
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Psychiatry, 05 July 2021
Sec. Child and Adolescent Psychiatry
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.668577
This article is part of the Research TopicMolecular and Genetic Mechanisms in Neurodevelopmental Disorders: From Bench to Bedside View all 10 articles
 David Q. Beversdorf1,2*
David Q. Beversdorf1,2* Ayten Shah3Allison Jhin4
Ayten Shah3Allison Jhin4 Janelle Noel-MacDonnell5Patrick Hecht2
Janelle Noel-MacDonnell5Patrick Hecht2 Bradley J. Ferguson2,6Danielle Bruce7
Bradley J. Ferguson2,6Danielle Bruce7 Michael Tilley7
Michael Tilley7 Zohreh Talebizadeh5*
Zohreh Talebizadeh5*Background: Genetics and environment both are critical in autism spectrum disorder (ASD), but their interaction (G × E) is less understood. Numerous studies have shown higher incidence of stress exposures during pregnancies with children later diagnosed with ASD. However, many stress-exposed mothers have unaffected children. The serotonin transporter (SERT) gene affects stress reactivity. Two independent samples have shown that the association between maternal stress exposure and ASD is greatest with maternal presence of the SERT short (S)-allele (deletion in the promoter region). MicroRNAs play a regulatory role in the serotonergic pathway and in prenatal stress and are therefore potential mechanistic targets in this setting.
Design/methods: We profiled microRNA expression in blood from mothers of children with ASD, with known stress exposure during pregnancy. Samples were divided into groups based on SERT genotypes (LL/LS/SS) and prenatal stress level (high/low).
Results: Two thousand five hundred mature microRNAs were examined. The ANOVA analysis showed differential expression (DE) of 119 microRNAs; 90 were DE in high- vs. low-stress groups (stress-dependent). Two (miR-1224-5p, miR-331-3p) were recently reported by our group to exhibit stress-dependent expression in rodent brain samples from embryos exposed to prenatal stress. Another, miR-145-5p, is associated with maternal stress. Across SERT genotypes, with high stress exposure, 20 significantly DE microRNAs were detected, five were stress-dependent. These microRNAs may be candidates for stress × SERT genotype interactions. This is remarkable as these changes were from mothers several years after stress-exposed pregnancies.
Conclusions: Our study provides evidence for epigenetic alterations in relation to a G × E model (prenatal maternal stress × SERT gene) in ASD.
The developmental origins of health and disease (DOHaD) hypothesis proposes that environment experiences during development in utero influence health after birth (1). Numerous studies demonstrate that adverse environmental exposures affect neurological development, including those which are salient to autism spectrum disorder (ASD). Genetics is a critical factor in ASD (2–6). However, importance of environmental factors is being increasingly recognized (7), with heritability estimated at 0.87 by the latest, more conservative analysis (8). While progress has been made toward the understanding of genetic factors, including development of animal models based on these genes (9–12), environmental factors are less understood.
Psychological stress during pregnancy impacts behavioral and developmental outcomes in humans (13). Early personality development in the child, schizophrenia, and emotional disturbances in offspring are all associated with maternal stress (14–17). Relationships between maternal stress and a range of adverse offspring behavioral outcomes are reported in animal models, including abnormal behavioral fear responses, as well as abnormal physiological stress reactivity in offspring, which lasts into adulthood (18, 19). Recent evidence suggests that among environmental factors, maternal stress exposure is important in ASD (20–22).
Early studies surveyed for history and timing of prenatal psychosocial stressors corresponding to major life events on the Social Readjustment Rating Scale in mothers of children with ASD, Down syndrome, and typically developing controls. A higher overall incidence of stressors among the mothers who had a child with ASD was found compared to other groups, with a peak in stressors among mothers with a child with ASD at 25–28 weeks of gestation, but not in the other groups (21). This has subsequently been supported by other studies, showing a relationship between the incidence and severity of tropical storms in the United States in Louisiana during the 5th to 6th months of gestation and the incidence of ASD births (22). Larger epidemiological studies support this relationship between prenatal stress and ASD. One Danish national registry study suggested against the presence of an association between maternal bereavement and ASD (23), but the association between maternal bereavement and ASD was present when maternal psychological conditions were included (23). Another Danish national registry study found that maternal psychological conditions were one of the strongest prenatal risk factors for ASD (24). A Swedish registry study also confirmed a relationship between third-trimester prenatal stress and ASD (25). Furthermore, results from the Nurses' Health Study, another large registry study, showed that maternal exposure to partner abuse during pregnancy is strongly associated with ASD (26). Children in utero in New York City during the September 11 terrorist attacks were also found to be 7–9% more likely to be in special education classes (27), although no specific data was available regarding ASD diagnoses. A recent meta-analysis has further supported the relationship between prenatal stress and ASD (28). Finally, a recent study reported that children with ASD that were exposed to prenatal stress present with a more severe condition than those with no history of prenatal stress exposure (29). Therefore, a better understanding of the relationship between prenatal stressors and gene × environment (G × E) interaction in ASD would represent a significant breakthrough, as reviewed recently (30, 31).
A significant proportion of stress-exposed mothers have unaffected children. Several factors could interact with stress exposure to increase impact on neurodevelopment. For example, prenatal exposure to air pollution, which is associated with ASD (32–35), can impact microglia–neuron interactions in a sex-specific manner (36), which may interact with prenatal stress exposure and further impact development. A potential reason why prenatal stressors might result in neurodevelopmental effects only in some cases could be a G × E interaction. One candidate is the serotonin transporter (SERT) gene, which is well-studied for its role in stress susceptibility. The SERT gene encodes for the SERT protein, which transports serotonin from the synaptic cleft back to the presynaptic neuron (37). Variations in this gene can alter aspects of its function (38–42). The most widely studied variation is an insertion or deletion of 44 base pairs within the promoter region of the SERT gene, SLC6A4, resulting in a long (L) or short (S) allele, respectively (37, 38, 40). The relationship between the S-allele (SS or LS genotype) and increased risk of depression after exposure to stress has not reliably been demonstrated (43–45). However, presence of the S-allele is also related to suicidality (46), alcoholism (47, 48), and susceptibility to anxiety (40), and greater activation of the amygdala, a brain region which is critical for fear reactions (49). The S-allele of the SERT gene has not been consistently linked to ASD itself (50–54), but its role as a gene mediating stress susceptibility remains of interest. It has also been recently demonstrated that maternal serotonin concentrations affect core symptoms and cognitive ability in ASD, with the lowest maternal serotonin levels associated with the greatest severity (55). Linkage studies have also associated rigid-compulsive behaviors in patients with ASD with the region of the genome containing SERT (56). A variation in a single nucleotide on the gene, Gly56Ala, is also linked to ASD (42). However, the SERT polymorphisms most reliably associated with ASD result in overactivity, rather than a loss of function. Therefore, for a potential role of the S-allele in a G × E interaction in ASD, the superimposed effect of prenatal stress on the maternal SERT genotype in this case may be distinct from the mechanism of the SERT polymorphisms directly associated with ASD.
The clinical salience of a G × E interaction was explored for SERT and prenatal stress exposure, demonstrating that the relationship between prenatal stress exposure and ASD appears to be mediated by maternal genetic susceptibility to stress, specifically by maternal presence of the S-allele, which has been shown in two independent patient populations (57). Stress surveys were administered to two independent samples of mothers of children with ASD. The SERT gene was genotyped for the L-alleles and S-alleles in the mothers. Those individuals that have the LS or SS genotypes (~64% of the population) are known from previous work (43, 45–49) to have increased stress reactivity and express lower levels of SERT than those individuals that have the L/L genotype. Thus, mothers were examined for the presence vs. absence of the S-allele as a genetic marker associated with stress reactivity and the presence vs. absence of prenatal stress according to stress surveys (57).
