 Whitney Worsham
Whitney Worsham Susan Dalton2
Susan Dalton2 Deborah A. Bilder
Deborah A. Bilder
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 01 July 2021
Sec. Molecular Psychiatry
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.655438
This article is part of the Research Topic Molecular and Genetic Mechanisms in Neurodevelopmental Disorders: From Bench to Bedside View all 10 articles
Though the etiology of autism spectrum disorder (ASD) remains largely unknown, recent findings suggest that hormone dysregulation within the prenatal environment, in conjunction with genetic factors, may alter fetal neurodevelopment. Early emphasis has been placed on the potential role of in utero exposure to androgens, particularly testosterone, to theorize ASD as the manifestation of an “extreme male brain.” The relationship between autism risk and obstetric conditions associated with inflammation and steroid dysregulation merits a much broader understanding of the in utero steroid environment and its potential influence on fetal neuroendocrine development. The exploration of hormone dysregulation in the prenatal environment and ASD development builds upon prior research publishing associations with obstetric conditions and ASD risk. The insight gained may be applied to the development of chronic adult metabolic diseases that share prenatal risk factors with ASD. Future research directions will also be discussed.
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by persistent deficits in social interaction and communication in addition to stereotyped, repetitive behaviors (1). While the etiology of ASD remains largely unknown, multifactorial contributors such as genetics, neuroanatomical abnormalities, and the environment likely play a role (2). Prenatal factors associated with elevated ASD risk include maternal conditions that contribute to a suboptimal prenatal environment, particularly as it relates to inflammation, metabolism, and steroid hormone regulation (3, 4).
Prenatal stress (5, 6), maternal immune dysfunction (5, 7–9), pre-existing/gestational diabetes (3, 10), pre-pregnancy obesity (11), weight gain during pregnancy (12), pre-existing/gestational hypertension (13, 14), polycystic ovarian syndrome (PCOS) (4, 15) and prenatal complications such as low birth weight (3) and pre-term birth (16) have all been associated with ASD in offspring. How these conditions may alter prenatal mechanisms promoting ASD pathogenesis has yet to be clearly established, though several physiological processes that link these conditions have been implicated including direct insults (e.g., oxidative stress, hypoxia, inflammation) and adaptive responses (e.g., epigenetic changes, fetal programming) (17–20).
Studying the relevance of prenatal risk factors in ASD pathogenesis inherently involves understanding the remarkable, well-coordinated interaction among the mother, fetus, and placenta—referred to as the maternofetoplacental unit (21, 22). The interdependence across the maternofetoplacental unit for steroid hormone production, immune response mediation, and nutrient transfer is particularly relevant to sustain pregnancy and ensure newborn viability. The placenta, together with the amnion and chorion, create a unique immune environment that permits co-existence of the fetal allograft within the mother while accessing her rich nutrient and oxygen supply for development, growth, and survival.
Immune response and steroid production are intrinsically linked during pregnancy with the placenta acting as the key mediator, particularly regarding estradiol and progesterone synthesis. Estradiol augments both cell- and antibody-mediated immune responses (23). Rising levels of estradiol during pregnancy promote an immunologic shift from an inflammatory state to a regulatory response (23–26). The dramatic increases in progesterone enhance maternal-fetal tolerance (27) and promote anti-inflammatory factors through progesterone-induced binding factor (PIBF) (28, 29). Placental progesterone, estradiol, and human chorionic gonadotropin (hCG) facilitate blastocyst implantation and spiral artery formation, which are essential early gestational events to sustain pregnancy. These hormones support implantation by recruiting immune cells to the maternal-fetal interface (30–33). Immune cells subsequently secrete angiogenic factors such as transforming growth factor beta (TGF-β) and vascular endothelial growth factor (VEGF) (31) to promote spiral artery formation (34) and placental vascularization.
The influence that placental steroid hormone production exerts across the maternofetoplacental unit—involving inflammatory responses, oxygen/nutrient exchange, and neuroendocrine functioning—cannot be overstated. This review article will describe the current understanding of the in utero steroid-related environment associated with ASD and explore its potential role in ASD pathogenesis. Existing literature that is relevant to the prenatal hormone milieu in ASD largely appears to fall within the following four domains: obstetric conditions, fetal programming, sex differential, and steroid-related biomarkers. The literature on each domain will be summarized.
Hypertensive disorders during pregnancy, regardless of subset (i.e., chronic, gestational, de novo or superimposed preeclampsia), have been correlated with elevated ASD incidence among offspring (13, 14, 35–37). Placental insufficiency is one of the most concerning complications of hypertension during pregnancy (22, 38, 39). Common indicators or complications of placental insufficiency that overlap with ASD risk factors include small for gestational age/in utero growth restriction, prematurity, maternal infection, and maternal metabolic syndrome (16, 38–42). Histologic signs of placental insufficiency, such as trophoblastic inclusions, are also more commonly found in placentas of children who develop ASD (43). Steroid hormone dysregulation, altered immune function, and placental insufficiency are intertwined, as maternal serum inflammatory markers (e.g., atypical cytokine profiles, leukocytosis, and elevated platelet counts) are associated with gestational hypertension (44), while the maternal cardiovascular adaption to pregnancy is influenced by placental estrogen and progesterone production (45).
Maternal diabetes, both pre-existing and gestational onset, is also an established ASD risk factor (46, 47). While the exact mechanism underlying the relationship between ASD and maternal diabetes is unknown, maternal diabetes can lead to several obstetric complications affecting the mother (e.g., gestational hypertension, pre-eclampsia) and baby (e.g., high or low birth weight, shoulder dystocia, hypoglycemia, hyperbilirubinemia, hypocalcemia, and respiratory distress) (48, 49). These conditions result from the amplification of the typical prenatal metabolic state characterized by hypercortisolemia (50) and insulin resistance (51, 52). During uncomplicated pregnancies, relative maternal hypercortisolemia and insulin resistance facilitate adequate transfer of nutritional resources from mother to baby. As pregnancy progresses, a developmental switch prompted by rising fetal cortisol synthesis shifts resource allocation from tissue proliferation to maturation (53). In maternal diabetes, excess glucose supply to the fetus causes higher fetal insulin production to maintain glucose homeostasis, stimulating fetal overgrowth and delaying lung maturation (54). Diabetes during pregnancy also leads to higher placental release of pro-inflammatory cytokines (e.g., leptin, tumor necrosis factor-α (TGF-α), and interleukins) (55), with the potential consequence of reducing oxygen diffusion across the placenta by enhancing placental thickening (48).
Epidemiologic studies identify increased ASD risk associated with pre-pregnancy obesity (46) and/or gestational weight gain (2, 12, 56). Obesity promotes both inflammation and endocrine dysfunction, perturbing the prenatal environment. As a pro-inflammatory state, obesity contributes to elevated lipids, leptin, and IL-6 during pregnancy (57). The bioavailability and synthesis of estrogens are impacted through the endocrine and metabolic function of adipose tissue (58). Furthermore, studies exploring the in utero steroid environment, primarily as it relates to hormone-sensitive cancer risk among offspring, have found elevated maternal serum estrogen, progesterone, and testosterone levels in pregnancies characterized by higher weight gain (58–61). Because the gestational weight gain associated with increased ASD risk (12) is not clinically relevant from an obstetric perspective (i.e., about three pounds), its link with ASD may relate to a shared etiology rather than a cause and effect relationship.
Epidemiologic studies have also found that heightened maternal stress during the 2nd trimester increases fetal vulnerability to adverse outcomes such as shortened gestational age, preterm birth, low birth weight, and small for gestational age (62). ASD has also been associated with intense maternal stress (e.g., life events and hurricanes) during this gestational window (5, 6, 63).
Fetal programming, a concept also referred to as the “developmental origins of health and disease” hypothesis, explains that in utero disruption during critical developmental periods can relay health consequences to the fetus that persist throughout adulthood (64–68). Originally bolstered by epidemiological findings describing regional overlap in areas with high infant mortality and coronary heart disease (69), there has been increased recognition that in utero phenomena can lead to a wide range of chronic conditions. Supporting evidence includes the well-established relationship between prenatal exposure to maternal metabolic conditions [e.g., hypertension (70), diabetes (71–73), obesity (74–76)] and chronic metabolic disorders that begin in adolescence and adulthood (77, 78). Pre-pregnancy maternal obesity has been linked to poorer metabolic, endocrine, cardiovascular, and neurodevelopmental outcomes in offspring (79). Even after adjusting for pre-pregnancy obesity, maternal metabolic conditions such as gestational diabetes remain associated with increased risk of cardiovascular disease (80), obesity (81), type 2 diabetes (82), and early childhood metabolic syndrome development (83) among offspring.
Fetal programming of adolescent/adult metabolic disorders may occur through epigenetic changes (84). Telomeres are repetitive DNA tracts that protect against excessive chromosomal degradation (85). Shortened telomeres occur in fetuses exposed to gestational diabetes and are associated with higher cardiometabolic disease risk in adults (86, 87). Pre-eclampsia has been shown to induce epigenetic changes in offspring through decreased DNA methylation of IGF2 (88), a mediator of cell proliferation and apoptosis (89). Aside from its influence on postnatal growth, aberrant IGF2 expression is linked to subsequent development of hypertension, diabetes, and other metabolic disorders (88).
The proposed basis for fetal programming includes calibration of fetal regulatory systems in response to intrauterine nutrient availability to optimize extra-uterine survival. The subsequent mismatch between pre- and postnatal resources—whether involving excess or scarcity—predisposes offspring to develop one or more metabolic disorders. Fetal programming serves as a potential mechanism through which stress, metabolic disturbances, inflammation, and steroid dysregulation during pregnancy could predispose offspring to ASD. Altered fetal hypothalamic-pituitary-adrenal (HPA) axis development falls within the concept of fetal programming (90–92).
The HPA axis modulates neural, endocrine, and immune responses to stress to maintain homeostasis (93). While HPA axis plays a critical role in coordinating short-term physiological stress responses (94), HPA axis dysregulation has been implicated in several psychological and physiological disorders (95–100). Some children with ASD demonstrate signs of HPA axis dysregulation (101–106) such as altered circadian rhythms (107) and abnormal cortisol stress responses (105). Collectively, these findings raise the possibility that aberrant HPA axis functioning in some ASD individuals may have originated during fetal life through fetal programming.
Understanding fetal HPA axis development in the 2nd trimester (i.e., 13th to 26th week gestation) provides a context in which to interpret the association between ASD and steroid hormone levels (whether measured in maternal serum or amniotic fluid) during this gestational window. By the 12th week of gestation, the fetal hypothalamus releases corticotropin-releasing hormone (CRH) to signal the anterior pituitary gland to release adrenocorticotropic hormone (ACTH) (108–110). ACTH stimulates the fetal zone of the adrenal gland to synthesize dehydroepiandrosterone (DHEA) (110) and its conjugated form, dehydroepiandrosterone sulfate (DHEAS) (111). Although the fetal HPA axis is active by 12–18 weeks gestation (112), fetal adrenal gland development lags behind that of the hypothalamus and anterior pituitary. The fetal adrenal gland does not typically develop de novo cortisol synthesis capacity—referred to as fetal HPA axis maturation—until around 23–24 weeks gestation (91, 113–115). Because cortisol promotes the physiologic shift from somatic growth to organ maturation [e.g., lungs (116), gut (117), liver (116, 118)], fetal HPA axis maturation is essential for extra-uterine survival (119). Under duress, fetal HPA axis maturation can occur as early as 20 weeks gestation (113). This may be induced by increased placenta estradiol production (120).
Placental estradiol synthesis promotes fetal HPA axis maturation through multiple mechanisms. Estradiol influences cortisol transfer from the mother to fetus to establish a maternal-fetal cortisol gradient through its actions on placental 11β-hydroxysteroid dehydrogenase (11β-HSD) activity (121). 11β-HSD converts active maternal cortisol into inert cortisone. This gradient serves to protect the fetus from maternal cortisol overexposure (122, 123). Maternal cortisol in the fetal compartment exerts negative feedback on the fetal hypothalamus and anterior pituitary gland. As pregnancy progresses, 11β-HSD activity increases, less maternal cortisol reaches the fetal compartment, and negative feedback to the fetal HPA axis is reduced (115, 122, 124–126). The subsequent increase in fetal HPA axis activation coincides with the emergence of the fetal adrenal's de novo cortisol synthesis capacity (125) leading to fetal HPA axis maturation (see Figure 1). The placenta synthesizes estradiol from its precursor DHEA(S) which is derived from both fetal and maternal adrenal glands (124, 140). Excess fetal DHEA(S) supply can drive increased placental estradiol synthesis (110, 124, 141, 142). Because the placenta shunts over 90% of the estradiol it produces into the maternal circulation, maternal serum estradiol levels reflect placental estradiol synthesis (130).
