 Qi Shao
Qi Shao Yiping Wu
Yiping Wu Tian Xu
Tian Xu Fafeng Cheng
Fafeng Cheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Psychiatry, 13 December 2021
Sec. Molecular Psychiatry
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.711835
Major depressive disorder (MDD), which is highly associated with non-alcoholic fatty liver disease (NAFLD), has complex pathogenic mechanisms. However, a limited number of studies have evaluated the mutual pathomechanisms involved in MDD and NAFLD development. Chronic stress-mediated elevations in glucocorticoid (GC) levels play an important role in the development of MDD-related NAFLD. Elevated GC levels can induce the release of inflammatory factors and changes in gut permeability. Elevated levels of inflammatory factors activate the hypothalamic–pituitary–adrenal (HPA) axis, which further increases the release of GC. At the same time, changes in gut permeability promote the release of inflammatory factors, which results in a vicious circle among the three, causing disease outbreaks. Even though the specific role of the thyroid hormone (TH) in this pathogenesis has not been fully established, it is highly correlated with MDD and NAFLD. Therefore, changing lifestyles and reducing psychological stress levels are necessary measures for preventing MDD-related NAFLD. Among them, GC inhibitors and receptor antagonists may be key in the alleviation of early and mid-term disease progression. However, combination medications may be important in late-stage diseases, but they are associated with various side effects. Traditional Chinese medicines have been shown to be potential therapeutic alternatives for such complex diseases.
Major depressive disorder (MDD) is characterized by loss of interest, difficulty in paying attention, decreased appetite, and suicidal ideation, among other abnormal cognitive, behavioral, and social functions, with low mood as the main symptom (1). Chronic stress is highly correlated with the onset of MDD (2). With societal developments and acceleration of the pace of life, stress due to various life pressures has significantly increased, leading to annually increasing MDD incidences. It is projected that by 2030, MDD will be the leading cause of the global disease burden (3). Long-term psychological stress plays an important role in initiating and mediating the occurrence and development of many major diseases (4–6). Non-alcoholic fatty liver (NAFL) disease (NAFLD), a clinicopathologic syndrome, is characterized by diffuse hepatic alveolar steatosis and fat storage in liver lobules, with the exception of alcohol and other definite liver-damaging factors, including NAFL and non-alcoholic steatohepatitis (NASH) (7, 8). However, the natural history of this disorder is unknown (9, 10). Studies have shown that NAFLD has the risk of progression to cirrhosis and cancer (11, 12). Not only is NAFLD the world's most common chronic liver disease, but it has also become a common cause of other liver diseases (8). Unfortunately, NAFLD and MDD mediate and promote the progression of each other (13, 14). Interactions between these diseases increase the complexity of pathomechanisms involved in the pathogenesis of these two kinds of diseases. By analyzing and summarizing social, clinical, and scientific literature, we elucidate on the relationship between MDD and NAFLD as well as the potential common pathogenic mechanisms. This review provides a basis for clinical prevention and treatment of NAFLD.
In a large study of relationships between psychological stress and NAFLD, elevated stress levels were associated with a high prevalence of NAFLD (15). This finding was confirmed in an assessment of U.S. adults from 2007 to 2016 by Kim et al., who found that MDD patients were 1.6 to 2.2 times more likely to have NAFLD, relative to those without MDD (16). In a study involving 567 biopsy-confirmed NAFLD patients, after adjusting for potential confounding factors, Youssef et al. found that there was a dose-dependent relationship between the severity of depressive symptoms and the degree of hepatocyte swelling. Compared with non-depressed patients, subclinical MDD patients were 2.1 times more likely to exhibit hepatocyte swelling, while clinical MDD patients were 3.6 times more likely (17). Meanwhile, mentally and intellectually impaired youth are at a greater risk of liver steatosis development (18), and more significantly, there is a significant correlation between psychological distress and liver disease-associated mortality. In their survey of 166,631 people, Russ et al. found that the risk of liver disease-associated mortality significantly increased with increasing General Health Questionnaire (GHQ) (a health questionnaire consisting of 12 items) score, and they divided the GHQ score into 0 (no pain), 1–3, 4–6, and 7–12 points. After adjustment for age and gender, the risk ratio for the 7–12 group compared with the 0 group was 3.48, while after adjustment of health behaviors, socioeconomic conditions, and so on, the hazard ratio reduced to 2.59 (19). Due to the complexities of human disease mechanisms and other uncontrollable factors, the processes involved in depression-induced NAFLD occurrence and development should be evaluated in animal experiments. Donkelaar et al. orally administered corticosterone in mice for a long time to simulate a physiological stress state. They found that the mice gained weight, their insulin sensitivity was decreased, and they also exhibited depressive-like behaviors (20). Liu et al. established chronic stress models by subjecting mice to electric foot shock and restraint stress. They found that chronic stress caused increased triglyceride (TG) and total cholesterol (TC) levels while decreasing body weight, visceral fat mass, microvesicular steatosis, lobular inflammation, and ballooning degeneration (21). These studies show that depression plays an important role in NAFLD occurrence and development.
Depressive states mediate NAFLD occurrence and development, while NAFLD aggravates depressive states. For example, in a study of a large primary care team in Germany, Labenz et al. found that NAFLD patients were more likely to suffer from MDD than the control group without NAFLD. Therefore, they considered NAFLD to be an independent risk factor for MDD and anxiety (22). Weinstein et al. reported that NAFLD patients had a significantly higher prevalence of depressive disorders (27.2%) than controls (2%−5%) (23). These findings have been validated by many studies. Filipović et al. found that compared with those of healthy patients, NAFLD patients' volumes of the brain, white, and gray matter reduced while lateral ventricles increased. Moreover, the risk of cognitive decline, cognitive impairment, and cognitive deficits was four times higher in patients without NAFLD (24). In their assessment of the relationship between MDDs, generalized anxiety disorders (GADs), and NASH, Elwing et al. found that MDD and GADs were more highly expressed in NASH patients than in the control group. Moreover, the degree of steatosis in MDD patients and inflammation grade as well as fibrosis stage in GAD patients were higher than those of patients without a diagnosis of mental illness (13). The hypothesis that NAFLD aggravates depression has been verified in animal experiments. To elucidate on the impact of liver disease on MDD, Higarza et al. conducted preclinical evaluations in NASH animal models to verify whether there were behavioral and emotional changes. One of the studies used a forced swimming test to evaluate depression-like rat behaviors; they found that NASH rat models exhibited a lack of escape struggle compared with the normal group, implying that NASH induces neurobehavioral dysfunctions (25).
The hypothalamic–pituitary–adrenal (HPA) axis is a neuroendocrine axis composed of the hypothalamus, pituitary gland, and adrenal gland. It plays a key role in regulating the response of organisms to environmental changes (26). Stress impulses are transmitted from the nerves to the brain and then to the hypothalamus of the central nervous system (CNS). They mainly act on the corticotropin-releasing factor (CRF) in the paraventricular nucleus (PVN) of the hypothalamus. CRF is synthesized by neurons dividing small cells in PVN (27). As the main neurohormone that regulates the HPA axis, it plays an important role in psychological stress (26). Synthetic CRF enters the anterior pituitary through the pituitary portal system (28), whereby it binds the CRF1 receptor and initiates signal transduction of protein kinase A (PKA) and protein kinase C (PKC) to promote the synthesis and secretion of the adrenocorticotropic hormone (ACTH) (26). ACTH stimulates the adrenal cortex to secrete cortisol and release it into the blood (28). Under normal physiological conditions, cortisol acts on the hippocampus, PVN, and anterior pituitary gland; then, through a negative feedback loop mediated by the glucocorticoid (GC) receptor (GR), it inhibits the HPA axis, thereby suppressing the synthesis and secretion of CRF as well as ACTH (29). However, long-term psychological stress can lead to continuous hyperactivity of the HPA axis and increases GC levels (30, 31). Excess GC can damage cortisol receptors in the hippocampus at the high position of the HPA axis, causing the HPA axis to lose negative feedback regulation and, ultimately, leading to a hyperactivated HPA axis that leads to abnormally high GC levels (27).
Monoamine hypothesis, the most common hypothesis in pathophysiology, describes the molecular mechanisms of MDD. Most commonly used antidepressants have been developed according to the monoamine hypothesis. This hypothesis describes changes in 5-hydroxytryptamine (5-HT), norepinephrine (NE), and dopamine (DA) in the synaptic cleft (32). However, the current antidepressants developed on the basis of this hypothesis have not achieved the desired results (33). Therefore, the inflammatory mediator hypothesis (34) and a series of neural-related hypotheses, such as the neuroplasticity hypothesis/neurogenesis hypothesis (32), the neurotrophic factor (NF) hypothesis (35), the neuron regeneration hypothesis (36), and the neurosynaptic hypothesis, have been developed (37). These hypotheses describe the complexity and heterogeneity of MDD mechanisms. GC may be key to proving the existing types of hypotheses and an important target for MDD treatment (29, 38, 39).
GC exerts both anti-inflammatory and proinflammatory actions. Body responses to GC depend on GC concentrations, physiological states of the body's immune systems, and duration of stimulation (acute or chronic), among others (40). GC regulates interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-6 (IL-8), nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3), tumor necrosis factor-α (TNF-α) and other inflammatory factors through the c-Jun N-terminal kinase (c-JNK), extracellular-regulated protein kinases (ERKs), phosphatidylinositol 3-kinase (PI3K), IκB kinase (IκK), nuclear factor kappa-B (NF-κB), and signal transducers and activators of transcription (STAT) pathways (41, 42). Meanwhile, elevated GC levels can suppress 5-HT synthesis by inhibiting the expressions of central tryptophan hydroxylase 2 (TPH2) (43) and can reduce NE synthesis (44). The HPA axis, GC, and hippocampal volumes have been shown to form a vicious circle in MDD patients: stress increases GC levels, and increased GC reduces the hippocampal volume by exerting negative effects on neuromorphologies (neuroplasticity hypothesis) and/or neurogenesis of dentate gyrus (neurogenesis hypothesis). The decrease in hippocampal volumes may lead to the failure of negative feedback of the HPA axis. Then, GC levels further increase, thereby aggravating the failure of the HPA axis, leading to MDD occurrence (32).
Second, GR can reduce the expressions of brain-derived NF (BDNF) by directly binding the restriction sequence upstream of exon IV (exon IV) (45). In addition, GC causes neuronal apoptosis and degeneration by affecting hippocampal neural stem cells (NSCs) and neural precursor cells (NPCs) (46). GC-regulated kinase 1 (SGK1) plays a key role in MDD. GC improves SGK1 activities by increasing the phosphorylation of SGK1 at serine 422 (Ser422) and threonine 256 (Thr256), leading to phosphorylation of GR at Ser203 and Ser211, thereby affecting its nuclear translocation and further leading to suppressed BDNF synthesis (33). In addition, SGK1 is involved in the regulation of the growth of dendrites (47), establishment of neuronal plasticity (48), and neuronal excitement (49), which may lead to MDD occurrence.
The pathogenesis of NAFLD has yet to be fully elucidated. The “second hit” theory is a classic theory to describe its pathogenesis (50). “The first hit” theory is centered on insulin resistance (IR), involving liver steatosis caused by the imbalance of fatty acid (FA) and TG metabolism in liver cells (51). “The second hit” theory is mainly centered on oxidative stress (OS), and it also involves liver inflammation, necrosis, and fibrosis that are a result of inflammatory cytokine production, lipid peroxidation, and mitochondrial dysfunction (52). Since the “second hit” theory does not fully explain NAFLD pathogenesis, studies have proposed the “multiple parallel hits” theory, which involves IR, lipotoxicity, inflammation, genetic polymorphism and epigenetics, adipokines, bile acid (BA), gut microbiota (GM), and environmental and dietary factors, among others (53). However, the “multiple parallel hits” theory cannot fully explain the interrelationship between complex mechanisms and is only considered to be a supplement to the original classic “second hit” theory. Among them, GC may be key in implementing these two theories (54).
First, GC is directly associated with IR. GC enhances the phosphorylations of p38MAPK and c-JNK via the mitogen-activated protein kinase (MAPK) pathway, induces the apoptosis of islet B cells, and destroys glucose sensitivity, thereby promoting gluconeogenesis. Its indirect products, ceramide and diglyceride, can directly affect signal transductions in pancreatic islets through PKC and PI3K pathways (55, 56). GC has also been shown to directly act on nuclear receptors to increase mRNA expressions of hormone-sensitive lipase (HSL) and fatty TG lipase (ATGL) to promote lipolysis (57) and increase concentrations of free FAs acids (FFAs) in the blood, leading to IR. In addition, GC can significantly reduce insulin-stimulated Akt phosphorylation by reducing protein expressions of insulin receptor substrate-1 (IRS-1), PI3K/Akt, and protein kinase B (PKB) (58). It also causes a decrease in expression levels of glucose transporter 1 (GLUT1) protein and reduces the translocation of insulin-stimulated GLUT4 (GLUT4) to the plasma membrane, thereby reducing insulin-stimulated glucose uptake (59), further leading to IR.