If the S-allele is a maternal risk factor for development of ASD with exposure to prenatal stress, one might expect that among mothers of children with ASD, a history of prenatal stress exposure during pregnancy would be observed more frequently in presence of the S-allele. In other words, if the association between prenatal stress and ASD was driven by the need for additional presence of maternal genetic susceptibility to stress, then this association between prenatal stress and ASD should be primarily observed in the pregnancies of mothers with this genetic susceptibility to stress. In both samples, the presence of the S-allele and the history of prenatal stress were found to significantly co-segregate in mothers of children with ASD within the critical period of pregnancy suggested in our previous work. To account for the possibility that the S-allele simply conferred increased recall of stress, or for some other reason greater exposure to during pregnancy, prenatal stress exposure history was obtained from the same mothers for the pregnancies of the proband's unaffected sibling. There was no increase in report of prenatal stress exposure regardless of genotype when these same mothers were queried about pregnancies of unaffected siblings. This suggests that the S-allele does not cause an overall increase in recall of stress or stress exposure during pregnancy. Rather, this provides support that the S-allele might serve as a genetic risk factor for increased maternal stress response in association with the development of ASD, and the effect is specific to exactly the same timeframe as reported by previous research using self-report measures and also the timeframe suggested by the Louisiana storms (21, 22, 57).
An animal model was developed to facilitate exploration of mechanism and experimental manipulations directed at developing treatment and prevention strategies. Social behavior was examined in the offspring of female SERT-heterozygous knockout (SERT-het) mice, whose SERT function is reduced 50%, comparable to that observed in humans with the S-allele and are known to have an increased susceptibility to stress (41, 58, 59), which are then exposed to chronic variable stress during gestation. In a 2 ×2 (stress × genotype) experimental design, SERT-wild-type (SERT-wt) and SERT-het dams were exposed to stress during gestation. A control group for each genotype had no stress exposure. This stress paradigm has previously been shown effective based on cortisol measurements but does not cause changes in feeding or body weight (60). Using the three-chamber social approach task (61), a significant maternal genotype × stress interaction was found, with unstressed offspring of wild-type mice spending significantly more time with the novel stranger than prenatally stressed offspring of SERT-het dams, supportive of a maternal gene/stress interaction in offspring behavior in the mouse model. These offspring of stressed heterozygous mothers did not demonstrate more general anxiety as assessed by elevated plus maze, suggesting specificity of this avoidance effect to the social domain (62).
To explore the mechanism of these effects, epigenetic markers were explored. Recent research revealed numerous gene expression changes associated with stress exposure. With maternal stress exposure in rodents, placental tissue showed increased expression in peroxisome proliferator-activated receptors α (PPARα), insulin-like growth factor-binding protein 1 (IGFBP-1), GLUT4, and HIF3α, specifically observed in placentas associated with male offspring (63), in addition to O-GlcNAc transferase (OGT) (64), of particular interest given the high percentage males with ASD. MicroRNAs (miRNAs) also play a significant regulatory role in serotonergic pathways (65, 66), immunity (67), and prenatal stress (68–70). Dysregulation of miR-103, miR-145, miR-219, miR-323, and miR-98 is associated with maternal stress (71). Inflammatory responses in the brain may be altered by miR-323 and miR-98 (71). MiR-135 regulates response to chronic stress through interaction with serotonergic activity (72). Furthermore, roles of specific miRNAs have been reported in regulating serotonergic genes (Let-7a) (66), SERT (miR-16 and miR-15a) (73, 74), and SLC6A4 (miR-325) (75). Recent work has shown that parental stress effects on offspring are also mediated by miRNA changes (76, 77).
Previous work explored the miRNA gene profile, expression, and methylation profile in the brains of the offspring of this SERT-het/stress model in mice, revealing a striking attenuation of the gene expression and miRNA changes in response to stress in the brains of the SERT-het/stress offspring mice in contrast to response to prenatal stress in the brains of SERT-wt mice (78). Significantly increased global methylation was observed in SERT-het/stress offspring brains, and there were more upregulated miRNA in stressed control mice as compared to wt, but not for SERT-het/stress compared to SERT-wt. Similarly, there were fewer upregulated genes when SERT-het/stress was compared to SERT-wt than when stressed control mice were compared to wt. Therefore, with increased methylation (generally suppressing gene expression), and decreased miRNA and overall expression, it appears that the typical epigenetic response to stress in offspring brains is blunted in the presence of maternal SERT-het (78).
Given the findings in the G × E mouse model, we began to explore the potential for detecting epigenetic changes in the clinical G × E setting. As an initial investigation in this direction, we examined the miRNA profile in the samples from the mothers in the previously described clinical G × E study to determine if significant changes were detectable in those mothers of children with ASD exposed to stress during pregnancy and whether these were further impacted by the maternal SERT genotype. Additionally, we performed exome sequencing of the maternal samples in an exploratory manner to determine whether there are other maternal factors that might be of interest for potential future exploration.
Thirty-four maternal blood samples from one of the sites in the previous G × E study were examined in this study. We did not have any samples with LS genotype in the low-stress group. Therefore, the samples were divided into five groups based on SERT genotypes and prenatal stress level, as shown in Table 1. Families with a child diagnosed with ASD under the age of 10 years were contacted from the University of Missouri Thompson Center for Autism & Neurodevelopmental Disorders database. All participants with ASD were below 10 years of age (average age = 6.8 ± 1.8) to maximize the parents' ability to recall information from the prenatal period. Families were invited to provide samples for genetic analysis and complete a questionnaire regarding the prenatal period. All ASD diagnoses were confirmed via Autism Diagnostic Interview-Revised (ADI-R) (79) and/or Autism Diagnostic Observation Scale (ADOS) (80) scores. Experimental procedures were approved and conducted in accordance with the University of Missouri Health Sciences Institutional Review Board. Blood was drawn via a standard venipuncture from the median cubital vein of the arm. Genomic DNA was obtained from the subjects' whole blood (FlexiGene Kit; Qiagen, Hilden, Germany) according to the manufacturer's instructions. PCR was performed as previously described (81). Briefly, the promoter region of the serotonin transporter gene was amplified using the Qiagen PCR kit from 25 ng genomic DNA to determine the SERT genotype, as described in a previous study (57), with 35% of mothers revealing the SS genotype, 32% the SL genotype, and 32% the LL genotype from the sample in the present study, where samples from mothers with at least three stressors were selected for the high-stress group, and samples from mothers with 0 or 1 stressor were selected for the low-stress group. At the time of the appointment with the experimenter, mothers completed questionnaires regarding their child with ASD as well as the gestational period in which the stressors occurred for that child. Details of the stress surveys to which the mothers replied are previously described (57). Survey questions obtained information on the child's birth date, pregnancy length, and the occurrence and subjective severity of major stressful events during the pregnancy. A list of common stressors was provided to the subjects to facilitate recall of events that may have occurred during the pregnancy. Severity of each stressor was also recorded using an established ranked scale of typical stressor intensities (21, 57).
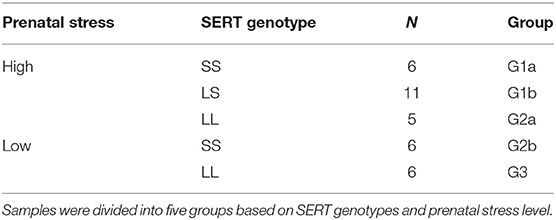
Table 1. Comparison groups.
Lymphocytes from all mothers, varying in SERT genotype and degree of stress exposure, were assessed for miRNA expression. Total RNAs were isolated using the mirVana kit (Ambion, Foster City, CA, USA) to capture small RNAs. miRNA profiling was conducted using a previously used service provider (LC Sciences, Houston, TX, USA), which applies an in-house-developed μParaflo® technology platform. Each region of chips used consists of miRNA probes, which detect miRNA transcripts listed in Sanger miRBase Release 21 (http://www.mirbase.org/). Multiple control probes were included on each chip for quality controls of chip production, sample labeling, and assay conditions. Image digitization was done using “Array-Pro Analyzer” (Media Cybernetics, Rockville, MD, USA). Normalization was carried out using LOWESS (locally weighted scatterplot smoothing), a pair-wise regression method, on background-subtracted data. The purpose of the experiment was to identify miRNAs that differ in expression levels (DE miRNAs) among the comparison groups. ANOVA was applied to detect DE miRNAs (p < 0.05), considering the variability of the expression levels across all samples (within and among the groups). miRDB was used to identify miRNA predicted target genes (82). Pathway analysis was conducted using the DAVID tool (83).
