
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Psychiatry, 28 October 2022
Sec. Addictive Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.1038122
This article is part of the Research TopicAppropriateness and Safety of using Cannabinoid and Psychedelic Medicines as Treatments for Psychiatric DisordersView all 7 articles
 Marco Colizzi1,2*
Marco Colizzi1,2* Riccardo Bortoletto1,2†
Riccardo Bortoletto1,2† Chiara Colli1†
Chiara Colli1† Enrico Bonomo1Daniele Pagliaro1Elisa Maso1
Enrico Bonomo1Daniele Pagliaro1Elisa Maso1 Gianfranco Di Gennaro3
Gianfranco Di Gennaro3 Matteo Balestrieri1
Matteo Balestrieri1Cognitive decline is believed to be associated with neurodegenerative processes involving excitotoxicity, oxidative damage, inflammation, and microvascular and blood-brain barrier dysfunction. Interestingly, research evidence suggests upregulated synthesis of lipid signaling molecules as an endogenous attempt to contrast such neurodegeneration-related pathophysiological mechanisms, restore homeostatic balance, and prevent further damage. Among these naturally occurring molecules, palmitoylethanolamide (PEA) has been independently associated with neuroprotective and anti-inflammatory properties, raising interest into the possibility that its supplementation might represent a novel therapeutic approach in supporting the body-own regulation of many pathophysiological processes potentially contributing to neurocognitive disorders. Here, we systematically reviewed all human and animal studies examining PEA and its biobehavioral correlates in neurocognitive disorders, finding 33 eligible outputs. Studies conducted in animal models of neurodegeneration indicate that PEA improves neurobehavioral functions, including memory and learning, by reducing oxidative stress and pro-inflammatory and astrocyte marker expression as well as rebalancing glutamatergic transmission. PEA was found to promote neurogenesis, especially in the hippocampus, neuronal viability and survival, and microtubule-associated protein 2 and brain-derived neurotrophic factor expression, while inhibiting mast cell infiltration/degranulation and astrocyte activation. It also demonstrated to mitigate β-amyloid-induced astrogliosis, by modulating lipid peroxidation, protein nytrosylation, inducible nitric oxide synthase induction, reactive oxygen species production, caspase3 activation, amyloidogenesis, and tau protein hyperphosphorylation. Such effects were related to PEA ability to indirectly activate cannabinoid receptors and modulate proliferator-activated receptor-α (PPAR-α) activity. Importantly, preclinical evidence suggests that PEA may act as a disease-modifying-drug in the early stage of a neurocognitive disorder, while its protective effect in the frank disorder may be less relevant. Limited human research suggests that PEA supplementation reduces fatigue and cognitive impairment, the latter being also meta-analytically confirmed in 3 eligible studies. PEA improved global executive function, working memory, language deficits, daily living activities, possibly by modulating cortical oscillatory activity and GABAergic transmission. There is currently no established cure for neurocognitive disorders but only treatments to temporarily reduce symptom severity. In the search for compounds able to protect against the pathophysiological mechanisms leading to neurocognitive disorders, PEA may represent a valid therapeutic option to prevent neurodegeneration and support endogenous repair processes against disease progression.
At the neurobiological level, neurocognitive disorders (NCDs) are characterized by reduced neuronal survival and increased neuronal death in the central nervous system (CNS), with a consequent progressive loss of neural function (1–3). Such damages are believed to underpin the cognitive deficits observed at the behavioral level, ranging from mild cognitive impairment to frank NCDs, previously classified as dementias (4). With the progressive increase in life expectancy, the prevalence and incidence of NCDs have dramatically increased, making them leading causes of disability. Specifically, both primary (e.g., Alzheimer's disease, AD) and secondary (e.g., Parkinson's disease, PD) cognitive decline associated with NCDs have increased exponentially during the last years (5), doubling from 1990 to 2016 (6), and are estimated of affecting around 100 million by 2050 (7). This implies a consistent burden for the health-care systems, considering the growing demand for treatments and support services.
Although the pathological mechanisms underlying neurodegenerative diseases are complex and not completely understood, neuroinflammation seems to play a crucial role in the neurodegenerative process (8). The neuroinflammatory response is a protective process that promote neuronal regeneration, however when sustained over time it may lead to neurodegeneration. The main cells involved in this process are microglia and astrocytes and their excessive and prolonged activation has been suggested to produce deleterious effects (9).
Currently, there are no therapeutic agents that effectively counteract the neurodegenerative damage or even slow the progression of these disorders (10). In this context, targeting and modulating neuroinflammation pathways seems to be a promising strategy to contrast neurodegeneration and cognitive symptoms (11). Consistently, endogenous lipids belonging to the N-acyl-ethanolamine (NAE) fatty acid amide family, such as anandamide (AEA), oleoylethanolamide (OEA), and palmitoylethanolamide (PEA), have shown the ability to mitigate pathogenetic mechanisms involved in the neurodegeneration process (12). PEA was initially discovered in egg yolk, soybean, and peanut oil and, later, in mammalian tissues. While exerting cannabimimetic action, it does not bind to cannabinoid receptors (13). Instead, it activates the peroxisome proliferator-activated receptor-α (PPAR-α) as well as its associated independent pathways, including ion channels involved in neuronal firing and the Transient Receptor Potential Vanilloid 1 (TRPV1) receptor (14). Such peculiar activity is believed to explain PEA potential anti-inflammatory, analgesic, and anti-epileptic effects (15–18). Interestingly, several PEA-containing products are licensed as nutraceuticals or food supplements for human use in different countries, at a recommended dose of 600–1,200 mg/day (19).
Accumulating evidence suggests that PEA may play a role in counteracting neurodegenerative processes (20), by modulating neuroinflammation pathways such as astrocyte and microglia proliferation and neuronal loss (21). Thus, PEA may be a promising therapeutic option to contrast inflammatory and oxidative stress, with potential effects in the treatment of neurodegeneration processes. Within this systematic review, we tried and better clarify the role of PEA in the context of NCDs and cognitive decline by gathering and discussing all available data from clinical and preclinical research, including both interventional and observational studies.
In order to bring together previous evidence on the topic, inclusion criteria were used as follows: (1) human or animal studies, (2) studies investigating palmitoylethanolamide (PEA) effects over primary (e.g., Alzheimer's disease), secondary (e.g., Parkinson's disease) or acquired (e.g., Traumatic Brain Injury) cognitive decline associated with neurocognitive disorders (NCDs), (3) studies investigating PEA effects over cognitive decline associated to conditions (e.g., neuropathic pain, obesity) other than NCDs, (4) studies investigating PEA modulatory effects over the biological underpinnings (e.g., neuroinflammation, gliosis, neuronal death) of cognitive decline in the context of NCDs, (5) studies investigating PEA and PEA signaling-related molecular markers (e.g., brain and/or other tissue quantitative alterations) of cognitive decline in the context of NCDs. Exclusion criteria were (1) studies investigating neither PEA as the intervention of interest (e.g., studies evaluating only exogenous cannabinoid agonists or antagonists) nor PEA or PEA signaling-related molecular markers, (2) studies where PEA bio-cognitive correlates were not investigated with reference to NCDs nor other conditions associated with cognitive decline, and (3) studies where PEA bio-cognitive correlates were not directly reported on.
A literature search was performed using electronic databases (Pubmed, Web of Science, and Scopus) for any published original study written in English, using a combination of terms concerning PEA (“palmitoylethanolamide,” “palmitylethanolamide,” “N-(2-hydroxyethyl)hexadecanamide,” “N-(2-hydroxyethyl)palmitate” and “N-palmitoylethanolamine”) and NCDs (“dementia,” “memory,” “cognit*”, “executive function,” “neurocognitive disorder,” “attenti*”, “learning,” “language,” “sensory-motor” and “neurodegenerati*”) on 25 May 2022. Broad-meaning terms were used to make the study search as inclusive as possible. Reference lists of eligible studies were screened to identify additional eligible research. Publication data screening and extraction were performed following a conventional double-screening process independently conducted by two reviewers (R.B. and C.C.).