Second, the increase in FFA caused by a high level of GC induces the production of CYP2E1, a major enzyme in the microsomal oxidation system. CYP2E1 is the main source of reactive oxygen species (ROS), which is significantly increased in NAFLD patients and is positively correlated with steatosis degree (60, 61). This process may aggravate OS in the mitochondrial environment and further damage organelle functions (62). In addition, excessive release of stress-related corticosteroids can promote peripheral blood leukocyte migration to liver tissues and elevate proinflammatory cytokine levels, such as TNF-α and IL-6. These activated inflammatory cells produce more ROS, thereby exacerbating OS, causing inflammation and liver necrosis (63). Increased ROS activates NF-κB by directly interacting with the small G protein p21ras, and then activating the signaling pathways involved in transforming growth factor-β-activated kinase-1 (TAK1), PI3K, and MAPK 1 (MEK1), which further aggravates liver damage (12).
In hepatocytes, accumulated FFAs act as a danger-associated molecular pattern (DAMP), which produces IL-1β and TNF-α among others by activating the NALP3 inflammasome and inducing lysosomal translocation of Bax (a porin that can trigger lysosomal instability) to promote and maintain inflammation. At the same time, excess FFA can mediate cell apoptosis via various pathways, such as the intrinsic mitochondrial apoptotic pathway, endoplasmic reticulum stress apoptotic pathway, lysosomal apoptotic pathway, and proinflammatory cytokine apoptotic pathway, further aggravating liver damage (62). In addition, under stress conditions, hypothalamus-secreted GC can increase gut permeability (64). Increased gut permeability is closely correlated with NAFLD (see below for details).
Human endogenous GM is an important “organ” that provides nutrition, regulates epithelial development, and guides innate immunity. Moreover, it plays an important role in health and disease (65). GM, which is associated with the brain and liver, has recently been correlated with theories of the gut–brain axis (66), gut–liver axis (67), and gut–liver–brain axis (68).
In MDD animal models, chronic stress affected psychological as well as physiological responses, disrupted the GM (69), and increased gut permeability (70). Overgrowth of gut bacteria, enhanced gut permeability, and bacterial translocation can all lead to elevated lipopolysaccharide (LPS) levels (71–73). Toll-like receptors (TLRs) are pattern recognition receptors for LPS, which are widely present in cell membranes, cytoplasm, and nucleus. TLRs play an important role as bridges in GM-mediated inflammation (74) and are involved in various immune response processes in the body, especially in inducing and promoting inflammation (75). Compared with other TLR-like receptors, activation of TLR4 triggers proinflammatory transcription via 2 adaptor proteins, myeloid differentiation factor 88 (MyD88), and β-interferon-containing TIR domain adaptor protein (TRIF), which induces the activation of NF-κB, AP-1, and IRF3 (76). Activation of these transcription factors causes the release of proinflammatory cytokines, including IL-1β, TNF-α, IL-6, and CXCL10, and elevates cyclooxygenase-2 (COX-2) levels, resulting in activated proinflammatory signals; please refer to the following text in section Release of Inflammatory Factors and references of the details for this content (76). These factors may directly stimulate the HPA axis or inhibit the negative feedback loop to make it upregulated, leading to the release of GC and CRF (77–79). TLRs are expressed in peripheral immune cells and in CNS cells, such as microglia, astrocytes, neurons, and oligodendrocytes (80). Activation of TLR4 can shift microglia polarization toward the M1 phenotype, thereby inducing proinflammatory responses in the CNS (76), which in turn leads to depressive symptoms (80). Astrocytes promote MDD pathophysiology by enhancing serotonin reuptake; however, their potential as TLR-expressing cells in MDD has not been elucidated. Moreover, expressions of TLR4 in astrocytes have not been conclusively determined (80).
IL-1, a GM-produced cytokine in the circulatory system, induces the expressions of COX-2 in cerebrovascular cells (81). It catalyzes the conversion of arachidonic acid (AA) into prostaglandin G2 (PGG2) and further into prostaglandin H2 (PGH2) and then generates prostaglandins (PGs) under the action of cell-specific synthetase (82). Due to their small size and strong lipophilicity, PGs are able to translocate to the brain parenchyma, and their induced synthesis in the brain is necessary for IL-1 to activate the HPA axis (83). At the same time, GM may also exert its effects on the brain through the tryptophan metabolic pathway (84), which may be associated with the gut inflammation state (85–87). When immune activation occurs, inflammatory factors induce the activation of indoleamine-2,3-dioxygenase (IDO), which exists in the periphery. Then, levels of peripheral transient receptor potential (TRP) decrease, and it crosses the blood–brain barrier (BBB) to enter the brain. Meanwhile, the presence of proinflammatory cytokines overactivates the IDO enzyme in the brain, leading to enhanced TRP metabolism, which in turn causes TRP levels in the brain to decrease (88, 89). TRP is converted to 5-hydroxytryptophan (5-HTP) by TPH, which is the rate-limiting step in this pathway. Aromatic amino acid decarboxylase (AAAD) then converts 5-HTP to 5-HT (90). This pathway is closely associated with the occurrence of depression (91, 92).
A high amount of serotonin is localized in the gut, where it is synthesized from tryptophan in the enterochromaffin cells (ECs) of the gastrointestinal tract. It is also present in enteric nerves (84). The vagus nerve can transmit signals to the brain via the paracrine mode (93), thereby triggering depression-like behaviors. However, the specific mechanisms involved in this process remain undefined.
First, GM participates in NAFLD pathogenesis by regulating BA metabolic pathways (94). PXR can be activated by lithocholic acid (LCA), a secondary metabolite of BAs. Transgenic mice expressing a constitutively activated PXR exhibited hepatomegaly and marked hepatic steatosis. Treatment of mice with a PXR agonist elicited a similar effect (95). PXR-induced hepatic steatosis has been attributed to a combination of several mechanisms. First, PXR activation increases FA influx by inducing the expressions of FA translocase (FAT/CD36) in the liver (95), while suppressing FA oxidation by preventing the binding of Forkhead box A2 (FoxA2) to its target genes, such as Cpt1a and Hmgcs2, which are involved in lipid oxidation (96). BA can also bind CAR, an orphan nuclear receptor that is mainly expressed in the liver and small intestines, but its effects contrast with those of PXR. CAR agonist administration significantly suppressed or reversed the adiposity gain induced by carbohydrate-free diet (HFD) feeding in C57BL/6J mice without affecting the lean mass. This reduced adiposity was associated with improved serum lipid profiles and insulin sensitivity (97); however, the specific mechanism has not been established (98). BA can also bind S1P2R. S1P2R-associated activation of neurons led to increased expressions of the proinflammatory chemokine, CCL2, which in turn activated the microglia to secrete more proinflammatory cytokines (99).
After BA stimulation, the L-cell membrane receptor, TGR-5, can activate the accumulation of cytochrome c oxidase (COX) and cAMP, increasing the ATP/ADP ratio and cell oxygen consumption. BA promotes glucagon-like peptide-1 (GLP-1) secretion by opening the voltage-gated calcium (CAV) and closing the ATP-sensitive potassium channel (KATP) (100). On the one hand, GLP-1 regulates liver lipid metabolism by enhancing the expressions of FA oxidation genes (acyl-CoA oxidase 1 (AOX1) and peroxisome proliferator-activated receptor alpha (PPARα)) while inhibiting the expressions of sterol regulatory element-binding protein-1c (SREBP-1C) as well as stearoyl–CoA desaturase (SCD1) (101). On the other hand, it may inhibit the release of inflammatory factors (102), reducing endoplasmic reticulum stress (103) and OS (104) to improve IR, inhibit lipid synthesis, promote FA oxidation, reduce liver cell apoptosis, and further improve NAFLD. In the liver, activation of FXR by BAs upregulates the short heterodimer partner (SHP) and PPARα while inhibiting SREBP-1C, thereby suppressing the synthesis of TGs and very-low-density lipoproteins (VLDLs), while enhancing FA oxidation (105, 106).
GM can also affect the BA reabsorption pathway. Most of the conjugated BA (taurocholic acid, glycocholic acid, etc.) is actively reabsorbed by the apical sodium ion-dependent BA transporter on the brush border side of the distal ileum gut epithelial cells (ASBT), combined with ileal BA-binding protein (IBABP), transported to the basement membrane, and reabsorbed by the organic solute transporter (OST) α/β on the basolateral membrane to the portal vein bloodstream, passing through the basolateral hepatocyte sodium ion/sodium taurocholate cotransport peptide (NTCP) uptake on the membrane (107). GM inhibits the expressions of ASBT by stimulating the GATA4 transcription factor (108) and Farnesoid X receptor–SHP–fetoprotein transcription factor (FXR–SHP–FTF) cascade reaction (109), thereby suppressing BA reabsorption. In adipose tissue cells, TGR-5 promotes the conversion of T4 to T3 by upregulating the expressions of type 2 deiodinase (DiO2), thus accelerating metabolic rates in adipose tissues (110).
Second, GM overgrowth, enhanced gut wall permeability, and bacterial translocation all lead to the secretion of large amounts of LPS, which then enters the liver via the portal vein (71–73). Secreted LPS binds LBP in serum, which transmits it to CD14 to form a complex that then causes TLR4 dimerization via MDD-2 and, finally, activates TLR4 (111–113). TLR4 is expressed in various liver cells, such as Kupffer cells, liver cells, hepatic stellate cells (HSCs), bile duct epithelial cells, hepatic sinusoidal endothelial cells, and hepatic dendritic cells. Its activation induces the activation of transcription factors, such as NF-κB, AP-1, and IRF3, leading to the secretion of proinflammatory cytokines, IL-1β, IL-6, and TNF-α, as well as CXCL10, and to the upregulation of COX-2, thereby generating proinflammatory signals and triggering a series of responses. The LPS/TLR4 signaling pathway has been shown to induce inflammation, mediate OS, participate in IR and liver inflammatory damage as well as fibrosis repair, and play a key role in NAFLD pathogenesis (114). Kupffer cells are a type of macrophage that are located in hepatic sinuses and have the ability to phagocytize, process, and present antigens. Due to their special locations, they play an important role in liver defenses. They can effectively phagocytize pathogens entering via the portal vein and act as the first barriers against bacteria and related toxins from the gastrointestinal tract (115, 116). LPS, TLR4, and Kupffer cells play a key role in NAFLD (73, 117). Multiple receptors are expressed on the surface of the plasma membrane of Kupffer cells. LPS binds its receptor, CD14, to activate Kupffer cells to release inflammatory mediators. LPS can also bind TLR4 to activate NF-κB by causing calcium overload in Kupffer cells (118). By stimulating Kupffer cells to activate the TLR4 signaling pathway, LPS induces the secretion of TNF-α, IL-1, IL-6, IL-12, and IL-18 in Kupffer cells (119, 120), thereby triggering hepatic steatosis, IR, and hepatocyte apoptosis, among others.
In addition, GM promotes the release of gastrointestinal hormones through its metabolites (short-chain Fas, SCFAs), thereby regulating IR and insulin sensitivity through its transmembrane receptor G protein-coupled receptor 43 (GPR43) and G protein-coupled receptor 41 (GPR41). These effects induce an increase in intracellular Ca2+ and a decrease in cAMP via the G protein pathway Gq/11 and Gi/o (121, 122). GM disorders can also enhance the conversion of choline to toxic methylamine and reduce phosphatidylcholine levels as well as choline bioavailability in the blood. The liver can also metabolize methylamine to trimethylamine N-oxide (TMAO), which exposes the host to inflammatory toxic metabolites, resulting in liver manifestations similar to those of a choline-deficient diet (123, 124). Its metabolite, ethanol, can also promote gut permeability and aggravate liver injury (125–127).
Inflammatory cytokines, which are elevated in many diseases, are involved in inflammatory responses. After treatment, TNF-α levels in MDD patients have been found to be significantly lower than before (128). Elevated IL-6 levels are significantly positively correlated with psychological stress (129). IL-8 levels have been positively correlated with depressive symptoms and perceived stress, but negatively correlated with social support (130). In a previous study, average plasma IL-18 levels for patients with mental disorders were found to be 3.7 times higher than those of the control group (131). In chronic liver disease, elevated systemic levels of TNF-α are highly correlated with NAFLD severity (132), IL-6 levels are correlated with the degree of liver inflammation (133), and IL-8 levels are significantly correlated with NAFLD disease activity and fibrosis severity (134). Significant correlations between IL-18 and alanine aminotransferase (ALT), gamma glutamyltransferase (GGT), TG, high-sensitivity C-reactive protein (hsCRP), and the degree of liver steatosis have been reported (135). These findings suggest that inflammatory factors in MDD and NAFLD have important mediating roles, and they may be important links between the two conditions. However, the current studies have mainly focused on TNF-α and IL-6, with relatively few studies evaluating IL-8 and IL-18.
