
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 26 July 2021
Sec. Molecular Psychiatry
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.677039
This article is part of the Research Topic Bridging Neuro-Immune System and Gut Microbiota in Neuropsychiatric Disorders View all 4 articles
 Peifen Zhang1
Peifen Zhang1 Huimin Huang2Xingle Gao1Jiajun Jiang1Caixi Xi1Lingling Wu1Yaoyang Fu1
Huimin Huang2Xingle Gao1Jiajun Jiang1Caixi Xi1Lingling Wu1Yaoyang Fu1 Jianbo Lai1,3,4
Jianbo Lai1,3,4 Shaohua Hu1,3,4*
Shaohua Hu1,3,4*Bipolar disorder (BD) is a severe affective disorder, mainly characterized by alternative depressive and manic or hypomanic episodes, yet the pathogenesis of BD has not been fully elucidated. Recent researches have implicated the altered kynurenine (KYN) metabolism involved in the neurobiology of BD. Excessive activation of the immune system also occurs in patients with BD, which further accelerates the KYN pathway for tryptophan metabolism. Changes of the KYN metabolites have effects on neuronal receptors and are involved in neuroendocrine transmissions. Interactions between KYN metabolism and the immune system may contribute to the neuropathogenesis of BD. Various studies have shown that alterations of the KYN metabolites were associated with mood, psychotic symptoms, and cognitive functions in patients with BD. In this review, we briefly introduce the KYN pathway and describe the immune dysregulation in BD as well as their interactions. We then focus on the research advances on the KYN metabolism in BD, which hold promise for identifying novel treatment targets in patients stricken with this disorder.
Bipolar disorder (BD) is a chronic and recurrent mental disorder, characterized by alternating episodes of depression and mania or hypomania, along with significant cognitive impairments (1). It is estimated that ~1% population worldwide is affected and the lifetime prevalence ranges from 0.6 to 2.4% (2, 3). The onset age of BD is mainly during the adolescence (4). Most patients with BD firstly present with depressive episodes during the illness course and its differential diagnosis with major depressive disorder (MDD) remains clinically challenging. Due to the lack of typical markers, the confirmed diagnosis of BD is largely based on clinical experience and may be delayed for 5 or even 10 years (5, 6), thus contributing to the unfavorable prognosis and suicidal risks (7). However, the pathogenesis of BD has not been fully elucidated yet.
Previous studies showed that immune dysfunctions, such as elevated inflammatory and proinflammatory factors, were accompanied by the progression of BD or even occurred before the onset of illness (8, 9). As indicated by two landmark studies, administration of lipopolysaccharide (LPS) failed to elicit depressive-like behaviors when the stimulation of the kynurenine (KYN) pathway was blocked, despite that the levels of proinflammatory cytokines were elevated (10, 11). Immune activation could contribute to the metabolism of tryptophan (TRP) by shifting catabolic routes toward the KYN pathway (12). Proinflammatory responses and the mood status-related immune stimulation also trigger the KYN pathway in BD (13), predominantly via activating the key enzymes, such as indoleamine 2,3-dioxygenase (IDO) (14). Subsequently, various metabolites act on downstream receptors, including the N-methyl-D aspartate (NMDA) receptor and non-competitively α7 nicotinic acetylcholine receptor (α7nAChR) in the brain, thus modulating neuroendocrine transmission and brain functions (15). Therefore, the KYN pathway may be a bridge that mediates immune and neuroendocrine dysregulations in the pathogenesis of BD.
The purpose of this review is to outline the KYN pathway, the immune dysfunction, and their interactions in BD, which may emerge as novel treatment targets for this intractable disease. In this review, we firstly introduce the KYN pathway in regard to its metabolism, relationship with immune dysfunction and involvement in the pathogenesis of BD. We also summarize the current findings of the alterations of the KYN pathway metabolites and their correlations with the clinical symptoms of BD as well as the potential targets aiming at the KYN pathway. Finally, we discuss the current dilemma, and potential measures to tackle with this disease.
As an essential amino acid in the human body, TRP is exclusively obtained by dietary intake. In addition to serotonin and indoles, ~95% of TRP is massively diverted into the primary intermediate metabolite- N-formyl-L-kynurenine, which can be further catabolized into KYN via two key enzymes, tryptophan 2,3-dioxygenase (TDO) and IDO (12). TDO exists in the intra-hepatic tissues (16), while IDO mainly resides in the extrahepatic tissues, especially in the brain and immune cells, such as astrocytes, microglia, macrophages and monocytes (17). Once KYN is produced, it is mainly converted into quinolinic acid (QA) and kynurenic acid (KYNA) along with two distinct metabolic branches. KYN can be decomposed into 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO). 3-HK is the precursor of 3-hydroxyanthranilic acid (3-HAA), which is further metabolized into an agonist of the NMDA receptor, QA. Along with another branch, KYN is metabolized into KYNA via kynurenine aminotransferase (KAT) enzyme, and KYNA is regarded as an antagonist of the NMDA receptor with neuroprotective effects (18). In physiological states, these two branches coordinate with each other and maintain normal brain functions (Figure 1).
