
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 30 September 2021
Sec. Child and Adolescent Psychiatry
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.722378
This article is part of the Research TopicFragile X Spectrum DisordersView all 24 articles
 Edgard Verdura1
Edgard Verdura1 Laura Pérez-Cano1*Rubén Sabido-Vera1Emre Guney1,2
Laura Pérez-Cano1*Rubén Sabido-Vera1Emre Guney1,2 Jean-Marc Hyvelin3Lynn Durham1,3
Jean-Marc Hyvelin3Lynn Durham1,3 Baltazar Gomez-Mancilla3,4*
Baltazar Gomez-Mancilla3,4*Fragile X syndrome (FXS) is the most frequent monogenic cause of autism or intellectual disability, and research on its pathogenetic mechanisms has provided important insights on this neurodevelopmental condition. Nevertheless, after 30 years of intense research, efforts to develop treatments have been mostly unsuccessful. The aim of this review is to compile evidence from existing research pointing to clinical, genetic, and therapeutic response heterogeneity in FXS and highlight the need of implementing precision medicine-based treatments. We comment on the high genetic and phenotypic heterogeneity present in FXS, as a contributing factor to the difficulties found during drug development. Given that several clinical trials have showed a non-negligeable fraction of positive responders to drugs targeting core FXS symptoms, we propose that success of clinical trials can be achieved by tackling the underlying heterogeneity in FXS by accurately stratifying patients into drug-responder subpopulations. These precision medicine-based approaches, which can be first applied to well-defined monogenic diseases such as FXS, can also serve to define drug responder profiles based on specific biomarkers or phenotypic features that can associate patients with different genetic backgrounds to a same candidate drug, thus repositioning a same drug for a larger number of patients with NDDs.
Neurodevelopmental disorders (NDDs) are a group of prevalent and highly heterogeneous conditions characterized by impairment in “personal, social, academic, or occupational functioning” with onset early in development, which include autism spectrum disorder (ASD), intellectual disability (ID), attention deficit hyperactivity disorder (ADHD), communication disorders, specific learning disorders, and motor disorders (1); moreover, the definition can also include some neuropsychiatric disorders such as schizophrenia and bipolar disorder, and other neurologic disorders such as cerebral palsy or epilepsy (2). Comorbidity of two or more of these disorders is observed at rates higher than what it would be expected by chance, suggesting the existence of clusters of shared biological mechanisms (3, 4). While cooccurrence of neurological features and conditions is frequently observed, very often, NDDs also include a variety of extra-neurological clinical signs such as hypotonia, dysmorphology, cardiologic or metabolic features, as well as gastrological problems such as constipation or diarrhea, which are specially frequent in ASD and ADHD (5–7), or immunological abnormalities (8, 9). The recent advances in genotyping and sequencing technologies have propelled the identification of risk/causal genes, which has pointed to remarkable genetic heterogeneity among and within specific NDDs; for intellectual disability alone, pathogenic mutations in more than 1,000 genes have been confirmed in the SysID database (a systematic and manually curated catalog of ID-associated genes) (10). While syndromic neurodevelopmental conditions associated to mutations in one single gene are by definition genetically more homogeneous, they still involve varying degrees of phenotype and genetic heterogeneity (11). Further complexifying matters: well-defined clinical entities can be caused by mutations in several genes (as it is the case for Noonan syndrome, linked to mutations in 14 different genes) (12), while mutations occurring in a same gene can result into a wide spectrum of symptoms, as exemplified by MECP2 mutations in Rett syndrome (13). Adjunctly, different kinds of mutations can result in different pathophysiological mechanisms, as recently exemplified by mutations in SATB1, in which three different kinds of variants were associated to distinct pathological consequences (14).
Fragile X syndrome (FXS) is a well-characterized NDD syndrome, caused by deficiency of FMRP (Fragile X Mental Retardation Protein, encoded by FMR1), an RNA-binding protein that regulates editing, translation, stability, and transport of a large number of neuronal mRNAs (15). FXS has been extensively studied in the last 30 years. It was first clinically described in 1943 as a form of X-linked inherited intellectual disability (16). In 1969, Lubs developed the chromosomal test for FXS (17), although the causal gene, FMR1 (Fragile X Mental Retardation 1), and the mutational basis of FXS, were not discovered until 1991 (18). FXS is classically caused by an expansion of >200 CGG repeats in the promoter region of FMR1 (hereinafter referred as full FMR1 mutation); this leads to the promoter methylation and transcriptional silencing of FMR1. Nowadays, FXS represents the most frequently identified monogenic cause of ASD and inherited intellectual disability (19). But despite being a genetically well-characterized syndrome, there is considerable heterogeneity within patients with FXS, and the condition remains a high unmet medical need. This supports the necessity for deeper characterization of the population with FXS to enable the development of efficient treatments.
A considerably high level of clinical and genetic heterogeneity can be found in FXS. Despite being a highly recognizable syndrome from the clinical point of view, patients with FXS manifest a wide spectrum of behavioral phenotypes, although some of them such as attention/hyperactivity, hyperarousal, anxiety, and aggression episodes are commonly seen. Females with FXS show a similar spectrum of behavioral difficulties compared to males with FXS, but with milder symptoms (20). Notably, there is a strong association between FXS and autism spectrum disorder (ASD), as ~50% of FXS male and 20% female patients meet DSM-5 criteria for this disorder, and FXS is the leading monogenic cause of ASD (21). Although autistic features are not present in all individuals it is highly speculated that FXS and some groups of patients with idiopathic ASD present with shared pathophysiology, as defects in many proteins that interact with FMRP have also been associated with idiopathic ASD (15). Importantly, greater severity and lower level of functioning is associated with ASD co-morbidity in FXS (22–24). A higher prevalence of seizures, sleep problems, and co-occurring problematic behaviors, especially aggressive/disruptive behavior, is found in the pediatric population with FXS and a diagnosis of ASD than in the FXS population without ASD (19). Individuals with FXS also widely differ in level of cognitive impairment, while some males with FXS function nearly normally, and others are profoundly disabled (25). Several studies have reported “high-functioning” males with intellectual ability in the normal to borderline range (26, 27). Besides, one-third to half of females with a full FMR1 mutation have intellectual functioning in the normal range, due to the masking effect of the normal X-chromosome FMR1 allele (28). Numerous additional associated conditions and symptoms of variable severity can occur such as sleep disturbance, seizures, frequent otitis media, strabismus, and joint hyperlaxity (29). Interestingly, a FXS subgroup has been reported, characterized by hyperphagia, lack of satiation after meals and extreme obesity with a full, round face, small, broad hands/feet, and regional skin hyperpigmentation, referenced in literature as “Prader-Willi-like” (30, 31). This particular subgroup might point to the existence of several subgroups of patients with FXS which can be grouped based on phenotypic features and treated by targeting the corresponding underlying molecular differences. Prevalence of several conditions or symptoms associated to FXS are shown in Tables 1, 2.