Currently, there are two types of inflammation hypotheses for MDD. One is that MDD is the product of neurotoxicity and neurodegeneration caused by inflammation, which is mainly related to the tryptophan degradation product (quinolinic acid, QUIN) and kynurenic acid caused by IDO activation. They can destroy post-process components and lead to serious nerve cell degeneration, including the death of hippocampal granular cells and cerebellum meridians selective necrosis (136, 137). Another hypothesis that emphasizes the power factor of stress states that it can micro-damage neurons, resulting in reduced NF release and enhancement of neuroinflammatory activities, and the induction of depressive symptoms (34).
TNF-α can directly stimulate CRF and overactivate the HPA axis, which in turn leads to the secretion of large amounts of ACTH and GC (138). Besides, it prevents the cortisol–GR complex from entering the nucleus by activating the JNK signaling pathway. It also induces the NF-κB signaling pathway to prevent the complex from binding to DNA, causing GR resistance, expression decrease, or functional decrease (139), which induces depression. Meanwhile, TNF-α affects CRF activity of the central amygdala, increases the uptake of 5-HT and NE in PVN (140), and causes the degradation of the 5-HT synthetic raw material-TRP by stimulating IDO (141, 142). At the same time, levels of the degradation product (QUIN) increase, which in turn suppresses NF while enhancing neurotoxicity (143). Moreover, it induces depression by activating the serotonin transporter (SERT) through the p38/MAPK pathway and by suppressing 5-HT (144). TNF-α affects synaptic plasticity and neuronal atrophy by inhibiting negative feedback regulation of the HPA axis through the JNK, P38, and NF-κB signaling pathways. However, the specific mechanisms related to apoptosis, survival, and MDD induced by these pathways and relationships with TNF-α concentrations should be evaluated further (145–147).
Since TNF-α and interferon-γ (IFN-γ) as well as other inflammatory factors can induce the synthesis and secretion of IL-6 (148), IL-6 can play a similar role to TNF-α by excessively activating the HPA axis (138) and stimulating IDO activation to degrade tryptophan and hinder the synthesis of N-acetyl-5-HT and melatonin (148). Detailed mechanisms have been previously described (149, 150). IL-6 can play both anti-inflammatory and proinflammatory functions. It exerts its anti-inflammatory effects by binding receptors on cell membrane surfaces. This process is referred to as classical signal transduction, but it only occurs on certain subsets of T cells, hepatocytes, megakaryocytes, neutrophils, and monocytes. When the soluble form of IL-6 receptor (sIL-6R) is detached from membrane-bound receptors and binds IL-6, it can be transported to any type of cells expressing membrane-bound glycoprotein 130 (gp130) to produce proinflammatory signals. This process is referred to as anti-signal transduction. In these two signal transduction processes, the IL-6/IL-6R/gp130 complex activates intracellular signal transduction through the JAK/STAT pathway and the MAPK pathway (151).
TNF-α is highly correlated with IR. It was the first proinflammatory factor to be associated with IR (152). It can inhibit IRS-1 tyrosine phosphorylation and downstream signal transduction by suppressing GKAP42 levels (153) and further inhibit GLUT4 translocation (154, 155), thereby inducing IR. It also induces the expression of Suppressor of Cytokine Signal Transduction 3 (SOCS3) to inhibit its downstream signals, JAK and STAT3, leading to IR (156, 157). In addition, it can damage adiponectin multimerization and, consequently, decrease adiponectin secretion by altering disulfide bond modification in the endoplasmic reticulum and increasing the sensitivity of hepatocytes to insulin (158).
Second, in addition to causing IR, TNF-α can also promote cell damage and apoptosis. After TNF-α binds its receptor (TNFR), it interacts with tumor necrosis factor receptor-related protein and death domain (TRADD), tumor necrosis factor receptor-related factor 2 (TRAF2), and receptor-interacting protein (RIPK). Together, they form TNF-α-induced signal complex I (complex I) and activate the NF-κB signaling pathway to form a proinflammatory and anti-apoptotic pathway (147), which may be affected by the IκK complex, pyruvate dehydrogenase kinase 4 (PDK4), and regulation of p65 to promote apoptosis (159, 160). Besides, if TRADD, TRAF2, and RIPK are separated from their receptors, they recruit fas-related death domain protein (FADD) and precaspase-8 (Caspase 8) to form a TNF-α-induced signal complex II (complex II), which in turn binds ROS regulatory factor 1 (Romo1) located in the mitochondria and recruits Bcl-xl protein to reduce the mitochondrial membrane potential and to activate ROS as well as JNK to promote cell apoptosis (147, 159). The mechanisms involved in the activation of the JNK signaling pathway and NF-κB pathway have been previously described (161).
In addition, TNF-α can also induce liver steatosis by inhibiting the activity of AMP-activated protein kinase (AMPK), activating SREBP-1c, and upregulating the expression of acetyl-Coenzyme A carboxylase (ACC) as well as FA synthase (FAS). These effects lead to increased FA synthesis and excessive accumulation of TG, which results in liver steatosis and direct induction of lipid accumulation in HepG2 cells (162, 163).
As mentioned earlier, TNF-α can induce the production and secretion of IL-6, which may associate with TNF-α to cause diseases and aggravate NAFLD (163). IL-6 has also been associated with phosphorylation of STAT3, ERK, and JNK to indirectly induce changes in AKT and mTOR-S6K signals. Suppressed IL-6 inhibits the phosphorylations of STAT3, ERK, and JNK and reverses the decline in p38 phosphorylation (164). Activated JNK, STAT3, ERK, and AKT signaling pathways are involved in IR and apoptosis (165–167). However, IL-6 has also been shown to play protective roles in chronic liver disease, and blocking IL-6 signaling can improve IR as well as liver damage (168, 169). These contradictory outcomes may be associated with the two contrasting pathways involved in IL-6-mediated anti-inflammatory and proinflammatory effects. However, studies should aim at elucidating these outcomes.
Alterations in the HPA axis and hypothalamic–pituitary–thyroid (HPT) axis are important neuroendocrine abnormalities in MDD. However, their associated mechanisms have not been established, and there is no unified theory on the relationship between MDD and thyroid hormone (TH) levels (170).
Most studies report that the prevalence of subclinical hypothyroidism (SCH) and obvious hypothyroidism in MDD patients is higher than that of the general population. About 4–40% of patients with affective disorders have SCH (171, 172), and the prevalence of SCH in female MDD patients is about twice that of males (173). However, some studies have reported contrasting conclusions to the effect that there is no correlation between SCH and depression (174, 175). Other studies have documented that the prevalence and incidence of hypothyroidism or hyperthyroidism in patients with severe MDD are higher than those of the general population (176). In addition, contradictory conclusions have been reported with regard to other aspects. For instance, some studies claim that the correlation between MDD and SCH risk is more obvious in people aged over 50 years (172), while another study reported that it is associated with younger patients (<60 years old), but not elderly patients (177).
Geography (178), diet (179), personal habits (180), depression scale scores, thyroid-stimulating hormone (TSH) ranges, and other indicators do not have a unified standard, which may lead to differences in results. Since 17.6% to 30% of patients with overt thyroid disease were reported to have SCH due to inadequate therapy, SCH or mild thyroid dysfunction in patients with MDD should be paid more attention to in clinical practice (171).
Hypothyroidism is closely associated with NAFLD (128, 181, 182). Hypothyroidism is a metabolic disease that is characterized by decreased serum TH and increased TSH levels (183), which may be related to lipid metabolism, OS, and mitochondrial dysfunction. Moreover, it mediates NAFLD occurrence and development.
TSH can bind TSH receptor (TSHR) on the surface of liver cells, mediate peripheral FFA into the cell through FA-binding proteins (L-FABPs), FA transfer proteins (FATPs), and FA translocators (FAT), and increase liver SREBP-1c activities through the cAMP/PKA/AMPK pathway. TSH has also been shown to increase liver gluconeogenesis and TG accumulation, inhibit BA synthesis, and cause high cholesterolemia by regulating the phosphorylation of HMG-CoA reductase (a cholesterol synthesis rate-limiting enzyme) and the activity of PPAR-α, thereby leading to NAFLD occurrence (184–187). The remaining FAs are released into the blood in the form of VLDLs and hydrolyzed by lipoprotein lipase (LPL) to become low-density lipoproteins (LDLs). LDL binds LDLR receptor to cell membranes and returns to the cytoplasm (188). TH can increase the expressions and enzyme activities of LPL. Suppressed TG clearance leads to accumulation of TG in the liver when peripheral blood TH decreases, which in turn leads to NAFLD (VLDL is rich in TG cholesterol, etc.) (188). In addition, TH promotes FA catabolism through liver lipid phagocytosis (autophagy of FAs in liver cells), transports lipids to lysosomes, and stimulates β-oxidation (189). Other hormones such as leptin, adiponectin, fibroblast growth factor-21, and sex hormones are all related to TH (188, 190).
Obesity is one of the most important public health challenges of the 21st century, reaching epidemic proportions. It affects adults, adolescents, and children of both genders (191). In addition to the established medical comorbidities, obesity is also associated with psychiatric challenges and the developments of many diseases, including cardiovascular diseases, diabetes, obstructive sleep apnea, NASH, osteoarthritis, and mobility-related disorders (192). Nevertheless, the pathogeneses for these diseases have not been fully established because these proximal mechanisms may be seriously affected by more distal behavioral and psychosocial factors (193).
With various studies establishing a link between obesity and MDD, these conditions are bidirectional. The presence of one increases the risk of developing the other. This may be related to genetics, alterations in systems involved in homeostatic adjustments (HPA axis, immuno-inflammatory activation, and neuroendocrine regulators of energy metabolism such as leptin, insulin, and microbiome), and brain circuitries integrating homeostatic and mood regulatory responses (193).
Long-term HPA axis hyperactivation has also been found in nearly half of adult obese persons (194). At a young age, an almost 10-fold increased risk of obesity has been observed in children with the highest long-term cortisol levels (195). This is a result of chronic inflammation that is typical of obesity, which may disrupt GR functioning. Proinflammatory cytokines activate elements of the cellular transduction cascades that impede GR nuclear translocation or interfere in GR interactions with response elements in gene promoters (196).
Chronic low-grade inflammation is a hallmark of obesity. White adipocytes infiltrated by macrophages and other immune cells produce proinflammatory cytokines (197). This peripheral immune activation can be translated via humoral, neural, and cellular pathways (198) into brain inflammation, as indicated by higher hippocampal and cortical expressions of cytokines in obesity animal models (199, 200).
The effects of leptin on moods may be exerted via different pathways: direct action on neurons via receptors expressed in the hippocampus and amygdala, enhancement of neurogenesis and neuroplasticity in the hippocampus and cortex, and modulation of the HPA axis and immune systems (201, 202). It has been hypothesized that leptin resistance (peripheral hyperleptinemia due to reduced central signaling) may constitute a phenotype risk of depression (203). Alterations in regional cerebral metabolism due to IR have been associated with memory and executive function impairments as well as neuronal damage in the hippocampus and medial prefrontal cortex (204, 205). Therefore, it has been postulated that insulin dysregulation plays a role in neuropsychiatric conditions such as dementia and depression (206).
Microbiota of the gastrointestinal tract is emerging as a key player in the pathophysiology of obesity. Alterations in microbiota are markers of local inflammation, which can increase gut permeability to bacteria that, in turn, contributes to the onset and progression of systemic inflammation.
Obesity is a primary antecedent to NAFLD, which makes them closely related (207). Up to 80% of NAFLD patients are obese, and only 16% of individuals with a normal body mass index (BMI) and without metabolic risk factors are obese (208, 209). Obesity is aggravated by lipid metabolism, systemic inflammation, intestinal microorganisms, and IR.
Fat accumulation in the liver is the first step in NAFLD development. The higher the visceral fat content, the greater the risk of metabolic abnormalities. Visceral adipose tissue (VAT) has an abundant vascular and neural distribution than in other parts, which makes its cell metabolism more active, sensitive to sympathetic nervous system regulation, and produce more FFAs (210). VAT is mostly found in mature and hypertrophic adipocytes. Compared with the small adipocytes, their FFA and TG uptake rates are low, which makes it easy to cause fat deposition (211).
Excess FFAs and chronic low-grade inflammation from VAT are two of the most important factors contributing to liver injury progression in NAFLD. In hepatocytes, FFAs are bound to coenzyme A as fatty acyl-CoAs to form hepatic TGs, which stimulate the reduction of insulin-induced glucose uptake as well as induce intracellular inflammation and IR (212, 213). The hyperinsulinemia and hyperglycemia that occur with IR create an imbalance in lipid input relative to output and promote hepatic steatosis (214).
The associations between GM and the pathogenesis of obesity as well as NAFLD have been reported (215–217). One mechanism is that colonic-derived SCFAs account for 10% of harvested energy from the diet, with acetate (being mainly obesogenic) being the main source of energy (218). Besides, acetate is the most substantially absorbed SCFA and is a substrate for lipogenesis and cholesterol synthesis in the liver and adipose tissue (219). GM can also decrease intestinal expressions of the adipose tissue LPL inhibitor fasting-induced adipose factor (FIAF). The net result is increased uptake of FAs in the adipose tissues and liver, favoring the expansion of the adipose tissue and hepatic steatosis (215).
