 Tangfeng Su
Tangfeng Su
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 22 October 2021
Sec. Autism
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.734837
This article is part of the Research TopicInsights in Autism: 2021View all 6 articles
Autism spectrum disorder (ASD) is a serious neurodevelopmental disorder and characterized by early childhood-onset impairments in social interaction and communication, restricted and repetitive patterns of behavior or interests. So far there is no effective treatment for ASD, and the pathogenesis of ASD remains unclear. Genetic and epigenetic factors have been considered to be the main cause of ASD. It is known that endocannabinoid and its receptors are widely distributed in the central nervous system, and provide a positive and irreversible change toward a more physiological neurodevelopment. Recently, the endocannabinoid system (ECS) has been found to participate in the regulation of social reward behavior, which has attracted considerable attention from neuroscientists and neurologists. Both animal models and clinical studies have shown that the ECS is a potential target for the treatment of autism, but the mechanism is still unknown. In the brain, microglia express a complete ECS signaling system. Studies also have shown that modulating ECS signaling can regulate the functions of microglia. By comprehensively reviewing previous studies and combining with our recent work, this review addresses the effects of targeting ECS on microglia, and how this can contribute to maintain the positivity of the central nervous system, and thus improve the symptoms of autism. This will provide insights for revealing the mechanism and developing new treatment strategies for autism.
Autism spectrum disorder (ASD) is a group of heterogeneous neurodevelopmental disorders characterized by language and social dysfunctions and repetitive stereotyped behavior. In addition to the core diagnostic features, ASD also has a variety of comorbid symptoms, including aggression, hyperactivity, epilepsy, anxiety, sleep disorders, gastrointestinal disorders and immune dysfunction. The global incidence rate is about 1% and a male to female ratio of about 3 to 4:1 (1).
Currently, there is no pharmaceutical compound available to alleviate the core symptoms of ASD. Risperidone and aripiprazole have been approved by the US Food and Drug Administration (FDA) to treat irritability and behavioral disorders in patients with ASD. Long-term use of these drugs may cause some side effects, including sedation and weight gain (2). At present, the most widely used treatment of ASD is behavior intervention, but it is usually time-consuming and unaffordable. In recent years, oxytocin (OT) has attracted wide attention because it plays an important role in regulating the function of social communication in the central nervous system (CNS), and has gradually become a potential treatment for autism. For example, recent evidence supports entry of intranasal oxytocin into the brain through the olfactory and trigeminal neural pathways (3), as well as the nasal blood stream (4). Intranasal administration of the OT can enter the CNS through the blood-brain barrier, but the OT molecule is very likely to enter the vein, that is, the peripheral nervous system, which can cause uterine contraction in women. Therefore, almost all nasal spray OT experimental studies on the treatment of ASD were conducted on males (5). There may be gender differences in the effect of OT on ASD, so it is questionable to extend the results to all ASD patients. The Endocannabinoid system (ECS) plays a putative role in the control of neural processes of controlling social anxiety and social reward, and has become a new target for autism intervention in the last decade (6). The ECS includes the two most widely studied endogenous lipid mediators [N-arachidonoylethanolamide (anandamide; AEA), and 2-arachidonoylglycerol (2-AG)], cannabinoid type 1 and type 2 receptors (CB1 and CB2), and endogenous lipid mediator synthesis and catabolism enzymes (Figure 1). AEA and 2-AG were identified in 1992 and 1995, respectively. CB1 and CB2 were cloned in 1990 and 1993, respectively. The amino acid sequence homology of CB1 across species is 97–99%, which is mainly found in the brain, especially in neuronal cells, while CB2 are mainly found in immune cells. The two receptors have 44% homology (7). Lines of evidence demonstrated that targeting different components of ECS may provide therapeutic strategies for the treatment of ASD (8), but the mechanism is unclear.
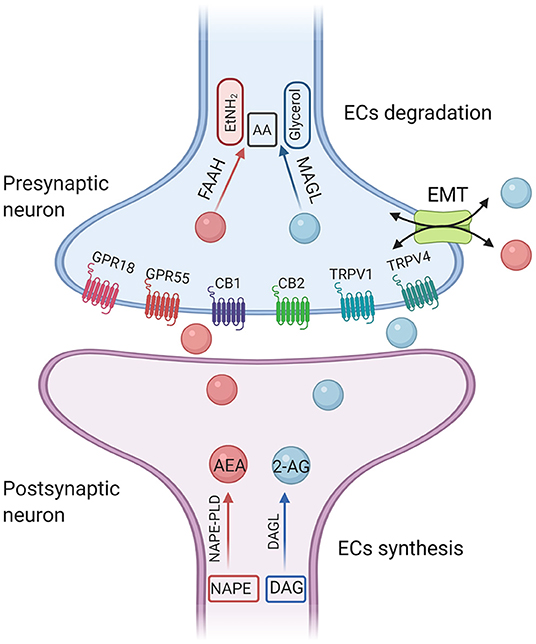
Figure 1. A simplified illustration of endocannabinoid synthesis and degradation pathway. The diacylglycerol lipases (DAGLα and DAGLβ) hydrolyze DAG to generate 2-AG. DAGL-α is expressed on postsynaptic neurons within various brain regions. In contrast, DAGL-β is most highly expressed on microglia. 2-AG is preferentially degraded by monoglyceride lipase (MAGL). The main synthetase of AEA is N-acyl-phosphatidylethanolamine-phospholipase D (NAPE-PLD). The degradation enzyme of AEA is fatty amide hydrolase (FAAH). AEA, anandamide; 2-AG, 2-arachidonoyl-glycerol; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; TRPV1, transient receptor potential cation channel subfamily V member 1; GPRs, orphan G-protein-coupled receptors; AA, arachidonic acid; EtNH2, ethanolamine; EMT, extraneuronal monoamine transporter.
It is reported that dysregulation of the immune system is a possible mechanism related to ASD pathogenesis and severity (9). Furthermore, microglia are the main producers of endocannabinoids under neuroinflammatory conditions (10). Here, we will review studies concerning the involvement of the ECS in the immuno-pathological mechanism and therapeutic strategies of ASD by summarizing preclinical and clinical studies to shed light on ECS and microglia as a potential target for the treatment of autism.
A Medline (PUBMED) search using the terms “microglia AND autism,” or “microglia AND autism spectrum disorder,” or “microglia, endocannabinoid AND autism,” or “microglia, cannabidiol AND autism,” or “microglia, endocannabinoid AND autism spectrum disorder,” or “microglia, cannabidiol AND autism spectrum disorder” was performed. A total of 339 articles with a publication date up to April 1st, 2021 were included. Articles related to clinical trials, preclinical studies, and animal studies were selected for this review.
