 Vikrant R. Mahajan1
Vikrant R. Mahajan1 Sophie K. Elvig2
Sophie K. Elvig2 Leandro F. Vendruscolo2
Leandro F. Vendruscolo2 George F. Koob2
George F. Koob2 Valerie L. Darcey3
Valerie L. Darcey3 M. Todd King4
M. Todd King4 Henry R. Kranzler5
Henry R. Kranzler5 Nora D. Volkow4
Nora D. Volkow4 Corinde E. Wiers5*
Corinde E. Wiers5*- 1Department of Pharmacology, Vanderbilt University, Nashville, TN, United States
- 2Integrative Neuroscience Research Branch, National Institute on Drug Abuse, Baltimore, MD, United States
- 3National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, United States
- 4National Institute on Alcohol Abuse and Alcoholism, Rockville, MD, United States
- 5Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
Alcohol use disorder (AUD) is a chronic, relapsing brain disorder, characterized by compulsive alcohol seeking and disrupted brain function. In individuals with AUD, abstinence from alcohol often precipitates withdrawal symptoms than can be life threatening. Here, we review evidence for nutritional ketosis as a potential means to reduce withdrawal and alcohol craving. We also review the underlying mechanisms of action of ketosis. Several findings suggest that during alcohol intoxication there is a shift from glucose to acetate metabolism that is enhanced in individuals with AUD. During withdrawal, there is a decline in acetate levels that can result in an energy deficit and could contribute to neurotoxicity. A ketogenic diet or ingestion of a ketone ester elevates ketone bodies (acetoacetate, β-hydroxybutyrate and acetone) in plasma and brain, resulting in nutritional ketosis. These effects have been shown to reduce alcohol withdrawal symptoms, alcohol craving, and alcohol consumption in both preclinical and clinical studies. Thus, nutritional ketosis may represent a unique treatment option for AUD: namely, a nutritional intervention that could be used alone or to augment the effects of medications.
Introduction
Alcohol use disorder (AUD) is a chronic, relapsing disorder characterized by disrupted function of brain circuits involved with reward, self-regulation, and emotion. During early abstinence or acute withdrawal, patients with AUD often exhibit signs and symptoms of the alcohol withdrawal syndrome (AWS), including intense alcohol craving, negative emotional states, restlessness, and in severe cases, seizures and delirium tremens. Treatment with benzodiazepines is currently the safest, most effective treatment for acute alcohol withdrawal, reducing the risk of serious symptoms such as seizures (1, 2). However, there is risk of dependence on benzodiazepines, particularly among patients with AUD, which precludes their use in this population beyond the period of acute withdrawal (3). Although anticonsulvants have also been shown to be efficacious in treating AWS and have less potential for dependence, these medications have a number of adverse effects (4, 5). Thus, additional efficacious treatments are needed that have less dependence potential and adverse effects than existing medications.
Recent preclinical and clinical studies show beneficial effects of a nutritional state of ketosis on alcohol withdrawal symptoms (6–8). Ketosis is characterized by elevated plasma and brain levels of ketone bodies (acetoacetate [AcAc], β-hydroxybutyrate [BHB] and acetone) that can be induced by prolonged or intermittent fasting, consumption of a low-carbohydrate, high-fat Ketogenic Diet (KD), a nutritional Ketone Ester (KE) supplement, Medium Chain Triglyceride (MCT) oils, or D-β-hydroxybutyrate (D-BHB) ketone salts. Here, we review the literature on the use of ketosis implemented using dietary interventions and the rationale for its potential use as a treatment for AUD. We also propose several mechanistic hypotheses based on the extant literature.
Nutritional Ketosis
Nutritional ketosis is a physiological state of energy consumption that relies primarily on elevated concentrations of ketone bodies. Ketone body concentrations can be elevated indirectly through ketogenic diets and prolonged fasts to promote fatty acid catabolism or directly through dietary supplementation with D-BHB ketone salts or esters. In addition, MCTs offer another potential avenue for supplementation via octanoic and decanoic acids, which produce more ketones per unit of energy than dietary fat (9, 10). A KD with a traditional 4:1 ratio of grams of fat to grams of carbohydrates/protein (i.e., 80% calories from fat, 15% calories from protein and 5% calories from carbohydrates) raises blood BHB levels up to 4.5 mM (8), while D-BHB salts and MCT oils elevate peak blood levels of BHB to around 0.5 mM (11, 12) and the D-BHB ketone ester raises BHB levels to ~3.2 mM (13–15).
In the presence of insufficient carbohydrates, hepatic catabolism of fatty acids from triglycerides increases ketone body levels in plasma and brain, inducing a state of metabolic ketosis. A KD shifts energy metabolism toward β-oxidation, the mitochondrial aerobic catabolism of fatty acids into acetyl-CoA (16), which can reduce the risk of seizures in patients with epilepsy (17–21). In addition, KDs have shown therapeutic effects in patients with Alzheimer's disease (22) and Parkinson's disease (23), and have been proposed as a potential therapeutic intervention for psychiatric disorders such as autism spectrum disorder (24, 25), major depressive disorder (26, 27), schizophrenia (28, 29), and bipolar disorder (30, 31). However, patient adherence to KDs, particularly those that most tightly restrict carbohydrate content (32), is limited by their poor palatability.
The nutritional supplement (R)-3-hydroxybutyl (R)-3-hydroxybutyrate (Ketone Ester; KE) is a safe (13, 33), effective, and commercially available method (e.g., DeltaG®, TdeltaS®, Orlando FL) for inducing ketosis. KE has been shown to stabilize brain networks, thereby protecting the hypometabolic, aging brain (34), increasing physical endurance in athletes (35) and improving indices of cognition in preclinical and clinical models of Alzheimer's Disease (36–38). Several advantages of ketone supplementation, specifically with D-BHB, over the traditionally used KD have been described. Within 30 min of its administration, KE (which is commercially available in a slightly bitter but palatable liquid) induces levels of plasma ketone bodies similar to those observed after 2 weeks of KD, with the effects maintained for 4–5 h with no further dietary manipulation (8, 15). Although KEs anecdotally are more effective in fasted states, their use, in contrast to KDs, does not require drastic carbohydrate restriction (39). Finally, KEs directly increase plasma ketone body levels, circumventing potential alcohol-induced inhibition of AMP-activated protein kinase (AMPK), a master regulator of ketogenesis (40, 41).
Effects of Nutritional Ketosis on Alcohol Withdrawal
Preclinical and clinical research provide evidence that KD-induced nutritional ketosis is a feasible strategy for mitigating the debilitating effects of alcohol withdrawal. Dencker et al. (6) measured the effect of a KD on signs of alcohol withdrawal in a rodent model of alcohol dependence. They found that, compared to regular chow, a KD attenuated muscular rigidity and irritability in alcohol-dependent rats during alcohol detoxification. However, despite previous evidence that exogenous ketone supplementation has anxiolytic effects in the elevated plus maze test (36, 42), this study showed no significant effect of the KD on anxiety-like behavior as measured either by the elevated plus maze test or locomotor activity (6). One potentially confounding factor in the study was that the KD decreased body weight, with the alcohol-dependent rats on the KD showing the greatest weight loss (6). The rats in the Dencker et al. (6) study were fed a KD or regular chow ad libitum. Therefore, the KD may have been less appetizing or more satiating then the regular chow, which could help to explain the greater weight loss in rats fed that diet. Studies that control for caloric intake are necessary to understand the interaction of KD with alcohol on weight loss.
In a randomized, blinded, placebo-controlled nutritional intervention in inpatients with AUD who were undergoing detoxification, during the first week of withdrawal a KD reduced benzodiazepine use more than a standard diet (50% calories from carbohydrates, 15% calories from protein, and 35% calories from fat) (8). Although withdrawal symptoms measured with the Clinical Institute Withdrawal Assessment—Alcohol revised did not differ between diet groups, patients in the standard American control diet received more benzodiazepines than patients treated with the KD. In the brain, the KD elevated levels of the metabolic markers acetone, AcAc, and glutamate and decreased choline and myo-inositol, metabolites linked to neuroinflammation (8). Correlations between low plasma BHB levels and greater social impairment, depression, and brain white matter alterations in patients with AUD also support the clinical relevance of BHB (43).
Patients who are seeking treatment for AUD often present with poor nutritional status and low appetite (44). Recently, Bornebusch et al. (7) retested the effect of a KD diet on alcohol withdrawal symptoms in mice, which included a KE-treated cohort. In two separate experiments, the researchers tested a “ketosis throughout” cohort, in which ketosis was induced during alcohol administration and abstinence, and a “ketosis after” cohort in which ketosis was induced only during abstinence. The KD diet reduced handling-induced convulsions and anxiety-like behaviors only in the ketosis throughout group, whereas a KE alleviated these withdrawal symptoms in both groups. Moreover, oral administration of 3-hydroxybutyrate alleviated tremor but not muscular rigidity in alcohol-dependent rats (45). This is important because adherence with a KE is greater than that observed with the KD. Although oral D-BHB supplements appear to have a positive therapeutic effect in alleviating withdrawal symptoms in animal models, studies are needed to elucidate the specific symptoms that are reduced and whether oral D-BHB supplements have similar effects on AWS in patients with AUD.
Effects of Nutritional Ketosis on Alcohol Craving, Consumption and Sensitivity
There is evidence that KD and KE reduce appetite and food intake (15, 46) and rodent studies have shown that nutritional ketosis reduces alcohol intake. Rats maintained on a 9-week KD followed by an 8-week regular chow diet self-administered less alcohol than those with no previous exposure to KD (i.e., mean history of KD = 30.8 ± 4.3 reinforcers/30 min vs. regular Chow = 48.3 ± 6.3) (8). Thus, a history of a KD deescalated alcohol consumption in alcohol-dependent rats (8). Although the authors initially aimed to study alcohol self-administration in rats on a current KD, the large group difference in blood alcohol levels as a function of the KD was a confounder. Specifically, rats on a current KD showed blood alcohol levels that were less than five-fold elevated following alcohol vapor exposure, an effect not seen with a regular chow diet (8). Although this suggests that a KD could interfere with alcohol metabolism, potentially due to altered activity of alcohol dehydrogenase (ADH) or aldehyde dehydrogenase (ALDH) enzymes in the liver, the hypothesis requires testing. Mice exposed to a 7-day KD showed a lower level of alcohol self-administration than those given a standard diet (i.e., mean KD = 0.51 ± 0.04 g/kg vs. standard diet = 1.04 ± 0.08 g/kg) (47). Together, these findings suggest that both current KD and a history of KD lower alcohol consumption in rodent models of alcohol drinking and dependence. However, more research is needed to investigate the effects of a KD on alcohol consumption when differences in blood alcohol levels are accounted for and to assess the effects of a KD and other means of inducing ketosis on acetaldehyde/acetate levels and ADH/ALDH enzyme activity.
