 Ajay Sarawagi
Ajay Sarawagi Narayan Datt Soni
Narayan Datt Soni Anant Bahadur Patel
Anant Bahadur Patel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 27 April 2021
Sec. Molecular Psychiatry
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.637863
This article is part of the Research TopicMagnetic Resonance Spectroscopy of GABA and Glutamate in Mental HealthView all 17 articles
Major depressive disorder (MDD) is a leading cause of distress, disability, and suicides. As per the latest WHO report, MDD affects more than 260 million people worldwide. Despite decades of research, the underlying etiology of depression is not fully understood. Glutamate and γ-aminobutyric acid (GABA) are the major excitatory and inhibitory neurotransmitters, respectively, in the matured central nervous system. Imbalance in the levels of these neurotransmitters has been implicated in different neurological and psychiatric disorders including MDD. 1H nuclear magnetic resonance (NMR) spectroscopy is a powerful non-invasive method to study neurometabolites homeostasis in vivo. Additionally, 13C-NMR spectroscopy together with an intravenous administration of non-radioactive 13C-labeled glucose or acetate provides a measure of neural functions. In this review, we provide an overview of NMR-based measurements of glutamate and GABA homeostasis, neurometabolic activity, and neurotransmitter cycling in MDD. Finally, we highlight the impact of recent advancements in treatment strategies against a depressive disorder that target glutamate and GABA pathways in the brain.
Major depressive disorder (MDD) is a neuropsychiatric condition, characterized by low mood, loss of interest in pleasurable activities, and suicidal ideation. It affects ~5% of the population worldwide (1). As per the WHO report (2020), around 0.8 million people commit suicide every year, and more than 90% of these had a psychiatric diagnosis (2, 3). MDD is one of the major contributors to chronic disease burden over the world population, and imparts a high socioeconomic impact (4). Despite several decades of research, there are no robust physiological and molecular markers for psychiatric disorders. Therefore, diagnosis of these disorders is achieved mostly by questionnaire-based psychiatric evaluation. The diagnostic criteria for psychiatric disorders have been evolving continuously. The diagnostic standards and specifiers of MDD as per the latest edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5, 2013) are described in Box 1 (5). Very often, the symptoms of different neuropsychiatric disorders overlap with each other and interfere in precise diagnosis. Hence, there is a need for extensive research on the identification of biomarkers for the development of novel diagnostic strategies for MDD. Depression is a highly variable disorder with multiple risk factors and causes that vary at the individual level. Certain environmental factors such as prematernal stress, childhood abuse, physical and sexual abuse, continuous failures, substance abuse, sadness and severe trauma increase the risk of depression (6, 7). Depression has been often seen to be associated with various neurodegenerative disorders (8) such as Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis, and systemic diseases like diabetes (9) and cancer (10).
Box 1. Symptoms of MDD: as per diagnostic and statistical manual of mental disorder (DSM-V) at least five of the following symptoms must be present during entire 2-week period (5).
➢ Consistently feeling sad, empty, and hopeless
➢ Markedly diminished interest in pleasurable activities
➢ Significant weight loss or weight gain
➢ Increased or decreased appetite
➢ Insomnia or hypersomnia
➢ Fatigue or loss of energy
➢ Feeling of worthlessness, feeling excessive, or inappropriate guilt
➢ Diminished ability to think or concentrate, or indecisiveness
➢ Recurrent thoughts of death and suicidal ideation without a specific plan
➢ Psychomotor agitation or retardation
These symptoms cause clinically significant distress or impairment in social, occupational, or other important areas of functioning. Moreover, the episode is not attributable to the physiological effects of a substance or another medical condition.
Despite enormous efforts made by the global psychiatric research community, the molecular mechanism of MDD is not yet very clear. Several neuroimaging and postmortem studies have shown a loss of neuronal and glial population in the cingulate cortex, prefrontal cortex (PFC) (11) and hippocampus (12, 13) of depressed subjects (Figure 1) (14, 15). Various genetic factors (16), epigenetic changes (17) and endocrine pathways (18) are believed to be involved in the pathophysiology of the disorder. The elevated activity of the hypothalamic–pituitary–adrenal (HPA) axis is at the heart of the neurobiological presumptions of depression (19). Higher activity of HPA axis increases levels of glucocorticoids in blood, plasma and cerebrospinal fluid (CSF), which are greatly associated with stress. Additionally, environmental factors and stress influence neuronal function epigenetically. These factors alter gene expression by histone acetylation or DNA methylation (20).
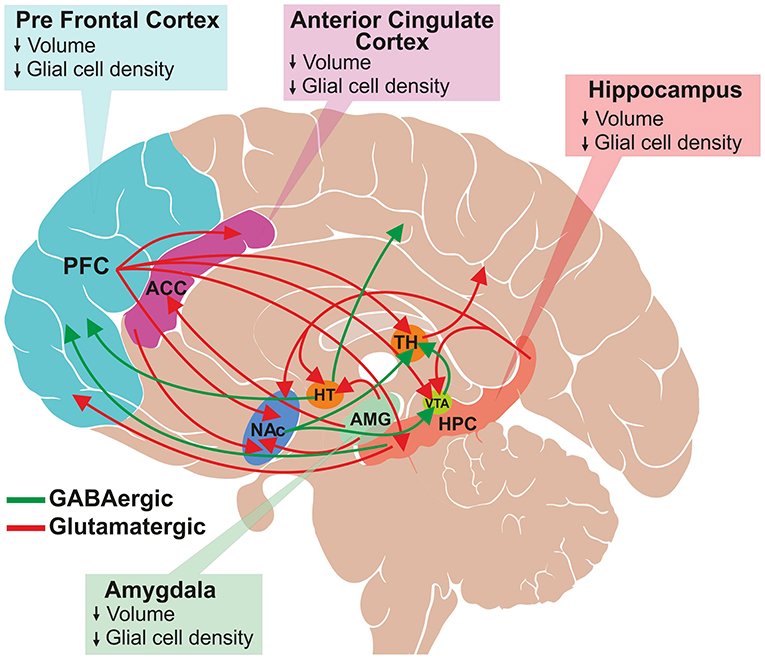
Figure 1. Schematic of glutamatergic and GABAergic projections involved in mood regulation and reward pathway. A subset of several known interconnections among different brain regions are shown. Major glutamatergic projections (red color) arise from the frontal cortex to the anterior cingulate cortex (ACC), thalamus (TH), ventral tegmental area (VTA), hippocampus (HPC) and nucleus accumbens (NAc). Additionally, glutamatergic neurons originate from hippocampus, and innervate into hypothalamus (HT), VTA, NAc and PFC and from amygdala to HT, ACC and NAc. The GABAergic projections (green color) are widely distributed throughout the brain. The major projections that are relevant to this review are from HT to the occipital and parietal cortex, HPC to PFC, and NAc to the thalamus and VTA. The structural changes observed in the brain regions of depressed subjects are shown in the respective boxes.
The role of epigenetics in depression is supported by studies reporting antidepressive effects of histone deacetylase inhibitors in rodent models of depression (20, 21). Additionally, a large number of studies have reported a reduced level of brain-derived neurotrophic factor (BDNF) in the hippocampus (HPC) and PFC of depressed subjects (22). BDNF is crucial for the activity-dependent formation and maintenance of synapses by regulating the activity of the mTORC1 complex. Activation of mTORC1 pathways promotes de novo synthesis of various synaptic proteins, including GluA1, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunits and postsynaptic density protein 95 (PSD95) (23). Interventions with different antidepressants have shown increased expression of BDNF in PFC of the rodent brain (24, 25).
Neurotransmitters are the chemical messengers present in presynaptic nerve terminals and are released into the synaptic cleft in response to the action potential (26). These neurotransmitters bind to specific receptors present on the postsynaptic membrane, and thus facilitate the transmission of the action potential across the synapse (26). Neurotransmitters are broadly classified into amino acids, peptides, and monoamines depending on their chemical properties. Amino acid neurotransmitters include glutamate, γ-aminobutyric acid (GABA), aspartate and glycine, which are abundant in the central nervous system (CNS). Substance P, cholecystokinin, opioids and neuropeptide Y belong to the peptide neurotransmitter category (27). In the monoamine category, several neurotransmitters including serotonin, dopamine, norepinephrine and epinephrine are well-studied, and are shown to be involved in various neuropsychiatric disorders (28, 29). Functionally, glutamate, aspartate, dopamine, epinephrine and norepinephrine are considered as excitatory neurotransmitters, while GABA, glycine and serotonin are the major inhibitory neurotransmitters in the matured mammalian CNS (30).
1H magnetic resonance spectroscopy (MRS) has emerged as a powerful non-invasive method for the measurement of levels of neurometabolites including glutamate and GABA in the brain (31). In addition, 13C-MRS in conjunction with administration of 13C-labeled respiratory substrates (glucose and acetate) allows analysis of the cell-specific metabolic activity in animals as well as in the human brain. This provides a non-invasive measurement of the cerebral metabolic rate of glucose oxidation, ATP production and neurotransmitter cycling (32, 33).
Depression is characterized by a deficit in various aspects of reward, which is defined as responses toward positive emotional stimuli such as food, sex and social interaction (34). Several brain regions such as prefrontal cortex (PFC), nucleus accumbens (NAc), ventral tegmental area (VTA), hippocampus (HPC) and amygdala are interconnected with each other via dopaminergic, serotonergic, glutamatergic and GABAergic neurons, which comprise the reward circuit (35, 36). The reward circuitry mainly includes dopaminergic projection from VTA to NAc, PFC, hippocampus, amygdala, as well as other brain regions. Additionally, glutamatergic and GABAergic projections interconnect these regions very densely (Figure 1). The cortical glutamate connections can be divided broadly into five major arcs that include PFC to the brainstem, PFC to the striatum and NAc, cerebral cortex to the thalamus, intracortical glutamate projections, and from the thalamus to the cerebral cortex (37, 38). Moreover, glutamatergic connections are found in subcortical regions: hippocampus to VTA, hypothalamus, NAc and PFC; and amygdala to NAc, hypothalamus and ACC. GABAergic neurons also make dense connections between brain regions that include projections from the striatum to substantia nigra (SN) and brainstem; thalamus to SN; HPC to occipital and parietal cortex; HPC to thalamus and striatum; NAc to VTA and thalamus; and VTA to PFC and NAc (Figure 1) (37, 39). The brain reward regions have been linked with specific behavioral functions, e.g., PFC for decision making and intelligence, HPC for emotional management, amygdala as the fear center, and NAc-VTA for motivation, pleasure and reward. These brain regions have broader functions in the management of emotional and cognitive behavior. Various imaging and postmortem studies have shown reduced volume and atrophy in these brain regions of depressed subjects and animal models of depression (11–15).
Neurometabolites homeostasis plays a very important role in brain function, and has been shown to be affected in animal models and human subjects of various neuropsychiatric disorders including MDD (40, 41). Several small molecules including N-acetyl-aspartate (NAA) (~9 μmol/g), alanine (~1 μmol/g), aspartate (~1.2 μmol/g), choline (~1.5 μmol/g), creatine (~7 μmol/g), GABA (~1.5 μmol/g), glutamate (~10 μmol/g), glutamine (~2.5 μmol/g), glycine (~1 μmol/g), myo-inositol (~6 μmol/g) and taurine (~1.2 μmol/g) contribute to major fraction of neurometabolites pool in healthy brain (42).
