptoms in patients with epilepsy. Epilepsia. (2010) 51:1139–45. doi: 10.1111/j.1528-1167.2009.02424.x
137. Spina E, Perugi G. Antiepileptic drugs: indications other than epilepsy. Epileptic Disord. (2004) 6:57–75.
138. Ettinger AB. Psychotropic effects of antiepileptic drugs. Neurology. (2006) 67:1916–25. doi: 10.1212/01.wnl.0000247045.85646.c0
139. Knott S, Forty L, Craddock N, Thomas RH. Epilepsy and bipolar disorder. Epilepsy Behav. (2015) 52 (Pt A):267–74. doi: 10.1016/j.yebeh.2015.07.003
140. Petty F. GABA and mood disorders: a brief review and hypothesis. J Affect Disord. (1995) 34:275–81. doi: 10.1016/0165-0327(95)00025-i
141. Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. Am J Med Genet C Semin Med Genet. (2003) 123C:48–58. doi: 10.1002/ajmg.c.20013
142. Gordovez FJA, McMahon FJ. The genetics of bipolar disorder. Mol Psychiatry. (2020) 25:544–59. doi: 10.1038/s41380-019-0634-7
143. Kuo CY, Chen T, Kao P, Huang W, Cho C, Lai Y, et al. Genetic pathways and functional subnetworks for the complex nature of bipolar disorder in genome-wide association study. Front Mol Neurosci. (2021) 14:772584. doi: 10.3389/fnmol.2021.772584
144. Harrison PJ, Geddes JR, Tunbridge EM. The emerging neurobiology of bipolar disorder. Trends Neurosci. (2018) 41:18–30. doi: 10.1016/j.tins.2017.10.006
145. Everett KV. Transient receptor potential genes and human inherited disease. Adv Exp Med Biol. (2011) 704:1011–32. doi: 10.1007/978-94-007-0265-3_52
146. Thapak P, Vaidya B, Joshi HC, Singh JN, Sharma SS. Therapeutic potential of pharmacological agents targeting TRP channels in CNS disorders. Pharmacol Res. (2020) 159:105026. doi: 10.1016/j.phrs.2020.105026
147. Jang Y, Lee S, Lee B, Jung S, Khalid A, Uchida K, et al. TRPM2, a susceptibility gene for bipolar disorder, regulates glycogen synthase Kinase-3 activity in the brain. J Neurosci. (2015) 35:11811–23. doi: 10.1523/JNEUROSCI.5251-14.2015
148. Hu H, Zhu T, Gong L, Zhao Y, Shao Y, Li S, et al. Transient receptor potential melastatin 2 contributes to neuroinflammation and negatively regulates cognitive outcomes in a pilocarpine-induced mouse model of epilepsy. Int Immunopharmacol. (2020) 87:106824. doi: 10.1016/j.intimp.2020.106824
149. Shrivastava AN, Triller A, Melki R. Cell biology and dynamics of neuronal na. Neuropharmacology. (2020) 169:107461. doi: 10.1016/j.neuropharm.2018.12.008
150. el-Mallakh RS, Li R. Is the Na(+)-K(+)-ATPase the link between phosphoinositide metabolism and bipolar disorder? J Neuropsychiatry Clin Neurosci. (1993) 5:361–8. doi: 10.1176/jnp.5.4.361
151. Lichtstein D, Ilani A, Rosen H, Horesh N, Singh S, Buzaglo N, et al. Na+, K+-ATPase signaling and bipolar disorder. Int J Mol Sci. (2018) 19:2314. doi: 10.3390/ijms19082314
152. Benedetti F, Aggio V, Pratesi ML, Greco G, Furlan R. Neuroinflammation in bipolar depression. Front Psychiatry. (2020) 11:71. doi: 10.3389/fpsyt.2020.00071
153. Russo M, Calisi D, De RM, Evangelista G, Consoli S, Dono F, et al. COVID-19 and first manic episodes: a systematic review. Psychiatry Res. (2022) 314:114677. doi: 10.1016/j.psychres.2022.114677