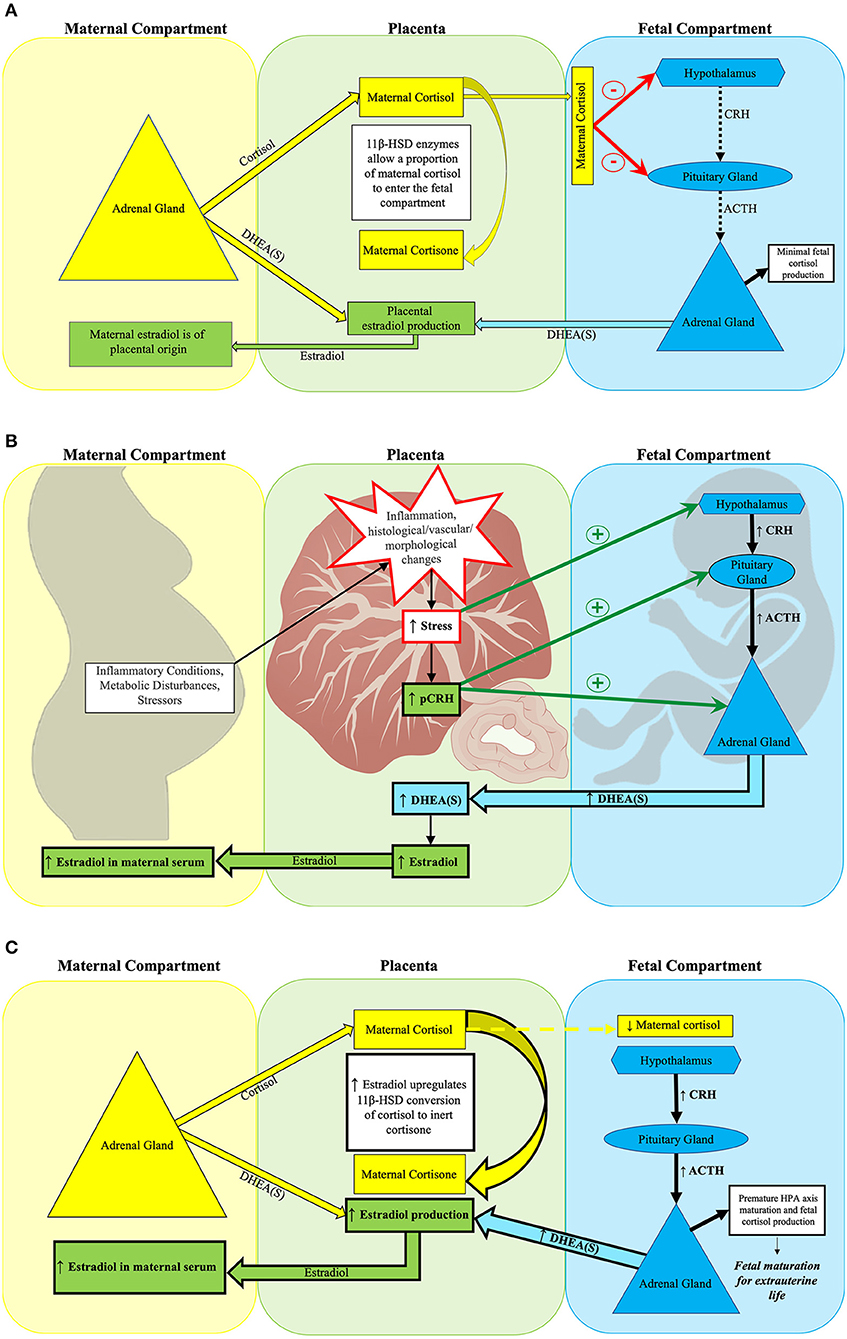
Figure 1. Placental estradiol and the fetal HPA axis at mid-gestation. (A) Normal fetal HPA axis functioning in the setting of typical placental estradiol activity at mid-gestation. (A) Depicts normal suppression of fetal HPA axis activity during mid-gestation by maternal cortisol. The placental glucocorticoid barrier contains 11β-HSD enzymes, which control fetal exposure to maternal cortisol (110) by converting most of the maternal cortisol entering the placenta to its inert form, cortisone. While fetal cortisol levels are 5–10 times lower than maternal cortisol levels (127), the maternal cortisol that enters the fetal compartment suppresses fetal HPA axis activity through negative feedback on the fetal hypothalamus and pituitary gland (112). Typically, the fetal adrenal gland has not yet developed de novo cortisol synthesis capacity at this point in gestation (124). Rather, the fetal adrenal gland primarily produces DHEA(S) when stimulated by ACTH or pCRH. Fetal DHEA(S), along with maternal DHEA(S), subsequently serves as the substrate for placental estradiol production (115, 128, 129). The placenta shunts over 90% of estradiol produced into the maternal circulation, thus maternal serum estradiol levels by mid-gestation reflect placental estradiol production (130). (B) Obstetrical adversity increases placental estradiol production at mid-gestation. Maternal adversity [e.g., stressors (131), inflammation (132–134), and metabolic disorders (135–138)] can increase placental estradiol production by stimulating pCRH release through disrupting placental structure and function. Subsequently, fetal stress increases, which activates the fetal HPA axis. Both pCRH and fetal HPA axis activation increase fetal adrenal DHEA(S) synthesis, leading to higher placental estradiol production. (C) Early fetal HPA axis maturation precipitated by excess placental estradiol activity at mid-gestation. Estradiol acts on 11β-HSD placental glucocorticoid barrier enzymes to increase conversion of maternal cortisol to inert cortisone, thereby reducing the amount of maternal cortisol entering the fetal compartment (139). Less maternal cortisol in the fetal compartment reduces its negative feedback on the fetal hypothalamus and pituitary gland. The relative absence of negative feedback precipitates fetal HPA axis maturation, which is characterized by the onset of de novo cortisol synthesis capacity by the fetal adrenal gland (125). Higher placental estradiol production leads to elevated maternal serum estradiol concentrations.
The placenta also influences fetal HPA axis maturation through its production of CRH (pCRH). pCRH stimulates fetal adrenal DHEA(S) production by (1) signaling fetal pituitary ACTH release (143), (2) heightening fetal adrenal gland sensitivity to ACTH (144), and (3) stimulating DHEA(S) producing fetal adrenal cells directly (145, 146). By increasing fetal DHEA(S) synthesis, pCRH facilitates increased placental estradiol production. As estradiol acts on placental barrier enzymes that modulate fetal cortisol exposure, pCRH can indirectly facilitate precocious HPA axis maturation by decreasing negative feedback from maternal cortisol. pCRH also directly promotes adrenal de novo cortisol synthesis (147, 148). Following its maturation, activation of the fetal HPA axis signals adrenal production of both DHEA(S) and de novo cortisol.
Excess inflammation triggered by infection, tissue injury, autoimmunity, or disruption in immunogenic tolerance to the fetus (149) can provoke early fetal HPA axis maturation through increased fetal HPA axis activity (115). Likewise, hypoxia can also stimulate fetal HPA axis maturation through a similar mechanism (150). Considering estradiol's role in strengthening the maternal-fetal cortisol gradient, estradiol serves as a feasible intermediary through which inflammation and hypoxia contribute to precocious fetal HPA axis maturation.
Furthermore, pCRH synthesis is upregulated in response to signifiers of an adverse intrauterine environment, such as elevated cortisol levels, pro-inflammatory cytokines, catecholamines, and decreased uterine blood flow (115, 151, 152). As pCRH promotes ACTH release from the fetal pituitary gland (143) and expression of cortisol synthesizing enzymes (147), elevated pCRH from in utero stress can serve as an additional mechanism through which the fetal HPA axis matures precociously (153).
One of the strongest risk factors for developing ASD is male sex. ASD prevalence in males is 3 to 4 fold higher than in females (154–156). In the absence of intellectual disability, this ratio increases further to 7:1 (157). Baron-Cohen et al. (158) has proposed the “extreme male brain” theory to explain the significant sex differential in ASD incidence. This theory suggests that excess prenatal androgen exposure contributes to the development of autistic traits in offspring by amplifying traits thought to be more typical of males, such as systemization, while diminishing traits commonly associated the femininity, such as empathy (158). Sex steroid hormones have long been thought to exert profound effects on the sexual dimorphism of the brain (159, 160); however, the exact mechanism has yet to be fully understood.
The “aromatization” hypothesis proposes that the roles of estradiol and testosterone in fetal neurodevelopment are intertwined (161), as masculinization of the rodent fetal brain is dependent upon the conversion of testosterone to estradiol by the enzyme aromatase (162, 163). In primates, reliance on this mechanism to explain masculinization of the human fetal brain remains unproven. Unlike primates, alpha-fetoprotein in rodents has a high binding affinity to circulating estradiol thereby sequestering estradiol and preventing it from entering and masculinizing the fetal rodent brain (164). Therefore, only the sex steroids produced by the fetal rodent influence brain masculinization. The assumption that estradiol's masculinizing role in the rodent fetal brain could be extrapolated to the human fetal brain is significantly flawed because it fails to account for the substantial difference between these mammals in regards to in utero estradiol sequestration by alpha-fetoprotein.
Zhao et al. (165) describes a multiple threshold liability model to explain the sex difference in ASD risk. This model theorizes that females require a higher genetic mutation load relative to their male counterparts to develop ASD thus contributing to ASD's male predominance. Werling and Geschwind (157) speculate that these mutations may interact with androgen-related mechanisms in families with affected females, as prior studies have found an association with ASD and genes that modulate sex-steroid function (166–168). As steroid hormone receptors exert epigenetic effects through DNA methylation and histone acetylation (169–172), it is possible that epigenetic modifications in response to in utero sex-steroid hormone exposure affect sex-specific neurodevelopmental processes.
Fetal growth, development, and HPA axis programming in the setting of obstetric adversity differ by fetal sex (173–175). These sex-specific responses serve as additional mechanisms in which to consider male ASD predominance (see Figure 2). As the placenta is derived from extra-embryonic tissues, the placenta has the same sex as the fetus (178). Evidence suggests that the placentas of male and female fetuses differ in response to adverse prenatal environments through modulation of steroid pathways, placental genes, and protein synthesis (176). Placental growth and structure differ by sex, with male placentas being smaller in size but more efficient at nutrient and oxygen delivery (179, 180). Fetal growth depends upon the limited capacity of the maternal-placental interface to deliver oxygen and nutrients. Thus, greater placental efficiency among males precipitates faster somatic growth while increasing vulnerability to in utero perturbations (179, 181). This may have deleterious neurodevelopmental consequences, as fetal brain development relies on the availability of oxygen and nutrients such as fatty acids, glucose, and amino acids (131, 182, 183). In contrast, female placentas may have superior ability to buffer and adapt to suboptimal prenatal conditions (180).
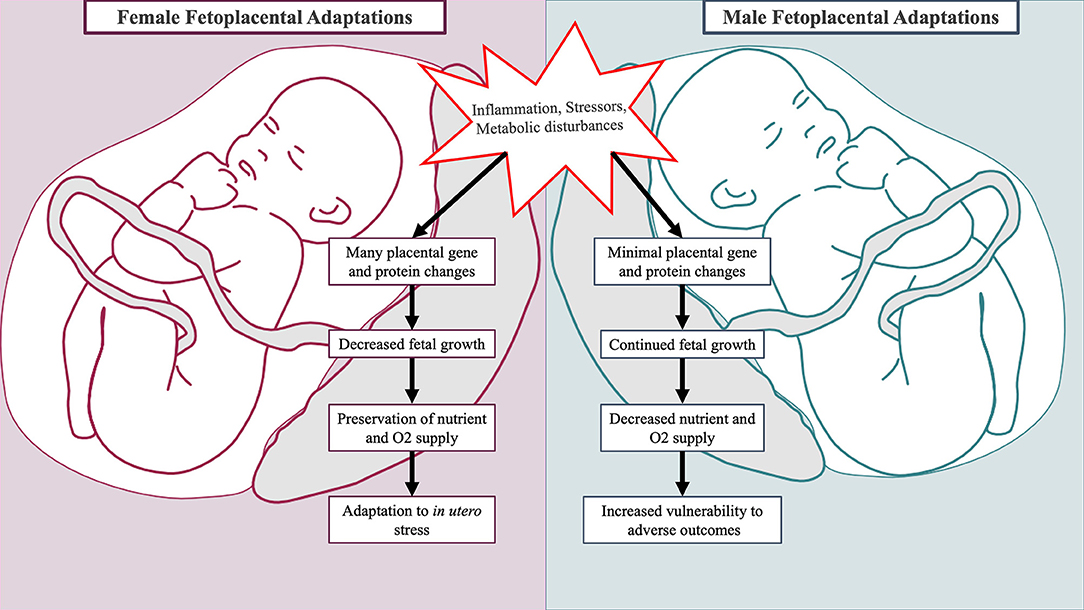
Figure 2. Sex-specific fetal and placental adaptations to maternal adversity. Placentas of male and female fetuses respond differently to mild forms of maternal adversity. In the placenta of female fetuses, multiple changes in glucocorticoid barrier enzyme activity, gene expression, and protein synthesis occur leading to decreased growth (176, 177). This is advantageous as it preserves fetal oxygen and nutrient delivery. In the placenta of male fetuses, minimal changes in gene and protein expression occur, and the male fetus continues to grow incurring increased vulnerability to adverse outcomes (176).
Newborns differ by sex in regards to birth weight, morbidity, and mortality. This is attributable at least in part to sex-specific adaptions that regulate the balance between fetal growth and extra-uterine survival (184, 185). In the setting of mild physical adversity (i.e., chronic maternal asthma), changes in placental 11β-HSD glucocorticoid barrier enzyme activity, gene expression, and protein synthesis occur and coincide with reduced female fetal growth (176, 177). This prepares the female fetus for future adverse events through preservation of oxygen and nutrient delivery. In pregnancies complicated by mild pre-eclampsia, male and female fetuses differ in growth progression mediated by differential fetal microvascular (186) and placental inflammatory cytokine responses (187). In the setting of maternal hyperglycemia, males demonstrate greater hyperglycemic growth stimulation (188) and higher incidences of respiratory distress (175). The sex hormones estradiol and testosterone stimulate opposing processes on fetal lung development: estradiol facilitates surfactant production while testosterone promotes lung tissue proliferation at the expense of lung maturation (189). Collectively, sex-specific responses to obstetrical adversity leave males less physiologically prepared for survival, particularly if birth were to occur prematurely (176).
A growing body of evidence demonstrates a link between placental pathology and ASD. Anderson et al. (43) found increased trophoblastic inclusions in the placentas of fetuses who later developed ASD. Trophoblastic inclusions, a histological finding that results from atypical growth and folding of the placenta (190), are more common in genetically atypical gestations (191–194). Furthermore, studies of high ASD risk cohorts (defined by having at least one older sibling with ASD) also found increased placental trophoblastic inclusions (195) in addition to altered placental morphology (196) and placental chorionic surface vascular networks (197). The specific morphological changes found, such as increased thickness and roundness, may reflect decreased ability to adapt to variations in the prenatal environment (196). Notably, variations within the placental chorionic surface vascular networks may be the result of atypical vasculogenesis and angiogenesis (197). Several conditions associated with ASD, such as pre-eclampsia (198), intrauterine growth restriction (41), and pre-term birth (16) are also attributed to placental vascular abnormalities (199–201). In ASD individuals, Straughen et al. demonstrated an association between placental inflammation, maternal vascular malperfusion, and ASD (9) that occurred more prominently among males.
Sex-specific placental signaling and epigenetic phenomena in fetal adaption to nutrient availability and maternal stress are exemplified by the X-linked gene O-linked N-acetylglucosamine transferase (OGT) (131, 202). OGT induces glucose-sensitive epigenetic changes that influence immune responses, steroidogenic activity, and fetal development. Because placental X inactivation spares OGT (20) such that the female placenta has two active gene copies while the male placenta has only one, OGT expression and interaction with steroid receptors demonstrate sex-specificity (203, 204). This provides an additional potential mechanism through which maternal metabolic conditions (i.e., hyperglycemia, insulin insensitivity) could influence the sex-differential observed in ASD (20).
In utero steroidogenesis occurs through the well-coordinated interactions among the mother, placenta, and fetus (21, 22). The placenta is the main orchestrator of adaptions by the maternofetoplacental unit of steroidogenic activity in response to in utero and external environmental cues (205). As the transcriptome of the placenta changes throughout pregnancy (206), expression of placental genes involved in steroid hormone regulation consequentially shift (205). This section is a review of steroid-related biomarkers that have been associated with ASD accompanied by a brief summary of their potential connection to ASD.
Multiple studies have demonstrated associations between ASD/ASD traits and amniotic fluid androgen levels (i.e., androstenedione and testosterone) (207–212) supporting the “extreme male brain” theory for ASD's etiology proposed by Baron-Cohen et al. (158). Amniocentesis is typically performed between the 15th and 20th week of pregnancy, overlapping the critical gestational window when the male fetus produces peak amount of androgens to promote genital differentiation (213). This testosterone surge occurs between 11 and 17 weeks gestation (214).