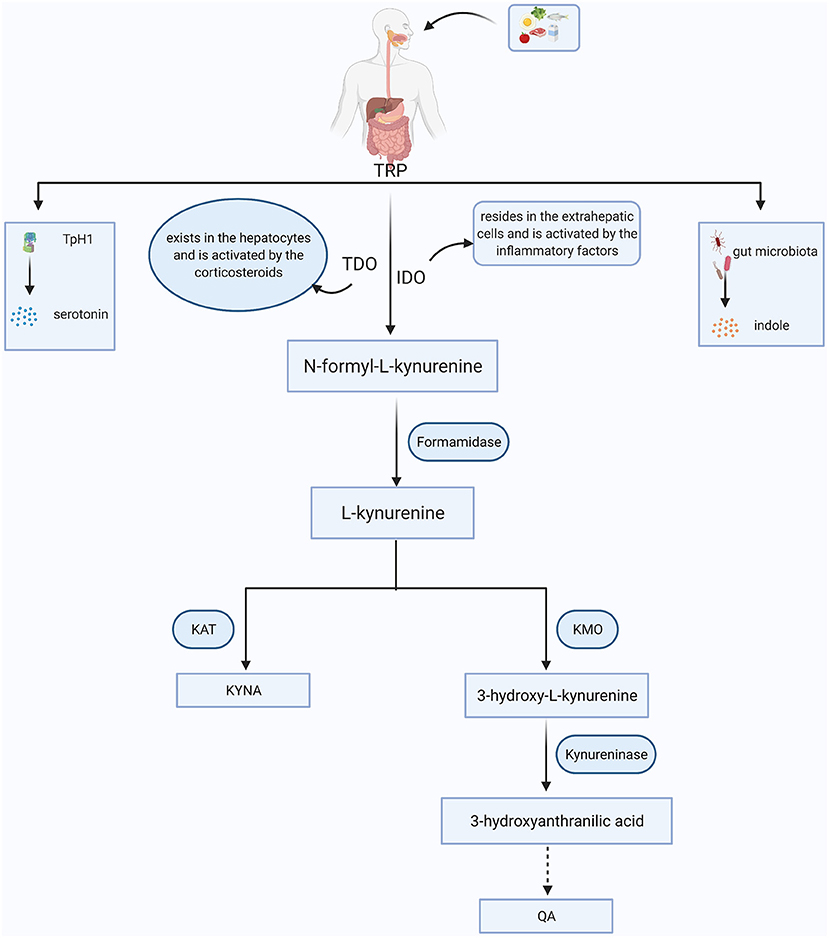
Figure 1. The simplified KYN metabolism pathways in general. Tryptophan (TRP) is obtained from food. Besides being converted into serotonin through tryptophan hydroxylase 1 (TpH1) and indoles via gut microorganism, more than 90% of TRP is broken down to the N-formyl-L-kynurenine with the aid of tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO). N-formyl-L-kynurenine is further degraded to kynurenine (KYN) via formamidase and the KYN can be converted into kynurenic acid (KYNA) and quinolinic acid (QA) mainly depending on the kynurenine aminotransferase (KAT) and kynurenine 3-monooxygenase (KMO), respectively.
Although KYNA and QA poorly cross the blood-brain barrier (BBB) under physiological processes, they also exist in the central nervous system (CNS) because nearly 60% of KYN and TRP can go through the BBB with the assistance of amino acid transporters (19). Moreover, these two main branches of the KYN metabolism are segregated in different cerebral glial cells. Specifically, QA is produced in microglia whereas KYNA is synthesized in astrocytes (20). The production of 3-HK generates free radicals and thus can induce oxidative stress. Overproduction of QA activates the NMDA receptor, causing synaptic dysregulation, oxidative stress and even neuronal apoptosis (21). KYNA acts on the NMDA receptor and the α7nAChR to regulate glutamate, synaptic plasticity and cognitive functions [(22); Figure 2]. Meanwhile, the normal functions of the KYN metabolites depend on the neuronal signals although existing evidence has shown that the KYN metabolism in the cerebra mainly occurs in the glial cells rather than in the neurons (23).
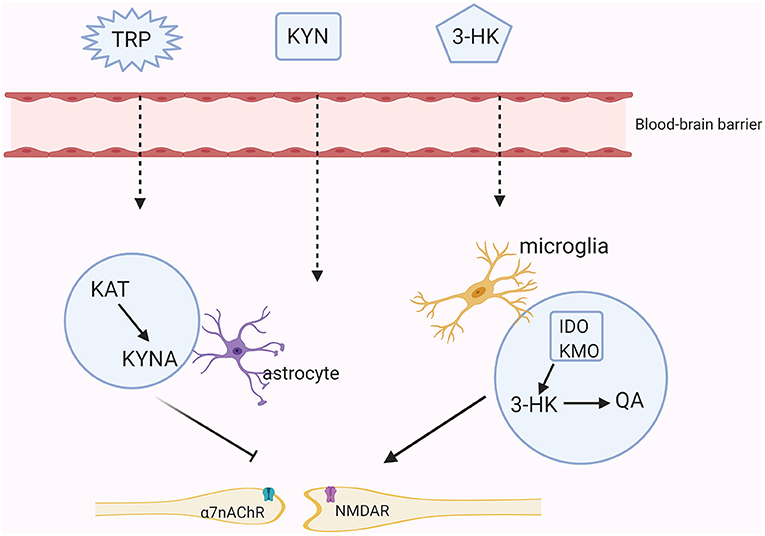
Figure 2. The KYN pathway in the brain. Tryptophan (TRP), kynurenine (KYN), and 3-hydroxykynurenine (3-HK) all can cross the brain-blood barrier (BBB). In astrocytes, KYN is mainly metabolized into the neuroprotective metabolite- kynurenic acid (KYNA) through the kynurenine aminotransferase (KAT). KYNA inhibits the activities of the N-methyl-D aspartate (NMDA) receptor and the non-competitively α7-nicotinic receptor (α7nAChR). While in microglia, KYN is further metabolized into an agonist of the NMDA receptor-quinolinic acid (QA) through indoleamine the 2,3-dioxygenase (IDO) and kynurenine monooxygenase (KMO).
Accumulating evidence has demonstrated that the immune system is dysregulated in BD. For example, the risk of developing BD is increased in patients experiencing autoimmune diseases (24). A high level of interleukin-6 (IL-6) was predominantly observed in mania, tumor necrosis factor-α (TNF-α) was elevated in both depression and mania and C-reactive protein (CRP) level was even increased in euthymia (25). In post-mortem BD brain samples, the microglia, astrocytes and macrophages were found to be involved in neuroinflammatory process (26). In addition, microglial activation was correlated with neuronal injury in the hippocampus (27). Reduced insula functional connectivity was related to the elevated level of IL-6 in drug- naïve BD patients (28). Elevated interferon-γ (IFN-γ) was related to the severity of mania while increased interleukin-1β (IL-1β) was associated with the severity of depression (9). All of these have indicated that the immune system plays a crucial role in BD. However, how immune dysregulation contributes to the onset of BD remains to be further elucidated.
Inflammatory factors can significantly shunt the TRP metabolism toward KYN pathway through upregulating the activity and expression of related rate-limiting enzymes. IFN-γ can independently induce the IDO activity. In addition, other inflammatory molecules, such as TNF-α and IL-1 can also synergistically enhance the IFN-induced IDO activity (29). The KMO transcript was increased via IL-1β stimulation and KMO expression was enhanced after LPS administration (30, 31). Furthermore, the activated immune state contributes to the impairment of BBB integrity (32), facilitating the transportation of TRP and KYN into the brain to provide original materials for the KYN pathway (18). Previous studies also showed that KYNA inhibited cytokine release to regulate the immune response through activating aryl hydrocarbon receptor while QA promoted the inflammatory response by inducing the production of proinflammatory mediators (33, 34). In this regard, the distinct metabolites of the KYN pathway may have opposites effects on immune responses.