154. Dargél AA, Godin O, Kapczinski F, Kupfer DJ, Leboyer M. C-reactive protein alterations in bipolar disorder: a meta-analysis. J Clin Psychiatry. (2015) 76:142–50. doi: 10.4088/JCP.14r09007
155. Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. (2010) 120:277–86. doi: 10.1007/s00401-010-0722-x
156. Ashok AH, Marques T, Jauhar S, Nour M, Goodwin G, Young A, et al. The dopamine hypothesis of bipolar affective disorder: the state of the art and implications for treatment. Mol Psychiatry. (2017) 22:666–79. doi: 10.1038/mp.2017.16
157. Milienne-Petiot M, Kesby J, Graves M, van Enkhuizen J, Semenova S, Minassian A, et al. The effects of reduced dopamine transporter function and chronic lithium on motivation, probabilistic learning, and neurochemistry in mice: modeling bipolar mania. Neuropharmacology. (2017) 113 (Pt A):260–70. doi: 10.1016/j.neuropharm.2016.07.030
158. Sun Z, Bo Q, Mao Z, Li F, He F, Pao C, et al. Reduced plasma dopamine-β-hydroxylase activity is associated with the severity of bipolar disorder: a pilot study. Front Psychiatry. (2021) 12:566091. doi: 10.3389/fpsyt.2021.566091
159. Jauhar S, Nour M, Veronese M, Rogdaki M, Bonoldi I, Azis M, et al. A test of the transdiagnostic dopamine hypothesis of psychosis using positron emission tomographic imaging in bipolar affective disorder and schizophrenia. JAMA Psychiatry. (2017) 74:1206–13. doi: 10.1001/jamapsychiatry.2017.2943
160. Sanacora G, Rothman DL, Mason G, Krystal JH. Clinical studies implementing glutamate neurotransmission in mood disorders. Ann N Y Acad Sci. (2003) 1003:292–308. doi: 10.1196/annals.1300.018
161. Bond DJ, da Silveira L, MacMillan E, Torres I, Lang D, Su W, et al. Relationship between body mass index and hippocampal glutamate/glutamine in bipolar disorder. Br J Psychiatry. (2016) 208:146–52. doi: 10.1192/bjp.bp.115.163360
162. Antunes F, Nunes C, Laranjinha J, Cadenas E. Redox interactions of nitric oxide with dopamine and its derivatives. Toxicology. (2005) 208:207–12. doi: 10.1016/j.tox.2004.11.033
163. Berk M, Kapczinski F, Andreazza A, Dean O, Giorlando F, Maes M, et al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev. (2011) 35:804–17. doi: 10.1016/j.neubiorev.2010.10.001
164. Scotti-Muzzi E, Chile T, Moreno R, Pastorello B, da Costa Leite C, Henning A, et al. ACC Glu/GABA ratio is decreased in euthymic bipolar disorder I patients: possible in vivo neurometabolite explanation for mood stabilization. Eur Arch Psychiatry Clin Neurosci. (2021) 271:537–47. doi: 10.1007/s00406-020-01096-0
165. Baumgarten HG, Grozdanovic Z. Psychopharmacology of central serotonergic systems. Pharmacopsychiatry. (1995) 28(Suppl. 2):73–9. doi: 10.1055/s-2007-979623
166. Berman ME, Tracy JI, Coccaro EF. The serotonin hypothesis of aggression revisited. Clin Psychol Rev. (1997) 17:651–65. doi: 10.1016/s0272-7358(97)00039-1
167. Salvadore G, Quiroz JA, Machado-Vieira R, Henter ID, Manji HK, Zarate CA. he neurobiology of the switch process in bipolar disorder: a review. J Clin Psychiatry. (2010) 71:1488–501. doi: 10.4088/JCP.09r05259gre