Elevated androgen levels (androstenedione and testosterone) in amniotic fluid collected from pregnancies with male offspring correlated with later ASD diagnoses (212). Decreased eye contact (207), poorer quality of social relationships (211), increased restricted interests (211), reduced empathy (210), and higher autism trait scores (208, 209) have also been associated with elevated amniotic fluid androgen levels. Researchers conducting these amniotic fluid studies applied ASD trait severity measures that they developed, including the Quantitative Checklist for Autism in Toddlers (Q-CHAT) (215), the Childhood Autism Spectrum Test (CAST) (216), and the Child Autism Spectrum Quotient (AQ-Child) (217). In contrast, Kung et al. (218) found no relationship between autistic traits and amniotic testosterone levels in pregnancies resulting in typically developing children using the CAST. Elevated amniotic androgen levels associated with ASD found by Baron-Cohen et al. (212) also did not appear to persist in a re-analysis published in Baron-Cohen et al. (219).
Maternal serum has also been analyzed to investigate the relationship between prenatal androgen levels and the emergence of ASD (19). Maternal serum drawn during early 2nd trimester did not identify significant associations between maternal androgen levels and ASD among offspring (19). Although gestational timing for the maternal serum and amniotic fluid collections overlap, differences in study findings between Baron-Cohen et al. (212) and Bilder et al. (19) may result in part from differences in study purposes and designs. For the maternal serum study, investigators were particularly interested in measuring steroid-related biomarkers as potential intermediaries between ASD outcome and the effects of obstetrical conditions associated with steroid dysregulation (i.e., hypertension and diabetes) which they referred to as “prenatal metabolic syndrome” (PNMS). As such, both the ASD case and non-ASD control groups were enriched (by 50%) for the presence of PNMS exposure. Extrinsic testosterone exposure to the fetus is measurable in maternal serum, though fetal testosterone production cannot be measured in maternal serum because androgen movement across the placenta is generally considered unidirectional from mother to fetus (220, 221). Testosterone levels in amniotic fluid, however, reflect both fetal testosterone exposure and production as the amniotic fluid collected is composed primarily of fetal urine.
In Park et al. (222), no association was found between androgen levels (testosterone, androstenedione, and DHEA) in umbilical cord blood and autistic traits at 12 and 36 months of age among the Early Autism Risk Longitudinal Investigation (EARLI) cohort using the Autism Observation Scale for Infants (AOSI) and Social Responsiveness Scale (SRS), respectively. The EARLI cohort were younger siblings of children with ASD. Several other studies have also failed to demonstrate an overall relationship between umbilical cord testosterone levels and ASD traits (223–225). Unlike the amniotic fluid and maternal serum studies described above, umbilical cord blood samples represent prenatal testosterone exposure at the time of parturition, far after the fetal testosterone surge occurs (214). Upon subdividing the sibling cohort into multiple groups based on proband and participant sex, umbilical cord blood testosterone levels correlated with ASD traits among siblings of female probands. The authors attributed their findings to the multiple threshold liability model (157, 165) suggesting a higher genetic load required for females to develop ASD.
Meconium androgen levels were also measured in the EARLI cohort (226). Findings demonstrated a positive correlation with some androgen levels (i.e., unconjugated testosterone and total DHEA) and SRS scores overall. Following stratification of the cohort by participant and proband sex, various associations were found between androgen levels and ASD traits at 12 and 36 months of age. Meconium begins accumulating in the 13th week of gestation, with the highest volume produced between 28 and 34 weeks gestation (227). As meconium represents cumulative exposures throughout gestation (228), it is difficult to link meconium steroid concentrations to a specific gestational time period of exposure.
Fetal testosterone in males is predominantly produced by the Leydig cells of the testes beginning around 8 weeks gestation (229). Placental human chorionic gonadotropin (hCG), in tandem with luteinizing hormone (LH) produced in the fetal pituitary gland, stimulates testosterone synthesis by the testes during early gestation (214, 230–233). Interestingly, LH and hCG share a common receptor transcribed from a single gene (234). Placental hCG production rapidly increases following implantation until it reaches its apex at the end of the first trimester (235). Following this, hCG levels progressively reach a nadir by 18–20 weeks gestation (236–238). As hCG levels decline, LH levels rise. From 8 to 24 weeks of gestation, the testosterone level in males substantially exceeds that of females (239), which is believed to impart neurodevelopmental effects resulting in sex-specific behavioral differences (240). However, the timing and mechanism in which this occurs in the human fetal brain has not been definitively determined. While the fetal adrenal gland can also synthesize testosterone, the amount is negligible (241, 242) and its contribution to amniotic fluid androgen levels is not clearly established.
Aside from stimulating testosterone synthesis, hCG serves a variety of functions, including pregnancy maintenance through stimulation of the corpus luteum to secrete progesterone (129, 243). Additionally, hCG has been associated with angiogenesis, mediation of immune tolerance, umbilical cord development, myometrial contraction suppression, and fetal organ growth and differentiation (244–246). To our knowledge, only one study has directly examined 2nd trimester maternal serum hCG levels in relation to ASD risk. Windham et al. (247) found a U-shaped relationship (i.e., both higher and lower levels) between hCG levels associated with increased ASD risk among offspring, particularly in males.
Elevated hCG levels during the 2nd trimester have also been linked to complications such as preeclampsia (248–250), intrauterine growth restriction (IUGR) (248), pre-term delivery (248, 250, 251), low birth weight (250), and fetal death (250, 251). As the placenta secretes hCG in response to stress hormones (252), an adverse prenatal environment may contribute to steroid dysregulation in ASD through elevated hCG exerting its influence on testosterone synthesis in male fetuses. This proposed mechanism of aberrations within the fetal steroid hormonal milieu aligns with findings of elevated 2nd trimester amniotic fluid testosterone levels associated with ASD described above. Interestingly, hCG can also stimulate fetal adrenal DHEA(S) secretion during the 2nd trimester (253). As DHEA(S) is a substrate for placental estradiol synthesis, abnormalities in 2nd trimester estradiol and testosterone linked to ASD development may share a common genesis involving the placental response to adversity.
While prior investigations have focused on androgens, recent research studies have explored the potential role that estrogen and progesterone may play in the development of ASD. Bilder et al. (19) found significantly higher estradiol levels in early 2nd trimester maternal serum associated with ASD among offspring. As described above, both ASD and comparison cohorts were enriched for PNMS exposure. Along with higher estradiol levels, lower SHBG levels were also identified, suggesting the potentiation of estradiol activity in pregnancies associated with ASD as SHBG binds biologically active estrogens rendering them inert (254). Estradiol, rather than estriol, was selected as the estrogen of interest for this study because maternal estradiol levels represent placental estrogen activity more accurately through estradiol's substantially higher potency (255), longer estrogen receptor binding duration (256), and higher maternal serum concentrations (257, 258). Because maternal serum estradiol levels result from, and reflect, placental estradiol production by this gestation window (130), Bilder et al. (19) interpreted these results as indicating increased ASD risk associated with elevated placental estradiol production and activity. Higher placental estradiol production could result from greater fetal steroidogenic activity, and increased placental estradiol activity could facilitate premature fetal HPA axis maturation in the early 2nd trimester. Bilder et al. (19) did not find a significant association between serum progesterone concentrations and ASD risk.
Baron-Cohen et al. (219) also identified higher estrogen levels (i.e., estradiol, estriol, and estrone), along with increased progesterone levels, in 2nd trimester amniotic fluid of male offspring with ASD relative to controls. Notably, elevated estradiol was the most significant predictor of subsequent ASD development. Baron-Cohen et al. (219) attributes the link between higher amniotic estrogens, progesterone, and ASD among offspring to increased fetal steroidogenic activity in ASD's etiologic pathway.
Elevated ASD risk associated with higher prenatal estrogen levels in maternal serum (19) and amniotic fluid (219) contrast with prior study results from Windham et al. (247), which demonstrated lower unconjugated estriol levels in 2nd trimester maternal serum. Estriol is a weak estrogen produced exclusively during pregnancy and requires fetal liver enzyme activity in its synthesis pathway (257, 258). Estriol levels in Windham et al. (247) were initially measured as a component of a prenatal integrated screening test for aneuploidy and neural tube defects. Low maternal serum estriol levels have been used for decades as a clinical indicator for obstetric complications (259), such as placental insufficiency (260), fetal growth restriction (261), pre-eclampsia (262), preterm birth (247), low birth weight (263), and pregnancy loss (260). Windham et al. (247) attributes study findings to the involvement of hormones in the development of ASD through their influence on fetal development and signaling activity in the CNS and immune system.
Baron-Cohen et al. (212) identified elevated cortisol levels in the amniotic fluid collected during the pregnancies of male offspring who developed ASD, though this finding was not replicated in Baron-Cohen et al. (219). However, Baron-Cohen and colleagues (212, 219) conclude that the findings from both studies support the presence of increase fetal steroidogenic activity in ASD. The significant diurnal variation in serum cortisol concentrations preempts its use as a biomarker in banked maternal serum samples from most large obstetric studies because standardization of serum collection times for these studies is typically not implemented.
Bilder et al. (19) found an inverse relationship between maternal serum SHBG levels and risk for diabetes/hypertension exposure and ASD. The highest SHBG levels were measured for the cohort with neither PNMS exposure nor ASD. Conversely, the lowest SHBG levels occurred in the cohort with both PNMS exposure and ASD. In addition to binding estrogens and testosterone, SHBG also serves as a serum biomarker for insulin sensitivity that supersedes its relationship with sex hormones regardless of sex, age, and pregnancy status (264). Even among pregnancies without clinical PNMS manifestations, SHBG levels were lower in the ASD cohort compared to either non-ASD cohorts, suggesting the presence of exposure to a subclinical metabolic condition during gestation in these offspring who developed ASD. Unlike most serum biomarkers for insulin sensitivity, SHBG measures require no specific collection time (e.g., time of day or proximity to caloric consumption) to ensure meaningful results interpretation. SHBG has been studied as a predictive 1st and early 2nd trimester biomarker for the emergence of gestational diabetes (265–267). Thus, SHBG is a useful biomarker for investigating maternal insulin sensitivity as a prenatal risk factor within maternal serum samples banked for non-specific purposes.
As a post hoc analysis, Bilder et al. (19) calculated a value from maternal serum measurements of estradiol, DHEA, and DHEAS to estimate the relative contribution by the fetal adrenal gland to the DHEA substrate supply for placental estradiol production (EF-DHEA). The area under the curve for EF-DHEA used to predict ASD among offspring exceeded those of its measured components. This was interpreted to indicate that the relationship between elevated maternal serum estradiol and increased ASD risk may in part be driven by excess fetal adrenal activity in early 2nd trimester. The EF-DHEA calculation was created specifically for this study and has not been validated in animal models nor have these findings been replicated. However, EF-DHEA findings are consistent with the amniotic fluid results, particularly elevated cortisol, from Baron-Cohen et al. (212) demonstrating an association between ASD among offspring and increased fetal steroidogenic activity during this gestational window.
Figure 3 links heightened in utero stress from inflammation, stressors, and metabolic disturbances to perturbation within the prenatal hormone milieu. Through pCRH stimulation, the placenta upregulates fetal HPA axis activity in response to in utero stress. Subsequently, the fetal adrenal glands increase DHEA(S) synthesis leading to elevated placental estradiol production. Higher placental estradiol and pCRH production promotes HPA axis maturation denoted by fetal adrenal de novo cortisol synthesis. In response to in utero stress, the placental also increases hCG production which stimulates fetal gonadal testosterone synthesis.
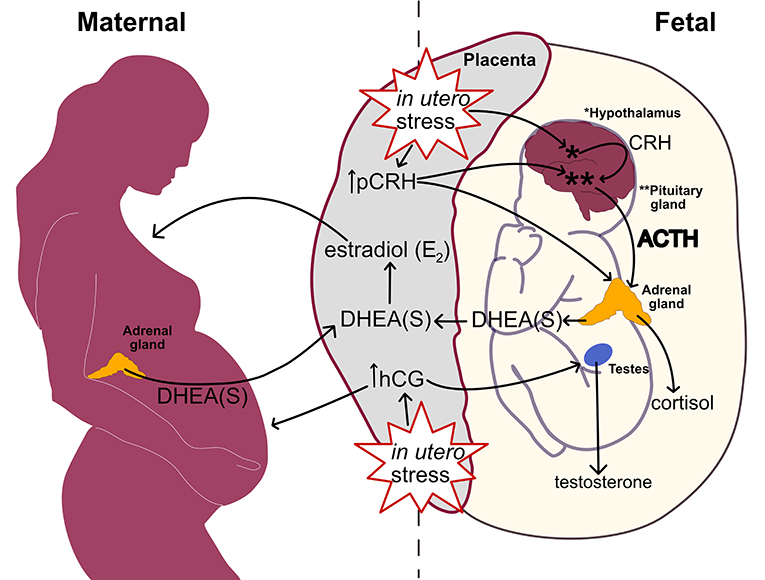
Figure 3. Linking in utero stress to increased fetal steroidogenic activity and ASD biomarkers at mid-gestation.
This review provides an up-to-date synopsis of the current evidence and supported theories regarding the role that the in utero steroid environment may play in ASD pathophysiology. Considering placental steroid hormone biosynthesis, metabolism, and transport (205), prenatal steroid dysregulation may be attributed to disruptions in vital components of placental structure and function in response to an adverse maternofetoplacental environment. In particular, stress, inflammation, and metabolic abnormalities can contribute to morphological and functional placental changes that affect nutrient and oxygen exchange, the protective transplacental barrier, and hormone synthesis (268, 269).
Obstetric conditions associated with ASD that may contribute to an adverse prenatal environment include hypertensive disorders of pregnancy (i.e., chronic, gestational, de novo or superimposed preeclampsia) (13, 14, 35–37), maternal immune dysfunction (5, 7–9), pre-existing/gestational diabetes (3, 10), pre-pregnancy obesity (11), gestational weight gain (12), and PCOS (4, 15). Notably, common indicators or complications of placental insufficiency also overlap with ASD risk factors, including small for gestational age/in utero growth restriction, prematurity, maternal infection, and maternal metabolic syndrome (16, 38–42). As the placenta is critical for fetal growth and development, adverse prenatal environments that impact the placenta may increase fetal stress and influence fetal programming, thereby contributing to ASD etiology.
The placenta shares the sex of the fetus as it is derived from extra-embryonic tissues (178). Numerous studies have documented that placentas of male and female fetuses differ in response to adverse prenatal environments (270–273). Specifically, placental signaling and gene expression influence sex-specific differences in fetal adaption to the in utero environment related to nutrient availability, maternal stress, and immune response (131, 202). In the setting of mild adversity, ongoing somatic growth of male fetuses confers increased vulnerability to subsequent adverse events (176, 177) that can influence neurodevelopment. Additionally, evidence of placental pathology appears more prominent from pregnancies of male offspring with ASD compared to those of their female counterparts. Further investigation of placental pathology in ASD is needed, particularly as it relates to fetal/placental sex differences.