In manic episodes, TNF-α level was positively connected with the IDO activity (represented by the KYN/TRP ratio) and neurotoxic production including 3-HK and QA. Besides, the KYNA/3-HK ratio was significantly reduced in and negatively correlated with IFN-γ in BD depressive subgroups (13). In patients with BD and/or schizoaffective disorder, Wurfel et al. found the KYNA/3-HK and KYNA/QA ratios were decreased and had negative correlations with CRP. However, the 3-HK, QA, 3-HK/KYN (a marker of KMO activity) and IDO activity were positively associated with CRP (35). As obesity is linked with chronic low-grade inflammation, an increased level of the KYN and a higher ratio of the KYN/TRP were also found in overweight, euthymic individuals with BD (36).
Therefore, immune dysfunctions in BD contribute to TRP metabolism toward the KYN pathway through activating activities of key enzymes, especially toward the neurotoxic metabolites.
Once the KYN pathway has been activated, the signal transmissions of monoamine neurotransmitters and neuronal functions would be directly affected. For example, Williams et al. found that maternal inflammation shunted TRP metabolism away from the serotonin to the KYNA pathway, and thus potentially impaired the development of thalamocortical fibers in the brain of a new-born rabbit model (37).
KYNA, an endogenous glutamate receptor antagonist, inhibits the glycine-binding site of the NMDA receptor. It is demonstrated that the levels of glutamate would be reduced by 30–40% following even low dose administration of KYNA into the brain (38). Glutamate release was enhanced by the expression of the α7nAchR in the glutamatergic axon terminals, which could be recognized by KYNA in physiological concentrations, causing lower expression and activity of the α7nAchR (39). Additionally, KYNA can also modulate the dopaminergic neurotransmission via acting on the α7nAchR (40). The suppression of the N-type Ca2+ channels in sympathetic neurons could be partly attributed to the agonistic role of KYNA on an orphan G-protein-coupled receptor (GPR35) (41). Even though KYNA has also been regarded as a neuroprotective metabolite against the neurotoxic NMDA receptor, excessive KYNA accumulation would cause glutamatergic hypofunction and was associated with the psychotomimetic effects (42, 43).
In addition, QA accelerates the synaptosomal glutamate release, while inhibits the glutamate uptake by astrocytes (44). QA also has excitotoxic effects on GABAergic neurons and activates the glycine site of the NMDA receptor, which in turn reduces the level of the extracellular dopamine (45, 46). The prefrontal cortex (PFC), amygdala, hippocampus, as well as ventral striatum of the brain, are the preferred locations for QA-binding NMDA receptor subunits, where a greater excitotoxic burden is presented (47).
The above evidence demonstrates that inflammatory processes accelerate the KYN pathway metabolism in the brain and its metabolites eventually act on the receptors that are associated with the signals of neurotransmitters, such as dopamine, GABA and glutamate. Besides, serotonin plays a vital role in the pathogenesis of depressive behaviors via its effects on the 5-HT1A receptor (48), and the release of the serotonin is reduced along with the KYN pathway activation Moreover, glutamatergic metabolism hypofunction may indicate an increased risk of depressed adolescents presenting mixed symptoms or developing BD (49). The high or low level of dopamine was thought to be related to the manic or depressive symptoms in BD, respectively (50). Therefore, we hypothesize that the KYN pathway may play a mediating role in the regulation of neuroendocrine signaling pathways and immune balance, which further participates in the pathogenesis of BD (Figure 3).
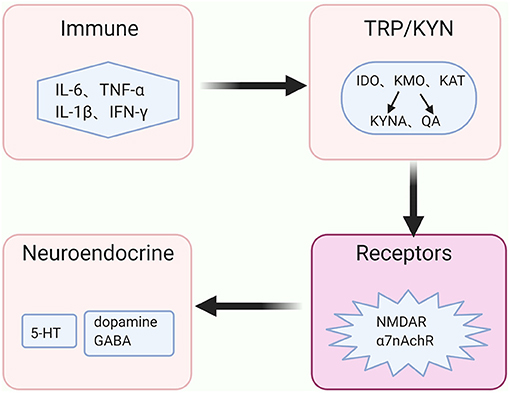
Figure 3. The immune-KYN-neuroendocrine network in BD. The abnormal immune system in bipolar disorder (BD) aberrantly activates the key enzymes' activities of the kynurenine (KYN) pathway, shifts the tryptophan (TRP) from serotonin (5-HT) toward kynurenine (KYN) pathway for metabolism, and causes the reduction of 5-HT. Levels and functions of other neurotransmitters, such as dopamine, γ-aminobutyric acid (GABA) as well as glutamate, rely on the metabolites of the KYN pathway in coordinating with distinct receptors, such as N-methyl-D aspartate (NMDA) receptor and non-competitively α7 nicotinic acetylcholine receptor (α7nAchR).
Up to present, alterations of the KYN pathway in BD have attracted adequate attention. Herein, we further reviewed current findings of the KYN metabolism dysfunctions in BD from perspectives of genetics, brain structural or functional imaging, as well as the relationships between the KYN metabolites and BD clinical symptoms (Table 1). In addition, some potential treatment strategies aiming at the KYN pathway have also been summarized.