We are not aware of human studies that show the effects of nutritional ketosis on alcohol metabolism, tolerance, or consumption. However, in an inpatient clinical trial testing the effects of KD on AWS signs and symptoms during detoxification, 3 weeks of KD were associated with lower subjective ratings of alcohol “wanting” and alcohol craving (at the level of a trend) than an isocaloric standard (control) diet (8). A functional magnetic resonance imaging component of the study also showed that during the 3-week treatment period, there were greater brain dorsal anterior cingulate cortex responses to alcohol visual cues in the KD group than the isocaloric control diet group, which may indicate enhanced control of alcohol craving in the KD group. In individuals with obesity, a 4-month KD lowered food craving and craving for alcohol (46). Interestingly, although alcohol alone did not increase plasma BHB in healthy volunteers, alcohol combined with a KD elevated BHB nearly 8 times more than the KD alone (48). A potential mechanism for these effects could be that elevated acetate concentrations resulting from alcohol catabolism compete with BHB as fuel for the tricarboxylic acid (TCA) cycle, resulting in higher BHB levels in plasma.
Clinical trials are currently underway (NCT04616781; NCT03255031; NCT03878225) that assess the effects of nutritional ketosis on alcohol consumption, metabolism, and tolerance in AUD and to explore potential mechanisms of action of the dietary manipulation.
Potential Mechanisms of Action for The therapeutic Effects of Ketosis in Alcohol Use Disorder
Low Glucose Utilization/High Acetate Metabolism
Glucose is the brain's primary fuel source in meeting its intensive energy demand. However, temporal variations in the brain's energy demand and supply necessitate alternative additional fuel sources to meet its metabolic and energy challenges. Circulating ketone bodies (AcAc, BHB and acetone) provide metabolic fuel the supply of which can be elevated through carbohydrate fasting-induced hepatic catabolism of fatty acids or exogenous supplementation. Passing through the brain blood barrier and entering the mitochondria of cells in the brain through monocarboxylate transporters, BHB is metabolized into AcAc and then into acetyl-CoA, which feeds into the TCA cycle (Figure 1). Studies of D-BHB supplementation have shown benefits of providing ketones as an alternative to glucose as an energy source for the brain. Some of these benefits include a elevation in the nicotinamide adenine dinucleotide (NAD) redox state (NAD+/NADH) (49, 50), which is important for mitochondrial function, an increase in the free energy for ATP synthesis in neurons (49, 51, 52), and furnishing the cell with acetyl-CoA and citric cycle intermediates (53). These findings lend support to the therapeutic potential of nutritional ketosis in pathologies characterized by glucose insensitivity by providing an alternative energy substrate.
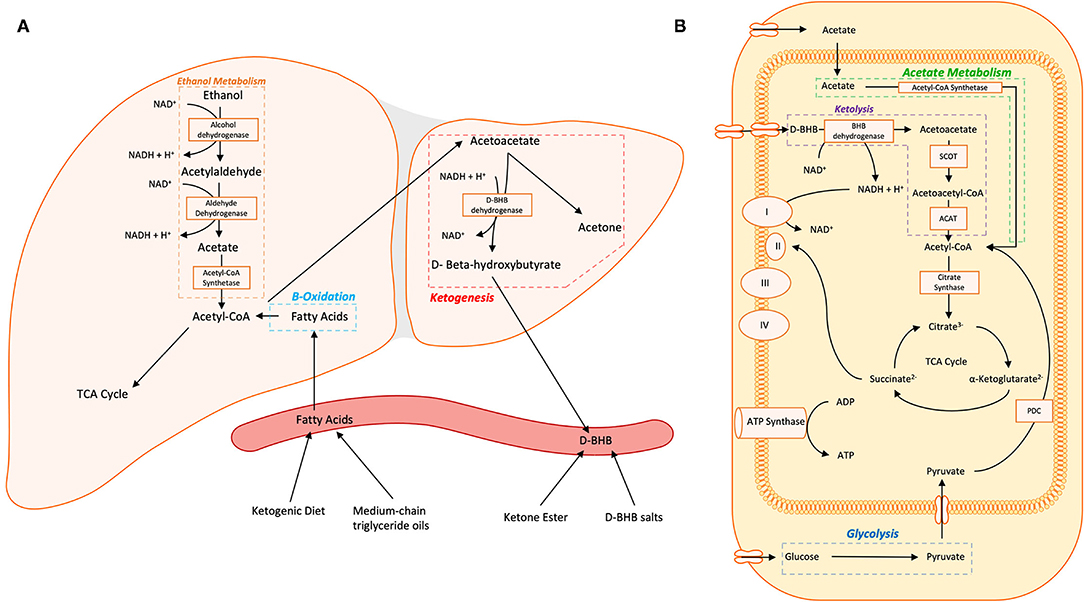
Figure 1. (A) Hepatic metabolism of ethanol and fatty acids and ketogenesis are shown. (B) Non-hepatic metabolism of D-BHB, acetate, and glucose in the cytosol and mitochondria converge on acetyl-CoA which enters the tricarboxylic acid (TCA) cycle. Within neurons, acetoacetate and D-BHB are transported into the mitochondria via monocarboxylate transporters. Abbreviations: TCA Cycle, tricarboxylic acid cycle/the citric acid cycle/Kreb's cycle; D-BHB, D-β-Hydroxybutyrate; NAD(H), nicotinamide adenine dinucleotide; SCOT, 3-ketoacid CoA transferase; ACAT, acetyl-CoA acetyltransferase; I, NADH ubiquinone oxireductase; II, succinate dehydrogenase; III, CoQH2-cytochrome c reductase; IV, cytochrome c oxidase; ATP Synthase, FOF1 ATP synthase; PDC, pyruvate dehydrogenase complex.
Substantial research has examined the metabolomic and bioenergetic effects of alcohol on the brain. Acute alcohol administration changes the brain's energetics, decreasing glucose metabolism while increasing the metabolism of acetate, a metabolite of alcohol (54). This alcohol-induced shift in brain energetics appears to be accentuated in AUD patients who, during sobriety, show higher brain acetate metabolism (55, 56) and lower brain glucose metabolism (54, 57) than non-alcohol dependent controls. These findings suggest that a shift from glucose to acetate metabolism persists beyond acute intoxication in individuals with AUD (Figure 2). During alcohol detoxification, when acetate supplies are low, this could lead to a central energy deficit that could contribute to the AWS and associated neurotoxicity (56) (Figure 2). The energy substrate deficit can be alleviated by increasing plasma ketone concentrations. Indeed, nutritional ketosis induced by a KD or oral D-BHB (ketone salts) decreased brain glucose metabolism, assessed with fludeoxyglucose ([18F]FDG-PET) and increased brain acetate metabolism, with [11C]acetoacetate binding in healthy controls (12, 58). However, aging may influence this effect, as Roy et al. (59) showed both elevated brain [18F]FDG and [11C]acetoacetate in aging rats after KD.
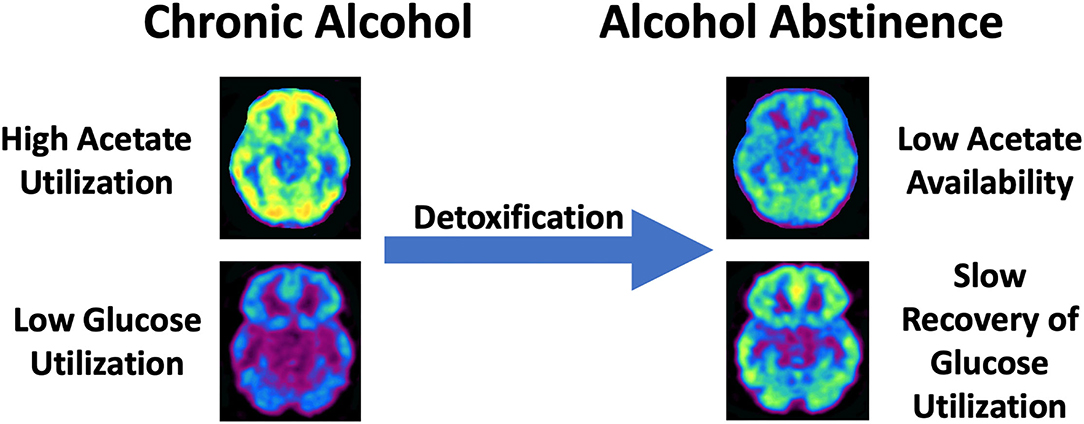
Figure 2. Schematic overview of the shift from high acetate utlization to low brain acetate avaliability with slow recovery of brain glucose metabolism in chronic AUD during detoxification. This shift is hypothesized to produce a central energy deficit that could contribute to alcohol withdrawal symptoms and associated neurotoxicity.
Brain studies in Alzheimer's disease can provide a useful parallel for AUD, as both diseases are associated with reductions in the global cerebral metabolic rate of glucose, which is estimated at 20–25% (60, 61). Reduced glycolytic flux and uptake (62) could help to explain this hypometabolism. Ketogenic diets and ketone supplementation have been shown to be protective in in vitro neuronal cell models (63) and benefits in clinical trials of Alzheimer's Disease (37, 64–66). Nutritional ketosis induced by the administration of MCT supplements has been shown to improve memory (67), and to double brain AcAc consumption in individuals with Alzheimer's disease, thereby increasing total brain energy metabolism without affecting brain glucose utilization (68). The relationship between plasma ketones and brain ketone uptake was the same in individuals with Alzheimer's Disease as in healthy young adults (58), indicating that there is intact AcAc utilization in Alzheimer's Disease. Thus, there is a potential for interventions that elevate circulating ketone bodies, primarily the administration of D-BHB, to be useful in treating pathologies characterized by impaired glucose metabolism and supply such as Alzheimer's Disease and AUD.
Imbalances in Glutamate and GABA
AWS is characterized by a general hyperexcitability of the central nervous system (69). The amino acids glutamate and γ-aminobutyric acid (GABA) are respectively the major excitatory and inhibitory neurotransmitters in the brain. Although alcohol initially inhibits excitatory effects by glutamate transmission and facilitates the inhibitory actions of GABA, chronic alcohol exposure results in compensatory changes in these amino acid transmitter systems that are opposite those seen with acute exposure and may contribute to alcohol withdrawal (70). There have been contradictory findings on brain glutamate concentrations in AUD from proton magnetic resonance spectroscopy (1H-MRS) studies. Glutamate levels in the nucleus accumbens (71) and thalamus (72) have been shown to be elevated in individuals with AUD compared to non-dependent controls. However, glutamate levels in the anterior cingulate cortex have been reported to be higher (73), lower (74, 75) or unchanged (71, 72, 76) in AUD individuals during early withdrawal compared to non-dependent controls.
Glutamate in the cingulate of AUD patients was inversely correlated with the number of heavy drinking days in the 14 days preceding the MRS scan (76). Additionally, the number of drinking years but not drinks per day was associated with higher concentrations of glutamate and glutamine (Glx) in AUD. Mon et al. (75) concluded that sobriety may normalize glutamate levels over the course of abstinence. GABA levels in plasma (70) and cingulate cortex (77) have been shown to be low during acute alcohol withdrawal. Moreover, initially low cingulate GABA levels may normalize within 3 days of last alcohol intake but only in treatment-naïve individuals with more severe AUD (77). Thus, more research is needed to better understand the dynamics of brain glutamate and GABA in individuals with AUD.