2-AG is the most abundant endogenous cannabinoid in the brain and its concentration is about 200 times that of AEA (11). The diacylglycerol lipases (DAGLα and DAGLβ) hydrolyze DAG to generate 2-AG. DAGL-α is expressed on postsynaptic neurons within various brain regions (12). In contrast, DAGL-β is most highly expressed on microglia (13). 2-AG is preferentially degraded by monoglyceride lipase (MAGL). Fragile X mental retardation 1 (FMR1) gene is one of the most common single-gene mutations associated with autism. It was reported that 2-AG-mediated decreased long term depression (LTD) in nucleus accumbens (NAc) and medial prefrontal cortex (mPFC) in FMR1 knockout mice. Administration of JZL184, a pharmacological inhibitor of MAGL, enhanced 2-AG signaling, corrected 2-AG-mediated LTD, and improved hyperactivity and anxiety-like responses in FMR1 knockout mice (14). In addition, a single mutation of Shank3 is sufficient to induce the typical autism syndrome (15). In terms of the neural circuits in the treatment of ASD, studies have shown photogenetic activation of the glutamatergic circuit in basolateral amygdala-nucleus accumbens (BLA-NAc) reduced social activity and increased social avoidance in Shank3B−/− mice. Systemic or local injection of 2-AG hydrolase inhibitor JZL184 into the brain area of NAc restored the social deficit of Shank3B−/− mice. At the same time, electrophysiological recordings in mouse brain slices in vitro showed that JZL184 corrected the abnormal excitatory and inhibitory neural transmission of NAc in Shank3B−/− mice, and reduced the feedforward inhibition of BLA on NAc neurons (16).
Compared with 2-AG, studies have shown that AEA is more specific in mediating social reward response (17). The main synthetase of AEA is the N-acyl-phosphatidylethanolamine-phospholipase D (NAPE-PLD), and the degradation enzyme of AEA is the fatty amide hydrolase (FAAH). Pharmacological inhibition of FAAH enhances AEA levels and reverses the behavioral deficits among different autistic animal models. FMR1 knockout mice displayed increased anxiety-related behaviors during social interaction. It was reported that modulation of AEA signaling can ameliorate some aspects of the behavioral phenotype in FMR1 knockout mice. Qin et al. used acute injection of FAAH inhibitor URB597 to improve aversion memory and relieve anxiety behavior in FMR1 knockout mice, but it did not affect social behavior (18). However, Wei et al. showed that blocking FAAH by acute administration of URB597 completely reversed the social impairment in FMR1 knockout mice (19), suggesting that increased AEA may play a prosocial role in ASD animal models. The different time points of pre-administration of URB597 [30 min (18) and 3 h (19), respectively] or the different mouse line [C57Bl/6J (18) and FVB/NJ (19), respectively] may give an explanation for the discrepancy reported in the two studies.
Both of the two FAAH inhibitors, PF3845 and URB597, can reverse the abnormal behaviors of male offspring of the valproic acid (VPA) model in SD rats (20, 21). It is noteworthy that prenatal exposure to VPA produced gender-specific differences in the rat model: the behavioral impairment of male rats exposed to VPA was more serious than that of female rats. Increasing AEA signaling by inhibiting AEA-degrading enzyme FAAH improved the behavioral impairment of both male and female offspring of the ASD model. Compared with male offspring VPA rats, female offspring were less vulnerable in social communication, emotional response and cognitive performance, while selective deficiencies in social games and stereotyped behavior were observed (22). This gender dichotomy is similar to the clinical phenotype of ASD in humans. The phenotypic differences of ASD between males and females may be partly related to abnormally activated microglia during early development, which leads to impaired microglia-mediated synaptic pruning, especially in males (23). A recent study found that URB597 could restore ECS-related synaptic plasticity by enhancing the AMPA receptors transportation in mPFC of the offspring of the VPA model, thus alleviating ASD-related behavioral symptoms (24).
In addition, autism has been associated with atypical sleep patterns and circadian rhythms (25). Polymorphisms in ASMT (involved in melatonin synthesis), paralleled by reduced levels of circulating melatonin, have been reported in autism (26). Interestingly, the EC system also plays a role in regulating circadian rhythms (27), thus making it a putative target to examine using animal models of autism-related phenotypes.
CB1 is widely distributed both in the central nervous system (CNS) and peripheral nervous system, participates in synaptogenesis, learning and memory, and regulates a variety of physiological activities by regulating neurohormone levels and signaling transduction. CB2 is mainly expressed in the immune system, such as the spleen margin and thymus of the peripheral immune organs. During inflammation or other pathological injuries, the expression of CB2 in the CNS, especially in glial cells, is significantly up-regulated (10). CB1 antagonist rimonabant can alleviate cognitive impairment, epileptic sensitivity, and hyperalgesia in FMR1 knockout mice. At the biochemical level, CB1 blockade corrected the excessive activation of mTOR signaling and restored the morphology of dendritic spines in the hippocampus of FMR1 knockout mice. It is worth noting that the treatment with CB2 antagonist AM630 can also reduce the sensitivity of anxiety-like behavior and audiogenic epilepsy, indicating that both CB1 and CB2 are involved in the behavior of Fragile X syndrome (FXS) (28). Another study also found that low doses of rimonabant and CB1 neutral antagonist NESS0327 prevented cognitive impairment in FMR1 knockout mice. The therapeutic effect of rimonabant on cognitive impairment is related to the functional recovery of LTD induced by metabotropic glutamate (mGlu) in the hippocampus of FMR1 knockout mice (29). In addition, the behavioral phenotype of inbred BTBR mice is consistent with the core symptoms of ASD. Multiple changes of neuroanatomy and histology of BTBR mice are similar to those of ASD, which make BTBR mice widely used in ASD study. Wei et al. reversed social impairment in BTBR mice by using FAAH inhibitor URB597 to increase AEA levels, which was not attributed to reduced anxiety. The reason is that FAAH inhibition did not change the performance of BTBR mice in elevated mazes. In addition, the effect of inhibition of FAAH on the social pathway in BTBR mice strictly depends on the enhancement of AEA in CB1 signaling pathway, since the CB1 antagonist AM251 can prevent the recovery of social ability mediated by URB597, which is not related to the alteration of 2-AG content (19).