Second trimester serves as a critical time for fetal steroidogenic activity in regards to fetal HPA axis maturation. Multiple ASD epidemiologic studies highlight the 2nd trimester as the period of greatest fetal vulnerability to stressful maternal life events (5, 6, 63). Bilder et al. (19) and Baron-Cohen et al. (212, 219) interpret the link between higher estrogen levels and ASD risk as an indicator of elevated fetal steroidogenic activity in the 2nd trimester of offspring who develop ASD. While Baron-Cohen et al. (212, 219) conceptualize fetal steroidogenic activity more broadly, particularly in regards to fetal androgen production, Bilder et al. (19) focuses on excess fetal adrenal activity and premature fetal HPA axis maturation. By strengthening the maternal-fetal cortisol gradient, placental estradiol reduces the amount of maternal cortisol in the fetal compartment (139) thereby easing maternal cortisol suppression of the fetal HPA axis and promoting its maturation. Fetal steroidogenic activity is enhanced by CRH released by the fetal hypothalamus and placenta (124, 145, 274–276). CRH production is stimulated by stress, inflammation, and hypoxia providing a mechanism through which obstetrical adversity could contribute to increased fetal steroidogenic activity and early HPA axis maturation (115, 150, 277).
The relationship between higher 2nd trimester estrogen levels and increased ASD risk is intriguing as estrogen is widely seen as an indicator of maternal and fetal health. Estradiol acts to preserve fetal viability during in utero stress through suppression of inflammation (278, 279) and uterine artery dilation that increases oxygen and nutrient supply to the fetus (280, 281). Estradiol also mediates several critical factors in neurodevelopment providing an overall neuroprotective benefit (162). From the perspective of fetal viability, excess estradiol in the early 2nd trimester can stimulate precocious fetal HPA axis maturation in preparation for extrauterine life. As described above, low—rather than high—maternal serum estrogen levels predict several conditions related to maternal and fetal adversity. Therefore, it is possible that elevated estradiol in early 2nd trimester maternal serum and amniotic fluid may be the result of a compensatory rather than primarily pathologic mechanism. However, it must be noted that the historical obstetric practice of using a synthetic estrogen to foster healthy pregnancies was quite misguided. The use of the synthetic estrogen diethylstilbestrol (DES) in pregnant women during the mid-1900's was halted when exposed adolescent and young adult female offspring were found to develop a rare type of cancer, vaginal adenocarcinoma (282). Subsequent studies have found transgenerational adverse effects of DES exposure, as reproductive tract and immunologic abnormalities have occurred in exposed male and female offspring (283). Grandchildren of exposed women were also found to have a higher incidence of ADHD (284). To our knowledge, there have been no studies published that report on an epidemiologic investigation of ASD risk associated with DES exposure. Although DES exposure differs in many respects from increased placental estrogen synthesis, such a study could improve understanding of estrogen's potential role in ASD's etiology.
An early theory connecting the prenatal steroid environment with ASD conceptualizes ASD as an extreme manifestation of male cognitive traits and implicates excess in utero androgen exposure in ASD's etiology (158). Although elevated amniotic testosterone levels were associated with the presence of ASD traits in most (207–211), but not all studies (218), results regarding ASD diagnosis and testosterone concentrations have been more variable (212, 219). Two amniotic fluid studies and a maternal serum study found no relationship between testosterone and ASD (19, 218, 219). Results from umbilical cord blood and meconium analyses were mixed (222, 226). Disparate findings on androgen biomarkers may be attributable to differences in sample type, gestational timing, ASD outcome measures, and cohort characteristics. Additional investigations are needed which implement multiple sampling strategies and gestational time points in a large obstetrical cohort to clarify the relationship between androgen levels throughout pregnancy and ASD risk.
Overall, prior research findings have established that an altered prenatal environment is a component of ASD pathogenesis. However, the exact process detailing the interaction between diverse prenatal factors and increased ASD risk remains unclear. While several studies link various aspects of perturbations in utero to ASD development, to our knowledge, there has been a lack of a unifying paradigm. Based on our current understanding, we propose that disruption within the maternofetoplacental unit initiates a causal sequence resulting in placental changes, thereby influencing fetal programming, steroid hormone modulation, and HPA axis development in ASD.
Factors that contribute to disruption within the maternofetoplacental unit include metabolic disturbances, inflammatory conditions, and stressors. These broad categories are manifested by several obstetric conditions that are associated with ASD development. The placenta reacts to changes in utero through adaptations that differ based on sex, which are disadvantageous to male fetuses. Fetal programming is affected, which may have neurodevelopmental implications. Considering the placenta's endocrine functions, steroid hormone modulation is also impacted which could alter HPA axis development and functioning. This may be relevant to HPA axis dysregulation found in some individuals with ASD.
Current literature findings support multidisciplinary research efforts to investigate the manner in which the in utero steroid milieu and fetal steroidogenic activity interact with genetic/epigenetic, environmental, inflammatory, and obstetrical ASD risk factors. Studies examining these interaction effects on fetal neuroendocrine development may identify feasible intervention targets that could reduce risk for neonatal morbidity/mortality and childhood neurodevelopmental disabilities. In particular, elucidating the link between the in utero steroid environment and immune response may provide an opportunity to optimize both immediate outcomes and long-term neurodevelopmental functioning.
WW conceptualized and designed all components of the paper, drafted the initial manuscript, reviewed, and revised the manuscript. SD conceptualized the obstetrical components of the paper, reviewed, and revised the manuscript. DB supported the conceptualization of all components of the paper, reviewed and revised the manuscript. All authors approve the final manuscript as submitted and agree to be accountable for all aspects of the work.
This scientific review was supported by the University of Utah Neuroscience Initiative.
DB was a consultant for BioMarin Pharmaceutical and Encoded Therapeutics and serves as a consultant and scientific advisory board member for Taysha GTx. The content of this manuscript does not overlap with her work for these companies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Michael Varner, MD, for providing an expert critique of this manuscript.
1. Association AP. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA American Psychiatric Association (2013).
2. Kheirouri S, Alizadeh M. Maternal excessive gestational weight gain as a risk factor for autism spectrum disorder in offspring: a systematic review. BMC Pregn Childbirth. (2020) 20:645. doi: 10.1186/s12884-020-03324-w
3. Gardener H, Spiegelman D, Buka SL. Perinatal and neonatal risk factors for autism: a Comprehensive meta-analysis. Pediatrics. (2011) 128:344–55. doi: 10.1542/peds.2010-1036
4. Hisle-Gorman E, Susi A, Stokes T, Gorman G, Erdie-Lalena C, Nylund CM. Prenatal, perinatal, and neonatal risk factors of autism spectrum disorder. Pediatric Res. (2018) 84:190–8. doi: 10.1038/pr.2018.23
5. Beversdorf DQ, Stevens HE, Margolis KG, Van De Water J. Prenatal stress and maternal immune dysregulation in autism spectrum disorders: potential points for intervention. Curr Pharm Design. (2020) 25:4331–43. doi: 10.2174/1381612825666191119093335
6. Kinney DK, Miller AM, Crowley DJ, Huang E, Gerber E. Autism prevalence following prenatal exposure to hurricanes and tropical storms in louisiana. J Autism Dev Disord. (2008) 38:481–8. doi: 10.1007/s10803-007-0414-0
7. Madore C, Leyrolle Q, Lacabanne C, Benmamar-Badel A, Joffre C, Nadjar A, et al. Neuroinflammation in autism: plausible role of maternal inflammation, dietary omega 3, and microbiota. Neural Plasticity. (2016) 2016:1–15. doi: 10.1155/2016/3597209
8. Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. (2011) 17:389–94. doi: 10.1016/j.molmed.2011.03.001
9. Straughen JK, Misra DP, Divine G, Shah R, Perez G, Vanhorn S, et al. The association between placental histopathology and autism spectrum disorder. Placenta. (2017) 57:183–8. doi: 10.1016/j.placenta.2017.07.006
10. Xu G, Jing J, Bowers K, Liu B, Bao W. Maternal diabetes and the risk of autism spectrum disorders in the offspring: a systematic review and meta-analysis. J Autism Dev Disord. (2014) 44:766–75. doi: 10.1007/s10803-013-1928-2
11. Sanchez CE, Barry C, Sabhlok A, Russell K, Majors A, Kollins SH, et al. Maternal pre-pregnancy obesity and child neurodevelopmental outcomes: a meta-analysis. Obesity Rev. (2018) 19:464–84. doi: 10.1111/obr.12643
12. Bilder DA, Bakian AV, Viskochil J, Clark EAS, Botts EL, Smith KR, et al. Maternal prenatal weight gain and autism spectrum disorders. Pediatrics. (2013) 132:e1276–e83. doi: 10.1542/peds.2013-1188
13. Dachew BA, Mamun A, Maravilla JC, Alati R. Pre-eclampsia and the risk of autism-spectrum disorder in offspring: meta-analysis. Br J Psychiatry. (2018) 212:142–7. doi: 10.1192/bjp.2017.27
14. Maher GM, McCarthy FP, McCarthy CM, Kenny LC, Kearney PM, Khashan AS, et al. A perspective on pre-eclampsia and neurodevelopmental outcomes in the offspring: does maternal inflammation play a role? Int J Dev Neurosci. (2019) 77:69–76. doi: 10.1016/j.ijdevneu.2018.10.004
15. Kosidou K, Dalman C, Widman L, Arver S, Lee BK, Magnusson C, et al. Maternal polycystic ovary syndrome and the risk of autism spectrum disorders in the offspring: a population-based nationwide study in sweden. Mol Psychiatry. (2016) 21:1441–8. doi: 10.1038/mp.2015.183
16. Agrawal S, Rao SC, Bulsara MK, Patole SK. Prevalence of autism spectrum disorder in preterm infants: a Meta-analysis. Pediatrics. (2018) 142:e20180134. doi: 10.1542/peds.2018-0134
17. Bölte S, Girdler S, Marschik PB. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol Life Sci. (2019) 76:1275–97. doi: 10.1007/s00018-018-2988-4
18. Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism. (2017) 8:13. doi: 10.1186/s13229-017-0121-4
19. Bilder DA, Esplin MS, Coon H, Burghardt P, Clark EAS, Fraser A, et al. Early second trimester maternal serum steroid-Related biomarkers associated with autism spectrum disorder. J Autism Dev Disord. (2019) 49:4572–83. doi: 10.1007/s10803-019-04162-2
20. Bale TL. The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialogues Clin Neurosci. (2016) 18:459–64. doi: 10.31887/DCNS.2016.18.4/tbale
21. Kallen CB. Steroid hormone synthesis in pregnancy. Obstet Gynecol Clin North Am. (2004) 31:795–816, x. doi: 10.1016/j.ogc.2004.08.009
22. Murphy VE, Smith R, Giles WB, Clifton VL. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocr Rev. (2006) 27:141–69. doi: 10.1210/er.2005-0011
23. Straub RH. The complex role of estrogens in inflammation. Endocr Rev. (2007) 28:521–74. doi: 10.1210/er.2007-0001
24. Marzi M, Vigano A, Trabattoni D, Villa ML, Salvaggio A, Clerici E, et al. Characterization of type 1 and type 2 cytokine production profile in physiologic and pathologic human pregnancy. Clin Exp Immunol. (1996) 106:127–33. doi: 10.1046/j.1365-2249.1996.d01-809.x
25. Matalka KZ. The effect of estradiol, but not progesterone, on the production of cytokines in stimulated whole blood, is concentration-dependent. Neuro Endocrinol Lett. (2003) 24:185–91.