Due to the methodological heterogeneity of the studies (Table 1) included in this review, risk of bias and study quality assessments were conducted with a reasonably inclusive and flexible approach, in line with previous research in the field (15, 53). To this extent, interventional and observational studies in humans were evaluated through an adapted set of criteria suggested by the Agency for Healthcare Research and Quality (AHRQ) guidance (54), and risk of systematic bias across human studies was further ruled out by screening all papers for potential confounding variables, such as patients' age and educational level (Table 2). Moreover, factors possibly accounting for similarities and differences between animal studies were assessed, extracting information about study characteristics, including animal model (e.g., mouse or rat), developmental stage (e.g., postnatal, adult, primary cultures of astrocytes or neurons), gender, PEA measure (e.g., PEA dosage and administration route, PEA assessment in tissues) and adequate PEA evaluation (e.g., time of exposure, single or multiple tissue assessments) (Table 2).
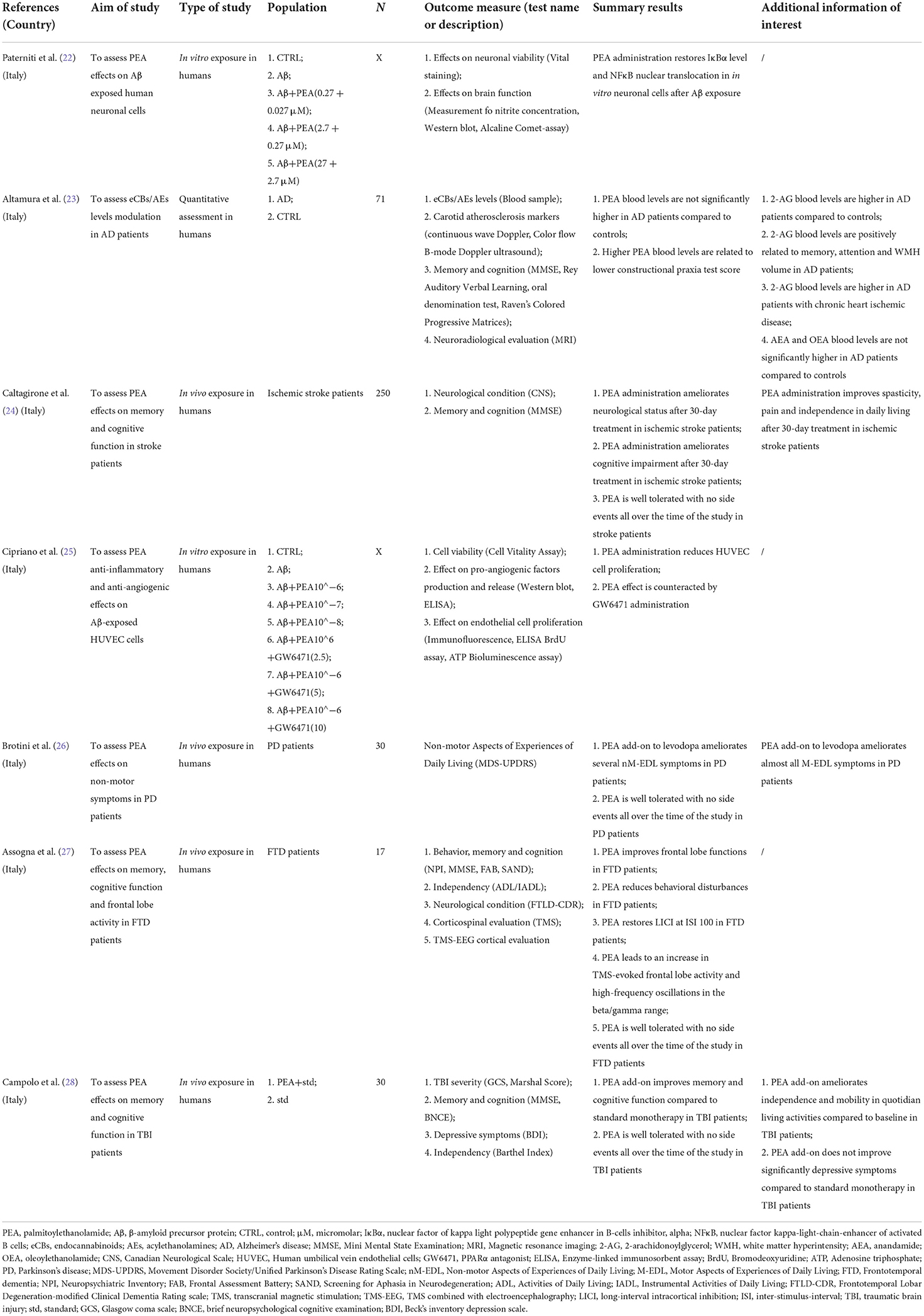
Table 1A. Summary of clinical studies investigating palmitoylethanolamide and its correlations to neurocognitive disorders (NCDs).
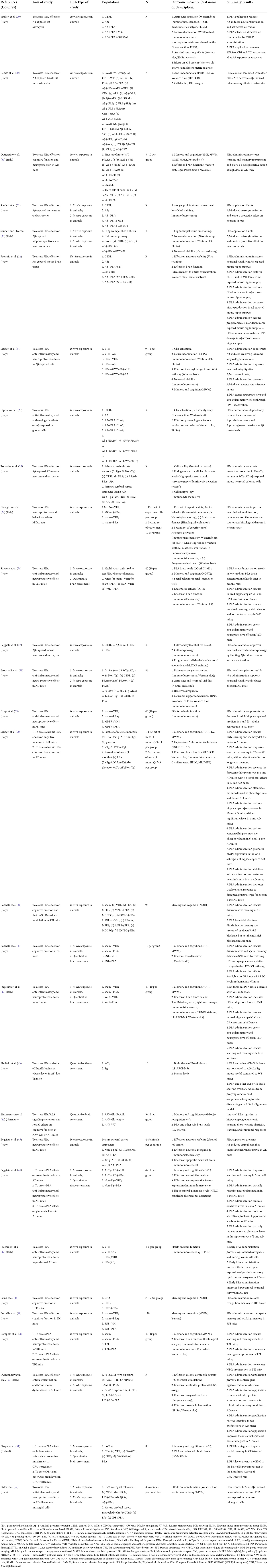
Table 1B. Summary of preclinical studies investigating palmitoylethanolamide and its correlations to neurocognitive disorders (NCDs).

Table 2A. Methodological quality of clinical studies investigating palmitoylethanolamide and its correlations to neurocognitive disorders (NCDs).
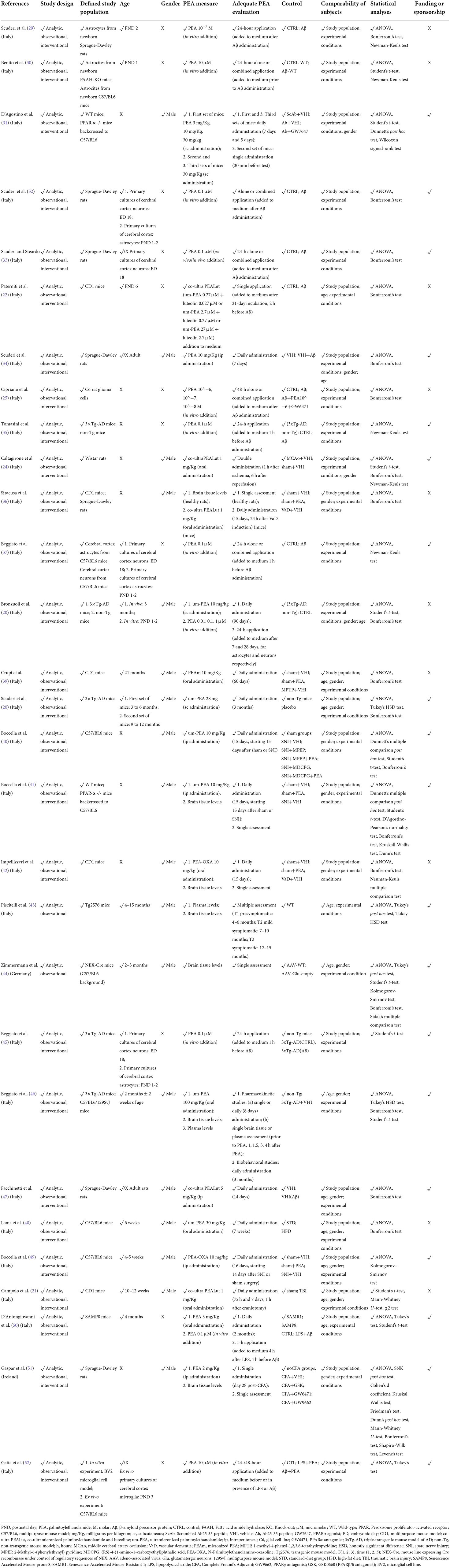
Table 2B. Methodological quality of preclinical studies investigating palmitoylethanolamide and its correlations to neurocognitive disorders (NCDs).