In addition to precursors for different metabolites, these molecules play various critical functions that include signal transduction, osmoregulation, cell growth and protein synthesis (42). Glutamate released into synaptic cleft increases the membrane potential of postsynaptic neurons, making them more likely to lead to an action potential. Moreover, it plays a critical role in long-term potentiation (43), synaptic plasticity (44), learning and memory (45), and various cognitive functions (46). Likewise, GABA, the major inhibitory neurotransmitter in the matured CNS, inhibits the propagation of action potential (47). Several studies have revealed the involvement of GABA in learning and memory (48–50), aggressive–defensive behavior, and impulsivity (51, 52). NAA is localized mostly in neurons, and is known to be a marker of neuronal viability and health. It acts as a precursor for NAAG, the storage form of aspartate, and serves a variety of other functions (53). Myo-inositol, mostly localized in astroglia, acts as an osmolite, plays an essential role in cell growth, and is believed to be a marker of the glial population (42). Moreover, it is considered an inflammatory marker in CNS (42). Nearly 20 vital metabolites, which include the above-mentioned molecules, can be detected and quantified in vivo by different MR spectroscopic approaches in human (54) and animal brains (55) (Figure 2). The most commonly used NMR methods for detection and quantification of brain metabolites are described in the subsequent section.
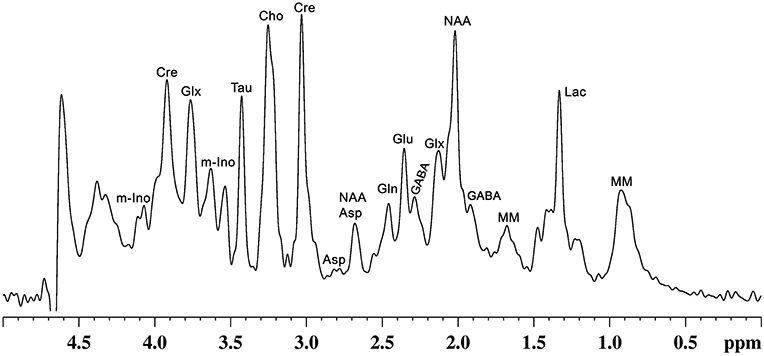
Figure 2. A representative localized in vivo 1H-NMR spectrum from mouse cerebral cortex. NMR spectrum was recorded using, a vertical wide bore magnet interfaced with 600 MHz MR spectrometer. 1H-MR spectroscopy was carried out using STEAM method in conjunction with outer volume suppression (OVS) and water suppression (VAPOR) from a voxel (4.0 × 1.2 × 2.5 mm3) with TE/TR = 4/4,000 ms with 512 averaging: Peak labels are Asp, aspartate; Cho, choline; Cr, creatine; GABA, γ-aminobutyric acid; Gln, glutamine; Glu, glutamate; Glx, glutamate + glutamine; Lac, lactate; m-Ino, myo-inositol; MM, macromolecule; NAA, N-acetyl aspartate; Tau, taurine.
The human brain accounts for 2% of the body weight, but it contributes to 20% of the total energy consumed, indicating the overwhelming energy demand of the brain (56, 57). In a matured brain, this energy requirement is majorly fulfilled by the oxidation of glucose. Most of the energy harvested in the brain is utilized for the processes associated with glutamatergic and GABAergic neurotransmission (57). The glutamate released from glutamatergic neurons into the synaptic cleft is taken up by astrocytes and converted to glutamine by glutamine synthetase. Glutamine is transported back to neurons, hydrolyzed to glutamate, and repackaged into vesicles for the next release. This process is referred as glutamate–glutamine neurotransmitter cycling (58). Similarly, substrate cycle involving GABA and glutamine (GABA–glutamine) occurs between GABAergic neurons and astrocytes (58). In this cycle, the released GABA into the synapse is taken up majorly by astrocytes, wherein it is metabolized to succinate by GABA-transaminase, and enters into the TCA cycle and ultimately converted to glutamine. The glutamine thus formed is further transported to GABAergic neurons and converted to GABA by the successive action of glutaminase and glutamate decarboxylase (59, 60). The rates of neuronal glucose oxidation and neurotransmitter cycling have been monitored by a tracer approach, wherein 13C-labeled glucose is administered intravenously, and labeling of brain amino acids is measured in vivo by 13C-NMR spectroscopy (61). The metabolism of [1,6-13C2]glucose via glycolysis followed by TCA cycle labels GluC4 in glutamatergic and GABAergic neurons (Figure 3). In GABAergic neurons, GluC4 is decarboxylated to GABAC2 by glutamate decarboxylase (GAD). GlnC4 gets labeled from GluC4 and GABAC2 through glutamate–glutamine and GABA-glutamine neurotransmitter cycling, respectively. Further metabolism of GluC4 and GABAC2 in the corresponding TCA cycle labels AspC2/C3, GluC2/C3, and GABAC3/C4. The kinetics of label incorporation in different amino acids is analyzed to determine the rate of glucose oxidation in the glutamatergic, GABAergic neurons, and rate of neurotransmitter cycling (59). Energy budget estimates for the cost of signaling based on anatomic and physiological data in the cerebral cortex indicated that most of the signaling energy is utilized on postsynaptic glutamate receptors, followed by action potentials and resting potentials. In the cerebellar cortex, glutamatergic neurons use 75%, while GABAergic neurons use 25% of the signaling energy (57). Hence, an estimate of the energy expenditure of the glutamatergic and GABAergic neurons using 13C-MRS approach directly reflects their functional status.
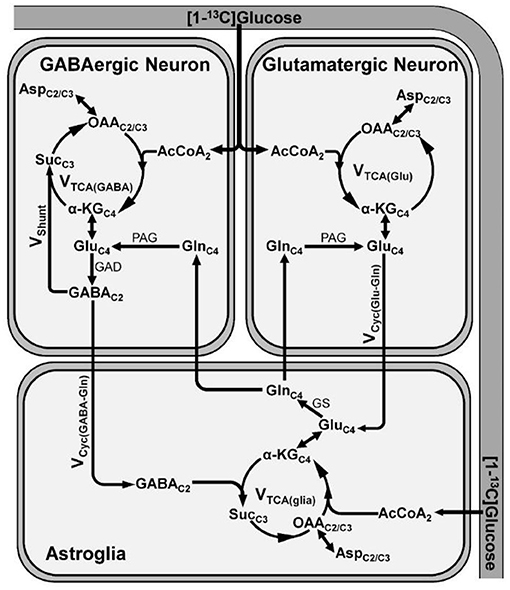
Figure 3. A schematic of three compartment metabolic model showing 13C labeling of amino acids from [1-13C]glucose. Metabolism of [1-13C]glucose via glutamatergic and GABAergic TCA cycle labels GluC4. In GABAergic neurons, GluC4 is further decarboxylated to GABAC2 by glutamate decarboxylase (GAD). The labeling of GlnC4 occurs by release and uptake of GluC4 and GABAC2 in astrocytes followed by transamination by glutamine synthetase (GS). Further metabolism of GluC4 and GABAC2 transfers the label into AspC2/C3. α-KGC4, α-ketoglutarate-C4; AcCoA2, acetyl co-enzymeA-C2; AspC2/C3, aspartate-C2/C3; GABAC2, γ-aminobutyric acid-C2; GluC4, glutamate-C4; GAD, glutamate decarboxylase; GlnC4, glutamine-C4; GS, glutamine synthetase; OAAC2/C3, oxaloacetate-C2/C3; PAG, phosphate activated glutaminase; SucC3, succinate-C3; Vcyc(GABA−Gln), GABA–glutamine cycling flux; Vcyc(Glu−Gln), glutamate–glutamine cycling flux; Vshunt, flux of GABA shunt; VTCA(glia), astroglial TCA cycle flux; VTCA(GABA), GABAergic TCA cycle flux; VTCA(Glu), glutamatergic TCA cycle flux.
The most prevalent hypothesis of depression posits that depletion in monoamine neurotransmitters level is the underlying cause of the disease (62, 63). Recent studies in animal models and human subjects have suggested an association of glutamatergic and GABAergic systems with the pathophysiology of depression (64–66). Reduced expression of receptor subunits, imbalances in their levels, decreased glutamatergic and GABAergic neurotransmission, and altered energy metabolism are known to play a critical role in the progression of depression (64, 65).
Glutamatergic neurons constitute approximately 80% of the synapses in the neocortex (67). Glutamate is released at synapses throughout the brain, and exerts changes in postsynaptic excitability and neuroplasticity (68). It activates various downstream pathways of nuclear genes by binding to a variety of membrane-bound receptors present on the postsynaptic membrane, which regulate secondary messenger systems. α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPAR), N-methyl-D-aspartate (NMDAR), and kainate are the fast-acting ionotropic receptors that get activated by glutamate binding (69). Glutamate also binds to G-protein-coupled receptors, known as metabotropic glutamate receptors, which mediate various cellular processes and slow-acting changes through secondary messengers such as cyclic adenosine monophosphate (cAMP), cyclic guanosine monophosphate (cGMP) and phosphatidylinositol (69). AMPA and kainate receptors help in the conduction of action potential primarily through the flux of Na+ ions, while NMDAR is distinguished by its more permeability to Ca2+ ions. NMDA receptor signaling promotes various responses such as excitation, neurotrophic function, and can even activate cell death pathways. Abnormal activity of NMDA receptor imparts harmful effects on neurons (69). Overexcitation of NMDAR by excessive glutamate release or impaired synaptic clearance leads to the death of neurons by excitotoxicity (70).
A large number of clinical as well as animal studies have reported impairment in the glutamatergic system in various limbic and cortical areas of the brain of depressed subjects (71, 72). Additionally, postmortem histopathology (73) and a number of 1H-MRS studies (74, 75) have shed light on the association of the aberrant glutamate system with maladaptive changes in the structure and function of excitatory circuitry. Several studies have reported decreased expression of NMDA (73, 76, 77) and AMPA receptor subunits (77, 78) in PFC of depressed individuals. Reduced expression of NMDA receptor subunits has also been seen in the postmortem brains of suicide victims (73, 79). Moreover, the decreased availability of metabotropic receptor mGluR5 in PFC, cingulate cortex, thalamus, hippocampus, and other cortical regions has been reported in depressed individuals (80, 81). Additionally, loss of glutamatergic neurons in the orbitofrontal cortex is associated with the pathophysiology of depression (82). These shreds of evidence suggest the involvement of glutamatergic system with the pathophysiology of MDD.
Glutamate acts as the precursor for GABA, the predominant inhibitory neurotransmitter in the matured brain (83). GABAergic neurons contribute to one-third of total synapses in the CNS and help in shaping the neural network dynamics (84). These inhibitory neurons are known to play a pivotal role in physiological processes that are often affected in psychiatric disorders such as neural plasticity, sensory processing, stress reactivity, memory formation, and attention (84, 85). GABA binds to two different classes of receptors, the fast-acting ligand gated or ionotropic receptor GABAA and GABAB. Activation of GABAA receptor leads to an influx of chloride ions, which inhibits the propagation of action potential. However, activation of GABAB receptors stimulates K+ channel opening, which helps in achieving a hyperpolarized state that leads to reduced transmission of action potential (86, 87).
GABAergic interneurons are identified by their expression of specific receptors for somatostatin (SST), parvalbumin (PV), and 5-HT3a. SST and PV interneurons make up to 30 and 40%, respectively, of the total GABAergic neuronal pool (88). Postmortem studies of depressed subjects have shown a reduced level of SST and PV interneurons in PFC as well as in other cortical areas (89). Additionally, a decrease in the level of SST messenger RNA (mRNA) has been reported in several brain regions, including dorsolateral PFC (90, 91), ACC (92) and amygdala (93) in depression (47). Moreover, multiple studies have reported reduced expression of GAD67 and GABA transporters in the brain of MDD subjects (90, 93, 94). In addition, genetically modified animals with deletion of specific GABA receptor subunits show depressive phenotypes (95, 96). Furthermore, treatment with various antidepressants (97), electroconvulsive therapy (ECT) (98) and cognitive behavioral therapy (74) tends to restore GABA level in depressed subjects (47). These multiple evidence suggest that impairment in GABAergic transmission plays a significant role in the pathophysiology of depression (99).