26. Sabahi F, Rola-Plesczcynski M, O'Connell S, Frenkel LD. Qualitative and quantitative analysis of t lymphocytes during normal human pregnancy. Am J Reprod Immunol. (1995) 33:381–93. doi: 10.1111/j.1600-0897.1995.tb00907.x
27. Rowe JH, Ertelt JM, Xin L, Way SS. Regulatory t cells and the immune pathogenesis of prenatal infection. Reproduction. (2013) 146:R191–R203. doi: 10.1530/REP-13-0262
28. Szekeres-Bartho J, Polgar B. PIBF: the double edged sword. Pregnancy and tumor. Am J Reprod Immunol. (2010) 64:77–86. doi: 10.1111/j.1600-0897.2010.00833.x
29. Szekeres-Bartho J, Faust Z, Varga P, Szereday L, Kelemen K. The immunological pregnancy protective effect of progesterone is manifested via controlling cytokine production. Am J Reprod Immunol. (1996) 35:348–51. doi: 10.1111/j.1600-0897.1996.tb00492.x
30. Moffett-King A. Natural killer cells and pregnancy. Nat Rev Immunol. (2002) 2:656–63. doi: 10.1038/nri886
31. Fuhler GM. The immune system and microbiome in pregnancy. Best Pract Res Clin Gastroenter. (2020) 44–45:101671. doi: 10.1016/j.bpg.2020.101671
32. Sentman CL, Meadows SK, Wira CR, Eriksson M. Recruitment of uterine nK cells: induction of cXC chemokine ligands 10 and 11 in human endometrium by estradiol and progesterone. J Immunol. (2004) 173:6760–6. doi: 10.4049/jimmunol.173.11.6760
33. Dekel N, Gnainsky Y, Granot I, Mor G. Review article: inflammation and implantation. Am J Reprod Immun. (2009) 63:17–21. doi: 10.1111/j.1600-0897.2009.00792.x
34. Ng S-W, Norwitz GA, Pavlicev M, Tilburgs T, Simón C, Norwitz ER. Endometrial decidualization: the primary driver of pregnancy health. Int J Mol Sci. (2020) 21:4092. doi: 10.3390/ijms21114092
35. Curran EA, O'Keeffe GW, Looney AM, Moloney G, Hegarty SV, Murray DM, et al. Exposure to hypertensive disorders of pregnancy increases the risk of autism spectrum disorder in affected offspring. Mol Neurobiol. (2018) 55:5557–64. doi: 10.1007/s12035-017-0794-x
36. Hultman CM, Sparén P, Cnattingius S. Perinatal risk factors for infantile autism. Epidemiology. (2002) 13:417–23. doi: 10.1097/00001648-200207000-00009
37. Walker CK, Krakowiak P, Baker A, Hansen RL, Ozonoff S, Hertz-Picciotto I. Preeclampsia, placental insufficiency, and autism spectrum disorder or developmental delay. JAMA Pediatrics. (2015) 169:154. doi: 10.1001/jamapediatrics.2014.2645
38. Limperopoulos C, Bassan H, Sullivan NR, Soul JS, Robertson RL, Moore M, et al. Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics. (2008) 121:758–65. doi: 10.1542/peds.2007-2158
39. Moore GS, Kneitel AW, Walker CK, Gilbert WM, Xing G. Autism risk in small- and large-for-gestational-age infants. Am J Obstetr Gynecol. (2012) 206:314.e1–e9. doi: 10.1016/j.ajog.2012.01.044
40. Krakowiak P, Walker CK, Bremer AA, Baker AS, Ozonoff S, Hansen RL, et al. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics. (2012) 129:e1121–e8. doi: 10.1542/peds.2011-2583
41. Lampi KM, Lehtonen L, Tran PL, Suominen A, Lehti V, Banerjee PN, et al. Risk of autism spectrum disorders in low birth weight and small for gestational age infants. J Pediatr. (2012) 161:830–6. doi: 10.1016/j.jpeds.2012.04.058
42. Zerbo O, Qian Y, Yoshida C, Grether JK, Van De Water J, Croen LA. Maternal infection during pregnancy and autism spectrum disorders. J Autism Dev Disord. (2015) 45:4015–25. doi: 10.1007/s10803-013-2016-3
43. Anderson GM, Jacobs-Stannard A, Chawarska K, Volkmar FR, Kliman HJ. Placental trophoblast inclusions in autism spectrum disorder. Biol Psychiatry. (2007) 61:487–91. doi: 10.1016/j.biopsych.2006.03.068
44. Mtali YS, Lyimo MA, Luzzatto L, Massawe SN. Hypertensive disorders of pregnancy are associated with an inflammatory state: evidence from hematological findings and cytokine levels. BMC Preg Childbirth. (2019) 19:237. doi: 10.1186/s12884-019-2383-7
45. Napso T, Yong HEJ, Lopez-Tello J, Sferruzzi-Perri AN. The role of placental hormones in mediating maternal adaptations to support pregnancy and lactation. Front Phys. (2018) 9:1091. doi: 10.3389/fphys.2018.01091
46. Li M, Fallin MD, Riley A, Landa R, Walker SO, Silverstein M, et al. The association of maternal obesity and diabetes with autism and other developmental disabilities. Pediatrics. (2016) 137:e20152206–e2015. doi: 10.1542/peds.2015-2206
47. Xiang AH, Wang X, Martinez MP, Page K, Buchanan TA, Feldman RK. Maternal type 1 diabetes and risk of autism in offspring. JAMA. (2018) 320:89. doi: 10.1001/jama.2018.7614
48. Vambergue A, Fajardy I. Consequences of gestational and pregestational diabetes on placental function and birth weight. World J Diabetes. (2011) 2:196–203. doi: 10.4239/wjd.v2.i11.196
49. Vignoles P, Gire C, Mancini J, Bretelle F, Boubli L, Janky E, et al. Gestational diabetes: a strong independent risk factor for severe neonatal respiratory failure after 34 weeks. Arch Gynecol Obstet. (2011) 284:1099–104. doi: 10.1007/s00404-010-1810-9
50. Mastorakos G, Ilias I. Maternal and fetal hypothalamic-Pituitary-Adrenal axes during pregnancy and postpartum. Ann NY Acad Sci. (2003) 997:136–49. doi: 10.1196/annals.1290.016
51. Kampmann U, Knorr S, Fuglsang J, Ovesen P. Determinants of maternal insulin resistance during pregnancy: an updated overview. J Diabetes Res. (2019) 2019:1–9. doi: 10.1155/2019/5320156
52. Baz B, Riveline JP, Gautier JF. ENDOCRINOLOGY oF pREGNANCY: gestational diabetes mellitus: definition, aetiological and clinical aspects. Eur J Endocrinol. (2016) 174:R43–51. doi: 10.1530/EJE-15-0378
53. Fowden AL, Li J, Forhead AJ. Glucocorticoids and the preparation for life after birth: are there long-term consequences of the life insurance? Proc Nutr Soc. (1998) 57:113–22. doi: 10.1079/PNS19980017
54. Gewolb IH. Effect of high glucose on fetal lung maturation at different times in gestation. Exp Lung Res. (1996) 22:201–11. doi: 10.3109/01902149609050847
55. Desoye G, Hauguel-De Mouzon S. The human placenta in gestational diabetes mellitus: the insulin and cytokine network. Diabetes Care. (2007) 30(Suppl. 2):S120–S6. doi: 10.2337/dc07-s203
56. Windham GC, Anderson M, Lyall K, Daniels JL, Kral TVE, Croen LA, et al. Maternal pre-pregnancy body mass index and gestational weight gain in relation to autism spectrum disorder and other developmental disorders in offspring. Autism Res. (2019) 12:316–27. doi: 10.1002/aur.2057
57. Kelly AC, Powell TL, Jansson T. Placental function in maternal obesity. Clin Sci (Lond). (2020) 134:961–84. doi: 10.1042/CS20190266
58. Lof M, Hilakivi-Clarke L, Sandin SS, De Assis S, Yu W, Weiderpass E. Dietary fat intake and gestational weight gain in relation to estradiol and progesterone plasma levels during pregnancy: a longitudinal study in swedish women. BMC Women's Health. (2009) 9:10. doi: 10.1186/1472-6874-9-10
59. Kinnunen TI, Luoto R, Gissler M, Hemminki E, Hilakivi-Clarke L. Pregnancy weight gain and breast cancer risk. BMC Womens Health. (2004) 4:7. doi: 10.1186/1472-6874-4-7
60. Petridou E, Katsouyanni K, Hsieh CC, Antsaklis A, Trichopoulos D. Diet, pregnancy estrogens and their possible relevance to cancer risk in the offspring. Oncology. (1992) 49:127–32. doi: 10.1159/000227025
61. Kallak TK, Hellgren C, Skalkidou A, Sandelin-Francke L, Ubhayasekhera K, Bergquist J, et al. Maternal and female fetal testosterone levels are associated with maternal age and gestational weight gain. Europ J Endocr. (2017) 177:379–88. doi: 10.1530/EJE-17-0207
62. Class QA, Lichtenstein P, Långström N, D'Onofrio BM. Timing of prenatal maternal exposure to severe life events and adverse pregnancy outcomes: a Population study of 2.6 million pregnancies. Psychosom Med. (2011) 73:234–41. doi: 10.1097/PSY.0b013e31820a62ce
63. Kinney DK, Munir KM, Crowley DJ, Miller AM. Prenatal stress and risk for autism. Neurosci Biobehav Rev. (2008) 32:1519–32. doi: 10.1016/j.neubiorev.2008.06.004
64. De Boo HA, Harding JE. The developmental origins of adult disease (Barker) hypothesis. Austr New Zeal J Obstetr Gynaecol. (2006) 46:4–14. doi: 10.1111/j.1479-828X.2006.00506.x
65. Barker DJ. The fetal and infant origins of adult disease. BMJ. (1990) 301:1111. doi: 10.1136/bmj.301.6761.1111
66. Barker DJP. Fetal origins of coronary heart disease. BMJ. (1995) 311:171–4. doi: 10.1136/bmj.311.6998.171
67. Barker DJP. The origins of the developmental origins theory. J Internal Med. (2007) 261:412–7. doi: 10.1111/j.1365-2796.2007.01809.x
68. Mandy M, Nyirenda M. Developmental origins of health and disease: the relevance to developing nations. Int Health. (2018) 10:66–70. doi: 10.1093/inthealth/ihy006
69. Barker D. Infant mortality, childhood nutrition, and ischaemic heart disease in england and wales. Lancet. (1986) 327:1077–81. doi: 10.1016/S0140-6736(86)91340-1
70. Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. (1989) 298:564–7. doi: 10.1136/bmj.298.6673.564
71. Dabelea D. The predisposition to obesity and diabetes in offspring of diabetic mothers. Diabetes Care. (2007) 30 (Suppl. 2):S169–74. doi: 10.2337/dc07-s211
72. Kelstrup L, Damm P, Mathiesen ER, Hansen T, Vaag AA, Pedersen O, et al. Insulin resistance and impaired pancreatic β-cell function in adult offspring of women with diabetes in pregnancy. J Clin Endocrinol Metab. (2013) 98:3793–801. doi: 10.1210/jc.2013-1536
73. Silverman BL, Metzger BE, Cho NH, Loeb CA. Impaired glucose tolerance in adolescent offspring of diabetic mothers. Relationship to fetal hyperinsulinism. Diabetes Care. (1995) 18:611–7. doi: 10.2337/diacare.18.5.611
74. Drake AJ, Reynolds RM. Impact of maternal obesity on offspring obesity and cardiometabolic disease risk. Reproduction. (2010) 140:387–98. doi: 10.1530/REP-10-0077
75. Eriksson JG, Sandboge S, Salonen MK, Kajantie E, Osmond C. Long-term consequences of maternal overweight in pregnancy on offspring later health: findings from the helsinki birth cohort study. Ann Med. (2014) 46:434–8. doi: 10.3109/07853890.2014.919728
76. Freeman DJ. Effects of maternal obesity on fetal growth and body composition: implications for programming and future health. Semin Fetal Neonatal Med. (2010) 15:113–8. doi: 10.1016/j.siny.2009.09.001
77. Barker DJP. Sir richard doll lecture. Developmental origins of chronic disease. Public Health. (2012) 126:185–9. doi: 10.1016/j.puhe.2011.11.014
78. Lau C, Rogers JM, Desai M, Ross MG. Fetal programming of adult disease: implications for prenatal care. Obstet Gynecol. (2011) 117:978–85. doi: 10.1097/AOG.0b013e318212140e
79. Neri C, Edlow AG. Effects of maternal obesity on fetal programming: molecular approaches. Cold Spring Harb Perspect Med. (2015) 6:a026591–a. doi: 10.1101/cshperspect.a026591
80. West NA, Crume TL, Maligie MA, Dabelea D. Cardiovascular risk factors in children exposed to maternal diabetes in utero. Diabetologia. (2011) 54:504–7. doi: 10.1007/s00125-010-2008-1
81. Tam WH, Ma RCW, Ozaki R, Li AM, Chan MHM, Yuen LY, et al. In utero exposure to maternal hyperglycemia increases childhood cardiometabolic risk in offspring. Diabetes Care. (2017) 40:679–86. doi: 10.2337/dc16-2397
82. Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, et al. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. (2000) 49:2208–11. doi: 10.2337/diabetes.49.12.2208
83. Yogev Y, Visser GH. Obesity, gestational diabetes and pregnancy outcome. Semin Fetal Neonatal Med. (2009) 14:77–84. doi: 10.1016/j.siny.2008.09.002
84. Ornoy A. Prenatal origin of obesity and their complications: gestational diabetes, maternal overweight and the paradoxical effects of fetal growth restriction and macrosomia. Reprod Toxic. (2011) 32:205–12. doi: 10.1016/j.reprotox.2011.05.002
85. Stroik S, Hendrickson EA. Telomere replication—When the going gets tough. DNA Repair. (2020) 94:102875. doi: 10.1016/j.dnarep.2020.102875
86. Ryckman K, Smith C. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diab Metab Syndr ObesityTargets Therapy. (2015) 2015:295. doi: 10.2147/DMSO.S61296
87. Xu J, Ye J, Wu Y, Zhang H, Luo Q, Han C, et al. Reduced fetal telomere length in gestational diabetes. PLoS ONE. (2014) 9:e86161. doi: 10.1371/journal.pone.0086161
88. He J, Zhang A, Fang M, Fang R, Ge J, Jiang Y, et al. Methylation levels at iGF2 and gNAS dMRs in infants born to preeclamptic pregnancies. BMC Genomics. (2013) 14:472. doi: 10.1186/1471-2164-14-472
89. Dynkevich Y, Rother KI, Whitford I, Qureshi S, Galiveeti S, Szulc AL, et al. Tumors, iGF-2, and hypoglycemia: insights from the clinic, the laboratory, and the historical archive. Endocr Rev. (2013) 34:798–826. doi: 10.1210/er.2012-1033
90. Braun T, Challis JR, Newnham JP, Sloboda DM. Early-life glucocorticoid exposure: the hypothalamic-pituitary-adrenal axis, placental function, and long-term disease risk. Endocr Rev. (2013) 34:885–916. doi: 10.1210/er.2013-1012
91. Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 1: outcomes. Nat Rev Endocrinol. (2014) 10:391–402. doi: 10.1038/nrendo.2014.73
92. Taylor JL, Corbett BA. A review of rhythm and responsiveness of cortisol in individuals with autism spectrum disorders. Psychoneuroendocrinology. (2014) 49:207–28. doi: 10.1016/j.psyneuen.2014.07.015
93. Fulford AJ, Harbuz MS. Chapter 1.3 - an introduction to the hPA axis. In: Steckler T, Kalin NH, Reul JMHM, editors. Techniques in the Behavioral and Neural Sciences. 15: Elsevier (2005). p. 43–65. doi: 10.1016/S0921-0709(05)80006-9
94. Herman J. Stress response: neural and feedback regulation of the hPA axis. Stress Sci Neuroendocr. (2010) 75:505–10. doi: 10.1016/B978-008045046-9.00097-8
95. Bruehl H, Rueger M, Dziobek I, Sweat V, Tirsi A, Javier E, et al. Hypothalamic-Pituitary-Adrenal axis dysregulation and memory impairments in type 2 diabetes. The Journal of Clinical Endocrinology & Metabolism. (2007) 92:2439–45. doi: 10.1210/jc.2006-2540
96. Chang L, Sundaresh S, Elliott J, Anton PA, Baldi P, Licudine A, et al. Dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis in irritable bowel syndrome. Neurogastroent Motility. (2009) 21:149–59. doi: 10.1111/j.1365-2982.2008.01171.x
97. Lopez-Duran NL, Kovacs M, George CJ. Hypothalamic–pituitary–adrenal axis dysregulation in depressed children and adolescents: a meta-analysis. Psychoneuroendocrinology. (2009) 34:1272–83. doi: 10.1016/j.psyneuen.2009.03.016
98. Pasquali R, Vicennati V, Cacciari M, Pagotto U. The hypothalamic-Pituitary-Adrenal axis activity in obesity and the metabolic syndrome. Ann N York Acad Sci. (2006) 1083:111–28. doi: 10.1196/annals.1367.009
99. Popp J, Wolfsgruber S, Heuser I, Peters O, Hüll M, Schröder J, et al. Cerebrospinal fluid cortisol and clinical disease progression in mCI and dementia of alzheimer's type. Neurobiol Aging. (2015) 36:601–7. doi: 10.1016/j.neurobiolaging.2014.10.031
100. Watson S, Mackin P. HPA axis function in mood disorders. Psychiatry. (2006) 5:166–70. doi: 10.1383/psyt.2006.5.5.166
101. Baker EK, Richdale AL, Hazi A, Prendergast LA. Assessing a hyperarousal hypothesis of insomnia in adults with autism spectrum disorder. Autism Res. (2019) 12:897–910. doi: 10.1002/aur.2094
102. Corbett BA, Simon D. Adolescence, stress and cortisol in autism spectrum disorders. OA Autism. (2014) 1:2. doi: 10.13172/2052-7810-1-1-348
103. Edmiston EK, Blain SD, Corbett BA. Salivary cortisol and behavioral response to social evaluative threat in adolescents with autism spectrum disorder. Autism Res. (2017) 10:346–58. doi: 10.1002/aur.1660
104. Muscatello RA, Corbett BA. Comparing the effects of age, pubertal development, and symptom profile on cortisol rhythm in children and adolescents with autism spectrum disorder. Autism Res. (2018) 11:110–20. doi: 10.1002/aur.1879
105. Spratt EG, Nicholas JS, Brady KT, Carpenter LA, Hatcher CR, Meekins KA, et al. Enhanced cortisol response to stress in children in autism. J Autism Dev Disord. (2012) 42:75–81. doi: 10.1007/s10803-011-1214-0
106. Sharpley CF, Bitsika V, Andronicos NM, Agnew LL. Further evidence of hPA-axis dysregulation and its correlation with depression in autism spectrum disorders: data from girls. Physiol Behav. (2016) 167:110–7. doi: 10.1016/j.physbeh.2016.09.003
107. Corbett BA, Mendoza S, Wegelin JA, Carmean V, Levine S. Variable cortisol circadian rhythms in children with autism and anticipatory stress. J Psychiatry Neurosci. (2008) 33:227–34.