Glucose metabolism through the TCA cycle is both the main source of energy to the brain and the main source of carbon for the synthesis of glutamate and GABA (78, 79). In a murine model with reduced brain-specific pyruvate dehydrogenase activity, reduced flux through the TCA cycle reduced the glutamate content of the brain and elicited epileptiform discharges, which were ameliorated by acetate administration (80). A recent study in a mouse model of Alzheimer's Disease (triple transgenic Alzheimer's 3xTgAD, which shows reduced brain glucose utilization) showed higher hippocampal glutamate and α-ketoglutarate (a precursor of glutamate) in animals who received a KE diet compared to regular chow and a positive correlation between glutamate and α-ketoglutarate levels in both groups (50). Thus, nutritional ketosis appears to furnish mitochondria with TCA cycle substrates (38), as evidenced by the finding that a 4-month KD elevated glutamate and glutamine in young adult rats (81). Although patients with epilepsy did not show differences from controls in posterior cingulate cortical glutamate measures, patients' elevated glutamate concentrations predicted short-term freedom from seizures, supporting the clinical relevance of glutamate concentrations in epilepsy (82). This underscores the need to elucidate the mechanism(s) underlying the association of KD-induced changes in glutamate with brain excitability.
Vesicular glutamate transporters (VGLUT) are required for packaging and exocytotic release of glutamate. VGLUT is inhibited by AcAc and BHB through a competitive interaction with the VGLUT allosteric activator Cl− (83). A decrease in the concentration of glutamate per vesicle from VGLUT inhibition reduces glutamatergic activation, thereby dampening excitation. BHB and AcAc may also dampen neuronal excitability via their effect on K+/ATP channels, having been shown to reduce the spontaneous firing rate of substantia nigra pars reticulata neurons in vitro (84). This effect was abolished by the genetic or metabolic elimination of metabolically sensitive K+/ATP channels (84). In a recent study, acetone and BHB acted as inhibitors of glutamate at NMDA receptors (85). In addition, D-BHB and acetoacetate reduced neuronal death and changes of neuronal membrane properties in rat neocortical neurons subjected to glutamate excitotoxicity (86). Further, calorically restricted KD increases the expression of glutamic acid decarboxylase, the enzyme responsible for the conversion of glutamate to GABA (87), in the brain which increases the conversion of glutamate and thereby reduces excitation. Thus, there is conflicting evidence and potential mechanistic roles of glutamate in the context of AUD and further elucidation is needed with a particular emphasis on intracellular versus extracellular changes.
The efficacy of a KD in preventing or reducing seizures in epilepsy (88) may have direct relevance to alcohol withdrawal, which can be complicated by seizures. The following mechanism(s) have been proposed for the reduction of seizures by a KD: (1) restoring glutamatergic neurotransmission and enhancement of GABA synthesis, (2) circumventing glycolysis and providing Acetyl-CoA for the TCA cycle through fatty acid oxidation, (3) stimulating ATP-sensitive K+ channels, and (4) inhibiting voltage-dependent Ca2+ channels (78, 89). However, seizures are uncommon in AUD patients undergoing detoxification, partly because benzodiazepines, which are widely use to manage the AWS, have anticonvulsant activity (90). A KD may reduce overall neuronal excitability, mitigating the severity of alcohol withdrawal symptoms and reducing the need for benzodiazepine treatment during acute withdrawal. Thus, rodent studies are needed to investigate the effect of a KD on alcohol-induced seizures, as these would inform efforts to prevent alcohol withdrawal-induced seizures in patients (90). However, it is unclear how the hypothesized reductions of neuronal excitability with KD would associate with brain glutamate concentrations.
Hormonal Regulation
Ghrelin is a homeostatic hormone that stimulates human appetite, having effects opposite to those of leptin (91, 92). Endogenous peripheral ghrelin levels decrease during alcohol drinking and increase during alcohol abstinence (93–99). Studies have shown that genetic or metabolic reductions in ghrelin levels decrease alcohol intake (100, 101). In addition, higher ghrelin levels are associated with greater self-reported craving (97, 98, 101, 102), longer and more intense subjective responses to alcohol (103), and activation of the bilateral insulae (104) and ventral striatum (105) as measured with functional magnetic resonance imaging during alcohol cue exposure. Higher levels of ghrelin and activation of the ghrelin receptor stimulate the cholinergic-dopaminergic reward link, which has implications for the reinforcing effects of ghrelin in AUD (106). In healthy volunteers, a single administration of a KE was associated with decreased self-reported hunger and plasma ghrelin levels than the ingestion of isocaloric dextrose (15).
Although there is some indication that KD may suppress ghrelin levels (see review by Roekenes and Martins (107)), there are some inconsistencies in the literature (108–111). Leptin and peptide YY have effects opposite to ghrelin, in that they promote satiety (112, 113). A KD has been shown to increase serum peptide YY levels (114), though it has also been shown to decrease leptin levels (115, 116).
Fibroblast growth factor 21 (FGF21) is a hormone of hepatic origin whose targets include white and brown adipose tissue, the hypothalamus, and the hindbrain (117, 118). A KD has been shown to increase the concentration of FGF21 in murine models (119, 120), but this effect was not seen in humans (121–123). Nevertheless, in humans, FGF21-based pharmacotherapy decreased body weight (124) and variation in the FGF21 gene has been associated with macronutrient preference (carbohydrate, fat, and protein) (125). Moreover, FGF21 administration reduced a preference for alcohol in mice and for sweets in mice and monkeys (126). Therefore, FGF21 may be a key factor involved in the effects of ketosis on alcohol preference and warrants further investigation.
Glucagon-like peptide 1 (GLP1) is an intestinal hormone that enhances insulin secretion, inhibits glucagon secretion, and decreases gastric motility (127). There is some clinical evidence that the concentration of GLP-1 is increased in response to high fat KDs (109, 128), although experiments in cell culture have yielded contradictory evidence (129). GLP-1 receptor activation by GLP-1 agonists suppresses the effects of alcohol on the mesolimbic dopamine system and decreases alcohol consumption and operant self-administration (130–135). However, the GLP-1 receptor agonist Exendin-4 failed to attenuate morphine conditioned place preference or remifentanil self-administration (132). In addition, there is limited evidence that GLP-1 receptor agonists affect cocaine consumption (136, 137). Taken together, GLP-1 receptor activation induced by increased GLP-1 levels produced by a KD could serve as a suppressor of alcohol intake. Further research is needed to establish the role of the KD effect on circulating GLP-1 levels.
Evidence suggests that alcohol dependence is associated with dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis and extrahypothalamic glucocorticoid signaling as well as other stress (e.g., corticotropin-releasing factor [CRF]) and anti-stress (e.g., neuropeptide Y) systems (138). However, the few available studies of the effects of ketosis on the HPA axis and other stress systems have yielded contradictory findings. For example, in one rat study, neither a KD nor a ketone supplementation diet affected plasma levels of adrenocorticotropic hormone or corticosterone (139). In another study, both KD and MCT increased HPA axis activity (140). Interestingly, in female but not male rats exposed to chronic mild stress, a KD prevented stress-related blood corticosterone and hypothalamic NPY expression; this effect was not accompanied by altered CRF mRNA expression (141). Furthermore, continuous microinjection of D-BHB into the prefrontal cortex attenuated the effects of a chronic unpredictable stress on depression-like behavior and HPA axis activity (142). More research on the effects of ketosis on stress systems is needed.
Nicotinamide Adenine Dinucleotide (NAD+)
NAD+ is present in all living cells and plays a vital role in cellular metabolism as a coenzyme for redox reactions, including those required for mitochondrial energy production. NAD+ decreases with age (143, 144) and lower NAD+ levels are associated with neurodegenerative and neuropsychiatric disorders including Alzheimer's Disease and schizophrenia (145). Although individuals with AUD have low liver concentrations of NAD+ (146), it remains to be determined whether their brain NAD+ concentrations are affected by chronic heavy alcohol consumption. Because NAD+ and pyruvate are implicated in both the oxidation of alcohol (147, 148) and in the metabolic effects of fasting (149), it is possible that these compounds mediate the clinical efficacy of nutritional ketosis in AUD. An intravenous infusion of NAD+ during alcohol or opioid withdrawal attenuated both craving and withdrawal symptoms (150). A 7-Tesla magnetic resonance spectroscopy study in healthy volunteers showed that ketone supplementation elevates the concentration of NAD+ in the brain (151). Mice who received dietary supplementation with KE had higher cortical and hippocampal free cytosolic [NAD+]/[NADH] than mice fed a control diet (50). A KD also increased cellular concentrations of NAD+ (152), along with concentrations of Sirt1, Parp-1, and 8-hydroxy-2′-deoxyguanosine, which could improve brain health by increasing resilience to DNA damage and oxidative stress (153). However, the effects of ketone modulation of NAD+ in patients with AUD and its clinical and cognitive effects are unstudied. Because nucleotide coenzymes and their corresponding oxidizing forms are compartmentalized and bind at a subcellular level, their measurement and the interpretation of the results require great care to ensure accuracy. For example, from fed, freeze-clamped, rat liver the calculated free cytoplasmic [NAD+]/[NADH] from lactate dehydrogenase was approximately 200 times higher than the ratio calculated measured using total concentrations of the coenzymes. Conversely, the free cytoplasmic [NADP+]/[NADPH] from isocitrate dehydrogenase was ~20 times lower than the ratio calculated from measured total respective amounts (149).
D-β-Hydroxybutyrate as a Signaling Molecule
In addition to its direct action in mitochondrial metabolism, D-BHB may exert therapeutic effects as a signaling molecule. D-BHB has been suggested to have direct involvement in epigenetic regulation due to its ability to act as an inhibitor of class 1 histone deacetylases (HDAC) that increases global acetylation levels in a dose-dependent manner (154). During withdrawal from chronic alcohol, anxiety-like behaviors were correlated with an increase in HDAC activity and a decrease in H3/H4 acetylation, but the behaviors could be reversed with the HDAC inhibitor trichostatin A (155). Furthermore, alcohol withdrawal-induced hyperalgesia was attenuated by the HDAC inhibitor suberoylanilide hydroxamic acid (156). In addition, the anxiolytic-like responses to acute alcohol administration were associated with increased histone acetylation and HDAC inhibition in the amygdala (155, 157). In vitro application of D-BHB also increased the expression of FOXO3, MnSOD, CAT, and MT2, genes that encode oxidative stress resistance factors (154). Bolstering this mechanism, various studies have demonstrated D-BHB's neuroprotective effect against oxidative stress (158–160). In humans, alcohol is processed by ADH enzymes into acetaldehyde, which produces unstable free radicals like hydrogen peroxide and superoxide (161, 162). Furthermore, chronic alcohol consumption depletes mitochondrial glutathione, a potent antioxidant (163). Thus, D-BHB may be unique in its capacity to respond to epigenetic and oxidative stress changes that occur during AUD.