When deemed appropriate, studies with similar methodologies and output measures were gathered to be further explored from a meta-analytic perspective. Specifically, baseline and post-treatment values were extracted. Change-from-baseline standard deviation was calculated when not reported by assuming a moderate pre-post correlation coefficient (r = 0.7) as suggested by Cochrane Handbook 5.1 (https://handbook-5-1.cochrane.org/chapter_16/16_1_3_2_imputing_standard_deviations_for_changes_from_baseline.htm). Data were pooled by using a DerSimonian and Laird random-effects model (55). A meta-regression model was developed to investigate the effects of patients' age and length of follow up. I-squared index was calculated to assess heterogeneity among studies. Publication bias was not investigated due to the low number of studies included. Data were analyzed by the statistical software STATA software, version 16.1.
In summary, 1914 records were identified through the initial data search. After excluding duplicates as well as articles owing to article type (systematic and non-systematic reviews), by using a three-step screening approach, titles, abstracts, or full texts of all records were screened against the inclusion and exclusion criteria (Figure 1). A final list of thirty-three studies was used for systematic analysis in this review, including 4 studies conducted only in human populations, 26 studies performed only in animal models, and 3 studies including both animal and human data, investigating different aspects of the palmitoylethanolamide (PEA) signaling pathway (Table 1). These include (i) in vivo PEA treatment exposure in humans with different neurocognitive disorders (NCDs) and related conditions (4 studies; Table 1A); (ii) in vitro PEA exposure in Amyloid-β (Aβ) exposed human cells (2 studies; Table 1A); (iii) PEA quantitative blood assessment in humans with Alzheimer's Disease (AD; 1 study; Table 1A); (iv) in vivo PEA exposure in animal models of NCDs and related conditions (17 studies; Table 1B); (v) in vitro PEA exposure in Aβ exposed animal cells (7 studies; Table 1B) and AD animal model cells (4 studies; Table 1B); (vi) ex vivo PEA exposure in in Aβ exposed animal cells (3 studies; Table 1B) and AD animal model cells (1 study; Table 1B); and (vii) PEA quantitative brain/tissue assessment in animal models of different NCDs (7 studies; Table 1B). Additional data on methodological quality of studies conducted in humans and animals are reported in Tables 2A,B. A detailed presentation of human and animal results is reported in Supplementary Tables 1A,B. A brief synthesis of the main results is presented below.
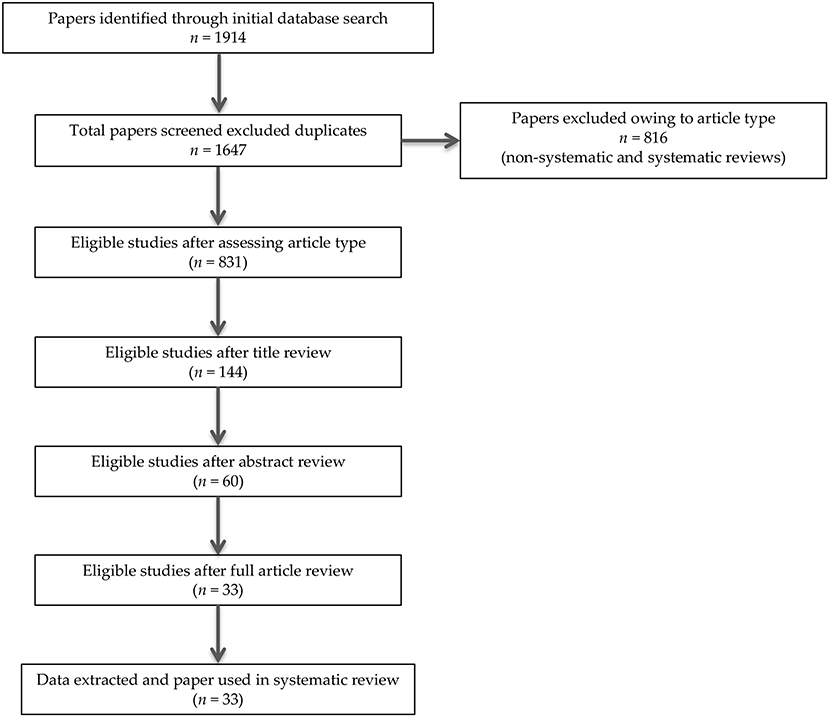
Figure 1. PRISMA flowchart of search strategy for systematic review.
Most human studies identified in this review addressed the effects of PEA exposure on cognitive function (Table 1A), using similar but not overlapping methodologies (Table 2A) in terms of disorder [stroke (24), Parkinson's disease (PD) (26), Frontotemporal dementia (FTD) (27), and traumatic brain injury (TBI) (28)], PEA dosage [600 mg bid/daily (26), 700 mg bid (24, 27, 28)], PEA formulation [alone (26), with luteolin (24, 27, 28)], PEA mode of administration [sublingual (24, 26), oral administration (27, 28)], and PEA period of exposure [4 weeks (27), 60 days (24), 180 days (28), 12 months (26)]. Apart from a single study that adopted a placebo-controlled design (28), all studies lacked a controlled condition (24, 26, 27). Nevertheless, results indicated a beneficial effect of PEA in ameliorating cognitive impairment following cerebral ischemia (24) and TBI (28) as well as non-motor aspects of experiences of daily living (nM-EDL; e.g., anxious-depressive symptoms, sleep problems, and fatigue) in PD (26) and frontal lobe disfunctions and behavioral disturbances in FTD (27). Noteworthy, PEA was well tolerated, in the absence of any relevant side effect across all the studies, and for the entire duration of the compound administration.
Out of the 4 studies, 3 (24, 27, 28) adopted the same tool to investigate impairment of cognitive abilities, the Mini Mental State Examination (MMSE), a widely accepted instrument to gather the cognitive state of patients suffering from NCDs (56), and were included in the meta-analysis. A total of 282 patients were considered. Weighted mean and standard deviation were 69.70 and 12.51 years respectively. The pooled change-from baseline was 3.80 points (95% C.I. −0.16–7.75; Figure 2). Meta-regression did not show a significant effect of patients' age (coefficient: 0.17; 95% CI: −0.28–0.61) or length of follow-up (coefficient: 0.06; 95% CI: −0.01–0.12). The heterogeneity was remarkable (I-squared: 98.40%, p < 0.001).
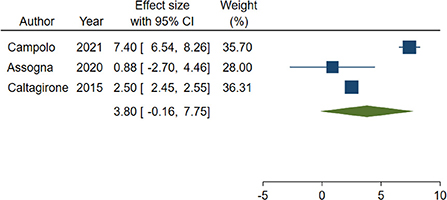
Figure 2. Forest plot showing the pooled Mini-Mental State Examination (MMSE) change from baseline. Homogeneity (I-squared): 98.40%, p < 0.001; Estimation by DerSimonian and Laird random-effects model.