Recently, imbalances in dopamine levels have been implicated in behavioral disorders such as schizophrenia, autism and depression. Neurochemical evidence shows that activation of CB1R expressed on Ventral tegmental area (VTA) GABA neurons inhibits GABAergic transmission and in turn stimulates dopaminergic neurotransmission in the NAc (30). Since the dopamine signaling abnormalities have been reported in both autistic patients (31) and ASD animal models (32), elucidating the relationship between ECS and DA in ASD will help to provide a better understanding of the etiopathogenesis of ASD along with new therapeutic strategies (33).
The changes of ECS in peripheral blood can be used as a new non-invasive diagnostic index for several neuroinflammatory diseases (34). Compared with healthy children, the expression of AEA synthase NAPE-PLD mRNA in peripheral monocytes of children with ASD was downregulated (35). Using improved liquid chromatography-tandem mass spectrometry, it was found that the AEA concentration in peripheral blood of children with ASD was lower than that of healthy children (36). The levels of endogenous cannabinoids AEA, OEA, and PEA in serum samples were lower than those in age-and sex-matched normal control groups, but there was no significant difference in 2-AG between the two groups (37). Cannabidiol (CBD) is a non-psychoactive component in the cannabis plant, which inhibits FAAH by activating peroxisome proliferator-activated receptor (PPAR) and transient receptor potential channels of vanilloid type-1 (TPRV1), and finally compensates for the decrease of AEA, OEA, and PEA levels in children with ASD. The Clinical Trials (Registration number: NCT02956226) conducted by an Israeli research team in 2017 for the first time investigated the effectiveness of cannabis drugs against behavioral problems in children with ASD. Nighty-three children with ASD and severe behavioral problems (age: 11.8 ± 3.5 years, 83% were boys) tolerated the use of drugs rich in CBD (the ratio of CBD to THC was 20:1). Sixty-three percentage of the children reported considerable amelioration in behavior problems associated with ASD. Improvement was also found in anxiety and communication problems in 39 and 47% of the children, respectively (38). Another clinical study reported that CBD treatment reduced comorbidities in multiple domains, including self-harm behavior, anger attacks, hyperactivity symptoms, sleep problems, and anxiety states (39). The average duration of CBD treatment for ASD children in the study was 66 days, with mild side effects, including mild drowsiness and appetite changes.
Genetics and early environmental factors play a key role in the etiology of ASD. Although disruptions of immune system, abnormal neurotransmission, mitochondrial deficiency, oxidative stress, aberrations of cell signaling transduction, and epigenetic alterations have been proven to be associated with ASD, the pathophysiological mechanisms of ASD remain poorly understood, and no single etiology is identified. Microglia have detrimental impacts on several key etiological factors of autism, such as the synaptic plasticity, neural circuitry, brain immune system, stem cell development, and the crosstalk between genetic and environmental factors. Postmortem studies found immune-related genes were upregulated, while the synaptic function-related genes in the brain of ASD patients were downregulated compared with the control group (40). Both genetic and environmental factors could contribute to abnormal synaptic pruning and immune responses mediated by microglia either in utero or during the early postnatal period, which may underlie the pathogenesis of ASD. An association between maternal immune activation (MIA) and ASD in offspring was reported in human epidemiological studies (41) and in rodent MIA models (42). Disruptions in the composition of the gut microbiota during early developmental stages due to perinatal events increase an individual's predisposition to autistic behavioral patterns (43).
A large number of studies have shown that immune abnormality is one of the most important factors in the occurrence of ASD (44). Microglia have long been thought to originate from peripheral macrophages that enter the brain after birth. However, it is now known that microglia develop from red marrow progenitor cells in the yolk sac of early embryos and migrate into the brain (45). Microglia are the innate immune effector cells in the CNS and are the first line of defense against CNS infection and injury. Microglia can be activated by any type of pathological events or changes in brain homeostasis and release cytokines, chemokines and growth factors to maintain normal brain function. Abnormal immune signaling and microglial function is consistently demonstrated in postmortem brain tissues from autistic individuals as well as in autism mouse models. Transcriptomic analyses of postmortem cortex tissue of ASD patients revealed that genes associated with activated microglia were upregulated with exhibited exaggerated M2 activation states (46). Early lipopolysaccharide (LPS) exposure rats showed impairment in communication and cognition, which is associated with M2-like microglia activation and enhanced neurogenesis in both the subgranular zone (SGZ) and the subventricular zone (SVZ) (47). Of note, a recent systematic review comprising 1,007 autism patients from 14 genetic or neuroimaging studies using PET/MRI collectively indicates that microglia mediated neuroinflammation plays a causative role in the pathogenesis of ASD. Those prominent manifestations of microglia in autistic patients include increased cell number or cell density, morphological alterations, and phenotypic shifts (48). Despite this, microglial activation may have diverse meanings in different patients, playing pathogenic roles among “dysimmune” patients (49) and leading downstream consequence in “neural disconnection” ASD patients (50).
Microglia play an important role in maintaining a healthy brain by engulfing improper and less active synapses. Thus, microglial dysfunction is hypothesized to be involved in the pathogenesis of ASD, and possibly through the mechanism of attenuated or excessive synaptic pruning. For example, microglia give priority to phagocytosis of weaker or less active synapses, promoting the development of functional neural circuits with stronger or more active synapses (51). Proper synaptic pruning is essential for the development of functional neural circuits. Defects in synaptic pruning disrupt the excitatory and inhibitory balance of synapses. For example, inhibition of microglia autophagy can lead to an increase in synaptic density and a decrease in social ability in mice (52). Chemokine CX3C receptor 1 (CX3CR1) is specifically expressed in microglia and mediates neuron-microglial interactions (53). In CX3CR1 knockout mice, the expression of IL-1β was robustly increased and the synaptic long-term potentiation (LTP) was significantly decreased (54). In addition, functional magnetic resonance imaging (fMRI) analysis showed that the functional connection between hippocampus and prefrontal cortex was impaired. It is noteworthy that those CX3CR1 knockout mice showed decreased social interaction and increased repetitive behavior similar to ASD (55).