168. Weilburg JB. An overview of SSRI and SNRI therapies for depression. Manag Care. (2004) 13(Suppl. 6):25–33.
169. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. (2008) 33:73–83. doi: 10.1038/sj.npp.1301571
170. Zhao Q, Xiang H, Cai Y, Meng SS, Zhang Y, Qiu P. Systematic evaluation of the associations between mental disorders and dementia: an umbrella review of systematic reviews and meta-analyses. J Affect Disord. (2022) 307:301–9. doi: 10.1016/j.jad.2022.03.010
171. Musat EM, Marlinge E, Leroy M, Olié E, Magnin E, Lebert F, et al. Characteristics of bipolar patients with cognitive impairment of suspected neurodegenerative origin: a multicenter cohort. J Pers Med. (2021) 11:1183. doi: 10.3390/jpm11111183
172. Diniz BS, Teixeira A, Cao F, Gildengers A, Soares J, Butters M, et al. History of bipolar disorder and the risk of dementia: a systematic review and meta-analysis. Am J Geriatr Psychiatry. (2017) 25:357–62. doi: 10.1016/j.jagp.2016.11.014
173. Kelly MJ, Baig F, Hu MT, Okai D. Spectrum of impulse control behaviours in Parkinson’s disease: pathophysiology and management. J Neurol Neurosurg Psychiatry. (2020) 91:703–11. doi: 10.1136/jnnp-2019-322453
174. Giovannoni G, O’Sullivan JD, Turner K, Manson AJ, Lees AJ. Hedonistic homeostatic dysregulation in patients with Parkinson’s disease on dopamine replacement therapies. J Neurol Neurosurg Psychiatry. (2000) 68:423–8. doi: 10.1136/jnnp.68.4.423
175. Pettorruso M, Fasano A, De Risio L, Ricciardi L, Di Nicola M, Martinotti G, et al. Punding in non-demented Parkinson’s disease patients: relationship with psychiatric and addiction spectrum comorbidity. J Neurol Sci. (2016) 362:344–7. doi: 10.1016/j.jns.2016.02.016
176. Fasano A, Petrovic I. Insights into pathophysiology of punding reveal possible treatment strategies. Mol Psychiatry. (2010) 15:560–73. doi: 10.1038/mp.2009.95
177. Pettorruso M, De Risio L, Di Nicola M, Martinotti G, Conte G, Janiri L. Allostasis as a conceptual framework linking bipolar disorder and addiction. Front Psychiatry. (2014) 5:173. doi: 10.3389/fpsyt.2014.00173
178. Wolters EC. Dopaminomimetic psychosis in Parkinson’s disease patients: diagnosis and treatment. Neurology. (1999) 52(Suppl. 3):S10–3.
179. Goetz CG, Vogel C, Tanner CM, Stebbins GT. Early dopaminergic drug-induced hallucinations in parkinsonian patients. Neurology. (1998) 51:811–4. doi: 10.1212/wnl.51.3.811
180. Uitti RJ, Tanner CM, Rajput AH, Goetz CG, Klawans HL, Thiessen B. Hypersexuality with antiparkinsonian therapy. Clin Neuropharmacol. (1989) 12:375–83. doi: 10.1097/00002826-198910000-00002
181. Celesia GG, Wanamaker WM. Psychiatric disturbances in Parkinson’s disease. Dis Nerv Syst. (1972) 33:577–83.
182. Moskovitz C, Moses H, Klawans HL. Levodopa-induced psychosis: a kindling phenomenon. Am J Psychiatry. (1978) 135:669–75. doi: 10.1176/ajp.135.6.669
183. Fernandez HH, Durso R. Clozapine for dopaminergic-induced paraphilias in Parkinson’s disease. Mov Disord. (1998) 13:597–8. doi: 10.1002/mds.870130338
184. Jiménez-Jiménez FJ, Sayed Y, García-Soldevilla MA, Barcenilla B. Possible zoophilia associated with dopaminergic therapy in Parkinson disease. Ann Pharmacother. (2002) 36:1178–9. doi: 10.1345/aph.1ph.1A277
185. Gschwandtner U, Aston J, Renaud S, Fuhr P. Pathologic gambling in patients with Parkinson’s disease. Clin Neuropharmacol. (2001) 24:170–2. doi: 10.1097/00002826-200105000-00009
187. Etminan M, Sodhi M, Samii A, Procyshyn RM, Guo M, Carleton BC. Risk of gambling disorder and impulse control disorder with aripiprazole, pramipexole, and ropinirole: a pharmacoepidemiologic study. J Clin Psychopharmacol. (2017) 37:102–4. doi: 10.1097/JCP.0000000000000634
188. Show LI. Il Morbo di Parkinson e il Gioco d’ Azzardo, Lega Nord: intervista a Riccardo Bossi. (2012). Available online at: https://www.ilsussidiario.net/news/cinema-televisione-e-media/2012/4/12/le-iene-show-il-morbo-di-parkinson-e-il-gioco-d-azzardo-lega-nord-intervista-a-riccardo-bossi-video/267210/ (accessed August 23, 2022).
189. Grow K. Ozzy Osbourne Reveals Parkinson’s Disease diagnosis. (2020). Available online at: https://www.rollingstone.com/music/music-news/ozzy-osbourne-parkinsons-disease-940329 (accessed August 23, 2022).
190. Roberts G. Parkinson’s Disease Drug’s Cause of High Sex Drive and Gambling Addiction Side Effect Revealed in Study. (2019). Available online at: https://www.abc.net.au/news/2019-10-28/parkinson-disease-side-effect-gambling-addiction-high-sex-drive/11645290 (accessed Aug 23, 2022).