In addition, GM contributes to the development and progression of NAFLD through several mechanisms: modulation of intestinal permeability by promoting endotoxemia as well as other microbe products that promote systemic and hepatic inflammation; modulation of choline metabolism (required for VLDL synthesis and export of lipids from the liver); generation of endogenous ethanol as well as other toxic products such as toxic compound TMAO; and modulation of BA homeostasis and FXR functions (220).
There is a significant correlation between MDD and NAFLD, and they mediate and promote each other, gradually forming a vicious circle, which is particularly obvious in patients with MDD-associated NAFLD. The most common biological feature for MDD patients is that long-term life pressure and psychological stress chronically elevated GC levels (33). Elevated GC can directly induce MDD; for another, the indirect products of inflammatory factors increase, and GM disturbance further promotes and aggravates MDD and NAFLD.
Therefore, we postulate that chronic elevations in GC levels may be key in the pathogenesis of MDD-related NAFLD. Long-term psychological stress can induce the increase and accumulation of GC. If not controlled, when GC concentrations reach a certain level, they can induce the release of inflammatory factors and change intestinal permeability (63). Elevated inflammatory cytokines activate the HPA axis (86), which elevates GC levels. Meanwhile, changes in gut permeability further promote the release of inflammatory factors (80), causing disease outbreaks. Changes in TH induce MDD and NAFLD, although interaction mechanisms and causality between GC and TH should be further studied (Figure 1).
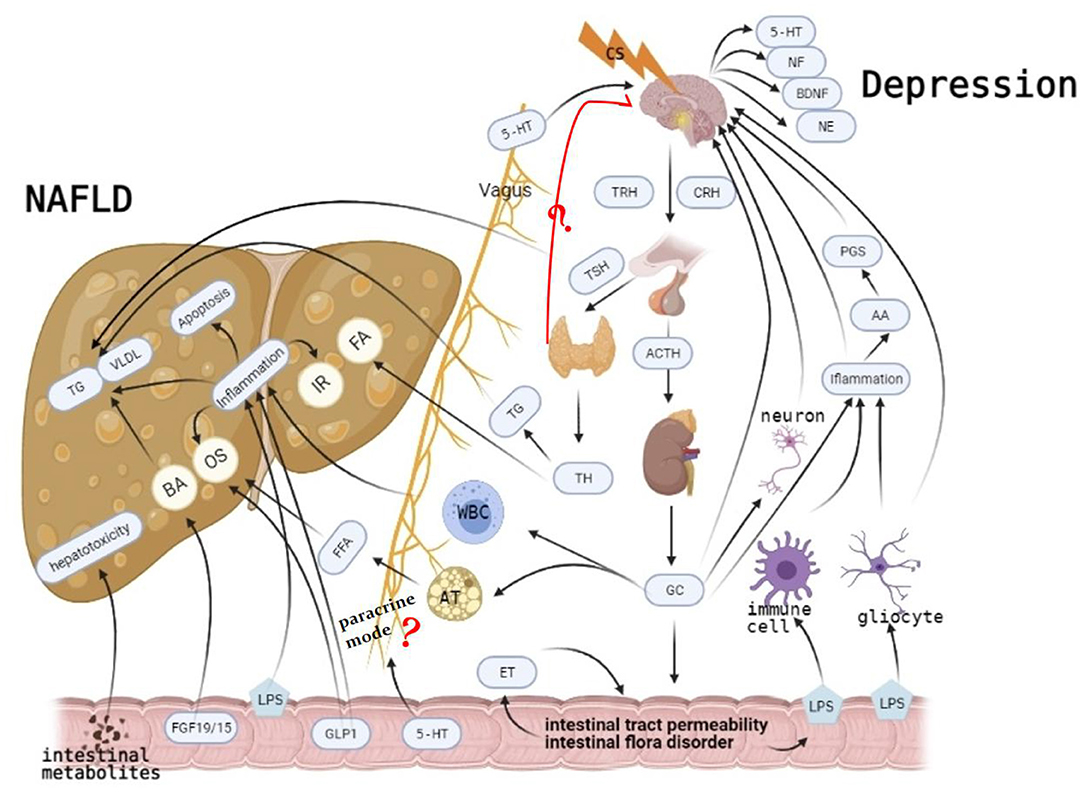
Figure 1. Long-term psychological stress induces the increase and accumulation of glucocorticoid (GC). On the one hand, it induces non-alcoholic fatty liver disease (NAFLD) by mediating liver transport of white blood cells and decomposition of fat cells. It also mediates major depressive disorder (MDD) occurrence by elevating inflammatory factors, damaging neurons, and inhibiting the negative feedback of the hypothalamic–pituitary–adrenal (HPA) axis. It also affects intestinal permeability and intestine flora, gradually leading to mutual promotions of NAFLD and MDD, causing disease outbreaks. Although more studies have shown that hypothyroidism is more related to MDD, there is still a big divergence between thyroid function and MDD. Its specific mechanism, which causes MD through paracrine mode, is still unclear. Similarly, the specific mechanism of MDD caused by intestinal flora through paracrine is not clear. Causal relationships between them should be further studied.
Not only is MDD related to NAFLD, but it is also associated with metabolic diseases such as diabetes. Since the concept of “microbe–gut–brain axis” was first put forward in 2012 (221), studies have begun to pay attention to the relationship between intestinal microbes and the brain. Mental disorders can lead to abnormal glucose metabolism and increase the risk of diabetes. However, diabetes can also directly or indirectly affect the function of the CNS, leading to the emergence of mental symptoms. Interactions between nervous system diseases and metabolic diseases increase the risk of complications and early death (222). This process may be highly related to the HPA axis (223, 224), monoamine neurotransmitters (225, 226), immune inflammation (227), cellular signaling pathway (228, 229), and the metabolic pathway (230, 231), among others. However, the specific mechanisms of action between the two systemic diseases have not been clarified.
Currently, there is no good management strategy for the cross talk between the two diseases. For example, the use of psychotropic drugs, including antidepressants, lithium salts, and antipsychotics, is associated with a higher prevalence of diabetes, among which clozapine, olanzapine, and quetiapine are correlated with the highest risks (232). If the patient has increased blood glucose levels, other antipsychotic drugs should be considered. In recent years, the integration of psychiatric nursing in diabetes education clinics has been considered as an effective management option for diabetes mellitus with depression (233). However, this mode of management needs further exploration.
In MDD-related NAFLD, we should prevent and treat from the source by improving our lifestyles and reducing our stress. Aerobic exercise is an effective way of stress reduction (234). People who exercise regularly have a higher level of mental health. Physical exercise can reduce work pressure and improve job satisfaction and happiness (235). Aerobic exercise has been shown to significantly improve serum ALT, aspartate aminotransferase (AST), and NAFLD activity scores in NAFLD patients and has a certain effect on TG (236, 237). In early and middle stages of the disease, GC inhibitors or antagonists may be key for the treatment of NAFLD. A few studies have investigated this perspective, although treatment of NAFLD from the perspective of GC has achieved significant effects (238). In the middle and late stages of the disease, GC, inflammatory factors, GM, and TH, among others, interact in a vicious circle. Combination medication may be an optional treatment. However, combined medications are associated with significant side effects. To reduce these risks, there is a need to ascertain the causal relationship between them as well as the dosage and order of medication.
The development of traditional Chinese medicine (TCM) has made it possible to treat complex diseases. In the TCM theory, the function of “liver governing free flow of qi” is an important theory for maintaining MDD and NAFLD, which involves the regulation of emotional response (nervous system) and digestive function (digestive system). The liver regulates the free flow of qi and prefers to act freely and dislikes depression, which will be destroyed by emotional dysfunction and cause qi stagnation, gradually leading to MDD. If liver qi stagnates for a long time, it induces liver inflammation, which leads to liver damage and then causes NAFLD development. In clinical medication, MDD and NAFLD can be treated with Sini powder (a prescription in TCM), which has been verified by relevant experiments and has achieved good therapeutic effects (239, 240). These outcomes may be highly related to its multi-target and multi-path characteristics (241–243). However, due to the complexities of TCM compounds and the possibility of chemical composition changes after processing (244), studies on Chinese medicine are challenging. We believe that this challenge will be easily solved with advances in science and technology.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
QS and YW: writing—original draft preparation. QS, TX, and QY: writing—article revision. CM, FC, and XW: writing—review and editing. XL and JJ: drawing—editing of picture. All authors have read and agreed to the published version of the manuscript.
This research was funded by the General Program of the National Natural Science Foundation of China (Nos. 81774122 and 82004327) and Key project of Beijing University of Chinese Medicine (No. 2020-JYB-ZDGG-004).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Epstein RM, Duberstein PR, Feldman MD, Rochlen AB, Bell RA, Kravitz RL, et al. “I didn't know what was wrong:” how people with undiagnosed depression recognize, name and explain their distress. J Gen Intern Med. (2010) 25:954–61. doi: 10.1007/s11606-010-1367-0
2. Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. (2008) 31:464–8. doi: 10.1016/j.tins.2008.06.006
3. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. (2006) 3:e442. doi: 10.1371/journal.pmed.0030442
4. Wang LJ, Chen CK, Hsu SC, Lee SY, Wang CS, Yeh WY. Active job, healthy job? Occupational stress and depression among hospital physicians in Taiwan. Ind Health. (2011) 49:173–184. doi: 10.2486/indhealth.MS1209
5. Cañas-González B, Fernández-Nistal A, Ramírez JM, Martínez-Fernández V. Influence of stress and depression on the immune system in patients evaluated in an anti-aging unit. Front Psychol. (2020) 11:1844. doi: 10.3389/fpsyg.2020.01844
6. Mizuno M, Siddique K, Baum M, Smith SA. Prenatal programming of hypertension induces sympathetic overactivity in response to physical stress. Hypertension. (2013) 61:180–6. doi: 10.1161/HYPERTENSIONAHA.112.199356
7. Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the United States and the rest of the world. Clin Liver Dis. (2016) 20:205–14. doi: 10.1016/j.cld.2015.10.001
8. Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of non-alcoholic fatty liver disease. World J Gastroenterol. (2017) 23:8263–76. doi: 10.3748/wjg.v23.i47.8263
9. Calzadilla Bertot L, Adams LA. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci. (2016) 17:774. doi: 10.3390/ijms17050774
10. Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of non-alcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. (1990) 11:74–80. doi: 10.1002/hep.1840110114
11. Adams LA, Roberts SK, Strasser SI, Mahady SE, Powell E, Estes C, et al. Non-alcoholic fatty liver disease burden: Australia, 2019-2030. J Gastroen Hepatol. (2020) 35:1628–35. doi: 10.1111/jgh.15009
12. Siddiqui MS, Carbone S, Vincent R, Patel S, Driscoll C, Celi FS, et al. Prevalence and severity of non-alcoholic fatty liver disease among caregivers of patients with non-alcoholic fatty liver disease cirrhosis. Clin Gastroenterol Hepatol. (2019) 17:2132–3. doi: 10.1016/j.cgh.2018.11.008
13. Elwing JE, Lustman PJ, Wang HL, Clouse RE. Depression, anxiety, and non-alcoholic steatohepatitis. Psychosom Med. (2006) 68:563–9. doi: 10.1097/01.psy.0000221276.17823.df
14. Macavei B, Baban A, Dumitrascu DL. Psychological factors associated with NAFLD/NASH: a systematic review. Eur Rev Med Pharmacol. (2016) 20:5081–97.