A number of studies have shown that there are abnormalities in the morphological characteristics of microglia in the brain of postmortem ASD patients. Tetreault et al. found that the density of microglia in two cortical regions (frontal lobe and visual cortex) of ASD patients was significantly higher than that in the normal brain (56). Tang et al. reported that the dendritic density of layer V pyramidal neurons in Brodmann 21 area of the middle temporal gyrus, which is mainly involved in social interaction and communication, is increased in postmortem ASD specimens (57). The density of dendritic spines in the childhood ASD group (2–9 years old) was similar to that in an age-matched control group, while the dendritic spine density in the adolescent ASD group (13–19 years old) was significantly higher than that in age-matched healthy controls (58). Recently, large-scale sequencing studies and transcriptomic genome-wide analyses have identified two broad categories of genes, neuronal genes and transcriptional regulation genes, which contains more than 100 de novo mutations or variants that encode proteins for synaptic formation, transcriptional regulation and chromatin-remodeling in ASD (59). Therefore, excessive microglial activation could be the consequence of abnormal neurodevelopment leading to unstable and malfunctioning long-range synaptic connections.
Besides being a key player in the brain's immune system, microglia also play a crucial role in synaptic pruning. Recently, studies have clarified complement-mediated synaptic pruning by microglia in the visual thalamus (60). In the dorsal lateral geniculate nucleus (LGN), the classical complement cascade mediates microglia's functions (61). Both the complement protein C1q, and the downstream complement protein C3 are highly localized to immature synapses and are required for synaptic pruning in the retinogeniculate system. C1q expression was increased in the peripheral serum of children with ASD (62). Alternatively, ubiquitously increased complement proteins may cover the specific localization of complement proteins on less active synapses, which may promote excessive synaptic pruning by microglia. Recently, Thomas and colleagues proposed the atypical synaptic over-pruning hypothesis by a neurocomputational model that ASD is caused by the exaggeration of a normal system-wide phase of brain development, elimination of excess connectivity. Normal individual differences in the onset or rate of this phase interact with the pathological pruning process to create different trajectories of atypical development. Individual differences in other neurocomputational parameters and in environmental stimulation operate as risk or protective factors. The atypical pruning is assumed to impact more on long-range connectivity, impairing integrative functions, which leads to the unique behavioral profile of ASD (63).
Environmental factors can also alter the functions of microglia, thereby affecting synaptic connections and development of the brain. Viral and bacterial infections during pregnancy are associated with an increased risk of autism in offspring (64). During neural tube closure, exposure to LPS induced maternal immune activation (MIA) in rodents seems to be associated with ASD-like behavior in offspring (65). Prenatal MIA and the subsequent increase in pro-inflammatory cytokines increase the risk of ASD and schizophrenia in the offspring (66).
Human epidemiological studies and MIA animal models have reported the association between MIA and offspring ASD. In MIA animal models, pregnant mice are immunologically activated by injection of polyinosine and polycytidine or LPS during embryonic development (E9-12). These MIA models will help to study the possible role of microglia in the pathogenesis of ASD, as offspring exhibit a range of ASD-like behavioral phenotypes, including impaired social interaction, changes in repetitive behavior, anxiety, and ultrasonic vocalization. LPS-exposed rodents at early postnatal days showed deficits in social interaction and induced a robust microglia activation, which is characterized by mixed microglial proinflammatory (M1) and anti-inflammatory (M2) phenotypes (47). A growing body of rodent studies have demonstrated that diverse inflammatory factors beyond infection, including stressors or environmental toxins, may similarly induce persistent effects on microglia, which in turn affect neurodevelopmental processes that lead to ASD-like phenotypes in offspring (67).
The MIA-induced ASD mouse model not only showed abnormal behavior, but also had changes in intestinal microflora (68). The communication between microbes in the gastrointestinal tract and the brain regulates mood and behavior through a two-way brain-gut pathway. It is known that gastrointestinal dysfunction affects inflammatory response and brain function through nerve, hormone and immune signaling, but the exact biological mechanism remains to be elucidated. Compared with healthy people, patients with ASD are more likely to develop gastrointestinal symptoms, including constipation, diarrhea, or abdominal pain, as well as other intestinal diseases, such as inflammatory bowel disease (IBD) (69).
Sharon et al. found that normal intestinal microbial population is necessary for the development of microglial structure and function to mature. Compared with mice fed in a specific pathogen free (SPF) environment, axenic mice showed significant differences in the microbial mRNA expression profile, microglial density in multiple brain regions and process branching complexity and process length of microglia (70). SPF mice are free of a specific list of pathogens, such as disease-causing pathogens. However, axenic mice are free of all microorganisms, including those that are typically found in the gut. After virus infection, the microglia in axenic mice showed a decreased innate immune response, which indicated that the host microbial population supported the resistance of microglia to bacterial and viral stimulation. Compared with SPF mice, aseptic mice showed lower social ability and more repetitive self-grooming behavior (71). At the same time, aseptic mice had more immature and malformed microglia and reduced immune response to LPS or virus infection (72). It is worth noting that the microglial phenotype and social impairment in aseptic mice were rescued after intestinal reproduction using microflora from typically developed mice. As the microglia activation and the subsequent inflammation in the CNS may partly result from microbial disorders, it is important to analyze the integrity of mucosal barrier and the changes of intestinal microbial population in those disease models.
It is known that CB1 is widely expressed in neurons, while CB2 is mainly expressed in immune cells and microglia. In the pathological state, the expression of CB2 in microglia significantly increased (10). The different activation states are considered to represent different functional states of microglia. Considerable studies have demonstrated that ECS may be a promising tool for changing the outcome of inflammation, especially by affecting the activity of microglia. Microglia contain a complete functional ECS signaling system, including synthesis and degradation components (Figure 2). The expression of cannabinoid receptors (mainly CB2) and the production of endogenous cannabinoids (eCB) are related to the activation characteristics of these cells. Previous study has shown that the ECS is closely associated with regulating polarization of microglia (73). Indeed, activation of CB2 receptors not only affects the migration, proliferation and releasing of proinflammatory cytokines of microglia, but also affects the phagocytosis and promotes the transformation of microglial cells to the anti-inflammatory M2 phenotype from M1 phenotype (10). Both CB1 and CB2 receptors are downregulated in M1 microglia, whereas the M2a and M2c microglia show phenotypic changes in the endocannabinoid machinery, such as M2a favors 2-AG synthesis and M2c favors AEA (10). CB1 is important for neuronal differentiation, normal axon migration and the establishment of neuronal connectivity. Interference in critical periods is considered to have adverse effects on brain development (74, 75). Overall, microglia in ASD are usually found more shifted toward the M2 state than M1 (76). This predominance of the M2 state would seem to point toward a compensatory role of microglial activation, not a pathogenic role, which would better explain the lack of efficacy of minocycline in ASD (77). Although stimulation of CB2 receptors promotes the shifting of microglia to the M2 state, it is still unclear whether overstimulating CB2 receptors will produce any further improvement. However, chronic CB2 activation has not been found to produce any tolerance, while CB1 activation is associated with withdrawal effects in the CNS (77). Understanding the effects of overstimulating CB2 receptors remains a topic of great interest. Hopefully, future research will address this issue.