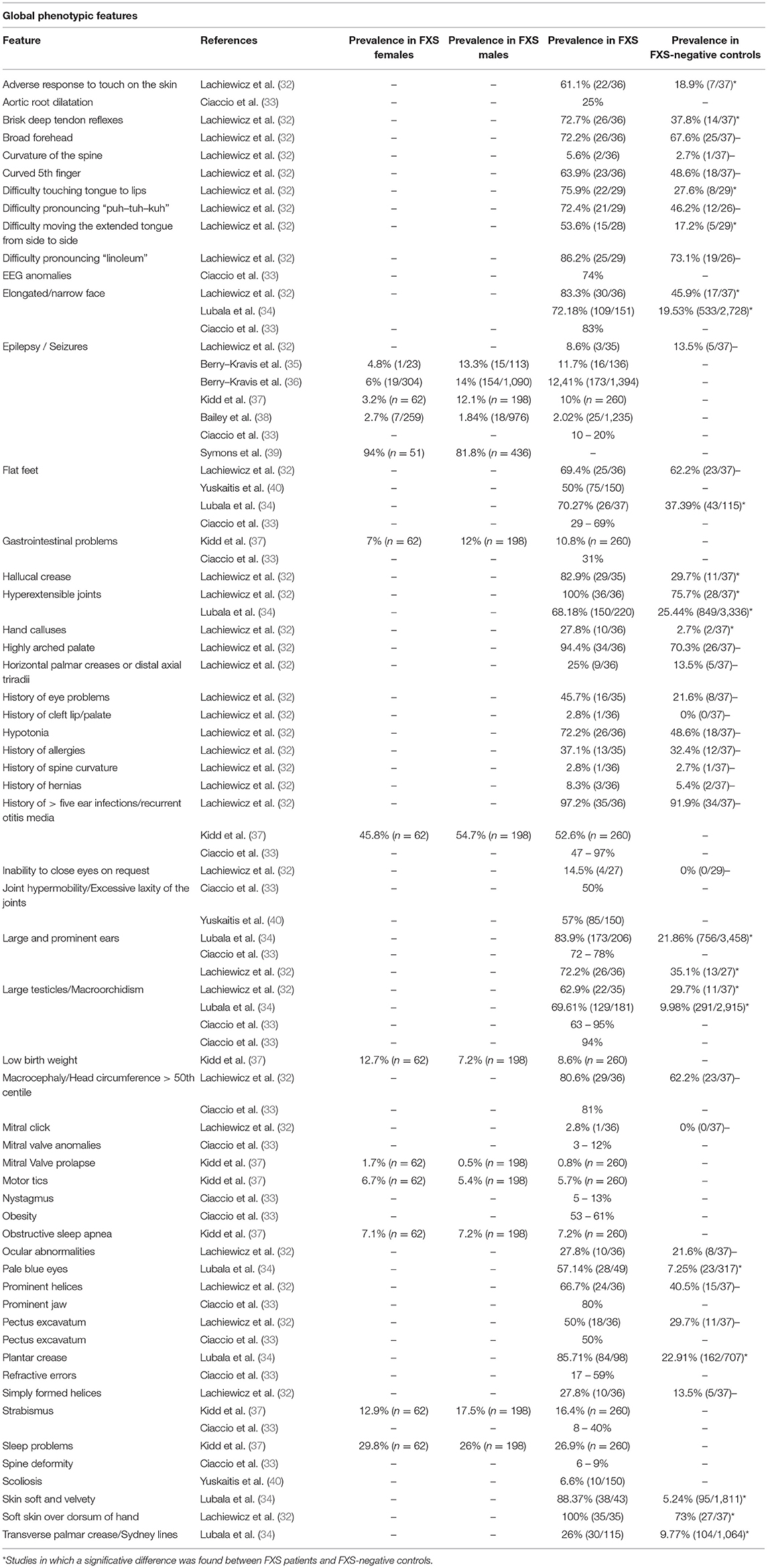
Table 1. Reported prevalence of heterogeneous phenotypic features in FXS patients in several studies.
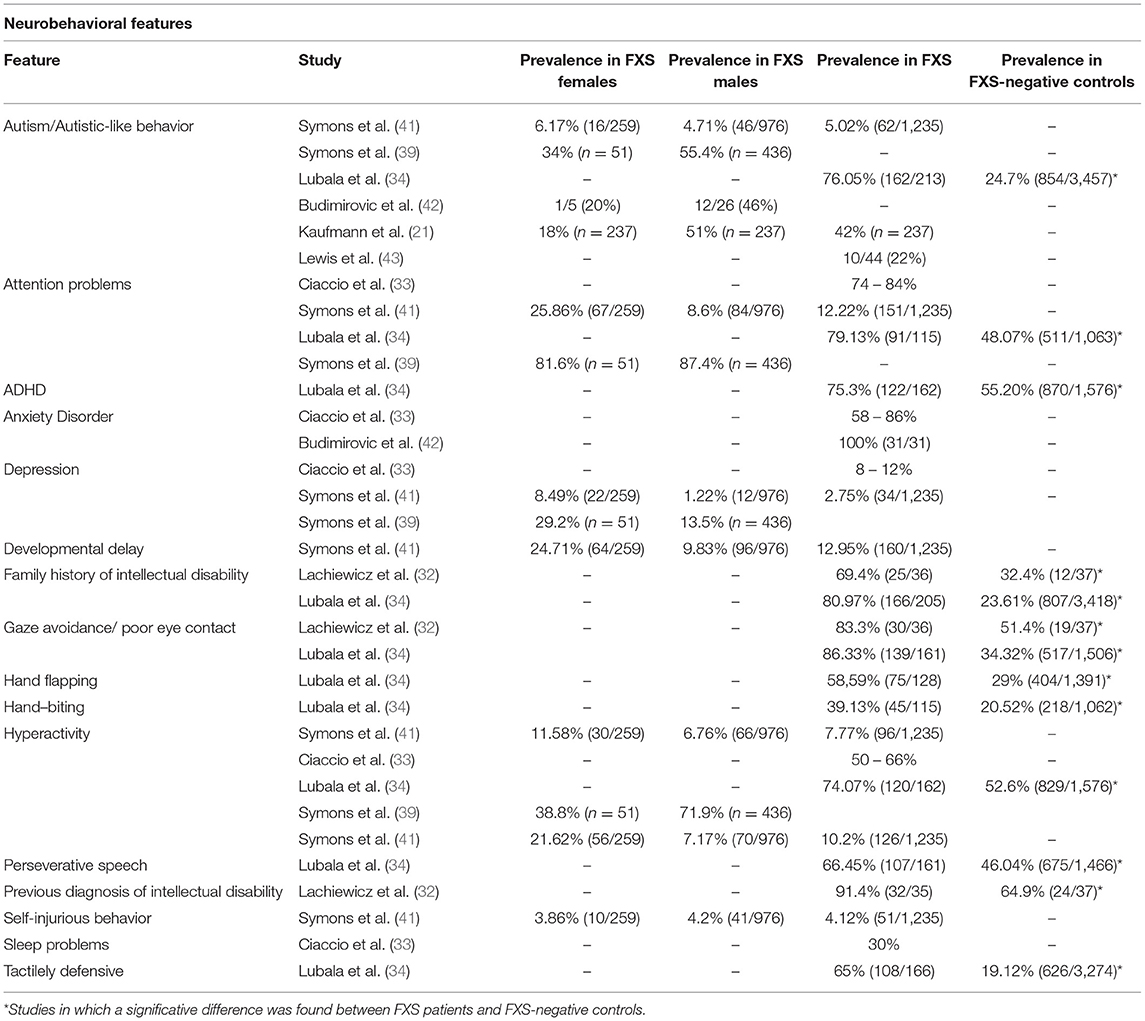
Table 2. Reported prevalence of heterogeneous neurobehavioral features in FXS patients in several studies.