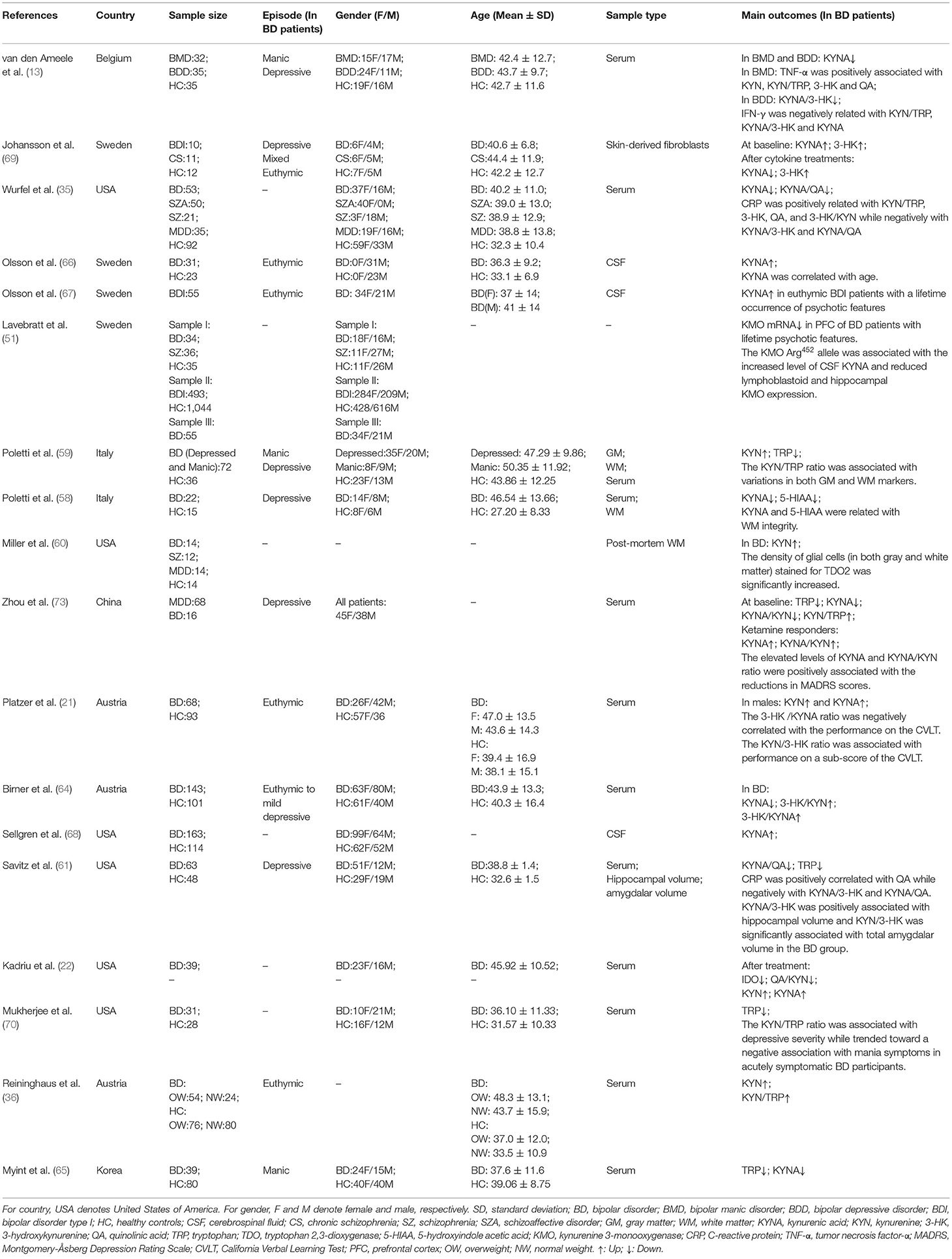
Table 1. Demographic characteristics and findings of included studies on the KYN pathway in BD.
KMO has a high affinity for KYNA, and metabolizes most of KYN into 3-HK (51). Genetic variations, such as KMO Arg452 allele, affected its enzyme expression or activity and the reduced KMO function was correlated with the higher KYNA level in the post-mortem PFC from manic patients with psychotic features (51). A genetic polymorphism, i.e., rs9657182 in the IDO1 gene enhanced the vulnerability to depressive symptoms along with the immune system activation in humans (52), whereas inhibition of IDO1 through genetic deletion or pharmacological manipulation could remove depressive phenotype in a mouse model (11).
IDO is vital for shunting KYN toward the microglial production of QA and may mediate depressive symptomatology (22). Excessive KYNA elevation was connected with the activation of the midbrain dopamine neurons (53, 54). Overactivation of the dopaminergic system may result in manic-like behaviors (55). Genetic variations affect the KYN metabolism, indicating the roles of genetic risk alleles in contributing to the vulnerability for BD.
Neuroimaging studies have revealed unspecific brain structural changes in BD patients. The gray matter (GM) volumes and thickness in the cortex, especially in prefrontal and temporal cortices, were reduced in BD individuals and the volume of white matter (WM) ranging from the posterior corpus callosum to posterior cingulate cortex was decreased (56, 57). Further studies showed that the reduced KYNA level was negatively associated with the WM integrity in BD (58). A higher KYN/TRP ratio was negatively correlated with the volume of the right amygdala in the manic group and the left fusiform gyrus in the depressive patients (59). The KYN level was increased, while the KYNA/KYN ratio was decreased in the anterior cingulate in post-mortem patients with BD (60). Furthermore, the KYNA/3-HK was significantly positively correlated with the volumes of the amygdala and hippocampus (61). All of these findings showed the close relationship between the abnormal KYN pathway metabolites and brain architectures in BD patients.
Therefore, the alterations in the brain structures reflected the underlying processes of neurotoxicity and immune activation (62, 63). The neuroinflammation existing in BD could facilitate the production of neurotoxic metabolites via KYN pathway, which further contributed to the brain dysfunction and neuropathogenesis in BD.
Changes in KYN metabolites were linked to the clinical symptoms in BD individuals. Specifically, the ratios of 3-HK/KYN and 3-HK/KYNA were higher, while the KYNA level was decreased in BD patients with euthymia to mild depression compared to healthy controls (64). Furthermore, the lower level of the KYNA was also observed in both manic and depressive patient groups (13, 65). However, the higher level of KYNA was found in BD patients with previous psychotic features and males in euthymic states (66–69). Mukherjee et al. found that the KYN/TRP ratio was positively correlated with depressive severity but trended toward a negative correlation with manic symptoms in BD (70). Platzer et al. found that the 3-HK/KYNA ratio was higher in male patients and negatively connected with poorer verbal memory performance, indicating potential correlations between the KYN pathway and executive functions (21).