Brain-derived neurotrophic factor (BDNF), a neurotrophin that helps control neurogenesis, has been implicated in the development of AUD (164–166). Although individuals with current AUD had lower overall serum BDNF levels than non-AUD controls (167), preclinical studies indicate that the directionality of the BDNF change is brain region-specific (168, 169). Further, BDNF levels raise during alcohol withdrawal in preclinical models (170), clinical populations (171–173), and raise during withdrawal from other addictive drugs (174, 175). Mechanistically, D-BHB enhances the expression of BDNF through downstream targeting of CREB and acetylation of BDNF promoters (176–178). While some clinical evidence points to serum BDNF being significantly increased following adherence to a KD (116, 179, 180), Vizuete et al. (181) found a KD decreased striatal BDNF levels and had no effect on hippocampal levels of BDNF in Wistar rats. The KD and D-BHB's effect on BDNF expression in the context of AUD warrants investigation.
D-BHB is a ligand of the hydroxyl carboxylic acid receptor type 2 (Hca2) (182), a GPCR encoded by the Hcar2 gene that mediates anti-inflammatory effects (183). In a rodent stroke model, a KD and D-BHB separately rescued stroke-induced neurological deficits but the effect was not seen in Hcar2 knockout mice (Hcar2−/−; (184). These findings reinforce the critical role of Hca2 as an intermediate for D-BHB's neuroprotective effects. In addition to lower hepatic D-BHB levels in humans with alcohol-associated hepatitis, D-BHB attenuated abnormalities in plasma ALT levels, steatosis, and hepatic trigylceride levels induced by the β-oxidation inhibitor etomoxir and alcohol (185). The protective effect of D-BHB was not seen in Hcar2−/− mice (185).
The NLR family pyrin domain containing 3 (NLRP3) inflammasome complex is a predominately macrophagic protein that mediates caspase-1 activation and the secretion of proinflammatory cytokines in response to mitochondrial dysfunction, ROS and more. Evidence suggests that the NLRP3 inflammasome complex is activated by alcohol consumption (186, 187) and inhibited by D-BHB supplementation (188). Although deficiencies of NLRP3 were shown to attenuate alcohol-associated steatosis (189), a study showed that this inhibition can increase the rate of hepatic damage (190), suggesting that the NLRP3 inflammasome complex may be protective during alcohol-induced hepatic damage. In vitro inhibition of the NLRP3 inflammasome by D-BHB was decreased by high insulin or high glucose, suggesting an influence of the metabolic state of the cell (191). Finally, a single dose of D-BHB clinically was shown to increase markers of NLRP3 inflammasome activation blood cells (192); however, this finding failed subsequent replication in patients with obesity (193). Further research is needed to elucidate the role of metabolic ketosis on the NLRP3 inflammasome in the context of AUD.
Conclusion and Future Directions
Preclinical and clinical research on the role of ketosis in the signs and symptoms of the AUD/AWS suggest that such an intervention could be useful as an adjunctive treatment. We reviewed potential mechanisms of clinical action of ketosis, with a particular emphasis on brain energy substrate utilization and the glutamatergic/GABAergic systems. An existing limitation of the proposed therapy is the potential of a KD to contribute to the development of alcohol-associated ketoacidosis, which occurs with some frequency in patients with AUD. Thus, clinical trials of ketosis as a treatment may need to exclude participants at increased risk of ketoacidosis and a history of ketoacidosis. Younger patients may be more susceptible to symptomatic hypoglycemia following adherence to a KD (194). The ingestion of ketone ester can also decrease blood glucose concentrations (14, 139). In addition, sex differences in alcohol metabolism and in the response to a KD warrant investigation (195–197), as does variation in genes involved in alcohol and fat metabolism (e.g., ADH, ALDH, FGF21) (120, 198–200). The existing literature supports further examination of nutritional ketosis as a therapeutic target for AWS and of the mechanistic underpinnings of its effects. Moreover, key questions as to the effects of nutritional ketosis on brain energetics in AWS, alcohol tolerance, and AUD-associated brain hypometabolism remain to be investigated.
Author Contributions
VM and CW drafted the first version of the manuscript. SE, LV, GK, VD, MK, HK, and NV provided critical input that significantly improved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
LV was supported by NIDA Intramural Research Program. HK is a member of an advisory board for Dicerna Pharmaceuticals and Sophrosyne Pharmaceuticals, a consultant for Sobrera Pharmaceuticals, and a member of the American Society of Clinical Psychopharmacology's Alcohol Clinical Trials Initiative, which was supported in the last three years by AbbVie, Alkermes, Dicerna, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer, Arbor, and Amygdala Neurosciences. CW was supported by a K99/R00 Pathway to Independence Award (AA026892) and a NARSAD Young Investigator Grant (28778), Brain & Behavior Research Foundation.
Conflict of Interest
HK is named as an inventor on PCT patent application #15/878,640 entitled: Genotypeguided dosing of opioid agonists, filed January 24, 2018.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the late Dr. Richard Veech for his lasting contributions to the field of nutritional ketosis.
References
1. Saitz R, O'malley SS. Pharmacotherapies for alcohol abuse. Withdrawal and treatment. Med Clin North Am. (1997) 81:881–907. doi: 10.1016/S0025-7125(05)70554-X
2. Day E, Daly C. Clinical management of the alcohol withdrawal syndrome. Addiction. (2021). doi: 10.1111/add.15647. [Epub ahead of print].
3. Morel A, Grall-Bronnec M, Bulteau S, Chauvin-Grelier P, Gailledrat L, Pinot ML, et al. Benzodiazepine dependence in subjects with alcohol use disorders: what prevalence? Expert Opin Drug Saf. (2016) 15:1313–9. doi: 10.1080/14740338.2016.1221922
4. Zindel LR, Kranzler HR. Pharmacotherapy of alcohol use disorders: seventy-five years of progress. J Stud Alcohol Drugs Suppl. (2014) 75:79–88. doi: 10.15288/jsads.2014.s17.79
5. Cheng YC, Huang YC, Huang WL. Gabapentinoids for treatment of alcohol use disorder: a systematic review and meta-analysis. Hum Psychopharmacol. (2020) 35:1–11. doi: 10.1002/hup.2751
6. Dencker D, Molander A, Thomsen M, Schlumberger C, Wortwein G, Weikop P, et al. Ketogenic diet suppresses alcohol withdrawal syndrome in rats. Alcohol Clin Exp Res. (2018) 42:270–7. doi: 10.1111/acer.13560
7. Bornebusch AB, Mason GF, Tonetto S, Damsgaard J, Gjedde A, Fink-Jensen A, et al. Effects of ketogenic diet and ketone monoester supplement on acute alcohol withdrawal symptoms in male mice. Psychopharmacology. (2021) 238:833–44. doi: 10.1007/s00213-020-05735-1
8. Wiers CE, Vendruscolo LF, Van Der Veen JW, Manza P, Shokri-Kojori E, Kroll DS, et al. Ketogenic diet reduces alcohol withdrawal symptoms in humans and alcohol intake in rodents Sci Adv. (2021) 7:eabf6780. doi: 10.1126/sciadv.abf6780
9. Freund G, Weinsier RL. Standardized ketosis in man following medium chain triglyceride ingestion. Metabolism. (1966) 15:980–91. doi: 10.1016/0026-0495(66)90046-1
10. Huttenlocher PR, Wilbourn AJ, Signore JM. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology. (1971) 21:1097–103. doi: 10.1212/WNL.21.11.1097
11. Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, et al. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol Aging. (2004) 25:311–4. doi: 10.1016/S0197-4580(03)00087-3
12. Cuenoud B, Hartweg M, Godin JP, Croteau E, Maltais M, Castellano CA, et al. Metabolism of exogenous D-Beta-Hydroxybutyrate, an energy substrate avidly consumed by the heart and kidney. Front Nutr. (2020) 7:13. doi: 10.3389/fnut.2020.00013
13. Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol. (2012) 63:401–8. doi: 10.1016/j.yrtph.2012.04.008
14. Myette-Cote E, Neudorf H, Rafiei H, Clarke K, Little JP. Prior ingestion of exogenous ketone monoester attenuates the glycaemic response to an oral glucose tolerance test in healthy young individuals. J Physiol. (2018) 596:1385–95. doi: 10.1113/JP275709
15. Stubbs BJ, Cox PJ, Evans RD, Cyranka M, Clarke K, De Wet H. A ketone ester drink lowers human ghrelin and appetite. Obesity. (2018) 26:269–73. doi: 10.1002/oby.22051
16. Sikder K, Shukla SK, Patel N, Singh H, Rafiq K. High fat diet upregulates fatty acid oxidation and ketogenesis via intervention of PPAR-gamma. Cell Physiol Biochem. (2018) 48:1317–31. doi: 10.1159/000492091
17. Kinsman SL, Vining EP, Quaskey SA, Mellits D, Freeman JM. Efficacy of the ketogenic diet for intractable seizure disorders: review of 58 cases. Epilepsia. (1992) 33:1132–6. doi: 10.1111/j.1528-1157.1992.tb01770.x
18. Vining EP, Freeman JM, Ballaban-Gil K, Camfield CS, Camfield PR, Holmes GL, et al. A multicenter study of the efficacy of the ketogenic diet. Arch Neurol. (1998) 55:1433–7. doi: 10.1001/archneur.55.11.1433
19. Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. (2008) 7:500–6. doi: 10.1016/S1474-4422(08)70092-9
20. Wheless JW. History of the ketogenic diet. Epilepsia. (2008) 49:3–5. doi: 10.1111/j.1528-1167.2008.01821.x
21. Liu H, Yang Y, Wang Y, Tang H, Zhang F, Zhang Y, et al. Ketogenic diet for treatment of intractable epilepsy in adults: a meta-analysis of observational studies. Epilepsia Open. (2018) 3:9–17. doi: 10.1002/epi4.12098
22. Phillips MCL, Deprez LM, Mortimer GMN, Murtagh DKJ, Mccoy S, Mylchreest R, et al. Randomized crossover trial of a modified ketogenic diet in Alzheimer's disease. Alzheimers Res Ther. (2021) 13:51. doi: 10.1186/s13195-021-00783-x