This important degree of phenotypic heterogeneity in FXS probably mirrors a heterogeneous genetic background and the cellular-level involvement of various signaling pathways co-regulated by FMRP, such as PI3K and mTOR pathways (44). Moreover, genetic background plays an important role as shown in animal models (which would result in different patterns of expression of other proteins, including FMRP-interacting proteins) (45, 46), as well as (a) genetic consequences of variation on FMRP function at different levels including FMRP expression, and (b) FMRP effect on other genes mRNA transcripts by regulation of splicing, translation (through ribosome stalling), and RNA stability through the recognition of mRNA codon bias and N6-methyladenosine (m6A) modifications (15). In addition, individuals with FXS might show mosaicism at two different levels: (1) CGG repeat lengths, with some cells harboring fully expanded mutation alleles and other cells harboring more benign alleles; and (2) methylation levels, with some cells containing methylated FMR1 alleles and other cells containing unmethylated FMR1 alleles. It is estimated that nearly half of individuals carrying the full FMR1 mutation exhibit some sort of size and/or methylation mosaicism (47) (Table 3). Novel methods have improved detection of these alleles, which previously could only be detected by Southern blot (50). Size mosaicism, which is thought to arise due to CGG repeat instability, is normally observed as a combination of full mutation (>200 repeats) alleles with premutation alleles (>55) or rarely, even normal alleles. Methylation mosaicism can be observed in the form of unmethylated alleles, either showing a full mutation or a premutation allele. Both types of mosaicism will support the production of some FMRP, so individuals with size and /or methylation mosaicism might have less severe cognitive and behavioral defects than a patient with a full mutation and a completely methylated FMR1 promoter, and in whom FMRP is markedly reduced or absent (19). Several authors have reported that male patients having full mutation with complete methylation had the lowest IQ scores and greatest physical involvement, in comparison to mosaic cases, although other studies have not observed this correlation (48, 51, 52). Correlation of degree of size and/or methylation mosaicism with other phenotypic features (seizures, hyperactivity, and autism) has been more difficult to establish. In a recent paper evaluating a cohort of male and female patients with FXS, male children carrying full FMR1 mutation and expressing some degree of FMR1 mRNA due to incomplete methylation had significantly higher Autism Diagnostic Observational Schedule (ADOS) severity scores, compared to individuals with FXS carrying full FMR1 mutation but with completely silenced FMR1. However, this association should be replicated in additional cohorts (53). In female premutation carriers, a study found that FMR1 mRNA levels increase as the number of CGG repeats increases, suggesting that due to skewed X inactivation, mRNA levels tend to normalize in females when the number of CGG repeats increases, making clinical-genetic correlations more difficult to establish (54). While larger studies are needed, the expression levels of FMRP and methylation status of the FMR1 gene have been correlated with cognitive ability (positive correlation for FMRP levels, negative correlation for methylation) (55), whereas little correlation between CGG repeat number and cognition is thought to exist. Given that residual levels of FMRP expression explain in part the heterogeneity in the FXS phenotype, the integration of diagnostic genomic data with FMR1 mRNA measuring assays and more accurate FMRP profiles could clarify the relationships between genotypes, mRNA/protein expression and patient phenotypes. Deciphering these links would both improve disease prognostics and be useful to stratify patients with FXS for clinical trials (56).
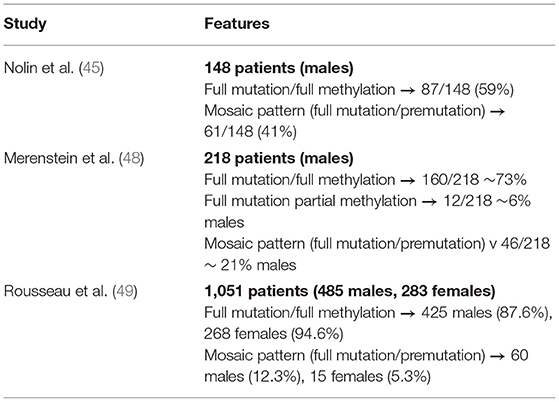
Table 3. Mosaicism features in FXS patient cohorts.
In a very recent study, which assessed quantification of methylation, DNA, RNA, and FMRP protein levels in buccal and blood samples, methylation mosaicism was estimated to be slightly over 50%. Molecular-neurobehavioral correlations confirmed the inverse relationship between overall severity of the FXS phenotype and decrease in FMRP levels. Co-occurrence of FXS with an autism diagnosis correlated significantly with 2-fold lower levels of FMRP specially in younger age- and IQ-adjusted males, compared to FXS without ASD, and patients with severe Intellectual Disability had even lower FMRP levels (42). In the same study, while Budimirovic et al. (42) also showed a high level of agreement in regard to FMRP protein levels between blood and buccal samples, but these findings have not always been replicated, and discordant presence of mutations between different tissues such as blood and skin have been reported (57–59). In parallel, it has been shown that repeat number can vary in different tissue types from the same individual carrying premutation alleles, suggesting that numbers obtained from blood mononuclear cells may not always directly translate to the brain. These results can complicate efforts to use blood results for clinical trial inclusion/exclusion criteria (60), as they suggest that FMRP levels in patients' cortical brain tissue might differ from levels observed in blood, at least for some FXS patients. Evidences of correlation between blood and brain FMRP levels in the individual subjects are limited; in particular, a study comparing brain and blood suggested that males with full mutations also have a certain level of mosaicism in brain tissue (61, 62). Further studies using blood and post-mortem brain samples will be required to shed light on this matter.