108. Rose J, Schwartz J, Green J, Kerr D. Development of the corticotropin-releasing factor adrenocorticotropic hormone/beta-endorphin system in the mammalian fetus. Fetal Neonatal Phys. (1998) 1998:2431–42.
109. Winter JS. Fetal and Neonatal Adrenocortical Physiology. Fetal and Neonatal Physiology. Philadelphia, PA: Elsevier (2004). p. 1915–25. doi: 10.1016/B978-0-7216-9654-6.50190-9
110. Mesiano S, Jaffe RB. Developmental and functional biology of the primate fetal adrenal cortex*. Endocr Rev. (1997) 18:378–403. doi: 10.1210/edrv.18.3.0304
111. Rainey WE, Nakamura Y. Regulation of the adrenal androgen biosynthesis. J Steroid Biochem Mol Biol. (2008) 108:281–6. doi: 10.1016/j.jsbmb.2007.09.015
112. Blackburn S. Maternal, Fetal, & Neonatal Physiology: A Clinical Perspective. 4th ed. Maryland Heights, MO: Elsevier Saunders (2013). P. 768.
113. Gitau R, Fisk NM, Teixeira JMA, Cameron A, Glover V. Fetal hypothalamic-Pituitary-Adrenal stress responses to invasive procedures are independent of maternal responses1. J Clin Endocr Metab. (2001) 86:104–9. doi: 10.1210/jc.86.1.104
114. Coulter CL, Jaffe RB. Functional maturation of the primate fetal adrenal in vivo: 3. Specific Zonal Localization and Developmental Regulation of CYP21A2 (P450c21) and CYP11B1/CYP11B2 (P450c11/Aldosterone Synthase) lead to integrated concept of zonal and temporal steroid biosynthesis 1. Endocrinology. (1998) 139:5144–50. doi: 10.1210/endo.139.12.6333
115. Ishimoto H, Jaffe RB. Development and function of the human fetal adrenal cortex: a Key component in the feto-Placental unit. Endocr Rev. (2011) 32:317–55. doi: 10.1210/er.2010-0001
116. Tegethoff M, Pryce C, Meinlschmidt G. Effects of intrauterine exposure to synthetic glucocorticoids on fetal, newborn, and infant hypothalamic-Pituitary-Adrenal axis function in humans: a Systematic review. Endocr Rev. (2009) 30:753–89. doi: 10.1210/er.2008-0014
117. Lebenthal A, Lebenthal E. The ontogeny of the small intestinal epithelium. JPEN J Parenter Enteral Nutr. (1999) 23(5 Suppl.):S3–6. doi: 10.1177/014860719902300502
118. Fowden AL, Forhead AJ. Adrenal glands are essential for activation of glucogenesis during undernutrition in fetal sheep near term. Am J Physiol Endocrinol Metab. (2011) 300:E94–102. doi: 10.1152/ajpendo.00205.2010
119. Wood CE, Keller-Wood M. The critical importance of the fetal hypothalamus-pituitary-adrenal axis. F1000Research. (2016) 5:115. doi: 10.12688/f1000research.7224.1
120. Pepe G. Central integrative role of oestrogen in the regulation of placental steroidogenic maturation and the development of the fetal pituitary-adrenocortical axis in the baboon. Hum Reprod Update. (1998) 4:406–19. doi: 10.1093/humupd/4.4.406
121. Green BB, Armstrong DA, Lesseur C, Paquette AG, Guerin DJ, Kwan LE, et al. The role of placental 11-Beta hydroxysteroid dehydrogenase type 1 and type 2 methylation on gene expression and infant birth weight1. Biol Reprod. (2015) 92:149. doi: 10.1095/biolreprod.115.128066
122. Burton PJ, Waddell BJ. Dual function of 11β-Hydroxysteroid dehydrogenase in placenta: modulating placental glucocorticoid passage and local steroid action1. Biol Reprod. (1999) 60:234–40. doi: 10.1095/biolreprod60.2.234
123. Lan N, Chiu MP, Ellis L, Weinberg J. Prenatal alcohol exposure and prenatal stress differentially alter glucocorticoid signaling in the placenta and fetal brain. Neuroscience. (2017) 342:167–79. doi: 10.1016/j.neuroscience.2015.08.058
124. Kaludjerovic J, Ward WE. The interplay between estrogen and fetal adrenal cortex. J Nutr Metab. (2012) 2012:1–12. doi: 10.1155/2012/837901
125. Ng PC. The fetal and neonatal hypothalamic-pituitary-adrenal axis. Arch Dis Childhood Fetal Neonatal Edition. (2000) 82:250F–4. doi: 10.1136/fn.82.3.F250
126. Albrecht ED, Pepe GJ. Central integrative role of oestrogen in modulating the communication between the placenta and fetus that results in primate fecal-placental development. Placenta. (1999) 20:129–39. doi: 10.1053/plac.1998.0359
127. Beitins IZ, Bayard F, Ances IG, Kowarski A, Migeon CJ. The metabolic clearance rate, blood production, interconversion and transplacental passage of cortisol and cortisone in pregnancy near term. Pediatric Res. (1973) 7:509–19. doi: 10.1203/00006450-197305000-00004
128. Rainey WE, Rehman KS, Carr BR. The human fetal adrenal: making adrenal androgens for placental estrogens. Semin Reprod Med. (2004) 22:327–36. doi: 10.1055/s-2004-861549
129. Tal R, Taylor HS, Burney RO, Mooney SB, Giudice LC. Endocrinology of pregnancy. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Stratakis CA, Trence DL, Wilson DP, editors. Endotext. South Dartmouth, MA: MDText.com, Inc. (2000). Available online at: https://europepmc.org/article/med/25905197#impact
130. Resko JA, Pleom JG, Stadelman HL. Estrogens in fetal and maternal plasma of the rhesus monkey. Endocrinology. (1975) 97:425–30. doi: 10.1210/endo-97-2-425
131. Bronson SL, Bale TL. The placenta as a mediator of stress effects on neurodevelopmental reprogramming. Neuropsychopharmacology. (2016) 41:207–18. doi: 10.1038/npp.2015.231
132. Redline RW. Placental inflammation. Semin Neonatol. (2004) 9:265–74. doi: 10.1016/j.siny.2003.09.005
133. Brien ME, Baker B, Duval C, Gaudreault V, Jones RL, Girard S. Alarmins at the maternal-fetal interface: involvement of inflammation in placental dysfunction and pregnancy complications (1). Can J Physiol Pharmacol. (2019) 97:206–12. doi: 10.1139/cjpp-2018-0363
134. Weckman AM, Ngai M, Wright J, McDonald CR, Kain KC. The impact of infection in pregnancy on placental vascular development and adverse birth outcomes. Front Microb. (2019) 10:1924. doi: 10.3389/fmicb.2019.01924
135. Huynh J, Dawson D, Roberts D, Bentley-Lewis R. A systematic review of placental pathology in maternal diabetes mellitus. Placenta. (2015) 36:101–14. doi: 10.1016/j.placenta.2014.11.021
136. Ray JG, Vermeulen MJ, Schull MJ, McDonald S, Redelmeier DA. Metabolic syndrome and the risk of placental dysfunction. J Obstetr Gynaeco Canada. (2005) 27:1095–101. doi: 10.1016/S1701-2163(16)30391-7
137. Salge AKM, Rocha KMN, Xavier RM, Ramalho WS, Rocha ÉL, Guimarães JV, et al. Macroscopic placental changes associated with fetal and maternal events in diabetes mellitus. Clinics (São Paulo). (2012) 67:1203–8. doi: 10.6061/clinics/2012(10)13
138. Wallace JG, Bellissimo CJ, Yeo E, Fei Xia Y, Petrik JJ, Surette MG, et al. Obesity during pregnancy results in maternal intestinal inflammation, placental hypoxia, and alters fetal glucose metabolism at mid-gestation. Scient Rep. (2019) 9:17621. doi: 10.1038/s41598-019-54098-x
139. Pepe GJ, Burch MG, Albrecht ED. Estrogen regulates 11β-Hydroxysteroid dehydrogenase-1 and−2 localization in placental syncytiotrophoblast in the second half of primate pregnancy. Endocrinology. (2001) 142:4496–503. doi: 10.1210/endo.142.10.8434
140. Bolté E, Gattereau D, Joly M, Lefebvre Y. PERFUSION oF hUMAN tERM pLACENTA iN sITU wITH c-19 aND c-18 sTEROIDS. Acta Endocrinol. (1969) 61:307–19. doi: 10.1530/acta.0.0610307
141. Baker BL, Jaffe RB. The genesis of cell types in the adenohypophysis of the human fetus as observed with immunocytochemistry. Am J Anatomy. (1975) 143:137–61. doi: 10.1002/aja.1001430202
142. Yen S. Endocrine-Metabolic Adaptations in Pregnancy. Reproductive Endocrinology. Philadelphia: WB Saunders. (1991). p.936–81.
144. Rehman KS, Sirianni R, Parker CR Jr, Rainey WE, Carr BR. The regulation of adrenocorticotrophic hormone receptor by corticotropin-releasing hormone in human fetal adrenal definitive/transitional zone cells. Reprod Sci. (2007) 14:578–87. doi: 10.1177/1933719107307908
145. Smith R, Mesiano S, Chan E-C, Brown S, Jaffe RB. Corticotropin-Releasing hormone directly and preferentially stimulates dehydroepiandrosterone sulfate secretion by human fetal adrenal cortical cells*. J Clin Endocr Metab. (1998) 83:2916–20. doi: 10.1210/jc.83.8.2916
146. Chakravorty A, Mesiano S, Jaffe RB. Corticotropin-Releasing hormone stimulates p450 17α-Hydroxylase/17,20-Lyase in human fetal adrenal cells via protein kinase c*. J Clin Endocr Metab. (1999) 84:3732–8. doi: 10.1210/jc.84.10.3732
147. Sirianni R, Rehman KS, Carr BR, Parker CR, Rainey WE. Corticotropin-Releasing hormone directly stimulates cortisol and the cortisol biosynthetic pathway in human fetal adrenal cells. J Clin Endocr Metab. (2005) 90:279–85. doi: 10.1210/jc.2004-0865
148. Parker JCR, Stankovic AM, Goland RS. Corticotropin-releasing hormone stimulates steroidogenesis in cultured human adrenal cells. Mol Cell Endocr. (1999) 155:19–25. doi: 10.1016/S0303-7207(99)00118-5
149. Chavan AR, Griffith OW, Wagner GP. The inflammation paradox in the evolution of mammalian pregnancy: turning a foe into a friend. Curr Opin Genet Dev. (2017) 47:24–32. doi: 10.1016/j.gde.2017.08.004
150. Newby EA, Myers DA, Ducsay CA. Fetal endocrine and metabolic adaptations to hypoxia: the role of the hypothalamic-pituitary-adrenal axis. Am J Phys Endocr Metab. (2015) 309:E429–E39. doi: 10.1152/ajpendo.00126.2015
151. Wadhwa PD, Culhane JF, Rauh V, Barve SS. Stress and preterm birth: neuroendocrine, immune/inflammatory, and vascular mechanisms. Matern Child Health J. (2001) 5:119–25. doi: 10.1023/A:1011353216619
152. Strauss JF III, Barbieri RL. Yen & Jaffe's Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management (Expert Consult-Online and Print). Philadelphia, PA: Elsevier Health Sciences (2013).