15. Kang D, Zhao D, Ryu S, Guallar E, Cho J, Lazo M, et al. Perceived stress and non-alcoholic fatty liver disease in apparently healthy men and women. Sci Rep. (2020) 10:38. doi: 10.1038/s41598-019-57036-z
16. Kim D, Yoo ER, Li AA, Tighe SP, Cholankeril G, Harrison SA, et al. Depression is associated with non-alcoholic fatty liver disease among adults in the United States. Aliment Pharm Ther. (2019) 50:590–598. doi: 10.1111/apt.15395
17. Youssef NA, Abdelmalek MF, Binks M, Guy CD, Omenetti A, Smith AD, et al. Associations of depression, anxiety and antidepressants with histological severity of non-alcoholic fatty liver disease. Liver Int. (2013) 33:1062–70. doi: 10.1111/liv.12165
18. Karaivazoglou K, Kalogeropoulou M, Assimakopoulos S, Triantos C. Psychosocial issues in pediatric non-alcoholic fatty liver disease. Psychosomatics. (2019) 60:10–7. doi: 10.1016/j.psym.2018.09.001
19. Russ TC, Kivimäki M, Morling JR, Starr JM, Stamatakis E, Batty GD. Association between psychological distress and liver disease mortality: a meta-analysis of individual study participants. Gastroenterology. (2015) 148:958–66. doi: 10.1053/j.gastro.2015.02.004
20. van Donkelaar EL, Vaessen KR, Pawluski JL, Sierksma AS, Blokland A, Cañete R, et al. Long-term corticosterone exposure decreases insulin sensitivity and induces depressive-like behaviour in the C57BL/6NCrl mouse. PLoS ONE. (2014) 9:e106960. doi: 10.1371/journal.pone.0106960
21. Liu YZ, Chen JK, Zhang Y, Wang X, Qu S, Jiang CL. Chronic stress induces steatohepatitis while decreases visceral fat mass in mice. BMC Gastroenterol. (2014) 14:106. doi: 10.1186/1471-230X-14-106
22. Labenz C, Huber Y, Michel M, Nagel M, Galle PR, Kostev K, et al. Non-alcoholic fatty liver disease increases the risk of anxiety and depression. Hepatol Commun. (2020) 4:1293–301. doi: 10.1002/hep4.1541
23. Weinstein AA, Kallman Price J, Stepanova M, Poms LW, Fang Y, Moon J, et al. Depression in patients with non-alcoholic fatty liver disease and chronic viral hepatitis B and C. Psychosomatics. (2011) 52:127–32. doi: 10.1016/j.psym.2010.12.019
24. Filipović B, Marković O, Ðurić V, Filipović B. Cognitive changes and brain volume reduction in patients with non-alcoholic fatty liver disease. Can J Gastroenterol Hepatol. (2018) 2018:9638797. doi: 10.1155/2018/9638797
25. Higarza SG, Arboleya S, Gueimonde M, Gómez-Lázaro E, Arias JL, Arias N. Neurobehavioral dysfunction in non-alcoholic steatohepatitis is associated with hyperammonemia, gut dysbiosis, and metabolic and functional brain regional deficits. PLoS ONE. (2019) 14:e223019. doi: 10.1371/journal.pone.0223019
26. Denver RJ. Structural and functional evolution of vertebrate neuroendocrine stress systems. Ann NY Acad Sci. (2009) 1163:1–16. doi: 10.1111/j.1749-6632.2009.04433.x
27. Thomson F, Craighead M. Innovative approaches for the treatment of depression: targeting the HPA axis. Neurochem Res. (2008) 33:691–707. doi: 10.1007/s11064-007-9518-3
28. Gragnoli C. Depression and type 2 diabetes: cortisol pathway implication and investigational needs. J Cell Physiol. (2012) 227:2318–22. doi: 10.1002/jcp.23012
29. Anacker C, Zunszain PA, Carvalho LA, Pariante CM. The glucocorticoid receptor: pivot of depression and of antidepressant treatment? Psychoneuroendocrinology. (2011) 36:415–25. doi: 10.1016/j.psyneuen.2010.03.007
30. Simpson EE, McConville C, Rae G, O'Connor JM, Stewart-Knox BJ, Coudray C, et al. Salivary cortisol, stress and mood in healthy older adults: the Zenith study. Biol Psychol. (2008) 78:1–9. doi: 10.1016/j.biopsycho.2007.12.001
31. Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, Kirschbaum C. HPA axis responses to laboratory psychosocial stress in healthy elderly adults, younger adults, and children: impact of age and gender. Psychoneuroendocrinology. (2004) 29:83–98. doi: 10.1016/S0306-4530(02)00146-4
32. Boku S, Nakagawa S, Toda H, Hishimoto A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiat Clin Neurosci. (2018) 72:3–12. doi: 10.1111/pcn.12604
33. Dattilo V, Amato R, Perrotti N, Gennarelli M. The emerging role of SGK1 (serum- and glucocorticoid-regulated kinase 1) in major depressive disorder: hypothesis and mechanisms. Front Genet. (2020) 11:826. doi: 10.3389/fgene.2020.00826
34. Wager-Smith K, Markou A. Depression: a repair response to stress-induced neuronal microdamage that can grade into a chronic neuroinflammatory condition? Neurosci Biobehav Rev. (2011) 35:742–64. doi: 10.1016/j.neubiorev.2010.09.010
35. Altar CA. Neurotrophins and depression. Trends Pharmacol Sci. (1999) 20:59–61. doi: 10.1016/S0165-6147(99)01309-7
36. Hanson ND, Owens MJ, Nemeroff CB. Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology. (2011) 36:2589–602. doi: 10.1038/npp.2011.220
37. Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. (2012) 338:68–72. doi: 10.1126/science.1222939
38. Herbert J. Cortisol and depression: three questions for psychiatry. Psychol Med. (2013) 43:449–69. doi: 10.1017/S0033291712000955
39. Fischer S, Strawbridge R, Vives AH, Cleare AJ. Cortisol as a predictor of psychological therapy response in depressive disorders: systematic review and meta-analysis. Brit J Psychiatry. (2017) 210:105–9. doi: 10.1192/bjp.bp.115.180653
40. Desmet SJ, De Bosscher K. Glucocorticoid receptors: finding the middle ground. J Clin Investig. (2017) 127:1136–45. doi: 10.1172/JCI88886
41. Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab. (2013) 24:109–19. doi: 10.1016/j.tem.2012.11.005
42. Zunszain PA, Anacker C, Cattaneo A, Carvalho LA, Pariante CM. Glucocorticoids, cytokines and brain abnormalities in depression. Prog Neuropsychophys. (2011) 35:722–9. doi: 10.1016/j.pnpbp.2010.04.011
43. Clark JA, Flick RB, Pai LY, Szalayova I, Key S, Conley RK, et al. Glucocorticoid modulation of tryptophan hydroxylase-2 protein in raphe nuclei and 5-hydroxytryptophan concentrations in frontal cortex of C57/Bl6 mice. Mol Psychiatr. (2008) 13:498–506. doi: 10.1038/sj.mp.4002041
44. Kvetnanský R, Fukuhara K, Pacák K, Cizza G, Goldstein DS, Kopin IJ. Endogenous glucocorticoids restrain catecholamine synthesis and release at rest and during immobilization stress in rats. Endocrinology. (1993) 133:1411–9. doi: 10.1210/endo.133.3.8396019
45. Chen H, Lombès M, Le Menuet D. Glucocorticoid receptor represses brain-derived neurotrophic factor expression in neuron-like cells. Mol Brain. (2017) 10:12. doi: 10.1186/s13041-017-0295-x
46. Kino T. Stress, glucocorticoid hormones, and hippocampal neural progenitor cells: implications to mood disorders. Front Physiol. (2015) 6:230. doi: 10.3389/fphys.2015.00230
47. Lang F, Artunc F, Vallon V. The physiological impact of the serum and glucocorticoid-inducible kinase SGK1. Curr Opin Nephrol Hypertens. (2009) 18:439–48. doi: 10.1097/MNH.0b013e32832f125e
48. Tai DJ, Su CC, Ma YL, Lee EH. SGK1 phosphorylation of IkappaB Kinase alpha and p300 Up-regulates NF-kappaB activity and increases N-Methyl-D-aspartate receptor NR2A and NR2B expression. J Biol Chem. (2009) 284:4073–89. doi: 10.1074/jbc.M805055200
49. Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. (2006) 86:1151–78. doi: 10.1152/physrev.00050.2005
50. Day CP, James OF. Steatohepatitis: a tale of two “hits?” Gastroenterology. (1998) 114:842–5. doi: 10.1016/S0016-5085(98)70599-2
51. Ganji SH, Kashyap ML, Kamanna VS. Niacin inhibits fat accumulation, oxidative stress, and inflammatory cytokine IL-8 in cultured hepatocytes: impact on non-alcoholic fatty liver disease. Metab Clin Exp. (2015) 64:982–90. doi: 10.1016/j.metabol.2015.05.002
52. Fang YL, Chen H, Wang CL, Liang L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: from “two hit theory” to “multiple hit model”. World J Gastroenterol. (2018) 24:2974–83. doi: 10.3748/wjg.v24.i27.2974
53. Thanapirom K, Tsochatzis EA. Non-alcoholic fatty liver disease (NAFLD) and the quest for effective treatments. Hepatobil Surg Nutr. (2019) 8:77–9. doi: 10.21037/hbsn.2018.11.06
54. Woods CP, Hazlehurst JM, Tomlinson JW. Glucocorticoids and non-alcoholic fatty liver disease. J Steroid Biochem Mol Biol. (2015) 154:94–103. doi: 10.1016/j.jsbmb.2015.07.020
55. Solas M, Gerenu G, Gil-Bea FJ, Ramírez MJ. Mineralocorticoid receptor activation induces insulin resistance through c-Jun N-terminal kinases in response to chronic corticosterone: cognitive implications. J Neuroendocrinol. (2013) 25:350–6. doi: 10.1111/jne.12006
56. Fransson L, Rosengren V, Saha TK, Grankvist N, Islam T, Honkanen RE, et al. Mitogen-activated protein kinases and protein phosphatase 5 mediate glucocorticoid-induced cytotoxicity in pancreatic islets and β-cells. Mol Cell Endocrinol. (2014) 383:126–36. doi: 10.1016/j.mce.2013.12.010
57. Bruder-Nascimento T, Da Silva MA, Tostes RC. The involvement of aldosterone on vascular insulin resistance: implications in obesity and type 2 diabetes. Diabetol Metab Syndr. (2014) 6:90. doi: 10.1186/1758-5996-6-90
58. Burén J, Liu HX, Jensen J, Eriksson JW. Dexamethasone impairs insulin signalling and glucose transport by depletion of insulin receptor substrate-1, phosphatidylinositol 3-kinase and protein kinase B in primary cultured rat adipocytes. Eur J Endocrinol. (2002) 146:419–29. doi: 10.1530/eje.0.1460419
59. Sakoda H, Ogihara T, Anai M, Funaki M, Inukai K, Katagiri H, et al. Dexamethasone-induced insulin resistance in 3T3-L1 adipocytes is due to inhibition of glucose transport rather than insulin signal transduction. Diabetes. (2000) 49:1700–8. doi: 10.2337/diabetes.49.10.1700
60. Chtioui H, Semela D, Ledermann M, Zimmermann A, Dufour JF. Expression and activity of the cytochrome P450 2E1 in patients with non-alcoholic steatosis and steatohepatitis. Liver Int. (2007) 27:764–71. doi: 10.1111/j.1478-3231.2007.01524.x
61. Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quiñones L, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci. (2004) 106:261–8. doi: 10.1042/CS20030285
62. Bessone F, Razori MV, Roma MG. Molecular pathways of non-alcoholic fatty liver disease development and progression. Cell Mol life Sci. (2019) 76:99–128. doi: 10.1007/s00018-018-2947-0
63. Joung JY, Cho JH, Kim YH, Choi SH, Son CG. A literature review for the mechanisms of stress-induced liver injury. Brain Behav. (2019) 9:e1235. doi: 10.1002/brb3.1235
64. Meddings JB, Swain MG. Environmental stress-induced gastrointestinal permeability is mediated by endogenous glucocorticoids in the rat. Gastroenterology. (2000) 119:1019–028. doi: 10.1053/gast.2000.18152
65. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. (2005) 308:1635–8. doi: 10.1126/science.1110591
66. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. (2013) 155:1451–63. doi: 10.1016/j.cell.2013.11.024
67. Miele L, Marrone G, Lauritano C, Cefalo C, Gasbarrini A, Day C, et al. Gut-liver axis and microbiota in NAFLD: insight pathophysiology for novel therapeutic target. Curr Pharm Design. (2013) 19:5314–24. doi: 10.2174/1381612811319290011
68. Victor DW, Quigley EM. Microbial therapy in liver disease: probiotics probe the microbiome-gut-liver-brain axis. Gastroenterology. (2014) 147:1216–8. doi: 10.1053/j.gastro.2014.10.023
69. Holdeman LV, Good IJ, Moore WE. Human fecal flora: variation in bacterial composition within individuals and a possible effect of emotional stress. Appl Environ Microb. (1976) 31:359–75. doi: 10.1128/aem.31.3.359-375.1976
70. Audet MC. Stress-induced disturbances along the gut microbiota-immune-brain axis and implications for mental health: does sex matter? Front Neuroendocrinol. (2019) 54:100772. doi: 10.1016/j.yfrne.2019.100772
71. Bergheim I, Weber S, Vos M, Krämer S, Volynets V, Kaserouni S, et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol. (2008) 48:983–92. doi: 10.1016/j.jhep.2008.01.035
72. Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, et al. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of non-alcoholic steatohepatitis. Am J Physiol. Gastrointestinal Liver Physiol. (2007) 292:G518–G25. doi: 10.1152/ajpgi.00024.2006
73. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
74. Velloso LA, Folli F, Saad MJ. TLR4 at the crossroads of nutrients, gut microbiota, and metabolic inflammation. Endocr Rev. (2015) 36:245–71. doi: 10.1210/er.2014-1100
75. Lorenz G, Anders HJ. Neutrophils, dendritic cells, toll-like receptors, and interferon-α in lupus nephritis. Semin Nephrol. (2015) 35:410–26. doi: 10.1016/j.semnephrol.2015.08.003