Despite two decades of preclinical work and several clinical trials targeting at least 10 different mechanisms associated with FXS pathogenesis (19), candidate drugs have been unsuccessful in reaching primary endpoints, and there are still no approved treatments to address the core symptoms of FXS. Well-studied pathophysiological mechanisms of FXS include excessive glutamatergic signaling, endocannabinoid system impaired signaling, voltage-gated ion channel dysfunction, GABAergic system inhibition, or excess of protein translation activity. While preclinical studies of drugs targeting these mechanisms in FXS mouse models (such as the widely used FMR1-KO model) provided positive results, translation to therapeutic use in human patients have not achieved the expected efficacy outcomes. Nevertheless, several clinical trials are ongoing, and several past clinical trials conducted in FXS did show clinical benefit in subsets of patients, providing hope for future drug development studies (63).
FMRP plays a critical role in mGluR (metabotropic glutamate receptor)-dependent long-term depression and its absence causes dysregulated synaptic function due to excessive and persistent protein synthesis in postsynaptic dendrites (64). Accordingly, in two clinical trials with mGlu5 receptor antagonists (fenobam, mavoglurant), which target an excessive activity of mGluR signaling downstream due to loss of inhibitory control by FMRP, improvement was observed in six and seven subjects out of 12 and 30 patients, respectively. In the fenobam study, a calmed behavior with improvement in eye contact, ability to interact, anxiety and/or motor overactivity was observed in nine out of 12 patients, pointing to an even higher rate, although further studies were not performed afterwards (65). In the initial mavoglurant clinical trial, seven out of 30 responder patients identified in a post-hoc analysis, had improved Aberrant Behavior Checklist—Community Edition scores (ABC-C) associated to complete FMR1 promoter methylation and no detectable FMR1 messenger RNA, while no improved response was shown in 18 patients with partial promoter methylation (66). These encouraging results prompted the design of two large IIb clinical trials (double—blind, placebo—controlled, performed in parallel, n = 175, 139), in which patients were also stratified by methylation status. Nevertheless, these studies did not report data on efficacy scores, nor evaluated the presence of positive responder patients (67). After negative results in the Phase III study, Novartis announced the discontinuation of the mavoglurant FXS development program (68). However, despite lack of success in past clinical trials, further studies continue to support a possible role of FMR1 promoter methylation to stratify patients (69, 70).
On the other hand, in 2012 a first phase II clinical trial with arbaclofen (a GABAB receptor agonist) was performed based on successful preclinical model studies (71). Although no differences from placebo on the primary endpoint (irritability scores) were found, secondary outcome measures were associated with significant improvement. Using a novel ABC—Social Avoidance (ABC-SA) scale validated for the assessment of FXS, arbaclofen treatment was also associated with an overall beneficial effect. Furthermore, a post-hoc analysis focused on a subgroup of 27 subjects with more severe social impairment showed improvements in several scales compared to placebo treatment. The results were also more robust among subjects who met Diagnostic and Statistical Manual of Mental Disorders-Fourth Edition (DSM-IV) and Autism Diagnostic Interview-Revised (ADI-R) criteria for autistic disorder. Significantly more subjects were responders on the CGI-I scale when receiving arbaclofen vs. placebo (35 vs. 18% overall; 50 vs. 6% autism) in the autism subgroup, although again the ABC-C Irritability was not sensitive to these effects. Arbaclofen was then tested in two parallel phase 3 studies (randomized, double—blind, placebo—controlled, n = 125 and 172) in adults and children (35). These two studies did not show benefit for arbaclofen over placebo for any measure (including the primary objective, showing efficacy reducing the ABC-CFX Social Avoidance score). Nevertheless, the child study showed that the highest dose group was associated to a benefit over placebo on ABC-CFX Irritability subscale and Parenting Stress Index, and results showed a trend toward improvement in social avoidance and hyperactivity subscales and CGI-I. Although additional studies with a larger cohort on higher doses would be required to confirm this finding, these results suggested potential dose- and age-related effects as well as a possible optimization of primary study endpoints, pointing toward a potential benefit of arbaclofen in future optimized clinical trials. Double-blind, randomized, placebo-controlled studies in FXS are also highly recommended to properly assess the sensitivity and specificity of clinical endpoints (63, 72, 73).
Clinical trials have also been performed for other drugs with limited success. Although not reaching primary outcomes, secondary endpoints or post-hoc analyses pointed to a considerable positive responder fraction of patients in several of them (Table 4), which suggest a potential benefit of these drugs that should be targeted and improved in future clinical trials. Only very recently, a promising phase-2 crossover study using phosphodiesterase-4D inhibitors (to increase cAMP production levels, which are reduced in patients with FXS) on a cohort of 30 FXS male adult patients showed improvement on cognition and daily function, while also meeting the primary objectives of safety and tolerability (78). Although these results shall be validated in future larger clinical trials, this study accounts for the importance of addressing cohort variability through the selection of meaningful endpoints capturing inter-individual variability.