In total, two studies did not evaluate the effect of PEA administration in humans suffering from NCDs and related conditions, while analyzing the biological effect of PEA in human cells from in vitro models of NCDs (22, 25). In such studies, AD features were induced by Aβ stimulation in either neuron-like cells (22) or Human Umbilical Vein Endothelial Cells (HUVEC) (25) (Tables 1A, 2A). When compared to control conditions, PEA alone (25) or combined with luteolin (22) was found to blunt Aβ-induced astrocyte activation and exert protective effects on glial cells (22) possibly via a peroxisome proliferator-activated receptor alpha (PPAR-α)-mediated reduction of the production of pro-inflammatory and pro-angiogenic markers (25).
This systematic review identified a single study specifically analyzing peripheral PEA levels in humans suffering from AD, as compared to healthy controls (23) (Tables 1A, 2A). Despite not significantly different between AD patients and controls, PEA levels appeared to be associated with cognitive performance among patients. Interestingly, 2-Arachidonoylglycerol (2-AG) levels also correlated with memory, attention, and underlying brain atrophy, suggesting an extensive role of the endocannabinoid system in the neuropathology of AD (23).
Most evidence regarding a potential therapeutic effect of PEA in NCDs was gathered from preclinical studies administering the compound to animal models of NCDs (Table 1B), including AD (20, 31, 38, 46, 47, 50), Aβ-exposed (34), middle cerebral artery occlusion (MCAo) (24), PD (39), vascular dementia (VaD) (36, 42), spared nerve injury (SNI) (40, 41, 49), TBI (28), Complete Freund's Adjuvant (CFA) (51), and high-fat diet (48) models. Despite the evidence of similar methodologies across the reviewed studies, a certain heterogeneity was found in terms of animal type [mice (20, 28, 31, 36, 38–42, 46, 48–50), rat (24, 34, 47, 51)], period of exposure [from 4 to 5 weeks old to 21 months old), PEA formulation [alone unspecified, PEA (31, 34, 50, 51); alone micronized, PEAm (39); alone ultra-micronized, um-PEA (20, 38, 40, 41, 46, 48); combined with oxazoline, PEA-OXA (42, 49); combined with luteolin, PEALut (24, 28, 36, 47)], PEA mode of administration [intraperitoneal (34, 40, 41, 47, 49, 51), subcutaneous (20, 31, 38), oral (24, 28, 36, 39, 42, 46, 48, 50)], dosage of PEA [2 to 10 mg/kg for intraperitoneal administration (34, 40, 41, 47, 49, 51), 3 to 30 mg/kg for subcutaneous (20, 31, 38), 1 to 100 mg/kg for oral administration (24, 28, 36, 39, 42, 46, 48, 50)], and duration of exposure (from single administration to 3 months) (Table 2B).
Studies conducted in experimental models of AD found a dose-dependent (31), early intervention (20), and chronic (46) effect of PEA in reducing highly representative features of AD such as working memory-like impairments (31) and learning and memory deficits (20, 31, 46) as well as the associated depressive-like and anhedonia-like phenotypes (20). Such effect was dependent of PPAR-α activation (31) and related to the ability of PEA of reducing AD-associated biomolecular mechanisms such as Aβ expression (20), abnormal hippocampal tau phosphorylation (20), lipid peroxidation (31), protein nytrosylation (31), inducible nitric oxide synthase induction (31) and reactive oxygen species production (46), and caspase3 activation (31). A role of PEA administration in restoring astrocyte (20, 38, 47) and glutamatergic (20, 46) functions, restraining neuroinflammation (20, 46, 47) and enteric inflammation and motor dysfunction (50), and promoting neuronal viability (38), hippocampal neuronal survival (47), and microtubule-associated protein 2 (MAP2) expression (20), in AD mice, was also found.
Similar findings were found among Aβ-exposed animals (Tables 1B, 2B) where PEA reduced memory impairments, reactive gliosis, amyloidogenesis, and neuroinflammation, and improved neuronal integrity, via PPAR-α activation (34). The same remarks suggesting an effect of PEA in rescuing memory deficits and injured hippocampal neurons, possibly by exerting anti-inflammatory and neuroprotective effects, were reported in preclinical models of VaD (36, 42). Finally, converging evidence for an improving effect of PEA administration on learning and memory and their biological underpinnings was also found in the context of brain ischemia reperfusion injury (24), PD (39), SNI (40, 41, 49), TBI (28), CFA (51), and high-fat diet (48), where cognitive decline is a common complication of the disease.
In total, 11 studies evaluated the effect of in vitro PEA exposure on several neurobiological mechanisms underlying NCDs (Tables 1B, 2B), using both Aβ exposed animal cells (25, 29, 30, 32, 33, 35, 37) and AD animal model cells (38, 45, 50, 52). PEA application was reported to reduce Aβ-induced neuroinflammation and astrocyte activation (25, 29, 30, 32, 33, 37) as well as angiogenesis (25), exerting a protective effect on neuronal cells (32, 33, 37). Such effect was dependent on PPAR-α and cannabinoid receptor type 1 (CB1) and 2 (CB2) activation (29). Also, PEA was found to exert protective properties in wild-type mouse cell cultures but not in AD mouse neuronal cultured cells overexpressing Aβ, suggesting its effectiveness in early AD or when Aβ is accumulating and initiating damage in the central nervous system (35). Similar findings were found among AD models where in vitro PEA application reduced neuroinflammation (52) and astrogliosis (38, 45), supporting neuronal viability and survival (38, 45), and also improving enteric inflammation (50).
To confirm the results obtained with the in vitro models, some studies made the same PEA treatment ex vivo (Tables 1B, 2B) with organotypic cultures challenged with Aβ (22, 32, 33) or lipopolysaccharide (LPS) (52). Converging evidence suggests that PEA may enhance neuroprotection against the neurodegenerative processes associated with Aβ deposition, including astrocyte activation (32, 33) and neuroinflammation (32, 33, 52). Also, PEA exposure showed specific effects in reducing inducible nitric oxide synthase (22), glial fibrillary acidic protein expression (22), and apoptosis (22, 52) as well as restoring neuronal nitric oxide synthase (22) and brain derived neurotrophic factor (BDNF) (22).
Seven studies analyzed PEA levels in the brain and tissues of animal models of NCDs (Tables 1B, 2B), including VaD (36, 42), SNI (41), CFA (51) and genic models of AD (43, 46) and related conditions (44). Genetically inducing a reduction in PEA levels resulted in changes in hippocampal synaptic activity and aberrant cognition (44). Consistently, PEA levels were reported to be reduced after VAD induction (42), but restored following exogenous PEA administration (36, 42), possibly accounting for the observed therapeutic effects (36, 42). Similarly, plasma and brain PEA levels were found to be slightly lower in a genic model of AD, despite not significantly (46). Another study in a genic model of AD revealed that changes in PEA levels may depend on the disease stage, from being relatively higher in the pre-symptomatic and mild symptomatic phases to being relatively lower in the symptomatic stage (43). Further, no differences in PEA levels were observed in CFA-injected models (51), neither PEA administration affected PEA levels in SNI models (41), warranting further investigation of a potential selective effect of PEA in primary cognitive decline.
This is the first systematic review of all studies investigating the biobehavioral effects of palmitoylethanolamide (PEA) with reference to cognitive decline, that is the core symptomatologic domain of neurocognitive disorders (NCDs) (4). Independently of potential effects of PEA on additional features of NCDs, such as motor impairments, pain, and overall disability [recently reviewed here (57)], disentangling whether PEA is effective in improving cognition, possibly corroborated by evidence of a restoring effect on its neurobiological underpinnings, is of paramount importance to tip the scales toward considering PEA an adjunctive therapeutic option for NCDs. Based on evidence that degeneration of basal forebrain neurons causes a loss of cholinergic tone in the basal forebrain cholinergic system, with implications for the development of cognitive decline (58), most research has focused on the role of acetylcholinesterase (AChE) inhibitors as a potential treatment for NCDs (59). Further, in the absence of other successful interventions, recent research is focusing on the possibility to refine AChE inhibitors to maximize their potential (10). However, growing evidence indicates a crucial role for neuroinflammation in neurodegeneration (8) and a potential therapeutic effect of neuroinflammation modulation in contrasting neurodegeneration at both the neurobiological and behavioral level (11). In this regard, recent research highlights the importance that cannabinoid-related compounds, whose actions depend on the interaction with non-CB receptors, may have in terms of anti-inflammatory properties (16–18), in turn accounting for their ability to mitigate biological mechanisms involved in neurodevelopmental disorders (53), epilepsy (15), and neurodegeneration (12).