23. Phillips MCL, Murtagh DKJ, Gilbertson LJ, Asztely FJS, Lynch CDP. Low-fat versus ketogenic diet in Parkinson's disease: a pilot randomized controlled trial. Mov Disord. (2018) 33:1306–14. doi: 10.1002/mds.27390
24. Evangeliou A, Vlachonikolis I, Mihailidou H, Spilioti M, Skarpalezou A, Makaronas N, et al. Application of a ketogenic diet in children with autistic behavior: pilot study. J Child Neurol. (2003) 18:113–8. doi: 10.1177/08830738030180020501
25. Lee RWY, Corley MJ, Pang A, Arakaki G, Abbott L, Nishimoto M, et al. A modified ketogenic gluten-free diet with MCT improves behavior in children with autism spectrum disorder. Physiol Behav. (2018) 188:205–11. doi: 10.1016/j.physbeh.2018.02.006
26. Wlodarczyk A, Cubala WJ. Mechanisms of action of the ketogenic diet in depression. Neurosci Biobehav Rev. (2019) 107:422–3. doi: 10.1016/j.neubiorev.2019.09.038
27. Ricci A, Idzikowski MA, Soares CN, Brietzke E. Exploring the mechanisms of action of the antidepressant effect of the ketogenic diet. Rev Neurosci. (2020) 31:637–48. doi: 10.1515/revneuro-2019-0073
28. Palmer CM, Gilbert-Jaramillo J, Westman EC. The ketogenic diet and remission of psychotic symptoms in schizophrenia: two case studies. Schizophr Res. (2019) 208:439–40. doi: 10.1016/j.schres.2019.03.019
29. Sarnyai Z, Kraeuter AK, Palmer CM. Ketogenic diet for schizophrenia: clinical implication. Curr Opin Psychiatry. (2019) 32:394–401. doi: 10.1097/YCO.0000000000000535
30. Phelps JR, Siemers SV, El-Mallakh RS. The ketogenic diet for type II bipolar disorder. Neurocase. (2013) 19:423–6. doi: 10.1080/13554794.2012.690421
31. Saraga M, Misson N, Cattani E. Ketogenic diet in bipolar disorder. Bipolar Disord. (2020) 22:765. doi: 10.1111/bdi.13013
32. Ye F, Li XJ, Jiang WL, Sun HB, Liu J. Efficacy of and patient compliance with a ketogenic diet in adults with intractable epilepsy: a meta-analysis. J Clin Neurol. (2015) 11:26–31. doi: 10.3988/jcn.2015.11.1.26
33. Soto-Mota A, Vansant H, Evans RD, Clarke K. Safety and tolerability of sustained exogenous ketosis using ketone monoester drinks for 28 days in healthy adults. Regul Toxicol Pharmacol. (2019) 109:104506. doi: 10.1016/j.yrtph.2019.104506
34. Mujica-Parodi LR, Amgalan A, Sultan SF, Antal B, Sun X, Skiena S, et al. Diet modulates brain network stability, a biomarker for brain aging, in young adults. Proc Natl Acad Sci USA. (2020) 117:6170–7. doi: 10.1073/pnas.1913042117
35. Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. (2016) 24:256–68. doi: 10.1016/j.cmet.2016.07.010
36. Kashiwaya Y, Bergman C, Lee JH, Wan R, King MT, Mughal MR, et al. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer's disease. Neurobiol Aging. (2013) 34:1530–9. doi: 10.1016/j.neurobiolaging.2012.11.023
37. Newport MT, Vanitallie TB, Kashiwaya Y, King MT, Veech RL. A new way to produce hyperketonemia: use of ketone ester in a case of Alzheimer's disease. Alzheimers Dement. (2015) 11:99–103. doi: 10.1016/j.jalz.2014.01.006
38. Pawlosky RJ, Kashiwaya Y, King MT, Veech RL. A Dietary Ketone ester normalizes abnormal behavior in a mouse model of Alzheimer's disease. Int J Mol Sci. (2020) 21:1044. doi: 10.3390/ijms21031044
39. Spoke C, Malaeb S. A case of hypoglycemia associated with the ketogenic diet and alcohol use. J Endocr Soc. (2020) 4:bvaa045. doi: 10.1210/jendso/bvaa045
40. You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. (2004) 127:1798–808. doi: 10.1053/j.gastro.2004.09.049
41. Liangpunsakul S, Wou SE, Zeng Y, Ross RA, Jayaram HN, Crabb DW. Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am J Physiol Gastrointest Liver Physiol. (2008) 295:G1173–81. doi: 10.1152/ajpgi.90349.2008
42. Ari C, Kovacs Z, Juhasz G, Murdun C, Goldhagen CR, Koutnik AP, et al. Exogenous ketone supplements reduce anxiety-related behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk rats. Front Mol Neurosci. (2016) 9:137. doi: 10.3389/fnmol.2016.00137
43. Leclercq S, Le Roy T, Furgiuele S, Coste V, Bindels LB, Leyrolle Q, et al. Gut microbiota-induced changes in beta-hydroxybutyrate metabolism are linked to altered sociability and depression in alcohol use disorder. Cell Rep. (2020) 33:108238. doi: 10.1016/j.celrep.2020.108238
44. Ross LJ, Wilson M, Banks M, Rezannah F, Daglish M. Prevalence of malnutrition and nutritional risk factors in patients undergoing alcohol and drug treatment. Nutrition. (2012) 28:738–43. doi: 10.1016/j.nut.2011.11.003
45. Derr RF, Derr MI. Separation of the tremorous and muscular rigidity components of the ethanol withdrawal syndrome in the rat. Physiol Behav. (1986) 38:1–3. doi: 10.1016/0031-9384(86)90123-X
46. Castro AI, Gomez-Arbelaez D, Crujeiras AB, Granero R, Aguera Z, Jimenez-Murcia S, et al. Effect of a very low-calorie ketogenic diet on food and alcohol cravings, physical and sexual activity, sleep disturbances, and quality of life in obese patients. Nutrients. (2018) 10:1348. doi: 10.3390/nu10101348
47. Blanco-Gandia MDC, Rodenas-Gonzalez F, Pascual M, Reguilon MD, Guerri C, Minarro J, et al. Ketogenic diet decreases alcohol intake in adult male mice. Nutrients. (2021) 13:2167. doi: 10.3390/nu13072167
48. Lefevre A, Adler H, Lieber CS. Effect of ethanol on ketone metabolism. J Clin Invest. (1970) 49:1775–82. doi: 10.1172/JCI106395
49. Marosi K, Kim SW, Moehl K, Scheibye-Knudsen M, Cheng A, Cutler R, et al. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem. (2016) 139:769–81. doi: 10.1111/jnc.13868
50. Pawlosky RJ, Kemper MF, Kashiwaya Y, King MT, Mattson MP, Veech RL. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer's disease. J Neurochem. (2017) 141:195–207. doi: 10.1111/jnc.13958
51. Sato K, Kashiwaya Y, Keon CA, Tsuchiya N, King MT, Radda GK, et al. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. (1995) 9:651–8. doi: 10.1096/fasebj.9.8.7768357
52. Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. (2003) 112:892–901. doi: 10.1172/JCI200318797
53. Suissa L, Kotchetkov P, Guigonis JM, Doche E, Osman O, Pourcher T, et al. Ingested ketone ester leads to a rapid rise of acetyl-CoA and competes with glucose metabolism in the brain of non-fasted mice. Int J Mol Sci. (2021) 22:524. doi: 10.3390/ijms22020524
54. Volkow ND, Wiers CE, Shokri-Kojori E, Tomasi D, Wang GJ, Baler R. Neurochemical and metabolic effects of acute and chronic alcohol in the human brain: Studies with positron emission tomography. Neuropharmacology. (2017) 122:175–88. doi: 10.1016/j.neuropharm.2017.01.012
55. Jiang L, Gulanski BI, De Feyter HM, Weinzimer SA, Pittman B, Guidone E, et al. Increased brain uptake and oxidation of acetate in heavy drinkers. J Clin Invest. (2013) 123:1605–14. doi: 10.1172/JCI65153
56. Volkow ND, Kim SW, Wang GJ, Alexoff D, Logan J, Muench L, et al. Acute alcohol intoxication decreases glucose metabolism but increases acetate uptake in the human brain. Neuroimage. (2013) 64:277–83. doi: 10.1016/j.neuroimage.2012.08.057
57. Tomasi DG, Wiers CE, Shokri-Kojori E, Zehra A, Ramirez V, Freeman C, et al. Association between reduced brain glucose metabolism and cortical thickness in alcoholics: evidence of neurotoxicity. Int J Neuropsychopharmacol. (2019) 22:548–59. doi: 10.1093/ijnp/pyz036
58. Courchesne-Loyer A, Croteau E, Castellano CA, St-Pierre V, Hennebelle M, Cunnane SC. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: a dual tracer quantitative positron emission tomography study. J Cereb Blood Flow Metab. (2017) 37:2485–93. doi: 10.1177/0271678X16669366
59. Roy M, Nugent S, Tremblay-Mercier J, Tremblay S, Courchesne-Loyer A, Beaudoin JF, et al. The ketogenic diet increases brain glucose and ketone uptake in aged rats: a dual tracer PET and volumetric MRI study. Brain Res. (2012) 1488:14–23. doi: 10.1016/j.brainres.2012.10.008
60. Rapoport SI. Positron emission tomography in normal aging and Alzheimer's disease. Gerontology. (1986) 32:6–13. doi: 10.1159/000212819
61. Mosconi L, Tsui WH, De Santi S, Li J, Rusinek H, Convit A, et al. Reduced hippocampal metabolism in MCI and AD: automated FDG-PET image analysis. Neurology. (2005) 64:1860–7. doi: 10.1212/01.WNL.0000163856.13524.08
62. An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, et al. Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers Dement. (2018) 14:318–29. doi: 10.1016/j.jalz.2017.09.011
63. Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. D-beta-hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc Natl Acad Sci USA. (2000) 97:5440–4. doi: 10.1073/pnas.97.10.5440
64. Rebello CJ, Keller JN, Liu AG, Johnson WD, Greenway FL. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: a randomized controlled trial. BBA Clin. (2015) 3:123–5. doi: 10.1016/j.bbacli.2015.01.001
65. Taylor MK, Sullivan DK, Mahnken JD, Burns JM, Swerdlow RH. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer's disease. Alzheimers Dement. (2018) 4:28–36. doi: 10.1016/j.trci.2017.11.002
66. Ota M, Matsuo J, Ishida I, Takano H, Yokoi Y, Hori H, et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer's disease. Neurosci Lett. (2019) 690:232–6. doi: 10.1016/j.neulet.2018.10.048
67. Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab. (2009) 6:31. doi: 10.1186/1743-7075-6-31
68. Croteau E, Castellano CA, Richard MA, Fortier M, Nugent S, Lepage M, et al. Ketogenic medium chain triglycerides increase brain energy metabolism in Alzheimer's disease. J Alzheimers Dis. (2018) 64:551–61. doi: 10.3233/JAD-180202
69. Becker HC, Mulholland PJ. Neurochemical mechanisms of alcohol withdrawal. Handb Clin Neurol. (2014) 125:133–56. doi: 10.1016/B978-0-444-62619-6.00009-4
70. Brousse G, Arnaud B, Vorspan F, Richard D, Dissard A, Dubois M, et al. Alteration of glutamate/GABA balance during acute alcohol withdrawal in emergency department: a prospective analysis. Alcohol Alcohol. (2012) 47:501–8. doi: 10.1093/alcalc/ags078