To explore other effects of maternal genotype, the same service provider was used to run whole-exome sequencing (WES). The Illumina HiSeq 2500/4000 platform was used with 50 × coverage depth. Data were analyzed using GATK (Genome Analysis Toolkit) and applying an analytical pipeline that included removing low-complexity sequences, aligning of sequence against reference human genome, excluding duplicate reads, sorting nucleotide sequence alignment, and base quality score and variant calling [INDEL and single-nucleotide variants (SNVs)]. Annotation and filtering of variants included minor allele frequency filtration (MAF < 0.05 using databases such as dbSNP, 1000 Genome, TopMed, and ExAC). False-positive variants commonly seen in WES were sorted out and removed, using recommendation by Fuentes Fajardo et al. (84). The functional effect of variants was assessed using prediction programs (SIFT and PolyPhen2), for retained variants with MAF < 5% and residing within the coding regions or 10 bases upstream/downstream from splicing junctions. Variants were divided into three categories based on their predicted effect on the protein function, with the following description: high-impact effects (splice site acceptor, splice site donor, start lost, exon deleted, frame shift, stop gained, stop lost), moderate-impact effects (non-synonymous coding, codon change/insertion/deletion, UTR 5′/UTR 3′ deletion), and low-impact effects (synonymous/non-synonymous start/stop, start gained, synonymous coding). We focused on high-impact variants.
MicroRNA expression in blood samples (n = 34) from mothers of children with ASD, with known pregnancy stress history, was profiled. Comparisons were conducted based on SERT genotypes (LL, LS, and SS) and prenatal stress level (high vs. low). Among the 2,500 mature miRNAs examined in all five groups, 119 miRNAs were found to be differentially expressed (DE). Ninety of the DE miRNAs (76%) showed a different pattern of expression in high vs. low stress-exposed groups (suggestive of being stress-dependent miRNAs); 77 (86%) of them were upregulated by stress and 13 (14%) were downregulated in the high- vs. low-stress groups, as shown in Supplementary Table 1A. Out of these DE miRNAs, the following three have been previously reported in association with stress: miR-1224-5p and miR-331-3p were found to be stress-dependent in offspring mouse brains by our group (78), and miR-145-5p has been reported in association with maternal stress (71, 85). Our previous work had suggested that prenatal stress exposure interacts with maternal stress susceptibility associated with the SERT genotype. To further explore this interaction, miRNA profiles were also assessed in the three groups exposed to a high level of prenatal stress, stratified by SERT genotypes. This analysis detected a smaller number of DE miRNAs (n = 20), as shown in Supplementary Table 1B. Moreover, five out of 20 (miR-663a, miR-664b-5p, miR-4787-5p, miR-6125, and miR-7704) were shared with the stress-dependent miRNAs, making them potential candidates for the SERT/stress mechanism.
For target predictions, we prioritized those DE miRNAs that are most likely to be associated with this G × E interaction model. To do so, we compiled a list of eight DE miRNAs (Table 2), including the five candidates for stress × SERT genotype interactions and the three that have been previously associated with stress, discussed above. A total of 1,074 genes were predicted for these eight DE miRNAs. Two hundred thirteen of them were annotated in the OMIM database. The leading functional target for these genes was the dopaminergic synapse, congruent with our effects on dopamine in the striatum described below in the gene × stress model (86), in addition to pathways associated with addiction and other neurotransmitter systems including glutamate, GABA, the serotonergic system, and the cholinergic system (Table 3). Additionally, DYRK1A was a predicted target for three of these miRNAs (miR-1224-5p, miR-145-5p, and miR-663a).
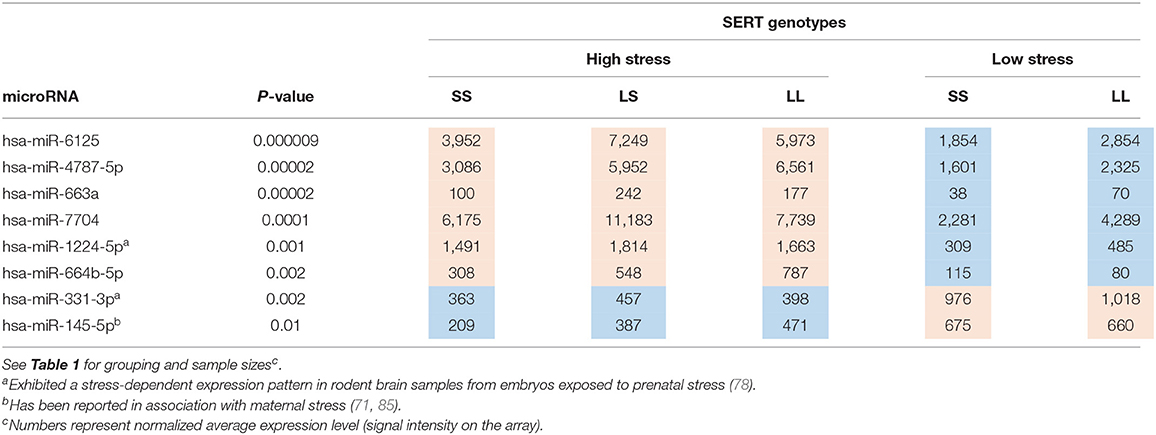
Table 2. Eight DE microRNAs candidate for stress × SERT interactions (stress upregulated = red, downregulated = bluec).
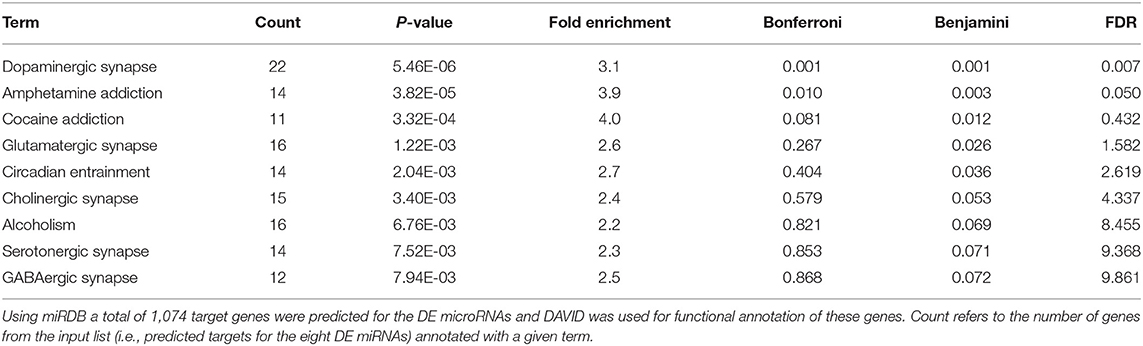
Table 3. Functional annotation of the predicted target genes for eight DE microRNAs listed in Table 2.
Furthermore, we found that high-stress groups exhibited a notably elevated level of high-impact SNVs compared with the low-stress groups, as shown in Supplementary Table 2. When comparing groups in the two extreme ends (G1a vs. G3), total number of SNVs per subjects were 4.9 times more in high-stress-exposure mothers with the SS genotype than those with low-stress-exposure and the LL genotype. SNVs seen in more than one subject in each group are listed in Table 4. In this list, we prioritized SNVs identified in G1a, the high-risk genotype (SS) group. Recurrent variants in four genes, AMY1C, CPA4, GC, and NOTCH2NL, were observed in this group. The NOTCH2NL and AMY1C variants were also detected in the LS and LL genotype groups, respectively.