Overall, this review indicated that PEA, whose biological effects are related to indirect activation of CB1 receptors as well as PPAR-α and Transient Receptor Potential Vanilloid 1 (TRPV1) modulation (14, 60), may be involved in NCDs and related conditions. With reference to human studies, evidence was obtained from interventional studies of the positive cognitive effects of PEA supplementation in humans, benefits of PEA at the neurobiological level in both in vivo and in vitro human studies, and a single observational study that changes in the PEA tone affect cognitive performance. Regarding animal studies, evidence was obtained from interventional studies of a PPAR-α-dependent, dose-dependent, and early intervention pro-cognitive effect of PEA, benefits of PEA on several biomolecular mechanisms in in vivo, in vitro, and ex vivo studies, and observational studies that a reduction in the PEA tone affects cognitive performance and related hippocampal activity, possibly specific to primary cognitive decline in the symptomatic stage.
Some important findings from this systematic review deserve to be highlighted. First, NCDs represent the group of conditions where the use of PEA seems to be the most supported by research studies, with an overwhelming convergence of evidence toward a therapeutic effect on core cognitive symptoms and underlying neurobiological underpinnings. Also, compared to other conditions, such as autism spectrum disorders (53) or epilepsy (15), where the evidence for a therapeutic potential of PEA is robust, but very limited (53) or absent (15) in humans, the present review identified 7 studies performed in humans. Such studies were either in vivo, in vitro, or observational studies, and a preliminary meta-analysis of studies assessing cognition before and after PEA administration (24, 27, 28) revealed an effect of PEA in partially reversing cognitive decline. Instead, studies of PEA in other neuropsychiatric disorders are still in their infancy, despite results seem promising. For instance, very recent clinical trials provided initial evidence that PEA may be a valid adjunctive treatment in acute mania (61) and schizophrenia (62). It is worth mentioning that, while not being the focus of this review, results presented here support a potential role of PEA also in depressive-like symptoms (20, 26).
Second, thanks to numerous preclinical studies, performed adopting different methodological strategies, as well as some in vitro human studies and an observational human study, this review was able to offer a sufficiently solid neurobiological explanation for the therapeutic effects of PEA. In fact, PEA was found to control Aβ expression (20), hippocampal tau phosphorylation (20), and associated astrocyte/glial dysfunction (20, 22, 25, 29, 30, 32, 33, 37, 38, 45, 47), resulting in increased neuronal viability (38, 45) and survival (22, 38, 45, 47, 52), MAP2 (20) and BDNF (22) expression, and overall neuroprotection (32, 33, 36, 37, 42). An effect of PEA in controlling glutamatergic function (20, 46) as well as neuroinflammation (20, 25, 32, 33, 36, 42, 46, 47, 52) and enteric inflammation (50) was observed, with specific modulation of lipid peroxidation (31), protein nytrosylation (31), inducible nitric oxide synthase induction (22, 31) and reactive oxygen species production (46), and caspase3 activation (31). PEA effects seemed to depend on PPAR-α activation (25, 34).
Third, brain PEA levels were found to be reduced in preclinical models of primary NCDs, that is VAD (42) and AD (46), the latter possibly as a result of the disease progression (43), further corroborating a potential need for its supplementation. Consistently, PEA level restoration via supplementation seemed to explain the therapeutic effect observed in VAD (36, 42). Instead, less clear appeared the role of PEA levels in other models of NCDs (41, 51). However, even when not different from a control group, animal brain (44) and human blood (23) PEA levels were found to be associated with modulation of cognitive function.
Fourth, some information was gathered in terms of PEA dosages and therapeutic window. Specifically, a a dose-dependent effect of PEA was revealed (31), with PEA exerting its maximal potential in the early stages of NCDs (20, 35). Interestingly, this may be due to PEA levels being still high in the early stages of NCDs, possibly reflecting a compensatory innate mechanism, before falling in the frank symptomatic stage (43).
Finally, the effect of PEA did not seem to be confined to the so-called primary dementias, such as AD or VAD. In fact, PEA ameliorated cognitive domains and associated symptoms also in patients with stroke (24), Parkinson's disease (PD) (26), and traumatic brain injury (TBI) (28), as well as learning and memory and underlying neurobiology in animal models of brain ischemia reperfusion injury (24), PD (39), spared nerve injury SNI (40, 41, 49), TBI (28), Complete Freund's Adjuvant (CFA) (51), and high-fat diet (48). Nevertheless, as for instance PEA levels were not altered in CFA-injected models (51), neither SNI models (41), further studies are needed to investigate potential differential mechanisms of action for PEA in primary vs. other NCDs.
The findings of this systematic review must be seen considering some strengths and limitations. Research in the field is quite advanced, even though especially in animal models. Despite being tested in different conditions, PEA effect needs to be further studied to fully address its relevance for the different clinical phenotypes of cognitive decline. In other words, evidence does not fully clarify whether PEA is useful only in primary cognitive decline associated with NCDs (e.g., AD) or also secondary (e.g., PD) and acquired (e.g., TBI) ones. While the beneficial effects of PEA in NCDs seem to be reasonably mediated by a protective role of the compound against altered neuroinflammation and related mechanisms, whether such effect is sustained in the longer-term remains to be tested. Longer-term studies are needed to support a potential effect of PEA as a disease modifying drug in blunting or halting the NCD course. Also, PEA levels seem to be altered in NCDs and differently depending on the phase of illness. However, whether this can be considered a biomarker for diagnosis and treatment response deserves additional studies. Finally, no study made a direct comparison of different PEA formulations in the same population, making it difficult to draw any conclusion about the potential superiority of any of such pharmaceutical forms.
In conclusion, this review revealed several experimental and observational investigations of PEA and its pathway in NCDs. Evidence discussed here converges in reporting alterations of the PEA signaling, implications for NCD-related biobehavioral manifestations, and benefits from PEA supplementation. In particular, PEA seems to be therapeutic in improving cognitive performance, whose decline is a characteristic manifestation of NCDs. Importantly, no serious adverse effects were reported across the in vivo PEA treatment exposure human studies, suggesting that PEA supplementation may represent not only an effective treatment strategy in NCDs but also exempt from major health risks.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
MC, RB, CC, EB, DP, EM, GD, and MB: conceptualization, methodology, resources, validation, and writing–review and editing. MC, RB, CC, EB, DP, and GD: data curation, investigation, and visualization. GD: formal analysis. MC and MB: project administration. MC: supervision. MC, RB, CC, and GD: writing–original draft preparation. All authors have read and agreed to the published version of the manuscript.
The authors would like to acknowledge infrastructure from the University of Udine.
Author MC has been a consultant/advisor to GW Pharma Limited, GW Pharma Italy SRL and F. Hoffmann-La Roche Limited, outside of this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2022.1038122/full#supplementary-material
Supplementary Table 1A. Results of clinical studies investigating palmitoylethanolamide and its correlations to neurocognitive disorders (NCDs). IκBα, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; Aβ, β-amyloid precursor protein; < , lower than; CTRL, control; PEA, palmitoylethanolamide; >, higher than; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; μM, micromolar; vs., compared to; NS, not significant; 2-AG, 2-arachidonoylglycerol; AD, Alzheimer's disease; AEA, anandamide; OEA, oleoylethanolamide; ↓, reduced; ↑, increased; CNS, Canadian Neurological Scale; T0, baseline; T30, after 30 days; MMSE, Mini Mental State Examination; p-p38, phospho-p38; MAPK, Mitogen-activated protein kinase; VEGF, Vascular endothelial growth factor; GW6471, PPARα antagonist; HUVEC, Human umbilical vein endothelial cells; ATP, Adenosine triphosphate; nM-EDL, Non-Motor Aspects of Experiences of Daily Living; NPI, Neuropsychiatric Inventory; FAB, Frontal Assessment Battery; SAND, Screening for Aphasia in Neurodegeneration; LICI, long-interval intracortical inhibition; ISI, inter-stimulus-interval; SICI, short-interval intracortical inhibition; ICF, intracortical facilitation; SAI, short-latency afferent inhibition; LTP, long-term potentiation; std, standard; BNCE, Brief Neuropsychological Cognitive Examination; BDI, Beck's inventory depression scale. Bold font emphasizes significant results.