71. Bauer J, Pedersen A, Scherbaum N, Bening J, Patschke J, Kugel H, et al. Craving in alcohol-dependent patients after detoxification is related to glutamatergic dysfunction in the nucleus accumbens and the anterior cingulate cortex. Neuropsychopharmacology. (2013) 38:1401–8. doi: 10.1038/npp.2013.45
72. Wiers CE, Cunningham SI, Tomasi DG, Ernst T, Chang L, Shokri-Kojori E, et al. Elevated thalamic glutamate levels and reduced water diffusivity in alcohol use disorder: association with impulsivity. Psychiatry Res Neuroimaging. (2020) 305:111185. doi: 10.1016/j.pscychresns.2020.111185
73. Hermann D, Weber-Fahr W, Sartorius A, Hoerst M, Frischknecht U, Tunc-Skarka N, et al. Translational magnetic resonance spectroscopy reveals excessive central glutamate levels during alcohol withdrawal in humans and rats. Biol Psychiatry. (2012) 71:1015–21. doi: 10.1016/j.biopsych.2011.07.034
74. Thoma R, Mullins P, Ruhl D, Monnig M, Yeo RA, Caprihan A, et al. Perturbation of the glutamate-glutamine system in alcohol dependence and remission. Neuropsychopharmacology. (2011) 36:1359–65. doi: 10.1038/npp.2011.20
75. Mon A, Durazzo TC, Meyerhoff DJ. Glutamate, GABA, and other cortical metabolite concentrations during early abstinence from alcohol and their associations with neurocognitive changes. Drug Alcohol Depend. (2012) 125:27–36. doi: 10.1016/j.drugalcdep.2012.03.012
76. Cheng H, Kellar D, Lake A, Finn P, Rebec GV, Dharmadhikari S, et al. Effects of alcohol cues on MRS glutamate levels in the anterior cingulate. Alcohol Alcohol. (2018) 53:209–15. doi: 10.1093/alcalc/agx119
77. Prisciandaro JJ, Schacht JP, Prescot AP, Brenner HM, Renshaw PF, Brown TR, et al. Intraindividual changes in brain GABA, glutamate, and glutamine during monitored abstinence from alcohol in treatment-naive individuals with alcohol use disorder. Addict Biol. (2020) 25:e12810. doi: 10.1111/adb.12810
78. Haslam RJ, Krebs HA. The metabolism of glutamate in homogenates and slices of brain cortex. Biochem J. (1963) 88:566–78. doi: 10.1042/bj0880566
79. Rothman DL, De Feyter HM, De Graaf RA, Mason GF, Behar KL. 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed. (2011) 24:943–57. doi: 10.1002/nbm.1772
80. Jakkamsetti V, Marin-Valencia I, Ma Q, Good LB, Terrill T, Rajasekaran K, et al. Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aan0457
81. Gzielo K, Janeczko K, Weglarz W, Jasinski K, Klodowski K, Setkowicz Z. MRI spectroscopic and tractography studies indicate consequences of long-term ketogenic diet. Brain Struct Funct. (2020) 225:2077–89. doi: 10.1007/s00429-020-02111-9
82. Gonen OM, Moffat BA, Desmond PM, Lui E, Kwan P, O'Brien TJ. Seven-tesla quantitative magnetic resonance spectroscopy of glutamate, gamma-aminobutyric acid, and glutathione in the posterior cingulate cortex/precuneus in patients with epilepsy. Epilepsia. (2020) 61:2785–2794. doi: 10.1111/epi.16731
83. Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, et al. Metabolic control of vesicular glutamate transport and release. Neuron. (2010) 68:99–112. doi: 10.1016/j.neuron.2010.09.002
84. Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci. (2007) 27:3618–25. doi: 10.1523/JNEUROSCI.0132-07.2007
85. Pflanz NC, Daszkowski AW, James KA, Mihic SJ. Ketone body modulation of ligand-gated ion channels. Neuropharmacology. (2019) 148:21–30. doi: 10.1016/j.neuropharm.2018.12.013
86. Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. (2007) 145:256–64. doi: 10.1016/j.neuroscience.2006.11.065
87. Cheng CM, Hicks K, Wang J, Eagles DA, Bondy CA. Caloric restriction augments brain glutamic acid decarboxylase-65 and−67 expression. J Neurosci Res. (2004) 77:270–6. doi: 10.1002/jnr.20144
88. Ulamek-Koziol M, Czuczwar SJ, Januszewski S, Pluta R. Ketogenic diet and epilepsy. Nutrients. (2019) 11. doi: 10.3390/nu11102510
89. Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. (2013) 36:32–40. doi: 10.1016/j.tins.2012.11.005
90. Mirijello A, D'angelo C, Ferrulli A, Vassallo G, Antonelli M, Caputo F, et al. Identification and management of alcohol withdrawal syndrome. Drugs. (2015) 75:353–65. doi: 10.1007/s40265-015-0358-1
91. Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. (2001) 86:5992. doi: 10.1210/jcem.86.12.8111
92. Yanagi S, Sato T, Kangawa K, Nakazato M. The homeostatic force of ghrelin. Cell Metab. (2018) 27:786–804. doi: 10.1016/j.cmet.2018.02.008
93. Kraus T, Schanze A, Groschl M, Bayerlein K, Hillemacher T, Reulbach U, et al. Ghrelin levels are increased in alcoholism. Alcohol Clin Exp Res. (2005) 29:2154–7. doi: 10.1097/01.alc.0000191753.82554.7e
94. Addolorato G, Capristo E, Leggio L, Ferrulli A, Abenavoli L, Malandrino N, et al. Relationship between ghrelin levels, alcohol craving, and nutritional status in current alcoholic patients. Alcohol Clin Exp Res. (2006) 30:1933–7. doi: 10.1111/j.1530-0277.2006.00238.x
95. Badaoui A, De Saeger C, Duchemin J, Gihousse D, De Timary P, Starkel P. Alcohol dependence is associated with reduced plasma and fundic ghrelin levels. Eur J Clin Invest. (2008) 38:397–403. doi: 10.1111/j.1365-2362.2008.01947.x
96. De Timary P, Cani PD, Duchemin J, Neyrinck AM, Gihousse D, Laterre PF, et al. The loss of metabolic control on alcohol drinking in heavy drinking alcohol-dependent subjects. PLoS ONE. (2012) 7:e38682. doi: 10.1371/journal.pone.0038682
97. Koopmann A, Von Der Goltz C, Grosshans M, Dinter C, Vitale M, Wiedemann K, et al. The association of the appetitive peptide acetylated ghrelin with alcohol craving in early abstinent alcohol dependent individuals. Psychoneuroendocrinology. (2012) 37:980–6. doi: 10.1016/j.psyneuen.2011.11.005
98. Leggio L, Ferrulli A, Cardone S, Nesci A, Miceli A, Malandrino N, et al. Ghrelin system in alcohol-dependent subjects: role of plasma ghrelin levels in alcohol drinking and craving. Addict Biol. (2012) 17:452–64. doi: 10.1111/j.1369-1600.2010.00308.x
99. Kim JH, Kim SJ, Lee WY, Cheon YH, Lee SS, Ju A, et al. The effects of alcohol abstinence on BDNF, ghrelin, and leptin secretions in alcohol-dependent patients with glucose intolerance. Alcohol Clin Exp Res. (2013) 37 Suppl 1:E52–8. doi: 10.1111/j.1530-0277.2012.01921.x
100. Bahi A, Tolle V, Fehrentz JA, Brunel L, Martinez J, Tomasetto CL, et al. Ghrelin knockout mice show decreased voluntary alcohol consumption and reduced ethanol-induced conditioned place preference. Peptides. (2013) 43:48–55. doi: 10.1016/j.peptides.2013.02.008
101. Cepko LC, Selva JA, Merfeld EB, Fimmel AI, Goldberg SA, Currie PJ. Ghrelin alters the stimulatory effect of cocaine on ethanol intake following mesolimbic or systemic administration. Neuropharmacology. (2014) 85:224–31. doi: 10.1016/j.neuropharm.2014.05.030
102. Akkisi Kumsar N, Dilbaz N. Relationship between craving and ghrelin, adiponectin, and resistin levels in patients with alcoholism. Alcohol Clin Exp Res. (2015) 39:702–9. doi: 10.1111/acer.12689
103. Ralevski E, Horvath TL, Shanabrough M, Hayden R, Newcomb J, Petrakis I. Ghrelin is supressed by intravenous alcohol and is related to stimulant and sedative effects of alcohol. Alcohol Alcohol. (2017) 52:431–8. doi: 10.1093/alcalc/agx022
104. Bach P, Bumb JM, Schuster R, Vollstadt-Klein S, Reinhard I, Rietschel M, et al. Effects of leptin and ghrelin on neural cue-reactivity in alcohol addiction: two streams merge to one river? Psychoneuroendocrinology. (2019) 100:1–9. doi: 10.1016/j.psyneuen.2018.09.026
105. Koopmann A, Bach P, Schuster R, Bumb JM, Vollstadt-Klein S, Reinhard I, et al. Ghrelin modulates mesolimbic reactivity to alcohol cues in alcohol-addicted subjects: a functional imaging study. Addict Biol. (2019) 24:1066–76. doi: 10.1111/adb.12651
106. Engel JA, Jerlhag E. Role of appetite-regulating peptides in the pathophysiology of addiction: implications for pharmacotherapy. CNS Drugs. (2014) 28:875–86. doi: 10.1007/s40263-014-0178-y
107. Roekenes J, Martins C. Ketogenic diets and appetite regulation. Curr Opin Clin Nutr Metab Care. (2021) 24:359–63. doi: 10.1097/MCO.0000000000000760
108. Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, et al. Ketosis and appetite-mediating nutrients and hormones after weight loss. Eur J Clin Nutr. (2013) 67:759–64. doi: 10.1038/ejcn.2013.90
109. Parvaresh Rizi E, Loh TP, Baig S, Chhay V, Huang S, Caleb Quek J, et al. A high carbohydrate, but not fat or protein meal attenuates postprandial ghrelin, PYY and GLP-1 responses in Chinese men. PLoS ONE. (2018) 13:e0191609. doi: 10.1371/journal.pone.0191609
110. De Amicis R, Leone A, Lessa C, Foppiani A, Ravella S, Ravasenghi S, et al. Long-term effects of a classic ketogenic diet on ghrelin and leptin concentration: a 12-month prospective study in a cohort of Italian children and adults with GLUT1-deficiency syndrome and drug resistant epilepsy. Nutrients. (2019) 11:1716. doi: 10.3390/nu11081716
111. Marchio M, Roli L, Lucchi C, Costa AM, Borghi M, Iughetti L, et al. Ghrelin plasma levels after 1 year of ketogenic diet in children with refractory epilepsy. Front Nutr. (2019) 6:112. doi: 10.3389/fnut.2019.00112
112. Woods SC, Seeley RJ, Porte D, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. (1998) 280:1378–83. doi: 10.1126/science.280.5368.1378
113. De Silva A, Bloom SR. Gut hormones and appetite control: a focus on PYY and GLP-1 as therapeutic targets in obesity. Gut Liver. (2012) 6:10–20. doi: 10.5009/gnl.2012.6.1.10
114. Hu T, Yao L, Reynolds K, Niu T, Li S, Whelton P, et al. The effects of a low-carbohydrate diet on appetite: a randomized controlled trial. Nutr Metab Cardiovasc Dis. (2016) 26:476–88. doi: 10.1016/j.numecd.2015.11.011
115. Fraser DA, Thoen J, Bondhus S, Haugen M, Reseland JE, Djoseland O, et al. Reduction in serum leptin and IGF-1 but preserved T-lymphocyte numbers and activation after a ketogenic diet in rheumatoid arthritis patients. Clin Exp Rheumatol. (2000) 18:209–14.