These findings provide valuable clues for the KYN pathway involved in the pathophysiology of BD, although the current results were inconsistent. Theoretically, KYN tends to be converted along the neurotoxic rather than the neuroprotective branch and the imbalance between these two branches plays a vital role in the etiology of BD. However, the mechanisms are complicated when it comes to the relationship between the KYN pathway metabolites and BD symptoms, predominantly including depressive or psychotic features, so that a concept of “double dissociation” has been proposed (35). Specifically, depression was associated with elevated levels of 3-HK and QA, which led to the inhibition of the NMDA receptor, and promoted glutamate release but reduced glutamate reuptake. The neuroprotective component KYNA was correlated with the psychotic features due to the striatal hyperdopaminergia and hypofunction of GABAergic interneurons. Considering the limitations of current studies, further researches are needed to fully clarify the pathophysiological mechanism of the KYN pathway on depressive, manic or psychotic features in BD.
Considering the agonistic and antagonistic roles of KYNA and QA on the NMDA receptor, respectively, it is intriguing to explore treatment targets toward the KYN pathway in BD. As a non-competitive NMDA receptor antagonist, ketamine could alter the LPS-induced depressive-like behaviors through blocking the NMDA receptor activity (71). A single intravenous infusion of ketamine rapidly exerted rapid anti-depressive effect and improved suicidal ideations in type I or II bipolar depression (72). After intravenous infusions of ketamine, increases in the KYNA/KYN ratio and KYNA level were observed in responders and both were correlated with the Montgomery-Åsberg Depression Rating Scale (MADRS) scores' reduction compared to non-responders at 24-h and 13-day periods (73). AV-101, also named 4-chlorokynurenine, is a KYNA analog and selective antagonist of the NMDA receptor glycine binding site. It is an undergoing preliminary clinical trial for mood improvement and has received fast track designation by the Food and Drug Administration (74). Oral administration of a KYNA synthesis (KAT II) inhibitor promoted cognitive functions (75). KMO inhibitor could moderately and persistently increase the KYNA level in the brain to play neuroprotective roles (76). Therefore, modulation of the KYN pathway may be a promising treatment target for BD patients.
Our current knowledge on the role of the KYN pathway in BD is rapidly increasing. Growing evidence has indicated that the KYN pathway serves as a crucial regulator between the immune balance and neurotransmitter signaling in the pathogenesis of BD.
However, some specific questions need special attention and further investigations are warranted. Firstly, factors that contribute to the heterogeneous results about the KYN pathway in BD need to be controlled, including gender, age, race groups, and study methodologies. Moreover, the mood episodes of enrolled patients and the specific analysis aiming at subgroup or general population also affect the final findings. Polycentric and large samples research may help to untangle this dilemma.
Secondly, existing clinical studies mainly focus on the connections between the KYN metabolites and BD, but they could not draw causal conclusions. At present, it is still difficult to apply all preclinical research methods to humans through experimental manipulation of the KYN pathway. Therefore, multi-omic analysis that integrates host genomics, metabonomics, single cell sequencing, cytology, and brain network will be helpful for uncovering the complicated communicating networks of the KYN pathway in BD.
A meta-analysis investigated the alterations of the KYN metabolites in patients with unipolar depression and BD and found different results in these cases (77). Hence, clinical applications of the KYN pathway-related biomarkers for BD diagnosis still need further verification. Besides, explorations of novel therapeutic interventions and prognostic biomarkers derived from the KYN metabolites in BD are also needed. Cross-sectional and longitudinal studies may provide additional help to advance this field.
In conclusion, convergent evidence has demonstrated that the immune system and the KYN pathway are changed in BD. Immune dysfunction could contribute to modulating the KYN pathway metabolism, which could further regulate the signals of the neurotransmitters, and finally affect the pathophysiological process of BD. The immune-KYN-neuroendocrine interaction in BD provides new clues for developing therapeutic targets.
PZ conducted literature searches and wrote the first draft of the manuscript. JL and SH gave substantial linguistic support. HH, XG, JJ, CX, LW, YF, and SH were involved in an intensive drafting and revision of the manuscript. All authors have read and approved the final manuscript.
This work was supported by the National Natural Science Foundation (No. 81971271 to SH), the Zhejiang Provincial Key Research and Development Program (No. 2021C03107 to SH), the Zhejiang Provincial Natural Science Foundation (No. LQ20H090013 to JL), and the Program from the Health and Family Planning Commission of Zhejiang Province (No. 2020KY548 to JL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vieta E, Berk M, Schulze TG, Carvalho AF, Suppes T, Calabrese JR, et al. Bipolar disorders. Nat Rev Dis Prim. (2018) 4:18008. doi: 10.1038/nrdp.2018.8
2. Bauer M, Andreassen OA, Geddes JR, Vedel Kessing L, Lewitzka U, Schulze TG, et al. Areas of uncertainties and unmet needs in bipolar disorders: clinical and research perspectives. Lancet Psychiatry. (2018) 5:930–9. doi: 10.1016/s2215-0366(18)30253-0