76. Liu J, Buisman-Pijlman F, Hutchinson MR. Toll-like receptor 4: innate immune regulator of neuroimmune and neuroendocrine interactions in stress and major depressive disorder. Front Neurosci. (2014) 8:309. doi: 10.3389/fnins.2014.00309
77. Kanczkowski W, Alexaki VI, Tran N, Großklaus S, Zacharowski K, Martinez A, et al. Hypothalamo-pituitary and immune-dependent adrenal regulation during systemic inflammation. Proc Natl Acad Sci USA. (2013) 110:14801–14806. doi: 10.1073/pnas.1313945110
78. Loum-Ribot E, Lafon P, Chaigniau M, Tramu G, Corio M. Glucocorticoids down-regulate lipopolysaccharide-induced de novo production of neurotensin mRNA in the rat hypothalamic, paraventricular, corticotrophin-releasing hormone neurons. Neuroimmunomodulatory. (2006) 13:170–8. doi: 10.1159/000098130
79. Goebel M, Stengel A, Wang L, Reeve J, Taché Y. Lipopolysaccharide increases plasma levels of corticotropin-releasing hormone in rats. Neuroendocrinology. (2011) 93:165–73. doi: 10.1159/000322590
80. Figueroa-Hall LK, Paulus MP, Savitz J. Toll-like receptor signaling in depression. Psychoneuroendocrinology. (2020) 121:104843. doi: 10.1016/j.psyneuen.2020.104843
81. Konsman JP, Vigues S, Mackerlova L, Bristow A, Blomqvist A. Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: relationship to patterns of inducible cyclooxygenase expression by peripheral inflammatory stimuli. J Comp Neurol. (2004) 472:113–29. doi: 10.1002/cne.20052
82. Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. (2004) 56:387–437. doi: 10.1124/pr.56.3.3
83. Schiltz JC, Sawchenko PE. Distinct brain vascular cell types manifest inducible cyclooxygenase expression as a function of the strength and nature of immune insults. J Neurosci. (2002) 22:5606–18. doi: 10.1523/JNEUROSCI.22-13-05606.2002
84. O'Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res. (2015) 277:32–48. doi: 10.1016/j.bbr.2014.07.027
85. Quak J, Doornbos B, Roest AM, Duivis HE, Vogelzangs N, Nolen WA, et al. Does tryptophan degradation along the kynurenine pathway mediate the association between pro-inflammatory immune activity and depressive symptoms? Psychoneuroendocrinology. (2014) 45:202–10. doi: 10.1016/j.psyneuen.2014.03.013
86. Dantzer R, O'Connor JC, Lawson MA, Kelley KW. Inflammation-associated depression: from serotonin to kynurenine. Psychoneuroendocrinology. (2011) 36:426–36. doi: 10.1016/j.psyneuen.2010.09.012
87. Christmas DM, Potokar J, Davies SJ. A biological pathway linking inflammation and depression: activation of indoleamine 2,3-dioxygenase. Neuropsychol Dis Treat. (2011) 7:431–9. doi: 10.2147/NDT.S17573
88. Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatr. (2002) 7:468–73. doi: 10.1038/sj.mp.4000995
89. Owe-Young R, Webster NL, Mukhtar M, Pomerantz RJ, Smythe G, Walker D, et al. Kynurenine pathway metabolism in human blood-brain-barrier cells: implications for immune tolerance and neurotoxicity. J Neurochem. (2008) 105:1346–57. doi: 10.1111/j.1471-4159.2008.05241.x
90. Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. (2009) 60:355–66. doi: 10.1146/annurev.med.60.042307.110802
91. Oxenkrug G. Serotonin-kynurenine hypothesis of depression: historical overview and recent developments. Curr Drug Targets. (2013) 14:514–21. doi: 10.2174/1389450111314050002
92. Maes M, Leonard BE, Myint AM, Kubera M, Verkerk R. The new '5-HT' hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry. (2011) 35:702–21. doi: 10.1016/j.pnpbp.2010.12.017
93. Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. (2007) 132:397–414. doi: 10.1053/j.gastro.2006.11.002
94. Wieland A, Frank DN, Harnke B, Bambha K. Systematic review: microbial dysbiosis and non-alcoholic fatty liver disease. Aliment Pharm Ther. (2015) 42:1051–63. doi: 10.1111/apt.13376
95. Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, et al. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem. (2006) 281:15013–20. doi: 10.1074/jbc.M511116200
96. Nakamura K, Moore R, Negishi M, Sueyoshi T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J Biol Chem. (2007) 282:9768–76. doi: 10.1074/jbc.M610072200
97. Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J Biol Chem. (2009) 284:25984–92. doi: 10.1074/jbc.M109.016808
98. Gao J, Xie W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol Sci. (2012) 33:552–8. doi: 10.1016/j.tips.2012.07.003
99. McMillin M, Frampton G, Grant S, Khan S, Diocares J, Petrescu A, et al. Bile acid-mediated sphingosine-1-phosphate receptor 2 signaling promotes neuroinflammation during hepatic encephalopathy in mice. Front Cell Neurosci. (2017) 11:191. doi: 10.3389/fncel.2017.00191
100. Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Bioph Res Commun. (2005) 329:386–90. doi: 10.1016/j.bbrc.2005.01.139
101. Ding X, Saxena NK, Lin S, Gupta NA, Gupta N, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. (2006) 43:173–81. doi: 10.1002/hep.21006
102. Zhang L, Yang M, Ren H, Hu H, Boden G, Li L, et al. GLP-1 analogue prevents NAFLD in ApoE KO mice with diet and Acrp30 knockdown by inhibiting c-JNK. Liver Int. (2013) 33:794–804. doi: 10.1111/liv.12120
103. Ao N, Yang J, Wang X, Du J. Glucagon-like peptide-1 preserves non-alcoholic fatty liver disease through inhibition of the endoplasmic reticulum stress-associated pathway. Hepatol Res. (2016) 46:343–53. doi: 10.1111/hepr.12551
104. Wang Z, Hou L, Huang L, Guo J, Zhou X. Exenatide improves liver mitochondrial dysfunction and insulin resistance by reducing oxidative stress in high fat diet-induced obese mice. Biochem Biophys Res Commun. (2017) 486:116–23. doi: 10.1016/j.bbrc.2017.03.010
105. Chow MD, Lee YH, Guo GL. The role of bile acids in non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Mol Aspects Med. (2017) 56:34–44. doi: 10.1016/j.mam.2017.04.004
106. Li Y, Tang R, Leung P, Gershwin ME, Ma X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun Rev. (2017) 16:885–96. doi: 10.1016/j.autrev.2017.07.002
107. Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. (2014) 66:948–83. doi: 10.1124/pr.113.008201
108. Out C, Patankar JV, Doktorova M, Boesjes M, Bos T, de Boer S, et al. Gut microbiota inhibit Asbt-dependent intestinal bile acid reabsorption via Gata4. J Hepatol. (2015) 63:697–704. doi: 10.1016/j.jhep.2015.04.030
109. Li H, Chen F, Shang Q, Pan L, Shneider BL, Chiang JY, et al. FXR-activating ligands inhibit rabbit ASBT expression via FXR-SHP-FTF cascade. Am J Physiol. (2005) 288:G60–6. doi: 10.1152/ajpgi.00170.2004
110. Chiang J, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol. (2020) 318:G554–73. doi: 10.1152/ajpgi.00223.2019
111. Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. (2007) 130:906–17. doi: 10.1016/j.cell.2007.08.002
112. Bryant CE, Spring DR, Gangloff M, Gay NJ. The molecular basis of the host response to lipopolysaccharide. Nat Rev Microbiol. (2010) 8:8–14. doi: 10.1038/nrmicro2266
113. Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. (2013) 45:e66. doi: 10.1038/emm.2013.97
114. Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol. (2013) 28:38–42. doi: 10.1111/jgh.12019
115. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. (2013) 3:785–97. doi: 10.1002/cphy.c120026
116. van der Heide D, Weiskirchen R, Bansal R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front Immunol. (2019) 10:2852. doi: 10.3389/fimmu.2019.02852
117. Wenfeng Z, Yakun W, Di M, Jianping G, Chuanxin W, Chun H. Kupffer cells: increasingly significant role in non-alcoholic fatty liver disease. Ann Hepatol. (2014) 13:489–95. doi: 10.1016/S1665-2681(19)31247-5
118. Jiang N, Zhang ZM, Liu L, Zhang C, Zhang YL, Zhang ZC. Effects of Ca2+ channel blockers on store-operated Ca2+ channel currents of Kupffer cells after hepatic ischemia/reperfusion injury in rats. World J Gastroenterol. (2006) 12:4694–8. doi: 10.3748/wjg.v12.i29.4694
119. Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, et al. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology. (2000) 31:932–6. doi: 10.1053/he.2000.5634
120. Seki E, Tsutsui H, Nakano H, Tsuji N, Hoshino K, Adachi O, et al. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J Immunol. (2001) 166:2651–7. doi: 10.4049/jimmunol.166.4.2651
121. Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. (2003) 278:25481–9. doi: 10.1074/jbc.M301403200
122. Ulven T. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front Endocrinol. (2012) 3:111. doi: 10.3389/fendo.2012.00111
123. Van Herck MA, Vonghia L, Francque SM. Animal models of non-alcoholic fatty liver disease-a starter's guide. Nutrients. (2017) 9:1072. doi: 10.3390/nu9101072
124. Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. (2014) 146:1513–24. doi: 10.1053/j.gastro.2014.01.020
125. Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. (2000) 32:742–7. doi: 10.1016/S0168-8278(00)80242-1
126. Farfán Labonne BE, Gutiérrez M, Gómez-Quiroz LE, Konigsberg Fainstein M, Bucio L, Souza V, et al. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol Toxicol. (2009) 25:599–609. doi: 10.1007/s10565-008-9115-5
127. Samak G, Gangwar R, Meena AS, Rao RG, Shukla PK, Manda B, et al. Calcium channels and oxidative stress mediate a synergistic disruption of tight junctions by ethanol and acetaldehyde in Caco-2 cell monolayers. Sci Rep. (2016) 6:38899. doi: 10.1038/srep38899
128. Lee BH, Park YM, Lee SH, Shim M. Serum levels of tumor necrosis factor-α and loudness dependence of auditory evoked potentials at pretreatment and posttreatment in patients with major depressive disorder. Brain Sci. (2019) 9:253. doi: 10.3390/brainsci9100253
129. Fumaz CR, Gonzalez-Garcia M, Borras X, Muñoz-Moreno JA, Perez-Alvarez N, Mothe B, et al. Psychological stress is associated with high levels of IL-6 in HIV-1 infected individuals on effective combined antiretroviral treatment. Brain Behav Immun. (2012) 26:568–572. doi: 10.1016/j.bbi.2012.01.001
130. Marsland AL, Sathanoori R, Muldoon MF, Manuck SB. Stimulated production of interleukin-8 covaries with psychosocial risk factors for inflammatory disease among middle-aged community volunteers. Brain Behav Immun. (2007) 21:218–28. doi: 10.1016/j.bbi.2006.07.006
131. Kokai M, Kashiwamura S, Okamura H, Ohara K, Morita Y. Plasma interleukin-18 levels in patients with psychiatric disorders. J Immunother. (2002) 25:S68–71. doi: 10.1097/00002371-200203001-00011
132. Paredes-Turrubiarte G, González-Chávez A, Pérez-Tamayo R, Salazar-Vázquez BY, Hernández VS, Garibay-Nieto N, et al. Severity of non-alcoholic fatty liver disease is associated with high systemic levels of tumor necrosis factor alpha and low serum interleukin 10 in morbidly obese patients. Clin Exp Med. (2016) 16:193–202. doi: 10.1007/s10238-015-0347-4
133. Leung C, Herath CB, Jia Z, Goodwin M, Mak KY, Watt MJ, et al. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J Hepatol. (2014) 60:832–8. doi: 10.1016/j.jhep.2013.11.033
134. Ajmera V, Perito ER, Bass NM, Terrault NA, Yates KP, Gill R, et al. Novel plasma biomarkers associated with liver disease severity in adults with non-alcoholic fatty liver disease. Hepatology. (2017) 65:65–77. doi: 10.1002/hep.28776
135. Flisiak-Jackiewicz M, Bobrus-Chociej A, Tarasów E, Wojtkowska M, Białokoz-Kalinowska I, Lebensztejn DM. Predictive role of interleukin-18 in liver steatosis in obese children. Can J Gastroenterol Hepatol. (2018) 2018:3870454. doi: 10.1155/2018/3870454
136. Maes M, Leonard B, Fernandez A, Kubera M, Nowak G, Veerhuis R, et al. (Neuro)inflammation and neuroprogression as new pathways and drug targets in depression: from antioxidants to kinase inhibitors. Prog Neuropsychopharmacol Biol Psychiatry. (2011) 35:659–63. doi: 10.1016/j.pnpbp.2011.02.019
137. Maes M, Yirmyia R, Noraberg J, Brene S, Hibbeln J, Perini G, et al. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. (2009) 24:27–53. doi: 10.1007/s11011-008-9118-1
138. Berthold-Losleben M, Himmerich H. The TNF-alpha system: functional aspects in depression, narcolepsy and psychopharmacology. Curr Neuropharmacol. (2008) 6:193–202. doi: 10.2174/157015908785777238
139. Postal M, Appenzeller S. The importance of cytokines and autoantibodies in depression. Autoimmun Rev. (2015) 14:30–5. doi: 10.1016/j.autrev.2014.09.001
140. Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci. (2009) 34:4–20.