191. Ghaemi N. “Not” Depression”: manic-Depression and Robin Williams. A Deadly Disease, Whether we Accept it or not. (2014). Available online at: https://www.psychologytoday.com/intl/blog/mood-swings/201408/not-depression-manic-depression-and-robin-williams (accessed August 23, 2022).
192. Ffytche DH, Creese B, Politis M, Chaudhuri K, Weintraub D, Ballard C, et al. The psychosis spectrum in Parkinson disease. Nat Rev Neurol. (2017) 13:81–95. doi: 10.1038/nrneurol.2016.200
193. de la Fuente-Fernández R, Schulzer M, Mak E, Calne DB, Stoessl AJ. Presynaptic mechanisms of motor fluctuations in Parkinson’s disease: a probabilistic model. Brain. (2004) 127:888–99. doi: 10.1093/brain/awh102
194. Cenci MA, Lundblad M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J Neurochem. (2006) 99:381–92. doi: 10.1111/j.1471-4159.2006.04124.x
195. Lewitt PA, Mouradian MM. Predicting the development of levodopa-induced dyskinesias: a presynaptic mechanism? Neurology. (2014) 82:1574–5. doi: 10.1212/WNL.0000000000000390
196. Leentjens AF. The role of dopamine agonists in the treatment of depression in patients with Parkinson’s disease: a systematic review. Drugs. (2011) 71:273–86. doi: 10.2165/11585380-000000000-00000
197. Picillo M, Rocco M, Barone P. Dopamine receptor agonists and depression in Parkinson’s disease. Parkinsonism Relat Disord. (2009) 15(Suppl. 4):S81–4. doi: 10.1016/S1353-8020(09)70841-7
198. Hori H, Kunugi H. Dopamine agonist-responsive depression. Psychogeriatrics. (2013) 13:189–95. doi: 10.1111/psyg.12014
199. Solla P, Fasano A, Cannas A, Marrosu F. Dopamine agonist withdrawal syndrome in Parkinson’s disease. J Neurol Sci. (2017) 382:47–8. doi: 10.1016/j.jns.2017.08.3263
200. Carrera E, Tononi G. Diaschisis: past, present, future. Brain. (2014) 137:2408–22. doi: 10.1093/brain/awu101
201. Vecchio F, Miraglia F, Piludu F, Granata G, Romanello R, Caulo M, et al. “Small World” architecture in brain connectivity and hippocampal volume in Alzheimer’s disease: a study via graph theory from EEG data. Brain Imaging Behav. (2017) 11:473–85. doi: 10.1007/s11682-016-9528-3
202. Aboraya A, France C, Young J, Curci K, Lepage J. The validity of psychiatric diagnosis revisited: the Clinician’s guide to improve the validity of psychiatric diagnosis. Psychiatry. (2005) 2:48–55.
203. Telles Correia D. Different perspectives of validity in psychiatry. J Eval Clin Pract. (2017) 23:988–93. doi: 10.1111/jep.12766
204. Zachar P, Stoyanov D, Aragona M, Jablensky A. Alternative Perspectives on Psychiatric Validation: Dsm, Icd, Rdoc, and Beyond. Oxford: Oxford University Press (2014).
205. Di Nicola V, Stoyanov D. Psychiatric Nosology Revisited: At the Crossroads of Psychology and Medicine,” in Psychiatry in Crisis: At the Crossroads of Social Sciences, the Humanities, and Neuroscience. Cham: Springer International Publishing (2021). p. 31–41.
Keywords: bipolar spectrum disorders, neurology, psychosis, dementia, immunity
Citation: Digiovanni A, Ajdinaj P, Russo M, Sensi SL, Onofrj M and Thomas A (2022) Bipolar spectrum disorders in neurologic disorders. Front. Psychiatry 13:1046471. doi: 10.3389/fpsyt.2022.1046471
Received: 16 September 2022; Accepted: 05 December 2022;
Published: 21 December 2022.
Edited by:
Carmine Tomasetti, Mental Health Center of Giulianova, ItalyReviewed by:
Michele Fornaro, University of Naples Federico II, ItalyCopyright © 2022 Digiovanni, Ajdinaj, Russo, Sensi, Onofrj and Thomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Digiovanni, ✉ YW5uYWRpZ2lvdmFubmk5M0BnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.