Proton (1H) is the most abundant and sensitive NMR active nucleus, and is an integral part of every neurometabolite. Due to the presence of different functional groups, 1H belonging to different molecules or attached to different carbon atoms within the same molecule experiences variation in the electronic environment. This results in differences in 1H frequencies, which is commonly known as chemical shift. This parameter is used for the distinction of metabolites by 1H-MR spectroscopy without administering any chemical agent.
The neurochemical profile provides valuable information when measured from a well-defined region/volume of the brain. This is measured using localized in vivo MR spectroscopy. The localization methods in MR spectroscopy are generally based on magnetic field gradients and radiofrequency pulses. A three dimensional voxel is selected by application of band selective radiofrequency (RF) pulses together with magnetic field gradient along X-, Y- and Z-axes. The most commonly used MR localization methods are described below.
This approach employs three frequency selective inversion pulses followed by non-selective excitation of the entire sample in the presence of three orthogonal magnetic field gradients. Image selected in vivo spectroscopy (ISIS) achieves complete 3D localization of voxel in eight scans (100).
This is referred as a double spin-echo localization method, wherein a 90° radiofrequency pulse is followed by two 180° pulses together with magnetic field gradients along three orthogonal axes (101). This produces signals exclusively from the desired volume of interest. Due to complete refocusing of the magnetization, the signal-to-noise ratio (SNR) is relatively higher in point-resolved spectroscopy (PRESS).
It is a single scan localization technique, which involves application of three 90° radiofrequency pulses together with magnetic field gradients along three orthogonal axes. Due to selection of stimulated echo using three slice-selective 90° radiofrequency pulses, stimulated echo acquisition mode (STEAM) provides signals from metabolites at a very short echo time (~5 ms) (102). Furthermore, as all the three pulses are 90° in STEAM, the amount of energy absorbed per mass of tissue is lower in this sequence as compared with PRESS. However, as STEAM focuses only 50% of the magnetization, the SNR of NMR signal in STEAM is 50% of that obtained in PRESS approach.
A combination of these localization methods together with outer volume suppression (103) provides better quality localization, especially when the voxel is relatively small to the entire excited volume. Furthermore, in vivo measurements of metabolites whose concentration is in the range of 1–30 μmol/g often encounter huge water signals (55,555 μmol/g), hence requires effective suppression of water for quantification. Various NMR characteristics like relaxation time, scalar coupling, chemical shift and diffusion have been exploited to develop several effective approaches for water suppression. Chemical shift selective (CHESS) (104) and variable pulse powers and optimized relaxation delay (VAPOR) (105) are commonly used approaches for water suppression during in vivo 1H-MR spectroscopy.
1H-MRS provides static information for metabolites from a given brain region. In contrast, 13C-MRS is very useful in monitoring the flow of labels from 13C-labeled substrates to different neurotransmitters such as GABA, glutamate and aspartate, similar to that is used in the tracer approach to evaluate the functional status of tissues and organs (Figure 4). The kinetics of 13C labeling of brain amino acids from 13C-labeled precursors (glucose/acetate) is useful to estimate the rates of synthesis and catabolism, and thus offer a measurement of neuroenergetics in a given brain region. 13C-NMR spectroscopy in the brain has been exploited extensively to understand brain energy metabolism in healthy and different neurological disorders (61).
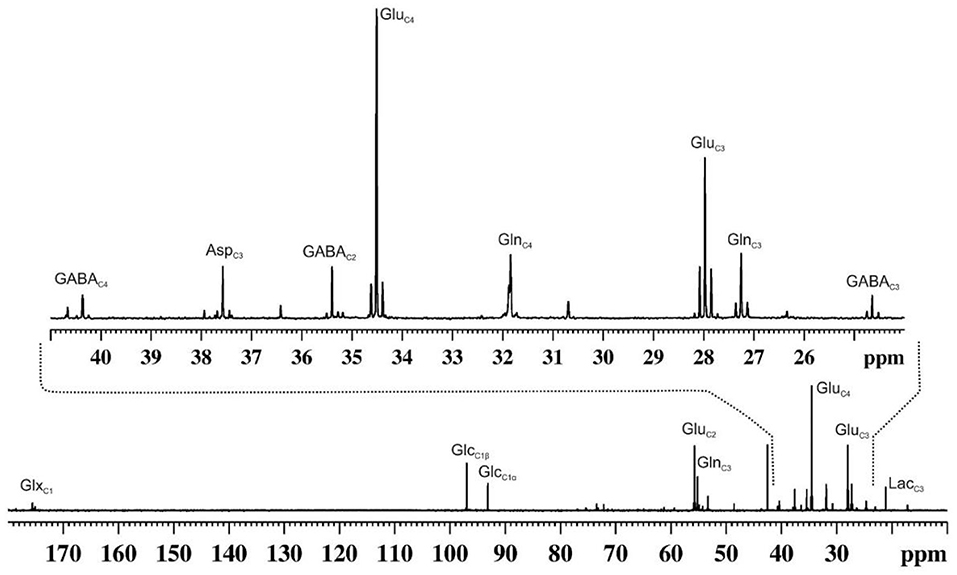
Figure 4. A representative 13C NMR spectrum of cortical extract of mouse brain showing labeling of various amino acids from [1,6-13C2]glucose. Urethane anesthetized mouse was infused with [1,6-13C2]glucose for 90 min. Brain metabolites were extracted from the cerebral cortex, and 13C NMR spectrum of the extract was recorded at 150 MHz NMR spectrometer using power gate decoupling. The spectrum shown in the upper panel is the expansion from 24 to 41 ppm. AspC3, aspartate-C3; GABAC2, γ-aminobutyric acid-C2; GABAC3, γ-aminobutyric acid-C3; GABAC4, γ-aminobutyric acid-C4; GluC2, glutamate-C2; GluC3, glutamate-C3; GluC4, glutamate-C4; GlnC2, glutamine-C2; GlnC3, glutamine-C3; GlnC4, glutamine-C4; GlxC1, (glutamate + glutamine)-C1; GlcC1β, β-D-glucose-C1; GlcC1α, α-D-glucose-C; LacC3, lactate-C3.
As mentioned earlier, the maintenance of neurometabolites homeostasis is critical for the proper functioning of a healthy brain. The changes in the levels of glutamate, GABA, and NAA are often reported under MDD. These are described in details in the following sections.
The 1H-MRS method has been used extensively for the assessment of glutamate and other metabolite levels in the brain of depressed subjects and rodent models of depression (Table 1). Reduced level of glutamate has been reported in PFC of mice in different models of depression such as chronic unpredictable mild stress (CUMS) (66), chronic social defeat stress (CSDS) (112, 116), and chronic forced swim stress (CFSS) (122). The decreased glutamate level in PFC has also been reported during the first episode of depression (107, 114). The progress of depression plays a crucial role in abnormalities in glutamate, e.g., chronic or remitted–recurrent MDD subjects showed further reduction in glutamate level in PFC as compared with the first episode depressed subjects (119). Antidepressive medication aids in the restoration of neurometabolite homeostasis to normal level. The unmedicated subjects exhibited lower levels of glutamate plus glutamine (Glx) in the dorsomedial/dorsal anterolateral prefrontal and ventromedial prefrontal cortex as compared with the medicated ones (71). However, there are few inconsistencies in the level of glutamate in depression, as some reports have shown increased glutamate in PFC of the postpartum depressed female subjects (117) and animal model of depression (125).
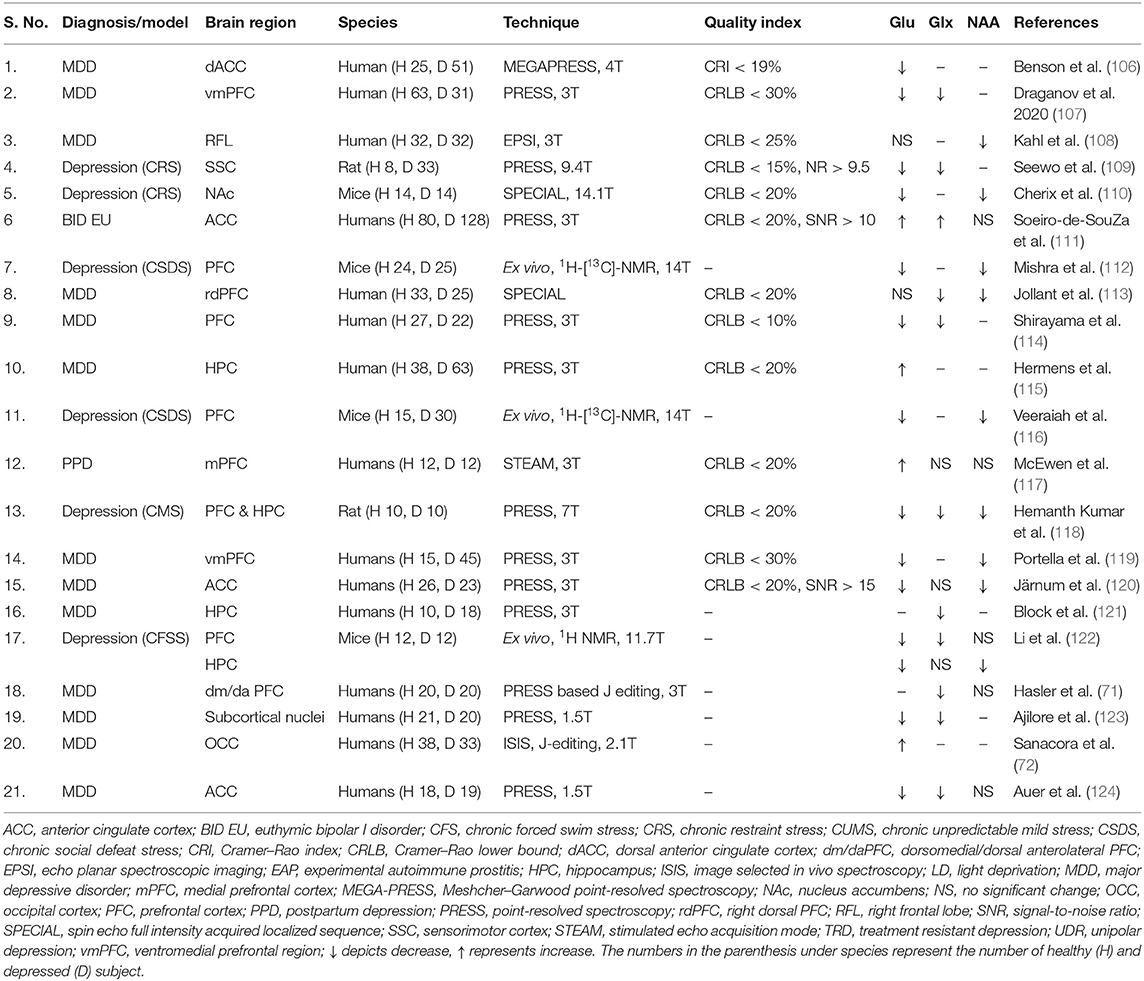
Table 1. Brain glutamate homeostasis in depression.
Glutamate level was reported to be decreased together with myo-inositol (a glial marker) and NAA in ACC of depressed subjects (120, 126). Reduced levels of Glx and glutamine have also been reported in the hippocampus of unipolar MDD subjects (121). In accordance with these findings, a reduction in the levels of Glu and NAA have been reported in the hippocampus of chronic mild stress (CMS) (118) and CFSS mouse models of depression (122). In a very recent study, levels of glutamate and glutamine have been reported to be reduced in the sensorimotor cortex of the chronic restraint stressed (CRS) rat model of depression. However, several studies have shown an increase in the level of glutamate in ACC of depressed subjects (106, 111), hippocampus of MDD subjects with alcoholic tendencies (115), and CSDS model of depression in mice.