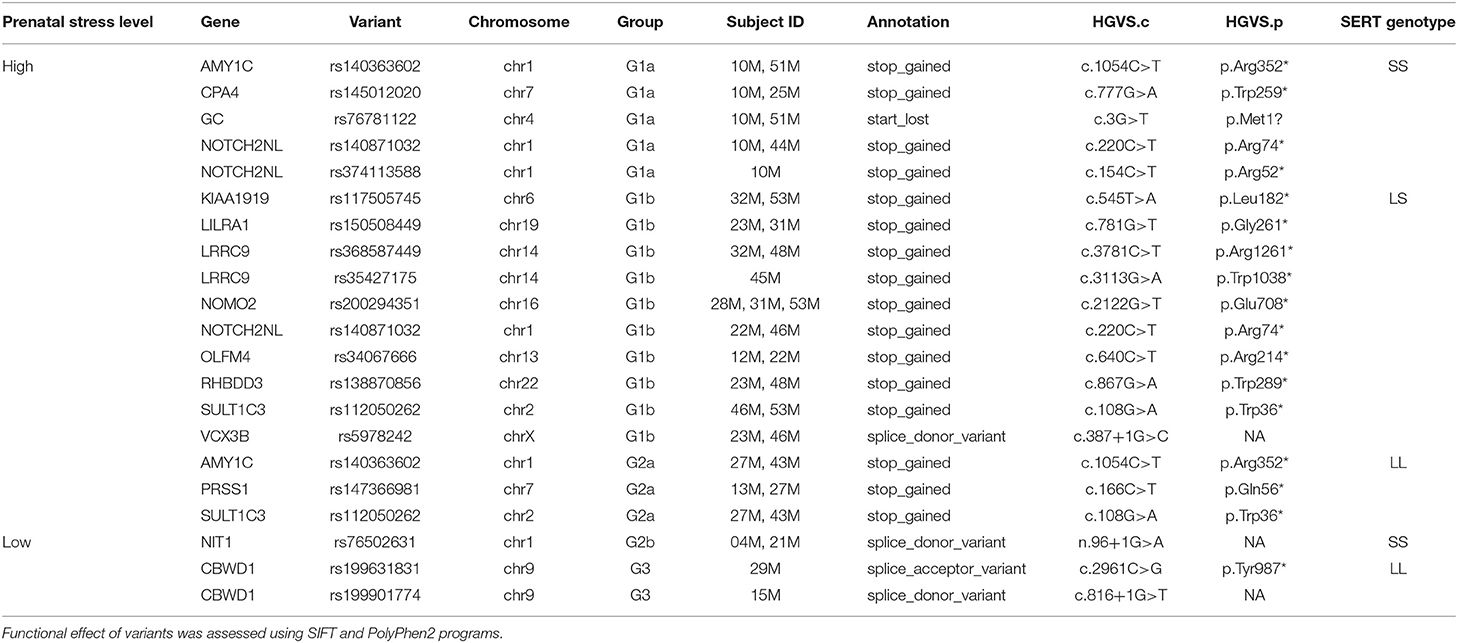
Table 4. High-impact recurrent SNVs identified in each group.
Our previous work supported a specific gene and stress interaction in the development of ASD (57). The present study provides evidence for epigenetic alterations in relation to a promising G × E model (prenatal maternal stress × SERT gene) in ASD detected in maternal blood samples. These findings are remarkable as these changes were detected in samples from mothers several years after stress-exposed pregnancies. Previous work has demonstrated an altered miRNA expression in the brains of offspring mice exposed to maternal stress and altered maternal serotonin transporter genotype (heterozygous KO) (78), with some of the same miRNA differentially expressed in the present study with human maternal blood. Additionally, DAVID revealed that the leading functional target of the miRNA differentially expressed by G × E in the maternal clinical samples was the dopaminergic synapse. This is of particular interest with the growing interest in the role of dopamine in social behavior (87), and the potential role for this as a treatment target in this subset of cases with ASD. The effects on dopaminergic synapse targets are also of particular interest given our recent finding that prenatal stress exposure in SERT-het mice resulted in increased striatal DA in offspring brains (86). Furthermore, these dopaminergic changes were reversed with DHA (86). The effects on glutamatergic and GABAergic targets are of particular interest in autism, given the importance of the excitatory/inhibitory imbalance in ASD (88), and the effects on serotonergic targets would be anticipated given the inclusion of maternal SERT genotype in the G × E interaction.
Persistent miRNA changes have been observed previously in other conditions, such as after cessation of smoking (89). Thus, the maternal miRNA changes observed after prenatal stress exposure associated with ASD appear to be long-lasting. This is an important step forward in our understanding of ASD, whose incidence appears to continue to rise, by identifying potential markers for an etiological subtype. Future work should explore these markers in children as well. Regulation of miRNA stability is currently an underappreciated area of research, but better understanding of its mechanism is likely to contribute to a broader range of regulation that can impact gene expression. New areas of investigation into how gene expression can be controlled by miRNA stability may provide novel advancements in therapeutic applications related to the fluctuations of gene expression associated with human disorders (90).
The finding that DYRK1A was targeted by three different DE miRNAs is of interest. DYRK1A was identified as a strong risk candidate gene for ASD based on a combination of recurrent de novo likely gene-disruptive mutations in affected individuals and their absence/very low frequency in controls (91). De novo dominant mutations in DYRK1A substantially reduce kinase function and account for ≈0.5% of severe developmental disorders (92). DYRK1A was found to be downregulated in samples from women exposed to Superstorm Sandy during pregnancy, across all trimesters (93). Overexpression of Dyrk1a causes a major deficit in the level of serotonin in the brain, as well as deficit of dopamine and adrenaline neurotransmitters in a transgenic mouse model for Down syndrome (94).
The exploratory exome sequencing revealed some findings of potential interest. First, the finding of NOTCH2NL variants in four individuals with high stress exposure is of particular interest due to its critical role in cortical development and radial glial stem cell proliferation, as well its association with the 1q21.1 distal deletion/duplication syndrome, where duplications are associated with autism (95). Additionally, the finding of AMY1C variants in four individuals with high stress exposure is of interest since salivary amylase is significantly correlated with repetitive behaviors in individuals with developmental disorders (96). Among the other SNVs, one variant, rs76781122 in GC, detected in two subjects from the high-stress-exposure group, in particular, caught our attention. This variant alters the start codon and creates a stop codon. The GC gene encodes a vitamin D-binding protein (VDBP), the major plasma carrier of vitamin D metabolites. A non-synonymous variant, R21L, in this gene has been reported in association with migraine, a condition often triggered by stress factors (97). Several SNPs in GC have been shown to effect vitamin D levels (98). The VDBP has also been associated with inflammatory-mediated conditions (99). Notably, vitamin D receptors are highly abundant in brain and involved in several key biological processes, including the serotonin-mediated pathways (100). One possible implication is that pregnant mothers, carriers of rs76781122, may have had decreased levels of VDBP, which further places them at risk for a compromised immune system in a stressful environment. When comparing the results from miRNAs profiling and WES, it was noted that six out of 1,047 predicted target genes (MADD, EPS15, PCDHGA10, WWOX, PGPEP1L, and NDUFA10) for the DE miRNAs (miR-1224-5p, miR-145-5p, miR-7704, miR-663a, miR-664b-5p, and miR-6125, respectively), also harbor SNVs in the high-stress groups. Further investigations are warranted to assess potential functional relation between these genes and the miRNAs.
Stress precipitates a systemic physiological response that involves inflammatory, cellular, and metabolic processes and their epigenetic regulation. Epidemiological studies have found increased susceptibility to schizophrenia, ASD, or ADHD to be associated with prenatal stress exposure. While exact pathophysiological mechanisms are still unknown, maternal immune activation, alteration of the hypothalamic–pituitary–adrenal axis, and epigenetic modifications regulating gene expression were proposed as potential causes of neuronal proliferation and migration disturbances in the developing fetus, which may lead to the increased susceptibility to these disorders (101, 102). Multiple studies using human whole blood reported that stress may cause shifts in the concentration levels of specific miRNAs and suggest their potential use as biomarkers in human whole blood (103–105).
While maternal exposure to stress during pregnancy is an elusive risk factor contributing to a wide range of ASD-like traits in offspring, a recent study involving children with ASD revealed that exposure to gestational stress can be used as a strong predictor of severity of ASD symptoms (p = 0.048) and communication abilities (p = 0.004), even after controlling for other variables. Moreover, significant increases in symptom severity were seen with multiple (two or more) prenatal stressful life events (29).