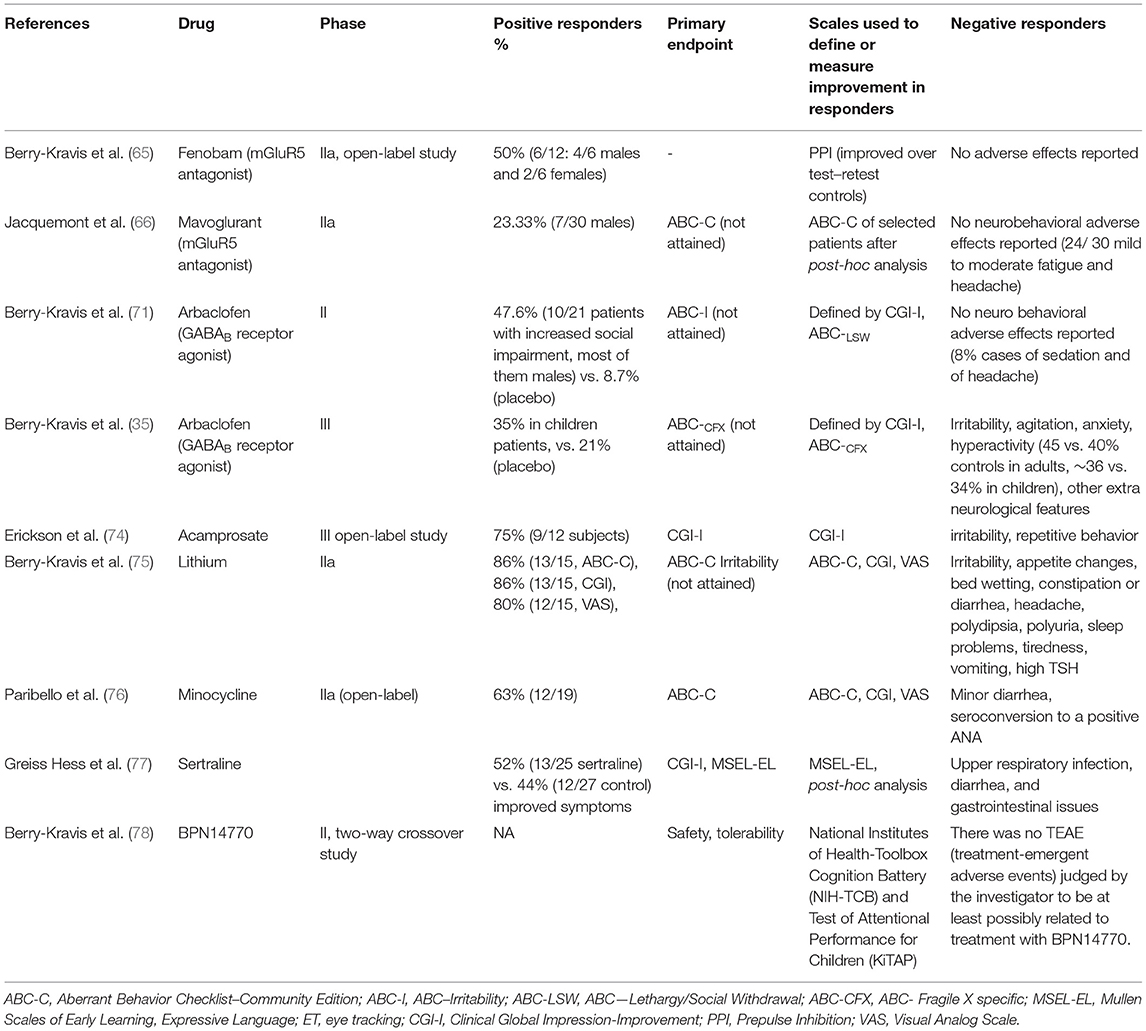
Table 4. FXS clinical trials showing positive responder results.
Although past efforts in clinical trials with FXS have mainly resulted in lack of success at meeting primary endpoints, these works have not been unfruitful. These large studies have built a base for future studies in FXS and other NDDs, in which several improvements should be incorporated, such as: use of optimized primary outcome measures (both neurobehavioral and related to cognition/language), discovery of novel prognosis and progression markers, administration of drugs from an earlier age and possibly longer times, performance of well-powered studies (as clinical trials with a lower number of participants were invariably positive in post-hoc analyses), and a better analysis and translation of preclinical mouse model studies to clinical studies in humans (63). A particular point of relevance for the future design of successful clinical trials in FXS is the characterization and further identification of subgroups of patients that respond to a specific drug treatment. While some studies have focused on establishing lists of minimal features to be screened to diagnose patients with FXS (32, 34), less focus has been put on the identification of subgroups of patients with FXS according to their phenotypic or molecular characteristics. Improved patient stratification would most likely help to pair pharmacologic agents with patients most likely to respond positively to such therapeutic treatments.
Besides FMR1 methylation levels and selection on isolated clinical features (such as high-functioning individuals or Prader-Willi-like subphenotype mentioned above), other stratification strategies based on precision medicine have been suggested and might be implemented in the future. A recent work reported the use of structural brain growth as a marker to identify clinically significative subgroups. Using topological data analysis on T1-weighted anatomical MRI data from 42 FXS children patients, researchers identified two previously unknown large subgroups of patients. Post-hoc analyses between these groups demonstrated that one group was consistently higher functioning on cognition, adaptive functioning, and autism severity scores. As pointed by the authors, anatomical MRI data analysis might become a useful method to define subtypes within other neuropsychiatric disorders (79). In another recent study, electronic health records from more than one million people were mined to investigate health characteristics of individuals clinically diagnosed with FXS. This resulted into (1) the identification of previously unnoticed significative co-occurring health conditions in patients with FXS (heart and circulatory disorders, medication side effects, and among others), and (2) the development of a predictive model to identify patients with FXS in the general population without using any genetic data, successfully identifying cases 5 years prior to clinical diagnosis of FXS (80). While this AI-assisted diagnosis method was instrumental to identify cases in the general population prior to the onset of more severe symptoms (80), no computational methods have been oriented toward stratifying patients with FXS into more homogenous subgroups, which is the first step needed to enrich future clinical trials in FXS with responder patients. Recently, the use of patient-derived induced pluripotent stem cells was proposed to model the disease in a patient-specific manner and to develop new therapeutic opportunities (81); nevertheless, to our knowledge, there is still no clinical trials in FXS involving therapeutic treatments developed using patient-derived induced pluripotent stem cells.
Accurate stratification of patients is expected to be crucial for the development of efficient drug treatments in FXS. Early applications of systems biology driven in silico drug repositioning in FXS were conducted without specific focus on genetic heterogeneity (HealX drove the advancement of a FXS repurposing discovery effort in collaboration with FRAXA) (82). In order to overcome this limitation, systems biology and precision-medicine based computational aided modeling are emerging in the NDD space both in academic and industry setting. These approaches offer new potential for novel subgroup characterization and further identification of FXS and other NDD patients with stronger biological potential to respond to specific drug candidates. For instance, STALICLA's DEPI platform was recently used to identify subsets of clinical features that significantly correlate with the molecular responses induced by arbaclofen in cellular models, which could support the identification of patients predicted to improve under arbaclofen's treatment. An observational study involving patients with FXS which had participated in previous clinical trials with arbaclofen is currently ongoing, with the main goal to provide clinical validation for this defined subgroup of patients with FXS. Importantly, other NDD patients (without a FXS diagnose) might qualify to fit into this target population and potentially benefit from the same compound.