A meta-analysis of 1H-MRS studies involving depressed subjects has revealed a decrease in levels of glutamate and glutamine primarily in ACC including the reduced level of Glx in other brain regions (127). Additionally, a very recent meta-analysis involving a greater number of participants concluded that lower levels of glutamatergic metabolites (glutamate and glutamine) in the medial frontal cortex are linked with the etiology of MDD (128). The reduced level of glutamate in MDD may be due to a lower supply of precursor glutamine by glutamate–glutamine, impaired glucose metabolism, and altered glial activity (64). The impaired functionality of glial cells in depression could lead to a reduction in synaptic glutamate uptake, which may result in elevated extracellular glutamate level that ultimately accelerates neuronal death by glutamate excitotoxicity (129). In fact, reduced expression of excitatory amino acid transporter (EAAT2) and glutamate synthetase (GS) transcripts, which are localized in glia, have been reported in CSDS mouse model of depression (116, 130). These studies support the hypo-glutamatergic hypothesis of depression and suggest that modulation of the glutamatergic system for remission of depression.
GABAergic system is involved in most psychiatric disorders including major depressive disorder (131), schizophrenia (132), bipolar disorder (133) and autism (134). Several approaches including epigenetics, postmortem studies, and measurement of GABA level in cerebrospinal fluid and plasma have been used to unravel the role of the GABA system in the pathophysiology of psychiatric disorders (131). Lower GABA levels in plasma (135) and cerebrospinal fluid (136) have been reported in depressed subjects. A summary of 1H MRS-based measure of GABA level in the depressed subjects as well as in animal models of depression is presented in Table 2. Several studies have shown a lower level of GABA in MDD subjects as compared with healthy controls (131, 154). These include lower GABA concentration in OCC (72, 75, 144), dorsomedial/dorsal anterolateral PFC (71) and ACC of depressed subjects (137, 139, 142, 143, 155). Moreover, a very recent report has shown reduced GABA level in ventromedial PFC (107) of depressed subjects. Additionally, reduced level of GABA has been reported in PFC of chronic stress model of depression in rodents (112, 118). Hence, lower GABA level is often considered as one of the most promising endophenotypes of MDD (156).
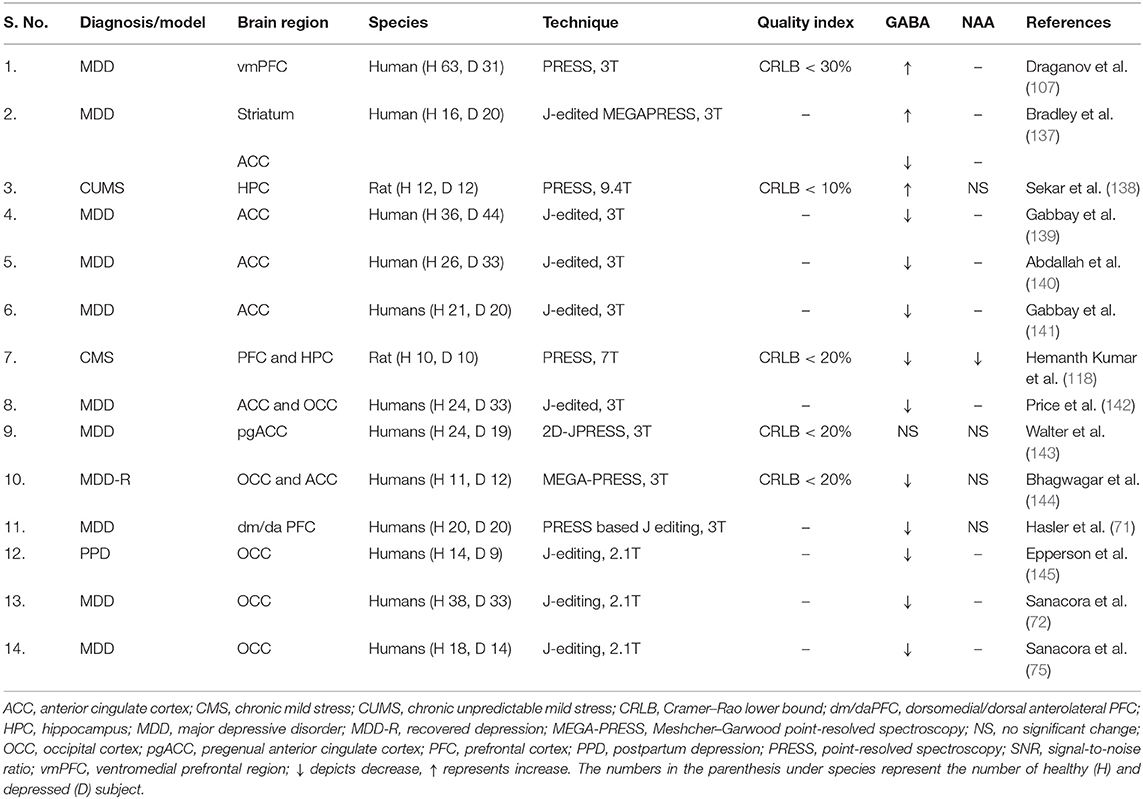
Table 2. Brain GABA homeostasis in depression.
In contrast to glutamate, whose level is independent of the mood of depressed subjects, the GABA level is state dependent, as its concentration in remitted MDD subjects is similar to healthy controls (40). It has been observed that unmedicated patients had reduced level of GABA in the dorsomedial/dorsal anterolateral PFC as compared with medicated subjects. Additionally, longitudinal 1H-MRS studies in MDD subjects have shown restoration of GABA level after electroconvulsive therapy (98), cognitive behavioral therapy (74), treatment with ketamine (150), and selective serotonin reuptake inhibitors (SSRIs) (97). Moreover, 1H-MRS measurements have shown lower OCC GABA level in treatment-resistant depressed subjects as compared with non-resistant depressed subjects and healthy volunteers (142, 157).
NAA is the strongest signal in 1H-MRS, and is exclusively localized in neurons. Although the physiological role of NAA in neural function is unclear, it is typically associated with neuronal integrity and mitochondrial health (158). Reduced level of NAA is reported in different brain regions of depressed subjects, including PFC (112, 113), ACC (120, 126), right frontal and parietal lobe (108), and in the hippocampus (122, 159) (Tables 1, 2). A lower level of NAA has also been seen in the hippocampus (122), nucleus accumbens (110) and PFC (112, 116) of rodent models of depression. Reduced levels of NAA along with glutamate suggest decrease in viability of glutamatergic neurons in depression.
Positron emission tomography (PET) (160, 161) and 13C-MRS are widely used techniques for evaluating brain energy metabolism (162) in humans and rodents. Neurometabolic activities have been investigated using 13C-MRS with an administration of 13C-labeled substrates (59, 61). As 13C-MRS can distinguish labeling of different carbon positions of glutamate, glutamine, GABA and aspartate, it is possible to measure TCA cycle fluxes separately for glutamatergic neurons, GABAergic neurons and astrocytes by appropriate modeling of the 13C turnover of neurometabolites (60, 163). Early 13C-MRS studies from Shulman et al. have led the foundation of quantitative measurement of rates of neuronal glucose oxidation and neurotransmitter cycling (164, 165). The 13C-NMR measurements together with the infusion of 13C-labeled substrates in mice (60), rats (163) and human (166) have shown that neuronal mitochondrial TCA cycle in the cerebral cortex contributes ~70–85% of the total energy produced and the remaining (~15–30%) by astroglia. The GABAergic mitochondrial TCA cycle contributes to ~20% of total neuronal TCA cycle in rats (59) and mice cerebral cortex (60). Most of the neuronal energy is used to support the processes associated with glutamate signaling such as postsynaptic glutamate receptors (50%) and action potential (20%) in the cerebral cortex (56, 57). Most importantly, 13C-NMR measurements have shown that rates of oxidative glucose metabolism in neurons and neurotransmitter cycling are stoichiometrically (1:1) coupled (165, 167), indicating that energy requirement for cycling of each glutamate molecule is powered by complete oxidation of one molecule of glucose in neurons (168, 169).
There is limited information about brain mitochondrial energetics in depressed subjects. A recent 13C-MRS study performed by the Yale Psychiatric group has reported a ~25% reduction in the mitochondrial energy production in glutamatergic neurons in the occipital cortex of depressed subjects (64). However, there was no change in the GABA synthesis rate and glutamate–glutamine neurotransmitter cycling flux. Using CSDS mouse model of depression, we have reported a reduction in the rate of glucose oxidation in glutamatergic and GABAergic neurons in PFC of C57BL6 mice (116). Additionally, glutamate–glutamine cycling was reduced in mice exhibiting depression-like phenotype (112). Moreover, a very recent measurement has revealed decreased glutamatergic (40%) and GABAergic (20%) neurometabolic activity in PFC of CUMS model of depression (66). These alterations were reflected in a large reduction in the rate of neuronal ATP synthesis. Additionally, excitatory and inhibitory synaptic transmissions were reduced by ~40% in these mice. The reduced synaptic transmission in CUMS mice was corroborated by decreased labeling of GABA-C2, Glu-C4, and Gln-C4 from [2-13C]acetate (66).
Antidepressants are categorized into different classes: selective serotonin reuptake inhibitors, serotonin–norepinephrine reuptake inhibitors, and selective norepinephrine reuptake inhibitors, which increase the level of synaptic monoamine neurotransmitters by blocking their reuptake in neurons. The antidepressants belonging to the monoamine oxidase inhibitors category increase tissue levels of monoamines by suppressing the activity of corresponding oxidases. These molecules increase synaptic plasticity, activate neurogenesis in the adult hippocampus, and enhance the expression of neurotrophic factors (170, 171). However, despite the increase in brain monoamine level with few doses of conventional antidepressants, the desired outcomes are usually achieved only after several weeks to months of continuous administration (172). Moreover, a significant fraction of subjects, commonly referred to as treatment-resistant, do not respond to these antidepressants despite the use of various therapeutic strategies (173).
Interestingly, a single subanesthetic dose of ketamine, a non-competitive NMDA channel blocker, produces rapid antidepressant actions within hours of administration, and the effects last for several days (150, 174) (Table 3). Although, the precise mechanism of ketamine action is elusive, various studies have reported that acute intervention with a low dose of ketamine increases glutamate efflux in PFC of mice and rats (112, 175, 176). These studies led to hypothesize that partial antagonism of NMDA receptor by a subanesthetic dose of ketamine may induce antidepressive effects by increasing neurotransmission and neurometabolism in PFC (175). Moreover, the antidepressive effects of ketamine could be related to the selective impact on GABAergic interneurons. Ketamine blocks the NMDA receptors of GABA interneurons, thus suppresses their ability to inhibit pyramidal neurons, thereby induces cortical excitation (11, 177).
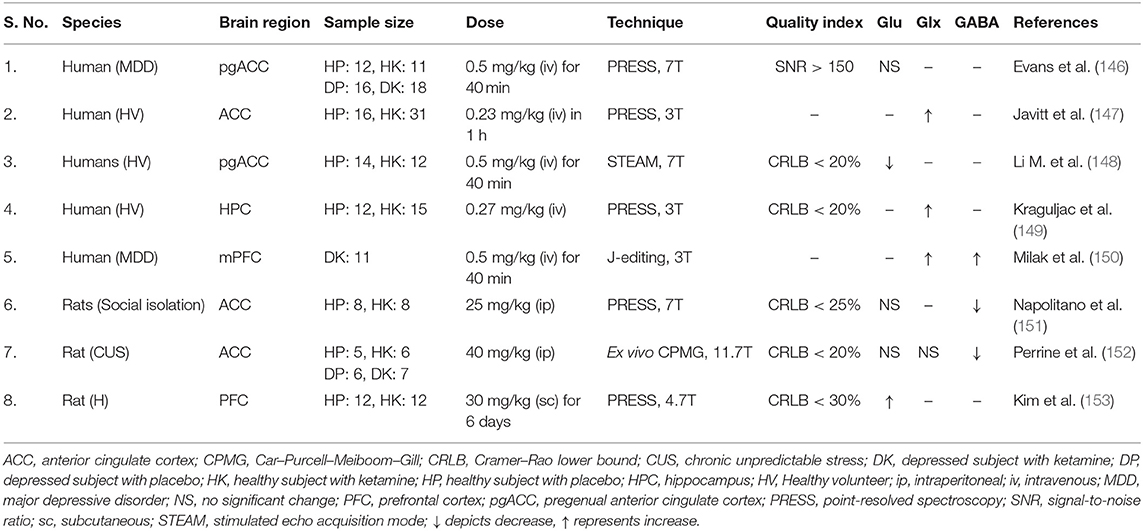
Table 3. Impact of ketamine on neurometabolites homeostasis in depression.