141. Sublette ME, Postolache TT. Neuroinflammation and depression: the role of indoleamine 2,3-dioxygenase (IDO) as a molecular pathway. Psychosom Med. (2012) 74:668–72. doi: 10.1097/PSY.0b013e318268de9f
142. Liu YN, Peng YL, Liu L, Wu TY, Zhang Y, Lian YJ, et al. TNFα mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur Cytokine Netw. (2015) 26:15–25. doi: 10.1684/ecn.2015.0362
143. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatr. (2009) 65:732–41. doi: 10.1016/j.biopsych.2008.11.029
144. Malynn S, Campos-Torres A, Moynagh P, Haase J. The pro-inflammatory cytokine TNF-α regulates the activity and expression of the serotonin transporter (SERT) in astrocytes. Neurochem Res. (2013) 38:694–704. doi: 10.1007/s11064-012-0967-y
145. Soczynska JK, Kennedy SH, Goldstein BI, Lachowski A, Woldeyohannes HO, McIntyre RS. The effect of tumor necrosis factor antagonists on mood and mental health-associated quality of life: novel hypothesis-driven treatments for bipolar depression? Neurotoxicology. (2009) 30:497–521. doi: 10.1016/j.neuro.2009.03.004
146. Hollos P, Marchisella F, Coffey ET. JNK Regulation of depression and anxiety. Brain Plasticity. (2018) 3:145–55. doi: 10.3233/BPL-170062
147. Kim JJ, Lee SB, Park JK, Yoo YD. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ. (2010) 17:1420–34. doi: 10.1038/cdd.2010.19
148. Anderson G, Kubera M, Duda W, Lasoń W, Berk M, Maes M. Increased IL-6 trans-signaling in depression: focus on the tryptophan catabolite pathway, melatonin and neuroprogression. Pharmacol Rep. (2013) 65:1647–54. doi: 10.1016/S1734-1140(13)71526-3
149. Ting EY, Yang AC, Tsai SJ. Role of interleukin-6 in depressive disorder. Int J Mol Sci. (2020) 21:2194. doi: 10.3390/ijms21062194
150. Roohi E, Jaafari N, Hashemian F. On inflammatory hypothesis of depression: what is the role of IL-6 in the middle of the chaos? J Neuroinflamm. (2021) 18:45. doi: 10.1186/s12974-021-02100-7
151. Hodes GE, Ménard C, Russo SJ. Integrating Interleukin-6 into depression diagnosis and treatment. Neurobiol Stress. (2016) 4:15–22. doi: 10.1016/j.ynstr.2016.03.003
152. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
153. Ando Y, Shinozawa Y, Iijima Y, Yu BC, Sone M, Ooi Y, et al. Tumor necrosis factor (TNF)-α-induced repression of GKAP42 protein levels through cGMP-dependent kinase (cGK)-Iα causes insulin resistance in 3T3-L1 adipocytes. J Biol Chem. (2015) 290:5881–92. doi: 10.1074/jbc.M114.624759
154. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of non-alcoholic steatohepatitis: the central role of non-triglyceride fatty acid metabolites. Hepatology. (2010) 52:774–88. doi: 10.1002/hep.23719
155. Tilg H, Moschen AR. Evolution of inflammation in non-alcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. (2010) 52:1836–46. doi: 10.1002/hep.24001
156. Chen S, Kang Y, Sun Y, Zhong Y, Li Y, Deng L, et al. Deletion of Gab2 in mice protects against hepatic steatosis and steatohepatitis: a novel therapeutic target for fatty liver disease. J Mol Cell Biol. (2016) 8:492–504. doi: 10.1093/jmcb/mjw028
157. Bi J, Sun K, Wu H, Chen X, Tang H, Mao J. PPARγ alleviated hepatocyte steatosis through reducing SOCS3 by inhibiting JAK2/STAT3 pathway. Biochem Biophys Res Commun. (2018) 498:1037–44. doi: 10.1016/j.bbrc.2018.03.110
158. He Y, Lu L, Wei X, Jin D, Qian T, Yu A, et al. The multimerization and secretion of adiponectin are regulated by TNF-alpha. Endocrine. (2016) 51:456–68. doi: 10.1007/s12020-015-0741-4
159. Khan HA, Ahmad MZ, Khan JA, Arshad MI. Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance. Hepatobil Pancreat Dis Int. (2017) 16:245–56. doi: 10.1016/S1499-3872(17)60014-6
160. Wu J, Zhao Y, Park YK, Lee JY, Gao L, Zhao J, et al. Loss of PDK4 switches the hepatic NF-κB/TNF pathway from pro-survival to pro-apoptosis. Hepatology. (2018) 68:1111–24. doi: 10.1002/hep.29902
161. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. (2003) 10:45–65. doi: 10.1038/sj.cdd.4401189
162. You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. (2004) 127:1798–808. doi: 10.1053/j.gastro.2004.09.049
163. Lv Q, Zhen Q, Liu L, Gao R, Yang S, Zhou H, et al. AMP-kinase pathway is involved in tumor necrosis factor alpha-induced lipid accumulation in human hepatoma cells. Life Sci. (2015) 131:23–9. doi: 10.1016/j.lfs.2015.03.003
164. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. (2010) 140:197–208. doi: 10.1016/j.cell.2009.12.052
165. Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. (2013) 339:218–22. doi: 10.1126/science.1227568
166. Nazarians-Armavil A, Menchella JA, Belsham DD. Cellular insulin resistance disrupts leptin-mediated control of neuronal signaling and transcription. Mol Endocrinol. (2013) 27:990–1003. doi: 10.1210/me.2012-1338
167. Tsuchiya H, Ikeda Y, Ebata Y, Kojima C, Katsuma R, Tsuruyama T, et al. Retinoids ameliorate insulin resistance in a leptin-dependent manner in mice. Hepatology. (2012) 56:1319–30. doi: 10.1002/hep.25798
168. Kroy DC, Beraza N, Tschaharganeh DF, Sander LE, Erschfeld S, Giebeler A, et al. Lack of interleukin-6/glycoprotein 130/signal transducers and activators of transcription-3 signaling in hepatocytes predisposes to liver steatosis and injury in mice. Hepatology. (2010) 51:463–73. doi: 10.1002/hep.23322
169. Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Umemura A, et al. Blockade of IL-6 signaling exacerbates liver injury and suppresses antiapoptotic gene expression in methionine choline-deficient diet-fed db/db mice. Lab Investig. (2011) 91:609–18. doi: 10.1038/labinvest.2011.2
170. Min W, Liu C, Yang Y, Sun X, Zhang B, Xu L, et al. Alterations in hypothalamic-pituitary-adrenal/thyroid (HPA/HPT) axes correlated with the clinical manifestations of depression. Prog Neuropsychophys. (2012) 39:206–11. doi: 10.1016/j.pnpbp.2012.06.017
171. Lang X, Hou X, Shangguan F, Zhang XY. Prevalence and clinical correlates of subclinical hypothyroidism in first-episode drug-naive patients with major depressive disorder in a large sample of Chinese. J Affect Disord. (2020) 263:507–15. doi: 10.1016/j.jad.2019.11.004
172. Tang R, Wang J, Yang L, Ding X, Zhong Y, Pan J, et al. Subclinical hypothyroidism and depression: a systematic review and meta-analysis. Front Endocrinol. (2019) 10:340. doi: 10.3389/fendo.2019.00340
173. Zhao S, Chen Z, Wang X, Yao Z, Lu Q. Increased prevalence of subclinical hypothyroidism in female hospitalized patients with depression. Endocrine. (2021) 72:479–485. doi: 10.1007/s12020-020-02490-3
174. Wildisen L, Del Giovane C, Moutzouri E, Beglinger S, Syrogiannouli L, Collet TH, et al. An individual participant data analysis of prospective cohort studies on the association between subclinical thyroid dysfunction and depressive symptoms. Sci Rep. (2020) 10:19111. doi: 10.1038/s41598-020-75776-1
175. Kim JS, Zhang Y, Chang Y, Ryu S, Guallar E, Shin YC, et al. Subclinical hypothyroidism and incident depression in young and middle-age adults. J Clin Endocrinol Metab. (2018) 103:1827–33. doi: 10.1210/jc.2017-01247
176. Wu EL, Chien IC, Lin CH, Chou YJ, Chou P. Increased risk of hypothyroidism and hyperthyroidism in patients with major depressive disorder: a population-based study. J Psychosom Res. (2013) 74:233–7. doi: 10.1016/j.jpsychores.2012.12.016
177. Zhao T, Chen BM, Zhao XM, Shan ZY. Subclinical hypothyroidism and depression: a meta-analysis. Transl Psychiatr. (2018) 8:239. doi: 10.1038/s41398-018-0283-7
178. van Tonder AJ, Bray JE, Jolley KA, Jansen Van Rensburg M, Quirk SJ, Haraldsson G, et al. Genomic analyses of >3,100 nasopharyngeal pneumococci revealed significant differences between pneumococci recovered in four different geographical regions. Front Microbiol. (2019) 10:317. doi: 10.3389/fmicb.2019.00317
179. Herrmann MJ, Tesar AK, Beier J, Berg M, Warrings B. Grey matter alterations in obesity: a meta-analysis of whole-brain studies. Obesity Rev. (2019) 20:464–71. doi: 10.1111/obr.12799
180. Al-Khelaifi F, Donati F, Botrè F, Latiff A, Abraham D, Hingorani A, et al. Metabolic profiling of elite athletes with different cardiovascular demand. Scand J Med Sci Sports. (2019) 29:933–43. doi: 10.1111/sms.13425
181. Kim D, Vazquez-Montesino LM, Escober JA, Fernandes CT, Cholankeril G, Loomba R, et al. Low thyroid function in non-alcoholic fatty liver disease is an independent predictor of all-cause and cardiovascular mortality. Am J Gastroenterol. (2020) 115:1496–504. doi: 10.14309/ajg.0000000000000654
182. Mantovani A, Nascimbeni F, Lonardo A, Zoppini G, Bonora E, Mantzoros CS, et al. Association between primary hypothyroidism and non-alcoholic fatty liver disease: a systematic review and meta-analysis. Thyroid. (2018) 28:1270–84. doi: 10.1089/thy.2018.0257
183. Ferrandino G, Kaspari RR, Spadaro O, Reyna-Neyra A, Perry RJ, Cardone R, et al. Pathogenesis of hypothyroidism-induced NAFLD is driven by intra- and extrahepatic mechanisms. Proc Natl Acad Sci USA. (2017) 114:E9172–E80. doi: 10.1073/pnas.1707797114
184. Yan F, Wang Q, Lu M, Chen W, Song Y, Jing F, et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity. J Hepatol. (2014) 61:1358–64. doi: 10.1016/j.jhep.2014.06.037
185. Iqbal A, Jorde R, Figenschau Y. Serum lipid levels in relation to serum thyroid-stimulating hormone and the effect of thyroxine treatment on serum lipid levels in subjects with subclinical hypothyroidism: the Tromsø Study. J Intern Med. (2006) 260:53–61. doi: 10.1111/j.1365-2796.2006.01652.x
186. Zhang X, Song Y, Feng M, Zhou X, Lu Y, Gao L, et al. Thyroid-stimulating hormone decreases HMG-CoA reductase phosphorylation via AMP-activated protein kinase in the liver. J Lipid Res. (2015) 56:963–71. doi: 10.1194/jlr.M047654
187. Wu Q, Wang Q, Fu J, Ren R. Polysaccharides derived from natural sources regulate triglyceride and cholesterol metabolism: a review of the mechanisms. Food Funct. (2019) 10:2330–9. doi: 10.1039/C8FO02375A
188. Sinha RA, Singh BK, Yen PM. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat Rev Endocrinol. (2018) 14:259–69. doi: 10.1038/nrendo.2018.10
189. Lonardo A, Ballestri S, Mantovani A, Nascimbeni F, Lugari S, Targher G. Pathogenesis of hypothyroidism-induced NAFLD: evidence for a distinct disease entity? Digest Liver Dis. (2019) 51:462–70. doi: 10.1016/j.dld.2018.12.014
190. Tanase DM, Gosav EM, Neculae E, Costea CF, Ciocoiu M, Hurjui LL, et al. Hypothyroidism-induced non-alcoholic fatty liver disease (HIN): mechanisms and emerging therapeutic options. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21165927
191. Jaacks LM, Vandevijvere S, Pan A, McGowan CJ, Wallace C, Imamura F, et al. The obesity transition: stages of the global epidemic. Lancet Diabet Endocrinol. (2019) 7:231–40. doi: 10.1016/S2213-8587(19)30026-9
192. Chrostowska M, Szyndler A, Hoffmann M, Narkiewicz K. Impact of obesity on cardiovascular health. Best Pract Res Clin Endocrinol Metab. (2013) 27:147–56. doi: 10.1016/j.beem.2013.01.004
193. Milaneschi Y, Simmons WK, van Rossum E, Penninx BW. Depression and obesity: evidence of shared biological mechanisms. Mol Psychiatr. (2019) 24:18–33. doi: 10.1038/s41380-018-0017-5
194. Wester VL, Staufenbiel SM, Veldhorst MA, Visser JA, Manenschijn L, Koper JW, et al. Long-term cortisol levels measured in scalp hair of obese patients. Obesity. (2014) 22:1956–8. doi: 10.1002/oby.20795
195. Noppe G, van den Akker EL, de Rijke YB, Koper JW, Jaddoe VW, van Rossum EF. Long-term glucocorticoid concentrations as a risk factor for childhood obesity and adverse body-fat distribution. Int J Obesity. (2016) 40:1503–9. doi: 10.1038/ijo.2016.113
196. Pace TW, Miller AH. Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann NY Acad Sci. (2009) 1179:86–105. doi: 10.1111/j.1749-6632.2009.04984.x
197. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. (2012) 18:363–74. doi: 10.1038/nm.2627
198. Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol therapeut. (2011) 130:226–38. doi: 10.1016/j.pharmthera.2011.01.014
199. Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, et al. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. (2010) 219:25–32. doi: 10.1016/j.jneuroim.2009.11.010
200. Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, et al. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci. (2014) 34:2618–31. doi: 10.1523/JNEUROSCI.4200-13.2014
201. Farr OM, Tsoukas MA, Mantzoros CS. Leptin and the brain: influences on brain development, cognitive functioning and psychiatric disorders. Metab Clin Exp. (2015) 64:114–30. doi: 10.1016/j.metabol.2014.07.004
202. Bouret SG. Neurodevelopmental actions of leptin. Brain Res. (2010) 1350:2–9. doi: 10.1016/j.brainres.2010.04.011
203. Lu XY. The leptin hypothesis of depression: a potential link between mood disorders and obesity? Curr Opin Pharmacol. (2007) 7:648–52. doi: 10.1016/j.coph.2007.10.010
204. Kenna H, Hoeft F, Kelley R, Wroolie T, DeMuth B, Reiss A, et al. Fasting plasma insulin and the default mode network in women at risk for Alzheimer's disease. Neurobiol Aging. (2013) 34:641–9. doi: 10.1016/j.neurobiolaging.2012.06.006
205. Rasgon NL, Kenna HA, Wroolie TE, Kelley R, Silverman D, Brooks J, et al. Insulin resistance and hippocampal volume in women at risk for Alzheimer's disease. Neurobiol Aging. (2011) 32:1942–8. doi: 10.1016/j.neurobiolaging.2009.12.005
206. Rasgon NL, McEwen BS. Insulin resistance-a missing link no more. Mol Psychiatr. (2016) 21:1648–52. doi: 10.1038/mp.2016.162
207. Willis SA, Bawden S, Malaikah S, Sargeant JA, Stensel DJ, Aithal GP, et al. The role of hepatic lipid composition in obesity-related metabolic disease. Liver Int. (2021). doi: 10.1111/liv.15059. [Epub ahead of print].
208. Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, et al. Prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. (2011) 140:124–31. doi: 10.1053/j.gastro.2010.09.038
209. Bellentani S, Saccoccio G, Masutti F, Crocè LS, Brandi G, Sasso F, et al. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med. (2000) 132:112–7. doi: 10.7326/0003-4819-132-2-200001180-00004
210. Kwon H, Kim D, Kim JS. Body fat distribution and the risk of incident metabolic syndrome: a longitudinal cohort study. Sci Rep. (2017) 7:10955. doi: 10.1038/s41598-017-09723-y
211. Vishvanath L, Gupta RK. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J Clin Investig. (2019) 129:4022–4031. doi: 10.1172/JCI129191
212. Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. (2006) 55:S9–15. doi: 10.2337/db06-S002
213. Stefan N, Kantartzis K, Häring HU. Causes and metabolic consequences of Fatty liver. Endocr Rev. (2008) 29:939–60. doi: 10.1210/er.2008-0009
214. Asaoka Y, Terai S, Sakaida I, Nishina H. The expanding role of fish models in understanding non-alcoholic fatty liver disease. Dis Model Mech. (2013) 6:905–14. doi: 10.1242/dmm.011981
215. Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. (2004) 101:15718–23. doi: 10.1073/pnas.0407076101
216. Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, Hooper LV. The intestinal microbiota regulates body composition through NFIL and the circadian clock. Science. (2017) 357:912–6. doi: 10.1126/science.aan0677
217. Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, et al. Intestinal microbiota in patients with non-alcoholic fatty liver disease. Hepatology. (2013) 58:120–7. doi: 10.1002/hep.26319
218. Brahe LK, Astrup A, Larsen LH. Is butyrate the link between diet, intestinal microbiota and obesity-related metabolic diseases? Obesity Rev. (2013) 14:950–9. doi: 10.1111/obr.12068
219. Syed-Abdul MM, Parks EJ, Gaballah AH, Bingham K, Hammoud GM, Kemble G, et al. Fatty acid synthase inhibitor TVB-2640 reduces hepatic de novo lipogenesis in males with metabolic abnormalities. Hepatology. (2020) 72:103–18. doi: 10.1002/hep.31000
220. Machado MV, Cortez-Pinto H. Diet, Microbiota, Obesity, and NAFLD: a dangerous quartet. Int J Mol Sci. (2016) 17:481. doi: 10.3390/ijms17040481
221. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. (2012) 13:701–12. doi: 10.1038/nrn3346
222. Booth G, Cheng AY. Canadian Diabetes Association 2013 clinical practice guidelines for the prevention and management of diabetes in Canada. Methods Can J Diabetes. (2013) 37:S4–7. doi: 10.1016/j.jcjd.2013.01.010
223. Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev. (2005) 4:141–94. doi: 10.1016/j.arr.2005.03.003
224. Luo Y, Zeng B, Zeng L, Du X, Li B, Huo R, et al. Gut microbiota regulates mouse behaviors through glucocorticoid receptor pathway genes in the hippocampus. Transl Psychiatr. (2018) 8:187. doi: 10.1038/s41398-018-0240-5
225. Pan JX, Deng FL, Zeng BH, Zheng P, Liang WW, Yin BM, et al. Absence of gut microbiota during early life affects anxiolytic Behaviors and monoamine neurotransmitters system in the hippocampal of mice. J Neurol Sci. (2019) 400:160–8. doi: 10.1016/j.jns.2019.03.027
226. Gardier AM. Antidepressant activity: contribution of brain microdialysis in knock-out mice to the understanding of BDNF/5-HT transporter/5-HT autoreceptor interactions. Front Pharmacol. (2013) 4:98. doi: 10.3389/fphar.2013.00098
227. Zhang Y, Huang R, Cheng M, Wang L, Chao J, Li J, et al. Gut microbiota from NLRP3-deficient mice ameliorates depressive-like behaviors by regulating astrocyte dysfunction via circHIPK2. Microbiome. (2019) 7:116. doi: 10.1186/s40168-019-0733-3
228. Liu L, Wang H, Yu Y, Zeng B, Rao X, Chen J, et al. Microbial regulation of a lincRNA-miRNA-mRNA network in the mouse hippocampus. Epigenomics. (2020) 12:1377–87. doi: 10.2217/epi-2019-0307
229. Chevalier G, Siopi E, Guenin-Macé L, Pascal M, Laval T, Rifflet A, et al. Effect of gut microbiota on depressive-like behaviors in mice is mediated by the endocannabinoid system. Nat Commun. (2020) 11:6363. doi: 10.1038/s41467-020-19931-2
230. Li B, Guo K, Zeng L, Zeng B, Huo R, Luo Y, et al. Metabolite identification in fecal microbiota transplantation mouse livers and combined proteomics with chronic unpredictive mild stress mouse livers. Transl Psychiatr. (2018) 8:34. doi: 10.1038/s41398-017-0078-2
231. Yang J, Zheng P, Li Y, Wu J, Tan X, Zhou J, et al. Landscapes of bacterial and metabolic signatures and their interaction in major depressive disorders. Sci Adv. (2020) 6:eaba8555. doi: 10.1126/sciadv.aba8555
232. Vancampfort D, Correll CU, Galling B, Probst M, De Hert M, Ward PB, et al. Diabetes mellitus in people with schizophrenia, bipolar disorder and major depressive disorder: a systematic review and large scale meta-analysis. World Psychiatry. (2016) 15:166–174. doi: 10.1002/wps.20309
233. Wong J, Mehta G. Efficacy of depression management in an integrated psychiatric-diabetes education clinic for comorbid depression and diabetes mellitus types 1 and 2. Can J Diabetes. (2020) 44:455–60. doi: 10.1016/j.jcjd.2020.03.013
234. O'Keefe EL, O'Keefe JH, Lavie CJ. Exercise counteracts the cardiotoxicity of psychosocial stress. Mayo Clin Proc. (2019) 94:1852–64. doi: 10.1016/j.mayocp.2019.02.022
235. Gil-Beltrán E, Meneghel I, Llorens S, Salanova M. Get vigorous with physical exercise and improve your well-being at work!. Int J Env Res Pub Health. (2020) 17:6384. doi: 10.3390/ijerph17176384
236. Katsagoni CN, Georgoulis M, Papatheodoridis GV, Panagiotakos DB, Kontogianni MD. Effects of lifestyle interventions on clinical characteristics of patients with non-alcoholic fatty liver disease: a meta-analysis. Metab Clin Exp. (2017) 68:119–32. doi: 10.1016/j.metabol.2016.12.006
237. Centis E, Moscatiello S, Bugianesi E, Bellentani S, Fracanzani AL, Calugi S, et al. Stage of change and motivation to healthier lifestyle in non-alcoholic fatty liver disease. J Hepatol. (2013) 58:771–7. doi: 10.1016/j.jhep.2012.11.031
238. Koorneef LL, van den Heuvel JK, Kroon J, Boon MR, T Hoen P, Hettne KM, et al. Selective glucocorticoid receptor modulation prevents and reverses non-alcoholic fatty liver disease in male mice. Endocrinology. (2018) 159:3925–36. doi: 10.1210/en.2018-00671
239. Cheng F, Ma C, Wang X, Zhai C, Wang G, Xu X, et al. Effect of traditional Chinese medicine formula Sinisan on chronic restraint stress-induced non-alcoholic fatty liver disease: a rat study. BMC Complem Altern Med. (2017) 17:203. doi: 10.1186/s12906-017-1707-2
240. Zhu F, Li YM, Feng TT, Wu Y, Zhang HX, Jin GY, et al. Freeze-dried Si-Ni-San powder can ameliorate high fat diet-induced non-alcoholic fatty liver disease. World J Gastroenterol. (2019) 25:3056–3068. doi: 10.3748/wjg.v25.i24.3056
241. Wang B, Lu S, Zhang C, Zhu L, Li Y, Bai M, et al. Quantitative proteomic analysis of the liver reveals antidepressant potential protein targets of Sinisan in a mouse CUMS model of depression. Biomed Pharmacother. (2020) 130:110565. doi: 10.1016/j.biopha.2020.110565
242. Li B, Rui J, Ding X, Chen Y, Yang X. Deciphering the multicomponent synergy mechanisms of SiNiSan prescription on irritable bowel syndrome using a bioinformatics/network topology based strategy. Phytomedicine. (2019) 63:152982. doi: 10.1016/j.phymed.2019.152982
243. Shu Z, He W, Shahen M, Guo Z, Shu J, Wu T, et al. Clarifying of the potential mechanism of Sinisan formula for treatment of chronic hepatitis by systems pharmacology method. Biomed Pharmacother. (2018) 100:532–50. doi: 10.1016/j.biopha.2018.02.047
244. Zhou J, Cai H, Tu S, Duan Y, Pei K, Xu Y, et al. Identification and analysis of compound profiles of sinisan based on 'individual herb, herb-pair, herbal formula' before and after processing using UHPLC-Q-TOF/MS coupled with multiple statistical strategy. Molecules. (2018) 23:3128. doi: 10.3390/molecules23123128
Keywords: chronic stress, MD, NAFLD, GC, inflammatory factors, gut permeability, TH
Citation: Shao Q, Wu Y, Ji J, Xu T, Yu Q, Ma C, Liao X, Cheng F and Wang X (2021) Interaction Mechanisms Between Major Depressive Disorder and Non-alcoholic Fatty Liver Disease. Front. Psychiatry 12:711835. doi: 10.3389/fpsyt.2021.711835
Received: 22 July 2021; Accepted: 11 November 2021;
Published: 13 December 2021.
Edited by:
Yi-Lang Tang, Emory University, United StatesReviewed by:
Matthew McMillin, University of Texas at Austin, United StatesCopyright © 2021 Shao, Wu, Ji, Xu, Yu, Ma, Liao, Cheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fafeng Cheng, ZmFmZW5nY2hlbmdAZ21haWwuY29t; Xueqian Wang, c2hpcmx5ZGluZ0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.