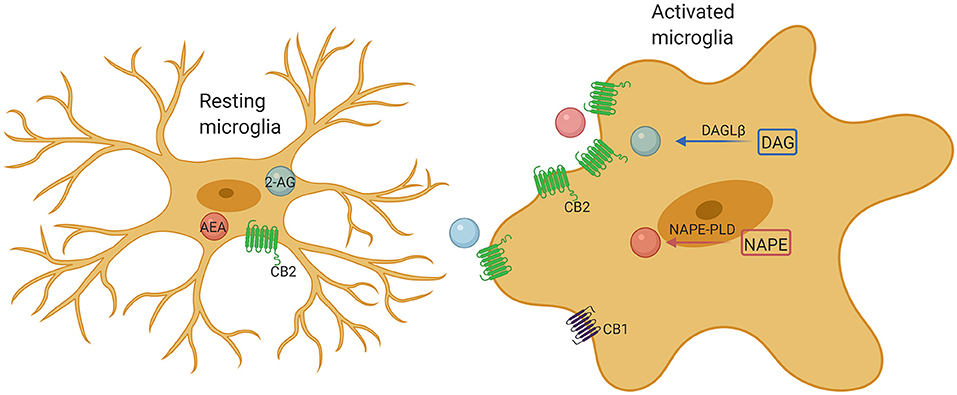
Figure 2. Microglia comprise a complete endocannabinoid signaling system. Microglia produce ~20-fold more endocannabinoids than neurons and other glial cells, and may be a major cellular source of endogenous cannabinoids under neuroinflammatory conditions. Upon activation, microglia markedly increase their endocannabinoids synthesis and upregulate their CB2 receptor expression, promoting protection of the microglia phenotype by enhancing their neuroprotective factor production and reducing their proinflammatory factor release.
Doenni et al. found that early inflammation caused by a single injection of LPS on postnatal day 14 reduced the social play and non-play behavior of adolescents in male and female rats. The social defect induced by LPS is related to the decreased binding of eCB with CB1, elevated AEA levels, and increased FAAH in the amygdala. Oral administration of FAAH inhibitor PF-04457845 (1 mg/kg) before social testing could normalize LPS-induced social behavior changes. A similar improvement was observed after direct injection of PF-04457845 into the basolateral amygdala, suggesting that changes in AEA signaling in this brain region play a key role in mediating LPS-induced social dysfunction (78).
Targeted activation of CB1 may bring some side effects to the CNS, such as depression, suicidal tendency, and other mental disorders (79). However, activation of CB2 receptor does not cause these central side effects. On the contrary, activation of CB2 can modulate inflammatory immune response and is becoming a potential intervention target for immune regulation, neuroinflammation and neurodegenerative diseases. Zamberletti et al. report that cannabidivarin (CBDV) treatment at two postnatal stages (postnatal day19–32 and postnatal day 34–58), respectively, bring beneficial therapeutic effect on the VPA autism rat model. Early preventive treatment relieves deficits in social novelty, impairment of short-term memory, and hyperactivity without affecting stereotyped behavior. Late rehabilitation therapy can improve social impairment, social novelty preferences, short-term memory deficit repetitive behavior and hyperactivity. CBDV treatment upregulated CB2 protein in the hippocampus, restored the expression of GFAP, CD11b, and TNF- α in the same brain region, promoted the activation of microglia, and further demonstrated its direct or indirect neuroprotective effect (80). Our previous work also found that CB2 activation reduced the release of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α in the peripheral skin of rats with inflammatory pain (81). Based on those studies, ECS intervention may regulate neuroimmune inflammation through CB2 to alleviate ASD symptoms.
The above animal and clinical studies provide solid evidence that unbalanced ECS signaling are involved in the occurrence of ASD. Therefore, modulating ECS signaling by targeting different components of ECS (2-AG, AEA, CB1, and CB2) may provide therapeutic strategies for the treatment of ASD (Figure 3). In the last decade, especially in 2018, FDA approved the application of CBD-rich Epidiolex in the treatment of refractory epilepsy, a common comorbid symptom of autism, and CBD and ECS have received extensive attention for the pathophysiological mechanism and therapeutic effect of neurological diseases in children. Currently, most of the studies on the relationship between ASD and ECS are still in the preclinical stage. Many histological and neurochemical abnormalities have been reported in ASD patients and animal models, reflecting the heterogeneity and complexity of this group of diseases, which poses a serious challenge to study on clinical treatment and hinders the identification of possible common pathophysiological mechanisms of ASD. Microglia are indispensable coordinators of CNS development and homeostasis, and possibly participated in the pathogenesis of ASD. ECS signaling can regulate the activity and functions of microglia (82). Both clinical and preclinical studies demonstrate that the neuropathology of ASD is characterized by an abnormal inflammatory status of the brain. Therefore, a detailed investigation of the roles of CB2 receptors and related ligands may shed new light on the pathophysiological mechanisms and therapeutic targets of autism.
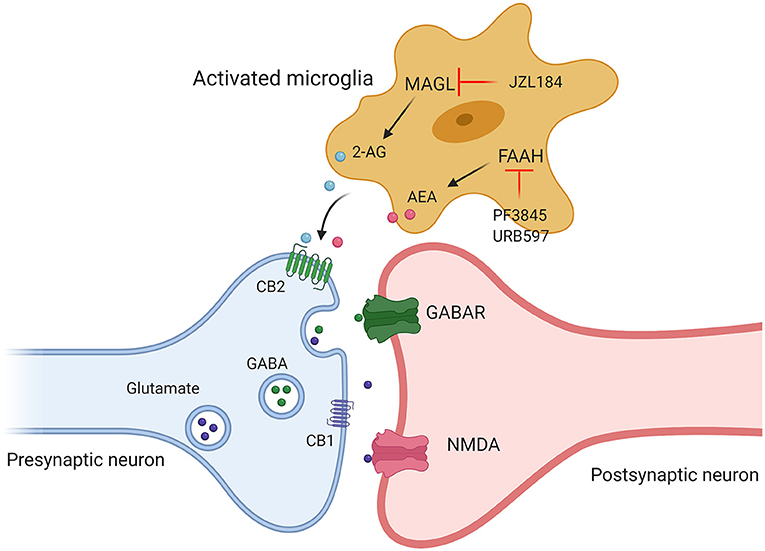
Figure 3. Targeting different components of ECS (2-AG, AEA, CB1, and CB2) may provide therapeutic strategies for the treatment of ASD. Administration of JZL184, a pharmacological inhibitor of MAGL, enhanced 2-AG signaling, by using FAAH inhibitor (PF3845, URB597) to increase AEA levels to promote the release of excitatory and inhibitory amino acid transmitters by acting on cannabinoid receptors.