A very recent pilot study has evaluated the impact of intravenous ketamine administration on neurotransmitter levels in the medial prefrontal cortex (mPFC) of MDD subjects (150). GABA/water and Glx/water peaked ~38% above baseline within 30 min of ketamine infusion (150) (Table 3). However, the majority of the studies reported insignificant changes in GABA and glutamate levels following ketamine treatment (178, 179). As mentioned above, the antidepressant effects of ketamine could be related to its impact on neurotransmitter cycling, oxidative energy metabolism, and neuronal–astroglial coupling. Very recently, we have shown that the subanesthetic dose of ketamine (10 mg/kg, intraperitoneal) increases 13C labeling of glutamate, GABA and glutamine from glucose and acetate in PFC of CSDS mice. These findings indicate that ketamine normalizes the neurometabolic activity of glutamatergic and GABAergic neurons along with astrocytes in depression (112, 175). Moreover, recent studies with ketamine in MDD subjects indicated an increase in the rate of glutamate–glutamine neurotransmitter cycling without any change in oxidative energy production in neurons (180, 181).
The homeostasis of tissue glutamate and GABA plays important role in neural activity. The GABAergic neurons are known to control the dopaminergic reward circuitry in the VTA (182, 183). Alteration in the GABAergic neurotransmission with defective GABAA receptor subunits (94, 95, 184) and GAD67 (90, 93) have been reported in depressed subjects. Moreover, modulation of GABAergic activity in mice using genetic and optogenetic approaches leads to anhedonia and neophobia, which are characteristics of depressive disorder (185, 186). The reduced regulatory inhibition on principal neurons may lead to the excessive release of excitatory neurotransmitters in the synapse. The elevated glutamate level in the synaptic cleft stimulates prolonged and excessive activation of NMDA receptors (187). This increased neural activity ultimately leads to atrophy of glutamatergic neurons by excitotoxicity. A homeostatic reduction in glutamate receptors and functional impairment of glutamatergic synapses in the hippocampus and medial prefrontal cortex have been reported in γ2-subunit of GABAA receptor knockout mice, which exhibit a modest defect in GABAergic transmission (188).
1H-MRS measures combined intracellular and extracellular glutamate and GABA pool in neurons and glia. The intracellular neurotransmitter pool dominates excessively with the extracellular (2,000–5,000:1) (189). Therefore, 1H-MRS measured changes in the levels of glutamate and GABA may not reflect the abnormalities in synaptic concentration and vice versa. Hence, the findings of 1H-MRS studies should be interpreted with great caution. 1H- and 13C-NMR measurements have revealed several vital information about depression. Limited measurements based on 13C-NMR spectroscopy together with administration of 13C precursor have suggested a reduced rate of glucose oxidation, neuronal and astroglial metabolic activity, and altered neurotransmitter trafficking in the prefrontal cortex in depression. However, there are some inconsistencies in the literature, which may be attributed to differences in the disease severity, age, gender, comorbidity, the investigated brain regions, the status and duration of medications in subjects. Hence, there is a further need for comprehensive large-scale collaborative analysis about neurotransmitter homeostasis and their energetics to better understand the etiology of depression similar to that proposed by the ENIGMA consortium for genetic and neuroimaging data.
AS: literature survey, preparation of figure, manuscript writing, and editing. NS: preparation of figure, manuscript writing, and editing. AP: conception of the idea, preparation of figures, manuscript writing and editing, supervised and directed the overall project. All authors contributed to the article and approved the submitted version.
This work was supported by the Council for Scientific and Industrial Research, Government of India (Health Care Theme FBR/MLP0150). AS thanks the Department of Biotechnology for the award of Junior Research fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Mr. Kamal Saba for his help with graphical works.
1. Bromet E, Andrade LH, Hwang I, Sampson NA, Alonso J, de Girolamo G, et al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med. (2011) 9:90. doi: 10.1186/1741-7015-9-90
2. Mullins N, Bigdeli TB, Borglum AD, Coleman JRI, Demontis D, Mehta D, et al. GWAS of suicide attempt in psychiatric disorders and association with major depression polygenic risk scores. Am J Psychiatry. (2019) 176:651–60. doi: 10.1176/appi.ajp.2019.18080957
3. Qin P. The impact of psychiatric illness on suicide: differences by diagnosis of disorders and by sex and age of subjects. J Psychiatr Res. (2011) 45:1445–52. doi: 10.1016/j.jpsychires.2011.06.002
4. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. (2017) 390:1211–59. doi: 10.1016/S0140-6736(17)32154-2
5. American Psychiatric Pub. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®). Washington, DC: American Psychiatric Pub (2013).
6. Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. (1997) 48:191–214. doi: 10.1146/annurev.psych.48.1.191
7. Schlossberg K, Massler A, Zalsman G. Environmental risk factors for psychopathology. Isr J Psychiatry Relat Sci. (2010) 47:139–43.
8. Galts CPC, Bettio LEB, Jewett DC, Yang CC, Brocardo PS, Rodrigues ALS, et al. Depression in neurodegenerative diseases: common mechanisms and current treatment options. Neurosci Biobehav Rev. (2019) 102:56–84. doi: 10.1016/j.neubiorev.2019.04.002
9. Semenkovich K, Brown ME, Svrakic DM, Lustman PJ. Depression in type 2 diabetes mellitus: prevalence, impact, and treatment. Drugs. (2015) 75:577–87. doi: 10.1007/s40265-015-0347-4
10. Szelei A, Dome P. Cancer and depression: a concise review. Orv Hetil. (2020) 161:908–16. doi: 10.1556/650.2020.31759
11. Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. (2016) 22:238–49. doi: 10.1038/nm.4050
12. Elbejjani M, Fuhrer R, Abrahamowicz M, Mazoyer B, Crivello F, Tzourio C, et al. Depression, depressive symptoms, and rate of hippocampal atrophy in a longitudinal cohort of older men and women. Psychol Med. (2015) 45:1931–44. doi: 10.1017/S0033291714003055
13. Kim K, Shin JH, Myung W, Fava M, Mischoulon D, Papakostas GI, et al. Deformities of the globus pallidus are associated with severity of suicidal ideation and impulsivity in patients with major depressive disorder. Sci Rep. (2019) 9:7462. doi: 10.1038/s41598-019-43882-4
14. Kempton MJ, Salvador Z, Munafo MR, Geddes JR, Simmons A, Frangou S, et al. Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Arch Gen Psychiatry. (2011) 68:675–90. doi: 10.1001/archgenpsychiatry.2011.60
15. Schmaal L, Pozzi E, T CH, van Velzen LS, Veer IM, Opel N, et al. ENIGMA MDD: seven years of global neuroimaging studies of major depression through worldwide data sharing. Transl Psychiatry. (2020) 10:172. doi: 10.1038/s41398-020-0842-6
16. Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. (2019) 22:343–52. doi: 10.1038/s41593-018-0326-7
17. Park C, Rosenblat JD, Brietzke E, Pan Z, Lee Y, Cao B, et al. Stress, epigenetics and depression: a systematic review. Neurosci Biobehav Rev. (2019) 102:139–52. doi: 10.1016/j.neubiorev.2019.04.010
18. Juruena MF, Bocharova M, Agustini B, Young AH. Atypical depression and non-atypical depression: is HPA axis function a biomarker? A systematic review. J Affect Disord. (2018) 233:45–67. doi: 10.1016/j.jad.2017.09.052
19. Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. (2008) 31:464–8. doi: 10.1016/j.tins.2008.06.006
20. Nestler EJ. Epigenetic mechanisms of depression. JAMA Psychiatry. (2014) 71:454–6. doi: 10.1001/jamapsychiatry.2013.4291
21. Menke A, Binder EB. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin Neurosci. (2014) 16:395–404. doi: 10.31887/DCNS.2014.16.3/amenke
22. Mondal AC, Fatima M. Direct and indirect evidences of BDNF and NGF as key modulators in depression: role of antidepressants treatment. Int J Neurosci. (2019) 129:283–96. doi: 10.1080/00207454.2018.1527328
23. Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2011) 35:1774–9. doi: 10.1016/j.pnpbp.2011.05.010
24. Li N, He X, Qi X, Zhang Y, He S. The mood stabilizer lamotrigine produces antidepressant behavioral effects in rats: role of brain-derived neurotrophic factor. J Psychopharmacol. (2010) 24:1772–8. doi: 10.1177/0269881109359102
25. Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. (2013) 74:742–9. doi: 10.1016/j.biopsych.2013.04.025
26. Sourkes TL. Chapter 54: the discovery of neurotransmitters, and applications to neurology. Handb Clin Neurol. (2010) 95:869–83. doi: 10.1016/S0072-9752(08)02154-4
27. Snyder SH, Innis RB. Peptide neurotransmitters. Annu Rev Biochem. (1979) 48:755–82. doi: 10.1146/annurev.bi.48.070179.003543
28. Conio B, Martino M, Magioncalda P, Escelsior A, Inglese M, Amore M, et al. Opposite effects of dopamine and serotonin on resting-state networks: review and implications for psychiatric disorders. Mol Psychiatry. (2020) 25:82–93. doi: 10.1038/s41380-019-0406-4
29. Hamon M, Blier P. Monoamine neurocircuitry in depression and strategies for new treatments. Prog Neuropsychopharmacol Biol Psychiatry. (2013) 45:54–63. doi: 10.1016/j.pnpbp.2013.04.009
31. Bertholdo D, Watcharakorn A, Castillo M. Brain proton magnetic resonance spectroscopy: introduction and overview. Neuroimaging Clin N Am. (2013) 23:359–80. doi: 10.1016/j.nic.2012.10.002
32. Rodrigues TB, Valette J, Bouzier-Sore AK. (13)C NMR spectroscopy applications to brain energy metabolism. Front Neuroenergetics. (2013) 5:9. doi: 10.3389/fnene.2013.00009
33. Rothman DL, de Graaf RA, Hyder F, Mason GF, Behar KL, De Feyter HM. In vivo (13) C and (1) H-[(13) C] MRS studies of neuroenergetics and neurotransmitter cycling, applications to neurological and psychiatric disease and brain cancer. NMR Biomed. (2019) 32:e4172. doi: 10.1002/nbm.4172
34. Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. (2013) 14:609–25. doi: 10.1038/nrn3381
35. Hoflich A, Michenthaler P, Kasper S, Lanzenberger R. Circuit mechanisms of reward, anhedonia, and depression. Int J Neuropsychopharmacol. (2019) 22:105–18. doi: 10.1093/ijnp/pyy081
36. Nestler EJ, Carlezon WA Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. (2006) 59:1151–9. doi: 10.1016/j.biopsych.2005.09.018