In summary, well-defined NDD syndromes such as FXS (the most frequent monogenic cause of intellectual disability and ASD among NDDs) constitute a first step to switch from a behavioral to a molecular-centered based diagnosis. Precision medicine in FXS will continue to be necessary to (1) define more precisely subgroups of patients for clinical trials, and (2) define drug responder profiles that can associate patients with different genetic backgrounds to a same candidate drug. Importantly, these targeted populations of patients might expand beyond the FXS indication.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
All authors work at Stalicla.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association, Michigan, Ann Arbor, MI, USA. (2013). doi: 10.1176/appi.books.9780890425596
2. Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. (2013) 12:406–14. doi: 10.1016/S1474-4422(13)70011-5
3. Hansen BH, Oerbeck B, Skirbekk B, Petrovski BÉ, Kristensen H. Neurodevelopmental disorders: prevalence and comorbidity in children referred to mental health services. Nord J Psychiatry. (2018) 72:285–91. doi: 10.1080/08039488.2018.1444087
4. Gillberg C. The ESSENCE in child psychiatry: early symptomatic syndromes eliciting neurodevelopmental clinical examinations. Res Dev Disabil. (2010) 31:1543–51. doi: 10.1016/j.ridd.2010.06.002
5. Wasilewska J, Klukowski M. Gastrointestinal symptoms and autism spectrum disorder: links and risks – a possible new overlap syndrome. Pediatr Heal Med Ther. (2015) 6:153. doi: 10.2147/PHMT.S85717
6. Kedem S, Yust-Katz S, Carter D, Levi Z, Kedem R, Dickstein A, et al. Attention deficit hyperactivity disorder and gastrointestinal morbidity in a large cohort of young adults. World J Gastroenterol. (2020) 26:6626. doi: 10.3748/wjg.v26.i42.6626
7. Kang V, Wagner GC, Ming X. Gastrointestinal dysfunction in children with autism spectrum disorders. Autism Res. (2014) 7:501–6. doi: 10.1002/aur.1386
8. Magalhães ES, Pinto-Mariz F, Bastos-Pinto S, Pontes AT, Prado EA, deAzevedo LC. Immune allergic response in Asperger syndrome. J Neuroimmunol. (2009) 216:108–12. doi: 10.1016/j.jneuroim.2009.09.015
9. Meltzer A, Van de Water J. The role of the immune system in autism spectrum disorder. Neuropsychopharmacol. (2016) 42:284–98. doi: 10.1038/npp.2016.158
10. Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, et al. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet. (2016) 98:149–64. doi: 10.1016/j.ajhg.2015.11.024
11. Parenti I, Rabaneda LG, Schoen H, Novarino G. Neurodevelopmental disorders: from genetics to functional pathways. Trends Neurosci. (2020) 43:608–21. doi: 10.1016/j.tins.2020.05.004
12. Allanson JE, Roberts AE. Noonan Syndrome. Seattle, WA: University of Washington (1993). Available online at: http://www.ncbi.nlm.nih.gov/pubmed/20301303 (accessed April 19, 2021).
13. Christodoulou J, Ho G. MECP2-Related Disorders. Seattle, WA: University of Washington (1993). Available online at: http://www.ncbi.nlm.nih.gov/pubmed/20301670 (accessed April 19, 2021).
14. den Hoed J, de Boer E, Voisin N, Dingemans AJM, Guex N, Wiel L, et al. Mutation-specific pathophysiological mechanisms define different neurodevelopmental disorders associated with SATB1 dysfunction. Am J Hum Genet. (2021) 108:346–56. doi: 10.1016/j.ajhg.2021.01.007
15. Richter JD, Zhao X. The molecular biology of FMRP: new insights into fragile X syndrome. Nat Rev Neurosci. (2021) 22:209–22. doi: 10.1038/s41583-021-00432-0
16. Martin JP, Bell J. A pedigree of mental defect showing. Sex-linkage. J Neurol Neurosurg Psychiatry. (1943) 6:154–7. doi: 10.1136/jnnp.6.3-4.154
17. Lubs HA. A marker X chromosome. Am J Hum Genet. (1969) 21:231–44. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1706424/ (accessed May 31, 2021).
18. Fu YH, Kuhl DPA, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. (1991) 67:1047–58. doi: 10.1016/0092-8674(91)90283-5
19. Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB, Moine H, Kooy RF, et al. Fragile X syndrome. Nat Rev Dis Prim. (2017) 3:17065. doi: 10.1038/nrdp.2017.65
20. Gross C, Hoffmann A, Bassell GJ, Berry-Kravis EM. Therapeutic strategies in fragile X syndrome: from bench to bedside and back. Neurotherapeutics. (2015) 12:584–608. doi: 10.1007/s13311-015-0355-9
21. Kaufmann WE, Kidd SA, Andrews HF, Budimirovic DB, Esler A, Haas-Givler B, et al. Autism spectrum disorder in fragile X syndrome: cooccurring conditions and current treatment. Pediatrics. (2017) 139:S194–206. doi: 10.1542/peds.2016-1159F
22. Niu M, Han Y, Dy ABC, Du J, Jin H, Qin J, et al. Hagerman RJ. Autism symptoms in fragile X syndrome. J Child Neurol. (2017) 32:903–9. doi: 10.1177/0883073817712875
23. Budimirovic DB, Kaufmann WE. What can we learn about autism from studying fragile X syndrome? Dev Neurosci. (2011) 33:379–94. doi: 10.1159/000330213
24. Budimirovic DB, Bukelis I, Cox C, Gray RM, Tierney E, Kaufmann WE. Autism spectrum disorder in Fragile X syndrome: differential contribution of adaptive socialization and social withdrawal. Am J Med Genet A. (2006) 140A:1814–1826. doi: 10.1002/ajmg.a.31405
25. Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. (2008) 16:666–72. doi: 10.1038/ejhg.2008.61
26. Hagerman RJ, Hull CE, Safanda JF, Carpenter I, Staley LW, O'Connor RA, et al. High functioning fragile X males: Demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am J Med Genet. (1994) 51:298–308. doi: 10.1002/ajmg.1320510404
27. Basuta K, Schneider A, Gane L, Polussa J, Woodruff B, Pretto D, et al. High functioning male with fragile X syndrome and fragile X-associated tremor/ataxia syndrome. Am J Med Genet A. (2015) 167:2154–61. doi: 10.1002/ajmg.a.37125
28. Chaste P, Betancur C, Gérard-Blanluet M, Bargiacchi A, Kuzbari S, Drunat S, et al. High-functioning autism spectrum disorder and fragile X syndrome: Report of two affected sisters. Mol Autism. (2012) 3:5. doi: 10.1186/2040-2392-3-5
29. Sherman SL, Kidd SA, Riley C, Berry-Kravis E, Andrews HF, Miller RM, et al. Forward: A registry and longitudinal clinical database to study fragile X syndrome. Pediatrics. (2017) 139:S183–93. doi: 10.1542/peds.2016-1159E
30. de Vries BBA, Fryns JP, Butler MG, Canziani F, Wesby-van Swaay E, van Hemel JO, et al. Clinical and molecular studies in fragile X patients with a Prader-Willi-like phenotype. J Med Genet. (1993) 30:761–6. doi: 10.1136/jmg.30.9.761
31. Muzar Z, Lozano R, Kolevzon A, Hagerman RJ. The neurobiology of the prader-willi phenotype of fragile x syndrome. Intractable Rare Dis Res. (2016) 5:255–61. doi: 10.5582/irdr.2016.01082