153. Challis JRG, Sloboda D, Matthews SG, Holloway A, Alfaidy N, Patel FA, et al. The fetal placental hypothalamic–pituitary–adrenal (HPA) axis, parturition and post natal health. Mol Cell Endocr. (2001) 185:135–44. doi: 10.1016/S0303-7207(01)00624-4
154. Halladay AK, Bishop S, Constantino JN, Daniels AM, Koenig K, Palmer K, et al. Sex and gender differences in autism spectrum disorder: summarizing evidence gaps and identifying emerging areas of priority. Mol Autism. (2015) 6:36. doi: 10.1186/s13229-015-0019-y
155. Kung KTF, Thankamony A, Ong KKL, Acerini CL, Dunger DB, Hughes IA, et al. No relationship between prenatal or early postnatal androgen exposure and autistic traits: evidence using anogenital distance and penile length measurements at birth and 3 months of age. J Child Psych Psychiatry. (2020) 90:279–85. doi: 10.1111/jcpp.13335
156. Loomes R, Hull L, Mandy WPL. What is the male-to-Female ratio in autism spectrum disorder? A systematic review and meta-analysis. J Am Acad Child Adol Psychiatry. (2017) 56:466–74. doi: 10.1016/j.jaac.2017.03.013
157. Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol. (2013) 26:146–53. doi: 10.1097/WCO.0b013e32835ee548
158. Baron-Cohen S. The extreme male brain theory of autism. Trends Cogn Sci. (2002) 6:248–54. doi: 10.1016/S1364-6613(02)01904-6
159. Arnold AP, Gorski RA. Gonadal steroid induction of structural sex differences in the central nervous system. Annu Rev Neurosci. (1984) 7:413–42. doi: 10.1146/annurev.ne.07.030184.002213
160. Arnold AP. Sex chromosomes and brain gender. Nat Rev Neurosci. (2004) 5:701–8. doi: 10.1038/nrn1494
161. Zuloaga DG, Puts DA, Jordan CL, Breedlove SM. The role of androgen receptors in the masculinization of brain and behavior: what we've learned from the testicular feminization mutation. Horm Behav. (2008) 53:613–26. doi: 10.1016/j.yhbeh.2008.01.013
162. McCarthy MM. Estradiol and the developing brain. Physiol Rev. (2008) 88:91–124. doi: 10.1152/physrev.00010.2007
163. Wu MV, Manoli DS, Fraser EJ, Coats JK, Tollkuhn J, Honda S-I, et al. Estrogen masculinizes neural pathways and sex-specific behaviors. Cell. (2009) 139:61–72. doi: 10.1016/j.cell.2009.07.036
164. Swartz SK, Soloff MS. The lack of estrogen binding by human alpha-fetoprotein. J Clin Endocrinol Metab. (1974) 39:589–91. doi: 10.1210/jcem-39-3-589
165. Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, Law K, et al. A unified genetic theory for sporadic and inherited autism. Proc Natl Acad Sci USA. (2007) 104:12831–6. doi: 10.1073/pnas.0705803104
166. Chakrabarti B, Dudbridge F, Kent L, Wheelwright S, Hill-Cawthorne G, Allison C, et al. Genes related to sex steroids, neural growth, and social-emotional behavior are associated with autistic traits, empathy, and asperger syndrome. Autism Res. (2009) 2:157–77. doi: 10.1002/aur.80
167. Sarachana T, Hu VW. Genome-wide identification of transcriptional targets of rORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism. (2013) 4:14. doi: 10.1186/2040-2392-4-14
168. Zettergren A, Jonsson L, Johansson D, Melke J, Lundström S, Anckarsäter H, et al. Associations between polymorphisms in sex steroid related genes and autistic-like traits. Psychoneuroendocrinology. (2013) 38:2575–84. doi: 10.1016/j.psyneuen.2013.06.004
169. McCarthy MM, Nugent BM. Epigenetic contributions to hormonally-mediated sexual differentiation of the brain. J Neuroendocr. (2013) 25:1133–40. doi: 10.1111/jne.12072
170. McCarthy MM, Auger AP, Bale TL, De Vries GJ, Dunn GA, Forger NG, et al. The epigenetics of sex differences in the brain. J Neurosci. (2009) 29:12815–23. doi: 10.1523/JNEUROSCI.3331-09.2009
171. Menger Y, Bettscheider M, Murgatroyd C, Spengler D. Sex differences in brain epigenetics. Epigenomics. (2010) 2:807–21. doi: 10.2217/epi.10.60
172. Nugent BM, Schwarz JM, McCarthy MM. Hormonally mediated epigenetic changes to steroid receptors in the developing brain: implications for sexual differentiation. Horm Behav. (2011) 59:338–44. doi: 10.1016/j.yhbeh.2010.08.009
173. Mayoral SR, Omar G, Penn AA. Sex differences in a hypoxia model of preterm brain damage. Pediatric research. (2009) 66:248–53. doi: 10.1203/PDR.0b013e3181b1bc34
174. Kapoor A, Dunn E, Kostaki A, Andrews MH, Matthews SG. Fetal programming of hypothalamo-pituitary-adrenal function: prenatal stress and glucocorticoids. J Phys. (2006) 572(Pt 1):31–44. doi: 10.1113/jphysiol.2006.105254
175. DiPietro JA, Voegtline KM. The gestational foundation of sex differences in development and vulnerability. Neuroscience. (2017) 342:4–20. doi: 10.1016/j.neuroscience.2015.07.068
176. Clifton VL. Review: sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta. (2010) 31 (Suppl.):S33–9. doi: 10.1016/j.placenta.2009.11.010
177. Murphy VE, Gibson PG, Giles WB, Zakar T, Smith R, Bisits AM, et al. Maternal asthma is associated with reduced female fetal growth. Am J Respir Crit Care Med. (2003) 168:1317–23. doi: 10.1164/rccm.200303-374OC
178. Dearden L, Bouret SG, Ozanne SE. Sex and gender differences in developmental programming of metabolism. Mol Metab. (2018) 15:8–19. doi: 10.1016/j.molmet.2018.04.007
179. Eriksson JG, Kajantie E, Osmond C, Thornburg K, Barker DJP. Boys live dangerously in the womb. Am J Hum Biol. (2010) 22:330–5. doi: 10.1002/ajhb.20995
180. Rosenfeld CS. Sex-Specific placental responses in fetal development. Endocrinology. (2015) 156:3422–34. doi: 10.1210/en.2015-1227
181. O'Tierney-Ginn P, Presley L, Minium J, Hauguel Demouzon S, Catalano PM. Sex-specific effects of maternal anthropometrics on body composition at birth. Am J Obstetr Gynec. (2014) 211:292.e1-.e9. doi: 10.1016/j.ajog.2014.05.031
182. Morgane PJ, Austin-LaFrance R, Bronzino J, Tonkiss J, Díaz-Cintra S, Cintra L, et al. Prenatal malnutrition and development of the brain. Neurosci Biobehav Rev. (1993) 17:91–128. doi: 10.1016/S0149-7634(05)80234-9
183. Innis SM. Essential fatty acid transfer and fetal development. Placenta. (2005) 26 (Suppl. A):S70–5. doi: 10.1016/j.placenta.2005.01.005
184. Melamed N, Ben-Haroush A, Pardo J, Chen R, Hadar E, Hod M, et al. Expectant management of preterm premature rupture of membranes: is it all about gestational age? Am J Obstet Gynecol. (2011) 204:48.e1–8. doi: 10.1016/j.ajog.2010.08.021
185. Stark MJ, Wright IM, Clifton VL. Sex-specific alterations in placental 11beta-hydroxysteroid dehydrogenase 2 activity and early postnatal clinical course following antenatal betamethasone. Am J Physiol Regul Integr Comp Physiol. (2009) 297:R510–4. doi: 10.1152/ajpregu.00175.2009
186. Stark MJ, Clifton VL, Wright IM. Neonates born to mothers with preeclampsia exhibit sex-specific alterations in microvascular function. Pediatr Res. (2009) 65:292–5. doi: 10.1203/PDR.0b013e318193edf1
187. Muralimanoharan S, Maloyan A, Myatt L. Evidence of sexual dimorphism in the placental function with severe preeclampsia. Placenta. (2013) 34:1183–9. doi: 10.1016/j.placenta.2013.09.015
188. Bahado-Singh RO, Mele L, Landon MB, Ramin SM, Carpenter MW, Casey B, et al. Fetal male gender and the benefits of treatment of mild gestational diabetes mellitus. Am J Obstet Gynecol. (2012) 206:422.e1-5. doi: 10.1016/j.ajog.2012.03.015
189. Seaborn T, Simard M, Provost PR, Piedboeuf B, Tremblay Y. Sex hormone metabolism in lung development and maturation. Trends Endocrinol Metab. (2010) 21:729–38. doi: 10.1016/j.tem.2010.09.001
190. Adler E, Madankumar R, Rosner M, Reznik SE. Increased placental trophoblast inclusions in placenta accreta. Placenta. (2014) 35:1075–8. doi: 10.1016/j.placenta.2014.09.014
191. Baergen RN. Hydatidiform Moles. Manual of Pathology of the Human Placenta. Boston, MA: Springer (2011). doi: 10.1007/978-1-4419-7494-5
192. Baergen RN. Abortion and the placenta in chromosomal anomalies. In: Baergen RN, editor. Manual of Pathology of the Human Placenta: Second Edition. Boston, MA: Springer US (2011). p. 165–83. doi: 10.1007/978-1-4419-7494-5_11
193. Silvestre E, Cusí V, Borrás M, Antich J. Cytogenetic and morphologic findings in chorionic villi from spontaneous abortions. Birth Defects Orig Artic Ser. (1996) 30:353–7.
194. Parazzini F, Mangili G, La Vecchia C, Negri E, Bocciolone L, Fasoli M. Risk factors for gestational trophoblastic disease: a separate analysis of complete and partial hydatidiform moles. Obstetrics and gynecology. (1991) 78:1039–45.
195. Walker CK, Anderson KW, Milano KM, Ye S, Tancredi DJ, Pessah IN, et al. Trophoblast inclusions are significantly increased in the placentas of children in families at risk for autism. Biological Psychiatry. (2013) 74:204–11. doi: 10.1016/j.biopsych.2013.03.006
196. Park BY, Misra DP, Moye J, Miller RK, Croen L, Fallin MD, et al. Placental gross shape differences in a high autism risk cohort and the general population. PLoS ONE. (2018) 13:e0191276. doi: 10.1371/journal.pone.0191276
197. Chang J-M, Zeng H, Han R, Chang Y-M, Shah R, Salafia CM, et al. Autism risk classification using placental chorionic surface vascular network features. BMC Med Inform Dec Making. (2017) 17:162. doi: 10.1186/s12911-017-0564-8
198. Jenabi E, Karami M, Khazaei S, Bashirian S. The association between preeclampsia and autism spectrum disorders among children: a meta-analysis. Kor J Pediatrics. (2019) 62:126–30. doi: 10.3345/kjp.2018.07010
199. Leach L, Taylor A, Sciota F. Vascular dysfunction in the diabetic placenta: causes and consequences. J Anatomy. (2009) 215:69–76. doi: 10.1111/j.1469-7580.2009.01098.x
200. Herr F, Baal N, Zygmunt M. Studies of placental vasculogenesis: a way to understand pregnancy pathology? Z Geburtshilfe Neonatol. (2009) 213:96–100. doi: 10.1055/s-0029-1224141
201. Barut F, Barut A, Gun BD, Kandemir NO, Harma MI, Harma M, et al. Intrauterine growth restriction and placental angiogenesis. Diag Pathol. (2010) 5:24. doi: 10.1186/1746-1596-5-24
202. Alexander J, Teague AM, Chen J, Aston CE, Leung YK, Chernausek S, et al. Offspring sex impacts dNA methylation and gene expression in placentae from women with diabetes during pregnancy. PLoS ONE. (2018) 13:e0190698. doi: 10.1371/journal.pone.0190698
203. Nugent BM, O'Donnell CM, Epperson CN, Bale TL. Placental h3K27me3 establishes female resilience to prenatal insults. Nat Commun. (2018) 9:2555. doi: 10.1038/s41467-018-04992-1
204. Pantaleon M, Steane SE, McMahon K, Cuffe JSM, Moritz KM. Placental o-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice. Sci Rep. (2017) 7:2017. doi: 10.1038/s41598-017-01666-8
205. Chatuphonprasert W, Jarukamjorn K, Ellinger I. Physiology and pathophysiology of steroid biosynthesis, transport and metabolism in the human placenta. Front Pharm. (2018) 9:1027. doi: 10.3389/fphar.2018.01027
206. Cox B, Leavey K, Nosi U, Wong F, Kingdom J. Placental transcriptome in development and pathology: expression, function, and methods of analysis. Am J Obstet Gynecol. (2015) 213 (4 Suppl.):S138–51. doi: 10.1016/j.ajog.2015.07.046
207. Lutchmaya S, Baron-Cohen S, Raggatt P. Foetal testosterone and eye contact in 12-month-old human infants. Infant Behav Dev. (2002) 25:327–35. doi: 10.1016/S0163-6383(02)00094-2
208. Auyeung B, Ahluwalia J, Thomson L, Taylor K, Hackett G, O'Donnell KJ, et al. Prenatal versus postnatal sex steroid hormone effects on autistic traits in children at 18 to 24 months of age. Molecular Autism. (2012) 3:17. doi: 10.1186/2040-2392-3-17
209. Auyeung B, Baron-Cohen S, Ashwin E, Knickmeyer R, Taylor K, Hackett G. Fetal testosterone and autistic traits. Br J Psych. (2009) 100:1–22. doi: 10.1348/000712608X311731
210. Chapman E, Baron-Cohen S, Auyeung B, Knickmeyer R, Taylor K, Hackett G. Fetal testosterone and empathy: evidence from the empathy quotient (EQ) and the “reading the mind in the eyes” test. Soc Neurosci. (2006) 1:135–48. doi: 10.1080/17470910600992239
211. Knickmeyer R, Baron-Cohen S, Raggatt P, Taylor K. Foetal testosterone, social relationships, and restricted interests in children. J Child Psych Psychiatry. (2005) 46:198–210. doi: 10.1111/j.1469-7610.2004.00349.x
212. Baron-Cohen S, Auyeung B, Nørgaard-Pedersen B, Hougaard DM, Abdallah MW, Melgaard L, et al. Elevated fetal steroidogenic activity in autism. Mol Psychiatry. (2015) 20:369–76. doi: 10.1038/mp.2014.48
213. McCarthy MM, Herold K, Stockman SL. Fast, furious and enduring: sensitive versus critical periods in sexual differentiation of the brain. Physiol Behav. (2018) 187:13–9. doi: 10.1016/j.physbeh.2017.10.030
214. Reyes FI, Boroditsky RS, Winter JS, Faiman C. Studies on human sexual development. II. Fetal and maternal serum gonadotropin and sex steroid concentrations. J Clin Endocrinol Metab. (1974) 38:612–7. doi: 10.1210/jcem-38-4-612
215. Allison C, Baron-Cohen S, Wheelwright S, Charman T, Richler J, Pasco G, et al. The q-CHAT (Quantitative cHecklist for autism in toddlers): a normally distributed quantitative measure of autistic traits at 18-24 months of age: preliminary report. J Autism Dev Disord. (2008) 38:1414–25. doi: 10.1007/s10803-007-0509-7
216. Williams J, Scott F, Stott C, Allison C, Bolton P, Baron-Cohen S, et al. The cAST (Childhood asperger syndrome test): test accuracy. Autism. (2005) 9:45–68. doi: 10.1177/1362361305049029
217. Auyeung B, Baron-Cohen S, Wheelwright S, Allison C. The autism spectrum quotient: children's version (AQ-Child). J Autism Dev Disord. (2008) 38:1230–40. doi: 10.1007/s10803-007-0504-z
218. Kung KTF, Spencer D, Pasterski V, Neufeld S, Glover V, O'Connor TG, et al. No relationship between prenatal androgen exposure and autistic traits: convergent evidence from studies of children with congenital adrenal hyperplasia and of amniotic testosterone concentrations in typically developing children. J Child Psych Psychiatry. (2016) 57:1455–62. doi: 10.1111/jcpp.12602
219. Baron-Cohen S, Tsompanidis A, Auyeung B, Norgaard-Pedersen B, Hougaard DM, Abdallah M, et al. Foetal oestrogens and autism. Mol Psychiatry. (2020) 25:2970–8. doi: 10.1038/s41380-019-0454-9
220. Rodeck CH, Gill D, Rosenberg DA, Collins WP. Testosterone levels in midtrimester maternal and fetal plasma and amniotic fluid. Prenat Diagn. (1985) 5:175–81. doi: 10.1002/pd.1970050303