37. Barth C, Villringer A, Sacher J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci. (2015) 9:37. doi: 10.3389/fnins.2015.00037
38. Schwartz TL, Sachdeva S, Stahl SM. Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Front Pharmacol. (2012) 3:195. doi: 10.3389/fphar.2012.00195
39. Fields HL, Hjelmstad GO, Margolis EB, Nicola SM. Ventral tegmental area neurons in learned appetitive behavior and positive reinforcement. Annu Rev Neurosci. (2007) 30:289–316. doi: 10.1146/annurev.neuro.30.051606.094341
40. Schur RR, Draisma LW, Wijnen JP, Boks MP, Koevoets MG, Joels M, et al. Brain GABA levels across psychiatric disorders: a systematic literature review and meta-analysis of (1) H-MRS studies. Hum Brain Mapp. (2016) 37:3337–52. doi: 10.1002/hbm.23244
41. Wenneberg C, Glenthoj BY, Hjorthoj C, Buchardt Zingenberg FJ, Glenthoj LB, Rostrup E, et al. Cerebral glutamate and GABA levels in high-risk of psychosis states: a focused review and meta-analysis of (1)H-MRS studies. Schizophr Res. (2020) 215:38–48. doi: 10.1016/j.schres.2019.10.050
42. Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. (2000) 13:129–53. doi: 10.1002/1099-1492(200005)13:3<129::AID-NBM619>3.0.CO
43. Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. (2006) 100:433–42. doi: 10.1254/jphs.CPJ06007X
44. Barnes JR, Mukherjee B, Rogers BC, Nafar F, Gosse M, Parsons MP. The relationship between glutamate dynamics and activity-dependent synaptic plasticity. J Neurosci. (2020) 40:2793–807. doi: 10.1523/JNEUROSCI.1655-19.2020
45. Balschun D, Wetzel W. Inhibition of mGluR5 blocks hippocampal LTP in vivo and spatial learning in rats. Pharmacol Biochem Behav. (2002) 73:375–80. doi: 10.1016/S0091-3057(02)00847-X
46. Peng S, Zhang Y, Zhang J, Wang H, Ren B. Glutamate receptors and signal transduction in learning and memory. Mol Biol Rep. (2011) 38:453–60. doi: 10.1007/s11033-010-0128-9
47. Fogaca MV, Duman RS. Cortical GABAergic dysfunction in stress and depression: new insights for therapeutic interventions. Front Cell Neurosci. (2019) 13:87. doi: 10.3389/fncel.2019.00087
48. Heaney CF, Kinney JW. Role of GABA(B) receptors in learning and memory and neurological disorders. Neurosci Biobehav Rev. (2016) 63:1–28. doi: 10.1016/j.neubiorev.2016.01.007
49. Kolasinski J, Hinson EL, Divanbeighi Zand AP, Rizov A, Emir UE, Stagg CJ. The dynamics of cortical GABA in human motor learning. J Physiol. (2019) 597:271–82. doi: 10.1113/JP276626
50. Sampaio-Baptista C, Filippini N, Stagg CJ, Near J, Scholz J, Johansen-Berg H. Changes in functional connectivity and GABA levels with long-term motor learning. Neuroimage. (2015) 106:15–20. doi: 10.1016/j.neuroimage.2014.11.032
51. Ende G, Cackowski S, Van Eijk J, Sack M, Demirakca T, Kleindienst N, et al. Impulsivity and aggression in female BPD and ADHD patients: association with ACC glutamate and GABA concentrations. Neuropsychopharmacology. (2016) 41:410–8. doi: 10.1038/npp.2015.153
52. Jager A, Amiri H, Bielczyk N, van Heukelum S, Heerschap A, Aschrafi A, et al. Cortical control of aggression: GABA signalling in the anterior cingulate cortex. Eur Neuropsychopharmacol. (2020) 30:5–16. doi: 10.1016/j.euroneuro.2017.12.007
53. Baslow MH. N-acetylaspartate in the vertebrate brain: metabolism and function. Neurochem Res. (2003) 28:941–53. doi: 10.1023/A:1023250721185
54. Tkac I, Oz G, Adriany G, Ugurbil K, Gruetter R. In vivo 1H NMR spectroscopy of the human brain at high magnetic fields: metabolite quantification at 4T vs. 7T. Magn Reson Med. (2009) 62:868–79. doi: 10.1002/mrm.22086
55. Pfeuffer J, Tkac I, Provencher SW, Gruetter R. Toward an in vivo neurochemical profile: quantification of 18 metabolites in short-echo-time (1)H NMR spectra of the rat brain. J Magn Reson. (1999) 141:104–20. doi: 10.1006/jmre.1999.1895
56. Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. (2001) 21:1133–45. doi: 10.1097/00004647-200110000-00001
57. Howarth C, Gleeson P, Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab. (2012) 32:1222–32. doi: 10.1038/jcbfm.2012.35
58. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. (2006) 98:641–53. doi: 10.1111/j.1471-4159.2006.03913.x
59. Patel AB, de Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc Natl Acad Sci USA. (2005) 102:5588–93. doi: 10.1073/pnas.0501703102
60. Tiwari V, Ambadipudi S, Patel AB. Glutamatergic and GABAergic TCA cycle and neurotransmitter cycling fluxes in different regions of mouse brain. J Cereb Blood Flow Metab. (2013) 33:1523–31. doi: 10.1038/jcbfm.2013.114
61. Gruetter R, Adriany G, Choi IY, Henry PG, Lei H, Oz G. Localized in vivo 13C NMR spectroscopy of the brain. NMR Biomed. (2003) 16:313–38. doi: 10.1002/nbm.841
62. Harmer CJ, Duman RS, Cowen PJ. How do antidepressants work? New perspectives for refining future treatment approaches. Lancet Psychiatry. (2017) 4:409–18. doi: 10.1016/S2215-0366(17)30015-9
63. Perez-Caballero L, Torres-Sanchez S, Romero-Lopez-Alberca C, Gonzalez-Saiz F, Mico JA, Berrocoso E. Monoaminergic system and depression. Cell Tissue Res. (2019) 377:107–13. doi: 10.1007/s00441-018-2978-8
64. Abdallah CG, Jiang L, De Feyter HM, Fasula M, Krystal JH, Rothman DL, et al. Glutamate metabolism in major depressive disorder. Am J Psychiatry. (2014) 171:1320–7. doi: 10.1176/appi.ajp.2014.14010067
65. Duman RS, Sanacora G, Krystal JH. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. (2019) 102:75–90. doi: 10.1016/j.neuron.2019.03.013
66. Mishra PK, Adusumilli M, Deolal P, Mason GF, Kumar A, Patel AB. Impaired neuronal and astroglial metabolic activity in chronic unpredictable mild stress model of depression: reversal of behavioral and metabolic deficit with lanicemine. Neurochem Int. (2020) 137:104750. doi: 10.1016/j.neuint.2020.104750
67. Douglas RJ, Martin KA. Mapping the matrix: the ways of neocortex. Neuron. (2007) 56:226–38. doi: 10.1016/j.neuron.2007.10.017
68. Ohgi Y, Futamura T, Hashimoto K. Glutamate signaling in synaptogenesis and NMDA receptors as potential therapeutic targets for psychiatric disorders. Curr Mol Med. (2015) 15:206–21. doi: 10.2174/1566524015666150330143008
69. Kew JN, Kemp JA. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology. (2005) 179:4–29. doi: 10.1007/s00213-005-2200-z
70. Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. (2010) 11:682–96. doi: 10.1038/nrn2911
71. Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry. (2007) 64:193–200. doi: 10.1001/archpsyc.64.2.193
72. Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. (2004) 61:705–13. doi: 10.1001/archpsyc.61.7.705
73. Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. (2009) 33:70–5. doi: 10.1016/j.pnpbp.2008.10.005
74. Sanacora G, Fenton LR, Fasula MK, Rothman DL, Levin Y, Krystal JH, et al. Cortical gamma-aminobutyric acid concentrations in depressed patients receiving cognitive behavioral therapy. Biol Psychiatry. (2006) 59:284–6. doi: 10.1016/j.biopsych.2005.07.015
75. Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, et al. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. (1999) 56:1043–7. doi: 10.1001/archpsyc.56.11.1043
76. Shatillo A, Salo RA, Giniatullin R, Grohn OH. Involvement of NMDA receptor subtypes in cortical spreading depression in rats assessed by fMRI. Neuropharmacology. (2015) 93:164–70. doi: 10.1016/j.neuropharm.2015.01.028
77. Treccani G, Gaarn du Jardin K, Wegener G, Muller HK. Differential expression of postsynaptic NMDA and AMPA receptor subunits in the hippocampus and prefrontal cortex of the flinders sensitive line rat model of depression. Synapse. (2016) 70:471–4. doi: 10.1002/syn.21918
78. Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. (2012) 73:962–77. doi: 10.1016/j.neuron.2011.12.033
79. Gray AL, Hyde TM, Deep-Soboslay A, Kleinman JE, Sodhi MS. Sex differences in glutamate receptor gene expression in major depression and suicide. Mol Psychiatry. (2015) 20:1057–68. doi: 10.1038/mp.2015.91
80. Esterlis I, DellaGioia N, Pietrzak RH, Matuskey D, Nabulsi N, Abdallah CG, et al. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: an [(11)C]ABP688 and PET imaging study in depression. Mol Psychiatry. (2018) 23:824–32. doi: 10.1038/mp.2017.58
81. Shin S, Kwon O, Kang JI, Kwon S, Oh S, Choi J, et al. mGluR5 in the nucleus accumbens is critical for promoting resilience to chronic stress. Nat Neurosci. (2015) 18:1017–24. doi: 10.1038/nn.4028
82. Rajkowska G, Miguel-Hidalgo JJ, Dubey P, Stockmeier CA, Krishnan KR. Prominent reduction in pyramidal neurons density in the orbitofrontal cortex of elderly depressed patients. Biol Psychiatry. (2005) 58:297–306. doi: 10.1016/j.biopsych.2005.04.013
83. Patel AB, Rothman DL, Cline GW, Behar KL. Glutamine is the major precursor for GABA synthesis in rat neocortex in vivo following acute GABA-transaminase inhibition. Brain Res. (2001) 919:207–20. doi: 10.1016/S0006-8993(01)03015-3
84. Mohler H. Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors. J Neurochem. (2007) 102:1–12. doi: 10.1111/j.1471-4159.2007.04454.x
85. Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. (2004) 27:569–75. doi: 10.1016/j.tins.2004.07.002
87. Mederos S, Perea G. GABAergic-astrocyte signaling: a refinement of inhibitory brain networks. Glia. (2019) 67:1842–51. doi: 10.1002/glia.23644
88. Fee C, Banasr M, Sibille E. Somatostatin-positive gamma-aminobutyric acid interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol Psychiatry. (2017) 82:549–59. doi: 10.1016/j.biopsych.2017.05.024
89. Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. (2007) 32:471–82. doi: 10.1038/sj.npp.1301234
90. Banasr M, Lepack A, Fee C, Duric V, Maldonado-Aviles J, DiLeone R, et al. Characterization of GABAergic marker expression in the chronic unpredictable stress model of depression. Chronic Stress. (2017) 1:2470547017720459. doi: 10.1177/2470547017720459
91. Sibille E, Morris HM, Kota RS, Lewis DA. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol. (2011) 14:721–34. doi: 10.1017/S1461145710001616
92. Tripp A, Kota RS, Lewis DA, Sibille E. Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis. (2011) 42:116–24. doi: 10.1016/j.nbd.2011.01.014
93. Guilloux JP, Douillard-Guilloux G, Kota R, Wang X, Gardier AM, Martinowich K, et al. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. (2012) 17:1130–42. doi: 10.1038/mp.2011.113
94. Klempan TA, Sequeira A, Canetti L, Lalovic A, Ernst C, ffrench-Mullen J, et al. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol Psychiatry. (2009) 14:175–89. doi: 10.1038/sj.mp.4002110
95. Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci USA. (2005) 102:15653–8. doi: 10.1073/pnas.0507901102
96. Shen Q, Lal R, Luellen BA, Earnheart JC, Andrews AM, Luscher B. gamma-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol Psychiatry. (2010) 68:512–20. doi: 10.1016/j.biopsych.2010.04.024
97. Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry. (2002) 159:663–5. doi: 10.1176/appi.ajp.159.4.663
98. Sanacora G, Mason GF, Rothman DL, Hyder F, Ciarcia JJ, Ostroff RB, et al. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry. (2003) 160:577–9. doi: 10.1176/appi.ajp.160.3.577
99. Mohler H. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology. (2012) 62:42–53. doi: 10.1016/j.neuropharm.2011.08.040
100. Ordidge RJ, Connelly A, Lohman JA. Image-selected in vivo spectroscopy (ISIS). A new technique for spatially selective NMR spectroscopy. J Magnet Reson. (1986) 66:283–94. doi: 10.1016/0022-2364(86)90031-4
101. Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann N Y Acad Sci. (1987) 508:333–48. doi: 10.1111/j.1749-6632.1987.tb32915.x
102. Frahm J, Bruhn H, Gyngell ML, Merboldt KD, Hanicke W, Sauter R. Localized high-resolution proton NMR spectroscopy using stimulated echoes: initial applications to human brain in vivo. Magn Reson Med. (1989) 9:79–93. doi: 10.1002/mrm.1910090110
103. Duyn JH, Gillen J, Sobering G, van Zijl PC, Moonen CT. Multisection proton MR spectroscopic imaging of the brain. Radiology. (1993) 188:277–82. doi: 10.1148/radiology.188.1.8511313
104. Haase A, Frahm J, Hanicke W, Matthaei D. 1H NMR chemical shift selective (CHESS) imaging. Phys Med Biol. (1985) 30:341–4. doi: 10.1088/0031-9155/30/4/008
105. Tkac I, Starcuk Z, Choi IY, Gruetter R. In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Magn Reson Med. (1999) 41:649–56. doi: 10.1002/(SICI)1522-2594(199904)41:4<649::AID-MRM2>3.0.CO;2-G
106. Benson KL, Bottary R, Schoerning L, Baer L, Gonenc A, Eric Jensen J, et al. (1)H MRS measurement of cortical GABA and glutamate in primary insomnia and major depressive disorder: relationship to sleep quality and depression severity. J Affect Disord. (2020) 274:624–31. doi: 10.1016/j.jad.2020.05.026
107. Draganov M, Vives-Gilabert Y, de Diego-Adelino J, Vicent-Gil M, Puigdemont D, Portella MJ. Glutamatergic and GABA-ergic abnormalities in First-episode depression. A 1-year follow-up 1H-MR spectroscopic study. J Affect Disord. (2020) 266:572–7. doi: 10.1016/j.jad.2020.01.138
108. Kahl KG, Atalay S, Maudsley AA, Sheriff S, Cummings A, Frieling H, et al. Altered neurometabolism in major depressive disorder: a whole brain (1)H-magnetic resonance spectroscopic imaging study at 3T. Prog Neuropsychopharmacol Biol Psychiatry. (2020) 101:109916. doi: 10.1016/j.pnpbp.2020.109916
109. Seewoo BJ, Hennessy LA, Feindel KW, Etherington SJ, Croarkin PE, Rodger J. Validation of chronic restraint stress model in young adult rats for the study of depression using longitudinal multimodal MR imaging. eNeuro. (2020) 7:1–22. doi: 10.1523/ENEURO.0113-20.2020
110. Cherix A, Larrieu T, Grosse J, Rodrigues J, McEwen B, Nasca C, et al. Metabolic signature in nucleus accumbens for anti-depressant-like effects of acetyl-L-carnitine. Elife. (2020) 9:e50631. doi: 10.7554/eLife.50631
111. Soeiro-de-Souza MG, Otaduy MCG, Machado-Vieira R, Moreno RA, Nery FG, Leite C, et al. Anterior cingulate cortex glutamatergic metabolites and mood stabilizers in euthymic bipolar i disorder patients: a proton magnetic resonance spectroscopy study. Biol Psychiatry Cogn Neurosci Neuroimaging. (2018) 3:985–91. doi: 10.1016/j.bpsc.2018.02.007
112. Mishra PK, Kumar A, Behar KL, Patel AB. Subanesthetic ketamine reverses neuronal and astroglial metabolic activity deficits in a social defeat model of depression. J Neurochem. (2018) 146:722–34. doi: 10.1111/jnc.14544
113. Jollant F, Near J, Turecki G, Richard-Devantoy S. Spectroscopy markers of suicidal risk and mental pain in depressed patients. Prog Neuropsychopharmacol Biol Psychiatry. (2016) 73:64–71. doi: 10.1016/j.pnpbp.2016.10.005
114. Shirayama Y, Takahashi M, Osone F, Hara A, Okubo T. Myo-inositol, glutamate, and glutamine in the prefrontal cortex, hippocampus, and amygdala in major depression. Biol Psychiatry Cogn Neurosci Neuroimaging. (2017) 2:196–204. doi: 10.1016/j.bpsc.2016.11.006
115. Hermens DF, Chitty KM, Lee RS, Tickell A, Haber PS, Naismith SL, et al. Hippocampal glutamate is increased and associated with risky drinking in young adults with major depression. J Affect Disord. (2015) 186:95–8. doi: 10.1016/j.jad.2015.07.009
116. Veeraiah P, Noronha JM, Maitra S, Bagga P, Khandelwal N, Chakravarty S, et al. Dysfunctional glutamatergic and gamma-aminobutyric acidergic activities in prefrontal cortex of mice in social defeat model of depression. Biol Psychiatry. (2014) 76:231–8. doi: 10.1016/j.biopsych.2013.09.024
117. McEwen AM, Burgess DT, Hanstock CC, Seres P, Khalili P, Newman SC, et al. Increased glutamate levels in the medial prefrontal cortex in patients with postpartum depression. Neuropsychopharmacology. (2012) 37:2428–35. doi: 10.1038/npp.2012.101
118. Hemanth Kumar BS, Mishra SK, Rana P, Singh S, Khushu S. Neurodegenerative evidences during early onset of depression in CMS rats as detected by proton magnetic resonance spectroscopy at 7 T. Behav Brain Res. (2012) 232:53–9. doi: 10.1016/j.bbr.2012.03.011
119. Portella MJ, de Diego-Adelino J, Gomez-Anson B, Morgan-Ferrando R, Vives Y, Puigdemont D, et al. Ventromedial prefrontal spectroscopic abnormalities over the course of depression: a comparison among first episode, remitted recurrent and chronic patients. J Psychiatr Res. (2011) 45:427–34. doi: 10.1016/j.jpsychires.2010.08.010
120. Jarnum H, Eskildsen SF, Steffensen EG, Lundbye-Christensen S, Simonsen CW, Thomsen IS, et al. Longitudinal MRI study of cortical thickness, perfusion, and metabolite levels in major depressive disorder. Acta Psychiatr Scand. (2011) 124:435–46. doi: 10.1111/j.1600-0447.2011.01766.x
121. Block W, Traber F, von Widdern O, Metten M, Schild H, Maier W, et al. Proton MR spectroscopy of the hippocampus at 3 T in patients with unipolar major depressive disorder: correlates and predictors of treatment response. Int J Neuropsychopharmacol. (2009) 12:415–22. doi: 10.1017/S1461145708009516
122. Li CX, Wang Y, Gao H, Pan WJ, Xiang Y, Huang M, et al. Cerebral metabolic changes in a depression-like rat model of chronic forced swimming studied by ex vivo high resolution 1H magnetic resonance spectroscopy. Neurochem Res. (2008) 33:2342–9. doi: 10.1007/s11064-008-9739-0
123. Ajilore O, Haroon E, Kumaran S, Darwin C, Binesh N, Mintz J, et al. Measurement of brain metabolites in patients with type 2 diabetes and major depression using proton magnetic resonance spectroscopy. Neuropsychopharmacology. (2007) 32:1224–31. doi: 10.1038/sj.npp.1301248
124. Auer DP, Putz B, Kraft E, Lipinski B, Schill J, Holsboer F. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biol Psychiatry. (2000) 47:305–13. doi: 10.1016/S0006-3223(99)00159-6
125. Yoo CH, Song KH, Lim SI, Woo DC, Choe BY. Metabolic effects of light deprivation in the prefrontal cortex of rats with depression-like behavior: in vivo proton magnetic resonance spectroscopy at 7T. Brain Res. (2018) 1687:95–103. doi: 10.1016/j.brainres.2018.02.047
126. Lewis CP, Port JD, Blacker CJ, Sonmez AI, Seewoo BJ, Leffler JM, et al. Altered anterior cingulate glutamatergic metabolism in depressed adolescents with current suicidal ideation. Transl Psychiatry. (2020) 10:119. doi: 10.1038/s41398-020-0792-z
127. Luykx JJ, Laban KG, van den Heuvel MP, Boks MP, Mandl RC, Kahn RS, et al. Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of (1)H-MRS findings. Neurosci Biobehav Rev. (2012) 36:198–205. doi: 10.1016/j.neubiorev.2011.05.014
128. Moriguchi S, Takamiya A, Noda Y, Horita N, Wada M, Tsugawa S, et al. Glutamatergic neurometabolite levels in major depressive disorder: a systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol Psychiatry. (2019) 24:952–64. doi: 10.1038/s41380-018-0252-9
129. Rajkowska G, Miguel-Hidalgo JJ. Gliogenesis and glial pathology in depression. CNS Neurol Disord Drug Targets. (2007) 6:219–33. doi: 10.2174/187152707780619326
130. Zhu X, Ye G, Wang Z, Luo J, Hao X. Sub-anesthetic doses of ketamine exert antidepressant-like effects and upregulate the expression of glutamate transporters in the hippocampus of rats. Neurosci Lett. (2017) 639:132–7. doi: 10.1016/j.neulet.2016.12.070
131. Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. (2011) 16:383–406. doi: 10.1038/mp.2010.120
132. Gonzalez-Burgos G, Fish KN, Lewis DA. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast. (2011) 2011:723184. doi: 10.1155/2011/723184
133. Brambilla P, Perez J, Barale F, Schettini G, Soares JC. GABAergic dysfunction in mood disorders. Mol Psychiatry. (2003) 8:721–37, 15. doi: 10.1038/sj.mp.4001362
134. Horder J, Petrinovic MM, Mendez MA, Bruns A, Takumi T, Spooren W, et al. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl Psychiatry. (2018) 8:106. doi: 10.1038/s41398-018-0155-1
135. Petty F, Kramer GL, Gullion CM, Rush AJ. Low plasma gamma-aminobutyric acid levels in male patients with depression. Biol Psychiatry. (1992) 32:354–63. doi: 10.1016/0006-3223(92)90039-3
136. Gold BI, Bowers MB Jr, Roth RH, Sweeney DW. GABA levels in CSF of patients with psychiatric disorders. Am J Psychiatry. (1980) 137:362–4. doi: 10.1176/ajp.137.3.362
137. Bradley KA, Alonso CM, Mehra LM, Xu J, Gabbay V. Elevated striatal gamma-aminobutyric acid in youth with major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 86:203–10. doi: 10.1016/j.pnpbp.2018.06.004
138. Sekar S, Grandjean J, Garnell JF, Willems R, Duytschaever H, Seramani S, et al. Neuro-metabolite profiles of rodent models of psychiatric dysfunctions characterised by MR spectroscopy. Neuropharmacology. (2019) 146:109–16. doi: 10.1016/j.neuropharm.2018.11.021
139. Gabbay V, Bradley KA, Mao X, Ostrover R, Kang G, Shungu DC. Anterior cingulate cortex gamma-aminobutyric acid deficits in youth with depression. Transl Psychiatry. (2017) 7:e1216. doi: 10.1038/tp.2017.187
140. Abdallah CG, Averill LA, Krystal JH. Ketamine as a promising prototype for a new generation of rapid-acting antidepressants. Ann N Y Acad Sci. (2015) 1344:66–77. doi: 10.1111/nyas.12718
141. Gabbay V, Mao X, Klein RG, Ely BA, Babb JS, Panzer AM, et al. Anterior cingulate cortex gamma-aminobutyric acid in depressed adolescents: relationship to anhedonia. Arch Gen Psychiatry. (2012) 69:139–49. doi: 10.1001/archgenpsychiatry.2011.131