116. Mohorko N, Cernelic-Bizjak M, Poklar-Vatovec T, Grom G, Kenig S, Petelin A, et al. Weight loss, improved physical performance, cognitive function, eating behavior, and metabolic profile in a 12-week ketogenic diet in obese adults. Nutr Res. (2019) 62:64–77. doi: 10.1016/j.nutres.2018.11.007
117. Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L, et al. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol. (2010) 24:2050–64. doi: 10.1210/me.2010-0142
118. Bookout AL, De Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL, et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med. (2013) 19:1147–52. doi: 10.1038/nm.3249
119. Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. (2009) 150:4931–40. doi: 10.1210/en.2009-0532
120. Song P, Zechner C, Hernandez G, Canovas J, Xie Y, Sondhi V, et al. The hormone FGF21 stimulates water drinking in response to ketogenic diet and alcohol. Cell Metab. (2018) 27:1338–47 e1334. doi: 10.1016/j.cmet.2018.04.001
121. Galman C, Lundasen T, Kharitonenkov A, Bina HA, Eriksson M, Hafstrom I, et al. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. (2008) 8:169–74. doi: 10.1016/j.cmet.2008.06.014
122. Christodoulides C, Dyson P, Sprecher D, Tsintzas K, Karpe F. Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J Clin Endocrinol Metab. (2009) 94:3594–601. doi: 10.1210/jc.2009-0111
123. Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. (2010) 139:456–63. doi: 10.1053/j.gastro.2010.04.054
124. Gimeno RE, Moller DE. FGF21-based pharmacotherapy—potential utility for metabolic disorders. Trends Endocrinol Metab. (2014) 25:303–11. doi: 10.1016/j.tem.2014.03.001
125. Hill CM, Qualls-Creekmore E, Berthoud HR, Soto P, Yu S, Mcdougal DH, et al. FGF21 and the physiological regulation of macronutrient preference. Endocrinology. (2020) 161:bqaa019. doi: 10.1210/endocr/bqaa019
126. Talukdar S, Owen BM, Song P, Hernandez G, Zhang Y, Zhou Y, et al. FGF21 regulates sweet and alcohol preference. Cell Metab. (2016) 23:344–9. doi: 10.1016/j.cmet.2015.12.008
127. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. (2007) 87:1409–39. doi: 10.1152/physrev.00034.2006
128. Nymo S, Coutinho SR, Jorgensen J, Rehfeld JF, Truby H, Kulseng B, et al. Timeline of changes in appetite during weight loss with a ketogenic diet. Int J Obes. (2017) 41:1224–31. doi: 10.1038/ijo.2017.96
129. Wallenius V, Elias E, Elebring E, Haisma B, Casselbrant A, Larraufie P, et al. Suppression of enteroendocrine cell glucagon-like peptide (GLP)-1 release by fat-induced small intestinal ketogenesis: a mechanism targeted by Roux-en-Y gastric bypass surgery but not by preoperative very-low-calorie diet. Gut. (2020) 69:1423–31. doi: 10.1136/gutjnl-2019-319372
130. Vallof D, Maccioni P, Colombo G, Mandrapa M, Jornulf JW, Egecioglu E, et al. The glucagon-like peptide 1 receptor agonist liraglutide attenuates the reinforcing properties of alcohol in rodents. Addict Biol. (2016) 21:422–37. doi: 10.1111/adb.12295
131. Thomsen M, Dencker D, Wortwein G, Weikop P, Egecioglu E, Jerlhag E, et al. The glucagon-like peptide 1 receptor agonist Exendin-4 decreases relapse-like drinking in socially housed mice. Pharmacol Biochem Behav. (2017) 160:14–20. doi: 10.1016/j.pbb.2017.07.014
132. Bornebusch AB, Fink-Jensen A, Wortwein G, Seeley RJ, Thomsen M. Glucagon-like peptide-1 receptor agonist treatment does not reduce abuse-related effects of opioid drugs. eNeuro. (2019) 6:ENEURO.0443-18.2019. doi: 10.1523/ENEURO.0443-18.2019
133. Thomsen M, Holst JJ, Molander A, Linnet K, Ptito M, Fink-Jensen A. Effects of glucagon-like peptide 1 analogs on alcohol intake in alcohol-preferring vervet monkeys. Psychopharmacology. (2019) 236:603–11. doi: 10.1007/s00213-018-5089-z
134. Vallof D, Kalafateli AL, Jerlhag E. Brain region specific glucagon-like peptide-1 receptors regulate alcohol-induced behaviors in rodents. Psychoneuroendocrinology. (2019) 103:284–95. doi: 10.1016/j.psyneuen.2019.02.006
135. Marty VN, Farokhnia M, Munier JJ, Mulpuri Y, Leggio L, Spigelman I. Long-acting glucagon-like peptide-1 receptor agonists suppress voluntary alcohol intake in male wistar rats. Front Neurosci. (2020) 14:599646. doi: 10.3389/fnins.2020.599646
136. Hernandez NS, Ige KY, Mietlicki-Baase EG, Molina-Castro GC, Turner CA, Hayes MR, et al. Glucagon-like peptide-1 receptor activation in the ventral tegmental area attenuates cocaine seeking in rats. Neuropsychopharmacology. (2018) 43:2000–8. doi: 10.1038/s41386-018-0010-3
137. Angarita GA, Matuskey D, Pittman B, Costeines JL, Potenza MN, Jastreboff AM, et al. Testing the effects of the GLP-1 receptor agonist exenatide on cocaine self-administration and subjective responses in humans with cocaine use disorder. Drug Alcohol Depend. (2021) 221:108614. doi: 10.1016/j.drugalcdep.2021.108614
138. Koob GF, Vendruscolo LF. The Oxford Handbook of Stress and Mental Health. New York, NY: Oxford University Press (2019).
139. Brownlow ML, Jung SH, Moore RJ, Bechmann N, Jankord R. Nutritional ketosis affects metabolism and behavior in Sprague-Dawley rats in both control and chronic stress environments. Front Mol Neurosci. (2017) 10:129. doi: 10.3389/fnmol.2017.00129
140. Ryan KK, Packard AEB, Larson KR, Stout J, Fourman SM, Thompson AMK, et al. Dietary manipulations that induce ketosis activate the HPA axis in male rats and mice: a potential role for fibroblast growth factor-21. Endocrinology. (2018) 159:400–13. doi: 10.1210/en.2017-00486
141. Sahagun E, Ward LM, Kinzig KP. Attenuation of stress-induced weight loss with a ketogenic diet. Physiol Behav. (2019) 212:112654. doi: 10.1016/j.physbeh.2019.112654
142. Kajitani N, Iwata M, Miura A, Tsunetomi K, Yamanashi T, Matsuo R, et al. Prefrontal cortex infusion of beta-hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, produces antidepressant-like effects in a rodent model of depression. Neuropsychopharmacol Rep. (2020) 40:157–65. doi: 10.1002/npr2.12099
143. Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. (2015) 350:1208–13. doi: 10.1126/science.aac4854
144. Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci USA. (2015) 112:2876–81. doi: 10.1073/pnas.1417921112
145. Kim SY, Cohen BM, Chen X, Lukas SE, Shinn AK, Yuksel AC, et al. Redox dysregulation in schizophrenia revealed by in vivo NAD+/NADH Measurement. Schizophr Bull. (2017) 43:197–204. doi: 10.1093/schbul/sbw129
146. Parker R, Schmidt MS, Cain O, Gunson B, Brenner C. Nicotinamide adenine dinucleotide metabolome is functionally depressed in patients undergoing liver transplantation for alcohol-related liver disease. Hepatol Commun. (2020) 4:1183–92. doi: 10.1002/hep4.1530
147. Cederbaum AI, Dicker E, Rubin E. Transfer and reoxidation of reducing equivalents as the rate-limiting steps in the oxidation of ethanol by liver cells isolated from fed and fasted rats. Arch Biochem Biophys. (1977) 183:638–46. doi: 10.1016/0003-9861(77)90398-8
148. Cederbaum AI. Alcohol metabolism. Clin Liver Dis. (2012) 16:667–85. doi: 10.1016/j.cld.2012.08.002
149. Veech RL, Todd King M, Pawlosky R, Kashiwaya Y, Bradshaw PC, Curtis W. The “great” controlling nucleotide coenzymes. IUBMB Life. (2019) 71:565–79. doi: 10.1002/iub.1997
150. O'hollaren P. Diphosphopyridine nucleotide in the prevention, diagnosis and treatment of drug addiction. A preliminary report. West J Surg Obstet Gynecol. (1961) 69:213–215.