Recent research evidence suggests that miRNAs are both responsive and susceptible to significant environmental insults such as gestational stress and may increase the offspring vulnerability to stress-related psychopathological conditions (85).
The exact route through which maternal stress affects gene expression in the offspring's brain are not yet known, but involvement of epigenetic changes may be one such mechanism (106). Epigenetic processes such as microRNAs are among gene regulatory mechanisms that are influenced by environmental factors such as stress, and identifying their potential mis-regulation will contribute to narrowing the existing gap in our understanding of the mechanism of maternal stress and ASD.
The findings from the exome sequencing, while exploratory in nature, deserve further investigation. It will be critical to determine whether aspects of vitamin D metabolism, known to have a range of effects on offspring during development (107), might also contribute to developmental susceptibility to the effects of prenatal stress, or if the other SNVs might have critical salience, such as the potential for altered stress reactivity among mothers with AMY1C variants (96).
To our knowledge, this study is the first to examine a potential mechanism for this specific interaction between genetics and stressors during a specific prenatal period. These findings may serve as evidence of a biomarker for this mechanism, or possibly a common biomarker for several etiologies, which warrants further investigation. As numerous genes as well as SERT can affect stress reactivity, exploring epigenetic pathways by which this occurs more broadly will help to identify pathways of action regardless of the specific gene associated with stress exposure. Additionally, it will be critical for subsequent studies to examine the impact of other SERT variants, including the L(A) allele (108, 109), as well as others (110, 111). It will also be critical to better understand the time course of development of these markers, from at the time of initial stress exposure through birth, to determine other such markers that might be present earlier in the course. While the apparent persistence of these stress-associated miRNA changes is remarkable, and there is precedent for persistent miRNA changes in other settings (89), one cannot be certain as to the relationship here, and future studies will need to reevaluate these findings at an earlier time point, and eventually during pregnancy in a longitudinal study. However, this would not appear to be due to changes simply resulting from raising a child with ASD, since all samples were collected from mothers of children with ASD, and the miRNA findings herein are due to the effects of the G × E interaction within the population of mothers of children with ASD. We cannot exclude, though, the possibility that other maternal factors, may have contributed such as those identified in the exome sequencing, or it might be due to stress resulting from phenotypical differences in the G × E-associated cases, since previous work suggests that prenatal stress-associated autism can be more severe (29). Thus, future work will be needed to determine whether these miRNA findings might serve as a biomarker that could be targeted in approaches to mitigate the effects of prenatal stress.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI Gene Expression Omnibus, accession no: GSE179222.
The studies involving human participants were reviewed and approved by University of Missouri Health Sciences Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
DQB and ZT conceptualized and designed the study, coordinated and supervised data management, drafted the initial manuscript, and reviewed and revised the manuscript. ZT led the epigenetics analysis. DQB led the sample and clinical aspects. AS implemented the epigenetics analysis with ZT. AJ assisted with WES data analysis and literature review. DB assisted MT and PH with the genotyping. JN-M assisted with statistical data analysis and interpretation. PH obtained the clinical samples, stress surveys, and with DQB. MT oversaw the genetics for the samples. PH and MT also reviewed and revised the manuscript. BF coordinated all aspects of the between-site collaboration and reviewed and revised the manuscript. All authors approved of the final manuscript as submitted.
This study was funded by an Interdisciplinary Intercampus (IDIC) Research Program grant from the University of Missouri system to DB and ZT.
DB has been a consultant or on the advisory board for Yamo Pharmaceuticals, Stalicla Biosciences, Impel Pharmaceuticals, MA Pharmaceuticals, and Quadrant Biosciences, which are all unrelated to this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Portions of this work were initially presented at the INSAR 2018 Annual Meeting.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2021.668577/full#supplementary-material
ANOVA, analysis of variance; ADHD, attention deficit hyperactivity disorder; ADI-R, Autism Diagnostic Interview-Revised; ADOS, Autism Diagnostic Observation Scale; ASD, autism spectrum disorder; DNA, deoxyribonucleic acid; DOHaD, developmental origins of health and disease; DE, differentially expressed; G x E, Gene x Environment; GATK, Genome Analysis Toolkit; IGFBP-1, insulin-like growth factor-binding protein 1; LOWESS, locally weighted scatterplot smoothing; L, long; miRNA and micro RNA, micro ribonucleic acid; MAF, minor allele frequency; PPARα, peroxisome proliferator-activated receptors α; OGT, O-GlcNAc transferase; PCR, polymerase chain reaction; SERT, serotonin transporter; S, short; SNP, single nucleotide polymorphisms; SNVs, Single Nucleotide Variants; WES, Whole Exome Sequencing; wt, wild type.
1. Gage SH, Munafò MR, Smith GD. Causal inference in developmental origins of health and disease (DOHaD) research. Annu Rev Psychol. (2016) 67:567–85. doi: 10.1146/annurev-psych-122414-033352
2. Folstein S, Rutter M. Infantile Autism: A Genetic Study of 21 twin pairs. J Child Psychol Psychiatry. (1977) 18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x
3. Maestrini E, Paul A, Monaco AP, Bailey A. Identifying autism susceptibility genes. Neuron. (2000) 28:19–24. doi: 10.1016/S0896-6273(00)00081-7
4. Ritvo ER, Freeman BJ, Mason-Brothers A, Mo A, Ritvo AM. Concordance for the syndrome of autism in 40 pairs of afflicted twins. Am J Psychiatry. (1985) 142:74–7. doi: 10.1176/ajp.142.1.74
5. Trottier G, Srivastava L, Walker CD. Etiology of infantile autism: a review of recent advances in genetic and neurobiological research. J Psychiatry Neurosci. (1999) 24:103–15.
6. The Autism Genome Project Consortium. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. (2007) 39:319–28. doi: 10.1038/ng1985
7. Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychaitry. (2011) 68:1095–102. doi: 10.1001/archgenpsychiatry.2011.76
8. Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. Heritability of autism spectrum disorder. J Am Med Assoc. (2017) 318:1182–4. doi: 10.1001/jama.2017.12141
9. Peça J, Feliciano C, Ting J, Wang W, Wells MF, Venkatraman TN, et al. Shank 3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. (2011) 472:437–42. doi: 10.1038/nature09965
10. Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. (2007) 318:71–6. doi: 10.1126/science.1146221
11. Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci. (2010) 30:2115–29. doi: 10.1523/JNEUROSCI.4517-09.2010
12. Etherton MR, Blaiss CA, Powell CM, Südhof TC. Mouse neurexin-1α deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci USA. (2009) 105:17998–8003. doi: 10.1073/pnas.0910297106
13. Dawson G, Ashman SB, Carver LJ. The role of early experience in shaping behavioral and brain development and its implications for social policy. Dev Psychopathol. (2000) 12:695–712. doi: 10.1017/S0954579400004089
14. Niederhofer H, Reiter A. Maternal stress during pregnancy, its objectivation by ultrasound observation of fetal intrauterine movements and child's temperament at 6 months and 6 years of age: A pilot study. Psychol Rep. (2000) 86:526–8. doi: 10.2466/pr0.2000.86.2.526
15. Van Os J, Selten JP. Prenatal exposure to maternal stress and subsequent schizophrenia – the May 1940 invasion of The Netherlands. Br J Psychiatry. (1998) 172:324–6. doi: 10.1192/bjp.172.4.324
16. Ward AJ. Prenatal stress and child psychopathology. Child Psychiatry Hum Dev. (1991) 22:97–110. doi: 10.1007/BF00707788
17. Ward AJ. A comparison and analysis of the presence of family problems during pregnancy of mothers of “autistic” children and mothers of typically developing children. Child Psychiatry Hum Dev. (1990) 20:279–88. doi: 10.1007/BF00706020
18. Ward HE, Johnson EA, Salm AK, Birkle DL. Effects of prenatal stress on defensive withdrawal behavior and corticotropin releasing factor systems in rat brain. Physiol Behav. (2000) 70:359–66. doi: 10.1016/S0031-9384(00)00270-5
19. Weinstock M. Does prenatal stress impair coping and regulation of hypothalamic-pituitary-adrenal axis? Neurosci Biobehav Rev. (1997) 21:1–10. doi: 10.1016/S0149-7634(96)00014-0
20. Roberts AL, Lyall K, Rich-Edwards JW, Ascherio A, Weisskopf MG. Maternal exposure to childhood abuse is associated with elevated risk of autism. JAMA-Psychiatry. (2013) 70:508–15. doi: 10.1001/jamapsychiatry.2013.447
21. Beversdorf DQ, Manning SE, Hillier A, Anderson SL, Nordgren RE, Walters SE, et al. Timing of prenatal stressors and autism. J Autism Dev Disord. (2005) 35:471–8. doi: 10.1007/s10803-005-5037-8
22. Kinney DK, Miller AM, Crowley DJ, Huang E, Gerber E. Autism prevalence following prenatal exposure to hurricanes and tropical storms in Louisiana. J Autism Devel Disord. (2008) 28:481–8. doi: 10.1007/s10803-007-0414-0
23. Li J, Vestergaard M, Obel C, Christensen J, Precht DH, Lu M, Olsen J. A nationwide study on the risk of autism after prenatal stress exposure to maternal bereavement. Pediatrics. (2009) 123:1102–7. doi: 10.1542/peds.2008-1734
24. Larsson JH, Eaton WW, Madsen KM, Vestergaard M, Olesen AV, Agerbo E, et al. Risk factors for autism: perinatal factors, parental psychiatric history, socioeconomic status. Am J Epidemiol. (2005) 161:916–25. doi: 10.1093/aje/kwi123
25. Class QA, Abel KM, Khashan AS, Rickert ME, Dalman C, Larsson H, et al. Offspring psychopathology following preconception, prenatal and postnatal maternal bereavement stress. Psychol Med. (2014) 44:71–84. doi: 10.1017/S0033291713000780
26. Roberts AL, Lyall K, Rich-Edwards JW, Ascherio A, Weisskopf MG. Maternal exposure to intimate partner abuse before birth is associated with risk of autism spectrum disorder in offspring. Autism. (2016) 20:26–36. doi: 10.1177/1362361314566049
27. Adelman M. In utero Exposure to Maternal Stress: Effects of the September 11th Terrorist Attacks in New York City on Birth and Early Schooling Outcomes. Dissertation: Harvard University (2012).