3. Merikangas KR, Jin R, He JP, Kessler RC, Lee S, Sampson NA, et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch Gen Psychiatry. (2011) 68:241–51. doi: 10.1001/archgenpsychiatry.2011.12
4. Vigo D, Thornicroft G, Atun R. Estimating the true global burden of mental illness. Lancet Psychiatry. (2016) 3:171–8. doi: 10.1016/s2215-0366(15)00505-2
5. Dagani J, Signorini G, Nielssen O, Bani M, Pastore A, Girolamo G, et al. Meta-analysis of the interval between the onset and management of bipolar disorder. Can J Psychiatry. (2017) 62:247–58. doi: 10.1177/0706743716656607
6. Carvalho AF, Firth J, Vieta E. Bipolar disorder. N Engl J Med. (2020) 383:58–66. doi: 10.1056/NEJMra1906193
7. Altamura AC, Dell'Osso B, Berlin HA, Buoli M, Bassetti R, Mundo E. Duration of untreated illness and suicide in bipolar disorder: a naturalistic study. Eur Arch Psychiatry Clin Neurosci. (2010) 260:385–91. doi: 10.1007/s00406-009-0085-2
8. Benedetti F, Aggio V, Pratesi ML, Greco G, Furlan R. Neuroinflammation in bipolar depression. Front Psychiatry. (2020) 11:71. doi: 10.3389/fpsyt.2020.00071
9. Anderson G, Maes M. Bipolar disorder: role of immune-inflammatory cytokines, oxidative and nitrosative stress and tryptophan catabolites. Curr Psychiatry Rep. (2015) 17:8. doi: 10.1007/s11920-014-0541-1
10. O'Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. (2009) 14:511–22. doi: 10.1038/sj.mp.4002148
11. Lawson MA, Parrott JM, McCusker RH, Dantzer R, Kelley KW, et al. Intracerebroventricular administration of lipopolysaccharide induces indoleamine-2,3-dioxygenase-dependent depression-like behaviors. J Neuroinflammation. (2013) 10:87. doi: 10.1186/1742-2094-10-87
12. Strasser B, Becker K, Fuchs D, Gostner JM. Kynurenine pathway metabolism and immune activation: peripheral measurements in psychiatric and co-morbid conditions. Neuropharmacology. (2017) 112:286–96. doi: 10.1016/j.neuropharm.2016.02.030
13. van den Ameele S, van Nuijs AL, Lai FY, Schuermans J, Verkerk R, van Diermen L, et al. A mood state-specific interaction between kynurenine metabolism and inflammation is present in bipolar disorder. Bipolar Disord. (2020) 22:59–69. doi: 10.1111/bdi.12814
14. Hunt C, Macedo ECT, Suchting R, de Dios C, Cuellar Leal VA, Soares JC, et al. Effect of immune activation on the kynurenine pathway and depression symptoms - a systematic review and meta-analysis. Neurosci Biobehav Rev. (2020) 118:514–23. doi: 10.1016/j.neubiorev.2020.08.010
15. Ramos-Chavez LA, Lugo Huitron R, Gonzalez Esquivel D, Pineda B, Rios C, Silva-Adaya D, et al. Relevance of alternative routes of kynurenic acid production in the brain. Oxid Med Cell Longev. (2018) 2018:5272741. doi: 10.1155/2018/5272741
16. Gibney SM, Fagan EM, Waldron AM, O'Byrne J, Connor TJ, Harkin A. Inhibition of stress-induced hepatic tryptophan 2,3-dioxygenase exhibits antidepressant activity in an animal model of depressive behaviour. Int J Neuropsychopharmacol. (2014) 17:917–28. doi: 10.1017/S1461145713001673
17. Guillemin GJ, Cullen KM, Lim CK, Smythe GA, Garner B, Kapoor V, et al. Characterization of the kynurenine pathway in human neurons. J Neurosci. (2007) 27:12884–92. doi: 10.1523/JNEUROSCI.4101-07.2007
18. Savitz J. The kynurenine pathway: a finger in every pie. Mol Psychiatry. (2020) 25:131–47. doi: 10.1038/s41380-019-0414-4
19. Wiedlocha M, Marcinowicz P, Janoska-Jazdzik M, Szulc A. Gut microbiota, kynurenine pathway and mental disorders - review. Prog Neuropsychopharmacol Biol Psychiatry. (2020) 106:110145. doi: 10.1016/j.pnpbp.2020.110145
20. Guillemin GJ, Kerr SJ, Smythe GA, Smith DG, Kapoor V, Armati PJ, et al. Kynurenine pathway metabolism in human astrocytes a paradox for neuronal protection. J Neurochem. (2001) 78:842–53. doi: 10.1046/j.1471-4159.2001.00498.x
21. Platzer M, Dalkner N, Fellendorf FT, Birner A, Bengesser SA, Queissner R, et al. Tryptophan breakdown and cognition in bipolar disorder. Psychoneuroendocrinology. (2017) 81:144–50. doi: 10.1016/j.psyneuen.2017.04.015
22. Kadriu B, Farmer CA, Yuan P, Park LT, Deng ZD, Moaddel R, et al. The kynurenine pathway and bipolar disorder: intersection of the monoaminergic and glutamatergic systems and immune response. Mol Psychiatry. (2019). doi: 10.1038/s41380-019-0589-8. [Epub ahead of print].
23. Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. (2012) 13:465–77. doi: 10.1038/nrn3257
24. Wang LY, Chen SF, Chiang JH, Hsu CY, Shen YC. Systemic autoimmune diseases are associated with an increased risk of obsessive-compulsive disorder: a nationwide population-based cohort study. Soc Psychiatry Psychiatr Epidemiol. (2019) 54:507–16. doi: 10.1007/s00127-018-1622-y
25. Rowland T, Perry BI, Upthegrove R, Barnes N, Chatterjee J, Gallacher D, et al. Neurotrophins, cytokines, oxidative stress mediators and mood state in bipolar disorder: systematic review and meta-analyses. Br J Psychiatry. (2018) 213:514–25. doi: 10.1192/bjp.2018.144
26. Giridharan VV, Sayana P, Pinjari OF, Ahmad N, da Rosa MI, Quevedo J. Postmortem evidence of brain inflammatory markers in bipolar disorder: a systematic review. Mol Psychiatry. (2020) 25:94–113. doi: 10.1038/s41380-019-0448-7
27. Haarman BC, Burger H, Doorduin J, Renken RJ, Sibeijn-Kuiper AJ, Marsman JB, et al. Volume, metabolites and neuroinflammation of the hippocampus in bipolar disorder - a combined magnetic resonance imaging and positron emission tomography study. Brain Behav Immun. (2016) 56:21–33. doi: 10.1016/j.bbi.2015.09.004
28. Chen P, Chen F, Chen G, Zhong S, Gong J, Zhong H, et al. Inflammation is associated with decreased functional connectivity of insula in unmedicated bipolar disorder. Brain Behav Immun. (2020) 89:615–22. doi: 10.1016/j.bbi.2020.07.004