151. Xin L, Ipek O, Beaumont M, Shevlyakova M, Christinat N, Masoodi M, et al. Nutritional ketosis increases NAD(+)/NADH ratio in healthy human brain: an in vivo study by (31)P-MRS. Front Nutr. (2018) 5:62. doi: 10.3389/fnut.2018.00062
152. Elamin M, Ruskin DN, Masino SA, Sacchetti P. Ketone-based metabolic therapy: is increased NAD(+) a primary mechanism? Front Mol Neurosci. (2017) 10:377. doi: 10.3389/fnmol.2017.00377
153. Elamin M, Ruskin DN, Masino SA, Sacchetti P. Ketogenic diet modulates NAD(+)-dependent enzymes and reduces DNA damage in hippocampus. Front Cell Neurosci. (2018) 12:263. doi: 10.3389/fncel.2018.00263
154. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. (2013) 339:211–4. doi: 10.1126/science.1227166
155. Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci. (2008) 28:3729–37. doi: 10.1523/JNEUROSCI.5731-07.2008
156. Pradhan AA, Tipton AF, Zhang H, Akbari A, Pandey SC. Effect of histone deacetylase inhibitor on ethanol withdrawal-induced hyperalgesia in rats. Int J Neuropsychopharmacol. (2019) 22:523–7. doi: 10.1093/ijnp/pyz031
157. Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC. Histone deacetylases (HDAC)-induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol Clin Exp Res. (2012) 36:61–71. doi: 10.1111/j.1530-0277.2011.01581.x
158. Kim DY, Davis LM, Sullivan PG, Maalouf M, Simeone TA, Van Brederode J, et al. Ketone bodies are protective against oxidative stress in neocortical neurons. J Neurochem. (2007) 101:1316–26. doi: 10.1111/j.1471-4159.2007.04483.x
159. Haces ML, Hernandez-Fonseca K, Medina-Campos ON, Montiel T, Pedraza-Chaverri J, Massieu L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp Neurol. (2008) 211:85–96. doi: 10.1016/j.expneurol.2007.12.029
160. Xie G, Tian W, Wei T, Liu F. The neuroprotective effects of beta-hydroxybutyrate on Abeta-injected rat hippocampus in vivo and in Abeta-treated PC-12 cells in vitro. Free Radic Res. (2015) 49:139–50. doi: 10.3109/10715762.2014.987274
161. Novitskiy G, Traore K, Wang L, Trush MA, Mezey E. Effects of ethanol and acetaldehyde on reactive oxygen species production in rat hepatic stellate cells. Alcohol Clin Exp Res. (2006) 30:1429–35. doi: 10.1111/j.1530-0277.2006.00171.x
162. Tamura M, Ito H, Matsui H, Hyodo I. Acetaldehyde is an oxidative stressor for gastric epithelial cells. J Clin Biochem Nutr. (2014) 55:26–31. doi: 10.3164/jcbn.14-12
163. Fernandez-Checa JC, Hirano T, Tsukamoto H, Kaplowitz N. Mitochondrial glutathione depletion in alcoholic liver disease. Alcohol. (1993) 10:469–75. doi: 10.1016/0741-8329(93)90067-X
164. Bolanos CA, Nestler EJ. Neurotrophic mechanisms in drug addiction. Neuromolecular Med. (2004) 5:69–83. doi: 10.1385/NMM:5:1:069
165. Davis MI. Ethanol-BDNF interactions: still more questions than answers. Pharmacol Ther. (2008) 118:36–57. doi: 10.1016/j.pharmthera.2008.01.003
166. Moonat S, Starkman BG, Sakharkar A, Pandey SC. Neuroscience of alcoholism: molecular and cellular mechanisms. Cell Mol Life Sci. (2010) 67:73–88. doi: 10.1007/s00018-009-0135-y
167. Joe KH, Kim YK, Kim TS, Roh SW, Choi SW, Kim YB, et al. Decreased plasma brain-derived neurotrophic factor levels in patients with alcohol dependence. Alcohol Clin Exp Res. (2007) 31:1833–8. doi: 10.1111/j.1530-0277.2007.00507.x
168. Jeanblanc J, He DY, Carnicella S, Kharazia V, Janak PH, Ron D. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J Neurosci. (2009) 29:13494–502. doi: 10.1523/JNEUROSCI.2243-09.2009
169. Raivio N, Miettinen P, Kiianmaa K. Innate BDNF expression is associated with ethanol intake in alcohol-preferring AA and alcohol-avoiding ANA rats. Brain Res. (2014) 1579:74–83. doi: 10.1016/j.brainres.2014.07.006
170. Schunck RV, Torres IL, Laste G, De Souza A, Macedo IC, Valle MT, et al. Protracted alcohol abstinence induces analgesia in rats: Possible relationships with BDNF and interleukin-10. Pharmacol Biochem Behav. (2015) 135:64–9. doi: 10.1016/j.pbb.2015.05.011
171. Huang MC, Chen CH, Chen CH, Liu SC, Ho CJ, Shen WW, et al. Alterations of serum brain-derived neurotrophic factor levels in early alcohol withdrawal. Alcohol Alcohol. (2008) 43:241–5. doi: 10.1093/alcalc/agm172
172. Lee BC, Choi IG, Kim YK, Ham BJ, Yang BH, Roh S, et al. Relation between plasma brain-derived neurotrophic factor and nerve growth factor in the male patients with alcohol dependence. Alcohol. (2009) 43:265–9. doi: 10.1016/j.alcohol.2009.04.003
173. Costa MA, Girard M, Dalmay F, Malauzat D. Brain-derived neurotrophic factor serum levels in alcohol-dependent subjects 6 months after alcohol withdrawal. Alcohol Clin Exp Res. (2011) 35:1966–73. doi: 10.1111/j.1530-0277.2011.01548.x
174. Kivinummi T, Kaste K, Rantamaki T, Castren E, Ahtee L. Alterations in BDNF and phospho-CREB levels following chronic oral nicotine treatment and its withdrawal in dopaminergic brain areas of mice. Neurosci Lett. (2011) 491:108–12. doi: 10.1016/j.neulet.2011.01.015
175. Ren Q, Ma M, Yang C, Zhang JC, Yao W, Hashimoto K. BDNF-TrkB signaling in the nucleus accumbens shell of mice has key role in methamphetamine withdrawal symptoms. Transl Psychiatry. (2015) 5:e666. doi: 10.1038/tp.2015.157
176. Sleiman SF, Henry J, Al-Haddad R, El Hayek L, Abou Haidar E, Stringer T, et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. Elife. (2016) 5. doi: 10.7554/eLife.15092.012
177. Hu E, Du H, Zhu X, Wang L, Shang S, Wu X, et al. Beta-hydroxybutyrate promotes the expression of BDNF in Hippocampal Neurons under adequate glucose supply. Neuroscience. (2018) 386:315–25. doi: 10.1016/j.neuroscience.2018.06.036
178. Hu E, Du H, Shang S, Zhang Y, Lu X. Beta-hydroxybutyrate enhances BDNF expression by increasing H3K4me3 and decreasing H2AK119ub in hippocampal neurons. Front Neurosci. (2020) 14:591177. doi: 10.3389/fnins.2020.591177
179. Gyorkos A, Baker MH, Miutz LN, Lown DA, Jones MA, Houghton-Rahrig LD. Carbohydrate-restricted diet and exercise increase brain-derived neurotrophic factor and cognitive function: a randomized crossover trial. Cureus. (2019) 11:e5604. doi: 10.7759/cureus.5604
180. Paoli A, Cenci L, Pompei P, Sahin N, Bianco A, Neri M, et al. Effects of two months of very low carbohydrate ketogenic diet on body composition, muscle strength, muscle area, and blood parameters in competitive natural body builders. Nutrients. (2021) 13. doi: 10.3390/nu13020374
181. Vizuete AF, De Souza DF, Guerra MC, Batassini C, Dutra MF, Bernardi C, et al. Brain changes in BDNF and S100B induced by ketogenic diets in Wistar rats. Life Sci. (2013) 92:923–8. doi: 10.1016/j.lfs.2013.03.004
182. Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. (2005) 280:26649–52. doi: 10.1074/jbc.C500213200
183. Graff EC, Fang H, Wanders D, Judd RL. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism. (2016) 65:102–13. doi: 10.1016/j.metabol.2015.10.001
184. Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Muller-Fielitz H, et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun. (2014) 5:3944. doi: 10.1038/ncomms4944
185. Chen Y, Ouyang X, Hoque R, Garcia-Martinez I, Yousaf MN, Tonack S, et al. beta-Hydroxybutyrate protects from alcohol-induced liver injury via a Hcar2-cAMP dependent pathway. J Hepatol. (2018) 69:687–96. doi: 10.1016/j.jhep.2018.04.004
186. Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt-Jones EA, et al. Alcohol-induced IL-1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J Leukoc Biol. (2013) 94:171–82. doi: 10.1189/jlb.1212659
187. Hoyt LR, Randall MJ, Ather JL, Depuccio DP, Landry CC, Qian X, et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. (2017) 12:883–96. doi: 10.1016/j.redox.2017.04.020
188. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. (2015) 21:263–9. doi: 10.1038/nm.3804
189. Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou R, et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1beta in mice. J Hepatol. (2015) 62:1311–8. doi: 10.1016/j.jhep.2014.12.027
190. Desantis DA, Ko CW, Liu Y, Liu X, Hise AG, Nunez G, et al. Alcohol-induced liver injury is modulated by Nlrp3 and Nlrc4 inflammasomes in mice. Mediators Inflamm. (2013) 2013:751374. doi: 10.1155/2013/751374
191. Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. (2020) 11:2127. doi: 10.1038/s41467-020-15983-6
192. Neudorf H, Durrer C, Myette-Cote E, Makins C, O'malley T, Little JP. Oral ketone supplementation acutely increases markers of NLRP3 inflammasome activation in human monocytes. Mol Nutr Food Res. (2019) 63:e1801171. doi: 10.1002/mnfr.201801171
193. Neudorf H, Myette-Cote E, P Little L. The impact of acute ingestion of a ketone monoester drink on LPS-stimulated NLRP3 activation in humans with obesity. Nutrients. (2020) 12:854. doi: 10.3390/nu12030854
194. Lin A, Turner Z, Doerrer SC, Stanfield A, Kossoff EH. Complications during ketogenic diet initiation: prevalence, treatment, and influence on seizure outcomes. Pediatr Neurol. (2017) 68:35–9. doi: 10.1016/j.pediatrneurol.2017.01.007
195. Wang GJ, Volkow ND, Fowler JS, Franceschi D, Wong CT, Pappas NR, et al. Alcohol intoxication induces greater reductions in brain metabolism in male than in female subjects. Alcohol Clin Exp Res. (2003) 27:909–17. doi: 10.1111/j.1530-0277.2003.tb04415.x
196. Erol A, Karpyak VM. Sex and gender-related differences in alcohol use and its consequences: contemporary knowledge and future research considerations. Drug Alcohol Depend. (2015) 156:1–13. doi: 10.1016/j.drugalcdep.2015.08.023
197. Durkalec-Michalski K, Nowaczyk PM, Siedzik K. Effect of a four-week ketogenic diet on exercise metabolism in CrossFit-trained athletes. J Int Soc Sports Nutr. (2019) 16:16. doi: 10.1186/s12970-019-0284-9
198. Bierut LJ, Goate AM, Breslau N, Johnson EO, Bertelsen S, Fox L, et al. ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Mol Psychiatry. (2012) 17:445–50. doi: 10.1038/mp.2011.124
199. Kranzler HR, Zhou H, Kember RL, Vickers Smith R, Justice AC, Damrauer S, et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun. (2019) 10:1499. doi: 10.1038/s41467-019-09480-8
Keywords: alcoholism, alcohol dependence, alcohol withdrawal, ketogenic diet, ketone ester, alcohol metabolism
Citation: Mahajan VR, Elvig SK, Vendruscolo LF, Koob GF, Darcey VL, King MT, Kranzler HR, Volkow ND and Wiers CE (2021) Nutritional Ketosis as a Potential Treatment for Alcohol Use Disorder. Front. Psychiatry 12:781668. doi: 10.3389/fpsyt.2021.781668
Received: 23 September 2021; Accepted: 08 November 2021;
Published: 30 November 2021.
Edited by:
Wendy J. Lynch, University of Virginia, United StatesReviewed by:
Dieter J. Meyerhoff, University of California, San Francisco, United StatesZoltan Sarnyai, James Cook University, Australia
Copyright © 2021 Mahajan, Elvig, Vendruscolo, Koob, Darcey, King, Kranzler, Volkow and Wiers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Corinde E. Wiers, Y29yaW5kZS53aWVycyYjeDAwMDQwO3Blbm5tZWRpY2luZS51cGVubi5lZHU=