28. Manzari N, Matvienko-Sikar K, Baldoni F, O'Keefe GW, Khashan AS. Prenatal maternal stress and risk of neurodevelopmental disorders in the offspring: a systematic review and meta-analysis. Soc Psychiatry Psychiatr Epidemiol. (2019) 54:1299–309. doi: 10.1007/s00127-019-01745-3
29. Vacrin KJ, Alvares GA, Uljarević M, Whitehouse AJO. Prenatal maternal stress and phenotypic outcomes in autism spectrum disorder. Autism Res. (2017) 10:1866–77. doi: 10.1002/aur.1830
30. Beversdorf DQ, Stevens HE, Jones KL. Prenatal stress, maternal immune dysregulation, and their association with autism spectrum disorders. Curr Psychiatry Rep. (2018) 20:76. doi: 10.1007/s11920-018-0945-4
31. Abbott PW, Gumusoglu SB, Bittle J, Beversdorf DQ, Stevens HE. Prenatal stress and genetic risk: how prenatal stress interacts with genetics to alter risk for psychiatric illness. Psychoneuroendocrinology. (2018) 90:9–21. doi: 10.1016/j.psyneuen.2018.01.019
32. Kalkbrenner AE, Windham GC, Serre ML, Akita Y, Wang X, Hoffman K, et al. Particulate matter exposure, prenatal and postnatal windows of susceptibility, and autism spectrum disorders. Epidemiology. (2015) 26:30–42. doi: 10.1097/EDE.0000000000000173
33. Raz R, Roberts AL, Lyall K, Hart JE, Just AC, Laden F, et al. Autism spectrum disorder and particulate matter air pollution before, during, and after pregnancy: a nested case-control analysis within the Nurses' Health Study II Cohort. Environ Health Perspect. (2015) 123:264–70. doi: 10.1289/ehp.1408133
34. Volk HE, Kerin T, Lurmann F, Hertz-Picciotto I, McConnell R, Campbell DB. Autism spectrum disorder: interaction of air pollution with the MET receptor tyrosine kinase gene. Epidemiology. (2014) 25:44–7. doi: 10.1097/EDE.0000000000000030
35. von Ehrenstein OS, Aralis H, Cockburn M, Ritz B. In utero exposure to toxic air pollutants and risk of childhood autism. Epidemiology. (2014) 25:851–8. doi: 10.1097/EDE.0000000000000150
36. Bolton JL, Marinero S, Hassanzadeh T, Natesan D, Le D, Belliveau C, et al. Gestational exposure to air pollution alters cortical volume, microglial morphology, and microglia-neuron interactions in a sex-specific manner. Front Synaptic Neurosci. (2017) 9:10. doi: 10.3389/fnsyn.2017.00010
37. Murphy DL, Lerner A, Rudnick G, Lesch KP. Serotonin transporter: Gene, genetic disorders, and pharmacogenomics. Mol Interv. (2004) 4:109–23. doi: 10.1124/mi.4.2.8
38. Heils A, Teufel A, Petri S, Stober G, Riederer P, Bengel D, et al. Allelic variation of human serotonin transporter gene expression. J Neurochem. (1996) 66:2621–4. doi: 10.1046/j.1471-4159.1996.66062621.x
39. Heinz A, Jones DW, Mazzanti C, Goldman D, Ragan P, Hommer D, et al., Weinberger DR. A relationship between serotonin transporter genotype and in vivo protein expression and alcohol neurotoxicity. Biol Psychiatry. (2000) 47:643–9. doi: 10.1016/S0006-3223(99)00171-7
40. Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. (1996) 274:1527–31. doi: 10.1126/science.274.5292.1527
41. Little KY, McLaughlin DP, Zhang L, Livermore CS, Dalack GW, McFinton PR, et al. Cocaine, ethanol, and genotype effects on human midbrain serotonin transporter binding sites and mRNA levels. Am J Psychiatry. (1998) 155:207–13.