29. Shirey KA, Jung JY, Maeder GS, Carlin JM. Upregulation of IFN-γ receptor expression by proinflammatory cytokines influences IDO activation in epithelial cells. J Interferon Cytokine Res. (2006) 26:53–62. doi: 10.1089/jir.2006.26.53
30. Zunszain PA, Anacker C, Cattaneo A, Choudhury S, Musaelyan K, Myint AM, et al. Interleukin-1beta: a new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology. (2012) 37:939–49. doi: 10.1038/npp.2011.277
31. Connor TJ, Starr N, O'Sullivan JB, Harkin A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: a role for IFN-gamma? Neurosci Lett. (2008) 441:29–34. doi: 10.1016/j.neulet.2008.06.007
32. Pollak TA, Drndarski S, Stone JM, David AS, McGuire P, Abbott NJ. The blood–brain barrier in psychosis. Lancet Psychiatry. (2018) 5:79–92. doi: 10.1016/s2215-0366(17)30293-6
33. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
34. Guillemin GJ, Croitoru-Lamoury J, Dormont D, Armati PJ, Brew BJ. Quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. Glia. (2003) 41:371–81. doi: 10.1002/glia.10175
35. Wurfel BE, Drevets WC, Bliss SA, McMillin JR, Suzuki H, Ford BN, et al. Serum kynurenic acid is reduced in affective psychosis. Transl Psychiatry. (2017) 7:e1115. doi: 10.1038/tp.2017.88
36. Reininghaus EZ, McIntyre RS, Reininghaus B, Geisler S, Bengesser SA, Lackner N, et al. Tryptophan breakdown is increased in euthymic overweight individuals with bipolar disorder: a preliminary report. Bipolar Disord. (2014) 16:432–40. doi: 10.1111/bdi.12166
37. Williams M, Zhang Z, Nance E, Drewes JL, Lesniak WG, Singh S, et al. Maternal inflammation results in altered tryptophan metabolism in rabbit placenta and fetal brain. Dev Neurosci. (2017) 39:399–412. doi: 10.1159/000471509
38. Muneer A. Kynurenine pathway of tryptophan metabolism in neuropsychiatric disorders: pathophysiologic and therapeutic considerations. Clin Psychopharmacol Neurosci. (2020) 18:507–26. doi: 10.9758/cpn.2020.18.4.507
39. Vecsei L, Szalardy L, Fulop F, Toldi J. Kynurenines in the CNS: recent advances and new questions. Nat Rev Drug Discov. (2013) 12:64–82. doi: 10.1038/nrd3793
40. Myint AM, Kim YK. Network beyond IDO in psychiatric disorders: revisiting neurodegeneration hypothesis. Prog Neuropsychopharmacol Biol Psychiatry. (2014) 48:304–13. doi: 10.1016/j.pnpbp.2013.08.008
41. Fallarini S, Magliulo L, Paoletti T, de Lalla C, Lombardi G. Expression of functional GPR35 in human iNKT cells. Biochem Biophys Res Commun. (2010) 398:420–5. doi: 10.1016/j.bbrc.2010.06.091
42. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. (1991) 148:1301–8. doi: 10.1176/ajp.148.10.1301
43. Moghaddam B, Krystal JH. Capturing the angel in “angel dust”: twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr Bull. (2012) 38:942–9. doi: 10.1093/schbul/sbs075
44. Guillemin GJ. Quinolinic acid, the inescapable neurotoxin. FEBS J. (2012) 279:1356–65. doi: 10.1111/j.1742-4658.2012.08485.x
45. Perez-De La Cruz V, Carrillo-Mora P, Santamaria A. Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int J Tryptophan Res. (2012) 5:1–8. doi: 10.4137/IJTR.S8158
46. Balla A, Nattini ME, Sershen H, Lajtha A, Dunlop DS, Javitt DC. GABAB/NMDA receptor interaction in the regulation of extracellular dopamine levels in rodent prefrontal cortex and striatum. Neuropharmacology. (2009) 56:915–21. doi: 10.1016/j.neuropharm.2009.01.021
47. Carvalho LP, Bochet P, Rossier J. The endogenous agonist quinolinic acid and the nonendogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem Int. (1996) 28:445–52. doi: 10.1016/0197-0186(95)00091-7
48. Haleem DJ. Glucocorticoids in the physiological and transcriptional regulation of 5-HT1A receptor and the pathogenesis of depression. Neuroscientist. (2020) 4:1–10. doi: 10.1177/1073858420975711
49. Sonmez AI, Lewis CP, Port JD, Cabello-Arreola A, Blacker CJ, Seewoo BJ, et al. Glutamatergic correlates of bipolar symptoms in adolescents. J Child Adolesc Psychopharmacol. (2020) 30:599–605. doi: 10.1089/cap.2020.0082
50. Ashok AH, Marques TR, Jauhar S, Nour MM, Goodwin GM, Young AH, et al. The dopamine hypothesis of bipolar affective disorder: the state of the art and implications for treatment. Mol Psychiatry. (2017) 22:666–79. doi: 10.1038/mp.2017.16
51. Lavebratt C, Olsson S, Backlund L, Frisen L, Sellgren C, Priebe L, et al. The KMO allele encoding Arg452 is associated with psychotic features in bipolar disorder type 1, and with increased CSF KYNA level and reduced KMO expression. Mol Psychiatry. (2014) 19:334–41. doi: 10.1038/mp.2013.11
52. Smith AK, Simon JS, Gustafson EL, Noviello S, Cubells JF, Epstein MP, et al. Association of a polymorphism in the indoleamine- 2,3-dioxygenase gene and interferon-alpha-induced depression in patients with chronic hepatitis C. Mol Psychiatry. (2012) 17:781–9. doi: 10.1038/mp.2011.67
53. Erhardt S, Oberg H, Mathé JM, Engberg G. Pharmacological elevation of endogenous kynurenic acid levels activates nigral dopamine neurons. Amino Acids. (2001) 20:353–62. doi: 10.1007/s007260170032
54. Olsson SK, Andersson AS, Linderholm KR, Holtze M, Nilsson-Todd LK, Schwieler L, et al. Elevated levels of kynurenic acid change the dopaminergic response to amphetamine: implications for schizophrenia. Int J Neuropsychopharmacol. (2009) 12:501–12. doi: 10.1017/S1461145708009383
55. Lopachev A, Volnova A, Evdokimenko A, Abaimov D, Timoshina Y, Kazanskaya R, et al. Intracerebroventricular injection of ouabain causes mania-like behavior in mice through D2 receptor activation. Sci Rep. (2019) 9:15627. doi: 10.1038/s41598-019-52058-z
56. Delvecchio G, Pigoni A, Bauer IE, Soares JC, Brambilla P. Disease-discordant twin structural MRI studies on affective disorders. Neurosci Biobehav Rev. (2020) 108:459–71. doi: 10.1016/j.neubiorev.2019.11.023