32. Lachiewicz AM, Dawson DV, Spiridigliozzi GA. Physical characteristics of young boys with fragile X syndrome: Reasons for difficulties in making a diagnosis in young males. Am J Med Genet. (2000) 92:229–36. doi: 10.1002/(SICI)1096-8628(20000605)92:4<229::AID-AJMG1>3.0.CO;2-K
33. Ciaccio C, Fontana L, Milani D, Tabano S, Miozzo M, Esposito S. Fragile X syndrome: a review of clinical and molecular diagnoses. Ital J Pediatr. (2017) 43:39. doi: 10.1186/s13052-017-0355-y
34. Lubala TK, Lumaka A, Kanteng G, Mutesa L, Mukuku O, Wembonyama S, et al. Fragile X checklists: A meta-analysis and development of a simplified universal clinical checklist. Mol Genet Genomic Med. (2018) 6:526–32. doi: 10.1002/mgg3.398
35. Berry-Kravis E, Hagerman R, Visootsak J, Budimirovic D, Kaufmann WE, Cherubini M, et al. Arbaclofen in fragile X syndrome: results of phase 3 trials. J Neurodev Disord. (2017) 9:6. doi: 10.1186/s11689-016-9181-6
36. Berry-Kravis E, Raspa M, Loggin-Hester L, Bishop E, Holiday D, Bailey DB. Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am J Intellect Dev Disabil. (2010) 115:461–72. doi: 10.1352/1944-7558-115.6.461
37. Kidd SA, Lachiewicz A, Barbouth D, Blitz RK, Delahunty C, McBrien D, et al. Fragile X syndrome: a review of associated medical problems. Pediatrics. (2014) 134:995–1005. doi: 10.1542/peds.2013-4301
38. Bailey DB, Raspa M, Olmsted M, Holiday DB. Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet A. (2008) 146A:2060–9. doi: 10.1002/ajmg.a.32439
39. Symons FJ, Byiers BJ, Raspa M, Bishop E, Bailey DB. Self-injurious behavior and fragile X syndrome: findings from the national fragile X survey. Am J Intellect Dev Disabil. (2010) 115:473–81. doi: 10.1352/1944-7558-115.6.473
40. Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of Fragile X syndrome. Biochem Pharmacol. 79, 632–646. doi: 10.1016/j.bcp.2009.09.023
41. Symons FJ, Shinde SK, Gilles E. Perspectives on pain and intellectual disability. J Intellect Disabil Res. (2008) 52:275–86. doi: 10.1111/j.1365-2788.2007.01037.x
42. Budimirovic DB, Schlageter A, Filipovic-Sadic S, Protic DD, Bram E, Mahone EM, et al. A genotype-phenotype study of high-resolution FMR1 nucleic acid and protein analyses in fragile X patients with neurobehavioral assessments. Brain Sci. (2020) 10:1–25. doi: 10.3390/brainsci10100694
43. Lewis P, Abbeduto L, Murphy M, Richmond E, Giles N, Bruno L, et al. Psychological well-being of mothers of youth with fragile X syndrome: syndrome specificity and within-syndrome variability. J Intellect Disabil Res. (2006) 50(Pt 12):894–904. doi: 10.1111/j.1365-2788.2006.00907.x
44. Wang T, Bray SM, Warren ST. New perspectives on the biology of fragile X syndrome. Curr Opin Genet Dev. (2012) 22:256–63. doi: 10.1016/j.gde.2012.02.002
45. Kennedy T, Rinker D, Broadie K. Genetic background mutations drive neural circuit hyperconnectivity in a fragile X syndrome model. BMC Biol. (2020) 18:817. doi: 10.1186/s12915-020-00817-0
46. Spencer CM, Alekseyenko O, Hamilton SM, Thomas AM, Serysheva E, Yuva-Paylor LA, et al. Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res. (2011) 4:40–56. doi: 10.1002/aur.168
47. Nolin SL, Glicksman A, Houck GE, Brown WT, Dobkin CS. Mosaicism in fragile X affected males. Am J Med Genet. (1994) 51:509–12. doi: 10.1002/ajmg.1320510444
48. Merenstein SA, Sobesky WE, Taylor AK, Riddle JE, Tran HX, Hagerman RJ. Molecular-clinical correlations in males with an expanded FMR1 mutation. Am J Med Genet. (1996) 64:388–94. doi: 10.1002/(SICI)1096-8628(19960809)64:2<388::AID-AJMG31>3.0.CO
49. Rousseau F, Heitz D, Tarleton, MacPherson J, Malmgren J, Dahl N, et al. A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: the first 2,253 cases. Am J Hum Genet. (1994) 55:225–37.
50. Aliaga SM, Slater HR, Francis D, Sart D, Du D, Li X, et al. Identification of males with cryptic fragile x alleles by methylation-Specific quantitative melt analysis. Clin Chem. (2016) 62:343–52. doi: 10.1373/clinchem.2015.244681
51. Pretto D, Yrigollen CM, Tang HT, Williamson J, Espinal G, Iwahashi CK, et al. Clinical and molecular implications of mosaicism in FMR1 full mutations. Front Genet. (2014) 5:1–11. doi: 10.3389/fgene.2014.00318
52. Jiraanont P, Kumar M, Tang HT, Espinal G, Hagerman PJ, Hagerman RJ, et al. Size and methylation mosaicism in males with Fragile X syndrome. Expert Rev Mol Diagn. (2017) 17:1023–32. doi: 10.1080/14737159.2017.1377612
53. Baker EK, Arpone M, Aliaga SM, Bretherton L, Kraan CM, Bui M, et al. Incomplete silencing of full mutation alleles in males with fragile X syndrome is associated with autistic features. Mol Autism. (2019) 10:1–13. doi: 10.1186/s13229-019-0271-7
54. García-Alegría E, Ibáñez B, Mínguez M, Poch M, Valiente A, Sanz-Parra A, et al. Analysis of FMR1 gene expression in female premutation carriers using robust segmented linear regression models. RNA. (2007) 13:756–62. doi: 10.1261/rna.206307
55. Loesch DZ, Huggins RM, Hagerman RJ. Phenotypic variation and FMRP levels in fragile X. Ment Retard Dev Disabil Res Rev. (2004) 10:31–41. doi: 10.1002/mrdd.20006
56. Budimirovic DB, Berry-Kravis E, Erickson CA, Hall SS, Hessl D, Reiss AL, et al. Updated report on tools to measure outcomes of clinical trials in fragile X syndrome. J Neurodev Disord. (2017) 9:14. doi: 10.1186/s11689-017-9193-x
57. Dobkin CS, Nolin SL, Cohen I, Sudhalter V, Bialer MG, Ding XH, et al. Tissue differences in fragile X mosaics: Mosaicism in blood cells may differ greatly from skin. Am J Med Genet. (1996) 64:296–301. doi: 10.1002/(SICI)1096-8628(19960809)64:2<296::AID-AJMG13>3.0.CO;2-A
58. Maddalena A, Yadvish KN, Spence WC, Howard-Peebles PN. A fragile X mosaic male with a cryptic full mutation detected in epithelium but not in blood. Am J Med Genet. (1996) 64:309–12. doi: 10.1002/(SICI)1096-8628(19960809)64:2<309::AID-AJMG15>3.0.CO;2-J
59. Tassone F, Hagerman RJ, Gane LW, Taylor AK. Strong similarities of theFMR1 mutation in multiple tissues: Postmortem studies of a male with a full mutation and a male carrier of a premutation. Am J Med Genet. (1999) 84:240–4. doi: 10.1002/(SICI)1096-8628(19990528)84:3<240::AID-AJMG15>3.0.CO;2-B
60. Schaefer TL, Davenport MH, Erickson CA. Emerging pharmacologic treatment options for fragile X syndrome. Appl Clin Genet. (2015) 8:75–93. doi: 10.2147/TACG.S35673
61. Pretto DI, Hunsaker MR, Cunningham CL, Greco CM, Hagerman RJ, Noctor SC, et al. Intranuclear inclusions in a fragile X mosaic male. Transl Neurodegener. (2013) 2:10. doi: 10.1186/2047-9158-2-10
62. De Graaff E, Willemsen R, Zhong N, De Die-Smulders CEM, Brown WT, Freling G, et al. Instability of the CGG repeat and expression of the FMRI protein in a male fragile X patient with a lung tumor. Am J Hum Genet. (1995) 57:609–18.