Supplementary Table 1B. Results of preclinical studies investigating palmitoylethanolamide and its correlations to neurocognitive disorders (NCDs). GFAP, Glial fibrillary acidic protein; S100B, S100 calcium-binding protein B; Aβ, β-amyloid precursor protein; >, higher than; CTRL, control; PEA, palmitoylethanolamide; GW9662, PPARγ antagonist; < , lower than; MK, MK886 (PPARα antagonist); vs., compared to; NS, not significant; iNOS, inducible nitric oxide synthase; COX-2; cyclooxygenase-2; PGE2, Prostaglandin E2; NO–, Nitrogen Dioxide; TNF-α, tumor necrosis factor alpha; IL-1β, Interleukin 1 beta; p-p38, pospho-p38; p-JNK, anti-phosphorylated Jun N-terminal kinase; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; AP-1, Activator protein 1; PPARs, Peroxisome proliferator-activated receptors; eCB, endocannabinoid; CB1, Cannabinoid receptor type 1; CB2, Cannabinoid receptor type 2; FAAH, Fatty acid amide hydrolase; CCL, C-C Motif Chemokine Ligand; WT, Wild-type; KO, Knock-out; IL-6, Interleukin 6; h, hours; SR1, SR141716A; SR2, SR144528; LDH-Abs, LDH activity; mRNA, messenger ribonucleic acid; AEA, anandamide; OEA, oleoylethanolamide; ERK1/2, Extracellular signal-regulated protein kinases 1 and 2; MAPK, Mitogen-activated protein kinase; TRPV1, transient receptor potential cation channel subfamily V member 1; WY, WY14643; TG, troglitazone; CPZ, capsazepine; YMT, Y-maze test; Ab, Ab25-35 peptide; VHI, vehicle; ScAb, Scrambled Ab25-35 peptide; PEA(3, 10, 30), PEA (3, 10, 30 mg/Kg); MWM, Morris Water Maze test; NORT, Novel Object Recognition test; MAP2, Microtubule-associated protein 2; GW6471, PPARα antagonist; BDNF, Brain-Derived Neurotrophic Factor; GDNF, Glial cell line-derived neurotrophic factor; nNOS, Neuronal nitric oxide synthase; AIF, Apoptosis-Inducing Factor; BACE1, Beta-secretase 1; APP, Amyloid precursor protein; WNT, Wingless and Int-1; pTau, phosphorylated Tau protein; pGSK3βB, phosphorylated-Glycogen synthase kinase 3-beta; Dkk1, Dickkopf WNT Signaling Pathway Inhibitor 1; DNA, Deoxyribonucleic acid; PARP1, Poly(ADP-Ribose)Polymerase1; C6, glial cell line; NO, Nitrogen Oxide; VEGF, Vascular endothelial growth factor; MMP-9, Matrix metallopeptidase 9; non-Tg, non-transgenic mouse model; 3xTg-AD, triple-transgenic mouse model of AD; MCAo, middle cerebral artery occlusion; Bax, Bcl-2 associated X-protein; Bcl-2, B-cell lymphoma 2; BrdU, Bromodeoxyuridine; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; VaD, vascular dementia; OFT, Open-field test; PAR, Poly(ADP-ribose); μm, micrometer; TST, Tail suspension test; FST, Forced swim test; SPT, Sucrose preference test; p[Thr308]Akt, Phospho-Akt(Threonine308) Antibody; p[Ser9]Gsκ3β, Phospho-Gsκ3β(Serine9); p[Ser396]tau, Phospho-tau(Serine396); p[Ser536]p65, Phospho-p65(Serine536); IL-16, Interleukin 16; IL-5, Interleukin 5; MCSF, Macrophage colony-stimulating factor; MCP5, Murine Monocyte Chemoattractant Protein; IL-10, Interleukin 10; Glx, Glutamine/glutamate; NAA, N-acetylaspartate; GLT1, Glutamate Transporter-1; RI, recognition index; SNI, spare nerve injury; MPEP, 2-Methyl-6-(phenylethynyl) pyridine; MDCPG, (RS)-4-(1-amino-1-carboxyethyl)phthalic acid; eCB, endocannabinoid; AE, acylethanolamine; IPSI, ipsilateral; CONTRA, contralateral; 2-AG, 2-arachidonoylglycerol; NAAA, N-acylethanolamine acid amide hydrolase; Iba1, Ionized calcium-binding adaptor molecule 1; IκBα, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha; NRF2, nuclear factor erythroid 2-related factor 2; MnSOD, Manganese superoxide dismutase; HO1, Heme oxygenase 1; T(1, 2, 3), time (1, 2, 3); Tg, transgenic; CX, cortex; HIPP, hippocampus; STR, striatum; CER, cerebellum; AAV, adeno-associated virus; Glu, glutamatergic neurons; AAV-Glu-FAAH, Animals overexpressing FAAH in glutamatergic neurons; PFC, prefrontal cortex; ROS, reactive oxygen species; CD11b, Integrin alpha M; STD, standard-diet group; HFD, high-fat diet; TBI, traumatic brain injury; DCX, doublecortine; d, days; NT3, neurotrophin 3; SAMP8, Senescence Accelerated Mouse-prone 8; SAMR1, Senescence-Accelerated Mouse-Resistant 1; SP, Stimulation protocol; α-syn, α-synuclein; TLR-4, Toll-like receptor 4; LPS, lipopolysaccharide; CFA, Complete Freund's Adjuvant; TG2, Tissue type 2 transglutaminase; TREM2, Triggering Receptor Expressed On Myeloid Cells 2; ARG1, Arginase 1. Bold font emphasizes significant results.
1. Shinosaki K, Nishikawa T, Takeda M. Neurobiological basis of behavioral and psychological symptoms in dementia of the Alzheimer type. Psychiatry Clin Neurosci. (2000) 54:611–20. doi: 10.1046/j.1440-1819.2000.00773.x
2. Halliday GM, Leverenz JB, Schneider JS, Adler CH. The neurobiological basis of cognitive impairment in Parkinson's disease. Mov Disord. (2014) 29:634–50. doi: 10.1002/mds.25857
3. Young JJ, Lavakumar M, Tampi D, Balachandran S, Tampi RR. Frontotemporal dementia: latest evidence and clinical implications. Ther Adv Psychopharmacol. (2018) 8:33–48. doi: 10.1177/2045125317739818
4. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5). Washington, DC: American Psychiatric Association (2013).
5. Fereshtehnejad SM, Vosoughi K, Heydarpour P, Sepanlou SG, Farzadfar F, Tehrani-Banihashemi A, et al. Burden of neurodegenerative diseases in the Eastern Mediterranean Region, 1990-2016: findings from the Global Burden of Disease Study 2016. Eur J Neurol. (2019) 26:1252–65. doi: 10.1111/ene.13972
6. Collaborators GD. Global, regional, and national burden of Alzheimer's disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) 18:88–106. doi: 10.1016/S1474-4422(18)30403-4
7. Collaborators GDF. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. (2022) 7:e105–e25. doi: 10.1016/S2468-2667(21)00249-8
8. Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, et al. Neuroinflammation Induces Neurodegeneration. J Neurol Neurosurg Spine. (2016) 1:1003–17.
9. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. (2020) 9:42. doi: 10.1186/s40035-020-00221-2
10. Moss DE. Improving Anti-Neurodegenerative Benefits of Acetylcholinesterase Inhibitors in Alzheimer's Disease: Are Irreversible Inhibitors the Future? Int J Mol Sci. (2020) 21:3438–55. doi: 10.3390/ijms21103438
11. Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front Pharmacol. (2019) 10:1008. doi: 10.3389/fphar.2019.01008
12. Kasatkina LA, Rittchen S, Sturm EM. Neuroprotective and immunomodulatory action of the endocannabinoid system under neuroinflammation. Int J Mol Sci. (2021) 22:5431. doi: 10.3390/ijms22115431
13. Di Marzo V. 'Endocannabinoids' and other fatty acid derivatives with cannabimimetic properties: biochemistry and possible physiopathological relevance. Biochim Biophys Acta. (1998) 1392:153–75. doi: 10.1016/S0005-2760(98)00042-3
14. Rankin L, Fowler CJ. The Basal Pharmacology of Palmitoylethanolamide. Int J Mol Sci. (2020) 21:7942–62. doi: 10.3390/ijms21217942
15. Bortoletto R, Balestrieri M, Bhattacharyya S, Colizzi M. Is It Time to Test the Antiseizure Potential of Palmitoylethanolamide in Human Studies? A systematic review of preclinical evidence. Brain Sci. (2022) 12:101. doi: 10.3390/brainsci12010101
16. Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. (2005) 67:15–9. doi: 10.1124/mol.104.006353
17. Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci U S A. (2009) 106:20966–71. doi: 10.1073/pnas.0907417106
18. Jaggar SI, Hasnie FS, Sellaturay S, Rice AS. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain. (1998) 76:189–99. doi: 10.1016/S0304-3959(98)00041-4
19. Pesce M, Seguella L, Cassarano S, Aurino L, Sanseverino W, Lu J, et al. Phytotherapics in COVID19: Why palmitoylethanolamide? Phytother Res. (2021) 35:2514–22. doi: 10.1002/ptr.6978
20. Scuderi C, Bronzuoli M, Facchinetti R, Pace L, Ferraro L, Broad K, et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer's disease by exerting anti-inflammatory and neuroprotective effects. Transl Psychiatry. (2018) 8:32. doi: 10.1038/s41398-017-0076-4
21. Petrosino S, Di Marzo V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br J Pharmacol. (2017) 174:1349–65. doi: 10.1111/bph.13580
22. Paterniti I, Cordaro M, Campolo M, Siracusa R, Cornelius C, Navarra M, et al. Neuroprotection by association of palmitoylethanolamide with luteolin in experimental Alzheimer's disease models: the control of neuroinflammation. CNS Neurol Disord Drug Targets. (2014) 13:1530–41. doi: 10.2174/1871527313666140806124322
23. Altamura C, Ventriglia M, Martini MG, Montesano D, Errante Y, Piscitelli F, et al. Elevation of Plasma 2-Arachidonoylglycerol Levels in Alzheimer's Disease Patients as a Potential Protective Mechanism against Neurodegenerative Decline. J Alzheimers Dis. (2015) 46:497–506. doi: 10.3233/JAD-142349
24. Caltagirone C, Cisari C, Schievano C, Di Paola R, Cordaro M, Bruschetta G, et al. Co-ultramicronized Palmitoylethanolamide/Luteolin in the Treatment of Cerebral Ischemia: from Rodent to Man. Transl Stroke Res. (2016) 7:54–69. doi: 10.1007/s12975-015-0440-8
25. Cipriano M, Esposito G, Negro L, Capoccia E, Sarnelli G, Scuderi C, et al. Palmitoylethanolamide Regulates Production of Pro-Angiogenic Mediators in a Model of beta Amyloid-Induced Astrogliosis In vitro. CNS Neurol Disord Drug Targets. (2015) 14:828–37. doi: 10.2174/1871527314666150317224155
26. Brotini S, Schievano C, Guidi L. Ultra-micronized palmitoylethanolamide: an efficacious adjuvant therapy for Parkinson's Disease. CNS Neurol Disord Drug Targets. (2017) 16:705–13. doi: 10.2174/1871527316666170321124949
27. Assogna M, Casula EP, Borghi I, Bonnì S, Samà D, Motta C, et al. Effects of palmitoylethanolamide combined with luteoline on frontal lobe functions, high frequency oscillations, and GABAergic transmission in patients with frontotemporal dementia. J Alzheimers Dis. (2020) 76:1297–308. doi: 10.3233/JAD-200426
28. Campolo M, Crupi R, Cordaro M, Cardali SM, Ardizzone A, Casili G, et al. Co-Ultra PEALut enhances endogenous repair response following moderate traumatic brain injury. Int J Mol Sci. (2021) 22:8717. doi: 10.3390/ijms22168717
29. Scuderi C, Esposito G, Blasio A, Valenza M, Arietti P, Steardo L, et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by β-amyloid peptide. J Cell Mol Med. (2011) 15:2664–74. doi: 10.1111/j.1582-4934.2011.01267.x
30. Benito C, Tolón RM, Castillo AI, Ruiz-Valdepeñas L, Martínez-Orgado JA, Fernández-Sánchez FJ, et al. β-Amyloid exacerbates inflammation in astrocytes lacking fatty acid amide hydrolase through a mechanism involving PPAR-α, PPAR-γ and TRPV1, but not CB1 or CB2 receptors. Br J Pharmacol. (2012) 166:1474–89. doi: 10.1111/j.1476-5381.2012.01889.x
31. D'Agostino G, Russo R, Avagliano C, Cristiano C, Meli R, Calignano A. Palmitoylethanolamide protects against the amyloid-β25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology. (2012) 37:1784–92. doi: 10.1038/npp.2012.25
32. Scuderi C, Valenza M, Stecca C, Esposito G, Carratù MR, Steardo L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-α. J Neuroinflammation. (2012) 9:49. doi: 10.1186/1742-2094-9-49
33. Scuderi C, Steardo L. Neuroglial roots of neurodegenerative diseases: therapeutic potential of palmitoylethanolamide in models of Alzheimer's disease. CNS Neurol Disord Drug Targets. (2013) 12:62–9. doi: 10.2174/1871527311312010011
34. Scuderi C, Stecca C, Valenza M, Ratano P, Bronzuoli MR, Bartoli S, et al. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer's disease. Cell Death Dis. (2014) 5:e1419. doi: 10.1038/cddis.2014.376
35. Tomasini MC, Borelli AC, Beggiato S, Ferraro L, Cassano T, Tanganelli S, et al. Differential Effects of Palmitoylethanolamide against Amyloid-β Induced Toxicity in Cortical Neuronal and Astrocytic Primary Cultures from Wild-Type and 3xTg-AD Mice. J Alzheimers Dis. (2015) 46:407–21. doi: 10.3233/JAD-143039
36. Siracusa R, Impellizzeri D, Cordaro M, Crupi R, Esposito E, Petrosino S, et al. Anti-Inflammatory and Neuroprotective Effects of Co-UltraPEALut in a Mouse Model of Vascular Dementia. Front Neurol. (2017) 8:233. doi: 10.3389/fneur.2017.00233
37. Beggiato S, Borelli AC, Ferraro L, Tanganelli S, Antonelli T, Tomasini MC. Palmitoylethanolamide blunts amyloid-β42-induced astrocyte activation and improves neuronal survival in primary mouse cortical astrocyte-neuron co-cultures. J Alzheimers Dis. (2018) 61:389–99. doi: 10.3233/JAD-170699
38. Bronzuoli M, Facchinetti R, Steardo L, Romano A, Stecca C, Passarella S, et al. Palmitoylethanolamide dampens reactive astrogliosis and improves neuronal trophic support in a triple transgenic model of Alzheimer's Disease: in vitro and in vivo evidence. Oxid Med Cell Longev. (2018) 2018:14. doi: 10.1155/2018/4720532
39. Crupi R, Impellizzeri D, Cordaro M, Siracusa R, Casili G, Evangelista M, et al. N-palmitoylethanolamide Prevents Parkinsonian Phenotypes in Aged Mice. Mol Neurobiol. (2018) 55:8455–72. doi: 10.1007/s12035-018-0959-2