LP and JY made manuscript planning and structuring. YY and QL did literature searching. TS drafted the manuscript. YY, QL, and JY revised the manuscript. TS and LP edited the final manuscript. All authors have read and approved the final manuscript.
This study was supported by grants from the National Natural Science Foundation of China (Nos. 81804188 and 81870932).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Dr. Daniel Fulton from University of Birmingham for the English language editing. Figures created with BioRender.com.
1. Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol. (2013) 26:146–53. doi: 10.1097/WCO.0b013e32835ee548
2. McCracken JT, McGough J, Shah B, Cronin P, Hong D, Aman MG, et al. Risperidone in children with autism and serious behavioral problems. N Engl J Med. (2002) 347:314–21. doi: 10.1056/NEJMoa013171
3. Yeomans DC, Hanson LR, Carson DS, Tunstall BJ, Lee MR, Tzabazis AZ, et al. Nasal oxytocin for the treatment of psychiatric disorders and pain: achieving meaningful brain concentrations. Transl Psychiatry. (2021) 11:388. doi: 10.1038/s41398-021-01511-7
4. Quintana DS, Lischke A, Grace S, Scheele D, Ma Y, Becker B. Advances in the field of intranasal oxytocin research: lessons learned and future directions for clinical research. Mol Psychiatry. (2021) 26:80–91. doi: 10.1038/s41380-020-00864-7
5. Ooi YP, Weng SJ, Kossowsky J, Gerger H, Sung M. Oxytocin and autism spectrum disorders: a systematic review and meta-analysis of randomized controlled trials. Pharmacopsychiatry. (2017) 50:5–13. doi: 10.1055/s-0042-109400
6. Fusar-Poli L, Cavone V, Tinacci S, Concas I, Petralia A, Signorelli MS, et al. Cannabinoids for people with ASD: a systematic review of published and ongoing studies. Brain Sci. (2020) 10:572. doi: 10.3390/brainsci10090572
7. Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. (1993) 365:61–5. doi: 10.1038/365061a0
8. Zamberletti E, Gabaglio M, Parolaro D. The endocannabinoid system and autism spectrum disorders: insights from animal models. Int J Mol Sci. (2017) 18:1916. doi: 10.3390/ijms18091916
9. Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun. (2012) 26:383–92. doi: 10.1016/j.bbi.2011.08.007
10. Mecha M, Feliu A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutierrez S, de Sola RG, et al. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immun. (2015) 49:233–45. doi: 10.1016/j.bbi.2015.06.002
11. Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. (1997) 388:773–8. doi: 10.1038/42015
12. Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, et al. Localization of diacylglycerol lipase-alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci. (2006) 26:4740–51. doi: 10.1523/JNEUROSCI.0054-06.2006
13. Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF. DAGLbeta inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol. (2012) 8:999–1007. doi: 10.1038/nchembio.1105
14. Jung KM, Sepers M, Henstridge CM, Lassalle O, Neuhofer D, Martin H, et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat Commun. (2012) 3:1080. doi: 10.1038/ncomms2045
15. Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Liu S, Powell CM. Altered striatal synaptic function and abnormal behaviour in Shank3 Exon4-9 deletion mouse model of autism. Autism Res. (2016) 9:350–75. doi: 10.1002/aur.1529
16. Folkes OM, Baldi R, Kondev V, Marcus DJ, Hartley ND, Turner BD, et al. An endocannabinoid-regulated basolateral amygdala-nucleus accumbens circuit modulates sociability. J Clin Invest. (2020) 130:1728–42. doi: 10.1172/JCI131752
17. Wei D, Allsop S, Tye K, Piomelli D. Endocannabinoid signaling in the control of social behavior. Trends Neurosci. (2017) 40:385–96. doi: 10.1016/j.tins.2017.04.005
18. Qin M, Zeidler Z, Moulton K, Krych L, Xia Z, Smith CB. Endocannabinoid-mediated improvement on a test of aversive memory in a mouse model of fragile X syndrome. Behav Brain Res. (2015) 291:164–71. doi: 10.1016/j.bbr.2015.05.003
19. Wei D, Dinh D, Lee D, Li D, Anguren A, Moreno-Sanz G, et al. Enhancement of anandamide-mediated endocannabinoid signaling corrects autism-related social impairment. Cannabis Cannabinoid Res. (2016) 1:81–9. doi: 10.1089/can.2015.0008
20. Kerr DM, Gilmartin A, Roche M. Pharmacological inhibition of fatty acid amide hydrolase attenuates social behavioural deficits in male rats prenatally exposed to valproic acid. Pharmacol Res. (2016) 113:228–35. doi: 10.1016/j.phrs.2016.08.033
21. Servadio M, Melancia F, Manduca A, di Masi A, Schiavi S, Cartocci V, et al. Targeting anandamide metabolism rescues core and associated autistic-like symptoms in rats prenatally exposed to valproic acid. Transl Psychiatry. (2016) 6:e902. doi: 10.1038/tp.2016.182
22. Melancia F, Schiavi S, Servadio M, Cartocci V, Campolongo P, Palmery M, et al. Sex-specific autistic endophenotypes induced by prenatal exposure to valproic acid involve anandamide signalling. Br J Pharmacol. (2018) 175:3699–712. doi: 10.1111/bph.14435
23. Xu ZX, Kim GH, Tan JW, Riso AE, Sun Y, Xu EY, et al. Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nat Commun. (2020) 11:1797. doi: 10.1038/s41467-020-15530-3
24. Wu HF, Lu TY, Chu MC, Chen PS, Lee CW, Lin HC. Targeting the inhibition of fatty acid amide hydrolase ameliorate the endocannabinoid-mediated synaptic dysfunction in a valproic acid-induced rat model of Autism. Neuropharmacology. (2020) 162:107736. doi: 10.1016/j.neuropharm.2019.107736
25. Glickman G. Circadian rhythms and sleep in children with autism. Neurosci Biobehav Rev. (2010) 34:755–68. doi: 10.1016/j.neubiorev.2009.11.017