221. Van De Beek C, Thijssen JHH, Cohen-Kettenis PT, Van Goozen SHM, Buitelaar JK. Relationships between sex hormones assessed in amniotic fluid, and maternal and umbilical cord serum: what is the best source of information to investigate the effects of fetal hormonal exposure? Horm Behav. (2004) 46:663–9. doi: 10.1016/j.yhbeh.2004.06.010
222. Park BY, Lee BK, Burstyn I, Tabb LP, Keelan JA, Whitehouse AJO, et al. Umbilical cord blood androgen levels and aSD-related phenotypes at 12 and 36 months in an enriched risk cohort study. Mol Autism. (2017) 8:3. doi: 10.1186/s13229-017-0118-z
223. Baron-Cohen S, Wheelwright S, Skinner R, Martin J, Clubley E. The autism-spectrum quotient (AQ): evidence from asperger syndrome/high-functioning autism, males and females, scientists and mathematicians. J Autism Dev Disord. (2001) 31:5–17. doi: 10.1023/a:1005653411471
224. Jamnadass ESL, Keelan JA, Hollier LP, Hickey M, Maybery MT, Whitehouse AJO. The perinatal androgen to estrogen ratio and autistic-like traits in the general population: a longitudinal pregnancy cohort study. J Neurodev Disord. (2015) 7:17. doi: 10.1186/s11689-015-9114-9
225. Whitehouse AJ, Mattes E, Maybery MT, Dissanayake C, Sawyer M, Jones RM, et al. Perinatal testosterone exposure and autistic-like traits in the general population: a longitudinal pregnancy-cohort study. J Neurodevelop Disord. (2012) 4:25. doi: 10.1186/1866-1955-4-25
226. Terloyeva D, Frey AJ, Park BY, Kauffman EM, Mathew L, Bostwick A, et al. Meconium androgens are correlated with aSD-related phenotypic traits in early childhood in a familial enriched risk cohort. Mol Autism. (2020) 11:93. doi: 10.1186/s13229-020-00395-6
227. Ramón y Cajal CL, Martínez RO. Defecation in utero: a physiologic fetal function. Am J Obstet Gynecol. (2003) 188:153–6. doi: 10.1067/mob.2003.107
228. Bearer CF. Meconium as a biological marker of prenatal exposure. Ambul Ped. (2003) 3:40–3. doi: 10.1367/1539-4409(2003)003<0040:MAABMO>2.0.CO;2
229. Word RA, George FW, Wilson JD, Carr BR. Testosterone synthesis and adenylate cyclase activity in the early human fetal testis appear to be independent of human chorionic gonadotropin control. J Clin Endocrinol Metab. (1989) 69:204–8. doi: 10.1210/jcem-69-1-204
230. Scott HM, Mason JI, Sharpe RM. Steroidogenesis in the fetal testis and its susceptibility to disruption by exogenous compounds. Endocr Rev. (2009) 30:883–925. doi: 10.1210/er.2009-0016
231. Clements JA, Reyes FI, Winter JS, Faiman C. Studies on human sexual development. III. Fetal pituitary and serum, and amniotic fluid concentrations of LH, CG, and FSH. J Clin Endocrinol Metab. (1976) 42:9–19. doi: 10.1210/jcem-42-1-9
232. Ahluwalia B, Williams J, Verma P. In vitro testosterone biosynthesis in the human fetal testis. II. Stimulation by Cyclic AMP and Human Chorionie Gonadotropin (hCG)*. Endocrinology. (1974) 95:1411–5. doi: 10.1210/endo-95-5-1411
233. Huhtaniemi T, Korenbrot C, Jaffe RB. hCG binding and stimulation of testosterone biosynthesis in the human fetal testis. J Clin Endocr Metab. (1977) 44:963–7. doi: 10.1210/jcem-44-5-963
234. Rousseau-Merck MF, Misrahi M, Atger M, Loosfelt H, Milgrom E, Berger R. Localization of the human luteinizing hormone/choriogonadotropin receptor gene (LHCGR) to chromosome 2p21. Cytogenet Cell Genet. (1990) 54:77–9. doi: 10.1159/000132962
235. Braunstein GD, Rasor J, Adler D, Danzer H, Wade ME. Serum human chorionic gonadotropin levels throughout normal pregnancy. Am J Obstetr Gynec. (1976) 126:678–81. doi: 10.1016/0002-9378(76)90518-4
236. Alfthan H, Schröder J, Fraser R, Koskimies A, Halila H, Stenman UH. Choriogonadotropin and its beta subunit separated by hydrophobic-interaction chromatography and quantified in serum during pregnancy by time-resolved immunofluorometric assays. Clin Chem. (1988) 34:1758–62. doi: 10.1093/clinchem/34.9.1753
237. Dugoff L. First- and second-trimester maternal serum markers for aneuploidy and adverse obstetric outcomes. Obstet Gynecol. (2010) 115:1052–61. doi: 10.1097/AOG.0b013e3181da93da
238. Yaron Y, Cherry M, Kramer RL, O'Brien JE, Hallak M, Johnson MP, et al. Second-trimester maternal serum marker screening: maternal serum alpha-fetoprotein, beta-human chorionic gonadotropin, estriol, and their various combinations as predictors of pregnancy outcome. Am J Obstet Gynecol. (1999) 181:968–74. doi: 10.1016/S0002-9378(99)70334-0
239. Smail PJRFI, Winter JSD, Faiman C. The fetal hormonal environment and its effect on the morphogenesis of the genital system. In: Kogan SJ, Hafez ESE, editor. Pediatric Andrology Clinics in Andrology. Dordrecht: Springer (1981). p. 7. doi: 10.1007/978-94-010-3719-8_2
240. Celec P, Ostatnã-Kovã¡ D, Hodosy Jl. On the effects of testosterone on brain behavioral functions. Front Neurosci. (2015) 9:12. doi: 10.3389/fnins.2015.00012
241. Melau C, Nielsen JE, Frederiksen H, Kilcoyne K, Perlman S, Lundvall L, et al. Characterization of human adrenal steroidogenesis during fetal development. J Clin Endocr Metab. (2018) 104:1802–12. doi: 10.1210/jc.2018-01759
242. Reyes FI, Winter JSD, Faiman C. Studies on human sexual development. I. Fetal Gonadal and Adrenal Sex Steroids1. J Clin Endocr Metab. (1973) 37:74–8. doi: 10.1210/jcem-37-1-74
243. Nwabuobi C, Arlier S, Schatz F, Guzeloglu-Kayisli O, Lockwood CJ, Kayisli UA. hCG: biological functions and clinical applications. Int J Mol Sci. (2017) 18:2037. doi: 10.3390/ijms18102037
244. Cole LA. Biological functions of hCG and hCG-related molecules. Reprod Biol Endocrinol. (2010) 8:102. doi: 10.1186/1477-7827-8-102
245. Korevaar TIM, Steegers EAP, De Rijke YB, Schalekamp-Timmermans S, Visser WE, Hofman A, et al. Reference ranges and determinants of total hCG levels during pregnancy: the generation r Study. Europ J Epid. (2015) 30:1057–66. doi: 10.1007/s10654-015-0039-0
246. Kayisli UA, Selam B, Guzeloglu-Kayisli O, Demir R, Arici A. Human chorionic gonadotropin contributes to maternal immunotolerance and endometrial apoptosis by regulating fas-Fas ligand system. J Immunol. (2003) 171:2305–13. doi: 10.4049/jimmunol.171.5.2305
247. Windham GC, Lyall K, Anderson M, Kharrazi M. Autism spectrum disorder risk in relation to maternal mid-Pregnancy serum hormone and protein markers from prenatal screening in california. J Autism Dev Disorders. (2016) 46:478–88. doi: 10.1007/s10803-015-2587-2
248. Onderoglu LS, Kabukçu A. Elevated second trimester human chorionic gonadotropin level associated with adverse pregnancy outcome. Int J Gynaecol Obstet. (1997) 56:245–9. doi: 10.1016/S0020-7292(96)02830-5
249. Olsen RN, Woelkers D, Dunsmoor-Su R, Lacoursiere DY. Abnormal second-trimester serum analytes are more predictive of preterm preeclampsia. Am J Obstet Gynecol. (2012) 207:228.e1-7. doi: 10.1016/j.ajog.2012.06.006
250. Benn PA, Horne D, Briganti S, Rodis JF, Clive JM. Elevated second-trimester maternal serum hcg alone or in combination with elevated alpha-fetoprotein. Obstetr Gynecol. (1996) 87:217–22. doi: 10.1016/0029-7844(95)00390-8
251. Wenstrom KD, Owen J, Boots LR, DuBard MB. Elevated second-trimester human chorionic gonadotropin levels in association with poor pregnancy outcome. Am J Obstet Gynecol. (1994) 171:1038–41. doi: 10.1016/0002-9378(94)90030-2
252. Tal J, Kaplan M, Sharf M, Barnea ER. Stress-related hormones affect human chorionic gonadotrophin secretion from the early human placenta in vitro. Hum Reprod. (1991) 6:766–9. doi: 10.1093/oxfordjournals.humrep.a137425
253. Serón-Ferré M, Lawrence CC, Jaffe RB. Role of hCG in regulation of the fetal zone of the human fetal adrenal gland*. J Clin Endocrin Metab. (1978) 46:834–7. doi: 10.1210/jcem-46-5-834
254. Hammond GL. Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. J Endocr. (2016) 230:R13–R25. doi: 10.1530/JOE-16-0070
255. Resnik R, Killam AP, Battaglia FC, Makowski EL, Meschia G. The stimulation of uterine blood flow by various estrogens. Endocrinology. (1974) 94:1192–6. doi: 10.1210/endo-94-4-1192
256. Anderson JN, Peck EJ, Clark SB. Estrogen-Induced uterine responses and growth: relationship to receptor estrogen binding by uterine nuclei11. Endocrinology. (1975) 96:160–7. doi: 10.1210/endo-96-1-160
257. Falah N, Torday JK. Quinney s, m. Haas D. Estriol review: clinical applications and potential biomedical importance. Clin Res Trials. (2015) 1:109. doi: 10.15761/CRT.1000109
258. Goebelsmann U, Jaffe RB. Oestriol metabolism in pregnant women. Acta Endocrinol (Copenh). (1971) 66:679–93. doi: 10.1530/acta.0.0660679
259. Gagnon A, Wilson RD. Obstetrical complications associated with abnormal maternal serum markers analytes. J Obstet Gynaecol Can. (2008) 30:918–32. doi: 10.1016/S1701-2163(16)32973-5
260. Schleifer RA, Bradley LA, Richards DS, Ponting NR. Pregnancy outcome for women with very low levels of maternal serum unconjugated estriol on second-trimester screening. Am J Obstet Gynecol. (1995) 173:1152–6. doi: 10.1016/0002-9378(95)91343-2
261. Kim SY, Kim SK, Lee JS, Kim IK, Lee K. The prediction of adverse pregnancy outcome using low unconjugated estriol in the second trimester of pregnancy without risk of down's syndrome. Yonsei Med J. (2000) 41:226. doi: 10.3349/ymj.2000.41.2.226
262. Taché V, Baer RJ, Currier RJ, Li C-S, Towner D, Waetjen LE, et al. Population-based biomarker screening and the development of severe preeclampsia in california. Am J Obstetr Gynec. (2014) 211:377.e1–e8. doi: 10.1016/j.ajog.2014.03.026
263. Kowalczyk TD, Cabaniss ML, Cusmano L. Association of low unconjugated estriol in the second trimester and adverse pregnancy outcome. Obstet Gynecol. (1998) 91:396–400. doi: 10.1016/S0029-7844(97)00677-7
264. Thaler MA, Seifert-Klauss V, Luppa PB. The biomarker sex hormone-binding globulin - from established applications to emerging trends in clinical medicine. Best Pract Res Clin Endocrinol Metab. (2015) 29:749–60. doi: 10.1016/j.beem.2015.06.005
265. Bartha JL, Comino-Delgado R, Romero-Carmona R, Gomez-Jaen MC. Sex hormone-binding globulin in gestational diabetes. Acta Obstet Gynecol Scand. (2000) 79:839–45. doi: 10.1034/j.1600-0412.2000.079010839.x
266. Jin Z, Guan X, Gao H, Shang L, Gao M, Su D, et al. The change in sex hormone binding globulin and the influence by gestational diabetes mellitus in fetal period. Gynecol Endocrinol. (2009) 25:647–52. doi: 10.1080/09513590903015437
267. Kopp HP, Festa A, Krugluger W, Schernthaner G. Low levels of sex-Hormone-Binding globulin predict insulin requirement in patients with gestational diabetes mellitus. Exp Clin Endocrinol Diabetes. (2001) 109:365–9. doi: 10.1055/s-2001-17408
268. Jansson T, Powell Theresa L. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci. (2007) 113:1–13. doi: 10.1042/CS20060339
269. Myatt L. Placental adaptive responses and fetal programming. J Phys. (2006) 572:25–30. doi: 10.1113/jphysiol.2006.104968
270. Misra DP, Salafia CM, Miller RK, Charles AK. Non-Linear and gender-Specific relationships among placental growth measures and the fetoplacental weight ratio. Placenta. (2009) 30:1052–7. doi: 10.1016/j.placenta.2009.09.008
271. van Abeelen AFM, de Rooij SR, Osmond C, Painter RC, Veenendaal MVE, Bossuyt PMM, et al. The sex-specific effects of famine on the association between placental size and later hypertension. Placenta. (2011) 32:694–8. doi: 10.1016/j.placenta.2011.06.012
272. Tarrade A, Panchenko P, Junien C, Gabory A. Placental contribution to nutritional programming of health and diseases: epigenetics and sexual dimorphism. J Exp Biol. (2015) 218:50–8. doi: 10.1242/jeb.110320
273. Sood R, Zehnder JL, Druzin ML, Brown PO. Gene expression patterns in human placenta. Proc Natl Acade Sci. (2006) 103:5478–83. doi: 10.1073/pnas.0508035103
274. Baulieu EE, Dray F. Conversion of h3-Dehydroisoandrosterone (3β-Hydroxy-Δ5-androsten-17-one) sulfate to h3-Estrogens in normal pregnant women. J Clin Endocrinol Metab. (1963) 23:1298–301. doi: 10.1210/jcem-23-12-1298
275. Pecks U, Rath W, Kleine-Eggebrecht N, Maass N, Voigt F, Goecke T, et al. Maternal serum lipid, estradiol, and progesterone levels in pregnancy, and the impact of placental and hepatic pathologies. Geburtshilfe Frauenh. (2016) 76:799–808. doi: 10.1055/s-0042-107078
276. Sandman CA. Fetal exposure to placental corticotropin-releasing hormone (pCRH) programs developmental trajectories. Peptides. (2015) 72:145–53. doi: 10.1016/j.peptides.2015.03.020
277. Thomson M. The physiological roles of placental corticotropin releasing hormone in pregnancy and childbirth. J Physiol Biochem. (2013) 69:559–73. doi: 10.1007/s13105-012-0227-2
278. Robinson DP, Klein SL. Pregnancy and pregnancy-associated hormones alter immune responses and disease pathogenesis. Horm Behav. (2012) 62:263–71. doi: 10.1016/j.yhbeh.2012.02.023
279. Magness RR, Rosenfeld CR. Local and systemic estradiol-17 beta: effects on uterine and systemic vasodilation. Am J Physiol. (1989) 256(4 Pt 1):E536–42. doi: 10.1152/ajpendo.1989.256.4.E536
280. Mandalà M. Influence of estrogens on uterine vascular adaptation in normal and preeclamptic pregnancies. Int J Mol Sci. (2020) 21:2592. doi: 10.3390/ijms21072592
281. Palmer SK, Zamudio S, Coffin C, Parker S, Stamm E, Moore LG. Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet Gynecol. (1992) 80:1000–6.
282. Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. (1971) 284:878–81. doi: 10.1056/NEJM197104222841604
283. Giusti RM, Iwamoto K, Hatch EE. Diethylstilbestrol revisited: a review of the long-term health effects. Ann Intern Med. (1995) 122:778–88. doi: 10.7326/0003-4819-122-10-199505150-00008
Keywords: autism, autism spectrum disorders, steroid hormone (progesterone, testosterone, estradiol), perinatal, prenatal, risk factors
Citation: Worsham W, Dalton S and Bilder DA (2021) The Prenatal Hormone Milieu in Autism Spectrum Disorder. Front. Psychiatry 12:655438. doi: 10.3389/fpsyt.2021.655438
Received: 19 January 2021; Accepted: 17 May 2021;
Published: 01 July 2021.
Edited by:
Jeremy Veenstra-VanderWeele, Columbia University Irving Medical Center, United StatesReviewed by:
Kurt Leroy Hoffman, Autonomous University of Tlaxcala, MexicoCopyright © 2021 Worsham, Dalton and Bilder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Whitney Worsham, d29yc2hhbS53aGl0bmV5QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.