40. Boccella S, Marabese I, Iannotta M, Belardo C, Neugebauer V, Mazzitelli M, et al. Metabotropic glutamate receptor 5 and 8 modulate the ameliorative effect of ultramicronized palmitoylethanolamide on cognitive decline associated with neuropathic pain. Int J Mol Sci. (2019) 20:1757–71. doi: 10.3390/ijms20071757
41. Boccella S, Cristiano C, Romano R, Iannotta M, Belardo C, Farina A, et al. Ultra-micronized palmitoylethanolamide rescues the cognitive decline-associated loss of neural plasticity in the neuropathic mouse entorhinal cortex-dentate gyrus pathway. Neurobiol Dis. (2019) 121:106–19. doi: 10.1016/j.nbd.2018.09.023
42. Impellizzeri D, Siracusa R, Cordaro M, Crupi R, Peritore AF, Gugliandolo E, et al. N-Palmitoylethanolamine-oxazoline (PEA-OXA): A new therapeutic strategy to reduce neuroinflammation, oxidative stress associated to vascular dementia in an experimental model of repeated bilateral common carotid arteries occlusion. Neurobiol Dis. (2019) 125:77–91. doi: 10.1016/j.nbd.2019.01.007
43. Piscitelli F, Coccurello R, Totaro A, Leuti A, Giacovazzo G, Verde R, et al. Targeted lipidomics investigation of N-acylethanolamines in a transgenic mouse model of AD: A longitudinal study. Eur J Lipid Sci Technol. (2019) 121:7. doi: 10.1002/ejlt.201900015
44. Zimmermann T, Bartsch JC, Beer A, Lomazzo E, Guggenhuber S, Lange MD, et al. Impaired anandamide/palmitoylethanolamide signaling in hippocampal glutamatergic neurons alters synaptic plasticity, learning, and emotional responses. Neuropsychopharmacology. (2019) 44:1377–88. doi: 10.1038/s41386-018-0274-7
45. Beggiato S, Cassano T, Ferraro L, Tomasini MC. Astrocytic palmitoylethanolamide pre-exposure exerts neuroprotective effects in astrocyte-neuron co-cultures from a triple transgenic mouse model of Alzheimer's disease. Life Sci. (2020) 257:118037. doi: 10.1016/j.lfs.2020.118037
46. Beggiato S, Tomasini MC, Cassano T, Ferraro L. Chronic oral palmitoylethanolamide administration rescues cognitive deficit and reduces neuroinflammation, oxidative stress, and glutamate levels in A transgenic murine model of Alzheimer's Disease. J Clin Med. (2020) 9:428–46. doi: 10.3390/jcm9020428
47. Facchinetti R, Valenza M, Bronzuoli M, Menegoni G, Ratano P, Steardo L, et al. Looking for a treatment for the early stage of alzheimer's disease: preclinical evidence with co-ultramicronized palmitoylethanolamide and luteolin. Int J Mol Sci. (2020) 21:3802–18. doi: 10.3390/ijms21113802
48. Lama A, Pirozzi C, Annunziata C, Morgese MG, Senzacqua M, Severi I, et al. Palmitoylethanolamide counteracts brain fog improving depressive-like behaviour in obese mice: Possible role of synaptic plasticity and neurogenesis. Br J Pharmacol. (2021) 178:845–59. doi: 10.1111/bph.15071
49. Boccella S, Guida F, Iannotta M, Iannotti FA, Infantino R, Ricciardi F, et al. 2-Pentadecyl-2-oxazoline ameliorates memory impairment and depression-like behaviour in neuropathic mice: possible role of adrenergic alpha2- and H3 histamine autoreceptors. Mol Brain. (2021) 14:28. doi: 10.1186/s13041-020-00724-z
50. D'Antongiovanni V, Pellegrini C, Antonioli L, Benvenuti L, Di Salvo C, Flori L, et al. Palmitoylethanolamide counteracts enteric inflammation and bowel motor dysfunctions in a mouse model of Alzheimer's Disease. Front Pharmacol. (2021) 12:748021. doi: 10.3389/fphar.2021.748021
51. Gaspar JC, Healy C, Ferdousi MI, Roche M, Finn DP. Pharmacological blockade of PPARα exacerbates inflammatory pain-related impairment of spatial memory in rats. Biomedicines. (2021) 9:610. doi: 10.3390/biomedicines9060610
52. Gatta N, Parente A, Guida F, Maione S, Gentile V. Neuronutraceuticals Modulate Lipopolysaccharide- or Amyloid-beta 1-42 Peptide-Induced Transglutaminase 2 Overexpression as a Marker of Neuroinflammation in Mouse Microglial Cells. Appl Sci. (2021) 11:5718–28. doi: 10.3390/app11125718
53. Colizzi M, Bortoletto R, Costa R, Zoccante L. Palmitoylethanolamide and Its biobehavioral correlates in autism spectrum disorder: a systematic review of human and animal evidence. Nutrients. (2021) 13:1346. doi: 10.3390/nu13041346
54. West S, King V, Carey TS, Lohr KN, McKoy N, Sutton SF, et al. Systems to rate the strength of scientific evidence. Evid Rep Technol Assess. (2002) 1–11.
55. DerSimonian R, Laird N. Meta-analysis in clinical trials revisited. Contemp Clin Trials. (2015) 45(Pt A):139–45. doi: 10.1016/j.cct.2015.09.002
56. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. (1975) 12:189–98. doi: 10.1016/0022-3956(75)90026-6
57. Landolfo E, Cutuli D, Petrosini L, Caltagirone C. Effects of palmitoylethanolamide on neurodegenerative diseases: a review from rodents to humans. Biomolecules. (2022) 12:667. doi: 10.3390/biom12050667
58. Hampel H, Mesulam MM, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. (2018) 141:1917–33. doi: 10.1093/brain/awy132
59. Yoshiyama Y, Kojima A, Ishikawa C, Arai K. Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J Alzheimers Dis. (2010) 22:295–306. doi: 10.3233/JAD-2010-100681
60. Citraro R, Russo E, Scicchitano F, van Rijn CM, Cosco D, Avagliano C, et al. Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-α receptor activation in a genetic model of absence epilepsy. Neuropharmacology. (2013) 69:115–26. doi: 10.1016/j.neuropharm.2012.11.017
61. Abedini T, Hosseyni R, Ghannadi F, Sanjari Moghaddam H, Khodaei Ardakani MR, Talaei A, et al. Efficacy and safety of palmitoylethanolamide as an adjunctive treatment for acute mania: a randomized, double-blind and placebo-controlled trial. Psychiatry Clin Neurosci. (2022) 76:505–11. doi: 10.1111/pcn.13441
62. Salehi A, Namaei P, TaghaviZanjani F, Bagheri S, Moradi K, Khodaei Ardakani MR, et al. Adjuvant palmitoylethanolamide therapy with risperidone improves negative symptoms in patients with schizophrenia: a randomized, double-blinded, placebo-controlled trial. Psychiatry Res. (2022) 316:114737. doi: 10.1016/j.psychres.2022.114737
Keywords: neurocognitive disorder, dementia, Alzheimer's disease, Parkinson's disease, cannabinoids, acylethanolamines, immune response
Citation: Colizzi M, Bortoletto R, Colli C, Bonomo E, Pagliaro D, Maso E, Di Gennaro G and Balestrieri M (2022) Therapeutic effect of palmitoylethanolamide in cognitive decline: A systematic review and preliminary meta-analysis of preclinical and clinical evidence. Front. Psychiatry 13:1038122. doi: 10.3389/fpsyt.2022.1038122
Received: 06 September 2022; Accepted: 30 September 2022;
Published: 28 October 2022.
Edited by:
Aisling O'Neill, Royal College of Surgeons in Ireland, IrelandReviewed by:
Paolo Tucci, University of Foggia, ItalyCopyright © 2022 Colizzi, Bortoletto, Colli, Bonomo, Pagliaro, Maso, Di Gennaro and Balestrieri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Colizzi, bWFyY28uY29saXp6aUB1bml1ZC5pdA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.