26. Melke J, Goubran Botros H, Chaste P, Betancur C, Nygren G, Anckarsater H, et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol Psychiatry. (2008) 13:90–8. doi: 10.1038/sj.mp.4002016
27. Vaughn LK, Denning G, Stuhr KL, de Wit H, Hill MN, Hillard CJ. Endocannabinoid signalling: has it got rhythm? Br J Pharmacol. (2010) 160:530–43. doi: 10.1111/j.1476-5381.2010.00790.x
28. Busquets-Garcia A, Gomis-Gonzalez M, Guegan T, Agustin-Pavon C, Pastor A, Mato S, et al. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med. (2013) 19:603–7. doi: 10.1038/nm.3127
29. Gomis-Gonzalez M, Busquets-Garcia A, Matute C, Maldonado R, Mato S, Ozaita A. Possible Therapeutic doses of cannabinoid type 1 receptor antagonist reverses key alterations in fragile X syndrome mouse model. Genes. (2016) 7:56. doi: 10.3390/genes7090056
30. Sperlagh B, Windisch K, Ando RD, Sylvester Vizi E. Neurochemical evidence that stimulation of CB1 cannabinoid receptors on GABAergic nerve terminals activates the dopaminergic reward system by increasing dopamine release in the rat nucleus accumbens. Neurochem Int. (2009) 54:452–7. doi: 10.1016/j.neuint.2009.01.017
31. Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D, Cohen RM. Low medial prefrontal dopaminergic activity in autistic children. Lancet. (1997) 350:638. doi: 10.1016/S0140-6736(05)63326-0
32. Pascucci T, Colamartino M, Fiori E, Sacco R, Coviello A, Ventura R, et al. P-cresol alters brain dopamine metabolism and exacerbates autism-like behaviors in the BTBR mouse. Brain Sci. (2020) 10:233. doi: 10.3390/brainsci10040233
33. Paval D. A dopamine hypothesis of autism spectrum disorder. Dev Neurosci. (2017) 39:355–60. doi: 10.1159/000478725
34. Centonze D, Battistini L, Maccarrone M. The endocannabinoid system in peripheral lymphocytes as a mirror of neuroinflammatory diseases. Curr Pharm Des. (2008) 14:2370–42. doi: 10.2174/138161208785740018
35. Siniscalco D, Sapone A, Giordano C, Cirillo A, de Magistris L, Rossi F, et al. Cannabinoid receptor type 2, but not type 1, is up-regulated in peripheral blood mononuclear cells of children affected by autistic disorders. J Autism Dev Disord. (2013) 43:2686–95. doi: 10.1007/s10803-013-1824-9
36. Karhson DS, Krasinska KM, Dallaire JA, Libove RA, Phillips JM, Chien AS, et al. Plasma anandamide concentrations are lower in children with autism spectrum disorder. Mol Autism. (2018) 9:18. doi: 10.1186/s13229-018-0203-y
37. Aran A, Eylon M, Harel M, Polianski L, Nemirovski A, Tepper S, et al. Lower circulating endocannabinoid levels in children with autism spectrum disorder. Mol Autism. (2019) 10:2. doi: 10.1186/s13229-019-0256-6
38. Aran A, Cassuto H, Lubotzky A, Wattad N, Hazan E. Brief Report: Cannabidiol-Rich Cannabis in Children with Autism Spectrum Disorder and Severe Behavioral Problems-A Retrospective Feasibility Study. J Autism Dev Disord. (2019) 49:1284–8. doi: 10.1007/s10803-018-3808-2
39. Barchel D, Stolar O, De-Haan T, Ziv-Baran T, Saban N, Fuchs DO, et al. Oral cannabidiol use in children with autism spectrum disorder to treat related symptoms and co-morbidities. Front Pharmacol. (2018) 9:1521. doi: 10.3389/fphar.2018.01521
40. Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. (2017) 79:619–43. doi: 10.1146/annurev-physiol-022516-034406
41. Lee BK, Magnusson C, Gardner RM, Blomstrom A, Newschaffer CJ, Burstyn I, et al. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav Immun. (2015) 44:100–5. doi: 10.1016/j.bbi.2014.09.001
42. Richetto J, Massart R, Weber-Stadlbauer U, Szyf M, Riva MA, Meyer U. Genome-wide DNA methylation changes in a mouse model of infection-mediated neurodevelopmental disorders. Biol Psychiatry. (2017) 81:265–76. doi: 10.1016/j.biopsych.2016.08.010
43. Eshraghi RS, Deth RC, Mittal R, Aranke M, Kay SS, Moshiree B, et al. Early disruption of the microbiome leading to decreased antioxidant capacity and epigenetic changes: implications for the rise in autism. Front Cell Neurosci. (2018) 12:256. doi: 10.3389/fncel.2018.00256
44. Matta SM, Hill-Yardin EL, Crack PJ. The influence of neuroinflammation in autism spectrum disorder. Brain Behav Immun. (2019) 79:75–90. doi: 10.1016/j.bbi.2019.04.037
45. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. (2017) 23:1018–27. doi: 10.1038/nm.4397
46. Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun. (2014) 5:5748. doi: 10.1038/ncomms6748
47. Pang Y, Dai X, Roller A, Carter K, Paul I, Bhatt AJ, et al. Early postnatal lipopolysaccharide exposure leads to enhanced neurogenesis and impaired communicative functions in rats. PLoS ONE. (2016) 11:e0164403. doi: 10.1371/journal.pone.0164403
48. Liao X, Yang J, Wang H, Li Y. Microglia mediated neuroinflammation in autism spectrum disorder. J Psychiatr Res. (2020) 130:167–76. doi: 10.1016/j.jpsychires.2020.07.013
49. Sacco R, Lenti C, Saccani M, Curatolo P, Manzi B, Bravaccio C, et al. Cluster analysis of autistic patients based on principal pathogenetic components. Autism Res. (2012) 5:137–47. doi: 10.1002/aur.1226
50. Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. (2009) 19:231–4. doi: 10.1016/j.conb.2009.06.003
51. Mallya AP, Wang HD, Lee HNR, Deutch AY. Microglial pruning of synapses in the prefrontal cortex during adolescence. Cereb Cortex. (2019) 29:1634–43. doi: 10.1093/cercor/bhy061
52. Kim HJ, Cho MH, Shim WH, Kim JK, Jeon EY, Kim DH, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry. (2017) 22:1576–84. doi: 10.1038/mp.2016.103
53. Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci. (2019) 22:1075–88. doi: 10.1038/s41593-019-0419-y
54. Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. (2011) 31:16241–50. doi: 10.1523/JNEUROSCI.3667-11.2011
55. Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. (2014) 17:400–6. doi: 10.1038/nn.3641
56. Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, et al. Microglia in the cerebral cortex in autism. J Autism Dev Disord. (2012) 42:2569–84. doi: 10.1007/s10803-012-1513-0
57. Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. (2014) 83:1131–43. doi: 10.1016/j.neuron.2014.09.001
58. Hong SJ, Vos de Wael R, Bethlehem RAI, Lariviere S, Paquola C, Valk SL, et al. Atypical functional connectome hierarchy in autism. Nat Commun. (2019) 10:1022. doi: 10.1038/s41467-019-08944-1
59. Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. (2020) 180:568–84.e23. doi: 10.1016/j.cell.2019.12.036
60. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
61. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
62. Corbett BA, Kantor AB, Schulman H, Walker WL, Lit L, Ashwood P, et al. A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol Psychiatry. (2007) 12:292–306. doi: 10.1038/sj.mp.4001943
63. Thomas MS, Davis R, Karmiloff-Smith A, Knowland VC, Charman T. The over-pruning hypothesis of autism. Dev Sci. (2016) 19:284–305. doi: 10.1111/desc.12303
64. Croen LA, Qian Y, Ashwood P, Zerbo O, Schendel D, Pinto-Martin J, et al. Infection and fever in pregnancy and autism spectrum disorders: findings from the study to explore early development. Autism Res. (2019) 12:1551–61. doi: 10.1002/aur.2175
65. Lammert CR, Lukens JR. Modeling autism-related disorders in mice with maternal immune activation (MIA). Methods Mol Biol. (2019) 1960:227–36. doi: 10.1007/978-1-4939-9167-9_20
66. Conway F, Brown AS. Maternal immune activation and related factors in the risk of offspring psychiatric disorders. Front Psychiatry. (2019) 10:430. doi: 10.3389/fpsyt.2019.00430
67. Bilbo SD, Block CL, Bolton JL, Hanamsagar R, Tran PK. Beyond infection - maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp Neurol. (2018) 299:241–51. doi: 10.1016/j.expneurol.2017.07.002
68. Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. (2017) 549:528–32. doi: 10.1038/nature23910
69. Lee M, Krishnamurthy J, Susi A, Sullivan C, Gorman GH, Hisle-Gorman E, et al. Association of autism spectrum disorders and inflammatory bowel disease. J Autism Dev Disord. (2018) 48:1523–9. doi: 10.1007/s10803-017-3409-5
70. Sharon G, Cruz NJ, Kang DW, Gandal MJ, Wang B, Kim YM, et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell. (2019) 177:1600–18.e17. doi: 10.1016/j.cell.2019.05.004
71. Desbonnet L, Clarke G, Shanahan F, Dinan TG, Cryan JF. Microbiota is essential for social development in the mouse. Mol Psychiatry. (2014) 19:146–8. doi: 10.1038/mp.2013.65
72. Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
73. Tanaka M, Sackett S, Zhang Y. Endocannabinoid modulation of microglial phenotypes in neuropathology. Front Neurol. (2020) 11:87. doi: 10.3389/fneur.2020.00087
74. Lisboa SF, Gomes FV, Guimaraes FS, Campos AC. Microglial cells as a link between cannabinoids and the immune hypothesis of psychiatric disorders. Front Neurol. (2016) 7:5. doi: 10.3389/fneur.2016.00005
75. Brigida AL, Schultz S, Cascone M, Antonucci N, Siniscalco D. Endocannabinod signal dysregulation in autism spectrum disorders: a correlation link between inflammatory state and neuro-immune alterations. Int J Mol Sci. (2017) 18:1425. doi: 10.3390/ijms18071425
76. Kalkman HO, Feuerbach D. Microglia M2A polarization as potential link between food allergy and autism spectrum disorders. Pharmaceuticals. (2017) 10:95. doi: 10.3390/ph10040095
77. Pardo CA, Buckley A, Thurm A, Lee LC, Azhagiri A, Neville DM, et al. A pilot open-label trial of minocycline in patients with autism and regressive features. J Neurodev Disord. (2013) 5:9. doi: 10.1186/1866-1955-5-9
78. Doenni VM, Gray JM, Song CM, Patel S, Hill MN, Pittman QJ. Deficient adolescent social behavior following early-life inflammation is ameliorated by augmentation of anandamide signaling. Brain Behav Immun. (2016) 58:237–47. doi: 10.1016/j.bbi.2016.07.152
79. Ettaro R, Laudermilk L, Clark SD, Maitra R. Behavioral assessment of rimonabant under acute and chronic conditions. Behav Brain Res. (2020) 390:112697. doi: 10.1016/j.bbr.2020.112697
80. Zamberletti E, Gabaglio M, Woolley-Roberts M, Bingham S, Rubino T, Parolaro D. Cannabidivarin treatment ameliorates autism-like behaviors and restores hippocampal endocannabinoid system and glia alterations induced by prenatal valproic acid exposure in rats. Front Cell Neurosci. (2019) 13:367. doi: 10.3389/fncel.2019.00367
81. Su TF, Zhao YQ, Zhang LH, Peng M, Wu CH, Pei L, et al. Electroacupuncture reduces the expression of proinflammatory cytokines in inflamed skin tissues through activation of cannabinoid CB2 receptors. Eur J Pain. (2012) 16:624–35. doi: 10.1002/j.1532-2149.2011.00055.x
Keywords: endocannabinoid system, microglia, autism spectrum disorder, immune, neurodevelopmental disorders
Citation: Su T, Yan Y, Li Q, Ye J and Pei L (2021) Endocannabinoid System Unlocks the Puzzle of Autism Treatment via Microglia. Front. Psychiatry 12:734837. doi: 10.3389/fpsyt.2021.734837
Received: 01 July 2021; Accepted: 27 September 2021;
Published: 22 October 2021.
Edited by:
David Quentin Beversdorf, University of Missouri, United StatesReviewed by:
Kathryn K. Chadman, Institute for Basic Research in Developmental Disabilities (IBR), United StatesCopyright © 2021 Su, Yan, Li, Ye and Pei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Pei, peilei@hust.edu.cn; Jiacai Ye, 21376689@qq.com
†These authors share first authorship
‡ORCID: Tangfeng Su orcid.org/0000-0002-1925-5519
Lei Pei orcid.org/0000-0002-6086-7779
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.