57. Pezzoli S, Emsell L, Yip SW, Dima D, Giannakopoulos P, Zarei M, et al. Meta-analysis of regional white matter volume in bipolar disorder with replication in an independent sample using coordinates, T-maps, and individual MRI data. Neurosci Biobehav Rev. (2018) 84:162–70. doi: 10.1016/j.neubiorev.2017.11.005
58. Poletti S, Myint AM, Schuetze G, Bollettini I, Mazza E, Grillitsch D, et al. Kynurenine pathway and white matter microstructure in bipolar disorder. Eur Arch Psychiatry Clin Neurosci. (2018) 268:157–68. doi: 10.1007/s00406-016-0731-4
59. Poletti S, Melloni E, Aggio V, Colombo C, Valtorta F, Benedetti F, et al. Grey and white matter structure associates with the activation of the tryptophan to kynurenine pathway in bipolar disorder. J Affect Disord. (2019) 259:404–12. doi: 10.1016/j.jad.2019.08.034
60. Miller CL, Llenos IC, Dulay JR, Weis S. Upregulation of the initiating step of the kynurenine pathway in postmortem anterior cingulate cortex from individuals with schizophrenia and bipolar disorder. Brain Res. (2006) 1073–74:25–37. doi: 10.1016/j.brainres.2005.12.056
61. Savitz J, Dantzer R, Wurfel BE, Victor TA, Ford BN, Bodurka J, et al. Neuroprotective kynurenine metabolite indices are abnormally reduced and positively associated with hippocampal and amygdalar volume in bipolar disorder. Psychoneuroendocrinology. (2015) 52:200–11. doi: 10.1016/j.psyneuen.2014.11.015
62. Petrik D, Lagace DC, Eisch AJ. The neurogenesis hypothesis of affective and anxiety disorders: are we mistaking the scaffolding for the building? Neuropharmacology. (2012) 62:21–34. doi: 10.1016/j.neuropharm.2011.09.003
63. Barichello T, Giridharan VV, Bhatti G, Sayana P, Doifode T, Macedo D, et al. Inflammation as a mechanism of bipolar disorder neuroprogression. Bipolar disorder: from neuroscience to treatment. Curr Top Behav Neurosci. (2020) 48:215–37. doi: 10.1007/7854_2020_173
64. Birner A, Platzer M, Bengesser SA, Dalkner N, Fellendorf FT, Queissner R, et al. Increased breakdown of kynurenine towards its neurotoxic branch in bipolar disorder. PLoS ONE. (2017) 12:e0172699. doi: 10.1371/journal.pone.0172699
65. Myint AM, Kim YK, Verkerk R, Park SH, Scharpe S, Steinbusch HW, et al. Tryptophan breakdown pathway in bipolar mania. J Affect Disord. (2007) 102:65–72. doi: 10.1016/j.jad.2006.12.008
66. Olsson SK, Samuelsson M, Saetre P, Lindstrom L, Jonsson EG, Nordin C, et al. Elevated levels of kynurenic acid in the cerebrospinal fluid of patients with bipolar disorder. J Psychiatry Neurosci. (2010) 35:195–9. doi: 10.1503/jpn.090180
67. Olsson SK, Sellgren C, Engberg G, Landén M, Erhardt S. Cerebrospinal fluid kynurenic acid is associated with manic and psychotic features in patients with bipolar I disorder. Bipolar Disorders. (2012) 14:719–26. doi: 10.1111/bdi.12009
68. Sellgren CM, Gracias J, Jungholm O, Perlis RH, Engberg G, Schwieler L, et al. Peripheral and central levels of kynurenic acid in bipolar disorder subjects and healthy controls. Transl Psychiatry. (2019) 9:37. doi: 10.1038/s41398-019-0378-9
69. Johansson AS, Owe-Larsson B, Asp L, Kocki T, Adler M, Hetta J, et al. Activation of kynurenine pathway in ex vivo fibroblasts from patients with bipolar disorder or schizophrenia: cytokine challenge increases production of 3-hydroxykynurenine. J Psychiatr Res. (2013) 47:1815–23. doi: 10.1016/j.jpsychires.2013.08.008
70. Mukherjee D, Krishnamurthy VB, Millett CE, Reider A, Can A, Groer M, et al. Total sleep time and kynurenine metabolism associated with mood symptom severity in bipolar disorder. Bipolar Disord. (2018) 20:27–34. doi: 10.1111/bdi.12529
71. Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology. (2013) 38:1609–16. doi: 10.1038/npp.2013.71
72. Zarate CA Jr., Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, et al. Replication of ketamine's antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. (2012) 71:939–46. doi: 10.1016/j.biopsych.2011.12.010
73. Zhou Y, Zheng W, Liu W, Wang C, Zhan Y, Li H, et al. Antidepressant effect of repeated ketamine administration on kynurenine pathway metabolites in patients with unipolar and bipolar depression. Brain Behav Immun. (2018) 74:205–12. doi: 10.1016/j.bbi.2018.09.007
74. Park LT, Kadriu B, Gould TD, Zanos P, Greenstein D, Evans JW, et al. A randomized trial of the N-methyl-d-aspartate receptor glycine site antagonist prodrug 4-chlorokynurenine in treatment-resistant depression. Int J Neuropsychopharmacol. (2020) 23:417–25. doi: 10.1093/ijnp/pyaa025
75. Bortz DM, Wu HQ, Schwarcz R, Bruno JP. Oral administration of a specific kynurenic acid synthesis (KAT II) inhibitor attenuates evoked glutamate release in rat prefrontal cortex. Neuropharmacology. (2017) 121:69–78. doi: 10.1016/j.neuropharm.2017.04.023
76. Zwilling D, Huang SY, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu HQ, et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. (2011) 145:863–74. doi: 10.1016/j.cell.2011.05.020
Keywords: bipolar disorder, kynurenine pathway, immune, pathogenesis, treatment
Citation: Zhang P, Huang H, Gao X, Jiang J, Xi C, Wu L, Fu Y, Lai J and Hu S (2021) Involvement of Kynurenine Metabolism in Bipolar Disorder: An Updated Review. Front. Psychiatry 12:677039. doi: 10.3389/fpsyt.2021.677039
Received: 07 March 2021; Accepted: 01 July 2021;
Published: 26 July 2021.
Edited by:
Tianhua Niu, Tulane University, United StatesReviewed by:
Shaolong Cao, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2021 Zhang, Huang, Gao, Jiang, Xi, Wu, Fu, Lai and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaohua Hu, ZG9yaHVzaGFvaHVhQHpqdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.