42. Prasad HC, Zhu CB, McCauley J, Samuvel DJ, Ramamoorthy S, Shelton RC, et al. Human serotonin transporter variants display altered sensitivity to protein kinase G and p38 mitogen-activated protein kinase. Proc Natl Acad Sci USA. (2005) 102:11545–50. doi: 10.1073/pnas.0501432102
43. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-htt gene. Science. (2003) 301:386–9. doi: 10.1126/science.1083968
44. Risch N, Herrell R, Lehner T, Liang KY, Eaves L. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression. A meta-analysis. J Am Med Assoc. (2009) 301:2462–71. doi: 10.1001/jama.2009.878
45. Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter (5-HTTLPR), stress, and depression meta-analysis revisited: evidence for genetic moderation. Arch Gen Psychiatry. (2011) 68:444–54. doi: 10.1001/archgenpsychiatry.2010.189
46. Bondy B, Buettner A, Zill P. Genetics of suicide. Mol Psychiatry. (2006) 11:336–51. doi: 10.1038/sj.mp.4001803
47. Feinn R, Nellissery M, Kranzler HR. Meta-analysis of the association of a functional serotonin transporter promoter polymorphism with alcohol dependences. Am J Med Genet B Neuropsychiatr Genet. (2005) 133:79–84. doi: 10.1002/ajmg.b.30132
48. Sander T, Harms H, Lesch KP, Dufeu P, Kuhn S, Hoehe M, et al. Association analysis of a regulatory variation of the serotonin transporter gene with severe alcohol dependence. Alcohol Clin Exp Res. (1997) 21:1356–9. doi: 10.1111/j.1530-0277.1997.tb04462.x
49. Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, et al. Serotonin transporter genetic variation and the response of the human amygdala. Science. (2002) 297:400–3. doi: 10.1126/science.1071829
50. Brune C, Kim S, Salt J, Leventhal B, Lord C, Cook E. 5-HTTLPR genotype-specific phenotype in children and adolescents with autism. Am J Psychiatry. (2006) 163:2148–56. doi: 10.1176/ajp.2006.163.12.2148
51. Cook E, Courchesne R, Lord C, Cox N, Yan S, Lincoln A, et al. Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiatry. (1997) 2:247–50. doi: 10.1038/sj.mp.4000266
52. Freitag CM. The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. (2007) 12:2–22. doi: 10.1038/sj.mp.4001896
53. Losh M, Sullivan P, Trembath D, Piven J. Current developments in the genetics of autism: from phenome to genome. J Neuropathol Exp Neurol. (2008) 67:829–37. doi: 10.1097/NEN.0b013e318184482d
54. Zhong N, Ye L, Ju W, Brown W, Tsiouris J, Cohen I. 5-HTTLPR variants not associated with autistic spectrum disorders. Neurogenetics. (1999) 2:129–31. doi: 10.1007/s100480050064
55. Montgomery AK, Shuffrey LC, Guter SJ, Anderson GM, Jacob S, Mosconi MW, et al. Maternal serotonin levels are associated with cognitive ability and core symptoms in autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. (2018) 57:867–75. doi: 10.1016/j.jaac.2018.06.025
56. McCauley JL, Olson LM, Dowd M, Amin T, Steele A, Blakely RD, et al. Linkage and association analysis at the serotonin transporter (SLC6A4) locus in a rigid compulsive subset of autism. Am J Med Genet B Neuropsychiatr Genet. (2004) 127:104–12. doi: 10.1002/ajmg.b.20151
57. Hecht PM, Hudson M, Connors SL, Tilley MR, Liu X, Beversdorf DQ. Maternal serotonin transporter genotype affects risk for ASD with exposure to prenatal stress. Autism Res. (2016) 9:1151–60. doi: 10.1002/aur.1629
58. Bengel D, Murphy DL, Andrews AM, Wichems, C H, Feltner D, et al. Altered brain serotonin homeostasis and locomotor insensitivity to 3,4-methylenedioxymethamphetamine (“Ecstasy”) in serotonin transporter-deficient mice. Mol Pharmacol. (1998) 53:649–55. doi: 10.1124/mol.53.4.649
59. Montanez S, Owens WA, Gould GG, Murphy DL, & Daws LC. Exaggerated effect of fluvoxamine in heterozygote serotonin transporter knockout mice. J Neurochem. (2003) 86:210–9. doi: 10.1046/j.1471-4159.2003.01836.x
60. Mueller BR, Bale TL. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol Behav. (2006) 88:605–14. doi: 10.1016/j.physbeh.2006.05.019
61. Nadler JJ, Moy SS, Dold G, Trang D, Simmons N, Perez A, et al. Automated apparatus for quantification of social approach behaviors in mice. Genes Brain Behav. (2004) 3:303–14. doi: 10.1111/j.1601-183X.2004.00071.x
62. Jones KL, Smith RM, Edwards KS, Givens B, Tilley MR, Beversdorf DQ. Combined effect of maternal serotonin transporter genotype and prenatal stress in modulating offspring social interaction. Int J Devel Neurosci. (2010) 28:529–36. doi: 10.1016/j.ijdevneu.2010.05.002
63. Bale TL. Sex differences in prenatal epigenetic programming of stress pathways. Stess. (2011) 14:348–56. doi: 10.3109/10253890.2011.586447
64. Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci USA. (2013) 110:5169–74. doi: 10.1073/pnas.1300065110
65. Millan MJ. MicroRNA in the regulation and expression of serotonergic transmission in the brain and other tissues. Curr Opin Pharmacol. (2011) 11:11–22. doi: 10.1016/j.coph.2011.01.008
66. Bai M, Zhu XZ, Zhang Y, Zhang S, Zhang L, Xue L, et al. Anhedonia was associated with the dysregulation of hippocampal HTR4 and microRNA Let-7a in rats. Physiol Behav. (2014) 129:135–41. doi: 10.1016/j.physbeh.2014.02.035
67. Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene. (2013) 532:1–12. doi: 10.1016/j.gene.2012.12.009
68. Monteleone MC, Adrover E, Pallarés ME, Antonelli MC, Frasch AC, Brocco MA. Prenatal stress changes the glycoprotein GPM6A gene expression and induces epigenetic changes in rat offspring brain. Epigenetics. (2014) 9:152–60. doi: 10.4161/epi.25925
69. Singh NP, Singh UP, Guan H, Nagarkatti P, Nagarkatti M. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS ONE. (2012) 7:e45054. doi: 10.1371/journal.pone.0045054
70. Babenko O, Kovalchuk I, Metz GA. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci Biobehav Rev. (2015) 48:70–91. doi: 10.1016/j.neubiorev.2014.11.013
71. Zucchi FC, Yao Y, Ward ID, Ilnytskyy Y, Olson DM, Benzies K, et al. Maternal stress induces epigenetic signatures of psychiatric and neurological diseases in the offspring. PLoS ONE. (2013) 8:e56967. doi: 10.1371/journal.pone.0056967
72. Issler O, Haramati S, Paul ED, Maeno H, Navon I, Zwang R, et al. MicroRNA 135 is essential for chronic stress resiliency, antidepressant efficacy, and intact serotonergic activity. Neuron. (2014) 83:344–60. doi: 10.1016/j.neuron.2014.05.042
73. Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O. miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science. (2010) 329:1537–41. doi: 10.1126/science.1193692
74. Moya PR, Wendland JR, Salemme J, Fried RL. Murphy DmiR-15a L, and miR-16 regulate serotonin transporter expression in human placental and rat brain raphe cells. Int J Neuropsychopharmacol. (2013) 16:621–9. doi: 10.1017/S1461145712000454
75. Arisawa T, Tahara T, Fukuyama T, Hayashi R, Matsunaga K, Hayashi N, et al. Genetic polymorphism of pri-microRNA 325, targeting SLC6A4 3'-UTR, is closely associated with the risk of functional dyspepsia in Japan. J Gastroenterol. (2012) 47:1091–8. doi: 10.1007/s00535-012-0576-1
76. Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci. (2013) 33:9003–12. doi: 10.1523/JNEUROSCI.0914-13.2013
77. Rodgers AB, Morgan CP, Leu NA, Bale TL. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci USA. (2015) 112:13699–704. doi: 10.1073/pnas.1508347112
78. Sjaarda CP, Hecht P, McNaughton AJM, Zhou A, Hudson ML, Will MJ, et al. Interplay between maternal Slc6a4 mutation and prenatal stress: a possible mechanism for autistic behavior development. Sci Rep. (2017) 7:8735. doi: 10.1038/s41598-017-07405-3
79. Lord C, Rutter N, Goode S, Heemsbergen J, Jordan H, Mawhood L, et al. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J Autism Dev Disord. (1989) 19:185–212. doi: 10.1007/BF02211841
80. Lord C, Rutter N, LeCouteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. (1994) 24:659–85. doi: 10.1007/BF02172145