142. Price RB, Shungu DC, Mao X, Nestadt P, Kelly C, Collins KA, et al. Amino acid neurotransmitters assessed by proton magnetic resonance spectroscopy: relationship to treatment resistance in major depressive disorder. Biol Psychiatry. (2009) 65:792–800. doi: 10.1016/j.biopsych.2008.10.025
143. Walter M, Henning A, Grimm S, Schulte RF, Beck J, Dydak U, et al. The relationship between aberrant neuronal activation in the pregenual anterior cingulate, altered glutamatergic metabolism, and anhedonia in major depression. Arch Gen Psychiatry. (2009) 66:478–86. doi: 10.1001/archgenpsychiatry.2009.39
144. Bhagwagar Z, Wylezinska M, Jezzard P, Evans J, Boorman E, P MM, et al. Low GABA concentrations in occipital cortex and anterior cingulate cortex in medication-free, recovered depressed patients. Int J Neuropsychopharmacol. (2008) 11:255–60. doi: 10.1017/S1461145707007924
145. Epperson CN, Gueorguieva R, Czarkowski KA, Stiklus S, Sellers E, Krystal JH, et al. Preliminary evidence of reduced occipital GABA concentrations in puerperal women: a 1H-MRS study. Psychopharmacology. (2006) 186:425–33. doi: 10.1007/s00213-006-0313-7
146. Evans JW, Lally N, An L, Li N, Nugent AC, Banerjee D, et al. 7T (1)H-MRS in major depressive disorder: a Ketamine Treatment Study. Neuropsychopharmacology. (2018) 43:1908–14. doi: 10.1038/s41386-018-0057-1
147. Javitt DC, Carter CS, Krystal JH, Kantrowitz JT, Girgis RR, Kegeles LS, et al. Utility of imaging-based biomarkers for glutamate-targeted drug development in psychotic disorders: a randomized clinical trial. JAMA Psychiatry. (2018) 75:11–9. doi: 10.1001/jamapsychiatry.2017.3572
148. Li M, Demenescu LR, Colic L, Metzger CD, Heinze HJ, Steiner J, et al. Temporal dynamics of antidepressant ketamine effects on glutamine cycling follow regional fingerprints of AMPA and NMDA receptor densities. Neuropsychopharmacology. (2017) 42:1201–9. doi: 10.1038/npp.2016.184
149. Kraguljac NV, Frolich MA, Tran S, White DM, Nichols N, Barton-McArdle A, et al. Ketamine modulates hippocampal neurochemistry and functional connectivity: a combined magnetic resonance spectroscopy and resting-state fMRI study in healthy volunteers. Mol Psychiatry. (2017) 22:562–9. doi: 10.1038/mp.2016.122
150. Milak MS, Proper CJ, Mulhern ST, Parter AL, Kegeles LS, Ogden RT, et al. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol Psychiatry. (2016) 21:320–7. doi: 10.1038/mp.2015.83
151. Napolitano A, Shah K, Schubert MI, Porkess V, Fone KC, Auer DP. In vivo neurometabolic profiling to characterize the effects of social isolation and ketamine-induced NMDA antagonism: a rodent study at 7.0 T. Schizophr Bull. (2014) 40:566–74. doi: 10.1093/schbul/sbt067
152. Perrine SA, Ghoddoussi F, Michaels MS, Sheikh IS, McKelvey G, Galloway MP. Ketamine reverses stress-induced depression-like behavior and increased GABA levels in the anterior cingulate: an 11.7 T 1H-MRS study in rats. Prog Neuropsychopharmacol Biol Psychiatry. (2014) 51:9–15. doi: 10.1016/j.pnpbp.2013.11.003
153. Kim SY, Lee H, Kim HJ, Bang E, Lee SH, Lee DW, et al. In vivo and ex vivo evidence for ketamine-induced hyperglutamatergic activity in the cerebral cortex of the rat: potential relevance to schizophrenia. NMR Biomed. (2011) 24:1235–42. doi: 10.1002/nbm.1681
154. Kalueff AV, Nutt DJ. Role of GABA in anxiety and depression. Depress Anxiety. (2007) 24:495–517. doi: 10.1002/da.20262
155. Abdallah CG, Jackowski A, Sato JR, Mao X, Kang G, Cheema R, et al. Prefrontal cortical GABA abnormalities are associated with reduced hippocampal volume in major depressive disorder. Eur Neuropsychopharmacol. (2015) 25:1082–90. doi: 10.1016/j.euroneuro.2015.04.025
156. Hasler G, Northoff G. Discovering imaging endophenotypes for major depression. Mol Psychiatry. (2011) 16:604–19. doi: 10.1038/mp.2011.23
157. Levinson AJ, Fitzgerald PB, Favalli G, Blumberger DM, Daigle M, Daskalakis ZJ. Evidence of cortical inhibitory deficits in major depressive disorder. Biol Psychiatry. (2010) 67:458–64. doi: 10.1016/j.biopsych.2009.09.025
158. Paslakis G, Traber F, Roberz J, Block W, Jessen F. N-acetyl-aspartate (NAA) as a correlate of pharmacological treatment in psychiatric disorders: a systematic review. Eur Neuropsychopharmacol. (2014) 24:1659–75. doi: 10.1016/j.euroneuro.2014.06.004
159. de Diego-Adelino J, Portella MJ, Gomez-Anson B, Lopez-Moruelo O, Serra-Blasco M, Vives Y, et al. Hippocampal abnormalities of glutamate/glutamine, N-acetylaspartate and choline in patients with depression are related to past illness burden. J Psychiatry Neurosci. (2013) 38:107–16. doi: 10.1503/jpn.110185
160. Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. (2015) 86:883–901. doi: 10.1016/j.neuron.2015.03.035
161. Jones T, Rabiner EA, Company PETRA. The development, past achievements, and future directions of brain PET. J Cereb Blood Flow Metab. (2012) 32:1426–54. doi: 10.1038/jcbfm.2012.20
162. Rothman DL, De Feyter HM, de Graaf RA, Mason GF, Behar KL. 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed. (2011) 24:943–57. doi: 10.1002/nbm.1772
163. Patel AB, de Graaf RA, Rothman DL, Behar KL, Mason GF. Evaluation of cerebral acetate transport and metabolic rates in the rat brain in vivo using 1H-[13C]-NMR. J Cereb Blood Flow Metab. (2010) 30:1200–13. doi: 10.1038/jcbfm.2010.2
164. Mason GF, Gruetter R, Rothman DL, Behar KL, Shulman RG, Novotny EJ. Simultaneous determination of the rates of the TCA cycle, glucose utilization, alpha-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J Cereb Blood Flow Metab. (1995) 15:12–25. doi: 10.1038/jcbfm.1995.2
165. Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci USA. (1998) 95:316–21. doi: 10.1073/pnas.95.1.316
166. Rothman DL, Behar KL, Hyder F, Shulman RG. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: implications for brain function. Annu Rev Physiol. (2003) 65:401–27. doi: 10.1146/annurev.physiol.65.092101.142131
167. Patel AB, de Graaf RA, Mason GF, Kanamatsu T, Rothman DL, Shulman RG, et al. Glutamatergic neurotransmission and neuronal glucose oxidation are coupled during intense neuronal activation. J Cereb Blood Flow Metab. (2004) 24:972–85. doi: 10.1097/01.WCB.0000126234.16188.71
168. Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG. Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J Cereb Blood Flow Metab. (2006) 26:865–77. doi: 10.1038/sj.jcbfm.9600263
169. Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. (1999) 283:496–7. doi: 10.1126/science.283.5401.496
170. Castren E, Hen R. Neuronal plasticity and antidepressant actions. Trends Neurosci. (2013) 36:259–67. doi: 10.1016/j.tins.2012.12.010
171. Krishnan V, Nestler EJ. Linking molecules to mood: new insight into the biology of depression. Am J Psychiatry. (2010) 167:1305–20. doi: 10.1176/appi.ajp.2009.10030434
172. Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. (2009) 60:1439–45. doi: 10.1176/ps.2009.60.11.1439
173. Mrazek DA, Hornberger JC, Altar CA, Degtiar I. A review of the clinical, economic, and societal burden of treatment-resistant depression: 1996-2013. Psychiatr Serv. (2014) 65:977–87. doi: 10.1176/appi.ps.201300059
174. Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. (2000) 47:351–4. doi: 10.1016/S0006-3223(99)00230-9
175. Chowdhury GM, Behar KL, Cho W, Thomas MA, Rothman DL, Sanacora G. (1)H-[(1)(3)C]-nuclear magnetic resonance spectroscopy measures of ketamine's effect on amino acid neurotransmitter metabolism. Biol Psychiatry. (2012) 71:1022–5. doi: 10.1016/j.biopsych.2011.11.006
176. Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. (1997) 17:2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997
177. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. (2007) 27:11496–500. doi: 10.1523/JNEUROSCI.2213-07.2007
178. Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. (2005) 162:394–6. doi: 10.1176/appi.ajp.162.2.394
179. Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, et al. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry Res. (2011) 191:122–7. doi: 10.1016/j.pscychresns.2010.10.009
180. Abdallah CG, De Feyter HM, Averill LA, Jiang L, Averill CL, Chowdhury GMI, et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology. (2018) 43:2154–60. doi: 10.1038/s41386-018-0136-3
181. Pothula S, Kato T, Liu RJ, Wu M, Gerhard D, Shinohara R, et al. Cell-type specific modulation of NMDA receptors triggers antidepressant actions. Mol Psychiatry. (2020). doi: 10.1038/s41380-020-0796-3
182. Brown MT, Tan KR, O'Connor EC, Nikonenko I, Muller D, Luscher C. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature. (2012) 492:452–6. doi: 10.1038/nature11657
183. Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouebe G, et al. GABA neurons of the VTA drive conditioned place aversion. Neuron. (2012) 73:1173–83. doi: 10.1016/j.neuron.2012.02.015
184. Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS ONE. (2009) 4:e6585. doi: 10.1371/journal.pone.0006585
185. Kolata SM, Nakao K, Jeevakumar V, Farmer-Alroth EL, Fujita Y, Bartley AF, et al. Neuropsychiatric phenotypes produced by GABA reduction in mouse cortex and hippocampus. Neuropsychopharmacology. (2018) 43:1445–56. doi: 10.1038/npp.2017.296
186. Luscher B, Fuchs T. GABAergic control of depression-related brain states. Adv Pharmacol. (2015) 73:97–144. doi: 10.1016/bs.apha.2014.11.003
187. Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. (1995) 15:961–73. doi: 10.1016/0896-6273(95)90186-8
188. Ren Z, Pribiag H, Jefferson SJ, Shorey M, Fuchs T, Stellwagen D, et al. Bidirectional homeostatic regulation of a depression-related brain state by gamma-aminobutyric acidergic deficits and ketamine treatment. Biol Psychiatry. (2016) 80:457–68. doi: 10.1016/j.biopsych.2016.02.009
Keywords: antidepressant, brain, 13C-NMR spectroscopy, glutamine, ketamine, neurocircuitry, neurometabolism, neurotransmitter
Citation: Sarawagi A, Soni ND and Patel AB (2021) Glutamate and GABA Homeostasis and Neurometabolism in Major Depressive Disorder. Front. Psychiatry 12:637863. doi: 10.3389/fpsyt.2021.637863
Received: 04 December 2020; Accepted: 09 March 2021;
Published: 27 April 2021.
Edited by:
Richard Edden, Johns Hopkins University, United StatesReviewed by:
Richard E. Harris, University of Michigan, United StatesCopyright © 2021 Sarawagi, Soni and Patel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anant Bahadur Patel, YWJwYXRlbEBjY21iLnJlcy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.