63. Berry-Kravis EM, Lindemann L, Jønch AE, Apostol G, Bear MF, Carpenter RL, et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat Rev Drug Discov. (2018) 17:280–98. doi: 10.1038/nrd.2017.221
64. Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol Mech Dis. (2012) 7:219–45. doi: 10.1146/annurev-pathol-011811-132457
65. Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. (2009) 46:266–71. doi: 10.1136/jmg.2008.063701
66. Jacquemont S, Curie A, Des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. (2011) 3:1708. doi: 10.1126/scitranslmed.3001708
67. Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J, et al. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med. (2016) 8:aab4109. doi: 10.1126/scitranslmed.aab4109
68. FRAXA Research Foundation. Fragile X Syndrome Research & Treatment. FRAXA Research Foundation - Finding a Cure for Fragile X. Available online at: https://www.fraxa.org/ (accessed April 23, 2021).
69. Baker EK, Arpone M, Kraan C, Bui M, Rogers C, Field M, et al. FMR1 mRnA from full mutation alleles is associated with ABc-c fX scores in males with fragile X syndrome. Sci Rep. (2020) 10:11701. doi: 10.1038/s41598-020-68465-6
70. Zynerba Pharmaceuticals. Zynerba Pharmaceuticals Provides Regulatory Update on ZygelTM in Fragile X Syndrome - Zynerba. Available online at: https://zynerba.com/zynerba-pharmaceuticals-provides-regulatory-update-on-zygel-in-fragile-x-syndrome/ (accessed April 23, 2021).
71. Berry-Kravis EM, Hessl D, Rathmell B, Zarevics P, Cherubini M, Walton-Bowen K, et al. Effects of STX209 (Arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. (2012) 4:152ra127. doi: 10.1126/scitranslmed.3004214
72. Luu S, Province H, Berry-Kravis E, Hagerman R, Hessl D, Vaidya D, et al. Response to placebo in fragile X syndrome clinical trials: an initial analysis. Brain Sci. (2020) 10:629. doi: 10.3390/brainsci10090629
73. Jacquemont S, Berry-Kravis E, Hagerman R, Von Raison F, Gasparini F, Apostol G, et al. The challenges of clinical trials in fragile X syndrome. Psychopharmacology. (2014) 231:1237–50. doi: 10.1007/s00213-013-3289-0
74. Erickson CA, Wink LK, Ray B, Early MC, Stiegelmeyer E, Mathieu-Frasier L, et al. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology. (2013) 228:75–84. doi: 10.1007/s00213-013-3022-z
75. Berry-Kravis E, Sumis A, Kim OK, Lara R, Wuu J. Characterization of potential outcome measures for future clinical trials in fragile X syndrome. J Autism Dev Disord. (2008) 38:1751–7. doi: 10.1007/s10803-008-0564-8
76. Paribello C, Tao L, Folino A, Berry-Kravis E, Tranfaglia M, Ethell IM, et al. Open-label add-on treatment trial of minocycline in fragile X syndrome. MC Neurol. (2010) 10:91. doi: 10.1186/1471-2377-10-91
77. Greiss Hess L, Fitzpatrick SE, Nguyen DV, Chen Y, Gaul KN, Schneider A, et al. A randomized, double-blind, placebo-controlled trial of low-dose sertraline in young children with fragile X syndrome. J Dev Behav Pediatr. (2016) 37:619–28. doi: 10.1097/DBP.0000000000000334
78. Berry-Kravis EM, Harnett MD, Reines SA, Reese MA, Ethridge LE, Outterson AH, et al. Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: a randomized, placebo-controlled, phase 2 clinical trial. Nat Med. (2021) 27:862–70. doi: 10.1038/s41591-021-01321-w
79. Bruno JL, Romano D, Mazaika P, Lightbody AA, Hazlett HC, Piven J, et al. Longitudinal identification of clinically distinct neurophenotypes in young children with fragile X syndrome. Proc Natl Acad Sci USA. (2017) 114:10767–72. doi: 10.1073/pnas.1620994114
80. Movaghar A, Page D, Scholze D, Hong J, Dawalt LS, Kuusisto F, et al. Artificial intelligence-assisted phenotype discovery of fragile X syndrome in a population-based sample. Genet Med. 23:1273–80. doi: 10.1038/s41436-021-01144-7
81. Mor-Shaked H, Eiges R. Modeling fragile? syndrome using human pluripotent stem cells. Genes. (2016) 7:100077. doi: 10.3390/genes7100077
Keywords: Fragile X syndrome, precision medicine-based treatments, autism spectrum disorders, biomarkers, monogenic disease, heterogeneity
Citation: Verdura E, Pérez-Cano L, Sabido-Vera R, Guney E, Hyvelin J-M, Durham L and Gomez-Mancilla B (2021) Heterogeneity in Fragile X Syndrome Highlights the Need for Precision Medicine-Based Treatments. Front. Psychiatry 12:722378. doi: 10.3389/fpsyt.2021.722378
Received: 08 June 2021; Accepted: 01 September 2021;
Published: 30 September 2021.
Edited by:
Randi Jenssen Hagerman, UC Davis, United StatesReviewed by:
William T. Brown, AOL, United StatesCopyright © 2021 Verdura, Pérez-Cano, Sabido-Vera, Guney, Hyvelin, Durham and Gomez-Mancilla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Pérez-Cano, bGF1cmEucGVyZXotY2Fub0BzdGFsaWNsYS5jb20=; Baltazar Gomez-Mancilla, YmFsdGF6YXIuZ29tZXotbWFuY2lsbGFAc3RhbGljbGEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.