81. Wendland JR, Martin BJ, Kruse MR, Lesch KP, Murphy DL. Simultaneous genotyping of four functional loci of human SLC6A4, with a reappraisal of 5-HTTLPR and rs25531. Mol Psychiatry. (2006) 11:224–6. doi: 10.1038/sj.mp.4001789
82. Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. (2008) 14:1012–7. doi: 10.1261/rna.965408
83. Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Re. (2007) 35:W169–75. doi: 10.1093/nar/gkm415
84. Fuentes Fajardo KV, Adams D, NISC Comparative Sequencing Program, Mason CE, Sincan M, Tifft C, et al. Detecting false-positive signals in exome sequencing. Hum Mutat. (2012) 33:609–13. doi: 10.1002/humu.22033
85. Hollins SL, Cairns MJ. MicroRNA: small RNA mediators of the brains genomic response to environmental stress. Prog Neurobiol. (2016) 143:61–81. doi: 10.1016/j.pneurobio.2016.06.005
86. Matsui F, Hecht P, Yoshimoto K, Watanabe Y, Morimoto M, Fritsche K, et al. DHA mitigates autistic behaviors accompanied by dopaminergic change on a gene/prenatal stress mouse model. Neuroscience. (2017) 371:407–19. doi: 10.1016/j.neuroscience.2017.12.029
87. Kopec AM, Smith CJ, Bilbo SD. Neuro-immune mechanisms regulating social behavior: dopamine as mediator? Trends Neurosci. (2019) 42:337–47. doi: 10.1016/j.tins.2019.02.005
88. Hegarty JP, Weber DJ, Cirstea CM, Beversdorf DQ. Cerebro-cerebellar functional connectivity is associated with cerebellar excitation-inhibition balance in autism spectrum disorder. J Autism Devel Disord. (2018) 48:3460–73. doi: 10.1007/s10803-018-3613-y
89. Wang G, Wang R, Strulovici-Barel Y, Salit J, Staudt MR, Ahmed J, et al. Persistence of smoking-induced dysregulation of miRNA expression in the small airway epithelium despite smoking cessation. PLoS ONE. (2015) 10:e0120824. doi: 10.1371/journal.pone.0120824
90. Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, et al. Differential regulation of microRNA stability. RNA. (2010) 16:1032–9. doi: 10.1261/rna.1851510
91. Iossifov I, Levy D, Allen J, Ye K, Ronemus M, Lee YH, et al. Low load for disruptive mutations in autism genes and their biased transmission. Proc Natl Acad Sci USA. (2015) 112:E5600–7. doi: 10.1073/pnas.1516376112
92. Evers JM, Laskowski RA, Bertolli M, Clayton-Smith J, Deshpande C, Eason J, et al. Structural analysis of pathogenic mutations in the DYRK1A gene in patients with developmental disorders. Hum Mol Genet. (2017) 26:519–26. doi: 10.1093/hmg/ddw409
93. Zhang W, Li Q, Deyssenroth M, Lambertini L, Finik J, Ham J, et al. Timing of prenatal exposure to trauma and altered placental expressions of HPA-Axis genes and genes driving neurodevelopment. J Neuroendocrinol. (2018) 30:e12581. doi: 10.1111/jne.12581
94. London J, Rouch C, Bui LC, Assayag E, Souchet B, Daubigney F, et al. Overexpression of the DYRK1A Gene (Dual-specificity tyrosine phosphorylation-regulated kinase 1A) induces alterations of the serotoninergic and dopaminergic processing in murine brain tissues. Mol Neurobiol. (2018) 55:3822–31. doi: 10.1007/s12035-017-0591-6
95. Fiddes IT, Lodewijk GA, Mooring M, Bosworth CM, Ewing AD, Mantalas GL. Human-specific NOTCH2NL genes affect notch signaling and coritcal neogenesis. Cell. (2018) 173:1356–69.e22. doi: 10.1016/j.cell.2018.03.051
96. Symons FJ, Wolff JJ, Stone LS, Lim TKY, Bodfish JW. Salivary biomarkers of HPA axis and autonomic activity in adults with intellectual disability with and without stereotyped and self-injurious behavior disorders. J Neurodev Disord. (2011) 3:144–51. doi: 10.1007/s11689-011-9080-9
97. Nagata E, Fujii N, Hosomichi K, Mitsunaga S, Suzuki Y, Mashimo Y, et al. Possible association between dysfunction of vitamin D binding protein (GC Globulin) and migraine attacks. PLoS ONE. (2014) 9:e105319. doi: 10.1371/journal.pone.0105319
98. Disanto G, Ramagopalan SV, Para AE, Handunnetthi L. The emerging role of vitamin D binding protein in multiple sclerosis. J Neurol. (2011) 258:353–8. doi: 10.1007/s00415-010-5797-8
99. Wilson RT, Bortner JD, Roff A, Das A, Battaglioli EJ, Richie JP, et al. Genetic and environmental influences on plasma vitamin D binding protein concentrations. Transl Res. (2015) 165:667–76. doi: 10.1016/j.trsl.2014.08.003
100. Huang JY, Arnold D, Qiu CF, Miller RS, Williams MA, Enquobahrie DA. Association of serum vitamin D with symptoms of depression and anxiety in early pregnancy. Womens Health. (2014) 23:588–95. doi: 10.1089/jwh.2013.4598
101. Udagawa J, Hino K. Impact of maternal stress in pregnancy on brain function of the offspring. Nihon Eiseigaku Zasshi. (2016) 71:188–94. doi: 10.1265/jjh.71.188
102. Khambadkone SG, Cordner ZA, Tamashiro KLK. Maternal stressors and the developmental origins of neuropsychiatric risk. Front Neuroendocrinol. (2020) 57:100834. doi: 10.1016/j.yfrne.2020.100834
103. Wiegand C, Savelsbergh A, Heusser P. MicroRNAs in psychological stress reactions and their use as stress-associated biomarkers, especially in human saliva. Biomed Hub. (2017) 10:1–15. doi: 10.1159/000481126
104. Beech RD, Leffert JJ, Lin A, Hong KA, Hansen J, Umlauf S, et al. Stress-related alcohol consumption in heavy drinkers correlates with expression of miR-10a, miR-21, and components of the TAR-RNA-binding protein-associated complex. Alcohol Clin Exp Res. (2014) 38:2743–53. doi: 10.1111/acer.12549
105. Vaisvaser S, Modai S, Farberov L, Lin T, Sharon H, Gilam A, et al. Neuro-epigenetic indications of acute stress response in humans: the case of MicroRNA-29c. PLoS ONE. (2016) 11:e0146236. doi: 10.1371/journal.pone.0146236
106. Bale TL. Epigenetic and transgenerational reprogramming of brain development. Nat Rev Neurosci. (2015) 16:332–44. doi: 10.1038/nrn3818
107. Belenchia AM, Jones KL, Will M, Beversdorf DQ, Vieria-Potter V, Rosenfeld CS, et al. Maternal vitamin D deficiency during pregnancy affects expression of adipogenic-regulating genes peroxisome proliferator-activated receptor gamma (PPAR?) and vitamin D receptor (VDR) in lean male mice offspring. Eur J Nutr. (2018) 57:723–30. doi: 10.1007/s00394-016-1359-x
108. Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD, et al. Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. (2006) 78:815–26. doi: 10.1086/503850
109. Kenna GA, Roder-hanna N, Leggio L, Zywiak WH, Clifford J, Edwards S, et al. Association of the 5-HTT gene-linked promoter region (5-HTTLPR) polymorphism with psychiatric disorders : review of psychopathology and pharmacotherapy. Pharmacogenom Personal Med. (2012) 5:19–35. doi: 10.2147/PGPM.S23462
110. Gyawali S, Subaran R, Weissman MM, Hershkowitz D, McKenna MC, Talati A, et al. Association of a polyadenylation polymorphism in the serotonin transporter and panic disorder. Biol Psychiatry. (2010) 67:33138. doi: 10.1016/j.biopsych.2009.10.015
Keywords: autism spectrum disorder, prenatal stress, miRNA, epigenetics, dopamine, gene x environment
Citation: Beversdorf DQ, Shah A, Jhin A, Noel-MacDonnell J, Hecht P, Ferguson BJ, Bruce D, Tilley M and Talebizadeh Z (2021) microRNAs and Gene–Environment Interactions in Autism: Effects of Prenatal Maternal Stress and the SERT Gene on Maternal microRNA Expression. Front. Psychiatry 12:668577. doi: 10.3389/fpsyt.2021.668577
Received: 16 February 2021; Accepted: 24 May 2021;
Published: 05 July 2021.
Edited by:
Jeremy Veenstra-VanderWeele, Columbia University Irving Medical Center, United StatesReviewed by:
Munis Dundar, Erciyes University, TurkeyCopyright © 2021 Beversdorf, Shah, Jhin, Noel-MacDonnell, Hecht, Ferguson, Bruce, Tilley and Talebizadeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Q. Beversdorf, YmV2ZXJzZG9yZmRAaGVhbHRoLm1pc3NvdXJpLmVkdQ==; Zohreh Talebizadeh, enRhbGViaUBjbWguZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.